
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle electron reduction of NHC–CO2–borane compounds†
Agustín
Morales
ab,
Caroline
Gonçalves
a,
Alix
Sournia-Saquet
a,
Laure
Vendier
a,
Agustí
Lledós
 *b,
Olivier
Baslé
*a and
Sébastien
Bontemps
*a
*b,
Olivier
Baslé
*a and
Sébastien
Bontemps
*a
aLCC-CNRS, Université de Toulouse, CNRS, 205 route de Narbonne, 31077 Toulouse Cedex 04, France. E-mail: olivier.basle@lcc-toulouse.fr; sebastien.bontemps@lcc-toulouse.fr
bDepartament de Química, Universitat Autonoma de Barcelona, 08193 Cerdanyola del Valles, Catalonia, Spain. E-mail: agusti.ledos@uab.cat
First published on 20th January 2024
Abstract
The carbon dioxide radical anion [CO2˙−] is a highly reactive species of fundamental and synthetic interest. However, the direct one-electron reduction of CO2 to generate [CO2˙−] occurs at very negative reduction potentials, which is often a limiting factor for applications. Here, we show that NHC–CO2–BR3 species – generated from the Frustrated Lewis Pair (FLP)-type activation of CO2 by N-heterocyclic carbenes (NHCs) and boranes (BR3) – undergo single electron reduction at a less negative potential than free CO2. A net gain of more than one volt was notably measured with a CAAC–CO2–B(C6F5)3 adduct, which was chemically reduced to afford [CAAC–CO2–B(C6F5)3˙−]. This room temperature stable radical anion was characterized by EPR spectroscopy and by single-crystal X-ray diffraction analysis. Of particular interest, DFT calculations showed that, thanks to the electron withdrawing properties of the Lewis acid, significant unpaired spin density is localised on the carbon atom of the CO2 moiety. Finally, these species were shown to exhibit analogous reactivity to the carbon dioxide radical anion [CO2˙−] toward DMPO. This work demonstrates the advantage provided by FLP systems in the generation and stabilization of [CO2˙−]-like species.
Introduction
The carbon dioxide radical anion [CO2˙−] is a very unstable molecule that was characterized from trapping experiments with 5,5-dimethyl-1-pyrroline N-oxide (DMPO) by EPR,1 using pulse radiolysis with time-resolved IR detection in acetonitrile,2 by IR (hydrated form,3 on surfaces4 or in Ne matrix5), and by mass spectrometry in the gas phase.6 As a classical distonic anion, the charge and radical sites of [CO2˙−] are separated.7 Notably, the radical is located on a σ-type orbital centred on the carbon atom.6 This highly reactive molecule was exploited in diverse synthetic approaches to produce carboxylic acid-containing molecules through C–C bond formation,8 or as a powerful single electron reductant in challenging reductive processes.8a,r,9The use of alkali formate compounds [MHCO2] has recently emerged as an efficient method to generate [CO2˙−] by Hydrogen Atom Transfer (HAT) (Chart 1a).8o–t As an alternative, the direct one-electron reduction of CO2 has been employed to generate [CO2˙−] in few synthetic transformations by photo- or electroreduction.8b–n However, such reduction is energy demanding since it requires very negative reduction potentials (e.g. E1/2 = −2.65 V in DMF, −2.70 V in CH3CN and −2.53 V in aqueous solution vs. Fc+/0),8b,10 which limits practical applications (Chart 1a). In the context of using CO2 as a sustainable source of carbon,11 it is therefore appealing to seek CO2 activation modes that would enable an easier one-electron reduction process to access [CO2˙−]. With this in mind, we decided to investigate the single electron reduction of FLP (Frustrated Lewis Pair)-type activated CO2 molecules, corresponding to a bifunctional activation of the molecule by Lewis basic and Lewis acidic entities. The ambiphilic nature of carbon dioxide makes it an ideal candidate for FLP-type activation.12 Quickly after the pioneering disclosure of the FLP concept on H2 activation, the FLP activation was explored for CO2.12a,c,d This process was then exploited for the trapping and reactivity of CO2. However, while the FLP concept was recently extended to single electron transfer between the frustrated donor and acceptor counterparts,13 the one-electron-reduction of FLP-activated CO2 has seldom been investigated. To the best of our knowledge, only the (t-Bu)3P–CO2–B(C6F5)3 adduct was subjected to one-electron reduction by Heiden et al., in a broader investigation on the role of main group elements in CO2 reduction.10 They studied the reduction of the (t-Bu)3P–CO2–B(C6F5)3 adduct by means of DFT, electro- and chemical-reduction (Chart 1b). The DFT study indicated that the monoreduced species is a minimum on the energy surface with an unpaired spin density of 16% on the carbon of the CO2 unit. However, the electroreduction of (t-Bu)3P–CO2–B(C6F5)3 gave rise to irreversible waves at rather low potential of −2.56 V and −2.61 V vs. Fc+/0 in THF and acetonitrile, respectively while the chemical reduction led to an ill-defined mixture of products.
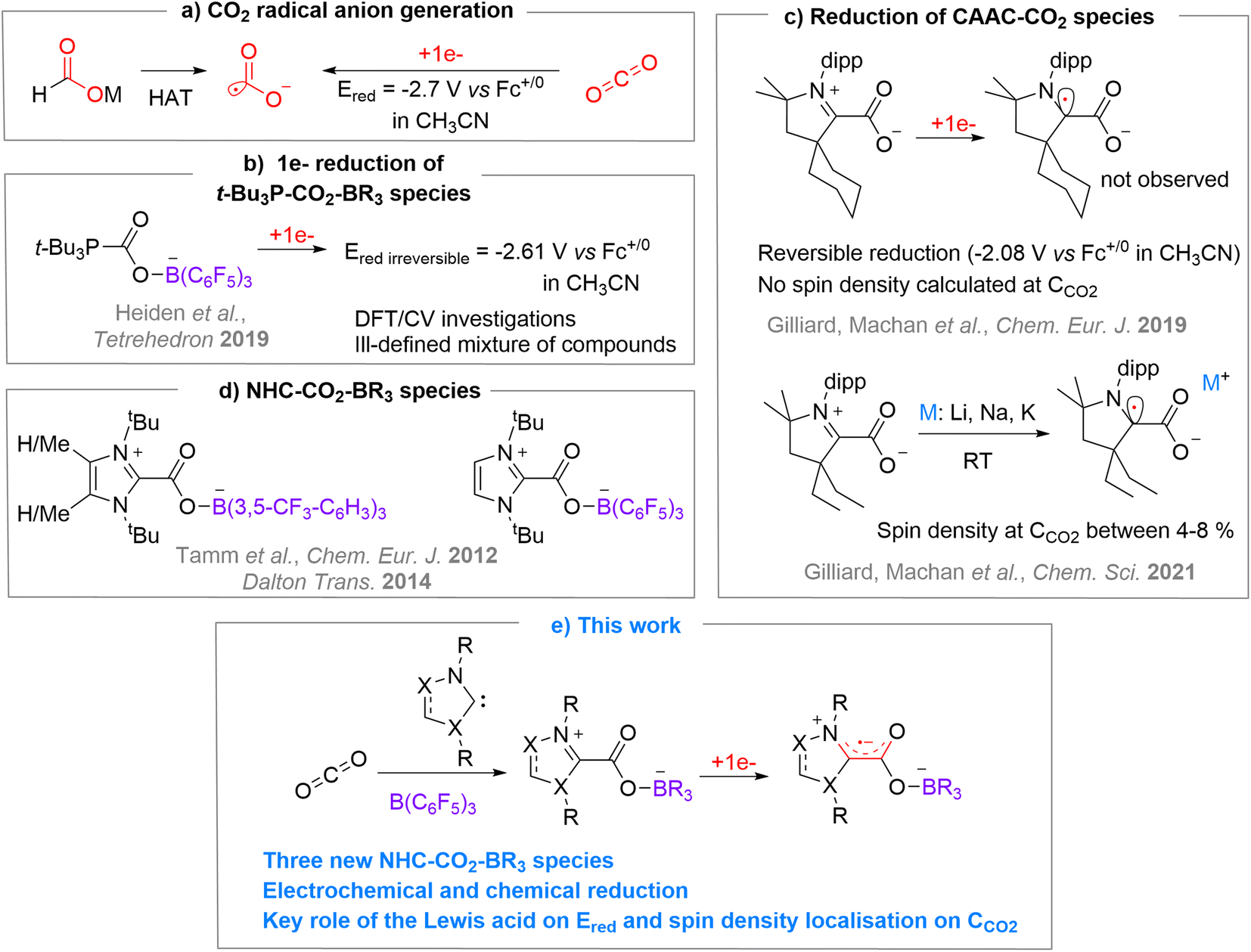 | ||
| Chart 1 (a) Common strategies to generate [CO2˙−], (b) study of the one-electron reduction of t-Bu3P–CO2–B(C6F5)3, (c) one-electron reduction of CAAC–CO2 species, (d) NHC–CO2–borane species, (e) synthesis and one-electron reduction of NHC–CO2–B(C6F5)3 compounds. | ||
In order to explore the monoelectronic reduction of CO2 when activated by an FLP system, we turned our attention to N-heterocyclic carbenes (NHCs) as Lewis bases because of their high electronic and steric modularity and also because of their known ability to stabilise radical species.14 Applied to CO2, Machan, Gilliard et al. have shown that CAAC–CO2 adducts could be reduced by one electron (Chart 1c), either by cyclic voltammetry in a reversible manner at −2.08 V vs. Fc+/0 in CH3CN and −2.15 V vs. Fc+/0 in THF for CyCAAC,15 or by alkali metals (Li, Na, K) for EtCAAC.16 Interestingly, the calculated unpaired spin density of [CAAC–CO2˙−] highlights the importance of the CAAC moiety in the reduction process, since most of the added density is located on the carbenic carbene and barely any density on the carbon atom of the CO2 moiety with a maximum of 8%.15,16 One can notice, though, that the evaluation of the single electron reduction of other NHC–CO2 adducts with more classical NHCs is lacking.17 On the other hand, very few NHCs have been used in FLP-CO2 activation. To the best of our knowledge, only three examples were reported by Tamm et al.18 in their study concerning the ability of NHC/borane systems to trap CO2 and N2O whether the Lewis pair was frustrated or not12d,18 (Chart 1d).
Herein, we report the expansion of the NHC–CO2–borane library and the exploration of their single electron reduction leading to reversible or irreversible reduction depending on the nature of the NHC (Chart 1e). Two radical anion FLP-CO2 systems could be characterized by EPR spectroscopy and one crystallized. The combined theoretical investigations enabled us to scrutinize the reduction potential of these species and the localisation of the unpaired spin density.
Results and discussion
We chose to use tris(pentafluorophenyl)borane (B(C6F5)3) as the Lewis acid and varied the NHC Lewis base to prepare four NHC–CO2–B(C6F5)3 molecules 2a–d. We reproduced the synthesis of the adduct 2a, described by Tamm et al., featuring 1,3-di-tert-butylimidazolin-2-ylidene (It-Bu) and B(C6F5)3 by reacting the ItBu-CO2 adduct 1a with the borane.18a Following a similar strategy, the NHC–CO2–B(C6F5)3 systems 2b–d were synthesized from the reaction between the NHC–CO2 adducts 1b–d of 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene (IPr, 1b), 1,3-bis(1-adamantyl)imidazol-2-ylidene (IAd, 1c),19 and cyclohexyl cyclic (alkyl)(amino)carbenes (cyCAAC, 1d) and an equimolar amount of B(C6F5)3 under a CO2 atmosphere (Scheme 1). The adducts 2b–d were fully characterized by NMR and IR spectroscopy. Overall, these compounds exhibit similar signatures compared to 2a. In the 13C{1H} NMR spectra, the resonances for the carbon atoms of the CO2 moiety appear at δ = 156.0, 152.8, 161.1 and 158.0 ppm for 2a–d, respectively, which represents a de-shielding of approximatively 30 ppm compared to free CO2. In the 11B NMR spectra, the tetrahedral boron atom displays characteristic resonance at δ = −3.9, −3.4, −3.8 and −2.9 ppm for 2a–d, respectively. In the solid-state, the IR analysis indicates C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching frequencies at 1714, 1717, 1715 and 1709 cm−1 for 2a–d, respectively (Table 1). These values are included in the large range of FLP-CO2 molecules (1645 < υ(CO) < 1742 cm−1).12b As a comparison, the antisymmetric OCO stretching frequency of CO2 appears at 2349 cm−1.20 Theoretical investigations revealed that the bands observed at slightly lower frequencies correspond to the υ(CC) and υ(C–F) (Table 1 and ESI†). In the case of 2a for example, these bands were observed at 1644 and 1513 cm−1, respectively.
O stretching frequencies at 1714, 1717, 1715 and 1709 cm−1 for 2a–d, respectively (Table 1). These values are included in the large range of FLP-CO2 molecules (1645 < υ(CO) < 1742 cm−1).12b As a comparison, the antisymmetric OCO stretching frequency of CO2 appears at 2349 cm−1.20 Theoretical investigations revealed that the bands observed at slightly lower frequencies correspond to the υ(CC) and υ(C–F) (Table 1 and ESI†). In the case of 2a for example, these bands were observed at 1644 and 1513 cm−1, respectively.
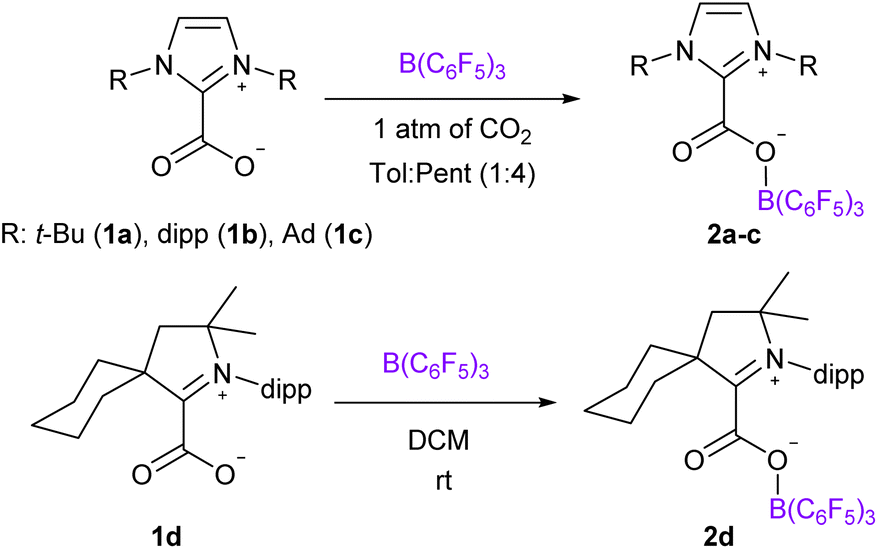 | ||
| Scheme 1 Synthesis of compounds 2a–d. | ||
The solid-state structures were further analysed by single crystal X-ray diffraction analysis (Fig. 1). Selected structural parameters of 2b–d as well as the comparison with 2a are reported in Table 2. In all four cases, the incorporation of the CO2 molecule in the FLP systems resulted in its bending with OCO angles comprised between 130.09(15)° for 2a and 128.78(13)° for 2b and with a significant lengthening of the carbon–oxygen bonds leading to an ester like trigonal planar geometry. The structures of 2a–d feature similar parameters, except for the dihedral angle between the plane of the CO2 and the plane of the NHC moieties, noted D(NHC–CO2) in Table 2. In fact, while structures 2a, 2c and 2d show torsion angles of 80.5(2), 76.7(3) and 79.2(2)°, respectively, the CO2 moiety and the imidazole ring are almost coplanar in 2b with a dihedral angle of 7.3(2)°. These data can be correlated with the CNHC–CCO2 bond distances, which are longer for 2a, 2c and 2d (1.516(2), 1.52071(19), and 1.5223(3) Å, respectively) than for the adduct 2b (1.5071(19) Å). This feature is in accordance with data gathered on NHC–CO2 adducts showing that the C–C bond distance between the carbenic carbon (CNHC) and the CO2 carbon (CCO2) is larger with a greater dihedral angle. These C–C distances and torsion angles have then been correlated with the temperature of decarboxylation which is lower with higher dihedral angles and longer C–C distances.21
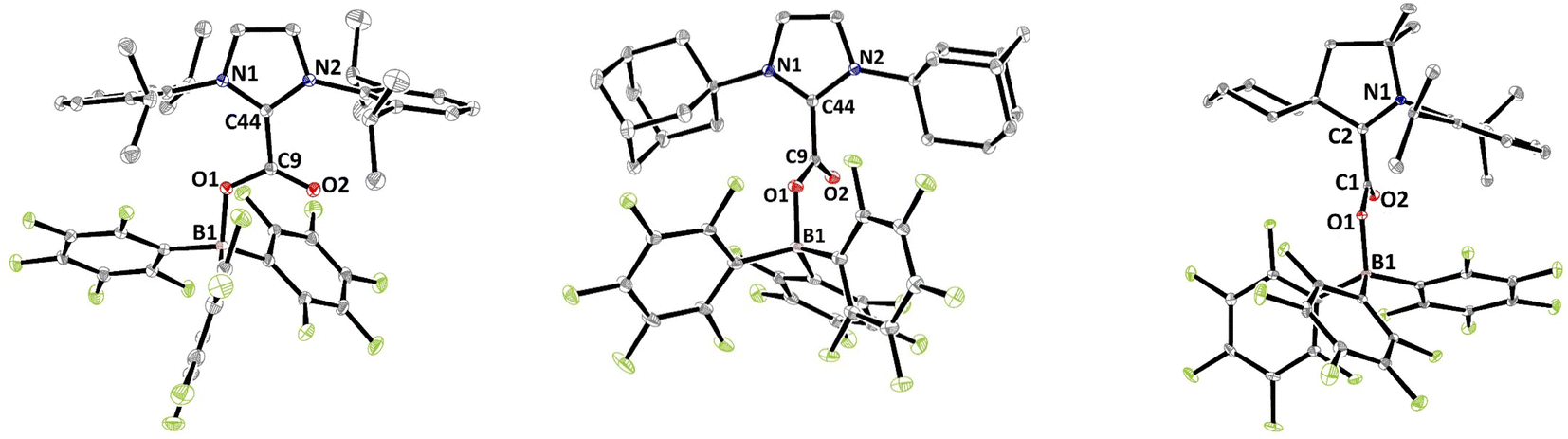 | ||
| Fig. 1 Structures of compounds 2b (left), 2c (middle) and 2d (right). Thermal ellipsoids drawn at 30% probability. | ||
| Structural parameters | 2a 18a | 2b | 2c | 2d |
|---|---|---|---|---|
|
d(CO)/Å |
1.2024(19) | 1.2074(18) | 1.207(3) | 1.2070(17) |
| d(C–O)/Å | 1.2972(19) | 1.2980(17) | 1.298(3) | 1.2977(17) |
| d(O–B)/Å | 1.535(2) | 1.5363(18) | 1.540(3) | 1.5492(18) |
| d(Ccarbenic–CO2)/Å | 1.516(2) | 1.5071(19) | 1.523(3) | 1.5247(19) |
| α(CO2)/° | 130.09(15) | 128.78(13) | 129.96(19) | 129.31(13) |
| D(NHC–CO2)/° | 80.5(2) | 7.3(2) | 76.7(3) | 79.2(2) |
The redox chemistry of species 2a–d was probed by cyclic voltammetry experiments at room temperature in dry dichloromethane (nBu4PF6, 0.1 M) to ensure good solubility. Satisfyingly, the reduction of adducts 2a–d led to clear waves depicted in Fig. 2 with reduction potentials expressed vs. Fc+/0. The cyclic voltammograms of 2a (orange wave) and 2c (blue wave) show irreversible reduction at Eredp = −2.30 V and −2.44 V, respectively. Although, upon reduction, the related (t-Bu)3P–CO2–B(C6F5)3 and CyCAAC–CO2 led to interesting CO2 disproportionation transformations under certain conditions,10,15 we did not explore further the fate of the reduced 2a and 2c. The cyclic voltammogram of 2b (brown wave) reveals a quasi-reversible reduction at E1/2 = −2.08 V (ΔEp = 150 mV). Finally, the quasi-reversible one-electron reduction of compound 2d (green wave) occurs at a potential of E1/2 = −1.34 V (ΔEp = 90 mV). The reduction potential of compound 1d was also recorded at E1/2 = −2.13 V (ΔEp = 132 mV), in accordance with the data reported in the literature.15 Overall, the recorded Ered data indicate that compounds 2a–d featuring an FLP-type activated molecule of CO2 are more easily reduced by one electron than free CO2. The most important gain is obtained with the combination of CAAC and B(C6F5)3 enabling the achievement of a remarkable reduction potential E1/2 of −1.34 V in CH2Cl2 compared to free CO2 (i.e., Eredp = −2.65 V in DMF, −2.70 V in CH3CN and −2.53 V in aqueous solution vs. Fc+/0).8b,10 The less negative reduction potential observed in the case of CAAC vs. the more classical NHC probably results from the higher π-accepting ability of CAAC, which is the consequence of a more accessible LUMO orbital.22 This feature would thus indicate that the reduction event is at least partially localised on the carbenic carbon, which is confirmed by the following experimental and theoretical investigations. The beneficial role of the borane – which was expected because it decreases the electronic density from the carbene–CO2 adduct – is quantified by a net gain of 0.79 V observed between the E1/2 of 2d compared to that of 1d.
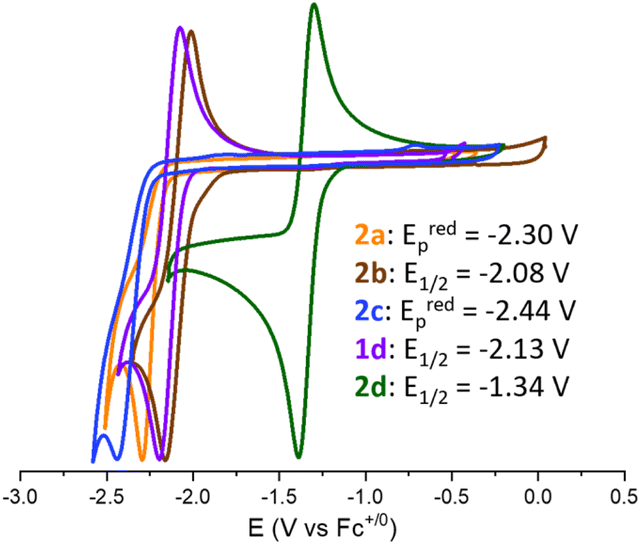 | ||
| Fig. 2 Normalized cyclic voltammograms of NHC–CO2–B(C6F5)3 adducts 1d and 2a–d on a GC microdisk in 0.1 M [nBu4N][PF6]/CH2Cl2 media under an Ar atmosphere (5 × 10−3 M of adduct, rt, scan rate: 0.2 V s−1). | ||
In order to gain further insights into the role of the NHC/borane pair in the activation of CO2, DFT calculations were undertaken using the M06-2X functional combined with Grimme's D3 correction to consider dispersion effects (see Computational details in the ESI†). The structures of 2a–d and of NHC–CO2 adducts 1a–d were computed. In addition, the structures of 2(a–d)-BH3, featuring BH3 in place of B(C6F5)3 were also computed as models for a less Lewis acidic borane. The calculated charges by Natural Population Analysis (NPA) on the CCO2 reflect the electron withdrawing effect of the borane to a moderate extent (Table S9†). For example, the NPA charge of 1a is 0.75e and is 0.81e for 2a, while the NPA charge of the CCO2 of free CO2 is significantly more positive (1.05). Similar variations of 0.05 to 0.09e are observed in the case of the other NHC. The LUMO orbitals of the series were also analysed (Fig. 3). In the adducts 1a–d the π-type LUMOs are mostly located on the carbenic centres, which is in line with the less negative reduction potentials observed for the CAAC adducts. The addition of borane (BH3 and B(C6F5)3) leads to the delocalisation of the LUMO over the CCO2 as well. This phenomenon is enhanced with the increased Lewis acidity of B(C6F5)3vs. BH3. The role of the dihedral angle D(NHC–CO2) was also explored. Relaxed potential energy scans were computed for the rotation, carrying out constrained optimisations for each value of the dihedral angle (see Fig. S76†). From them, a minimum and rotational transition states were located for each structure. In each case, the minimum corresponds to the relative orientation of the NHC and CO2 moieties observed in the crystal structures: orthogonal in the case of 2a, 2c–d, and coplanar for 2b. Computations also evidenced these preferences in the absence of borane and with BH3, since the NHC and CO2 moieties are also found orthogonal for 1a, 1c–d, as well as for the corresponding BH3 adducts and coplanar for 1b and 2b-BH3. These features suggest that the observed variations between 2 and 2-BH3 stem mostly from the difference in the electronic properties between B(C6F5)3 and BH3 of lower Lewis acidity and not from steric differences. The barriers of rotation of 2a–d were evaluated to be accessible at room temperature since the highest is calculated at 16.5 kcal mol−1. The barrier for 2b (4.6 kcal mol−1) is significantly smaller compared to the one for 2a, 2c and 2d (13.5, 16.5 and 11.8 kcal mol−1, respectively).
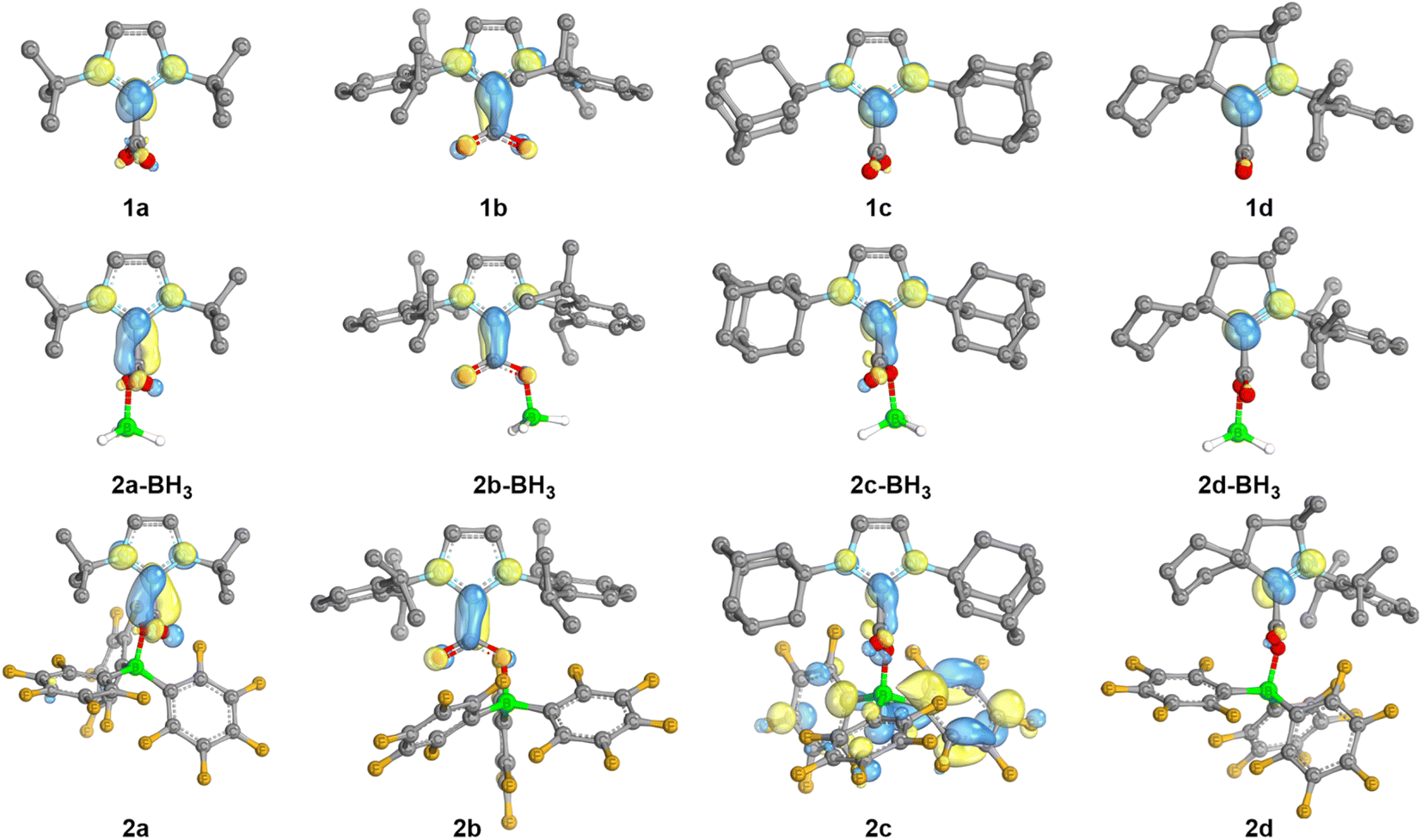 | ||
| Fig. 3 LUMO orbitals of the neutral NHC-based adducts. Visualization (IBOview,23 threshold = 45) with colour code: carbon (grey), nitrogen (light blue), boron (green), fluorine (dark yellow), oxygen (red). | ||
Reduction potentials of compounds 2a–d were computed accurately with maximum deviation from the experimental values of 0.14 V. However, the calculated potential for 1d is off by 0.25 V (vide infra for the proposed explanation). We then explored the effect of the borane on the reduction potential. As for 1dvs.2d, the absence of borane in 1a–c has a significant impact with calculated potentials of −3.42, −3.00 and −3.44 V vs. Fc+/0, respectively. These values are more negative than that of free CO2, explaining the absence of any reduction potential values for the classical NHC–CO2 adduct in the literature.17a
Having calculated that the relative rotations of NHC and CO2–B(C6F5)3 moieties are accessible at room temperature, we wondered if the reduction potentials vary with the dihedral angle. Transition states [1d]‡ and [2a–d]‡ were found resulting from the rotation of the NHC and CO2 moieties in 1d and 2a–d, respectively. The calculated values indicate that indeed the reduction potential is sensitive to the dihedral angle since much less negative potentials were calculated for these TS (Table 3), in line with the fact that the LUMOs are lower in energy after rotation (Fig. S76†). Nonetheless, the values calculated for [2a–d]‡ do not correspond to the experimental values suggesting that it is indeed 2a–d that are reduced in the CV and not [2a–d]‡. However, in the case of [1d]‡, the calculated potential is significantly closer to the experimental value (Δ = 0.04 vs. 0.25 V), which could indicate that it is in fact the reduction of [1d]‡ that is measured experimentally. Further exploration aiming at controlling the degree of rotation would nonetheless be necessary to confirm this hypothesis.
In addition to electrochemical studies, we investigated the chemical reduction of adducts 2a–d with one equivalent of potassium graphite (KC8) as the chemical reductant in THF under argon. In accordance with the irreversible waves observed for 2a and 2c, no radical species could be detected upon chemical reduction. However, the reduction of 2b and 2d at 298 K led to a deep red and yellow solution, respectively, exhibiting EPR signals. As shown in Fig. 4b, five (g = 2.0026) and three (g = 2.0026) hyperfine lines were observed for [2b˙−] and [2d˙−], respectively, indicative of a radical electron coupling with the two equivalent nitrogen nuclei (aN = 4.6 G) for [2b˙−] and one nitrogen nucleus (aN = 6.1 G) for [2d˙−]. DFT calculation reproduced the EPR signals accurately (Fig. S66 and S67†). In accordance with the reduction potential measured at −1.34 V for 2d, [2d˙−] could also be generated using Co(Cp*)2 as a milder reductant (Ered = −1.94 V vs. Fc+/0 in CH2Cl2), as proved by the observation of a very similar EPR signal (Fig. S69†). These radical species are stable in solution under an inert argon atmosphere at room temperature, at least for two days as shown by EPR analyses (Fig. S66 and S67†). IR frequency involving the CO bond was measured at 1596 cm−1 in the solid state for the isolated [2d˙−], while only the solution IR frequency was recorded for the in situ generated [2b˙−]. DFT calculations indicate that these frequencies correspond to the combination of the stretching of the CO and C–CNHC bonds (ESI†). While attempts to obtain crystals were unsuccessful with the radical [2b˙−], single crystals of [2d˙−][K+].(THF)2, suitable for X-ray diffraction, were grown from a concentrated THF/pentane solution at −35 °C. In comparison with the structural parameters of the parent neutral form 2d, the additional electron in [2d˙−] is responsible for important changes. The most striking difference concerns the torsion between the plane of the CO2 moiety and the plane of the CAAC ring, which, from a dihedral angle of 79.2(2)° in 2d, reaches a minimum in [2d˙−] with a dihedral angle of 0.1(3)°. This phenomenon was also observed in the absence of borane by Machan, Gilliard et al. and highlights the increased resonance between the NHC and the CO2 fragments in the reduced form. In line, important shortening of the CNHC–CCO2 bond [1.433(3) A°] and the O–B bond [1.486(3) A°] is witnessed in [2d˙−], along with lengthening of the N–CNHC [1.374(3) Å], CO [1.241(3) Å] and C–O [1.331(2) Å] bonds. In addition, the bending of the CO2 moiety is enhanced reaching an angle of 122.26(18)° (vs. 129.31(13) for 2d, Fig. 4d). This bending is very similar to the bending observed in the reduction of the CAAC–CO2 adduct with Li, Na and K leading to a bending of 122.9(2)° with Na to 123.71(9)° for Li.16 DFT calculations were conducted on [NHC–CO2–BR3˙−] and [NHC–CO2˙−] adducts and indicate that the coplanar rearrangement of the NHC and CO2 planes is a general phenomenon since it is predicted for the all series of [NHC–CO2–BR3˙−] and [NHC–CO2˙−] adducts (Fig. 4c and ESI†). All cases feature separated anionic and radical sites and can thus be described as distonic anions. While as in [CO2˙−] the anionic charge is mostly located on the oxygen atoms, the radical charge is however mostly located on the CNHC in the case of [1a–d˙−] compounds and not on the CCO2. Moreover, the radical resides in a π-type orbital instead of a σ-type orbital in [CO2˙−]. Interestingly, the addition of a borane enables transferring of a part of the unpaired spin density to the CCO2 in [2(a–d)-BH3˙−] and [2a–d˙−], which is slightly closer to the [CO2˙−] situation. In more detail, the localized Mulliken atomic spin density of [2b˙−] and [2d˙−] indicates a large distribution on the CNHC (0.57 and 0.69, respectively) and on the CCO2 (0.42 and 0.33, respectively), while lower spin density is found on the nitrogen (0.17, 0.23, respectively) and on the oxygen (0.14, 0.12, respectively) atoms. The portion of spin density found on the CCO2 of [2b˙−] and [2d˙−] is remarkable when taking into account the absence of any calculated spin density on the CCO2 of the mono-reduced form of the cyCAAC–CO2 adduct [1d˙−] (Table S8†).15 It is in line with the spin density calculation of Heiden et al. on the parent [tBu3P–CO2–B(C6F5)3˙−] adduct with 16% on CCO2, but to a larger extent.10
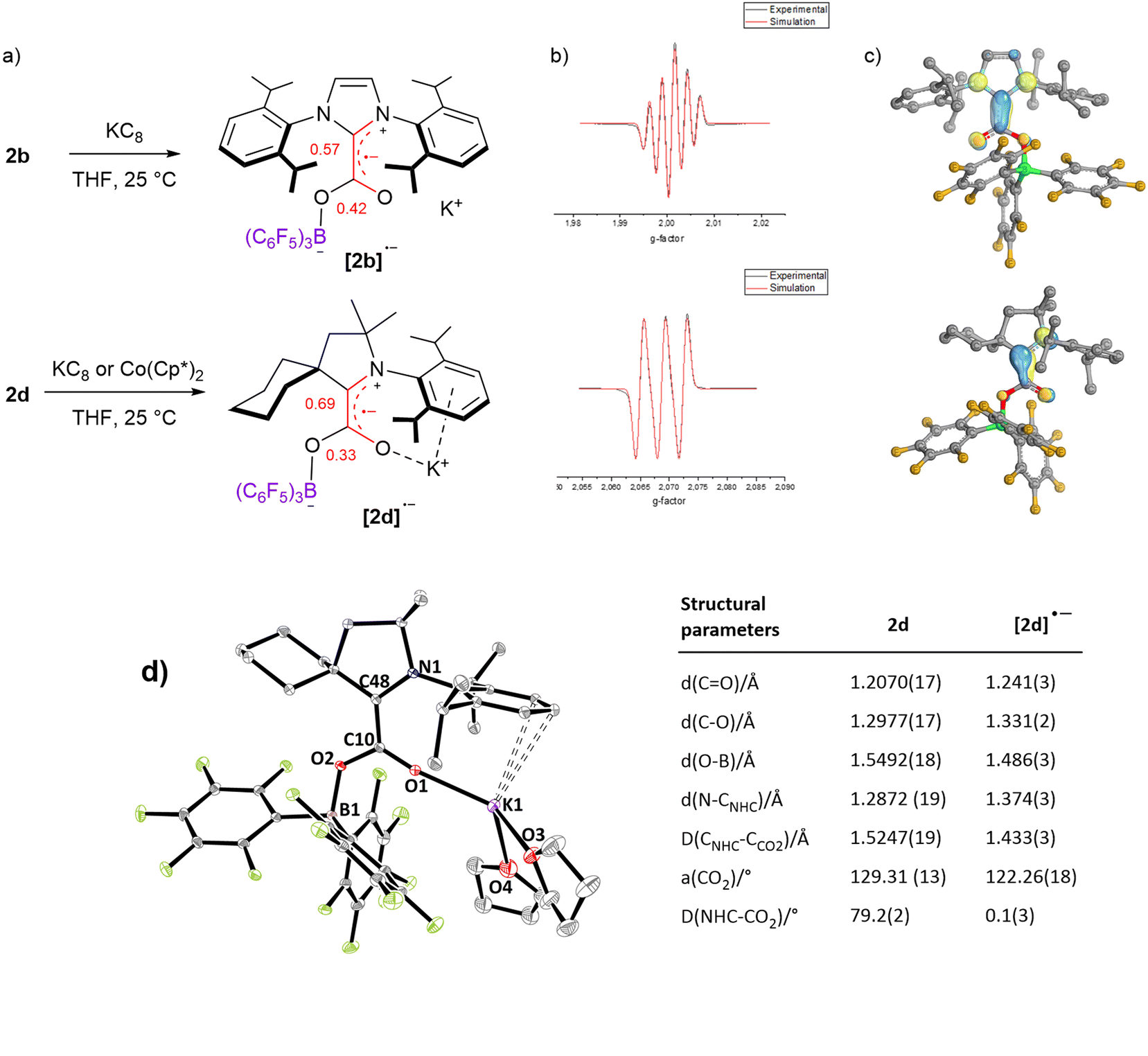 | ||
| Fig. 4 Characterization of the radical anions [2b˙−] and [2d˙−]: (a) mono-reduction of 2b and 2d into [2b˙−] and [2d˙−], (b) experimental (black) and simulated (red) EPR spectra, (c) calculated SOMO orbitals, (d) solid-state structure of [2d˙−][K+]·(THF)2 and selected structural parameters of [2d˙−]. | ||
As a preliminary evaluation of the reactivity of the radical anionic species [2b˙−] and [2d˙−], we wished to probe their reactivity with DMPO since this is a classical trapping agent for [CO2˙−], giving rise to a very characteristic EPR signal featuring six bands at g = 2.0058 (aN = 15.8 G, aβ-H = 19.1 G) in aqueous media (Table 4, entry 1).1 Satisfyingly, when [2b˙−] and [2d˙−] were reacted with five equivalents of DMPO, the EPR signals of [2b˙−] and [2d˙−] were replaced by a similar sextet at g = 2.0052 (aN = 14.0 G and aβ-H = 18.6 G) and g = 2.0050 (aN = 14.4 G and aβ-H = 20.0 G), respectively (Table 4, entries 2 and 3, Fig. S71†). A loss of signal intensity is however observed, presumably due to stability issues under these conditions. In the absence of borane, the chemically reduced compound [1d˙−] also reacts like [CO2˙−] toward DMPO as indicated by a similar EPR signal at g = 2.0050 (aN = 15.0 G and aβ-H = 20.1 G), (Fig. S71†). This is in line with the observation that when [1d˙−] was generated electrochemically under a CO2 atmosphere by Gilliard, Machan et al., a disproportionation reaction was observed, which is a known reactivity of [CO2˙−].15 At this stage, it is difficult to assess whether or not the borane moiety is maintained in the products of these spin trapping experiments. As indicated by DFT calculations, [DMPO-CO2˙−] and [DMPO-CO2-B(C6F5)3˙−] would afford similar EPR signals (Table 4, entries 5 and 6). Further studies will be necessary to understand and exploit the reactivity of the monoreduced FLP-type activated CO2 molecules in valuable synthetic strategies.
Conclusions
Three new NHC–CO2–B(C6F5)3 adducts 2b–d are reported. The one-electron reduction of these adducts and of the known adduct It–Bu–CO2–B(C6F5)32a, shows reversible and irreversible waves depending on the nature of the NHC. This combined experimental and theoretical study highlights the beneficial role of the FLP-type activation of CO2 with NHC/BR3 pair for the one electron redox event. The LUMOs of the NHC–CO2–B(C6F5)3 adducts are mostly located on the CNHC which was shown to be the main site of reduction. The adduct 2d featuring a π-accepting CAAC exhibits less negative reduction potential than adducts with more classical NHCs. DFT calculations highlighted the role of the dihedral angle between the carbene and CO2 moieties and of the borane in the reduction potential. The withdrawing effect of the Lewis acidic borane was indeed shown to lead to a less negative reductive potential (E1/2 = −1.34 V vs. Fc+/0 for 2d). Furthermore, the two species exhibiting reversible waves in the CV experiment were shown to afford stable anionic radical species by chemical reduction, which could be characterised by EPR for [2b˙−] and [2d˙−] and X-ray diffraction analysis for [2d˙−]. In these distonic species, the radical site was shown to be located not only on the CNHC but also on the CCO2 with a maximum spin density on the CCO2 of 0.42 for [2b˙−]. Finally, [2b˙−] and [2d˙−] were shown to exhibit a reactivity similar to [CO2˙−] toward DMPO. Future studies will further explore the reactivity of the stable and unstable radical anions of type [2˙−], as well as the broader use of FLP-type activation of carbon dioxide in single electron reduction transformations.Data availability
Raw data of NMR, CV, EPR and X-ray crystallography are available upon reasonable request.Author contributions
AM conducted the theoretical and experimental work. CG conducted the experimental work. AS-S and LV conducted CV and X-ray diffraction analyses, respectively. AL, OB and SB conceptualised the project and supervised the experimental and theoretical work. All authors participated in writing, reviewing and editing the original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 860322. We acknowledge the valuable support of the NMR service and EPR platform at LCC-CNRS. Calculations on this project were carried out with the use of CSUC supercomputing resources. We thank Vincent César and Nicolas Queyriaux for fruitful discussions.References
- (a) F. A. Villamena, E. J. Locigno, A. Rockenbauer, C. M. Hadad and J. L. Zweier, Theoretical and Experimental Studies of the Spin Trapping of Inorganic Radicals by 5,5-Dimethyl-1-Pyrroline N-Oxide (DMPO). 1. Carbon Dioxide Radical Anion, J. Phys. Chem. A, 2006, 110, 13253–13258 CrossRef CAS PubMed; (b) J. R. Harbour and M. L. Hair, Spin trapping of the •CO2− radical in aqueous medium, Can. J. Chem., 1979, 57, 1150–1152 CrossRef CAS.
- D. C. Grills and S. V. Lymar, Radiolytic formation of the carbon dioxide radical anion in acetonitrile revealed by transient IR spectroscopy, Phys. Chem. Chem. Phys., 2018, 20, 10011–10017 RSC.
- A. Herburger, M. Ončák, C.-K. Siu, E. G. Demissie, J. Heller, W. K. Tang and M. K. Beyer, Infrared Spectroscopy of Size-Selected Hydrated Carbon Dioxide Radical Anions CO2.−(H2O)n (n=2–61) in the C−O Stretch Region, Chem. – Eur. J., 2019, 25, 10165–10171 CrossRef CAS PubMed.
- N. J. Firet and W. A. Smith, Probing the Reaction Mechanism of CO2 Electroreduction over Ag Films via Operando Infrared Spectroscopy, ACS Catal., 2017, 7, 606–612 CrossRef CAS.
- M. E. Jacox and W. E. Thompson, The vibrational spectra of molecular ions isolated in solid neon. I. CO2+ and CO2−, J. Chem. Phys., 1989, 91, 1410–1416 CrossRef CAS.
- D. Schröder, C. A. Schalley, J. N. Harvey and H. Schwarz, On the formation of the carbon dioxide anion radical CO2− · in the gas phase, Int. J. Mass Spectrom., 1999, 185–187, 25–35 CrossRef.
- S. J. Yu, C. L. Holliman, D. L. Rempel and M. L. Gross, The β-distonic ion from the reaction of pyridine radical cation and ethene: a demonstration of high-pressure trapping in Fourier transform mass spectrometry, J. Am. Chem. Soc., 1993, 115, 9676–9682 CrossRef CAS.
-
(a) A. Malandain, M. Molins, A. Hauwelle, A. Talbot, O. Loreau, T. D'Anfray, S. Goutal, N. Tournier, F. Taran, F. Caillé and D. Audisio, Carbon Dioxide Radical Anion by Photoinduced Equilibration between Formate Salts and [11C, 13C, 14C]CO2: Application to Carbon Isotope Radiolabeling, J. Am. Chem. Soc., 2023, 145, 16760–16770 CrossRef CAS PubMed;
(b) Y. You, W. Kanna, H. Takano, H. Hayashi, S. Maeda and T. Mita, Electrochemical Dearomative Dicarboxylation of Heterocycles with Highly Negative Reduction Potentials, J. Am. Chem. Soc., 2022, 144, 3685–3695 CrossRef CAS PubMed;
(c) L. Song, W. Wang, J.-P. Yue, Y.-X. Jiang, M.-K. Wei, H.-P. Zhang, S.-S. Yan, L.-L. Liao and D.-G. Yu, Visible-light photocatalytic di- and hydro-carboxylation of unactivated alkenes with CO2, Nat. Catal., 2022, 5, 832–838 CrossRef CAS;
(d) L.-L. Liao, Z.-H. Wang, K.-G. Cao, G.-Q. Sun, W. Zhang, C.-K. Ran, Y. Li, L. Chen, G.-M. Cao and D.-G. Yu, Electrochemical Ring-Opening Dicarboxylation of Strained Carbon–Carbon Single Bonds with CO2: Facile Synthesis of Diacids and Derivatization into Polyesters, J. Am. Chem. Soc., 2022, 144, 2062–2068 CrossRef CAS PubMed;
(e) J.-H. Ye, T. Ju, H. Huang, L.-L. Liao and D.-G. Yu, Radical Carboxylative Cyclizations and Carboxylations with CO2, Acc. Chem. Res., 2021, 54, 2518–2531 CrossRef CAS PubMed;
(f) T. Ju, Y.-Q. Zhou, K.-G. Cao, Q. Fu, J.-H. Ye, G.-Q. Sun, X.-F. Liu, L. Chen, L.-L. Liao and D.-G. Yu, Dicarboxylation of alkenes, allenes and (hetero)arenes with CO2 via visible-light photoredox catalysis, Nat. Catal., 2021, 4, 304–311 CrossRef CAS;
(g) H. Huang, J.-H. Ye, L. Zhu, C.-K. Ran, M. Miao, W. Wang, H. Chen, W.-J. Zhou, Y. Lan, B. Yu and D.-G. Yu, Visible-Light-Driven Anti-Markovnikov Hydrocarboxylation of Acrylates and Styrenes with CO2, CCS Chem., 2021, 3, 1746–1756 CrossRef CAS;
(h) R. Mello, J. C. Arango-Daza, T. Varea and M. E. González-Núñez, Photoiodocarboxylation of Activated CC Double Bonds with CO2 and Lithium Iodide, J. Org. Chem., 2018, 83, 13381–13394 CrossRef CAS PubMed;
(i) J.-H. Ye, M. Miao, H. Huang, S.-S. Yan, Z.-B. Yin, W.-J. Zhou and D.-G. Yu, Visible-Light-Driven Iron-Promoted Thiocarboxylation of Styrenes and Acrylates with CO2, Angew. Chem., Int. Ed., 2017, 56, 15416–15420 CrossRef CAS PubMed;
(j) H. Seo, A. Liu and T. F. Jamison, Direct β-Selective Hydrocarboxylation of Styrenes with CO2 Enabled by Continuous Flow Photoredox Catalysis, J. Am. Chem. Soc., 2017, 139, 13969–13972 CrossRef CAS PubMed;
(k) H. Seo, M. H. Katcher and T. F. Jamison, Photoredox activation of carbon dioxide for amino acid synthesis in continuous flow, Nat. Chem., 2017, 9, 453–456 CrossRef CAS PubMed;
(l) A. Akhgarnusch, R. F. Höckendorf, Q. Hao, K. P. Jäger, C.-K. Siu and M. K. Beyer, Carboxylation of Methyl Acrylate by Carbon Dioxide Radical Anions in Gas-Phase Water Clusters, Angew. Chem., Int. Ed., 2013, 52, 9327–9330 CrossRef CAS PubMed;
(m) M. D. Otero, B. Batanero and F. Barba, CO2 anion–radical in organic carboxylations, Tetrahedron Lett., 2006, 47, 2171–2173 CrossRef CAS;
(n) D. A. Morgenstern, R. E. Wittrig, P. E. Fanwick and C. P. Kubiak, Photoreduction of carbon dioxide to its radical anion by nickel cluster [Ni3(μ3-I)2(dppm)3]: formation of two carbon-carbon bonds via addition of carbon dioxide radical anion to cyclohexene, J. Am. Chem. Soc., 1993, 115, 6470–6471 CrossRef CAS;
(o) M. Mikhael, S. N. Alektiar, C. S. Yeung and Z. K. Wickens, Translating Planar Heterocycles into Three-Dimensional Analogs by Photoinduced Hydrocarboxylation, Angew. Chem., Int. Ed., 2023, 62, e202303264 CrossRef CAS PubMed;
(p) S. N. Alektiar, J. Han, Y. Dang, C. Z. Rubel and Z. K. Wickens, Radical Hydrocarboxylation of Unactivated Alkenes via Photocatalytic Formate Activation, J. Am. Chem. Soc., 2023, 145, 10991–10997 CrossRef CAS PubMed;
(q) S. N. Alektiar and Z. K. Wickens, Photoinduced Hydrocarboxylation via Thiol-Catalyzed Delivery of Formate Across Activated Alkenes, J. Am. Chem. Soc., 2021, 143, 13022–13028 CrossRef CAS PubMed;
(r) C. M. Hendy, G. C. Smith, Z. Xu, T. Lian and N. T. Jui, Radical Chain Reduction via Carbon Dioxide Radical Anion (CO2˙−), J. Am. Chem. Soc., 2021, 143, 8987–8992 CrossRef CAS PubMed;
(s) J. Majhi and G. A. Molander, Recent Discovery, Development, and Synthetic Applications of Formic Acid Salts in Photochemistry, Angew. Chem., Int. Ed., 2023, e202311853 Search PubMed;
(t) H. Wang, Y. Gao, C. Zhou and G. Li, Visible-Light-Driven Reductive Carboarylation of Styrenes with CO2 and Aryl Halides, J. Am. Chem. Soc., 2020, 142, 8122–8129 CrossRef CAS PubMed;
(u) S. Wang, P. Xu and X. Zhu, CO2 Radical Anion in Photochemical Dicarboxylation of Alkenes, ChemCatChem, 2023, 15, e202300695 CrossRef CAS.
- (a) P. Xu, X.-Y. Wang, Z. Wang, J. Zhao, X.-D. Cao, X.-C. Xiong, Y.-C. Yuan, S. Zhu, D. Guo and X. Zhu, Defluorinative Alkylation of Trifluoromethylbenzimidazoles Enabled by Spin-Center Shift: A Synergistic Photocatalysis/Thiol Catalysis Process with CO2˙−, Org. Lett., 2022, 24, 4075–4080 CrossRef CAS PubMed; (b) A. F. Chmiel, O. P. Williams, C. P. Chernowsky, C. S. Yeung and Z. K. Wickens, Non-innocent Radical Ion Intermediates in Photoredox Catalysis: Parallel Reduction Modes Enable Coupling of Diverse Aryl Chlorides, J. Am. Chem. Soc., 2021, 143, 10882–10889 CrossRef CAS PubMed; (c) S.-S. Yan, S.-H. Liu, L. Chen, Z.-Y. Bo, K. Jing, T.-Y. Gao, B. Yu, Y. Lan, S.-P. Luo and D.-G. Yu, Visible-light photoredox-catalyzed selective carboxylation of C(sp3)−F bonds with CO2, Chem, 2021, 7, 3099–3113 CrossRef CAS; (d) O. P. Williams, A. F. Chmiel, M. Mikhael, D. M. Bates, C. S. Yeung and Z. K. Wickens, Practical and General Alcohol Deoxygenation Protocol, Angew. Chem., Int. Ed., 2023, 62, e202300178 CrossRef CAS PubMed.
- B. L. Thompson and Z. M. Heiden, Investigation of main group promoted carbon dioxide reduction, Tetrahedron, 2019, 75, 2099–2105 CrossRef CAS PubMed.
- (a) G. Prieto, Carbon Dioxide Hydrogenation into Higher Hydrocarbons and Oxygenates: Thermodynamic and Kinetic Bounds and Progress with Heterogeneous and Homogeneous Catalysis, ChemSusChem, 2017, 10, 1056–1070 CrossRef CAS PubMed; (b) J. Luo and I. Larrosa, C−H Carboxylation of Aromatic Compounds through CO2 Fixation, ChemSusChem, 2017, 10, 3317–3332 CrossRef CAS PubMed; (c) J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Selective Catalytic Synthesis Using the Combination of Carbon Dioxide and Hydrogen: Catalytic Chess at the Interface of Energy and Chemistry, Angew. Chem., Int. Ed., 2016, 55, 7296–7343 CrossRef CAS PubMed; (d) Q. Liu, L. Wu, R. Jackstell and M. Beller, Using carbon dioxide as a building block in organic synthesis, Nat. Commun., 2015, 6, 1–15 Search PubMed; (e) A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Frontiers, Opportunities, and Challenges in Biochemical and Chemical Catalysis of CO2 Fixation, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed; (f) M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Transformation of Carbon Dioxide with Homogeneous Transition-Metal Catalysts: A Molecular Solution to a Global Challenge?, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed.
- (a) M. Pérez-Jiménez, H. Corona, F. de la Cruz-Martínez and J. Campos, Donor-Acceptor Activation of Carbon Dioxide, Chem. – Eur. J., 2023, e202301428 CrossRef PubMed; (b) S. Bontemps, Boron-Mediated Activation of Carbon Dioxide, Coord. Chem. Rev., 2016, 308(2), 117–130 CrossRef CAS; (c) D. W. Stephan and G. Erker, Frustrated Lewis Pair Chemistry: Development and Perspectives, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed; (d) E. L. Kolychev, E. Theuergarten and M. Tamm, in Frustrated Lewis Pairs II: Expanding the Scope, ed. G. Erker and D. W. Stephan, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 121–155 Search PubMed.
- (a) L. J. C. van der Zee, S. Pahar, E. Richards, R. L. Melen and J. C. Slootweg, Insights into Single-Electron-Transfer Processes in Frustrated Lewis Pair Chemistry and Related Donor–Acceptor Systems in Main Group Chemistry, Chem. Rev., 2023, 123, 9653–9675 CrossRef CAS PubMed; (b) Z. Lu, M. Ju, Y. Wang, J. M. Meinhardt, J. I. Martinez Alvarado, E. Villemure, J. A. Terrett and S. Lin, Regioselective aliphatic C–H functionalization using frustrated radical pairs, Nature, 2023, 619, 514–520 CrossRef CAS PubMed; (c) A. Dasgupta, E. Richards and R. L. Melen, Frustrated Radical Pairs: Insights from EPR Spectroscopy, Angew. Chem., Int. Ed., 2021, 60, 53–65 CrossRef CAS PubMed; (d) F. Holtrop, A. R. Jupp, N. P. van Leest, M. Paradiz Dominguez, R. M. Williams, A. M. Brouwer, B. de Bruin, A. W. Ehlers and J. C. Slootweg, Photoinduced and Thermal Single-Electron Transfer to Generate Radicals from Frustrated Lewis Pairs, Chem. – Eur. J., 2020, 26, 9005–9011 CrossRef CAS PubMed; (e) F. Holtrop, A. R. Jupp, B. J. Kooij, N. P. van Leest, B. de Bruin and J. C. Slootweg, Single-Electron Transfer in Frustrated Lewis Pair Chemistry, Angew. Chem., Int. Ed., 2020, 59, 22210–22216 CrossRef CAS PubMed; (f) W. E. Piers, A. J. V. Marwitz and L. G. Mercier, Mechanistic Aspects of Bond Activation with Perfluoroarylboranes, Inorg. Chem., 2011, 50, 12252–12262 CrossRef CAS PubMed.
- (a) A. Merschel, D. Rottschäfer, B. Neumann, H.-G. Stammler, M. Ringenberg, M. van Gastel, T. I. Demirer, D. M. Andrada and R. S. Ghadwal, Crystalline Anions Based on Classical N-Heterocyclic Carbenes, Angew. Chem., Int. Ed., 2023, 62, e202215244 CrossRef CAS PubMed; (b) J. Fan, A.-P. Koh, J. Zhou, Z.-F. Zhang, C.-S. Wu, R. D. Webster, M.-D. Su and C.-W. So, Tetrakis(N-heterocyclic Carbene)-Diboron(0): Double Single-Electron-Transfer Reactivity, J. Am. Chem. Soc., 2023, 145, 11669–11677 CrossRef CAS PubMed; (c) S. Choe, H. Song, H. Choi, S. Yoo, J. Kim, Y. H. Ko and E. Lee, Formation of Highly Stable 1,2-Dicarbonyl Organic Radicals from Cyclic (Alkyl)(amino)carbenes, Org. Lett., 2023, 25, 4292–4297 CrossRef CAS PubMed; (d) J. L. Peltier, M. R. Serrato, V. Thery, J. Pecaut, E. Tomás-Mendivil, G. Bertrand, R. Jazzar and D. Martin, An air-stable radical with a redox-chameleonic amide, Chem. Commun., 2023, 59, 595–598 RSC; (e) K. Breitwieser, H. Bahmann, R. Weiss and D. Munz, Gauging Radical Stabilization with Carbenes, Angew. Chem., 2022, 134, e202206390 CrossRef; (f) M. M. Hansmann, M. Melaimi and G. Bertrand, Crystalline Monomeric Allenyl/Propargyl Radical, J. Am. Chem. Soc., 2017, 139, 15620–15623 CrossRef CAS PubMed; (g) J. K. Mahoney, D. Martin, F. Thomas, C. E. Moore, A. L. Rheingold and G. Bertrand, Air-Persistent Monomeric (Amino)(carboxy) Radicals Derived from Cyclic (Alkyl)(Amino) Carbenes, J. Am. Chem. Soc., 2015, 137, 7519–7525 CrossRef CAS PubMed; (h) N. M. Gallagher, H.-Z. Ye, S. Feng, J. Lopez, Y. G. Zhu, T. Van Voorhis, Y. Shao-Horn and J. A. Johnson, An N-Heterocyclic-Carbene-Derived Distonic Radical Cation, Angew. Chem., Int. Ed., 2020, 59, 3952–3955 CrossRef CAS PubMed.
- L. E. Lieske, L. A. Freeman, G. Wang, D. A. Dickie, R. J. Gilliard Jr and C. W. Machan, Metal-Free Electrochemical Reduction of Carbon Dioxide Mediated by Cyclic(Alkyl)(Amino) Carbenes, Chem. – Eur. J., 2019, 25, 6098–6101 CrossRef CAS PubMed.
- L. A. Freeman, A. D. Obi, H. R. Machost, A. Molino, A. W. Nichols, D. A. Dickie, D. J. D. Wilson, C. W. Machan and R. J. Gilliard, Soluble, crystalline, and thermally stable alkali CO2− and carbonite (CO22−) clusters supported by cyclic(alkyl)(amino) carbenes, Chem. Sci., 2021, 12, 3544–3550 RSC.
- (a) D. M. Denning, M. D. Thum and D. E. Falvey, Photochemical Reduction of CO2 Using 1,3-Dimethylimidazolylidene, Org. Lett., 2015, 17, 4152–4155 CrossRef CAS PubMed; (b) O. R. Luca, C. C. L. McCrory, N. F. Dalleska and C. A. Koval, The Selective Electrochemical Conversion of Preactivated CO2 to Methane, J. Electrochem. Soc., 2015, 162, H473–H476 CrossRef CAS.
- (a) E. Theuergarten, T. Bannenberg, M. D. Walter, D. Holschumacher, M. Freytag, C. G. Daniliuc, P. G. Jones and M. Tamm, Computational and experimental investigations of CO2 and N2O fixation by sterically demanding N-heterocyclic carbenes (NHC) and NHC/borane FLP systems, Dalton Trans., 2014, 43, 1651–1662 RSC; (b) E. L. Kolychev, T. Bannenberg, M. Freytag, C. G. Daniliuc, P. G. Jones and M. Tamm, Reactivity of a Frustrated Lewis Pair and Small-Molecule Activation by an Isolable Arduengo Carbene-B{3,5-(CF3)2C6H3}3 Complex, Chem. – Eur. J., 2012, 18, 16938–16946 CrossRef CAS PubMed.
- 1c was generated in situ by applying 1 atm of CO2 to IAd, before adding (BC6F5)3.
- (a) B. J. Knurr and J. M. Weber, Infrared Spectra and Structures of Anionic Complexes of Cobalt with Carbon Dioxide Ligands, J. Phys. Chem. A, 2014, 118, 4056–4062 CrossRef CAS PubMed; (b) G. Herzberg, Molecular spectra and molecular structure, R. E. Krieger Publishing Co., Malabar, FL, Malabar, Fla, 1991, vol. III, p. 598 Search PubMed.
- B. R. Van Ausdall, J. L. Glass, K. M. Wiggins, A. M. Aarif and J. Louie, A Systematic Investigation of Factors Influencing the Decarboxylation of Imidazolium Carboxylates, J. Org. Chem., 2009, 74, 7935–7942 CrossRef CAS PubMed.
- O. Back, M. Henry-Ellinger, C. D. Martin, D. Martin and G. Bertrand, 31P NMR Chemical Shifts of Carbene–Phosphinidene Adducts as an Indicator of the π-Accepting Properties of Carbenes, Angew. Chem., Int. Ed., 2013, 52, 2939–2943 CrossRef CAS PubMed.
- IboView – program for analysing molecular electronic structure, based on Intrinsic Atomic Orbitals (IAOs). http://www.iboview.org, G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834–4843.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2306774–2306777. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc06325a |
| This journal is © The Royal Society of Chemistry 2024 |