
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceShining light on tryptamine-derived isocyanides: access to constrained spirocylic scaffolds†
Minghui
Wu
 a,
Jordy M.
Saya
a,
Peiliang
Han
b,
Rajat
Walia
c,
Bapi
Pradhan
d,
Maarten
Honing
b,
Prabhat
Ranjan
*a and
Romano V. A.
Orru
*a
a,
Jordy M.
Saya
a,
Peiliang
Han
b,
Rajat
Walia
c,
Bapi
Pradhan
d,
Maarten
Honing
b,
Prabhat
Ranjan
*a and
Romano V. A.
Orru
*a
aAachen Maastricht Institute for Biobased Materials (AMIBM), Maastricht University, Urmonderbaan 22, 6167 RD Geleen, The Netherlands. E-mail: r.orru@maastrichtuniversity.nl; pranjan057@gmail.com
bMaastricht MultiModal Molecular Imaging Institute (M4i), Division of Imaging Mass Spectrometry, Maastricht University, Universiteitssingel 50, 6229 ER Maastricht, The Netherlands
cDepartment of Chemistry, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong SAR
dMolecular Imaging and Photonics, Department of Chemistry, KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium
First published on 8th April 2024
Abstract
Dearomatization of indoles through a charge transfer complex constitutes a powerful tool for synthesizing three-dimensional constrained structures. However, the implementation of this strategy for the dearomatization of tryptamine-derived isocyanides to generate spirocyclic scaffolds remains underdeveloped. In this work, we have demonstrated the ability of tryptamine-derived isocyanides to form aggregates at higher concentration, enabling a single electron transfer step to generate carbon-based-radical intermediates. Optical, HRMS and computational studies have elucidated key aspects associated with the photophysical properties of tryptamine-derived isocyanides. The developed protocol is operationally simple, robust and demonstrates a novel approach to generate conformationally constrained spirocyclic scaffolds, compounds with high demand in various fields, including drug discovery.
Introduction
Isocyanides are important and versatile building blocks in organic chemistry, well appreciated for their role in multicomponent reactions (MCRs; e.g. Passerini and Ugi reactions).1 Their distinctive ability to engage in the simultaneous α-addition of a potent electrophile and nucleophile has led to the development of numerous innovative isocyanide-based MCRs, resulting in the rapid generation of structurally diverse complex scaffolds. These one-pot synthetic sequences have found widespread application in combinatorial synthesis, medicinal chemistry and catalysis. Isocyanides also show unique reactivity in radical reactions, engaging efficiently with electron-rich as well as electron-deficient radical partners (Fig. 1A).2 This unique reactivity has been extensively utilized in radical cascade reactions, generating a diverse range of N-heterocyclic molecules.3 Recent developments in visible-light-mediated photochemistry have further broadened the use of isocyanides as geminal radical acceptors in organic synthesis,4 with a rising but still limited interest in applying their potent reactivity in charge transfer complexes.5,6 For instance, Wang and co-workers reported the formation of an EDA complex involving a biaryl isocyanide and in situ generated arylsulfinate anions leading to the formation of a sulfonyl radical and an imidoyl radical anion (Fig. 1B(i)).5 | ||
| Fig. 1 (a) Reactivity of isocyanides;2 (b) previous work;5,6 (c) this work: aggregation-induced charge transfer. | ||
Similarly, Giustiniano and co-workers reported the formation of an EDA complex between tertiary amines as electron donors and alkyl isocyanides as electron acceptors (Fig. 1B(ii)).6 The formation of an EDA-complex triggers the single-electron transfer (SET) from the tertiary amine to the isocyanide under visible light, leading to the generation of an imidoyl radical anion.
In light of these results and with the aim to extend this reactivity pattern to the synthesis of complex molecular scaffolds, we wondered if it could be possible to engage tryptamine-derived isocyanides in an intramolecular charge transfer complex or in the formation of an EDA complex with electron-deficient phenacyl bromides.7,8 The choice of tryptamine-derived isocyanides was dictated by their versatility. Containing an electron-rich indole ring and an electron-deficient isocyanide group, their reactivity has been often exploited in the generation of several naturally occurring and structurally complex alkaloids.9–11 The isocyanide group electronically resembles a terminal alkyne, with the additional benefit of higher polarizability of the π*-orbital of the isocyanide group.2 This feature could in principle favour an intramolecular charge transfer with the electron-rich indole ring in tryptamine-derived isocyanides.8 As a proof of concept, we decided to attempt the synthesis of spirocyclic scaffolds through indole dearomatization.
In recent years, spirocylic scaffolds have garnered tremendous attention in the field of medicinal chemistry due to their rigid three-dimensional structure, which can reduce the number of possible conformations, leading to enhanced potency and selectivity for target binding in drug discovery.12,13 In this respect, we envisioned the dearomatization of tryptamine-derived isocyanides as a straightforward way to generate these complex three-dimensional structures in a selective and efficient manner. In general, strategies to form spirocyclic scaffolds are based on metal catalysed insertion reactions14,15 or electrophilic activation of the isocyanide group9,16 followed by nucleophilic attack of the indole group, with scarce reports employing a radical approach.17–19 The EDA-mediated dearomatization of tryptamine-derived isocyanides has not been realized yet and despite its convenience, this kind of reactivity can be in principle hindered by the presence of an easily oxidizable indole ring that can lead to unwanted side products.
Results and discussion
We set out to probe our initial hypothesis as delineated above employing tryptamine-derived isocyanide 1a, phenacyl bromide 1b as a radical source and 2,6-lutidine as a base in dry DCE at ambient temperature. We were pleased to see that when the reaction mixture was irradiated with 2*40 W blue LEDs under nitrogen atmosphere, we obtained 70% of the desired spiroindolenine product 2 (see ESI, Table S1†). Cognizant of the role of the solvent in charge transfer complexes, a variety of polar and non-polar solvents were screened (see ESI, Table S1†) to understand if further improvement of the reaction performance could be achieved.20,21 Among the solvents tested, the use of toluene proved optimal compared to more polar solvents (Table 1, entry 1). We also evaluated the efficiency of transition metal-based as well as organic photocatalysts for this dearomative spirocyclization process (see ESI, Table S3†).|
|
||
|---|---|---|
| Entry | Deviations from the optimized conditions | Yieldb (%) |
| a All reactions were performed using 1a (0.2 mmol, 1 equiv.) and 1b (0.3 mmol, 1.5 equiv.). b Yields were determined by 1H NMR using CH2Br2 as an internal standard. c 2,4,5,6-Tetrakis(diphenylamino)isophthalonitrile (4DPAIPN) (5 mol%), 2 h. d Ir(ppy)3 (2 mol%), 2 h. e 12 h reaction time. | ||
| 1 | None | 82 |
| 2 | No light | 0 |
| 3 | No light, 50 °C | 0 |
| 4 | Air instead of N2 | 0 |
| 5c | 4DPAIPN, DCE | 67 |
| 6d | Ir(ppy)3, DCE | 72 |
| 7 | 10% H2O | 70 |
| 8e | 50% H2O | 64 |
| 9 | Et3N instead of 2,6 lutidine | 72 |
| 10 | Morpholine instead of 2,6 lutidine | 16 |
| 11 | No 2,6-lutidine | 6 |
Interestingly, in the presence of 4DPAIPN or Ir(ppy)3, 1b was fully consumed within 2 h, but the reaction resulted overall in a lower yield of spiroindolenine 2 (67% and 72% yield respectively), due to competitive reductive debromination of 1b (Table 1, entries 5 and 6).22 In light of a recent work that explores the possibility of forming EDA complexes in water,23 we performed the reaction in a mixture of water and toluene. To our delight, we observed 64% of the desired product after 12 h, without observing any concomitant side product generation, providing the possibility to exploit this methodology in the future in a more sustainable and biocompatible manner (Table 1, entries 7 and 8). Control reactions substantiated the necessity of light in the formation of spiroindolenine 2 and 2,6-lutidine as a base to prevent the decomposition of 1a (Table 1, entries 2–4 and 11).24
In light of these results, we started investigating the nature of the observed reactivity. In contrast to the reported literature where red colour appeared upon mixing electron-rich indoles and electron-deficient phenacyl bromides7 as an indication of ground state EDA complex formation, in our case such a colour change was not perceived upon mixing 1a and 1b.
To gain more insights and analyse the actual active photo-absorbing species in our reaction, we recorded the individual UV-Vis absorption spectra of the reaction components and the absorption spectrum of the reaction mixture itself (Fig. 2a). As shown in Fig. 2a, no significant bathochromic shift upon mixing 1a and 1b in toluene could be detected. On the other hand, the absorption of 1a in the visible region was at first surprising. From a structural point of view, there is no extended conjugation present in 1a, which can account for the large bathochromic shift of 1a compared to the parent indole (2-methyl indole, Fig. S6†). To explain this, we initially hypothesized the formation of an intramolecular charge transfer complex between the electron-rich indole ring and the tethered isocyanide group via through space or through bond interactions.25–29
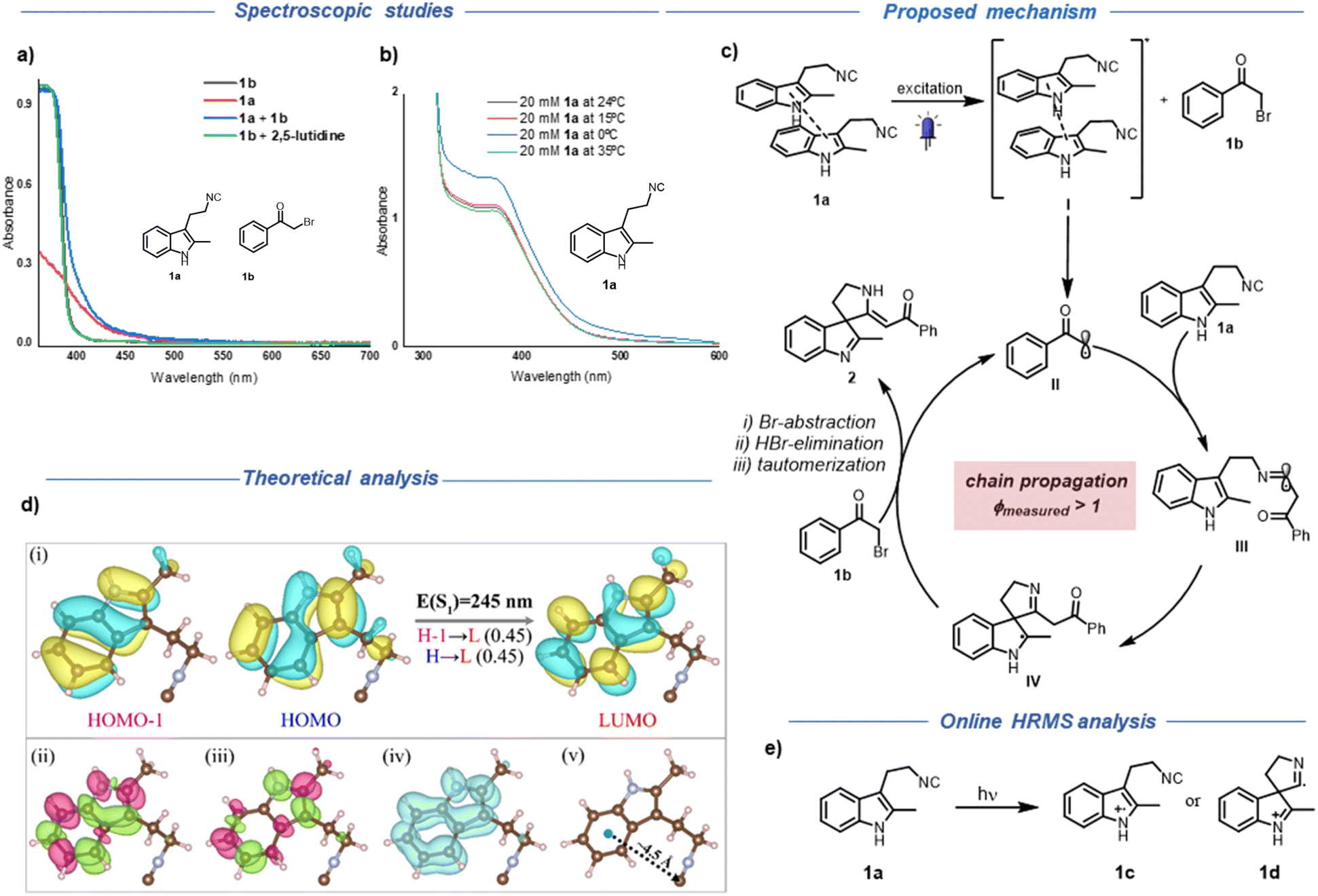 | ||
| Fig. 2 (a) Absorption spectra of individual components and reaction mixture, recorded in toluene: [1b] = 0.15 M; [1a] = 0.1 M; [1a+1b] = solution of 0.15 mmol of 1b and 0.1 mmol of 1a in toluene (0.1 M); [1b+2,6-lutidine] = solution of 0.1 mmol of 1b and 0.13 mmol of 2,6-lutidine in toluene (0.1 M); (b) absorption spectra of 1a (20 mM) in MeCN at different temperatures; (c) proposed reaction mechanism; (d) (i) the composition of S1 state with the CI coefficients for an orbital-pair given in bracket; (ii) transition density; (iii) difference density and (iv) electron–hole overlap distribution of S1 state at wb97x-d/cc-pVTZ level of theory (v) distance between isonitrile group and the indole ring in the ground-state equilibrium structure of 1a; (e) on-line HRMS analysis. | ||
To analyse the possibility of intramolecular charge transfer, time-dependent density functional theory (TD-DFT) calculations were performed.30 The calculations were carried out at the wb97x-d/cc-pVTZ level of theory using ORCA package.31 The wave-function analysis revealed that the first excited singlet state (S1) is constructed by equal contributions of HOMO−1→LUMO and HOMO→LUMO configurations as shown in Fig. 2d(i). Interestingly, these three orbitals are primarily located on the indole fragment, thus ruling out the possibility of intramolecular charge-transfer in the S1 state.8 To further substantiate this observation, we visualised the transition density, difference density and electron–hole overlap of S1 state, as shown in Fig. 2d(ii)–(iv). It is evident that the electronic transition is originated only from the indole moiety. Moreover, our DFT calculations revealed a significant distance of 4.50 Å between the isonitrile group and the indole ring in the ground-state equilibrium structure of 1a, surpassing distances commonly associated with through-space CT as reported in literature (Fig. 2d(v)).25,27 Next, we measured the absorption spectra of a mixture of 2-methyl indole and butyl isocyanide and we did not observe any bathochromic shift.8 However, a mixture of 3-methyl indole and tert-butyl isocyanide showed a slight bathochromic shift at higher concentration but still no appreciable absorption in the visible region compared to 1a (Fig. S6b†).
In light of these findings and since the absorption peak of 1a in the visible region only appears at higher concentrations (Fig. S7†), we suspected the formation of aggregates. We explored a slip-stacked model-dimer system with an interdimer separation of ∼3.50 Å (see ESI, Fig. S8†).32,33 As expected, we found a bathochromic shift in the absorption of the dimer [E(S1) = 256 nm] compared to the monomer [E(S1) = 245 nm], that should be more prominent in the case of higher-order oligomers. These effects are more enhanced in a similarly constructed model-trimer [E(S1) = 263 nm] (see ESI, Table S9†). A similar TD-DFT based wave-function analysis suggests the presence of inter-fragment charge-transfer in the S1 state of this model dimer, while the excitation remains localised on indole moieties in both fragments (see ESI, Fig. S9†). More details of TD-DFT calculations are provided in Section 5 of ESI.† As a further prove to the aggregation dependent absorption, UV-Vis absorption spectra of 1a at various temperatures showed an increment in the absorption intensity of 1a at 0 °C in comparison with room temperature (Fig. 2b).34
After having identified the main photo-absorbing species under our reaction conditions, we turned our attention to delineate how radical generation from 1b happens. Based on the photophysical properties of 1a, we speculated that the SET could be initiated from the excited state of 1a (see Section 6 of ESI† for more information). Despite the short lifetime, we assumed that the pre-association between the excited state of 1a and 1b could facilitate the electron transfer to generate the radical from 1b.35 Next, we investigated the presence of a radical chain mechanism, since the α-amino radical (intermediate IV, Fig. 2c) can undergo bromine abstraction leading to a chain propagation pathway.18,36,37 As expected and similar to the reported literature, a quantum yield measurement (ϕmeasured > 1) showed that a radical chain process is operative under our reaction conditions.18,36,37 At this stage we decided to analyse the charge transfer in isocyanide 1a through online HRMS since this type of charge transfer can generate an indole radical cation intermediate. Unfortunately, we could not observe such intermediate, possibly due to a fast back electron transfer, which would lead back to the neutral molecule. In order to confirm this hypothesis, we introduced 4DPAIPN, that can oxidize the indole unit of isocyanide 1a.38 As expected, we observed a decrease in the peak intensity of the starting material 1a and an increase in the intensity of a peak with m/z 184 by 40 times (Fig. S12†). The structure of this intermediate can be either 1c or 1d (Fig. 2e). However, at this stage we cannot assign the exact structure only based on the MS analysis. In addition, a peak of m/z 367 also became apparent. This could be assigned to a dimer derived from intermediate 1c or 1d (Fig. S12†). The enhancement of the side product with m/z 367 may be due to the consumption of the key intermediate with m/z 184. We also observed the peak with m/z 367 through LC-MS during our optimization studies, supporting that there is a possibility of the formation of a similar intermediate under our reaction conditions through aggregation based charge transfer.39
Taking into account all these observations, we propose the following mechanism for the dearomative spirocyclization of tryptamine derived isocyanides (Fig. 2c). When subjected to excitation with blue LEDs, 1a undergoes charge transfer to form intermediate I, which further interacts with the ground state of 1b, facilitating the pivotal single electron transfer to generate the transient radical intermediate II. The ensuing phenacyl radical attack on isocyanide 1a generates the persistent imidoyl radical intermediate III, which then undergoes a rapid dearomatization process to generate the spirocyclic intermediate IV. At this point, an intriguing radical chain process can be operative, where intermediate IV can abstract the bromine atom from 1b, effectively setting in motion a chain process. At last, the elimination of hydrogen bromide followed by tautomerization generates the final product 2 (for alternative mechanistic pathway and further discussion see Section 8 of ESI†).
Having established the optimal reaction conditions and mechanistic rationale, the scope and limitations of our photocatalyzed dearomative spirocylization were established (Scheme 1). Pleasingly, the optimized conditions provided the desired products with a wide variety of electron-poor and electron-rich phenacyl bromides (Scheme 1, 2–12). We observed higher yield of the desired product in the case of phenacyl bromide-bearing electron-withdrawing groups as compared to electron-donating groups. Surprisingly, the phenacyl bromide-bearing a nitro group led to depreciated yield of the desired product (Scheme 1, 6). In this particular case, we observed the appearance of a bright red colour upon mixing 1a and 2-bromo-1-(4-nitrophenyl)ethan-1-one, an indication of ground state EDA-formation. This result indicates that strong ground state EDA-complex formation can lead to lower yield presumably due to back electron transfer from the phenacyl bromide radical anion intermediate to the isocyanide 1a. Moreover, the higher yield obtained when adding a photocatalyst (outer-sphere electron transfer) also abets this hypothesis.20 Next, we employed aliphatic bromides under our optimized reaction conditions to push the boundaries of this reactivity. To our delight, we observed desired product formation, albeit in lower yield (Scheme 1, 13–14). Alkyl partners such as dimethyl 2-bromomalonate, which cannot form a preassociated complex with the excited state of 1a, gave lower yield (44%) with 84% conversion of isocyanide 1a.40 This result could be caused by inefficient bimolecular single electron transfer due to the short lifetime of the excited state of 1a. To further show the versatility of the developed protocol, we employed diaryl iodonium, trifluoromethyl thianthrenium triflate and pyridinium salts under our optimized conditions (see also ESI, Tables S4–S6†). Pleasingly, we observed the formation of the desired product in good to moderate yields (Scheme 1, 15–19). We next turned our attention to delineate the scope of isocyanides.
 | ||
| Scheme 1 Scope of spiroindolenines: aconditions unless otherwise noted: all reactions were performed using isocyanide (0.2 mmol, 1 equiv.), bromide (0.3 mmol, 1.5 equiv.), 2,6-lutidine (2 equiv.), dry toluene (0.1 M), 6 h–12 h, irradiating with Kessil lamp 456 nm (2*40W). b1a (0.3 mmol, 1.5 equiv.) and bromide (0.2 mmol, 1 equiv.), 4DPAIN (5 mol%), dry DCE (0.1 M), 2 h, irradiating with Kessil lamp 456 nm (2*40W). c 48 h. d1a (0.2 mmol) and CF3 source (0.3 mmol, 1.5 equiv.). e1a (0.2 mmol) and diaryl iodonium salt (0.3 mmol, 1.5 equiv.). f1a (0.2 mmol) and pyridinium salt (0.3 mmol, 1.5 equiv.), morpholine (0.4 mmol, 2 equiv.), 48 h. | ||
Halogen-substituted tryptamine-derived isocyanides smoothly underwent the dearomative spirocyalization, delivering the desired product in good yields (Scheme 1, 20–23). A variety of substituents at the C-2 position of tryptamine-derived isocyanides such as –Ph, -tBu and p-FPh also delivered the desired product in high yield, except for the Br-substituted isocyanide at the C-2 position (Scheme 1, 24–28). In this particular case, we observed the decomposition of the isocyanide. Moreover, 3-(2-isocyanobenzyl)-indole was also reactive under our optimized conditions and delivered the 6-membered spiro ring in high yield (Scheme 1, 29).41 At last, we further compared the efficiency of our optimized reaction conditions with a photocatalytic pathway. With most of the substrates, the addition of a photocatalyst resulted in lower yield (Scheme S3†).
Conclusions
In conclusion, we have developed a novel catalyst-free dearomatization strategy to generate constrained spiroindolenines under mild reaction conditions. The ability of tryptamine-derived isocyanides to generate radical intermediates through an aggregation-based charge transfer was presented for the first time. The presented methodology involves a radical cascade reaction with the formation of two concomitant C–C bonds without the need for any type of catalyst, further streamlining the synthetic process. Due to the generation of closed-shell radical intermediates, the developed protocol showed higher selectivity and efficiency than the photocatalytic pathway. The on-line HRMS analysis further shed a light on the possible radical intermediates involved in the synthesis of the desired spiroindolenines.Data availability
Detailed synthetic procedures, complete characterization and spectroscopic data can be found in the ESI.† Raw spectroscopic and NMR data are available from the authors upon request.Author contributions
The authors confirm contribution to the paper as follows: study conception: P. R.; design and methodology: M. W., J. M. S., R. V. A. O. and P. R.; experimentation: M. W. and P. R.; theoretical studies: R. W.; fluorescence lifetime measurement: B. P.; on-line MS and HRMS analysis: P. H. and M. H.; draft manuscript preparation: P. R., M. W., J. M. S. and R. V. A. O.; supervision: R. V. A. O.; acquiring funding: P. R. and R. V. A. O.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union’s Horizon 2021 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 101063688 (project name: SusIsocyanide). This publication reflects only the author's view, exempting the Community from any liability. Neither the European Union nor the granting authority can be held responsible for them. P. R. is thankful to the Marie Skłodowska-Curie Postdoctoral Fellowship (Grant Agreement No. 101063688). M. W. is thankful to China Scholarship Council (No. 202207720033) for providing a doctoral scholarship. R. W. wishes to thank the Computer Service Centre (CSC) at the City University of Hong Kong for its High-Performance Computing (HPC) facilities. P. R. is thankful to Dr Rishabh Saxena (Soft Matter Optoelectronics, University of Bayreuth, Germany) and Serena Pillitteri (KU Leuven, Belgium) for their valuable suggestions. P. R. is also thankful to Prof. Burkhard König for allowing to perform UV-Vis and fluorescence measurements at the University of Regensburg.References
- E. Ruijter, R. Scheffelaar and R. V. A. Orru, Angew. Chem., Int. Ed., 2011, 50, 6234–6246 CrossRef CAS PubMed.
- G. dos P. Gomes, Y. Loginova, S. Z. Vatsadze and I. V. Alabugin, J. Am. Chem. Soc., 2018, 140, 14272–14288 CrossRef CAS PubMed.
- S. Sharma, A. P. Pandey and A. Sharma, Adv. Synth. Catal., 2020, 362, 5196–5218 CrossRef CAS.
- C. Russo, F. Brunelli, G. Cesare Tron and M. Giustiniano, Chem.–Eur. J., 2023, 29, e202203150 CrossRef CAS PubMed.
- Y. Li, T. Miao, P. Li and L. Wang, Org. Lett., 2018, 20, 1735–1739 CrossRef CAS PubMed.
- C. Russo, J. Amato, G. C. Tron and M. Giustiniano, J. Org. Chem., 2021, 86, 18117–18127 CrossRef CAS PubMed.
- S. R. Kandukuri, A. Bahamonde, I. Chatterjee, I. D. Jurberg, E. C. Escudero-Adán and P. Melchiorre, Angew. Chem., Int. Ed., 2015, 54, 1485–1489 CrossRef CAS PubMed.
- H. E. Ho, A. Pagano, J. A. Rossi-Ashton, J. R. Donald, R. G. Epton, J. C. Churchill, M. J. James, P. O'Brien, R. J. K. Taylor and W. P. Unsworth, Chem. Sci., 2020, 11, 1353–1360 RSC.
- J. M. Saya, B. Oppelaar, R. C. Cioc, G. van der Heijden, C. M. L. Vande Velde, R. V. A. Orru and E. Ruijter, Chem. Commun., 2016, 52, 12482–12485 RSC.
- L.-R. Yuan, S.-J. Ji and X.-P. Xu, Org. Lett., 2023, 25, 7858–7862 CrossRef CAS PubMed.
- J. M. Saya, E. Ruijter and R. V. A. Orru, Chem.–Eur. J., 2019, 25, 8916–8935 CrossRef CAS PubMed.
- K. Hiesinger, D. Dar'in, E. Proschak and M. Krasavin, J. Med. Chem., 2021, 64, 150–183 CrossRef CAS PubMed.
- A. Sveiczer, A. J. P. North, N. Mateu, S. L. Kidd, H. F. Sore and D. R. Spring, Org. Lett., 2019, 21, 4600–4604 CrossRef CAS PubMed.
- T. R. Roose, H. D. Preschel, H. Mayo Tejedor, J. C. Roozee, T. A. Hamlin, B. U. W. Maes, E. Ruijter and R. V. A. Orru, Chem.–Eur. J., 2023, 29, e202203074 CrossRef CAS PubMed.
- G. Chen, S. Chen, J. Luo, X. Mao, A. S. Chan, R. W. Sun and Y. Liu, Angew. Chem., Int. Ed., 2020, 59, 614–621 CrossRef CAS PubMed.
- J. M. Saya, T. R. Roose, J. J. Peek, B. Weijers, T. J. S. de Waal, C. M. L. Vande Velde, R. V. A. Orru and E. Ruijter, Angew. Chem., Int. Ed., 2018, 57, 15232–15236 CrossRef CAS PubMed.
- S. Jiang, Y. X. Huang, X. F. Wang, X. P. Xu and S. J. Ji, Org. Chem. Front., 2023, 10, 1660–1668 RSC.
- N. Inprung, H. E. Ho, J. A. Rossi-Ashton, R. G. Epton, A. C. Whitwood, J. M. Lynam, R. J. K. Taylor, M. J. James and W. P. Unsworth, Org. Lett., 2022, 24, 668–674 CrossRef CAS PubMed.
- M. Zhu, K. Zhou, X. Zhang and S.-L. You, Org. Lett., 2018, 20, 4379–4383 CrossRef CAS PubMed.
- C. G. S. Lima, T. de M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389–1407 CrossRef CAS.
- T. Mori and Y. Inoue, Chem. Soc. Rev., 2013, 42, 8122 RSC.
- M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951–954 CrossRef CAS PubMed.
- Y. Tian, E. Hofmann, W. Silva, X. Pu, D. Touraud, R. M. Gschwind, W. Kunz and B. König, Angew. Chem., Int. Ed., 2023, 62, e202218775 CrossRef CAS PubMed.
- P. Patil, Q. Zheng, K. Kurpiewska and A. Dömling, Nat. Commun., 2023, 14, 5807 CrossRef CAS PubMed.
- Z.-Y. Cao, T. Ghosh and P. Melchiorre, Nat. Commun., 2018, 9, 3274 CrossRef PubMed.
- J. W. Verhoeven, Pure Appl. Chem., 1986, 58, 1285–1290 CrossRef CAS.
- T. Ito, E. Nishiuchi, G. Fukuhara, Y. Inoue and T. Mori, Photochem. Photobiol. Sci., 2011, 10, 1405–1414 CrossRef CAS PubMed.
- H. E. Ho, A. Pagano, J. A. Rossi-Ashton, J. R. Donald, R. G. Epton, J. C. Churchill, M. J. James, P. O'Brien, R. J. K. Taylor and W. P. Unsworth, Chem. Sci., 2020, 11, 1353–1360 RSC.
- S. Kumar, L. G. Franca, K. Stavrou, E. Crovini, D. B. Cordes, A. M. Z. Slawin, A. P. Monkman and E. Zysman-Colman, J. Phys. Chem. Lett., 2021, 12, 2820–2830 CrossRef CAS PubMed.
- L. J. C. van der Zee, S. Pahar, E. Richards, R. L. Melen and J. C. Slootweg, Chem. Rev., 2023, 123, 9653–9675 CrossRef CAS PubMed.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- W. Wang, X. Sun, J. Qu, X. Xie, Z. H. Qi, D. Hong, S. Jing, D. Zheng, Y. Tian, H. Ma, S. Yu and J. Ma, Phys. Chem. Chem. Phys., 2017, 19, 31443–31451 RSC.
- R. Walia, Z. Deng and J. Yang, Chem. Sci., 2021, 12, 12928–12938 RSC.
- X. Yang, X. Xu and H.-F. Ji, J. Phys. Chem. B, 2008, 112, 7196–7202 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- A. Bahamonde and P. Melchiorre, J. Am. Chem. Soc., 2016, 138, 8019–8030 CrossRef CAS PubMed.
- R. K. Neff, Y.-L. Su, S. Liu, M. Rosado, X. Zhang and M. P. Doyle, J. Am. Chem. Soc., 2019, 141, 16643–16650 CrossRef CAS PubMed.
- X. Liu, D. Yang, Z. Liu, Y. Wang, Y. Liu, S. Wang, P. Wang, H. Cong, Y.-H. Chen, L. Lu, X. Qi, H. Yi and A. Lei, J. Am. Chem. Soc., 2023, 145, 3175–3186 CrossRef CAS PubMed.
- In order to confirm the structure of 367, we tried to run the reaction without 1b to favor the homocoupling product. However, we did not see any peak corresponding to 367. This peak only appeared in LC-MS when we had an electron acceptor molecule, either 1b or TEMPO in the reaction mixture.
- E. Arceo, E. Montroni and P. Melchiorre, Angew. Chem., Int. Ed., 2014, 53, 12064–12068 CrossRef CAS PubMed.
- J. Zhang, W. Xu, Y. Qu, Y. Liu, Y. Li, H. Song and Q. Wang, Chem. Commun., 2020, 56, 15212–15215 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06304f |
| This journal is © The Royal Society of Chemistry 2024 |