
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBorylative transition metal-free couplings of vinyl iodides with various nucleophiles, alkenes or alkynes†
Gesa
Seidler‡
 ,
Max
Schwenzer‡
,
Florian
Clausen
,
Constantin G.
Daniliuc
and
Armido
Studer
*
,
Max
Schwenzer‡
,
Florian
Clausen
,
Constantin G.
Daniliuc
and
Armido
Studer
*
Organisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, Corrensstraße 40, 48149 Munster, Germany. E-mail: studer@uni-muenster.de
First published on 29th December 2023
Abstract
Alkyl boronic esters are highly valuable compounds in organic chemistry and related fields due to their good stability and highly versatile reactivity. In this edge article, stereoselective borylative couplings of vinyl iodides with various nucleophiles, alkenes or alkynes is reported. These coupling reactions proceed through stereospecific hydroboration and subsequent stereospecific 1,2-metallate rearrangement. The cascades utilize readily available reagents and proceed without the need of a transition metal catalyst.
Introduction
Alkyl and aryl boronic esters are highly valuable building blocks in organic synthesis due to their stability and versatile reactivity.1 Various reactions have been developed that allow the transformation of the boronic ester moiety into a variety of other functional groups, in many cases in a stereoselective manner.2–6 Moreover, boronates can be protodeboronated7–10 or be used as nucleophiles in C–C bond forming reactions, for example in the highly valuable Suzuki–Miyaura cross coupling.11–13 In addition, boron containing organic compounds can also be found in medicinal chemistry or materials science.1,14 As a result, organic chemists are greatly interested in advancing novel reactions that facilitate the preparation of boronic esters or their chemical modification while preserving the valuable boron containing moiety.5,6,15 In this regard, hydroboration reactions16,17 and 1,2-metallate rearrangements such as the Matteson reaction18–20 have been established. Both reactions are highly stereospecific with the hydroboration proceeding through a concerted syn-addition of a B–H compound to a double bond16 and the Matteson reaction comprising the rearrangement of a readily generated boron ate complex resulting in the substitution of an anionic leaving group in the α-position of an alkyl boronic ester (Scheme 1A). This substitution occurs with inversion of the stereogenic center next to the boron atom.20,21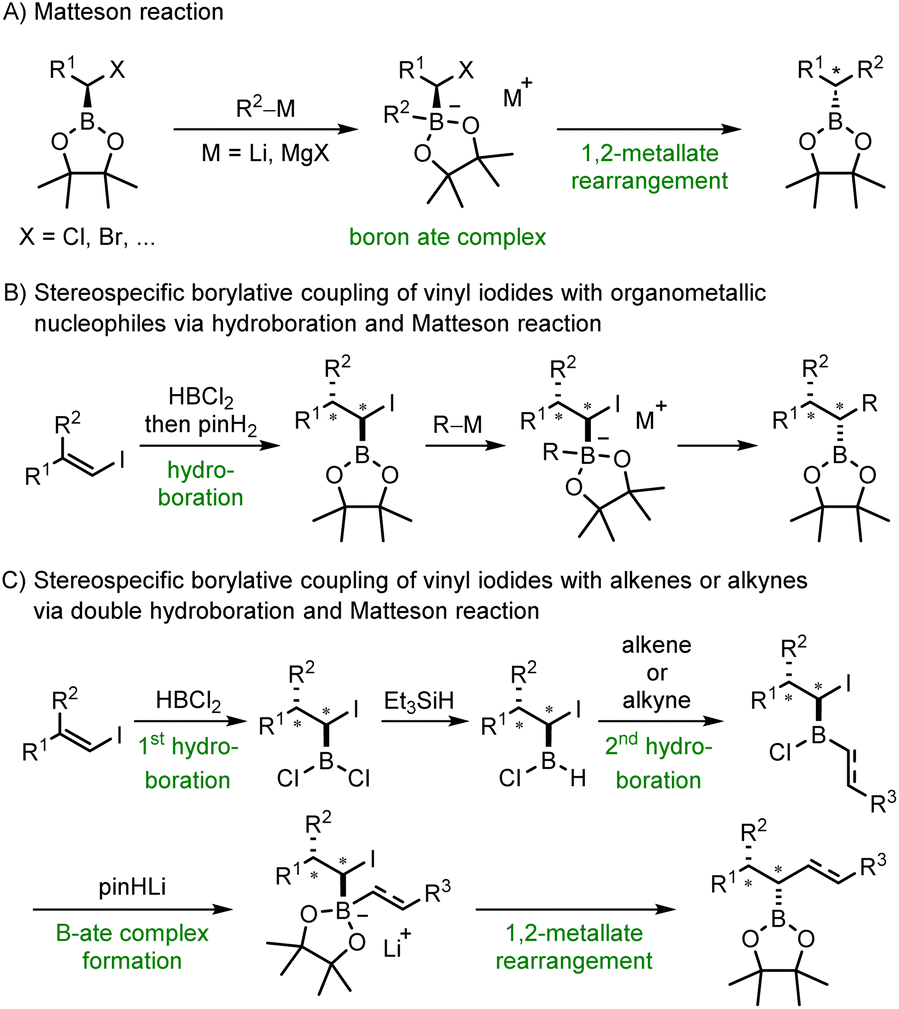 | ||
| Scheme 1 Alkyl boronic esters via 1,2-metallate rearrangement of boron ate complexes – Matteson reaction and beyond. | ||
We opted to combine these two highly valuable reactions with the aim of devising a practical procedure that facilitates the direct generation of substituted alkyl boronic esters from vinyl iodides and organometallic nucleophiles (Scheme 1B). Initial regioselective hydroboration of an alkenyl iodide with HBCl2 and subsequent treatment with pinacol (pinH2) should lead to the corresponding α-iodoalkyl pinacol boronic ester. Coupling with an organometallic reagent would provide through its boron ate complex the targeted alkyl boronic ester with complete diastereoselectivity. HBCl2 was chosen as the hydroboration reagent due to its increased reactivity.
Further, HBCl2 unveils an additional reaction pathway subsequent to the initial hydroboration. Instead of esterifying the intermediate α-iodoalkyldichloroborane with pinacol, we also intended to selectively mono-reduce this dichloroborane. This reduction would yield an alkylchloroborane, primed for a subsequent hydroboration reaction with a second alkene or alkyne (Scheme 1C). Boron ate complex formation should then be achieved by simple treatment with deprotonated pinacol and a 1,2-metallate rearrangement should deliver the coupling product with high diastereoselectivity, retaining the valuable boron moiety in the final compound. To our knowledge, boron ate complex formation using such a mild strategy not requiring a reactive organometallic species has not been reported to date.22 Of note, new C–R, C–B and C–H bonds are formed in both sequences and two stereocenters are constructed. The high diastereoselectivity is inherited from the stereospecifity both of the hydroboration and the subsequent 1,2-metallate rearrangement step. The relative configuration of the new stereocenters will be set by the alkene geometry of the starting vinyl iodide, which is directly accessible from a ketone or an alkyne. Both reductive coupling reactions proceed in the absence of any transition metal catalyst, utilize readily available reagents and can be conducted in one-pot only requiring a single purification step at the very end of the sequence.
Results and discussion
We commenced the investigations by first addressing the stepwise coupling of vinyl iodides with organometallic compounds (see Scheme 1B). The hydroboration of halogen substituted alkenes has turned out to be challenging for organic chemists in the past and is scarcely reported. Often, a dehalogenated product is obtained23,24 or the α-halogen bearing targeted boron compounds readily decompose.25 An additional challenge is the regioselectivity of the hydroboration reaction. To our knowledge, the only practical hydroboration procedure for haloalkenes was reported in 1994 by Deadman utilizing catecholborane and proceeding either in the presence of a rhodium catalyst or catalyst-free at high temperatures.26,27 However, these rather inconvenient conditions seemed unsuitable for our envisioned cascades, in particular considering the borylative vinyl iodide/alkene coupling proposed in Scheme 1C. As discussed above, HBCl2 which can be readily formed in situ from boron trichloride and triethylsilane seemed to be a promising alternative.28 The vinyl iodide 1a directly accessible from acetophenone via boron-Wittig reaction/iodinolysis29,30 (one-pot, see ESI†) was selected as the test substrate for our initial experiments (Scheme 2).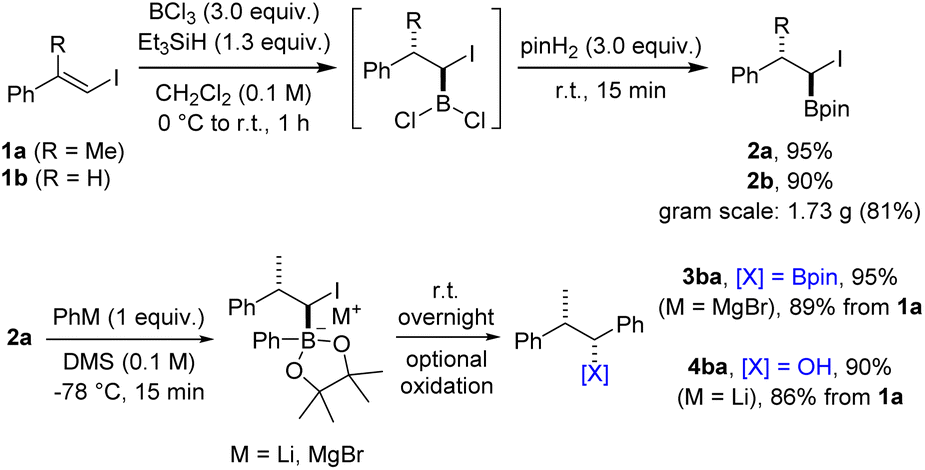 | ||
| Scheme 2 Optimization of the hydroboration and 1,2-metallate rearrangement. | ||
Hydroboration with HBCl2 proceeded smoothly in CH2Cl2 (0 °C, 30 min to room temperature for 30 min or several hours at −78 °C). The resulting dichloroalkylborane was then directly treated with pinacol to provide the corresponding α-iodoboronic ester 2a, which was isolated in 95% yield by distillation. The structure and relative configuration of an analogue of 2a was confirmed by single-crystal X-ray diffraction analysis (see ESI†).§ It should be mentioned that the order of addition of the hydroboration components is essential for the success of the transformation. BCl3 needs to be placed in the reaction flask first followed by addition of a solution containing iodoalkene and triethylsilane. Of note, hydroboration of vinyl chlorides and bromides under these conditions did not work well. While the reaction with the chloride or bromide analogues of vinyl iodide 1a did not lead to any product formation, a modest yield was obtained at full conversion using β-bromostyrene. In contrast, hydroboration of the iodoalkene 1b proceeded well (2b, 90%) even on gram-scale (81%), demonstrating the robustness of the protocol.
Typically, 1,2-metallate rearrangements involving α-halo boronic esters are accomplished by initiating the formation of the boron ate complex at low temperatures. Subsequently, the reaction mixture is allowed to gradually reach room temperature, promoting the 1,2-migration process. In some cases, this is conducted in the presence of a Lewis acid.31 Et2O and THF are the typically employed solvents. α-Iodoboronate 2a was selected as the test substrate in combination with PhLi as the coupling partner. In THF or Et2O, the targeted 3ba was formed as a minor product and side reactions were observed, which we attribute to a lithium iodine exchange on substrate 2a and subsequent coupling of the lithiated boronate with unreacted starting material 2a (see ESI†). However, using dimethyl sulfide (DMS) as the solvent, the undesired side reaction could be fully suppressed and the boronic ester 3ba could be obtained in high efficiency. Due to observed instability of 3ba during purification on silica gel, the boronic ester was oxidized to give the benzylic alcohol 4ba in 90% isolated yield with excellent diastereoselectivity. The same sequence was also realized with PhMgBr, and the benzylic boronic ester 3ba could be isolated in 95% yield. Subsequent investigations revealed that the reaction with PhMgBr is also compatible with THF or Et2O as solvents. Additionally, benzylic boronic esters can be more conveniently isolated through chromatography on NaOAc-deactivated silica gel, resulting in only negligible losses in yield (see ESI†). Driven by the consistently high yields achieved at each stage, we considered carrying out the transformation as a one-pot process. For this variation, isolation of intermediate 2 is replaced by a solvent exchange and an excess of the organometallic reagent is used in the following rearrangement step. Preliminary outcomes of this one-pot procedure were found to be quite satisfying with a yield of 87% for boronic ester 3ba. We consider the one-pot process to be particularly advantageous in scenarios where the organometallic reagent is inexpensive or when rapid access to the boronic ester is desired. On the other hand, the concomitantly developed and scalable two-step sequence might be most suitable for diversification originating from a single parent vinyl iodide or for reactions that are sensitive to an excess of reagent.
The transformation of vinyl iodide 1a into various substituted boronic esters was subsequently examined using a range of diversely hybridized nucleophiles, predominantly carbon-centered (Scheme 3). Alkyl Grignard reagents are tolerated, and reaction with MeMgBr in Et2O yielded the alkyl boronic ester 3aa in 76% yield. The corresponding alcohol 4aa could be isolated in 72% yield by the same one-pot approach, appended with an additional oxidation step. Similar yields were noted for the ethyl, benzyl, cyclopropyl or cyclobutyl Grignard reagents (3ab–3ae). Treatment with AllylMgBr allowed the synthesis of the homoallylic boronic ester 3af (82%).
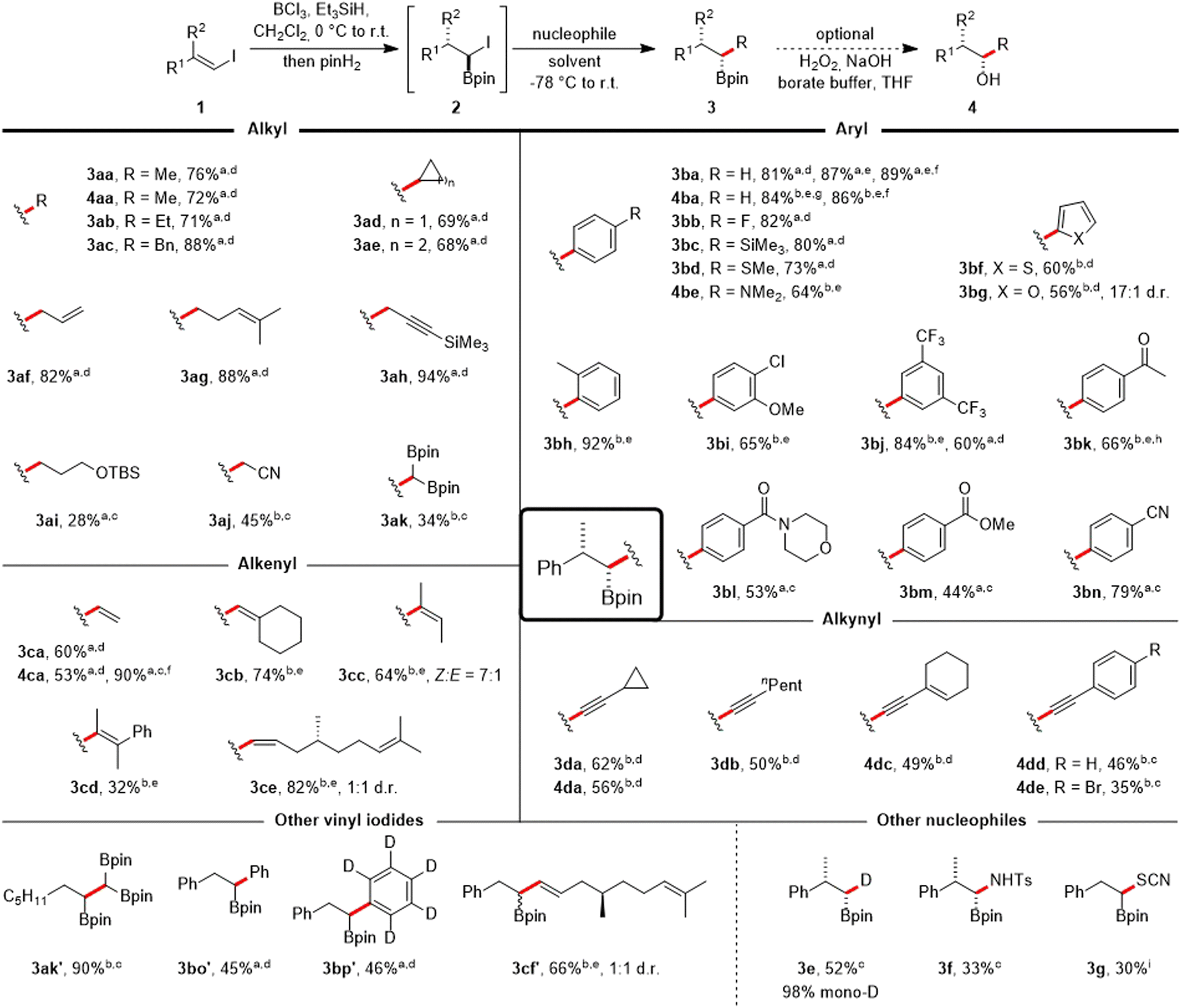 | ||
Scheme 3 Scope of the one-pot coupling of vinyl iodides with organometallic compounds. All reactions were run on a 0.25 mmol scale using 1.1 equiv. of BCl3, Et3SiH and pinH2 respectively in CH2Cl2 (0.1 M) at 0 °C (30 min) to r.t. (30 min). For information on the nucleophile stoichiometry (usually 1.5–3.0 equiv.) and reaction times of the Matteson step (usually 1 h) see ESI.† Products were obtained in diastereomeric ratios > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 if not noted otherwise. aGrignard reagent used. bOrganolithium reagent used. cTHF as solvent. dEt2O as solvent. eDMS as solvent. fOverall yield when running the sequence stepwise with isolation of the α-iodoboronic ester 2a. g1.0 equiv. BCl3, pinH2 and Et3SiH. hHydrolysis with HCl in H2O/THF after the reaction. iNucleophile = KSCN (2.0 equiv.), DMF, 60 °C, 4 h. :1 if not noted otherwise. aGrignard reagent used. bOrganolithium reagent used. cTHF as solvent. dEt2O as solvent. eDMS as solvent. fOverall yield when running the sequence stepwise with isolation of the α-iodoboronic ester 2a. g1.0 equiv. BCl3, pinH2 and Et3SiH. hHydrolysis with HCl in H2O/THF after the reaction. iNucleophile = KSCN (2.0 equiv.), DMF, 60 °C, 4 h. | ||
Homoallylic and protected propargylic Grignard reagents were also well tolerated, providing the boronic esters 3ag (88%) and 3ah (94%, see ESI† for its X-ray structure).§ We also investigated the incorporation of more complex alkyl groups.
Consequently, the boronic ester 3ai, which contains a silyl ether group, was produced. Additionally, the β-cyano boronic ester 3aj and the 1,1,2-tris(boronate) 3ak were synthesized from stabilized alkyl lithium reagents, achieving moderate yields when using THF as the solvent.
Aryl Grignard compounds with F, SiMe3 (see ESI† for its X-ray structure),§ SMe and NMe2 substituents in the para position are suitable reagents to afford the benzylic boronic esters 3bb–3be in 64–82% yields. Lithiated heteroarenes derived from thiophene and furan provided the boronic esters 3bf and 3bg in 60% and 56% yield, respectively. The sterically more hindered ortho-methyl phenyllithium worked very well to give 3bh in excellent 92% yield. Likewise, a meta-anisole moiety could be introduced (3bi, 65%). Aryllithium reagents bearing CF3-substituents also worked, as evidenced by the synthesis of 3bj with an 84% yield in just one hour. In contrast, using the corresponding ArMgI species necessitated a prolonged reaction time (overnight) and more reagent (3.0 equiv.) giving a lower yield of 60%. Subsequently, we examined the applicability of the protocol to coupling reactions involving Grignard reagents that carry more delicate and synthetically valuable functional groups, such as carbonyls. A ketal may be used as a protecting group for this purpose and acetophenone 3bk (66%) could be isolated after deprotection. Certain carbonyl containing aryl Grignard reagents can be formed at reduced temperatures through the reaction of the corresponding aryl iodides with iPrMgBr,32 which allowed their use for the synthesis of the morpholine benzamide 3bl (53%). Along these lines, the methyl benzoate 3bm (44%, see ESI† for its X-ray structure)§ and the benzonitrile 3bn (79%) were successfully prepared.
The utilization of vinyl magnesium bromide enabled the preparation of allylboronic ester 3ca (60%) and the corresponding allylic alcohol 4ca (53%). Running the cascade in two steps with a stoichiometric amount of the Grignard reagent improved the overall yield for alcohol 4ca to 90%. Vinyllithium reagents, readily generated from vinyl halides, could also be employed to the coupling of vinyl iodide 1a in DMS, providing trisubstituted (3cb, 74%; 3cc, 64%, E:Z = 1:7) and even tetrasubstituted (3cd, 32%) allylboronic esters,33 which are commonly employed in the allylboration of carbonyls34 or in radical chemistry.35,36 Lastly, a more complex scaffold derived from (+)-citronellal could be introduced in good yield (3ce, 82%).
The coupling could also be achieved with alkynyllithium reagents (in Et2O or THF) that are relatively less basic, enabling the isolation of propargylic boronic esters (3da, 62%; 3db, 50%), and an enyne could be coupled (4dc, 49%). Phenylacetylene and (p-bromophenyl)acetylene yielded the propargylic alcohols 4dd (46%) and 4de (35%).
Next, a brief exploration of the vinyl iodide coupling component was undertaken. Vinyl iodides derived from aldehydes took part in the cascade reaction. This resulted in the preparation of 1,1,2-tris(boronate) 3ak′ (90%), along with α-arylated boronic esters 3bo′ (45%) and 3bp′ (46%). Additionally, the incorporation of a scaffold originating from (+)-citronellal was accomplished while retaining the E-geometry from the vinyl iodide 3cf′ (66%).
The viability of incorporating non-carbon-based nucleophiles into the protocol was investigated in short. Notably, selective introduction of deuterium was achieved using LiDBEt3,37 resulting in the formation of α-monodeuterated alkyl boronic ester 3e with a 52% yield and an excellent degree of monodeuteration (98%). Coupling with the N-centered nucleophile LiHMDS, followed by derivatization with TsCl,38 enabled the isolation of sulfonamide-protected aminoboronic acid 3f in 33% yield. Even the heteroatom-centered, weakly nucleophilic potassium thiocyanate39 was applicable under more rigorous reaction conditions, yielding the α-borylated thiocyanate 3g.
Our investigations were then continued by addressing the challenging transition metal-free stereoselective borylative vinyl iodide/alkene coupling sequence suggested in Scheme 1C. Hydroboration with in situ generated HBCl2 was conducted under the above optimized conditions in dichloromethane. The intermediate alkyldichloroborane was then treated with a mixture containing an additional portion of Et3SiH and the second alkene component, leading to the corresponding chloroborane that engages in a renewed hydroboration reaction. Boron ate complex formation could be achieved by treatment of the intermediate dialkylchloroborane with a THF solution of monodeprotonated pinacol (pinHLi) at −78 °C. The reaction mixture was then allowed to slowly warm to room temperature to induce the final 1,2-metallate rearrangement. The clean formation of both the alkyldichloroborane and the dialkylchloroborane throughout this sequence could be verified by NMR analysis under inert conditions (see ESI†).
Studies on the compatibility of different alkenes for this reaction sequence were conducted using vinyl iodide 1a as the coupling partner (Scheme 4). Different styrene derivatives proved to be suitable and the boronic esters 5aa–5ad were obtained in moderate to very good yields (54–87%) with complete diastereoselectivity. The expected relative configuration of the stereocenters was confirmed by single-crystal X-ray diffraction of product 5aa.§ Considering aliphatic alkenes, both 1,2- and 1,1-disubstituted congeners could be employed in the reaction. While boronic ester 5ae was again obtained as a single diastereomer in a good yield (61%), the coupling product 5af was isolated as a mixture of the two diastereomers (75%). We assume a partial epimerization of the intermediate boronate complex to be the cause for this diminished diastereoselectivity. The use of different monosubstituted aliphatic alkenes allowed the introduction of a cyclohexyl group (5ag, 83%), a silane (5ah, 67%) and an ether moiety (5ai, 55%) into the coupling products. Notably, a tetrasubstituted alkene was found to be an eligible coupling partner, albeit reaction occurred with lower efficiency (5aj, 41%). The reaction with vinylferrocene led to iron complex 5ak (54%), again as a mixture of diastereomers. However, recrystallization provided crystals of the expected major diastereoisomer.§ Importantly, alkynes were also tolerated as coupling partners and through that approach, the allylic boronic esters 6aa and 6ab were successfully prepared in 52% and 25% yield, respectively. We attribute the reduced yield of product 6ab to steric repulsion of the coupling partners during the second hydroboration step.
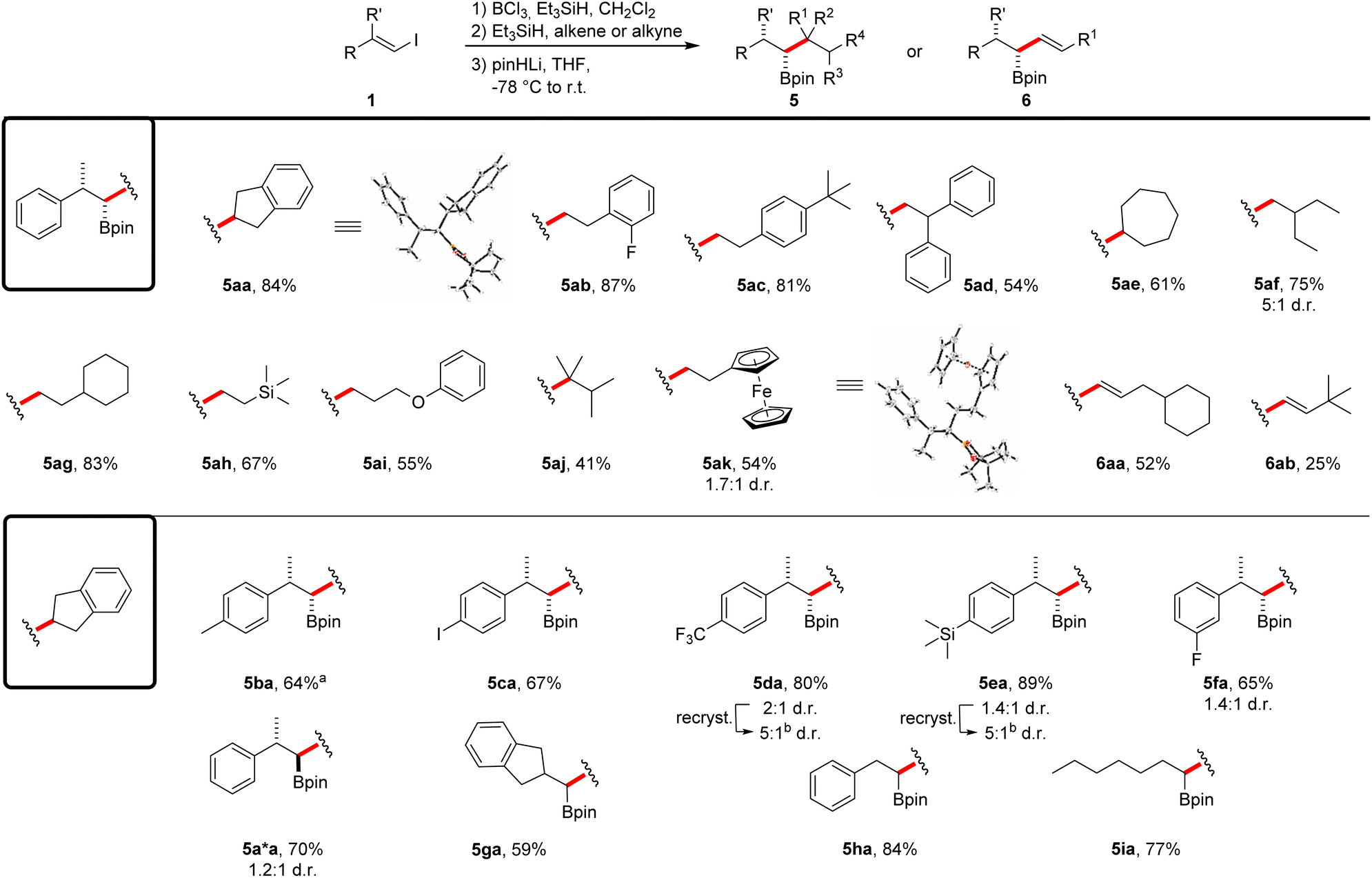 | ||
| Scheme 4 Scope of the borylative vinyl iodide–alkene/alkyne coupling reaction. All reactions were run on a 0.25 mmol scale using 1.1 equiv. of BCl3 and Et3SiH in CH2Cl2 (0.13 M) at 0 °C (30 min) to r.t. The second hydroboration step was run with Et3SiH (1.1 equiv.) and alkene/alkyne (1.0 equiv.) in CH2Cl2 (0.13 M) overnight. For details on the use of pinHLi and the rearrangement step see ESI.† Products were obtained in diastereomeric ratios > 20:1 if not noted otherwise. aBoth hydroborations were carried out at −78 °C. bRecrystallization from Et2O. For details see ESI.† | ||
We then investigated the scope with respect to the vinyl iodide component by using indene as the coupling partner. Methyl- and iodo-substituted styrene derivatives were well tolerated and the boronic esters 5ba and 5ca were obtained in good yields (64%, 67%). In case of the methyl derivative, the reaction had to be conducted at lower temperature in order to ensure a high diastereoselectivity. The CF3-, TMS- and fluoro-substituted vinyl iodides gave the corresponding boronic esters in good yields; however, a poor diastereoselectivity resulted even upon running these cascades at −78 °C (5da–5fa). Notably, recrystallization of these solid compounds led to improved diastereomeric ratios, as demonstrated for the boronic esters 5da–5fa. Low diastereoselectivity was also observed for the reaction of vinyl iodide (Z)-1a and the product 5aa was formed along with the expected anti-substituted boronic ester 5a*a. The vinyl iodides derived from 2-indanone, benzaldehyde and heptyne provided the corresponding boronic esters 5ga–5ia in good yields (59–84%).
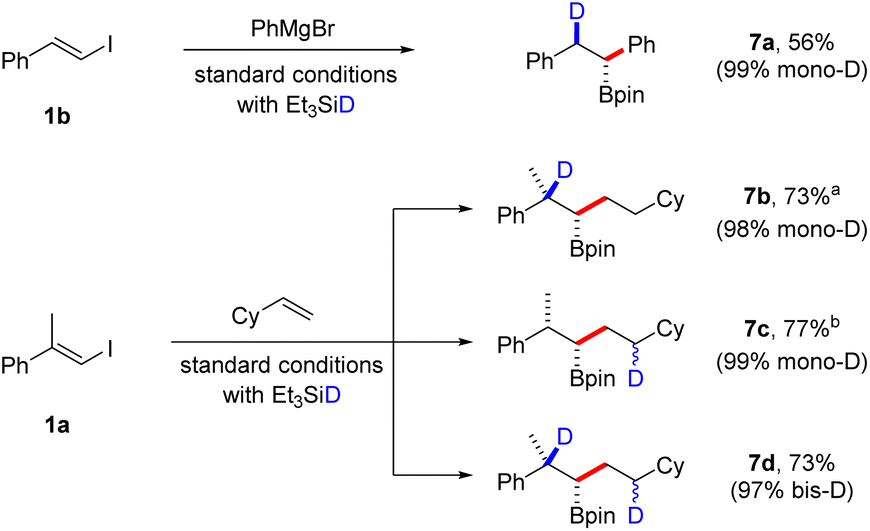
We also envisioned that our borylative protocols would facilitate practical stereoselective deuteroborations by substituting Et3SiH with Et3SiD, aiming to prepare β-deuterated and/or γ-deuterated boronic esters (Scheme 5). Consequently, when we conducted a coupling reaction with β-iodostyrene 1b and PhMgBr using deuterated silane under standard conditions, we were delighted to achieve a 55% yield of boronic ester 7a, with an excellent 99% incorporation of deuterium. Additionally, the coupling reaction between vinyl iodide and a second alkene offers the possibility of monodeuteration at two distinct positions in the product, as well as the formation of a bisdeuterated product. The site-selective incorporation of deuterium at these various positions was showcased in the coupling of vinyl iodide 1a with vinyl cyclohexane. These reactions displayed remarkable efficiency, yielding a mono-D-incorporation of 98% for β-deuterated 7b, 99% for γ-deuterated 7c, and a bisdeuteration degree of 97% for boronic ester 7d. Since deuteration during the second hydroboration step introduces an additional stereocenter in the product, we anticipate that compounds 7c and 7d exist as a mixture of diastereomers. However, NMR analysis could not discern between different isomers.
 | ||
| Scheme 5 Application of Et3SiD for D-labeling. aEt3SiH is substituted with Et3SiD (1.1 equiv.) only in the first hydroboration step. bEt3SiH is substituted with Et3SiD (1.1 equiv.) only in the second hydroboration step. | ||
Conclusions
In summary, we developed two methods that allow the formation of a broad range of substituted boronic esters by the coupling of vinyl iodides with either organometallic reagents, alkenes or alkynes. Reactions proceed with good to complete diastereoselectivity in the absence of a transition metal and can be conducted as one-pot processes. These cascades comprise stereospecific hydroborations, or optionally deuteroborations, and 1,2-metallate rearrangements. Both processes utilize simple starting materials and stereoselectively lead to rather complex product structures, plenty of which are not accessible by established hydroboration/oxidation sequences from alkenes. We are confident these methods will find applications in organic synthesis and related fields.Data availability
The data that support the findings of this study are available in the ESI.†Author contributions
G. S., M. S. and F. C. conducted all experiments and characterized the novel compounds. C. G. D. conducted the single-crystal X-ray diffraction analysis. G. S., M. S., F. C. and A. S. designed the experiments and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Fonds der Chemischen Industrie (doctoral fellowship to G. S.) and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – GRK 2678-437785492 for supporting this work.Notes and references
- D. G. Hall, in Boronic Acids, ed. D. G. Hall, Wiley-VCH-Verl., Weinheim, 2011, pp. 1–133 Search PubMed.
- H. C. Brown and B. Singaram, Acc. Chem. Res., 1988, 21, 287–293 CrossRef CAS.
- H. K. Scott and V. K. Aggarwal, Chem.–Eur. J., 2011, 17, 13124–13132 CrossRef CAS PubMed.
- C. Sandford and V. K. Aggarwal, Chem. Commun., 2017, 53, 5481–5494 RSC.
- C. M. Crudden and D. Edwards, Eur. J. Org Chem., 2003, 4695–4712 CrossRef CAS.
- C. M. Crudden, B. W. Glasspoole and C. J. Lata, Chem. Commun., 2009, 6704–6716 RSC.
- D. Pozzi, E. M. Scanlan and P. Renaud, J. Am. Chem. Soc., 2005, 127, 14204–14205 CrossRef CAS.
- S. Nave, R. P. Sonawane, T. G. Elford and V. K. Aggarwal, J. Am. Chem. Soc., 2010, 132, 17096–17098 CrossRef CAS PubMed.
- S. Roesner, D. J. Blair and V. K. Aggarwal, Chem. Sci., 2015, 6, 3718–3723 RSC.
- F. Clausen, M. Kischkewitz, K. Bergander and A. Studer, Chem. Sci., 2019, 10, 6210–6214 RSC.
- N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 20, 3437–3440 CrossRef.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 RSC.
- E. Hey-Hawkins and C. V. Teixidor, Boron-Based Compounds. Potential and Emerging Applications in Medicine, John Wiley & Sons, Ltd., Oxford, 2018 Search PubMed.
- K. K. Das, S. Paul and S. Panda, Org. Biomol. Chem., 2020, 18, 8939–8974 RSC.
- H. C. Brown and G. Zweifel, J. Am. Chem. Soc., 1961, 83, 2544–2551 CrossRef CAS.
- K. Burgess and M. J. Ohlmeyer, Chem. Rev., 1991, 91, 1179–1191 CrossRef CAS.
- D. S. Matteson and R. W. H. Mah, J. Am. Chem. Soc., 1963, 85, 2599–2603 CrossRef CAS.
- M. Burns, S. Essafi, J. R. Bame, S. P. Bull, M. P. Webster, S. Balieu, J. W. Dale, C. P. Butts, J. N. Harvey and V. K. Aggarwal, Nature, 2014, 513, 183–188 CrossRef CAS PubMed.
- D. S. Matteson, J. Org. Chem., 2013, 78, 10009–10023 CrossRef CAS PubMed.
- D. S. Matteson, Chem. Rev., 1989, 89, 1535–1551 CrossRef CAS.
- Formation of boron ate complexes via electrophilic boryla-tion, see: C. You, M. Sakai, C. G. Daniliuc, K. Bergander, S. Yamaguchi and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 21697–21701 CrossRef CAS PubMed.
- G. Xu, M. Lüthy, J. Habegger and P. Renaud, J. Org. Chem., 2016, 81, 1506–1519 CrossRef CAS PubMed.
- S. Colin, L. Vaysse-Ludot, J.-P. Lecouvé and J. Maddaluno, J. Chem. Soc., Perkin Trans. 1, 2000, 4505–4511 RSC.
- D. J. Pasto, J. Hickman and T.-C. Cheng, J. Am. Chem. Soc., 1968, 90, 6259–6260 CrossRef.
- S. Elgendy, G. Claeson, V. V. Kakkar, D. Green, G. Patel, C. A. Goodwin, J. A. Baban, M. F. Scully and J. Deadman, Tetrahedron, 1994, 50, 3803–3812 CrossRef CAS.
- S. Elgendy, G. Patel, V. V. Kakkar, G. Claeson, D. Green, E. Skordalakes, J. A. Baban and J. Deadman, Tetrahedron Lett., 1994, 35, 2435–2436 CrossRef CAS.
- R. Soundararajan and D. S. Matteson, Organometallics, 1995, 14, 4157–4166 CrossRef CAS.
- S. Namirembe, C. Gao, R. P. Wexler and J. P. Morken, Org. Lett., 2019, 21, 4392–4394 CrossRef CAS PubMed.
- C. Wang, T. Tobrman, Z. Xu and E. Negishi, Org. Lett., 2009, 11, 4092–4095 CrossRef CAS PubMed.
- D. S. Matteson, Tetrahedron, 1998, 54, 10555–10607 CrossRef CAS.
- L. Boymond, M. Rottländer, G. Cahiez and P. Knochel, Angew. Chem., Int. Ed., 1998, 37, 1701–1703 CrossRef CAS PubMed.
- C. Diner and K. Szabó, J. Am. Chem. Soc., 2017, 139, 2–14 CrossRef CAS PubMed.
- P. Barrio, E. Rodríguez and S. Fustero, Synthesis, 2018, 50, 1935–1957 CrossRef.
- K. Jana and A. Studer, Org. Lett., 2022, 24, 1100–1104 CrossRef CAS PubMed.
- K. Jana, A. Bhunia and A. Studer, Chem, 2020, 6, 512–522 CAS.
- D. S. Matteson, A. A. Kandil and R. Soundararajan, J. Am. Chem. Soc., 1990, 112, 3964–3969 CrossRef CAS.
- O. Eidam, C. Romagnoli, E. Caselli, K. Babaoglu, D. T. Pohlhaus, J. Karpiak, R. Bonnet, B. K. Shoichet and F. Prati, J. Med. Chem., 2010, 53, 7852–7863 CrossRef CAS PubMed.
- L. Yang, D.-H. Tan, W.-X. Fan, X.-G. Liu, J.-Q. Wu, Z.-S. Huang, Q. Li and H. Wang, Angew. Chem., Int. Ed., 2021, 60, 3454–3458 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2167695 and 2293538–2293542. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc06131k |
| ‡ These authors contributed equally. |
| § Deposition numbers 2167695 (for analogue of 2a), 2293538 (for 3ah), 2293539 (for 3bc), 2293540 (for 3bm), 2293541 (for 5aa) and 2293542 (for 5ak) contain the supplementary crystallographic data for this paper. |
| This journal is © The Royal Society of Chemistry 2024 |