DOI:
10.1039/D3SC05997A
(Edge Article)
Chem. Sci., 2024,
15, 271-277
Photocatalytic (3 + 2) dipolar cycloadditions of aziridines driven by visible-light†
Received
8th November 2023
, Accepted 29th November 2023
First published on 29th November 2023
Abstract
Herein, we document the design and development of a novel (3 + 2) cycloaddition reaction aided by the activity of an organic photocatalyst and visible light. The process is extremely fast, taking place in a few minutes, with virtually complete atom economy. A large variety of structurally diverse aziridines were used as masked ylides in the presence of different types of dipolarophiles (28 examples with up to 94% yield and >95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 dr). Mechanistic insights obtained from photophysical, electrochemical and experimental studies highlight that the chemistry is driven by the in situ generation of the reactive ylide through two consecutive electron-transfer processes. We also report an aerobic cascade process, where an additional oxidation step grants access to a vast array of pyrrole derivatives (19 examples with up to 95% yield). Interestingly, the extended aromatic core exhibits a distinctive absorption and emission profile, which can be easily used to tag the effectiveness of this covalent linkage.
:5 dr). Mechanistic insights obtained from photophysical, electrochemical and experimental studies highlight that the chemistry is driven by the in situ generation of the reactive ylide through two consecutive electron-transfer processes. We also report an aerobic cascade process, where an additional oxidation step grants access to a vast array of pyrrole derivatives (19 examples with up to 95% yield). Interestingly, the extended aromatic core exhibits a distinctive absorption and emission profile, which can be easily used to tag the effectiveness of this covalent linkage.
Introduction
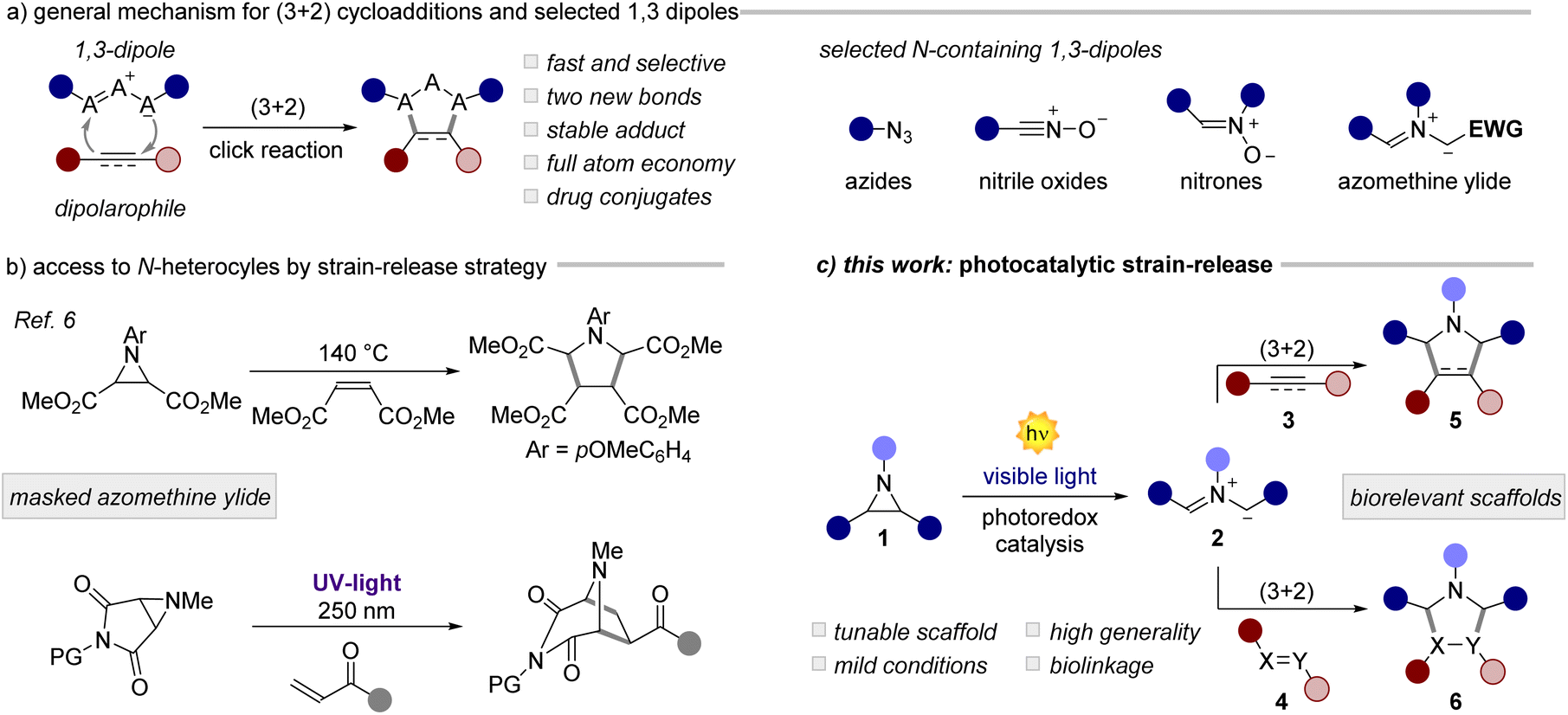
Since its discovery in the 1960s,1 the Huisgen 1,3-dipolar cycloaddition (1,3-DPC) has become one of the most studied and useful reactions in synthetic and medicinal chemistry (Fig. 1a).2 Through its unique mechanistic pathway, it is possible to access in a rapid and atom-economical manner biorelevant natural products, pharmaceuticals, materials, and bioconjugates. The 1,3-DPC is the prototypical click reaction, recently further popularized by a widespread use under bioorthogonal settings.3 Owing to the excellent synthetic potential of the 1,3-DPC, extensive research has been made towards the identification of versatile 1,3-dipoles.2 Across these scaffolds (Fig. 1a, right), azomethine ylides are particular appealing due to (i) their structural diversity and (ii) the ability to form two new C–C bonds in a single step, while (iii) accessing synthetically relevant cores, such as pyrrolidine derivatives.4 Azomethine ylides can be generated in situ from stable precursors such as esters/malonates or acids (EWG in Fig. 1a) upon deprotonation or decarboxylation, respectively. This approach has been largely explored in organocatalysis,5 although it restricts the generality of the process to tailored substrates bearing electron-withdrawing groups (EWGs). A more general approach, pioneered by Heine and Huisgen,6 involves the use of strained aziridines, as masked azomethine ylides (Fig. 1b, top). Interestingly, the 1,3-DPC for small ring expansion via the generation of an ylide intermediate allows the straightforward installation of multiple C–C or C-heteroatom bonds into complex structural units, which is difficult to achieve by other means. On the other hand, this strain-release strategy involves the use of harsh reaction conditions with temperatures spanning from 140 to 220 °C. To circumvent this issue, the authors have also investigated a photochemical variant, although involving the use of highly energetic UV-light sources (Fig. 1b, bottom). Due to these limitations and the low generality of the process, the community has gradually forsaken this synthetic strategy. Over the last few decades, it has been demonstrated that photoredox catalysis7 can grant access to structurally complex scaffolds by the activation of small molecules under very mild reaction conditions.8 Thus, many historical photoreactions9 have been revisited using milder and more general conditions. In this scenario, the activation of small rings is a promising area of research.10 In fact, the visible-light-driven activation of cyclopropylamines,11 cyclopropanols,12 cyclopropanes13 and many others has led to a substantial synthetic and mechanistic progress. However, the use of aziridines as azomethine ylide precursors for cycloadditions under photoredox conditions has been long overlooked.14
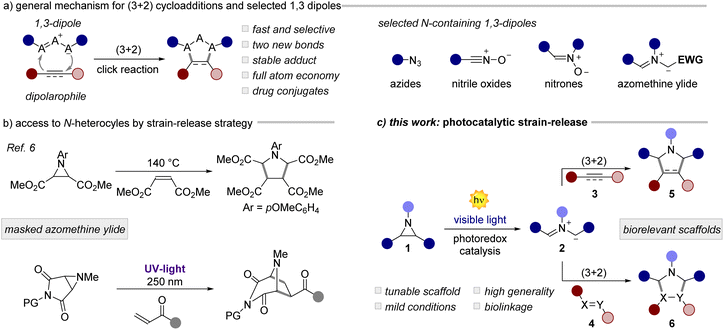 |
| | Fig. 1 (a) General mechanism for (3 + 2) cycloadditions and an array of 1,3-dipoles containing a nitrogen atom. (b) Use of aziridines to access azomethine ylides. (c) This work: visible-light photocatalytic reactions of aziridines. PG: protecting group. | |
Herein, we documented the design and development of a general and mild methodology for the generation of azomethine ylides by means of photoredox catalysis (Fig. 1c). The visible light mediated ring-opening of aziridines, as strained 3-membered N-heterocycles, occurs rapidly with a wide variety of dipolarophiles, including olefins, aldehydes, azodicarboxylates, and Schiff bases, yielding relevant structural targets with high diastereoselectivity. In the presence of alkynes, a photocatalytic cascade process takes place leading to densely functionalized pyrroles in high yield, in a single synthetic operation. Finally, we applied this method to the conjugation of biologically relevant molecules including dehydroabietylamine, glucose and phenylglycine derivatives.15
Results and discussion
Optimization and scope of the reaction
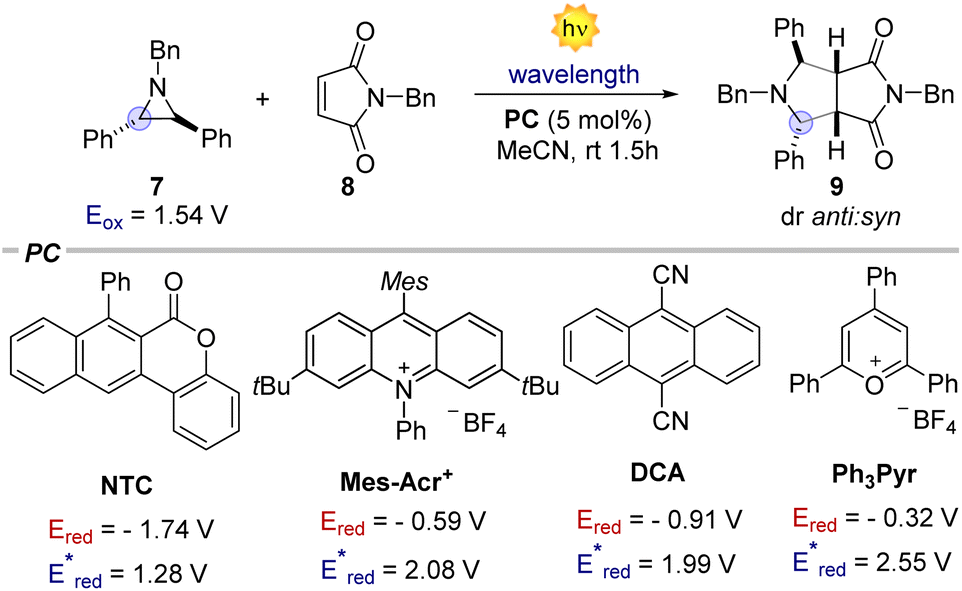
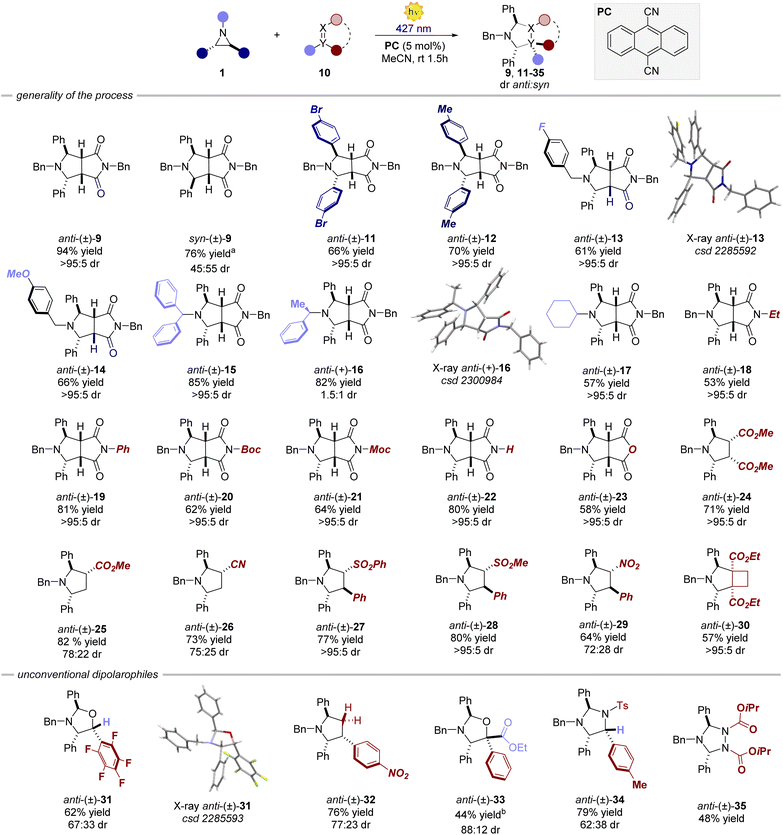
We initiated our investigation using aziridine anti-7 and maleimide 8. We initially evaluated the possibility of engaging anti-7 into an EDA complex.16 However, no new band was observed upon mixing of anti-7 and 8 (see ESI†). Consistently, under 400 nm irradiation the (3 + 2) cycloaddition product 9 was obtained only in traces (Table 1, entry 1). We thus evaluated diverse organic PCs with various excited state redox potentials (entries 2–6), spanning from 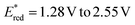 (vs. SCE in MeCN).7a,17 Dicyanoanthracene (DCA) outperformed the other catalysts, furnishing product 9 in 94% yield as the single anti-diastereomer (entry 5). Using syn-7, a modest diastereomeric ratio (dr) was observed, slightly favouring the syn-product 9 (entry 7). Furthermore, this photocatalytic reaction is very fast (see ESI† for the kinetic profile of the reaction), delivering product 9 after only 10 minutes of irradiation (entry 8), establishing its place among other photocatalytic click reactions.18 Finally, without irradiation, no reaction was observed (entry 9). With the optimized reaction conditions in hand, we next explored the generality of the process (Fig. 2). Albeit the reaction showed fast kinetics, we decided to keep the reaction time at 1.5 hours in order to have more general conditions regardless of the nature of the acceptor.
(vs. SCE in MeCN).7a,17 Dicyanoanthracene (DCA) outperformed the other catalysts, furnishing product 9 in 94% yield as the single anti-diastereomer (entry 5). Using syn-7, a modest diastereomeric ratio (dr) was observed, slightly favouring the syn-product 9 (entry 7). Furthermore, this photocatalytic reaction is very fast (see ESI† for the kinetic profile of the reaction), delivering product 9 after only 10 minutes of irradiation (entry 8), establishing its place among other photocatalytic click reactions.18 Finally, without irradiation, no reaction was observed (entry 9). With the optimized reaction conditions in hand, we next explored the generality of the process (Fig. 2). Albeit the reaction showed fast kinetics, we decided to keep the reaction time at 1.5 hours in order to have more general conditions regardless of the nature of the acceptor.
Table 1 Optimization of the reaction conditionsa
 |
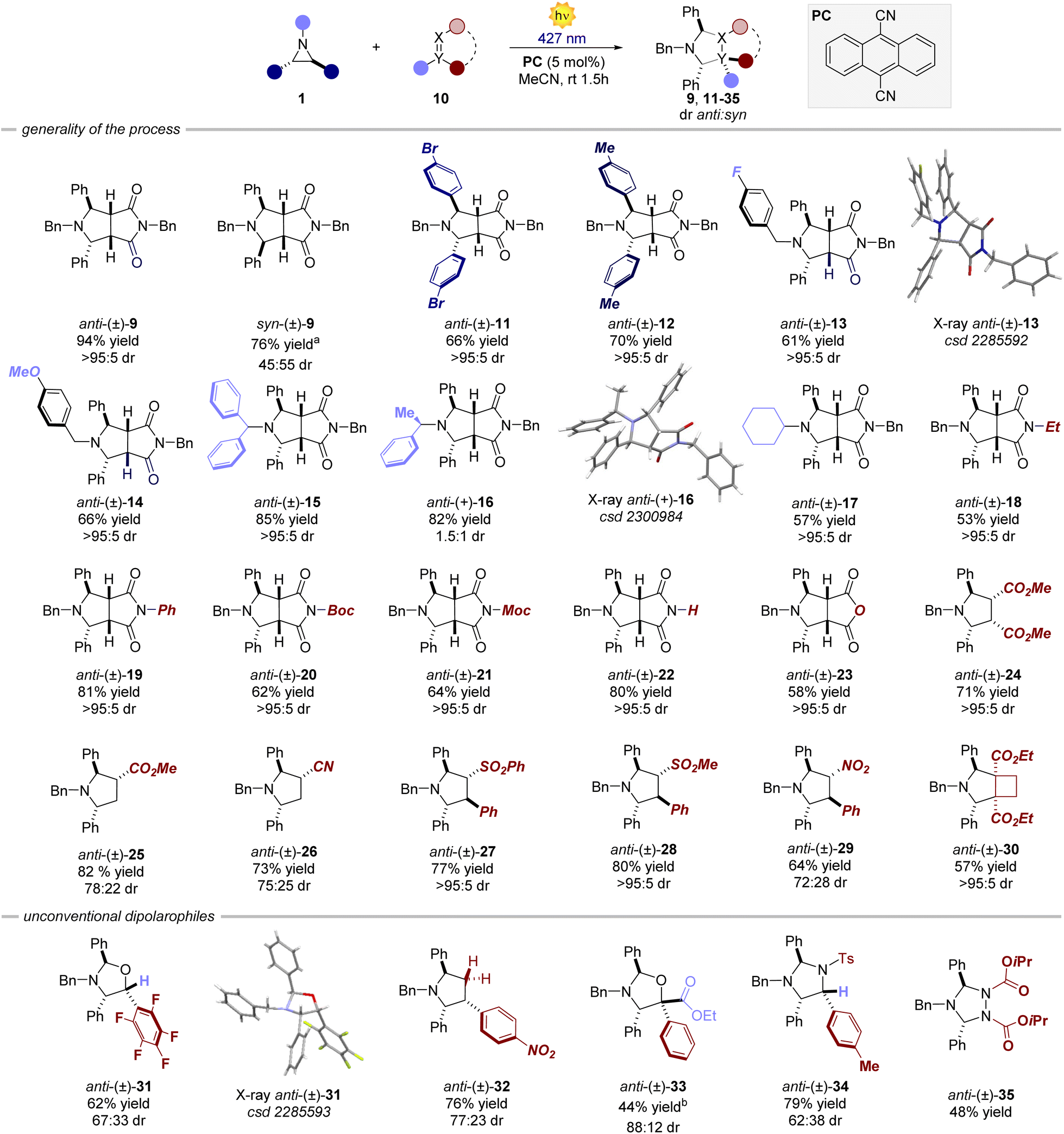
| | Fig. 2 Survey of the aziridines and dipolarophiles that can participate in the process. Reaction performed on a 0.2 mmol scale, using 1.2 equiv. of dipolarophile in 4 mL of MeCN. Yields refer to isolated products. a The syn aziridine was used. bYield measured by 1H NMR. Boc: tert-butyloxycarbonyl. Moc: methoxycarbonyl. Ts: tosyl. | |
Variations of the steric and electronic properties of the aziridine, involving both the aryl rings and the N-protecting group, were well tolerated affording the corresponding cycloaddition products 9, 11–17 in high yields from 61% to 85% and a diastereomeric ratio depending on the stereoisomer of the parent aziridine (vide infra). The process is amenable to a wide variety of dipolarophiles. Differently substituted maleimides and maleic anhydride (18–23) delivered the corresponding products in high yield (53–81%) and virtually complete diastereoselectivity. Interestingly, also non-cyclic precursors, including maleate, acrylate, acrylonitrile, sulphone and nitrostyrene derivatives, were found to be competent dipolarophiles, resulting in the formation of products 24–29 in high yield and dr. A strained cyclobutene derivative also afforded the corresponding bicyclic product 30 in 57% yield as a single diastereomer. Subsequently, we turned our attention to other non-classical dipolarophiles such as aldehydes, imines and other nitrogen-containing molecules. In all the cases, the reactions furnished the desired cycloaddition products with moderate to high diastereoselectivity (up to >20:1 dr) and 40–79% yield. When employing non-cyclic precursors, the reaction displayed lower diastereocontrol arising from the possible exo and endo attacks of the azomethine ylide.19
The only limitation was found for aliphatic aldehydes. Octanal furnished the cycloaddition product in 10% yield.
Mechanistic investigations
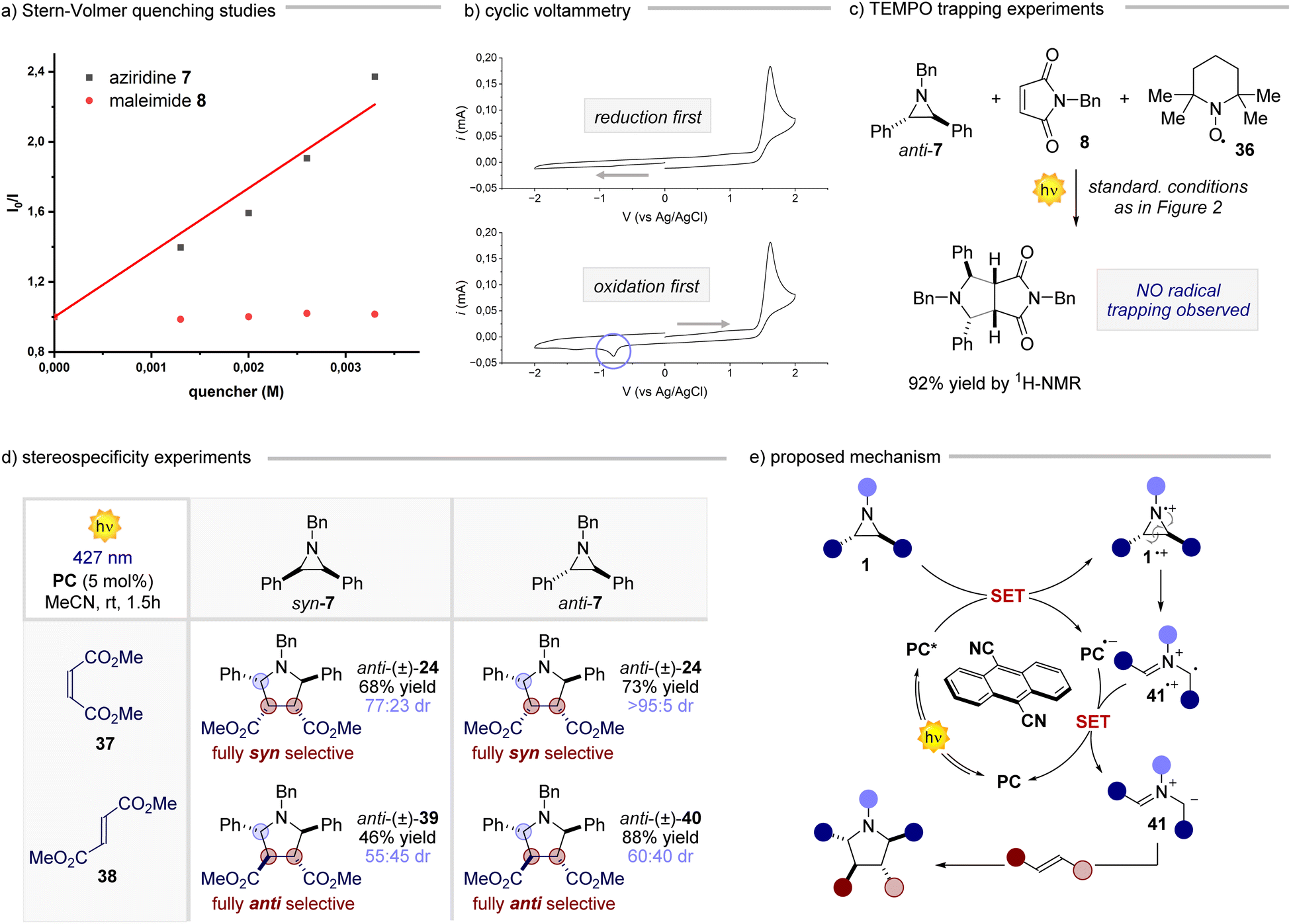
After having assessed the generality of this novel reaction, we delved into understanding its underlying mechanism.20 Our investigation began by exploring the interactions of the reaction components with the PC (Fig. 3a). While maleimide 8 did not significantly quench the excited state of the PC, the addition of aziridine anti-7 led to a significant reduction of the PC emission. This observation prompted us to gather electrochemical evidence regarding the feasibility of the single electron transfer (SET) step between the photoexcited PC (Eox 1.99 V vs. SCE in MeCN) and aziridine anti-7 (Fig. 3b). Upon subjecting a solution of the aziridine anti-7 to cyclic voltammetry during a reductive scan to −2 V, followed by an oxidative scan to 2 V, we observed a single irreversible anodic peak at approximately 1.54 V (vs. Ag/AgCl). This peak was ascribed to the oxidation of the aziridine. The obtained electrochemical data support the energetic feasibility of the SET from anti-7 to the excited state of the PC. Furthermore, the non-reversible behaviour of the aziridine during the CV analysis suggests that, upon SET, the resulting radical cation rapidly transforms into another species. This notion was further reinforced by conducting the cyclic voltammetry analysis in the reverse order (i.e., an initial oxidative scan followed by a reductive scan), which revealed a new cathodic peak around −0.8 V. These results collectively indicate that the aziridine radical cation promptly undergoes a ring-opening process upon single electron oxidation, followed by a single electron reduction to form the corresponding azomethine ylide.21 This SET-ring opening-SET sequence occurs at a remarkably fast rate, as evidenced by the rapid kinetics observed for the model reaction (see ESI† for further details). Additionally, when conducting the reaction in the presence of 2 equivalents of TEMPO (Fig. 3c), product 9 was obtained in almost quantitative yield, corroborating the fast reaction kinetics. This result gives insight into the polar nature of the cycloaddition process, ruling out the hypothesis of any radical addition of 41˙+ to the olefin. Subsequently, a set of experiments where both syn and anti aziridine 7 were reacted with maleate 37 and fumarate 38 derivatives were performed (Fig. 3d). The reaction displayed a remarkable stereospecificity, preserving the relationship between the substituents on the double bond of the acceptor in the corresponding product. The reaction outcome aligns well with the rapid formation of the ylide, as discussed earlier, since an addition involving a radical ion would result in a non-stereospecific stepwise process. Based on all the gathered data, we propose the following mechanistic scenario (Fig. 3e). Upon light absorption, the PC captures one electron from aziridine 1, generating the radical anion of the PC and aziridine radical cation 1˙+. The latter undergoes a rapid ring-opening process, giving rise to radical cation 41˙+, which promptly engages in a SET process to regenerate the PC and form azomethine ylide 41. This reactive intermediate engages the chosen olefin in a cycloaddition step, yielding the desired 5-membered cyclic product. Overall, these mechanistic studies highlight how this process makes use of photocatalysis to convert aziridines very rapidly into azomethine ylides, which react in a (3 + 2) cycloaddition.
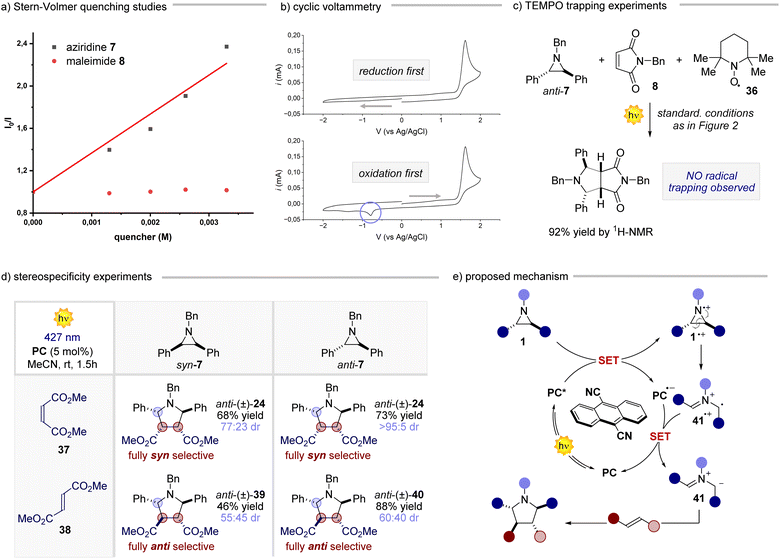 |
| | Fig. 3 Mechanistic studies for the photocatalytic reaction of aziridines. (a) Stern–Volmer quenching studies for aziridine 7 and maleimide 8. (b) Electrochemical analyses of aziridine 7. (c) TEMPO trapping experiments for the model reaction involving compounds 7 and 8. (d) Stereospecificity experiments between syn-7, anti-7 maleate 37 and fumarate 38. (e) Proposed mechanism for the photocatalytic reaction of aziridines with olefins. | |
Cascade process for the photocatalytic synthesis of pyrroles
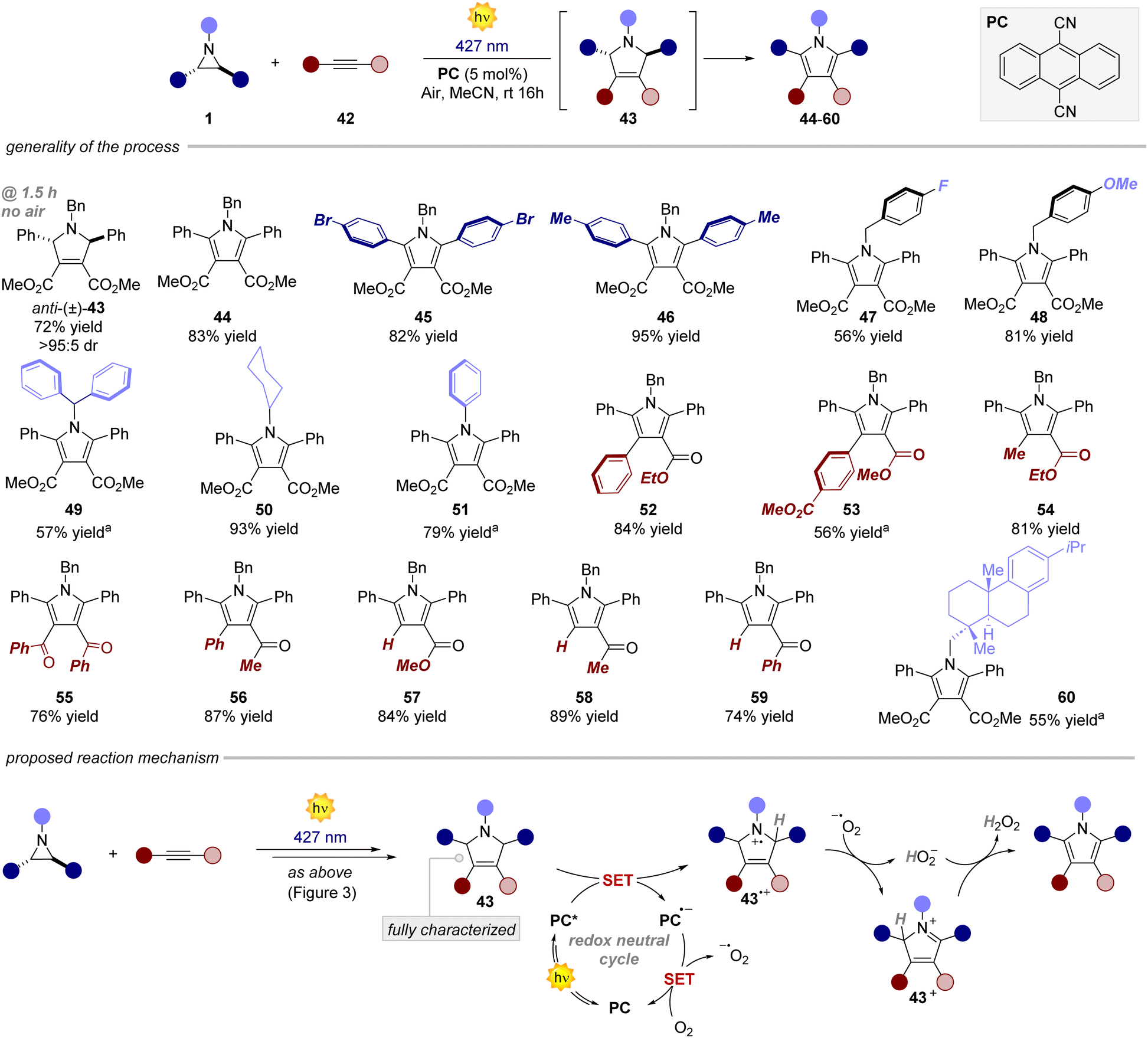
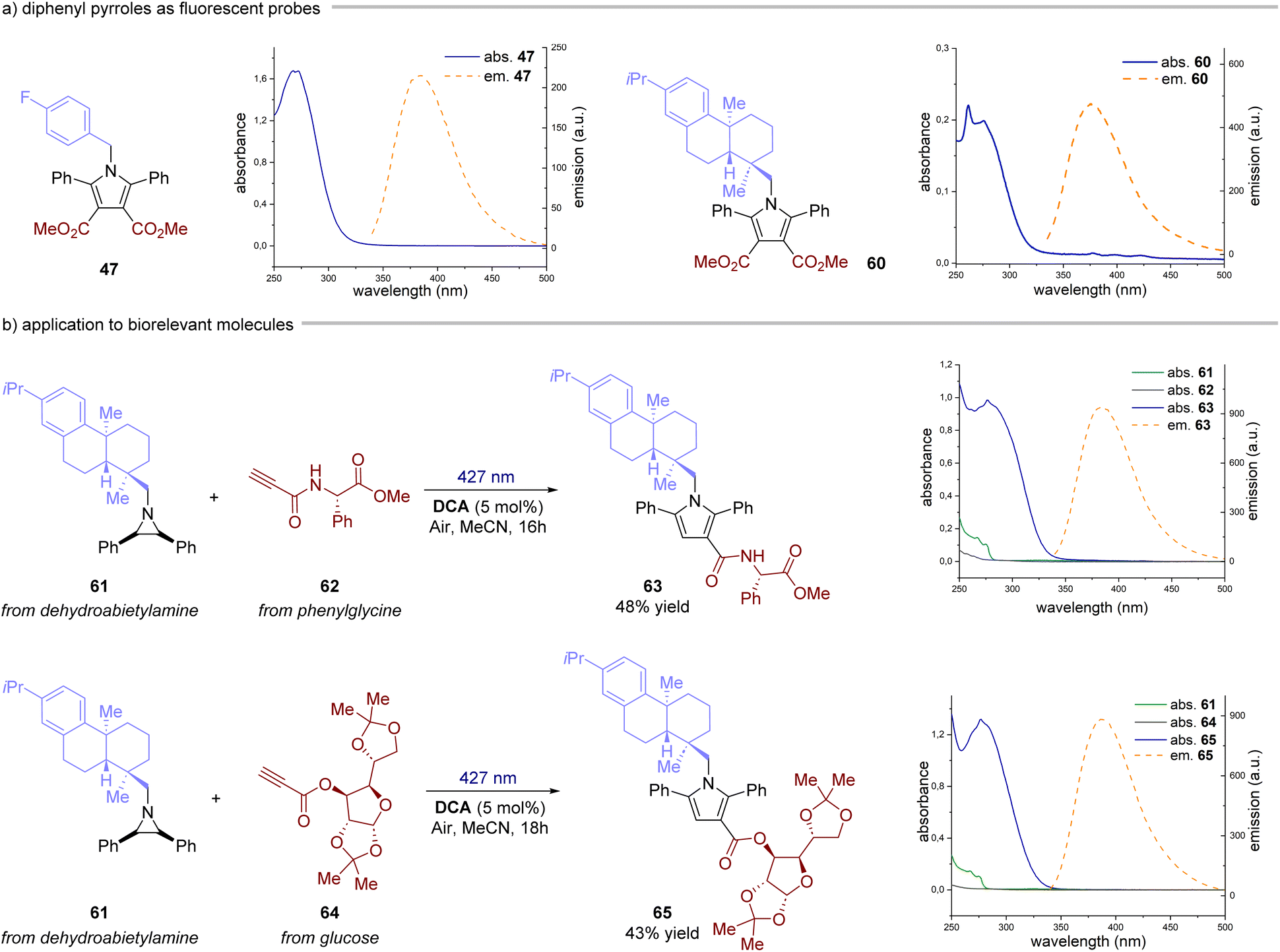
Next, we investigated the reaction with alkynes as dipolarophiles (Fig. 4). As expected, when conducting the reaction with dimethyl acetylenedicarboxylate, we observed the formation of dihydropyrrole 43, which was isolated as a single diastereomer. Careful analysis of the reaction crude, however, revealed traces of the corresponding pyrrole 44. We thus reasoned that compound 44 could have arisen from an oxidative pathway from 43 (vide infra), due to the possible presence of adventitious oxygen in the reaction mixture.22 Indeed, when extending the reaction time to 16 hours under an aerobic atmosphere, pyrrole 44 was formed as the only product in very good yields (83%). Next, we tested the generality of this light-driven process for differently substituted aziridines. Variations at the aromatic rings (45, 46) as well as at the N-protecting group (47–51) were well tolerated, furnishing the products in 56–95% yield. Also, structurally diverse alkynes bearing esters and both aromatic and aliphatic ketones were found to be synthetically useful precursors (52–59), leading to the corresponding pyrroles with yields spanning from 56% to 87%. Notably, an aziridine obtained from naturally abundant dehydroabietylamine was also successfully converted into the corresponding pyrrole 60 in good yield. Remarkably, these products exhibit distinctive absorption and emission profiles resulting from the formation of the 2,5-diarylpyrrole core, making them potentially valuable molecular probes for bioconjugation reactions (Fig. 5a).15,23 Intrigued by these findings, we decided to delve deeper into this area by combining an aziridine derived from dehydroabietyl amine (Fig. 5b, compound 61) with alkynes bearing bio-relevant fragments such as an amino acid and a sugar (62 and 64). In both cases, the resulting products 63 and 65 displayed similar absorption and emission profiles compared to the model compound 47, further confirming that the photophysical properties are indeed attributed to the pyrrole core.
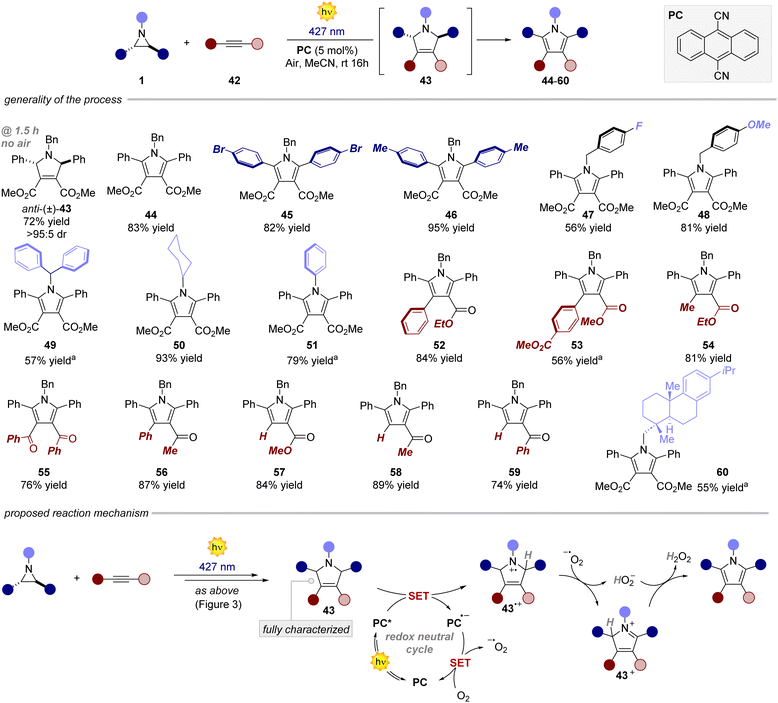 |
| | Fig. 4 Survey of the aziridines and alkynes that can participate in the process. Reaction performed on a 0.2 mmol scale, using 1.2 equiv. of dipolarophile in 4 mL of MeCN. Yields refer to isolated products. a Reaction performed with syn-aziridine. | |
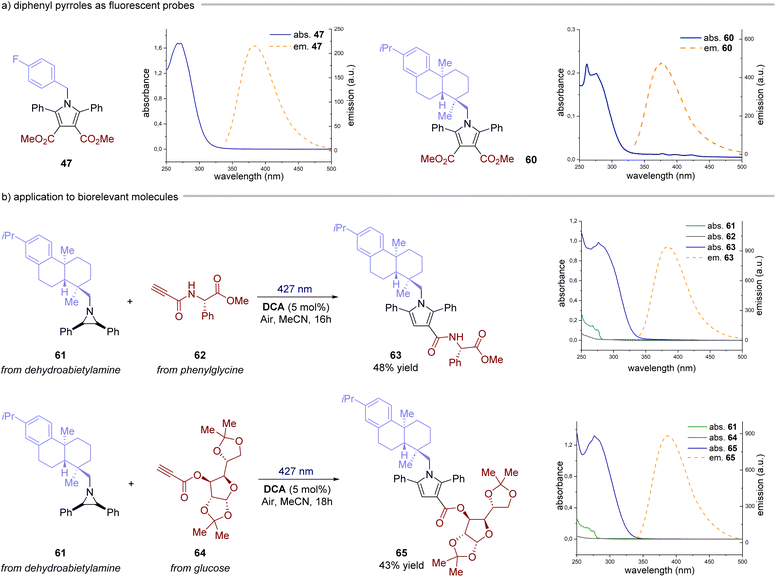 |
| | Fig. 5 Diphenyl pyrroles as fluorescent probes. (a) Absorption and emission of compounds 47 and 60. (b) Synthesis and photophysical characterization of compounds 63 and 65. | |
Conclusions
In summary, we have developed a versatile synthetic strategy that harnesses the power of visible light to trigger the formation of reactive azomethine ylides through two consecutive SET events. The developed method is highly versatile, leading access to pyrrolidines and dihydropyrroles with high chemo-, regio- and diastereoselectivity (28 examples with up to 94% yield and >95:5 dr). Noteworthy, when the reaction is performed in the presence of air, a variety of densely functionalized pyrroles were accessed (19 examples with up to 95% yield), in a peculiar light-driven cascade process. Due to the robustness and operational simplicity of this new (3 + 2) cycloaddition platform, we foresee its potential application under biological settings.
Data availability
Experimental procedures and spectral data can be found in the ESI.†
Author contributions
L. D. conceived and directed the project. D. M. and L. D have written the manuscript with contributions from all authors. D. M and T. B. have performed all the experiments. G. P. has performed the X-ray analyses. All authors have given approval to the final version of the manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by MUR (Ministero dell’Università) PRIN 2020927WY3_002, and (European Research Council) ERC-Starting Grant 2021 SYNPHOCAT 101040025 (L. D.). Chiesi Farmaceutici SpA and Dr Davide Balestri are acknowledged for the support with the D8 Venture X-ray equipment. Dr Deepak Singh is thanked for initial studies on the photo-catalytic reactivity of aziridines.
Notes and references
-
(a)
R. Huisgen, in Zehnjahresfeier des Fonds der Chemischen Industrie, Düsseldorf, 1960, pp. 73–102 Search PubMed;
(b) R. Huisgen, Proc. Chem. Soc., London, 1961, 357 Search PubMed;
(c) R. Huisgen, Angew Chem. Int. Ed. Engl., 1963, 2, 565 CrossRef.
- M. Breugst and H.-U. Reissig, Angew. Chem., Int. Ed., 2020, 59, 12293 CrossRef CAS.
-
(a) A. Lauria, R. Delisi, F. Mingoia, A. Terenzi, A. Martorana, G. Barone and A. M. Almerico, Eur. J. Org Chem., 2014, 16, 3289 CrossRef;
(b) R. J. Pieters, D. T. S. Rijkers and R. M. J. Liskamp, QSAR Comb. Sci., 2007, 26, 1181 CrossRef CAS;
(c) J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272 RSC;
(d) D. A. Bilodeau, K. D. Margison, M. Serhan and J. P. Pezacki, Chem. Rev., 2021, 121, 6699 CrossRef CAS.
-
(a)
O. Tsuge and S. Kanemasa, in Recent Advances in Heterocyclic Chemistry, ed. A. R. Katritzky, Academic Press, New York, 1989, vol. 45, pp. 231–349 Search PubMed;
(b) I. Coldham and R. Hufton, Chem. Rev., 2005, 105, 2765 CrossRef CAS;
(c) C. Najera and J. M. Sansano, Curr. Org. Chem., 2003, 7, 1105 CrossRef CAS.
- R. Dalpozzo, G. Bartoli and G. Bencivenni, Chem. Soc. Rev., 2012, 41, 7247 RSC.
-
(a) H. W. Heine and R. Peavy, Tetrahedron Lett., 1965, 6, 3123 CrossRef;
(b) R. Huisgen, W. Scheer and H. Huber, J. Am. Chem. Soc., 1967, 89, 1753 CrossRef CAS.
-
(a) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS;
(b) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS;
(c) J.-P. Goddard, C. Ollivier and L. Fensterbank, Acc. Chem. Res., 2016, 49, 1924 CrossRef CAS PubMed;
(d) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC;
(e) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898 CrossRef CAS PubMed;
(f) T. Bortolato, S. Cuadros, G. Simionato and L. Dell'Amico, Chem. Commun., 2022, 58, 1263 RSC.
- T. P. Nicholls, D. Leonori and A. C. Bissember, Nat. Prod. Rep., 2016, 33, 1248 RSC.
-
M. Oelgemöller and N. Hoffman, Photoreactions in Encyclopedia of Physical Organic Chemistry, ed. Z. Wang, U. Wille and E. Juaristi, Wiley, 2017, pp. 1–65 Search PubMed.
- P. Bellotti and F. Glorius, J. Am. Chem. Soc., 2023, 145, 20716 CrossRef CAS PubMed.
- For selected examples:
(a) S. Maity, M. Zhu, R. S. Shinabery and N. Zheng, Angew. Chem., Int. Ed., 2012, 51, 222 CrossRef CAS PubMed;
(b) D. H. White, A. Noble, K. I. Booker-Milburn and V. K. Aggarwal, Org. Lett., 2021, 23, 3038 CrossRef CAS PubMed;
(c) Y. Zheng, W. Huang, R. K. Dhungana, A. Granados, S. Keess, M. Makvandi and G. A. Molander, J. Am. Chem. Soc., 2022, 144, 23685 CrossRef CAS PubMed.
- For selected examples:
(a) J. Wang and X. Li, Chem. Sci., 2022, 13, 3020 RSC;
(b) N. Varabyeva, M. Barysevich, Y. Aniskevich and A. Hurski, Org. Lett., 2021, 23, 5452 CrossRef CAS;
(c) C. Jiang, P. Chen and G. Liu, Sci. China: Chem., 2023, 66, 2858 CrossRef CAS.
- For selected examples:
(a) Z. Zuo, C. G. Daniluc and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 25252 CrossRef CAS PubMed;
(b) L. Ge, D.-X. Wang, R. Xing, D. Ma, P. J. Walsh and C. Feng, Nat. Commun., 2019, 10, 4367 CrossRef;
(c) Y. Xu, H.-X. Gao, C. Pan, Y. Shi, C. Zhang, G. Huang and C. Feng, Angew. Chem., Int. Ed., 2023, 62, e202310671 CrossRef CAS;
(d) L. Ge, C. Zhang, C. Pan, D.-X. Wang, D.-Y. Liu, Z.-Q. Li, P. Shen, L. Tian and C. Feng, Nat. Commun., 2022, 13, 5938 CrossRef CAS.
-
(a) A. P. Schaap, G. Prasad and S. Siddiqui, Tetrahedron Lett., 1984, 25, 3035 CrossRef CAS;
(b) V. Caër, A. Laurent, É. Laurent and R. Tardivel, New J. Chem., 1987, 11, 351 Search PubMed . For an example involving the use of aziridines that does not involve azomethine ylide intermediates see:;
(c) Y. Li, F. Chen, S. Zhu and L. Chu, Org. Chem. Front., 2021, 8, 2196 RSC.
-
(a) K. L. Holmes and L. M. Lantz, Methods Cell Biol., 2001, 63, 185 CrossRef CAS PubMed;
(b) E. Gallo, Bioconjugate Chem., 2020, 31, 16 CrossRef CAS PubMed.
-
(a) G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461 CrossRef CAS PubMed;
(b) C. G. S. Lima, T. de M. Lima, M. Duarte, I. D. Jurberg and W. Paixão, ACS Catal., 2016, 6, 1389 CrossRef CAS.
- A. Vega-Peñaloza, J. Mateos, X. Companyó, M. Escudero-Casao and L. Dell'Amico, Angew. Chem., Int. Ed., 2021, 60, 1082 CrossRef.
- M. A. Tasdelen and Y. Yagci, Angew. Chem., Int. Ed., 2013, 52, 5930 CrossRef CAS.
-
(a) K. Matsumoto, H. Iida, T. Uchida, Y. Yabe, A. Kakehi and J. W. Lown, Can. J. Chem., 1994, 72, 2108 CrossRef CAS;
(b) C. Wittland, M. Arend and N. Risch, Synthesis, 1996, 367 CrossRef CAS.
- L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 3730 CrossRef CAS PubMed.
- This scenario is in agreement with the behaviour of radical cations generated from different phenyl-substituted aziridines by pulse radiolysis, please see: C. Gaebert, J. Mattay, M. Toubartz, S. Steenken, B. Müller and T. Bally, Chem.–Eur. J., 2005, 11, 1294 CrossRef CAS.
-
(a) X. Ju, D. Li, W. Li, W. Yu and F. Bian, Adv. Synth. Catal., 2012, 354, 3561 CrossRef CAS;
(b) S. Zhu, A. Das, L. Bui, H. Zhou, D. P. Curran and M. Rueping, J. Am. Chem. Soc., 2013, 135, 1823 CrossRef CAS PubMed;
(c) D. Chandrasekhar, S. Borra, J. S. Kapure, G. S. Shivaji, G. Srinivasulu and R. A. Maurya, Org. Chem. Front., 2015, 2, 1308 RSC;
(d) H. Liu, L. Ma, R. Zhou, X. Chen, W. Fang and J. Wu, ACS Catal., 2018, 8, 6224 CrossRef CAS.
- P. Pfaff, F. Anderl, M. Fink, M. Balkenhohl and E. Carreira, J. Am. Chem. Soc., 2021, 143, 14495 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Tommaso
Bortolato‡
a,
Tommaso
Bortolato‡
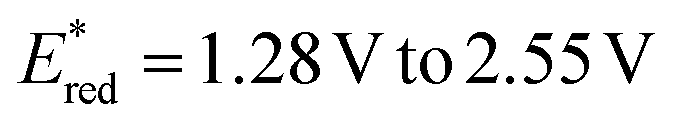 (vs. SCE in MeCN).7a,17 Dicyanoanthracene (DCA) outperformed the other catalysts, furnishing product 9 in 94% yield as the single anti-diastereomer (entry 5). Using syn-7, a modest diastereomeric ratio (dr) was observed, slightly favouring the syn-product 9 (entry 7). Furthermore, this photocatalytic reaction is very fast (see ESI† for the kinetic profile of the reaction), delivering product 9 after only 10 minutes of irradiation (entry 8), establishing its place among other photocatalytic click reactions.18 Finally, without irradiation, no reaction was observed (entry 9). With the optimized reaction conditions in hand, we next explored the generality of the process (Fig. 2). Albeit the reaction showed fast kinetics, we decided to keep the reaction time at 1.5 hours in order to have more general conditions regardless of the nature of the acceptor.
(vs. SCE in MeCN).7a,17 Dicyanoanthracene (DCA) outperformed the other catalysts, furnishing product 9 in 94% yield as the single anti-diastereomer (entry 5). Using syn-7, a modest diastereomeric ratio (dr) was observed, slightly favouring the syn-product 9 (entry 7). Furthermore, this photocatalytic reaction is very fast (see ESI† for the kinetic profile of the reaction), delivering product 9 after only 10 minutes of irradiation (entry 8), establishing its place among other photocatalytic click reactions.18 Finally, without irradiation, no reaction was observed (entry 9). With the optimized reaction conditions in hand, we next explored the generality of the process (Fig. 2). Albeit the reaction showed fast kinetics, we decided to keep the reaction time at 1.5 hours in order to have more general conditions regardless of the nature of the acceptor.