
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical assembly of isoxazoles via a four-component domino reaction†
Yuanyuan
Zhao‡
a,
Xinyue
Li‡
a,
Simon L.
Homölle
 b,
Bin
Wang
*abc and
Lutz
Ackermann
*b
b,
Bin
Wang
*abc and
Lutz
Ackermann
*b
aKey Laboratory of Xin'an Medicine of the Ministry of Education, School of Pharmacy, Anhui University of Chinese Medicine, Hefei, 230038, P. R. China. E-mail: bw5654@ahtcm.edu.cn
bInstitut für Organische und Biomolekulare Chemie and Wöhler Research Institute for Sustainable Chemistry(WISCh), Georg-August-Universität Göttingen, Tammannstraße 2, 37077, Göttingen, Germany. E-mail: Lutz.Ackermann@chemie.uni-goettingen.de
cInstitute of Pharmaceutical Chemistry, Anhui Academy of Chinese Medicine, Hefei, 230038, P. R. China
First published on 11th December 2023
Abstract
Multicomponent domino reactions via electrochemical annulations have emerged as a robust strategy for the rapid assembly of heterocyclics. Herein, an electrochemical annulation via a [1 + 2 + 1 + 1] four-component domino reaction was accomplished in a user-friendly undivided cell setup to assemble valuable five-membered isoxazole motifs. Our approach is characterized by a high level functional group tolerance and operational simplicity, avoiding the tedious and time-consuming preparation of pre-functionalized substrates. Detailed mechanistic studies were conducted including isotopic labeling, kinetic studies, cyclic voltammetry (CV) analysis, and intermediate characterization, providing support for a radical pathway.
Introduction
Domino reactions1 have emerged as powerful transformative strategies for the highly efficient construction of five-, six-, and seven-membered heterocycles.2 Despite undeniable progress, the presence of sacrificial chemical oxidants (e.g., Ag2O) and tedious operating procedures are generally indispensable in these processes. However, since the success of Kolbe electrolysis in 1847,3 electricity has consistently been recognized as a green, cost-effective, and sustainable redox equivalent, and it has found widespread use in accessing desired targets, particularly in their renaissance during the last few years.4 Notably, electro-organic synthesis, particularly the electrochemical annulation strategy, holds distinctive advantages such as operational simplicity and the utilization of sustainable electricity. This avenue in organic chemistry shows promise, offering abundant opportunities for the development of novel organic reaction methods, encompassing the synthesis of C–O, C–N, and C–C bonds.4f,gIsoxazoles represent important structural motifs.5 These five-membered heterocyclic compounds containing a weak N–O bond have gained recognition as relevant building blocks in approved pharmaceutical drugs,6a bioactive molecules,6b crop protection,6c and natural products (Fig. 1).6d As such, several strategies have been developed for the successful synthesis of isoxazoles, for instance the Claisen condensation (Scheme 1a),7 cyclo-isomerization (Scheme 1b),8 and 1,3-dipolar cycloaddition (Scheme 1c).9 These approaches have benefitted from significant contributions by the research groups of Claisen,7 Willis,8 Kittakoop,9a Fokin,9b and others.10 However, these strategies have considerable limitations, such as the use of expensive and environmentally toxic transition metal catalysts (e.g. rhodium, and palladium), the requirement of pre-functionalized substrates, and having a low functional group tolerance. Recently, several innovative multicomponent strategies leading to substituted isoxazoles have been reported. These approaches involve diverse intermediates such as α-alkynyl ketoximes11a and nitrostyrolenes.11b These consecutive transformations within these multicomponent systems simplify the process of creating intricate heterocyclic compounds from simple starting materials, highlighting the potential of one-pot multicomponent reactions.
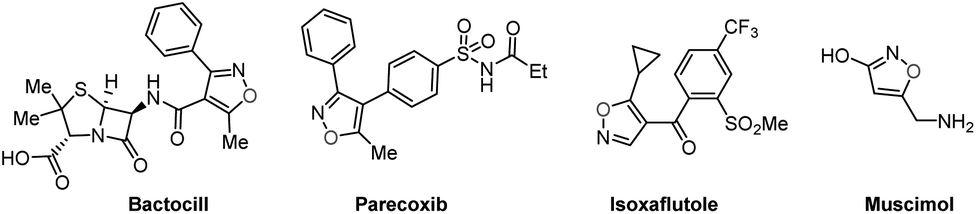 | ||
| Fig. 1 Examples of bearing isoxazoles with versatile functions. | ||
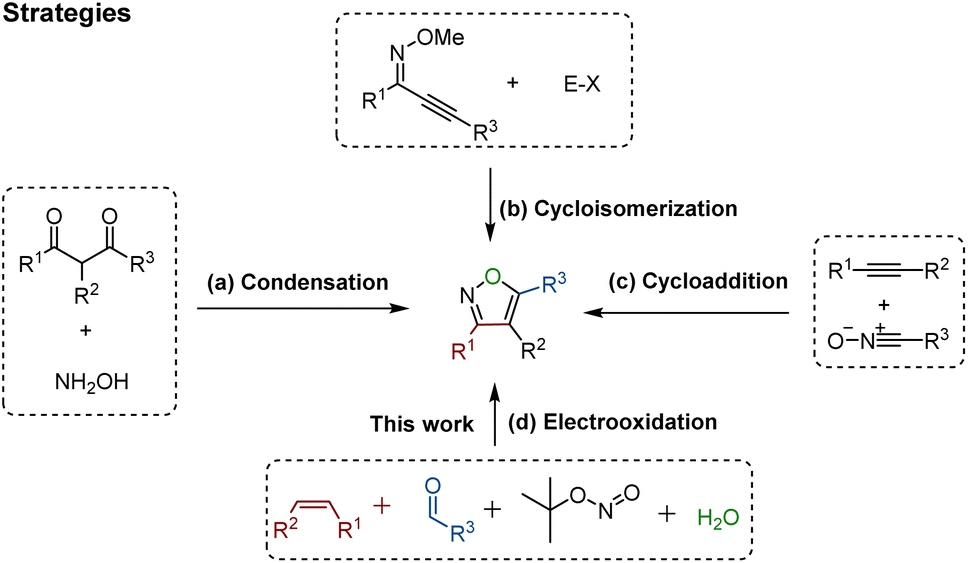 | ||
| Scheme 1 Synthetic methods of isoxazoles. | ||
Inspired by our previously successful approach for alkene cycloaddition,12 we herein present a novel four-component domino reaction utilizing electricity as an environmentally friendly oxidant for the efficient assembly of valuable five-membered isoxazole skeletons (Scheme 1d). The distinctive features of this strategy include: (a) the first electro-oxidative [1 + 2 + 1 + 1] cycloaddition, (b) identification and characterization of key β-carbonyl ketoxime intermediates, (c) use of H2O as an unusual oxygen source, and (d) detailed mechanistic insights through isotopic labelling, kinetic investigations, CV analysis, and intermediate reactivity studies.
Results and discussion
Optimization of the reaction conditions
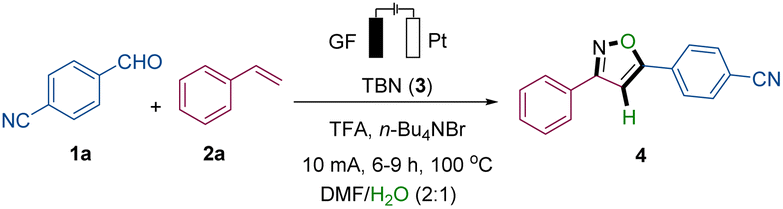
We started our studies with 4-cyanobenzaldehyde (1a) and styrene (2a), in the presence of tert-butyl nitrite (TBN, 3), as model substrates, and H2O in a user-friendly undivided cell setup using a graphite felt (GF) anode and a platinum plate (Pt) cathode (refer to Table 1 and S1 in the ESI for details). The solvent of the reaction has great influence on the yield and selectivity of the target. For instance, when employing solvents containing hydroxyl groups like hexafluoroisopropanol or tert-butanol, the desired product cannot be obtained. Instead, the system primarily facilitates the oxidation of the substrate, styrene, to form benzonitrile (Table S1†). The best solvent system was found to be a mixture of DMF and H2O (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (entries 1 and 2). Interestingly, LiBr as an electrolyte exhibited better electron transfer efficiency than n-Bu4NPF6 but was, however, slightly inferior to n-Bu4NBr, suggesting the potential significance of the bromine anion in the synthesis (entry 3).13 The introduction of acid into the reaction system proved to be advantageous for the reaction efficiency, with trifluoroacetic acid (TFA) being optimal (entry 4).14 Control experiments confirmed the indispensable role of electricity while also suggesting the dual functionality of TBN as both the substrate and oxidation agent (entries 5–6). Through electrode optimization experiments, it has been demonstrated that utilizing platinum as a cathode yields superior outcomes (entry 7). Under constant current conditions, The reaction time significantly influences the formation of the desired target product 4 (entry 8 and Fig. S12, see ESI†). Broadly, the reaction progress can be categorized into three distinct stages. Initially, within the first 2 hours, the reaction system predominantly experiences an induction period, characterized by a product concentration below 0.02 M. Subsequently, spanning from 2 to 6 hours, the system enters a developmental phase, witnessing an increase in product concentration from 0.02 M to 0.155 M. Finally, beyond 6 hours, the system stabilizes into an equilibrium period. Moreover, altering the reaction temperature within certain limits did not significantly compromise the efficacy of the reaction 1a (entry 9).
:1) (entries 1 and 2). Interestingly, LiBr as an electrolyte exhibited better electron transfer efficiency than n-Bu4NPF6 but was, however, slightly inferior to n-Bu4NBr, suggesting the potential significance of the bromine anion in the synthesis (entry 3).13 The introduction of acid into the reaction system proved to be advantageous for the reaction efficiency, with trifluoroacetic acid (TFA) being optimal (entry 4).14 Control experiments confirmed the indispensable role of electricity while also suggesting the dual functionality of TBN as both the substrate and oxidation agent (entries 5–6). Through electrode optimization experiments, it has been demonstrated that utilizing platinum as a cathode yields superior outcomes (entry 7). Under constant current conditions, The reaction time significantly influences the formation of the desired target product 4 (entry 8 and Fig. S12, see ESI†). Broadly, the reaction progress can be categorized into three distinct stages. Initially, within the first 2 hours, the reaction system predominantly experiences an induction period, characterized by a product concentration below 0.02 M. Subsequently, spanning from 2 to 6 hours, the system enters a developmental phase, witnessing an increase in product concentration from 0.02 M to 0.155 M. Finally, beyond 6 hours, the system stabilizes into an equilibrium period. Moreover, altering the reaction temperature within certain limits did not significantly compromise the efficacy of the reaction 1a (entry 9).
|
|
||
|---|---|---|
| Entry | Deviation and standard conditions | Yield [4]b |
| a Reaction conditions: 1 (0.75 mmol), 2 (2.25 mmol), 3 (2.25 mmol), n-Bu4NBr (0.75 mmol), TFA (1.50 mmol), solvent (3.0 mL), 100 °C, 6–9 h, constant current electrolysis (CCE) at 10 mA, GF electrodes (10 mm × 10 mm × 6 mm), Pt electrodes (10 mm × 0 mm × 0.25 mm). b Yield of the isolated product, TFA = trifluoroacetic acid. | ||
| 1 | None | 62 |
| 2 | CH3CN instead of DMF | 21 |
| 3 | n-Bu4NPF6/LiBr instead of n-Bu4NBr | 25/61 |
| 4 | AcOH instead of TFA | 40 |
| 5 | No current | 8 |
| 6 | CCE = 5 mA | 35 |
| 7 | Pt(+) I GF(−) | 54 |
| 8 | t = 4 h/10 h | 38/61 |
| 9 | T = 80 °C | 52 |
Mechanistic studies
Several meticulously designed experiments were conducted to elucidate the potential reaction mechanism. These were subsequently monitored by nuclear magnetic resonance spectroscopy (NMR spectroscopy), High Resolution Mass Spectrometry (HR-MS), and Gas Chromatography-Mass Spectrometry (GC-MS) (for detailed information, refer to Fig. 2, S1–S8, Schemes S1 and S2 in the ESI†). Under standard conditions, isoxazole 4 was obtained with a yield of 62% (Table 1, entry 1). However, when 1.0 equivalent of 2,2,6,6-tetramethylpiperidoxyl (TEMPO) or 2,6-di-tert-butyl-4-methylphenol (BHT) was added to the reaction system, the yield significantly decreased, indicating that TEMPO or BHT had inhibitory effects on the formation of the desired product 4 (Fig. 2a). In addition, BHT adduct 6 (see Fig. S3†) was successfully isolated and confirmed using HR-MS, further supporting the occurrence of N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O radicals in the system. Intermolecular competition experiments involving various substituted benzaldehydes under standard conditions demonstrated that the reaction exhibited higher efficiency when using benzaldehydes containing electron-deficient substituents (Fig. 2b).
O radicals in the system. Intermolecular competition experiments involving various substituted benzaldehydes under standard conditions demonstrated that the reaction exhibited higher efficiency when using benzaldehydes containing electron-deficient substituents (Fig. 2b).
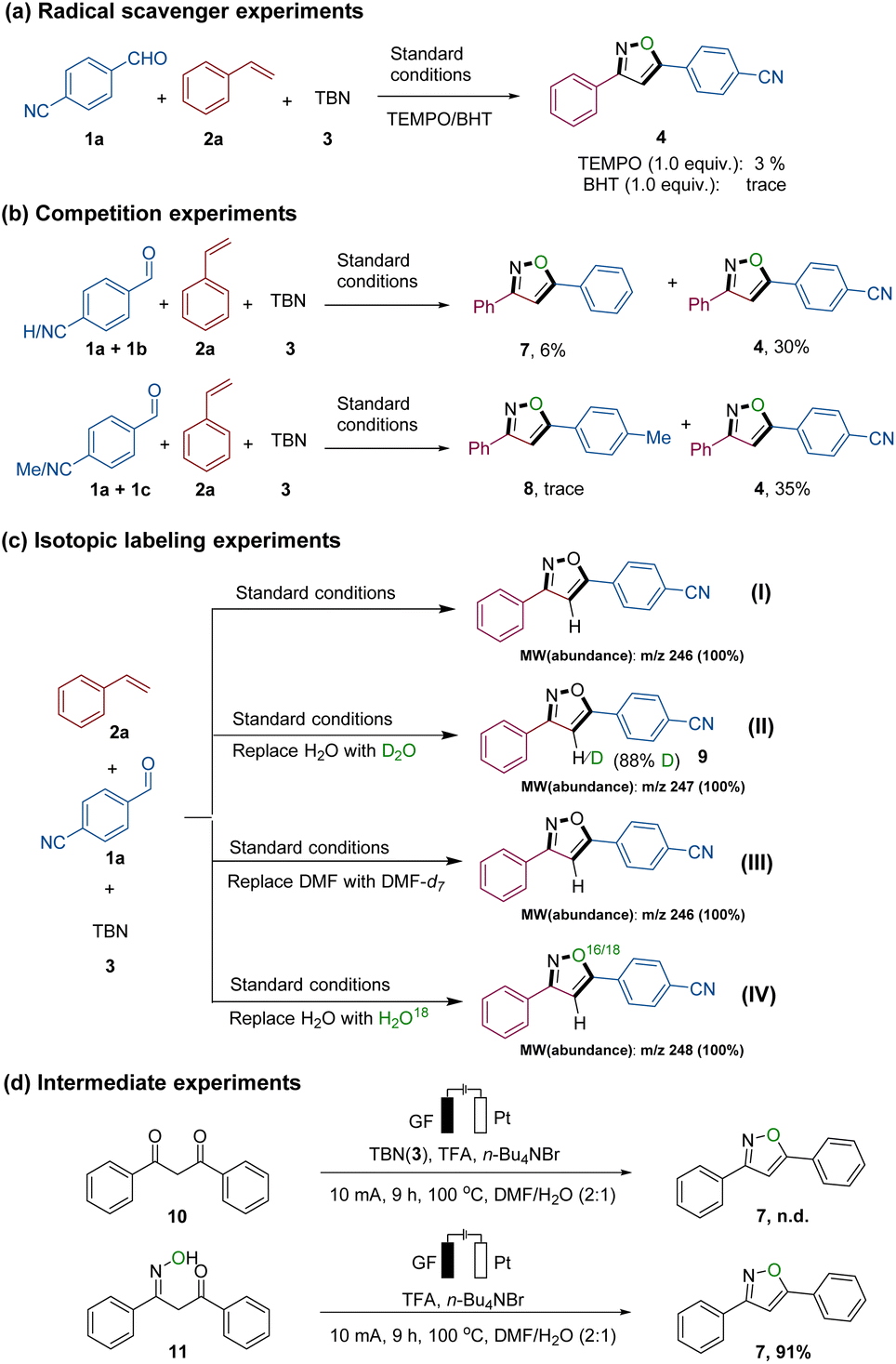 | ||
| Fig. 2 Mechanistic studies: (a) the effect of radical scavengers TEMPO and BHT on the domino reaction; (b) competition reaction of 1a/1b, and 1a/1c, respectively, with 2a; (c) isotope experiments: using DMF and H2O (I), DMF and D2O (II), DMF-d7 and H2O (III) and DMF and H2O18 (IV) as solvents; (d) experiments for yielding isoxazole 7via presumed intermediates 10 and 11. | ||
Afterwards, we conducted experiments in the presence of DMF-d7, D2O, and H2O18 to investigate whether solvent molecules participate in the formation of the isoxazole (refer to Fig. 2c, Schemes S1 and S2†). When employing DMF/H2O (2:1) or DMF-d7/H2O (2:1) as the solvent system, GC-MS analysis indicated that isoxazole 4 exhibited 100% abundance at m/z 246 (isoxazole 4, MW = 246.08). However, when DMF/D2O (2:1) was used, 100% abundance was observed at m/z 247. Furthermore, 1H NMR results indicated that the deuteration ratio of the hydrogen atom at position 4 of the isoxazole 4 skeleton was 88% (see NMR analysis in Fig. S4†). Notably, when H2O was replaced with H2O,18 GC-MS analysis showed an unexpected 100% abundance at m/z 248. This observation may be attributed to the interaction of TBN with water molecules, resulting in the in situ generation of a new NO species (IV). Subsequently, the oxygen atom from the water was successfully incorporated into the isoxazole molecule. Therefore, based on our observations, there are two main sources of nitric oxide species involved in isoxazole generation: Pathway one involves the release of NO species (III) from TBN, while pathway two entails the interaction of TBN with water to produce the molecule HNO2, which further generates the NO species (IV). These findings imply that (a) the solvent water actively participates in the formation of the isoxazole, (b) the hydrogen atom at position 4 of the isoxazole 4 skeleton partly originates from the H2O molecule, and (c) the oxygen atom of the isoxazole skeleton is derived not only from TBN but also from H2O (Scheme S2†).
Drawing upon our previous discoveries and relevant reports,7b,12b,15 we introduced two presumed intermediates, 10 and 11, individually into the reaction (Fig. 2d). When we changed the mixture of 1a and 2a to 1,3-dicarbonyl compound 10, the isoxazole 4 was not detected. However, when 11 was used as a substrate, the product 4 was formed in excellent yield (91%). This leads us to believe that compound 11 is a plausible intermediate in the generation of 4. Moreover, kinetic studies (refer to Fig. S6–S8†) indicated that the reaction exhibited a first-order dependence on the concentration of alkene 2 as well as TBN (3), while a zero-order dependence on the concentration of aldehyde 1 was observed. These findings strongly suggest that in the rate-limiting step of the transformation solely alkene 2 and TBN (3) are involved.
Subsequently, we conducted cyclic voltammetry studies to assess the oxidative electrochemical transformation (see Fig. 3). Substrates 1a and 3 exhibited irreversible oxidation peaks at 0.99 and 1.13 V vs. saturated calomel electrode (SCE), respectively. Substrate 2a did not exhibit an obvious oxidation peak between the potential window used. Interestingly, when a mixture of substrates 1a, 2a, and 3 was used, the oxidation potential of 1a was shifted to 0.73 V vs. SCE, indicating crucial anodic oxidation of the substrate. Additionally, irreversible oxidation of product 4 was observed at potentials beyond 1.25 V vs. SCE, ensuring that the isoxazole-formation can be carried out under shown mild constant-potential electrocatalysis.
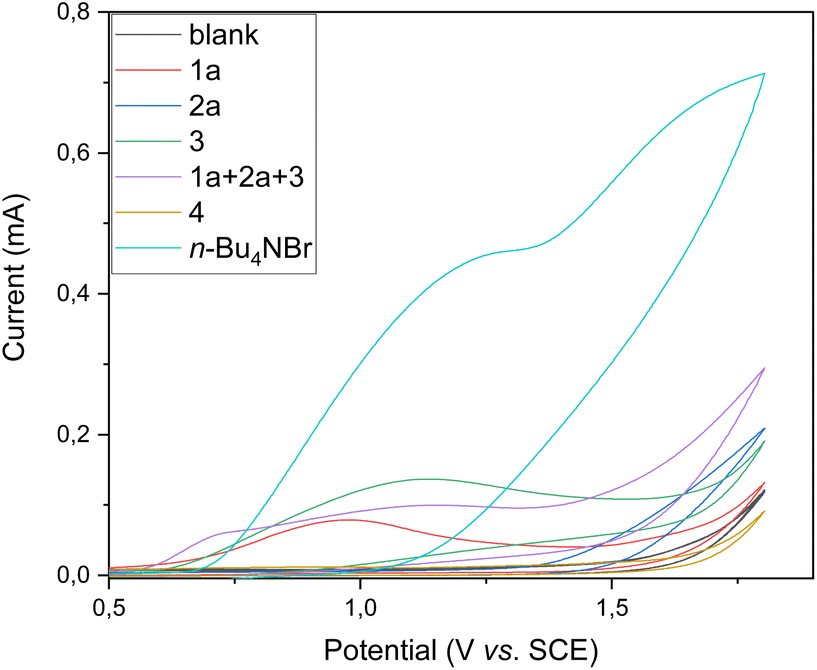 | ||
| Fig. 3 Cyclic voltammetry studies at 100 mV s−1 in DMF/H2O (2:1) using tetrabutylammonium hexafluorophosphate (TBAPF6, 100 mM) as the electrolyte. | ||
Based on the aforementioned findings and existing reports,16 we propose a plausible reaction mechanism, which is depicted in Scheme 2. Initially, bromine anions from the electrolyte undergo oxidation at the anode,13 leading to the formation of bromine radical I. Simultaneously, under thermal conditions, TBN undergoes homolysis, generating radicals II and III.15 The reaction commences with the formation of acyl radical A, which is generated in situ through the reaction of aldehyde 1 with radical I or II. Subsequently, radical A attacks the double bond of alkene, forming a carbon-central radical B. Concurrently, with the assistance of NO radical III or IV, compound C is generated. This is then followed by an isomerization process, rapidly yielding the crucial intermediate β-carbonyl ketoxime D. The addition of TFA promotes the dehydration of intermediate D facilitating the formation of the desired isoxazole F from E. Simultaneously, F has the potential to undergo hydrogen loss at site 4 of the isoxazole skeleton when influenced by a carboxylic acid anion. Subsequently, it can abstract a proton from deuterated water molecules to obtain deuterium-substituted moiety G.
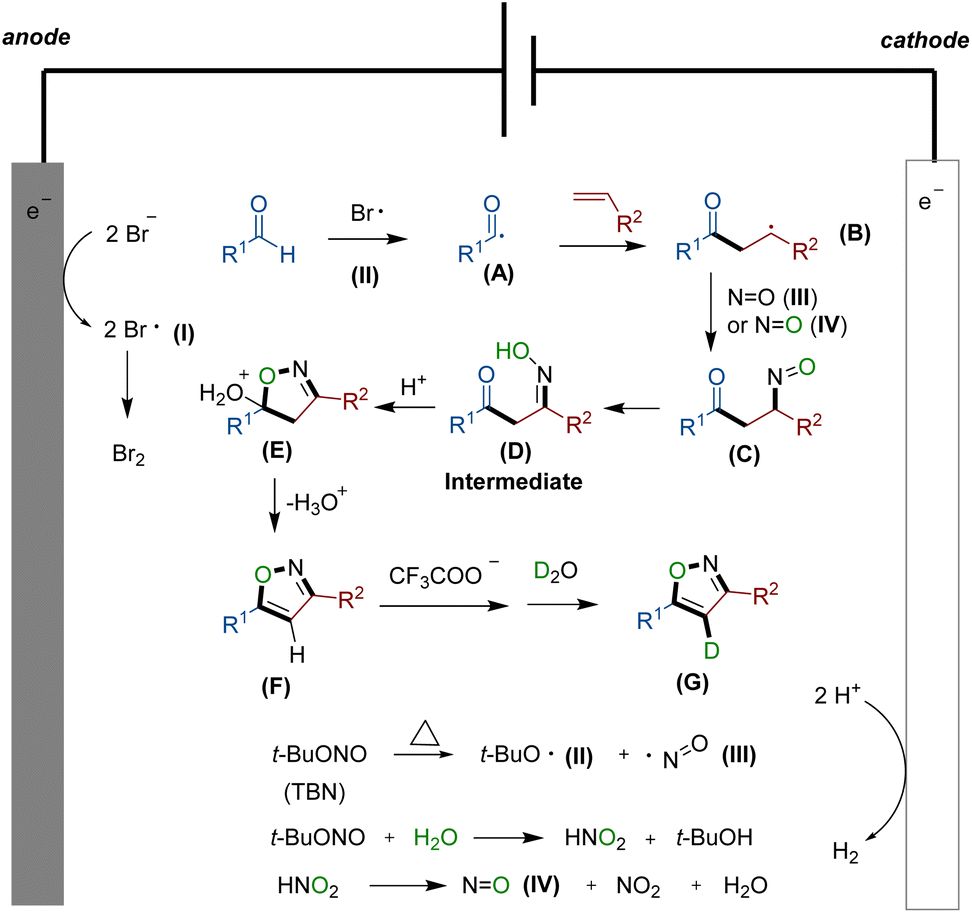 | ||
| Scheme 2 Plausible reaction mechanism. | ||
Substrate scope
With the optimized experimental conditions in hand, we proceeded to explore the viable substrate scope of the four-component domino reaction using variously substituted aldehydes 1 (refer to Scheme 3). The reaction demonstrated high efficacy in the selective electrochemical alkene annulations, accommodating aldehydes bearing both electron-donating and electron-withdrawing groups (4, 7–8, 12–27). A wide range of functional groups, including cyano (4), alkyl (8, 12–13), ether (14), chloro (15), nitryl (16), fluoro (17), bromo (18–20), trifluoromethyl (21–23), and hydroxy (27), were well tolerated within the four-component domino reaction. Notably, even challenging substrates such as heterocyclic substrate 4-formyl pyridine, aliphatic substrate cyclohexane carboxaldehyde, and condensed ring substrate 2-naphthaldehyde could be efficiently transformed into the desired products 24, 25, and 26, respectively, with moderate to good yields. Subsequently, the scope was further extended to various tethered styrene substrates (refer to Scheme 3). Styrenes bearing tert-butyl (30), nitryl (33), and chloro (34) substituents effectively furnished the desired products with good yields. Furthermore, various functional groups, including alkyl (28), ether (29), bromo (31), and fluoro (32), were also well tolerated in the electrochemical annulation system. The unexpectedly relatively lower yields can be attributed to the fact that the substrate styrene may undergo conversion to both the corresponding major product, isoxazole, and the by-product 5, as evidenced by the results of electrochemical annulation and previous findings.17 Notably, when α-methyl styrene was used as a substrate, no desired product was observed, revealing that steric effects are significantly important in this transformation. Remarkably, the electrochemical annulation system exhibited excellent functional group tolerance, enabling the facile assembly of di-substituted isoxazole products, namely 36–40. Moreover, the configuration of product 40 was further confirmed by X-ray crystal diffraction (Scheme 3 and ESI†). In our protocol, the electronic properties of the substrate play a crucial role in product formation. For instance, employing electron-rich benzaldehyde substrates and electron-deficient olefin substrates often results in yields below 40%. Additionally, ortho-substituted substrates typically yield only moderate yields, underscoring the substantial impact of steric effects in this transformation. Conversely, choosing an electron-deficient benzaldehyde and an electron-rich olefin substrate with reduced steric hindrance can enhance the efficiency of this reaction.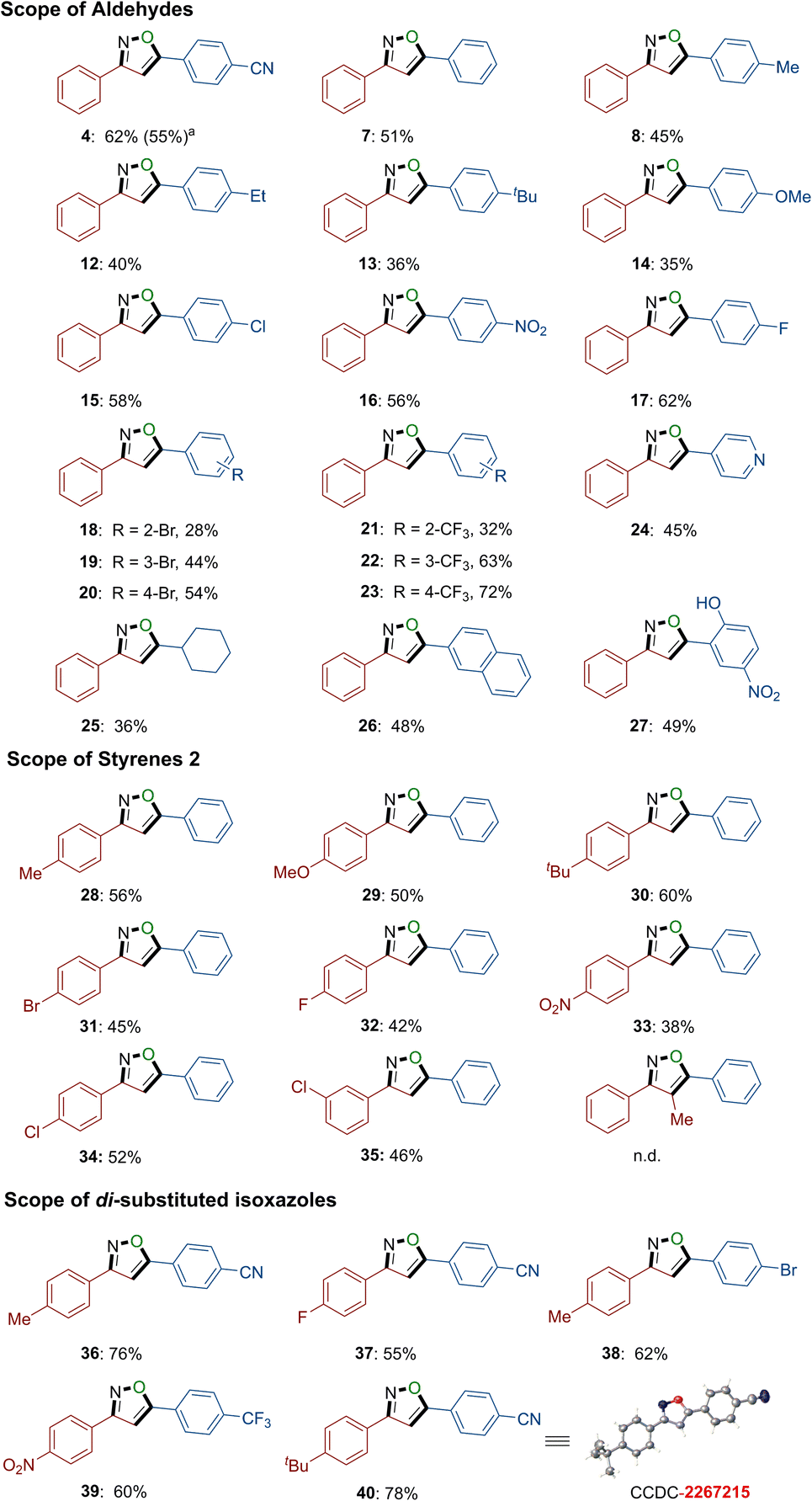 | ||
| Scheme 3 Electrochemical synthesis of isoxazoles via a four-component domino reaction. Reaction conditions: 1 (0.75 mmol), 2 (2.25 mmol), 3 (2.25 mmol), n-Bu4NBr (0.75 mmol), TFA (1.50 mmol), DMF/H2O (2:1, 3.0 mL), 100 °C, 6–9 h, CCE @ 10 mA, GF electrodes (10 mm × 10 mm × 6 mm), Pt electrodes (10 mm × 10 mm × 0.25 mm). Yield of the isolated product. aGram-scale synthesis. | ||
Late-stage diversification
The exceptional synthetic applicability of the electrochemical annulation approach was further extended to late-stage diversification (Scheme 4), leading to the formation of synthetically valuable building blocks and biologically significant motifs. Treating isoxazole 4 with 4-bromobenzonitrile in the presence of a palladium chloride catalyst resulted in the formation of a particularly noteworthy 3,4,5-trisubstituted isoxazole 41.18 Additionally, isoxazole 4 underwent reaction with hydrogen peroxide, generating a drug-like compound containing an amide group (45). Furthermore, the N–O bond within the isoxazole skeleton was utilized as a synthon and successfully cleaved using a copper catalyst, yielding a drug-like molecule, the β-aminoketone derivative (42).19 Moreover, product isoxazole 7 served as a versatile precursor of two synthetically useful isoxazoles: one possessing a formyl group (43) and another a phenyl pyridine derivative (44).20 These transformations were achieved through a ruthenium-catalyzed coupling reaction involving vinylene carbonate and a copper-catalyzed cyclization reaction involving dimethylsulfoxide (DMSO), respectively.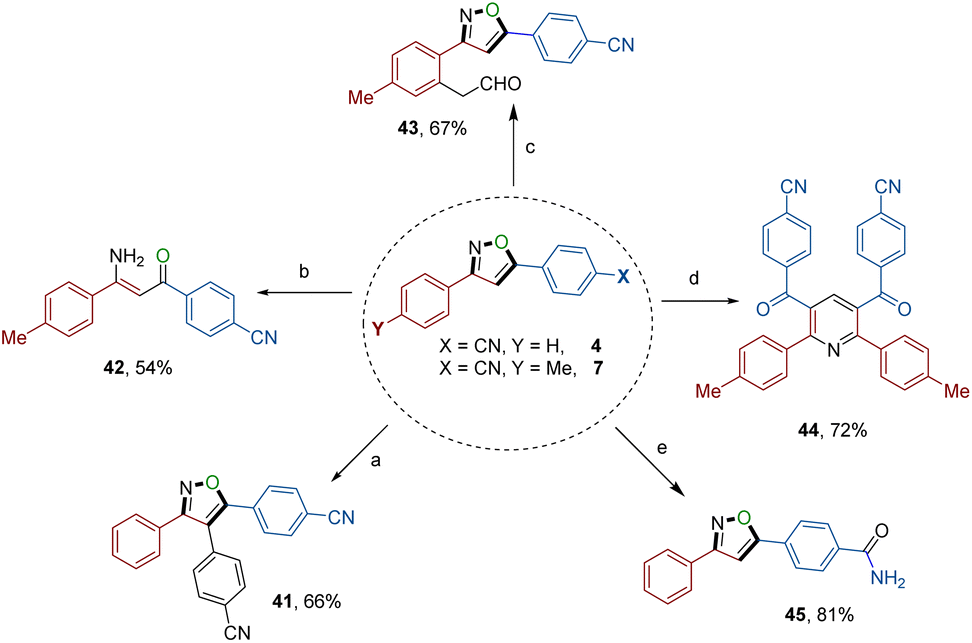 | ||
| Scheme 4 Late-stage diversification to access 3,4,5-trisubstituted isoxazole 41, aminoketone 42, and isoxazoles possessing formyl (43), phenyl pyridine (44), and amide (45) groups. (For detailed information, see the ESI†). | ||
Conclusion
In conclusion, we have introduced an innovative electrochemical annulation strategy for the cost-effective assembly of 3,5-di-substituted isoxazoles, which can be successfully accomplished by harnessing readily available substrates. The exceptional versatility of this approach is underscored by its capacity for late-stage diversification, exemplified through the synthesis of drug-like molecules and valuable synthons such as aminoketones, phenyl pyridines, amides, and 3,4,5-tri-substituted isoxazoles. This four-component domino reaction stands as a powerful and adaptable route for the construction of isoxazoles, showcasing significant potential for the creation of diverse heterocyclic compounds. Applications of this four-multicomponent domino reaction hold promising implications in drug discovery and materials science domains.Data availability
All experimental data, procedures for data analysis, and pertinent data sets are provided in the ESI.†Author contributions
Y. Z and Y. L. conducted the experiments; Y. Z did the CCDC; S. L. H. carried out the cyclic voltammetry studies; B. W. and L. A. conceived the project; B. W. and L. A. wrote, reviewed, and edited the manuscript; All authors discussed the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the support from the Overseas Study Project (gxgwfx2020039), the Natural Science Foundation of Anhui province (2008085MH271), the ERC Advanced Grant (101021358) and the DFG Gottfried Wilhelm Leibniz award (L. A.).Notes and references
- L. F. Tietze, Chem. Rev., 1996, 96, 115–136 CrossRef CAS PubMed.
- (a) Y. Liu, D. Ni, B. G. Stevenson, V. Tripathy, S. E. Braley, K. Raghavachari, J. R. Swierk and M. K. Brown, Angew. Chem., Int. Ed., 2022, 61, e202200725 CrossRef CAS; (b) D. M. Flores, M. L. Neville and V. A. Schmidt, Nat. Commun., 2022, 13, 2764 CrossRef CAS PubMed; (c) T. Fujii, S. Gallarati, C. Corminboeuf, Q. Wang and J. Zhu, J. Am. Chem. Soc., 2022, 144, 8920–8926 CrossRef CAS; (d) X. Abel-Snape, G. Wycich and M. Lautens, ACS Catal., 2022, 12, 3291–3301 CrossRef CAS; (e) J. Gong, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2022, 61, e202211470 CrossRef CAS; (f) Y. Wang, J. C. A. Oliveira, Z. Lin and L. Ackermann, Angew. Chem., Int. Ed., 2021, 60, 6419–6424 CrossRef CAS.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS.
- (a) T. Münchow, S. Dana, Y. Xu, B. Yuan and L. Ackermann, Science, 2023, 379, 1036–1042 CrossRef; (b) S. Ji, L. Zhao, B. Miao, M. Xue, T. Pan, Z. Shao, X. Zhou, A. Fu and Y. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202304434 CrossRef CAS; (c) S. L. Homölle, M. Stangier, E. Reyes and L. Ackermann, Precis. Chem., 2023, 1, 382–387 CrossRef PubMed; (d) L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC; (e) L. Ackermann, Acc. Chem. Res., 2020, 53, 84–104 CrossRef CAS PubMed; (f) T. Ali, H. Wang, W. Iqbal, T. Bashir, R. Shah and Y. Hu, Adv. Sci., 2023, 10, 2205077 CrossRef CAS PubMed; (g) Y. A. Wu, R. A. Wang, S. Y. Jiang, T. B. Jiang, J. R. Song, J. Shi, W. Wu, W. D. Pan and H. Ren, Green Chem., 2022, 24, 6720–6726 RSC.
- (a) C. P. Pandhurnekar, H. C. Pandhurnekar, A. J. Mungole, S. S. Butoliya and B. G. Yadao, J. Heterocycl. Chem., 2023, 60, 537–565 CrossRef CAS; (b) F. Hu and M. Szostak, Adv. Synth. Catal., 2015, 357, 2583–2614 CrossRef CAS.
- (a) G. C. Arya, K. Kaur and V. Jaitak, Eur. J. Med. Chem., 2021, 221, 113511 CrossRef CAS PubMed; (b) B. Heasley, Angew. Chem., Int. Ed., 2011, 50, 8474–8477 CrossRef CAS; (c) X. L. Sun, Z. M. Ji, S. P. Wei and Z. Q. Ji, J. Agric. Food Chem., 2020, 68, 15107–15114 CrossRef CAS PubMed; (d) J. Li, Z. Lin, W. Wu and H. Jiang, Org. Chem. Front., 2020, 7, 2325–2348 RSC.
- (a) R. Harigae, K. Moriyama and H. Togo, J. Org. Chem., 2014, 79, 2049–2058 CrossRef CAS PubMed; (b) L. Claisen and O. E. Lowman, Ber. Dtsch. Chem. Ges., 1888, 21, 1149–1157 CrossRef.
- (a) F. Zhou, C. Li, M. Li, Y. Jin, H. Jiang, Y. Zhang and W. Wu, Chem. Commun., 2021, 57, 4799–4802 RSC; (b) M. Hu, Z. Lin, J. Li, W. Wu and H. Jiang, Green Chem., 2020, 22, 5584–5588 RSC; (c) R. N. Straker, M. K. Majhail and M. C. Willis, Chem. Sci., 2017, 8, 7963–7968 RSC; (d) K. N. Tu, J. J. Hirner and S. A. Blum, Org. Lett., 2016, 18, 480–483 CrossRef CAS PubMed.
- (a) C. Kesornpun, T. Aree, C. Mahidol, S. Ruchirawat and P. Kittakoop, Angew. Chem., Int. Ed., 2016, 55, 3997–4001 CrossRef CAS; (b) S. Grecian and V. V. Fokin, Angew. Chem., Int. Ed., 2008, 47, 8285–8287 CrossRef CAS.
- (a) W. Wu, Q. Chen, Y. Tian, Y. Xu, Y. Huang, Y. You and Z. Weng, Org. Chem. Front., 2020, 7, 1878–1883 RSC; (b) W. Wei, Y. Tang, Y. Zhou, G. Deng, Z. Liu, J. Wu, Y. Li, J. Zhang and S. Xu, Org. Lett., 2018, 20, 6559–6563 CrossRef CAS PubMed; (c) P. A. Allegretti and E. M. Ferreira, Chem. Sci., 2013, 4, 1053–1058 RSC; (d) D. Roy, S. Mom, S. Royer, D. Lucas, J. C. Hierso and H. Doucet, ACS Catal., 2012, 2, 1033–1041 CrossRef CAS; (e) J. A. Burkhard, B. H. Tchitchanov and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 5379–5382 CrossRef CAS; (f) F. Gasparrini, M. Giovannoli, D. Misiti, G. Natile, G. Palmieri and L. Maresca, J. Am. Chem. Soc., 1993, 115, 4401–4402 CrossRef CAS.
- (a) X. W. Zhang, W. L. Hu, S. Chen and X. G. Hu, Org. Lett., 2018, 20, 860–863 CrossRef CAS PubMed; (b) M. Fu, H. Li, M. Su, Z. Cao, Y. Liu, Q. Liu and C. Guo, Adv. Synth. Catal., 2019, 361, 3420–3429 CrossRef CAS.
- (a) J. Loup, V. Müller, D. Ghorai and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 1749–1753 CrossRef CAS PubMed; (b) L. Chen, Z. Wang, H. Liu, X. Li and B. Wang, Chem. Commun., 2022, 58, 9152–9155 RSC; (c) H. Wang, N. Kaplaneris and L. Ackermann, Cell Rep. Phys. Sci., 2020, 1, 100178 CrossRef.
- (a) L. G. Gombos and S. R. Waldvogel, Sustainable Chem., 2022, 3, 430–454 CrossRef CAS; (b) Z. X. Luo, M. Liu, T. Li, D. C. Xiong and X. S. Ye, Front. Chem., 2021, 9, 796690 CrossRef CAS PubMed.
- E. Trogu, C. Vinattieri, F. D. Sarlo and F. Machetti, Chem. – Eur. J., 2012, 18, 2081–2093 CrossRef CAS.
- (a) B. Wang, Z. Yan, L. Liu, J. Wang, Z. Zha and Z. Wang, Green Chem., 2019, 21, 205–212 RSC; (b) Y. Liu, J. L. Zhang, R. J. Song, P. C. Qian and J. H. Li, Angew. Chem., Int. Ed., 2014, 53, 9017–9020 CrossRef CAS PubMed.
- B. Wang, L. Tang, L. Liu, Y. Li, Y. Yang and Z. Wang, Green Chem., 2017, 19, 5794–5799 RSC.
- U. Dutta, D. W. Lupton and D. Maiti, Org. Lett., 2016, 18, 860–863 CrossRef CAS PubMed.
- S. U. Dighe, S. Mukhopadhyay, S. Kolle, S. Kanojiya and S. Batra, Angew. Chem., Int. Ed., 2015, 54, 10926–10930 CrossRef CAS PubMed.
- C. Wan, J. Pang, W. Jiang, X. W. Zhang and X. G. Hu, J. Org. Chem., 2021, 86, 4557–4566 CrossRef CAS PubMed.
- J. Li, K. Korvorapun, S. D. Sarkar, T. Rogge, D. J. Burns, S. Warratz and L. Ackermann, Nat. Commun., 2017, 8, 15430 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2267215. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05946d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |