
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModular synthesis of functional libraries by accelerated SuFEx click chemistry†
Joshua A.
Homer
 a,
Rebecca A.
Koelln
a,
Andrew S.
Barrow
b,
Timothy L.
Gialelis
b,
Zlata
Boiarska
cd,
Nikita S.
Steinohrt
ef,
Erinna F.
Lee
efg,
Wen-Hsuan
Yang
a,
Robert M.
Johnson
a,
Taemoon
Chung
a,
Amber N.
Habowski
a,
Dharmendra S.
Vishwakarma
a,
Debmalya
Bhunia
a,
Charlotte
Avanzi
h,
Adam D.
Moorhouse
a,
Mary
Jackson
h,
David A.
Tuveson
a,
Scott K.
Lyons
a,
Michael J.
Lukey
a,
W. Douglas
Fairlie
efg,
Shozeb M.
Haider
i,
Michel O.
Steinmetz
cj,
Andrea E.
Prota
c and
John E.
Moses
*a
a,
Rebecca A.
Koelln
a,
Andrew S.
Barrow
b,
Timothy L.
Gialelis
b,
Zlata
Boiarska
cd,
Nikita S.
Steinohrt
ef,
Erinna F.
Lee
efg,
Wen-Hsuan
Yang
a,
Robert M.
Johnson
a,
Taemoon
Chung
a,
Amber N.
Habowski
a,
Dharmendra S.
Vishwakarma
a,
Debmalya
Bhunia
a,
Charlotte
Avanzi
h,
Adam D.
Moorhouse
a,
Mary
Jackson
h,
David A.
Tuveson
a,
Scott K.
Lyons
a,
Michael J.
Lukey
a,
W. Douglas
Fairlie
efg,
Shozeb M.
Haider
i,
Michel O.
Steinmetz
cj,
Andrea E.
Prota
c and
John E.
Moses
*a
aCancer Center, Cold Spring Harbor Laboratory, 1 Bungtown Rd, Cold Spring Harbor, NY 11724, USA. E-mail: moses@cshl.edu
bLa Trobe Institute for Molecular Science, La Trobe University, Melbourne, VIC 3086, Australia
cLaboratory of Biomolecular Research, Division of Biology and Chemistry, Paul Scherrer Institut, Villigen PSI, 5232, Switzerland
dDepartment of Chemistry, Università degli Studi di Milano, Via Golgi 19, 20133 Milan, Italy
eOlivia Newton-John Cancer Research Institute, Heidelberg, Victoria 3084, Australia
fSchool of Cancer Medicine, La Trobe University, Melbourne, Victoria 3086, Australia
gDepartment of Biochemistry and Chemistry, La Trobe Institute for Molecular Science, La Trobe University, Melbourne, Victoria 3086, Australia
hMycobacteria Research Laboratories, Department of Microbiology, Immunology and Pathology, Colorado State University, Fort Collins, CO 80523, USA
iSchool of Pharmacy, University College London, 29-39 Brunswick Square, London, WC1N 1AX, UK
jBiozentrum, University of Basel, 4056 Basel, Switzerland
First published on 4th March 2024
Abstract
Accelerated SuFEx Click Chemistry (ASCC) is a powerful method for coupling aryl and alkyl alcohols with SuFEx-compatible functional groups. With its hallmark favorable kinetics and exceptional product yields, ASCC streamlines the synthetic workflow, simplifies the purification process, and is ideally suited for discovering functional molecules. We showcase the versatility and practicality of the ASCC reaction as a tool for the late-stage derivatization of bioactive molecules and in the array synthesis of sulfonate-linked, high-potency, microtubule targeting agents (MTAs) that exhibit nanomolar anticancer activity against multidrug-resistant cancer cell lines. These findings underscore ASCC's promise as a robust platform for drug discovery.
Introduction
Click chemistry1–9 is a democratizing style of synthesis that offers modularity and dependability in crafting functional molecules. Click reactions, exemplified by the Copper-Catalyzed Azide-Alkyne Cycloaddition (CuAAC)3,4 and thiol–ene coupling,10–12 have advanced numerous fields, from drug discovery13–17 and materials science18 to bioconjugation and chemical biology.19,20The introduction of Sulfur(VI) Fluoride Exchange (SuFEx)5,8 technology marked a significant milestone in the field of click chemistry, bringing forth an innovative approach to forming stable sulfur-based linkages under metal-free conditions.5,8,17,21 This advancement in SuFEx chemistry harnesses the distinct properties of high oxidation sulfur–fluoride compounds, as depicted in Fig. 1A.8,22,23 Key examples of these compounds include sulfuryl fluoride (SO2F2), ethenesulfonyl fluoride (ESF),5,17,22,24 and polyvalent thionyl tetrafluoride (SOF4).25 The sulfur–fluorine (S–F) bonds in these compounds are characterized by their inherent, ‘spring-loaded’ reactivity, which can be selectively activated,26–29 embodying the essence of click reactivity.
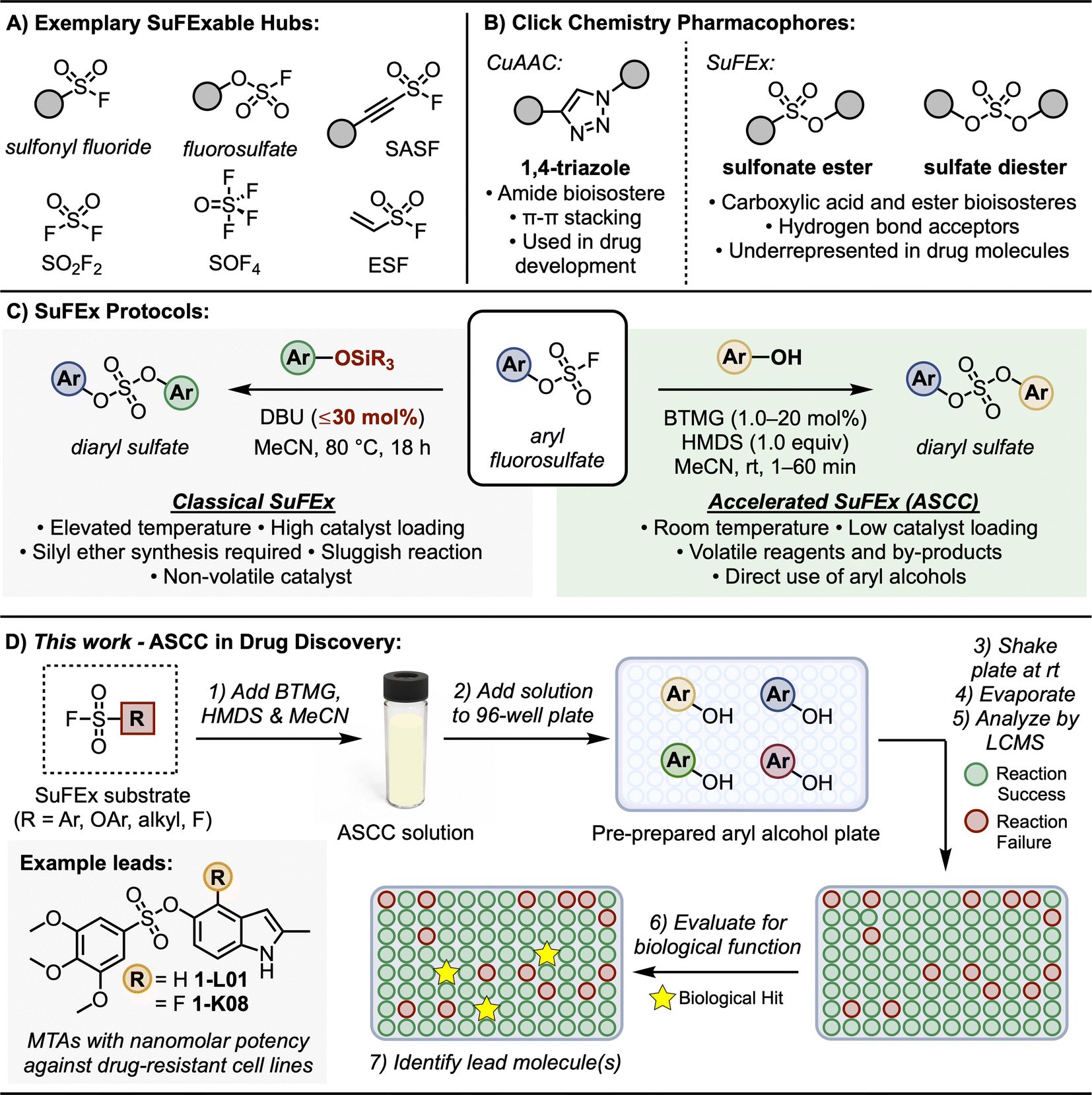 | ||
| Fig. 1 SuFEx click chemistry. (A) SuFExable hubs. (B) Click chemistry derived pharmacophores. (C) Classical and accelerated SuFEx protocols. (D) This work: ASCC in drug discovery. | ||
A notable aspect of SuFEx chemistry is the use of catalysts like DBU and Et3N.5 These catalysts are crucial in initiating the S–F exchange reaction with various nucleophiles. This process leads to the formation of diverse and complex molecular structures, characterized by stable sulfur-to-oxygen,5,30–34 sulfur-to-nitrogen,35–37 or sulfur-to-carbon38,39 bonds. This methodology underscores SuFEx click chemistry's role in enhancing synthetic capabilities, allowing for the efficient creation of intricate molecular architectures in a controlled, metal-free environment. This breakthrough in click chemistry has found broad applications, including in the development of pharmaceuticals, polymers, and other advanced materials, reflecting its versatility and importance in modern chemical synthesis. For example, as SuFEx was designed to help expedite the drug discovery of functional molecules, it was an essential component in shaping the development of Diversity Oriented Clicking (DOC)40–42 – a lead discovery platform that blends contemporary and traditional click reactions to probe chemical space.
The classical SuFEx reaction, as illustrated in Fig. 1C (grey box), while robust and reliable, still presents opportunities for optimization. For example, conditions conducive to catalyst degradation, notably the hydrolysis of DBU,43,44 sometimes necessitate elevated catalyst loading of up to 30 mol%,5 which can complicate the purification process and influence downstream biological assays. The rates of SuFEx reactions can also be notably affected by the steric bulk at the silicon center of the silyl ether substrate. While smaller groups like trimethylsilyl (TMS) ether exhibit rapid reactivity, the more stable tert-butyldimethylsilyl (TBS) groups may require extended reaction times.5
Building upon the foundational principles of SuFEx to optimize reaction efficiency and application, we recently introduced the Accelerated SuFEx Click Chemistry (ASCC) reaction (Fig. 1C, green box).32 This advanced SuFEx protocol enhances reaction kinetics without compromising the inherent advantages such as high yields, robustness, and specificity. The innovation behind ASCC lies in the synergistic interplay between the 2-tert-butyl-1,1,3,3-tetramethylguanidine base (Barton's base, BTMG)45,46 and hexamethyldisilazane (HMDS).47 The ASCC reaction process frequently achieves completion within just a few minutes and, in some cases, this method requires as little as 1 mol% of the catalyst, underscoring the minimal catalyst quantity needed for effective reaction progression.
One of the most significant advantages of the ASCC method is its ability to directly couple aryl and alkyl alcohol modules with SuFExable hubs.32 This approach effectively bypasses the previously necessary step of synthesizing silyl ether substrates, streamlining the process and enhancing overall efficiency. These features, combined with the enhanced volatility of ASCC reagents and by-products (specifically the volatility of BTMG relative to DBU), simplify the purification process and potentially allow for the immediate evaluation of the biological activity of the reaction product.
Furthermore, we,40,41,48 and others,49,50 have demonstrated that the motifs created through click reactions are not merely passive connectors; they can actively interact with biological targets due to their intrinsic hydrogen-bonding and dipole interactions (Fig. 1B). This interaction potential was first demonstrated by the stable 1,4-triazole products of CuAAC reactions – these planar compounds offer π–π stacking interactions vital for pharmacophore structures,48,49,51,52 while also serving as amide bioisosteres.53 Similarly, SuFEx connectors are equally viable as pharmacophores. For instance, sulfonamides (R–SO2–NR2) are a common feature in drug structures and are the primary sulfur-based motif in clinically approved drugs, especially in antibiotics and diuretics.54,55 Sulfonate esters (R–SO2–OR) and sulfate diesters (R–OSO2–OR) are excellent bioisosteric substitutes for carboxylic acids and esters56 and, despite being more lipophilic, they maintain a polarized S–O bond that can form robust electrostatic interactions with target proteins.56 Yet despite their stability, ease of synthesis, and potential as a pharmacophore, sulfur-ester linkages remain notably limited in drug molecules.57,58
Given their efficient, accessible preparation afforded by the ASCC protocol and the prevalent commercial accessibility of aryl and alkyl alcohol substrates,59 we assert that sulfonate esters and sulfate diesters merit broader integration as connectors and pharmacophores in drug discovery programs. In this study, we highlight the effectiveness of ASCC as a practical means of crafting modular libraries in a 96-well plate array format optimized for direct functional screening and lead discovery (Fig. 1D). We showcase this methodology in the fluorosulfonation of commercially available aryl alcohols, the late-stage functionalization (LSF) of bioactive compounds like the hormone estrone and the antibiotic dapsone, and the systematic creation of a library of sulfonate-linked combretastatin A-4 analogs that show promising results as microtubule targeting agents.
Results and discussion
ASCC reaction array development in 96-well plate format
Transitioning reactions to array format is essential for library synthesis.60 This not only refines the synthetic workflow but also dovetails with contemporary drug discovery methodologies that prioritize the rapid generation and assessment of compound libraries, subsequently hastening the identification of prospective drug candidates. Numerous plate-based methodologies have been devised to expedite the process of lead generation. These include techniques like activity-directed synthesis,61–63 synthetic fermentation,64 and direct-to-biology workflows.65 Each of these approaches offers a unique strategy to streamline the discovery and development of new leads in chemical and pharmaceutical research.Several criteria should be met when performing reactions in an array format: reactions should be high-yielding, selective, proceed under mild reaction conditions, and screening for biological function should be possible directly on the unpurified reaction mixture. As such, the evolution of click transformations to a microplate setup has precedence.66–69 A salient example is the work of Sharpless and Dong, who adeptly converted 1224 amines to their azide counterparts within microplates.66 These azides were subsequently linked with an alkyne module through CuAAC reactions, resulting in an extensive array of 1,4-triazole-tethered products.
We adapted the ASCC protocol32 to an array format, specifically harnessing 96-well polypropylene microplates for this purpose (see Table S1† for a complete list of equipment used). We identified a selection of commercially available aryl alcohol fragments suitable for the ASCC reaction; of these, 171 of the 203 chosen fragments (84%) met the rule of three criteria for fragment-based drug discovery.70,71 In essence, these criteria guide users to select fragments with a high probability of being developed into successful lead-like candidates and, thus, drug molecules. Fragments that align with the rule of three guidelines should have a molecular weight of less than 300 Da while incorporating less than three hydrogen bond donors and three hydrogen bond acceptors.70
To ensure the ASCC array protocol would remain effective over a range of substrate combinations, the selected aryl alcohol collection incorporated varied physicochemical properties (e.g., molecular weight, polarizability, lipophilicity). Additionally, the collection included potentially labile functional groups (e.g., amides, esters) and groups that added significant steric bulk to the reacting hydroxy center (e.g., 2,6-di-tert-butyl-4-methylphenol). The remaining aryl alcohol fragments were chosen to represent more structurally intricate drug compounds or natural products, justifying their selection due to their complexity and relevance to the study.
Stock solutions of the aryl alcohols were prepared using acetonitrile (MeCN), dimethyl sulfoxide (DMSO), or a combination thereof, as detailed in Table S2,† ensuring precise allocation of the components to the 96-well plates. In the subsequent step, 3 μmol of aryl alcohol was dispensed into individual wells of a 96-well plate. The solvents were removed by evaporation in a vacuum oven (2 mbar and 50 °C conditions for 3 hours), and the plates were sealed with a polyolefin film under a nitrogen atmosphere. When refrigerated at 4 °C, these sample plates were stable for up to six months.72
The optimal conditions for the ASCC array protocol were identified by reacting the fluorosulfate of p-methoxyphenol (2) with 20 aryl alcohols, as detailed in Fig. 2A and S1.† Changes to reaction concentration, reaction time, and the oscillation speed used to agitate the reaction plates were varied. LCMS analysis was utilized to determine the conversion of each reaction, effectively differentiating between the aryl alcohol starting materials and the resultant sulfate diester products (3). Fig. 2B depicts selected LC traces for the ASCC reaction between 2 and 2-((p-tolylamino)methyl)phenol (D08, please note aryl alcohols are numbered according to Table S2†) to give the corresponding sulfate-linked product 3-D08 (refer to Fig. S2 and S3† for additional information). The almost guaranteed product outcome of SuFEx reactions – a hallmark of click chemistry – allows straightforward thin-layer chromatography (TLC) to be employed to monitor the course of the reaction array (e.g., see Fig. 2C).73
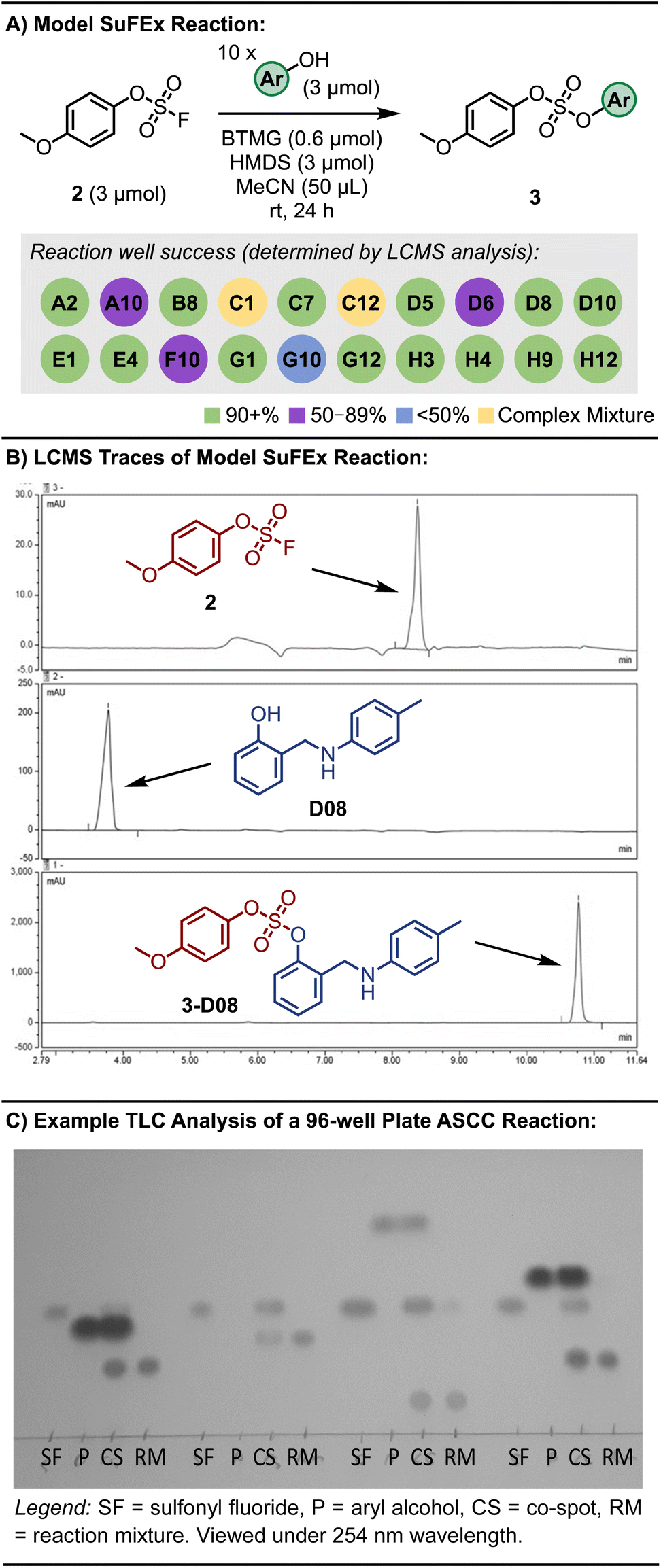 | ||
| Fig. 2 (A) Model 96-well plate ASCC reaction scheme and outcome. (B) Representative LC trace used to monitor reaction completion (see ESI† for LC method). (C) Representative TLC for reaction monitoring of the ASCC reaction. Color removed to improve clarity. Refer to Fig. S4† for full plate. | ||
Following reaction completion, the 96-well plates were placed in a vacuum oven at 50 °C for 2 h to remove the solvent, reagents, and by-products to afford the target products. A representative workflow is illustrated in Fig. 1D.
In a reaction setup using a volume of 50 μL at a concentration of 0.06 M, effective solubilization was achieved for both the aryl alcohol substrates and the SuFEx-compatible reactants. The use of 20 mol% of the BTMG catalyst, which is compatible with a diverse range of SuFEx substrates, ranging from less reactive types like aryl fluorosulfates to highly reactive ones such as aryl sulfonyl fluorides, significantly improved the reaction outcomes. Additionally, the incorporation of 1 equivalent of HMDS contributed to pronounced reactivity in this system. Specifically, we observed reaction conversion rates in 14 out of 20 plate wells surpassing 90%, with an additional 3 wells displaying at least 50% conversion within 24 hours at ambient conditions.
While extending the reaction duration to accommodate the SuFEx coupling of more inert substrate combinations is feasible, the 24 hours timeframe is a pragmatic balance between conversion efficiency and time expenditure, underlining the kinetic favorability of the reaction conditions. This approach underscores a methodological compromise, optimizing reaction efficacy without unnecessary prolongation, thereby enhancing the overall throughput of compound synthesis.
ASCC coupling of aryl alcohols with sulfuryl fluoride
Sulfuryl fluoride (SO2F2) is an important SuFEx hub for modular click chemistry and one of the most efficient reagents for synthesizing aryl fluorosulfates32 – a group with immense potential in drug discovery. Wu and co-workers first demonstrated that converting a panel of anticancer agents to fluorosulfates afforded several compounds with enhanced anti-proliferation activities compared to their aryl alcohol precursors, including the fluorosulfate derivatives of fulvestrant, combretastatin A4 (CA-4), and ABT-751.69 Specifically, the fluorosulfate of CA-4 was found to be 70-fold more active against drug-resistant colon cancer cell line HT-29.69 Additionally, the transformation of aryl alcohols into their corresponding aryl fluorosulfates has been effectively employed in the development of powerful antibiotic agents. These agents have demonstrated efficacy against multidrug-resistant bacterial strains, notably including the highly dangerous methicillin-resistant Staphylococcus aureus (MRSA).74 This strategy highlights the potential of aryl fluorosulfates in addressing critical challenges in antibiotic resistance.To harness the ASCC array protocol's potential for SuFEx reactions with sulfuryl fluoride, we transformed our assortment of aryl alcohols into the corresponding fluorosulfates (4) through direct interaction with the SO2F2 gas (refer to Fig. 3A). Whereas Wu and colleagues previously utilized a saturated acetonitrile (MeCN) solution of SO2F2 to facilitate SuFEx reactions for their synthetic library,69 we opted to use gastight zip lock bags inundated with SO2F2 gas, simplifying the reaction setup process (see Fig. 3A inset). This technique enhanced the protocol's practicality and streamlined the experimental procedure.
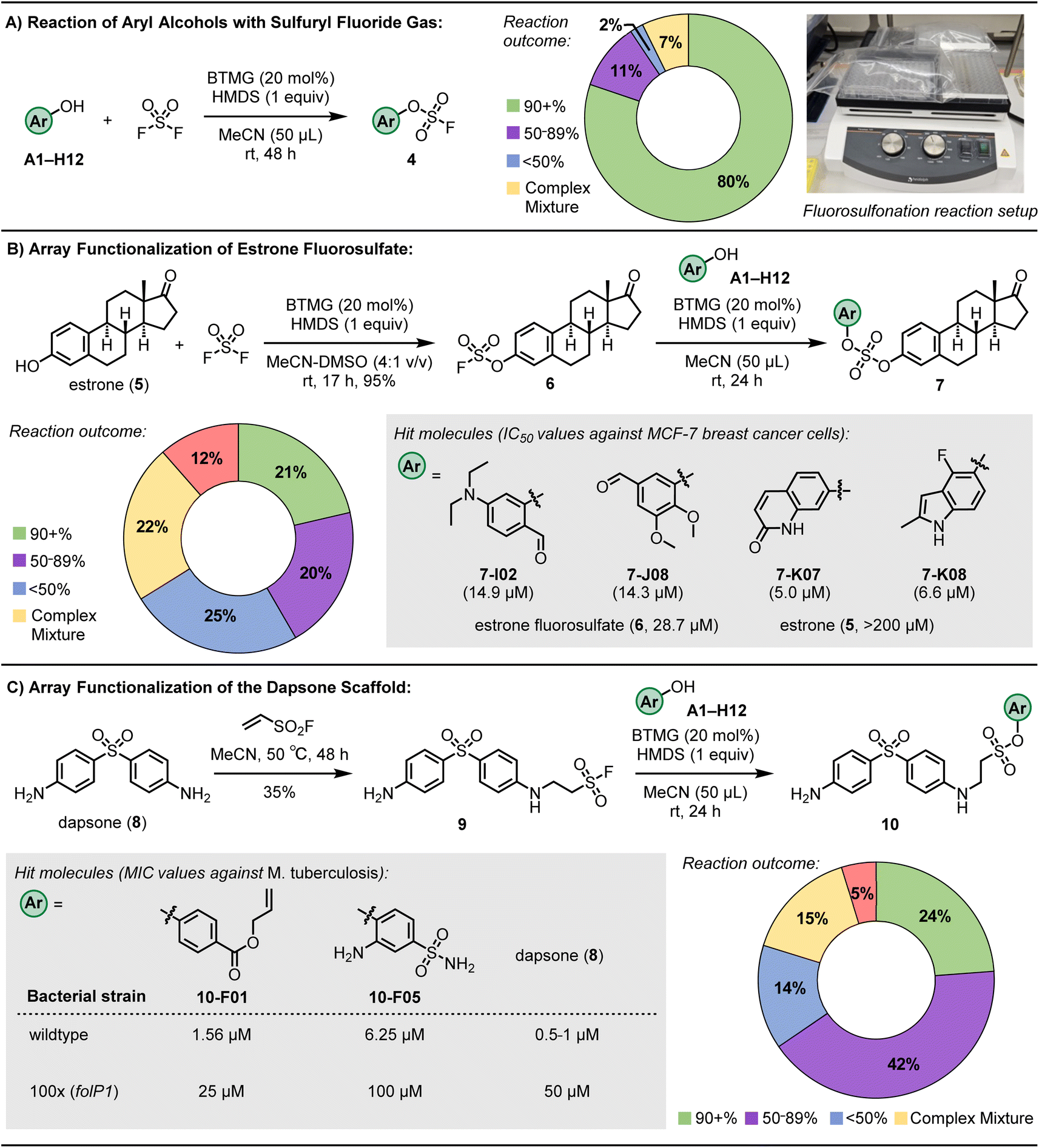 | ||
| Fig. 3 Results of 96-well plate ASCC reactions. (A) Reaction of aryl alcohols with sulfuryl fluoride gas. (B) Functionalization of estrone fluorosulfate (6). Refer to ESI† for associated confidence intervals. (C) Functionalization of the antimicrobial dapsone (8). | ||
Upon conducting the reaction over an extended agitation interval of 48 hours at ambient conditions, utilizing the BTMG catalyst and HMDS reagent in MeCN, subsequent LCMS analyses of the individual wells revealed an 80% success rate (quantified as >90% conversion) in the fluorosulfonation process (as depicted in Fig. 3A). This data represents an improvement relative to the performance benchmarks established by prior 96-well plate-based fluorosulfonation methodologies.69 This enhancement in reaction efficiency underscores the strategic refinements integrated into our experimental design, contributing to advancing high-throughput synthetic chemistry protocols.
ASCC functionalization of estrone fluorosulfate
Building upon the capabilities of the ASCC array format in these model reactions, we next advanced to the functionalization of more complex molecules, electing to explore the endogenous hormone estrone as an illustrative application of ASCC in LSF. This strategic maneuver in chemical synthesis facilitates introducing or modifying functional groups on sophisticated molecular frameworks, typically proximal to the culmination of a synthetic sequence. Such modifications optimize synthetic routes and enable the judicious introduction of labile groups. Given the role of estrogen mimetics as therapeutic agents for particular breast cancers,75,76 the derivatization of the estrone (5) core via the ASCC process holds the potential for generating novel bioactive entities against estrogen receptor (ER)-positive breast cancer phenotypes.We first prepared the estrone fluorosulfate (6) using conventional ASCC conditions in a 95% isolated yield.32 Subsequently, this product reacted with various aryl alcohol fragments, utilizing the reaction parameters optimized in the previous array synthesis experiments. Following evaporation of the solvent and reaction by-products, analysis by LCMS revealed selective conversion to the target product 7 in 21% of the reaction wells, with conversions surpassing 90%. Moreover, 20% of wells displayed conversions reaching a minimum threshold of 50%, as documented in Fig. 3B. The observed success rates indicate that in 41% of the conducted reactions, we successfully synthesized a novel sulfate diester derivative as the predominant product, achieving a purity level suitable for biological evaluation.
After the solvents used for LCMS analysis were evaporated, DMSO at a concentration of 0.02 M was added to each well of a 96-well plate. This step was crucial for preparing stock solutions, which were then tested for their antiproliferative effects against the ER-positive MCF-7 breast cancer cell line, as detailed in Table S3.† The experimental setup included incubating the cells with either unpurified reaction wells containing the sulfate diester products (7) or control fragments (such as estrone (5), estrone fluorosulfate (6), and aryl alcohol substrates) at four distinct concentrations. After a 72 hours period, the cell viability was assessed using a luminescence-based method, with further experimental specifics provided in the ESI.†
Of the evaluated compounds, four sulfate diester derivatives (7-I02, 7-J08, 7-K07, and 7-K08) had more significant cytotoxic activity than estrone fluorosulfate (6) against MCF-7 breast cancer cells, see Fig. 3B for structures. Refined cell viability assays with purified samples, as shown in Fig. S5,† identified more precise IC50 values (the concentration required to inhibit cell growth by 50%) for the sulfate derivatives, ranging from 5.0 to 14.9 μM. These values are significantly lower than those for estrone (compound 5, with an IC50 exceeding 200 μM) and the fluorosulfate (compound 6, IC50 = 28.7 μM). In comparison, the aryl alcohol substrates I02, J08, K07, and K08 exhibited minimal cytotoxicity within the tested concentration range, up to 200 μM.
In the context of identifying the therapeutic targets that are critical to the bioactivity of the identified sulfate diester derivatives, the estrogen receptor alpha (ERα) emerges as a key molecular target. The rationale for this focus is based on the known pharmacological profile of ERα antagonists, which have demonstrated substantial efficacy as antitumor agents, particularly against estrogen-receptor-positive breast cancer cell lines, exemplified by MCF-7.77 This insight aligns with the strategic targeting of ERα in the development of novel chemotherapeutic agents for estrogen-dependent malignancies.
To assess the antagonistic effects of the estrone derivatives, we conducted a Human ERα Luciferase Reporter Assay (INDIGO Biosciences). The provided reporter cells contain a luciferase reporter gene functionally linked to an ERα-responsive promoter. Quantifying changes in luciferase expression following treatment provides a surrogate measurement of the change in ERα activity. We found that while 17-β-estradiol, a potent agonist included as a positive control, enhanced luciferase expression (an agonistic effect), the diaryl sulfate estrone derivative 7-J08 inhibited luciferase expression (Fig. S6†).
Moreover, we observed that co-treatment of estrone fluorosulfate (6) or 7-J08 at increasing concentrations (6 pM to 100 nM) with a fixed concentration of the 17-β-estradiol (1 nM) decreased the relative luminescence expression in a concentration-dependent manner (Fig. 4A). These findings suggest that while the sulfate-linked derivatives have no significant agonistic effect on the ERα, they may exhibit receptor-specific antagonistic effects. Molecular modeling of 7-I02, 7-J08, and 7-K07 in the binding site of the ERα revealed lower binding scores (−37.68 to −35.46) relative to estrone (−33.99), suggesting a strong affinity for the receptor (Fig. 4B and S7†).
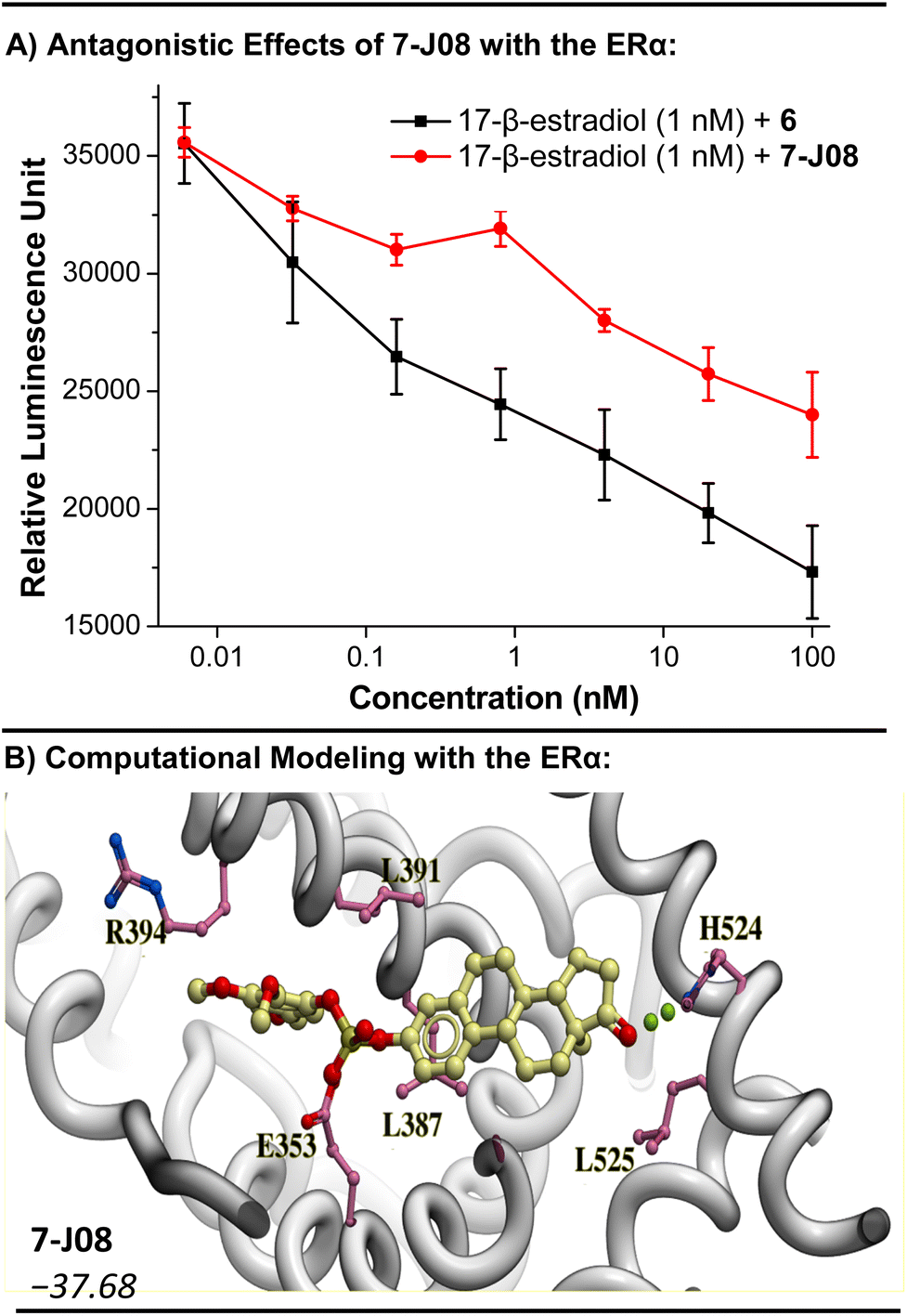 | ||
| Fig. 4 Investigation into the antagonism of ERα as a mode of action for the sulfate diester derivatives 7. (A) Antagonistic effect of 6 and 7-J08 in the presence of a known agonist 17-β-estradiol. Dose response curve was plotted using concentration against luminescence response after 24 h of incubation where n = 3. Curve was plotted by using OriginPro 8.5 software and the Savitzky–Golay smoothing method (5-point window) was applied to enhance clarity. A decrease in relative luminescence indicates receptor antagonism. (B) Computational modeling of 7-J08 with the structure of the ERα (PDB = 1ERE). RTCNN score shown in italics. | ||
The 17-β-hydroxysteroid dehydrogenase type 1 (17β-HSD1) enzyme, a key regulator of genes implicated in cellular proliferation and growth, is another plausible target for these diaryl sulfate estrone derivatives. Estrone (5) is a substrate of 17β-HSD1, and inhibitors of 17β-HSD1 have been explored as treatments for steroid-dependent breast cancers.78,79 Molecular modeling of the active diaryl sulfate derivatives 7-I02, 7-J08, and 7-K07, when aligned with the 17β-HSD1 structure, revealed a binding orientation akin to that of the known inhibitor STX1040![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 79 (refer to Fig. S8†). Furthermore, these three derivatives exhibited relatively stronger binding scores compared to STX1040, with 7-I02 showing a binding score of −42.14, in contrast to −33.38 for STX1040.
79 (refer to Fig. S8†). Furthermore, these three derivatives exhibited relatively stronger binding scores compared to STX1040, with 7-I02 showing a binding score of −42.14, in contrast to −33.38 for STX1040.
These results underscore the efficacy of late-stage functionalization in a 96-well plate format, enabling the rapid identification of potent hit compounds. This approach notably transforms the breast cancer-promoting hormone estrone (5) into cytotoxic agents through merely two click chemistry steps, exemplifying the function-driven philosophy of this method. Although a definitive mode of action remains to be clarified, preliminary evidence pointing to two potential targets, ERα and 17β-HSD1, warrants further investigation.
ASCC functionalization of the antibiotic dapsone
Dapsone (8) is a bacteriostatic drug that is included in the prescribed multidrug therapy (MDT) regimens80 for treating leprosy (Hansen's disease), a chronic and disfiguring infection caused by Mycobacterium leprae and the recently discovered Mycobacterium lepromatosis.81 Dapsone is a sulfone drug that disrupts bacterial folic acid synthesis, acting as a competitive inhibitor of the dihydropteroate synthase (DHPS) enzyme.82 Side effects associated with dapsone can be significant and include hemolytic anemia, hepatitis, photo-dermatitis, agranulocytosis, sulphone allergy, and loss of appetite.82 In addition to this, clinical resistance to dapsone by genetic mutations that convey drug resistance (e.g., missense mutations affecting Thr53 or Pro55 in dihydropteroate synthase that reduce the binding affinity, and thus efficacy, of dapsone83,84) is rising.85To develop dapsone derivatives with enhanced side effect profiles and sustained effectiveness against resistant strains, our approach involved the ASCC array functionalization methodology. Initially, dapsone (8) was combined with the ESF hub (see Fig. 1A), producing the SuFEx-compatible conjugate addition product 9.86 This compound was then reacted with a diverse library of aryl alcohols, employing the optimized ASCC conditions, to generate a suite of alkyl sulfonate esters (10). Among the 84 wells analyzed, 20 showed a reaction efficiency of 90% or higher, with an additional 35 wells achieving at least a 50% conversion rate (Fig. 3C).
The inherently sluggish replication of M. leprae, characterized by a doubling time of approximately 12–14 days, combined with its incapacity for propagation in axenic culture, significantly impedes the pace and efficiency of biological assays assessing antibiotic efficacy. The limitations of traditional methods lead to time-consuming and expensive procedures, which are impractical for screening large libraries of compounds. To overcome this, our research assessed dapsone derivatives using a specially engineered Mycobacterium tuberculosis strain (mc27000 ΔpabC), modified to be more sensitive to dapsone.87 This novel method enables quicker and more cost-effective screening, effectively bypassing the challenges posed by the stringent growth conditions required for M. leprae.
The activities of drugs against the M. tuberculosis mc27000 ΔpabC strain were tested using the pellet reading method in 96-well plates as previously described88 with minor modifications. Reaction wells with greater than 50% purity, as determined by LCMS analysis, were subjected to biological testing. The corresponding wells were resuspended in DMSO to achieve a stock solution with a concentration of 400 μM. Two-fold serial dilutions of each drug were prepared and added to the wells. Additionally, each plate included dapsone (8) as a positive control. After adding a mid-logarithmic-phase culture of M. tuberculosis, the plates were incubated for 12 days at 37 °C. After this time, the plates were visually scanned for bacterial growth. When inhibition was observed via optical density readings at 600 nm (OD600), minimum inhibitory concentrations (MICs) were determined using a subsequent 10 mM DMSO stock solution. Each compound was diluted 16 times in a two-fold dilution, and OD600 measurements determined the MIC90 (concentration of compound leading to 90% inhibition of bacterial growth).
Among the novel sulfonate ester dapsone derivatives (10), two compounds displayed activity against a dapsone-sensitive strain M. tuberculosis (mc27000 ΔpabC): compounds 10-F01 and 10-F05 (Fig. 3C). The MICs of compounds 10-F01 and 10-F05 were established at 1.56 μM and 6.25 μM, respectively. These values are in the same range as that of dapsone, which has an MIC between 0.5 and 1 μM. Notably, the control compounds, specifically the starting materials used to synthesize these dapsone derivatives, exhibited minimal activity against the mycobacteria, as detailed in Table S4.†
Next, 10-F01 and 10-F05 were tested against an M. tuberculosis strain that exhibited dapsone resistance due to a point mutation (P55L) in the folP1 gene encoding the DHPS enzyme (100×). Compound 10-F01 was found to maintain activity against the resistant bacteria, with slightly enhanced activity over dapsone against the 100× strain. These results identify derivative 10-F01 as a lead compound with activity against dapsone-sensitive and -resistant strains of M. tuberculosis comparable to or greater than that of dapsone.
Array synthesis of sulfonate ester-linked microtubule targeting agents (MTAs)
To further explore the ASCC reaction as a tool for the modular discovery of potent lead molecules, we finally sought to prepare a library of MTAs. Microtubules play a pivotal role during cell replication and thus are prime targets in cancer therapy.89 Disruption of microtubule function by binding small molecule MTAs leads to mitotic arrest and cell death.90 Consequentially, MTAs are often cornerstone drugs used in chemotherapy regimens – for example, paclitaxel (i.e., Taxol) is a standard-of-care drug for countless malignancies.90Numerous binding sites are present in the structure of microtubules,91 offering a significant opportunity to develop novel inhibitors. Although colchicine (11, Fig. 5A), the first known MTA, is not clinically used as an anticancer agent, efforts are being made to develop colchicine binding site inhibitors (CBSIs) that exploit their alternate action of targeting essential tumor vasculature, preventing new blood vessel formation and destroying existing tumor vasculature.92–94 Additionally, CBSIs often do not suffer from multidrug resistance issues,92 a common problem with MTAs that limits their effectiveness.
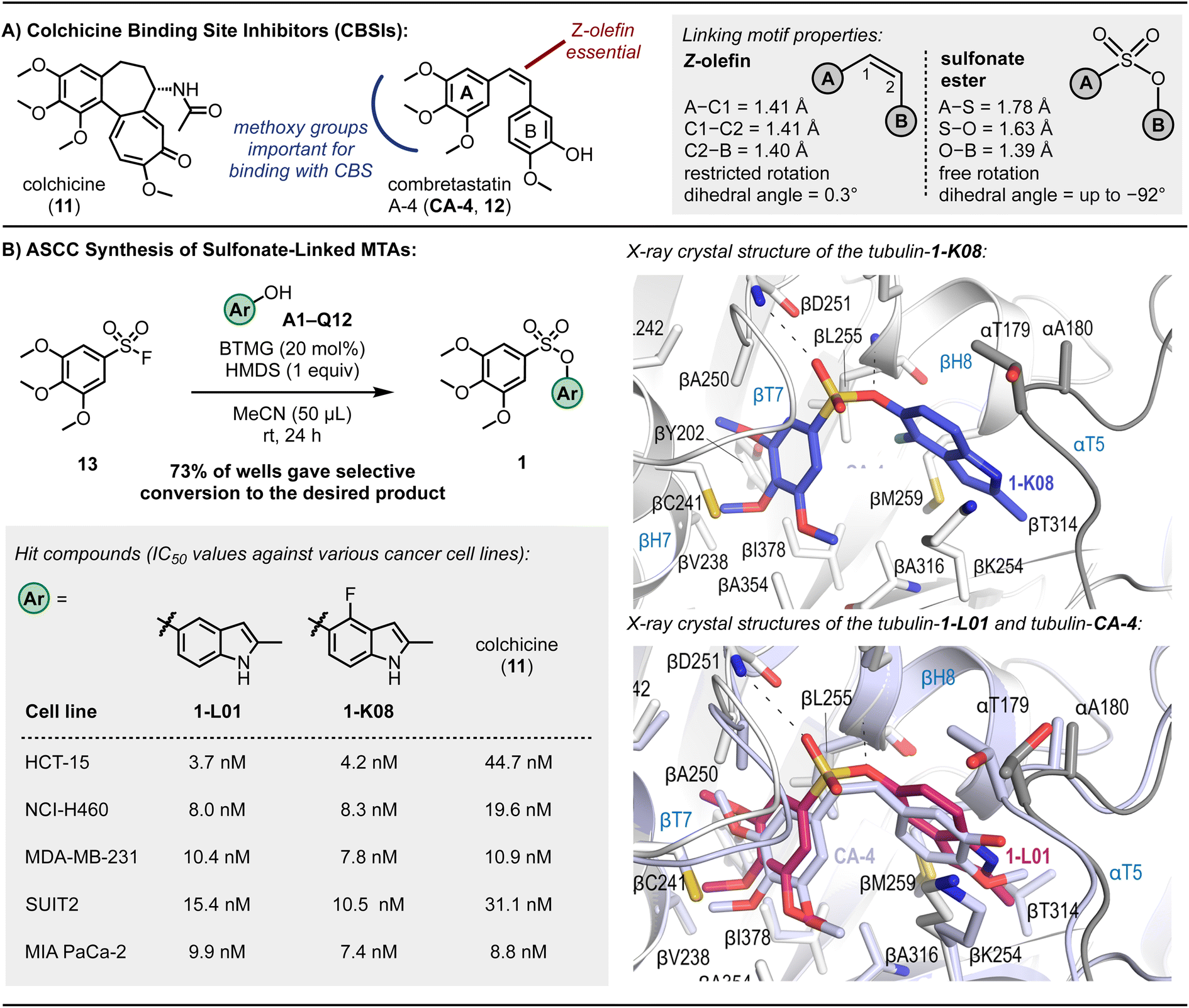 | ||
| Fig. 5 Microtubule targeting agents (MTAs). (A) Exemplary colchicine binding site inhibitors. (B) Sulfonate CA-4 mimetics, cytotoxic potencies against various cancer cell lines, and X-ray crystal structures of 1-K08 (dark blue) and overlay of 1-L01 (pink) and CA-4 (light purple) in the colchicine binding site. Reaction outcome determined by TLC analysis. Refer to ESI† for experimental details and associated SEM and standard deviation values. | ||
CA-4 (12, Fig. 5A) is a naturally occurring CBSI that destabilizes the growing microtubule.89,95CA-4 comprises a trimethoxybenzene A-ring and 3-hydroxy-4-methoxyphenyl B-ring linked together by a Z-olefin89 – a structural architecture reminiscent of colchicine. Despite promising anticancer activity, CA-4's in vivo efficacy is constrained by its limited water solubility and poor oral bioavailability.95
Efforts have been made to develop CA-4-inspired inhibitors to overcome some of these shortcomings.89,92 Structure-activity relationship studies have identified that the trimethoxybenzene motif (indicated in blue, Fig. 5A) of CA-4 is critical for a strong binding interaction between the ligand and the colchicine binding site (CBS), notably forming a hydrogen bond with Cys241.89 The Z-olefin is also essential for activity (indicated in red, Fig. 5A).
The pioneering research by Gwaltney et al.96 focused on investigating CA-4-inspired CBSIs where the sulfonate ester linkage was utilized as a replacement for the Z-olefin. Considering the elongated S–O bond length relative to C–O (e.g., 1.649 Å in propynyl tosylate vs. 1.414 Å in ethynyl benzoate97) and the absence of pi-resonance, substituting the Z-olefin of CA-4 with a sulfonate ester linkage, which exhibits reduced rotational barriers,98 offers mimetics with enhanced flexibility (Fig. 5A). Such ligands can exhibit full rotation around the A-ring–B-ring connection, potentially improving binding interactions with the CBS. Fortin et al.99 also prepared several sulfonate-linked CBSIs, identifying a molecule with IC50 values ranging from 2.5–9.4 nM against a panel of cancer cell lines, including those with drug resistance to standard-of-care MTAs such as paclitaxel and vinblastine. Access to both sets of compounds involved the base-mediated reaction of aryl sulfonyl chlorides with aryl alcohols, and, although useful on a larger scale, the use of stoichiometric base reagents and the limited stability of many sulfonyl chlorides100,101 likely precludes the reliable adaption of this approach to an array format.102
The 3,4,5-trimethoxybenzenesulfonyl fluoride (13) was selected as a starting fragment for building out using ASCC. Derivatization of this molecule via the 96-well plate ASCC protocol was monitored by TLC analysis as, in this instance, most starting materials and products were visible under a 254 nm UV lamp (see Fig. 2C and S3,† for example, TLC plates). Following complete evaporation of the reagents and by-products, out of 110 reaction wells, 80 sulfonate-linked compounds were generated, representing a 73% success rate (Fig. 5B).
In the preliminary investigation into antiproliferative properties, a series of tests were conducted on the HCT-15 human colon carcinoma cell line, known for its high expression of P-glycoprotein, which confers multidrug resistance.103 Among the compounds evaluated, eight exhibited superior potency in inhibiting the growth of these cells compared to colchicine (11), a well-established reference agent. This enhanced effectiveness was evident in both the graphical data presented in Fig. S9† and the detailed quantitative results listed in Table S5.† This finding is significant as it highlights the potential of these eight compounds as promising candidates in overcoming the challenge of multidrug resistance in cancer treatment, a critical hurdle in current oncological therapies.
In a subsequent phase of screening focused on the NCI-H460 lung cancer cell line characterized by the KRASQ61H mutation – a common oncogenic point mutation in lung cancers – two compounds, designated as 1-L01 and 1-K08, emerged as prominent leads. These findings are detailed in Fig. 5B. Notably, these compounds, which contain indole structures, demonstrated exceptional efficacy against both the NCI-H460 and the previously tested HCT-15 cell lines, with IC50 values ranging from 3.7 to 8.3 nM, indicating nanomolar potency. For comparison, CA-4 displayed low nanomolar IC50 values against these cell lines, with 1.7 nM for HCT-15 and 3.0 nM for NCI-H460.104 The performance of 1-L01 and 1-K08 is particularly noteworthy in this context, underscoring their potential as effective anticancer agents.
These lead compounds were then subjected to comprehensive biological evaluations to assess the extent of their antiproliferative effects across a range of challenging cancer types and to investigate their underlying mechanisms of action. This step is crucial for understanding the full potential and applicability of these compounds in cancer therapy, especially in scenarios involving resistant cancer cell lines and mutations like KRASQ61H.
The sulfonate ester CA-4 analogs 1-L01 and 1-K08 demonstrated high efficacy against pancreatic ductal adenocarcinoma (PDAC) cell lines SUIT2 and MIA PaCa-2, with IC50 values between 7.4 and 15.4 nM, as well as against triple-negative breast cancer cell line MDA-MB-231, with IC50 values between 7.8 and 10.4 nM. These potencies are similar to the IC50 values of CA-4, which range from 2.4 to 8.7 nM (detailed IC50 values can be found in Fig. S11 and S12†). In contrast, the sulfate-linked derivatives 14-L01 and 14-K08, synthesized via the ASCC method (details and structures in the ESI†), exhibited negligible antiproliferative activity. This disparity in activity underscores the critical role of the sulfonate ester pharmacophore in 1-L01 and 1-K08, as the main structural difference between these compounds and their sulfate counterparts 14-L01 and 14-K08 is an additional oxygen atom in the sulfur-containing linker.
Strikingly, the lead sulfonate ester mimetics maintained nanomolar antiproliferative activity comparable to colchicine (11) against a panel of patient-derived PDAC organoids (Fig. 6 and S12†). This is notable as the anticancer activity of molecules often decreases when moving from 2-dimensional to 3-dimensional models of cancer.105 The control compounds, namely sulfonyl fluoride 13, 5-hydroxy-2-methylindole (L01), and 4-fluoro-5-hydroxy-2-methylindole (K08), exhibited no antiproliferative activity over the evaluated concentration range.
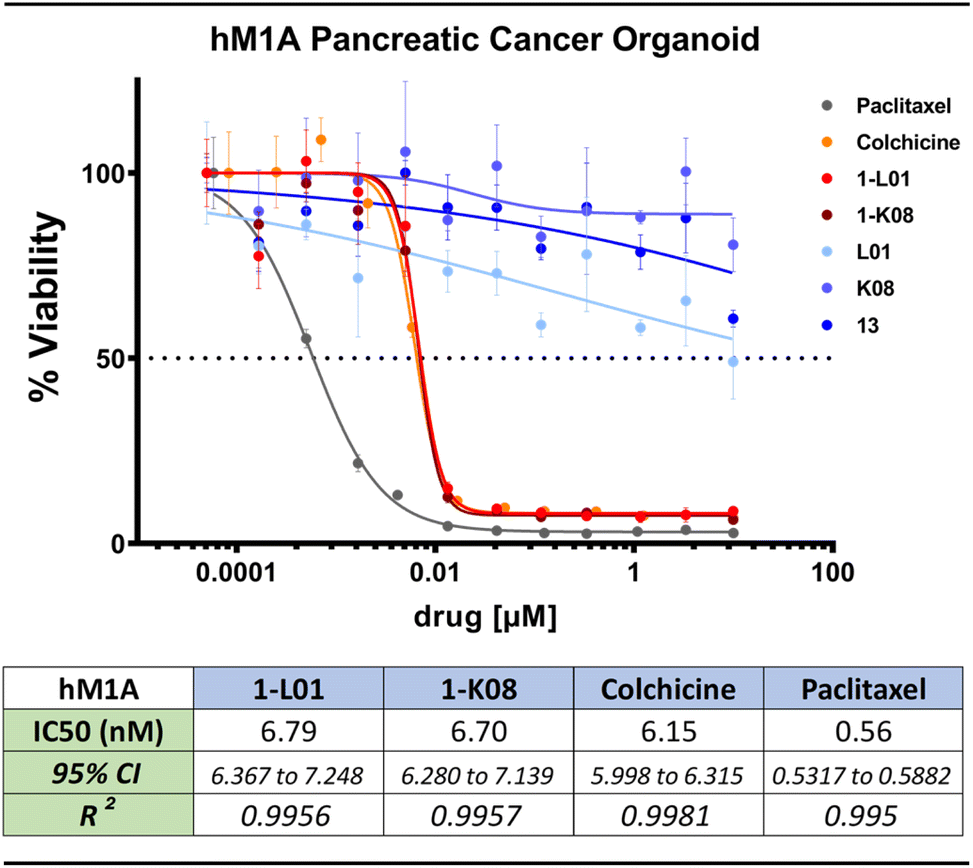 | ||
| Fig. 6 Growth inhibitory effect of 1-L01, 1-K08, and relevant control molecules against the hM1A patient-derived PDAC organoid line. All compounds were dissolved in DMSO and assayed in triplicates in a 12-point dose response. Data was analysed in Graphpad Prism to determine IC50 values using a non-linear regression. | ||
Investigation into the mode of action showed that compounds 1-L01 and 1-K08 effectively inhibit microtubule polymerization, similar to colchicine (11), as depicted in Fig. S13.† Additionally, X-ray crystallography experiments established that this inhibition stems from the binding of these ligands to the CBS at the interface between α- and β-tubulin. This interaction is illustrated in Fig. 4B (inset) and further detailed in Fig. S15 and S16.†89,91 This finding provides a clear understanding of the molecular basis of their activity.
The sulfonate ester analogs 1-L01 and 1-K08 crystal structures with the T2R-TTL complex106,107 were determined to 2.6–2.8 Å resolution (Table S6†). As with other CBSIs, binding induced a conformational change in the βT7 loop that prevents the curved-to-straight transition of β-tubulin that is required during microtubule assembly. Our results show that the 3,4,5-trimethoxyphenyl moiety of the sulfonate ester ligands occupied a hydrophobic pocket shaped by the side chains of βTyr202, βCys241, βLeu242, βLeu248, βAla250, βIle318, βAla354, and βIle378 (Fig. 4). The superimposition of CA-4 over the binding modes of the sulfonate ester mimetic 1-L01 highlights the remarkable similarities (Fig. 4B inset). A binding contact between the oxygens of the sulfonate ester linkage and the backbone amide of aspartic acid residue (βAsp251) was also noted (indicated with a dashed line in the inset of Fig. 4B); the ablation of this interaction when considering sulfate-linked compounds could go some way to explain the drastic loss of activity of 14-L01 and 14-K08 and positions the sulfonate ester connector as an essential pharmacophore of the nanomolar MTAs 1-L01 and 1-K08.
Next, the binding affinities between the lead sulfonate ester mimetics and β-tubulin were determined using a previously described competition assay using 2-methoxy-5-(2,3,4-trimethoxyphenyl)-2,4,5-cycloheptatrien-1-one (MTC).108MTC is a fluorescent analog of colchicine that, upon complexation with tubulin, emits at 421 nm upon excitation at 350 nm.109 Nocodazole, another CBSI,110 was used as a positive control molecule. Changes to the UV absorbance spectrum of the lead sulfonate ester compounds were determined, followed by their ability to displace MTC at different concentrations. For the compounds 1-L01 and 1-K08, binding constants of 1.7 × 106 and 3.0 × 106 were calculated, respectively, an order of magnitude stronger than colchicine (KD = 1.16 × 107 (at 37 °C)111) (Tables 1 and S7†).
| Compound | Nocodazole | 1-L01 | 1-K08 |
|---|---|---|---|
| K D (M−1) | 2.0 × 106 | 1.7 × 106 | 3.0 × 106 |
Finally, by applying live-cell imaging assays using bioluminescent cell cycle reporters, we confirmed the dramatic effects of 1-L01 and 1-K08 on the cell cycle of MIA PaCa-2 PDAC cells (Fig. 7). Cells were transfected with a single lentiviral vector (GemLuc) constructed to produce both red (S/G2/M Phase) and green (internal control color) colored bioluminescence.112 Sulfonate esters 1-L01 and 1-K08 were then dosed with increasing concentrations (from 0–100 nM) for 24 hours. The treatment of 1-L01 and 1-K08 resulted in a substantial increase in the red, indicative of cell cycle arrest, while the controls 13, L01, and K08 failed to elicit the same effect over the evaluated concentration range.
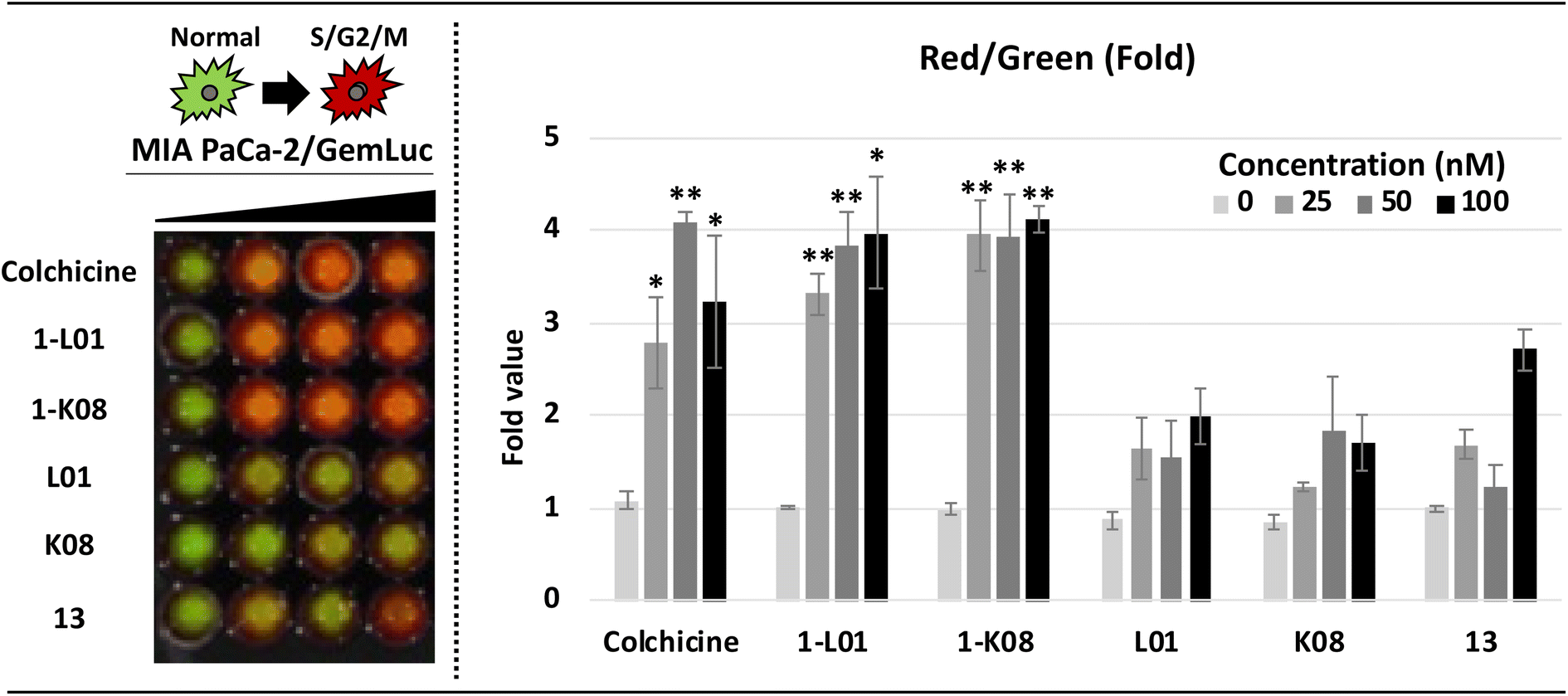 | ||
| Fig. 7 In vitro imaging assays detecting cell cycle arrest. Cells were plated day −1 on a black-walled 96-well plate and treated with escalating doses in triplicate of each compound. After 24 hours of drug treatment, multiple bioluminescent images were acquired 2 minutes after adding D-luciferin and images of the cell cycle were taken with an IVIS Spectrum scanner. An increase in red colour indicates S/G2/M phase arrest. Comparisons between groups were conducted using a two-tailed Student's t-test. Associated p-values of less than 0.05 and 0.01, indicated with * and ** respectively, are considered as statistically significant. | ||
These data position the SuFEx-derived sulfonate ester MTAs 1-L01 and 1-K08 as versatile and potent anticancer agents that operate via high affinity binding with the CBS. These compounds were found to have broad-spectrum activity against breast, pancreatic, colorectal, and lung cancer cells (in both 2D and 3D models of cancer) and demonstrate the ASCC-based array synthesis as a powerful tool for lead discovery. Additionally, the sulfonate ester pharmacophore was essential for the pronounced antiproliferative activity of these MTAs.
Conclusions
In this work, we have demonstrated the potential of the ASCC reaction as a practical method for discovering functional molecules – one of the overarching goals of click chemistry. The application of the ASCC reaction in array synthesis is effectively demonstrated using a 96-well plate format, hence seamlessly facilitating direct biological screening (due to the volatile nature of the reagents and by-products) and prioritizing the agnostic elucidation of function.By initiating a reaction between commercially available aryl alcohols and a range of SuxFExable fragments (including aryl and alkyl sulfonyl fluorides, aryl fluorosulfates, and sulfuryl fluoride gas), we have utilized ASCC to synthesize discrete libraries of molecules methodically. This approach guarantees experimental consistency and significantly enhances the efficiency in identifying potent hit molecules. Through this methodology, we have synthesized and subsequently evaluated numerous lead molecules, including compounds with both anticancer and antibiotic properties. A subset of these has demonstrated marked activity against cell lines representing particularly deadly cancers such as PDAC and triple-negative breast cancer.
Collectively, these results emphasize the inherent capability of unguided click derivatization in the rapid identification of biologically active molecules, facilitating the accelerated identification of lead-like candidates. The culmination of our findings reinforces the pivotal role of the ASCC reaction in advancing the field of drug discovery.
Data availability
Coordinates and structure factors of the T2R-TTL-1-K08 and -1-L01 complexes have been deposited at the Protein Data Bank (https://www.rcsb.org/) under accession numbers PDB 8RIV and 8RIW, respectively. The authors will release the atomic coordinates and experimental data upon publication of the article.Author contributions
Conceptualization, J. E. M.; methodology, J. A. H., R. A. K., A. S. B., T. L. G.; investigation, J. A. H., R. A. K., A. S. B., T. L. G., Z. B., N. S. S., E. F. L., W.-H. Y., R. M. J., T. C., A. N. H., D. V., D. B., C. A., S. M. H.; supervision, J. A. H., M. J., D. A. T., S. K. L., M. J. L., W. D. F., M. S., A. P., J. E. M.; writing, J. A. H., R. A. K., A. D. M., J. E. M.; funding acquisition, M. J., D. A. T., S. K. L., M. J. L., W. D. F., S. M. H., M. S., A. P., J. E. M.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. E. M. is grateful to the NCI Cancer Center (support grant 5P30CA045508), the Cold Spring Harbor Laboratory (CSHL) Northwell Health Affiliation, the F. M. Kirby Foundation, the Sunshine Foundation, S. J. Edwards, the Starr Foundation, Swim Across America Inc., the Wasily Family Foundation, the Australian Research Council (Future Fellowship; FT170100156), and La Trobe University for generous support. This work was performed with assistance from the CSHL Organoid Shared Resource and the CSHL Mass Spectrometry and Metabolomics Shared Resource, both partly supported by Cancer Center Support Grant 5P30CA045508 from the National Cancer Institute. M. J. L. is supported by METAvivor (and W.-H. Y.), the Department of Defense Breast Cancer Research Program (BC200599), and the National Institutes of Health (R01GM149957 and 5P30CA045508). The Lustgarten Foundation supported D. A. T, A. N. H, and all organoid work. A. N. H is also supported by a Postdoctoral Fellowship (PF-23-1036459-01-ET) from the American Cancer Society. E. F. L. is supported by a Victorian Cancer Agency Mid-Career Fellowship (MCRF19045). J. A. H. and C. A. are funded by The New York Community Trust's Heiser Program. Z. B. and A. E. P. are supported by the European Union H2020-MSCA-ITN-2019 860070 TUBINTRAIN. M. O. S. is supported by the Swiss National Science Foundation (310030_192566). C. A. and M. J. thank Dr Anthony Baughn from the University of Minnesota for providing the M. tuberculosis strain mc27000 ΔpabC. D. B. is funded by Prof. Michael Wigler. The authors would also like to thank Dr Qinheng Zheng for insightful discussions and Dr Christopher Smedley for supervising T. L. G. in the laboratory. While preparing this work, the authors used ChatGPT 4.0 and Grammarly to improve readability. After using this tool/service, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.Notes and references
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- The term click chemistry was first coined by K. Barry Sharpless in 1998 and was later published as a concept in 2001; see ref. 1.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430–9448 CrossRef CAS PubMed.
- S. Sun, J. A. Homer, C. J. Smedley, Q.-Q. Cheng, K. B. Sharpless and J. E. Moses, Chem, 2023, 9, 1–16 Search PubMed.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- A. S. Barrow, C. J. Smedley, Q. Zheng, S. Li, J. Dong and J. E. Moses, Chem. Soc. Rev., 2019, 48, 4731–4758 RSC.
- A. D. Moorhouse, J. A. Homer and J. E. Moses, Chem, 2023, 9, 2063–2077 CAS.
- L. M. Campos, K. L. Killops, R. Sakai, J. M. J. Paulusse, D. Damiron, E. Drockenmuller, B. W. Messmore and C. J. Hawker, Macromolecules, 2008, 41, 7063–7070 CrossRef CAS.
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A-1: Polym. Chem., 2010, 48, 743–750 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS PubMed.
- X. Jiang, X. Hao, L. Jing, G. Wu, D. Kang, X. Liu and P. Zhan, Expert Opin. Drug Discovery, 2019, 14, 779–789 CrossRef CAS PubMed.
- T. Abdul Fattah, A. Saeed and F. Albericio, J. Fluorine Chem., 2018, 213, 87–112 CrossRef CAS.
- S. N. Carneiro, S. R. Khasnavis, J. Lee, T. W. Butler, J. D. Majmudar, C. W. am Ende and N. D. Ball, Org. Biomol. Chem., 2023, 21, 1356–1372 RSC.
- J. A. Homer, L. Xu, N. Kayambu, Q. Zheng, E. J. Choi, B. M. Kim, K. B. Sharpless, H. Zuilhof, J. Dong and J. E. Moses, Nat. Rev. Methods Primers, 2023, 3, 59 CrossRef.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS.
- S. I. Presolski, V. P. Hong and M. g. Finn, Curr. Protoc. Chem. Biol., 2011, 3, 153–162 CrossRef PubMed.
- C. S. McKay and M. Finn, Cell Chem. Biol., 2014, 21, 1075–1101 CAS.
- D. Zeng, W.-P. Deng and X. Jiang, Natl. Sci. Rev., 2023, 10, nwad123 CrossRef CAS.
- M.-C. Giel, C. J. Smedley and J. E. Moses, in Science of Synthesis: Click Chemistry, ed. F. P. J. T. Rutjes, Thieme Chemistry, 2021, pp. 435–484 Search PubMed.
- A. Narayanan and L. H. Jones, Chem. Sci., 2015, 6, 2650–2659 RSC.
- C. J. Smedley, M.-C. Giel, T. Fallon and J. E. Moses, Angew. Chem., Int. Ed., 2023, 62, e202303916 CrossRef CAS PubMed.
- F. Liu, H. Wang, S. Li, G. A. Bare, X. Chen, C. Wang, J. E. Moses, P. Wu and K. B. Sharpless, Angew. Chem., Int. Ed., 2019, 58, 8029–8033 CrossRef CAS PubMed.
- Q. Zheng, J. L. Woehl, S. Kitamura, D. Santos-Martins, C. J. Smedley, G. Li, S. Forli, J. E. Moses, D. W. Wolan and K. B. Sharpless, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 18808–18814 CrossRef CAS PubMed.
- D. E. Mortenson, G. J. Brighty, L. Plate, G. Bare, W. Chen, S. Li, H. Wang, B. F. Cravatt, S. Forli, E. T. Powers, K. B. Sharpless, I. A. Wilson and J. W. Kelly, J. Am. Chem. Soc., 2018, 140, 200–210 CrossRef CAS PubMed.
- G. J. Brighty, R. C. Botham, S. Li, L. Nelson, D. E. Mortenson, G. Li, C. Morisseau, H. Wang, B. D. Hammock, K. B. Sharpless and J. W. Kelly, Nat. Chem., 2020, 12, 906–913 CrossRef CAS PubMed.
- G. M. Kline, K. Nugroho and J. W. Kelly, Curr. Opin. Chem. Biol., 2022, 67, 102113 CrossRef CAS PubMed.
- D.-D. Liang, D. E. Streefkerk, D. Jordaan, J. Wagemakers, J. Baggerman and H. Zuilhof, Angew. Chem., Int. Ed., 2020, 59, 7494–7500 CrossRef CAS PubMed.
- S. Li, G. Li, B. Gao, S. P. Pujari, X. Chen, H. Kim, F. Zhou, L. M. Klivansky, Y. Liu, H. Driss, D.-D. Liang, J. Lu, P. Wu, H. Zuilhof, J. Moses and K. B. Sharpless, Nat. Chem., 2021, 13, 858–867 CrossRef CAS PubMed.
- C. J. Smedley, J. A. Homer, T. L. Gialelis, A. S. Barrow, R. A. Koelln and J. E. Moses, Angew. Chem., Int. Ed., 2022, 61, e202112375 CrossRef CAS PubMed.
- A. F. J. van den Boom, M. Subramaniam and H. Zuilhof, Org. Lett., 2022, 24, 8621–8626 CrossRef CAS PubMed.
- D.-D. Liang, S. P. Pujari, M. Subramaniam, M. Besten and H. Zuilhof, Angew. Chem., Int. Ed., 2022, 61, e202116158 CrossRef CAS PubMed.
- M. Wei, D. Liang, X. Cao, W. Luo, G. Ma, Z. Liu and L. Li, Angew. Chem., Int. Ed., 2021, 60, 7397–7404 CrossRef CAS PubMed.
- J.-N. Luy and R. Tonner, ACS Omega, 2020, 5, 31432–31439 CrossRef CAS PubMed.
- S. Mahapatra, C. P. Woroch, T. W. Butler, S. N. Carneiro, S. C. Kwan, S. R. Khasnavis, J. Gu, J. K. Dutra, B. C. Vetelino, J. Bellenger, C. W. am Ende and N. D. Ball, Org. Lett., 2020, 22, 4389–4394 CrossRef CAS PubMed.
- B. Gao, S. Li, P. Wu, J. E. Moses and K. B. Sharpless, Angew. Chem., Int. Ed., 2018, 57, 1957–1961 CrossRef.
- C. J. Smedley, Q. Zheng, B. Gao, S. Li, A. Molino, H. M. Duivenvoorden, B. S. Parker, D. J. D. Wilson, K. B. Sharpless and J. E. Moses, Angew. Chem., Int. Ed., 2019, 58, 4552–4556 CrossRef CAS PubMed.
- C. J. Smedley, G. Li, A. S. Barrow, T. L. Gialelis, M.-C. Giel, A. Ottonello, Y. Cheng, S. Kitamura, D. W. Wolan, K. B. Sharpless and J. E. Moses, Angew. Chem., Int. Ed., 2020, 59, 12460–12469 CrossRef CAS PubMed.
- Y. Cheng, G. Li, C. J. Smedley, M.-C. Giel, S. Kitamura, J. L. Woehl, G. Bianco, S. Forli, J. A. Homer, J. R. Cappiello, D. W. Wolan, J. E. Moses and K. B. Sharpless, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2208540119 CrossRef PubMed.
- C. J. Smedley, Chem. Commun., 2022, 58, 11316–11319 RSC.
- H. Vorbrüggen, Synthesis, 2008, 2008, 1165–1174 CrossRef.
- A. M. Hyde, R. Calabria, R. Arvary, X. Wang and A. Klapars, Org. Process Res. Dev., 2019, 23, 1860–1871 CrossRef CAS.
- D. H. R. Barton, J. D. Elliott and S. D. Géro, J. Chem. Soc., Perkin Trans. 1, 1982, 2085–2090 RSC.
- D. H. R. Barton, B. Charpiot and W. B. Motherwell, Tetrahedron Lett., 1982, 23, 3365–3368 CrossRef CAS.
- Niu and co-workers also developed a one-pot SuFEx O-sulfation reaction that used HMDS as an in situ silylating agent in the presence of DBU. See C. Liu, C. Yang, S. Hwang, S. L. Ferraro, J. P. Flynn and J. Niu, Angew. Chem., Int. Ed., 2020, 59, 18435–18441 CrossRef CAS PubMed.
- A. D. Moorhouse, A. M. Santos, M. Gunaratnam, M. Moore, S. Neidle and J. E. Moses, J. Am. Chem. Soc., 2006, 128, 15972–15973 CrossRef CAS PubMed.
- S. G. Agalave, S. R. Maujan and V. S. Pore, Chem.–Asian J., 2011, 6, 2696–2718 CrossRef CAS PubMed.
- M. G. Finn, H. C. Kolb and K. B. Sharpless, Nat., Synth., 2022, 1, 8–10 CrossRef.
- A. D. Moorhouse, S. Haider, M. Gunaratnam, D. Munnur, S. Neidle and J. E. Moses, Mol. BioSyst., 2008, 4, 629 RSC.
- D. Lengerli, K. Ibis, Y. Nural and E. Banoglu, Expert Opin. Drug Discovery, 2022, 17, 1209–1236 CrossRef CAS PubMed.
- S. Kumari, A. V. Carmona, A. K. Tiwari and P. C. Trippier, J. Med. Chem., 2020, 63, 12290–12358 CrossRef CAS PubMed.
- M. Feng, B. Tang, S. H. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200–1216 CrossRef CAS PubMed.
- Highlighting the potential of SuFEx, Tang et al. reported a synthesis of sulfonamides using a 96-well plate SuFEx-based array, leading to the identification of powerful nanomolar acetylcholinesterase inhibitors. This reaction leveraged the reactivity of amines with sulfonyl fluorides, a reaction that proceeds smoothly in phosphate buffer solution, and generated compounds on a picomole scale for direct screening for biological activity. Refer to K. Tang, H.-H. Li, C. Wu, S.-L. Zhang, J.-G. Yang, W. Tang and H.-L. Qin, J. Enzyme Inhib. Med. Chem., 2023, 38, 2237213 CrossRef PubMed.
- B.-T. Hong, Y.-S. E. Cheng, T.-J. Cheng and J.-M. Fang, Eur. J. Med. Chem., 2019, 163, 710–721 CrossRef CAS PubMed.
- K. A. Scott and J. T. Njardarson, in Sulfur Chemistry, ed. X. Jiang, Springer International Publishing, Cham, 2019, pp. 1–34 Search PubMed.
- Contrary to these beliefs, our observations show organic sulfonate esters and sulfate diesters exhibit notable stability and resistance to hydrolysis. For instance, the sulfonate ester-linked compounds 1-L01 and 1-K08 (refer to Fig. 1D for structures) remain stable in phosphate buffers across a range of pH levels for up to two weeks (further Experimental details are provided in the ESI†).
- Over 1.6 million aryl alcohol-containing compounds are available from commercial sources. Number obtained from a SciFinder substance search of the drawn structure of phenol (C6H5OH) using the substructure feature on 07/24/2023. Filter parameters: commercial availability – available; number of components – 1; molecular weight – no. min to 300; functional group – phenol; isotopes – not containing isotopes.
- T. W. J. Cooper, I. B. Campbell and S. J. F. Macdonald, Angew. Chem., Int. Ed., 2010, 49, 8082–8091 CrossRef CAS PubMed.
- G. Karageorgis, S. Warriner and A. Nelson, Nat. Chem., 2014, 6, 872–876 CrossRef CAS PubMed.
- G. Karageorgis, S. Liver and A. Nelson, ChemMedChem, 2020, 15, 1776–1782 CrossRef CAS PubMed.
- A. I. Green, F. Hobor, C. P. Tinworth, S. Warriner, A. J. Wilson and A. Nelson, Chem.–Eur. J., 2020, 26, 10682–10689 CrossRef CAS PubMed.
- I. A. Stepek and J. W. Bode, Curr. Opin. Chem. Biol., 2018, 46, 18–24 CrossRef CAS PubMed.
- R. P. Thomas, R. E. Heap, F. Zappacosta, E. K. Grant, P. Pogány, S. Besley, D. J. Fallon, M. M. Hann, D. House, N. C. O. Tomkinson and J. T. Bush, Chem. Sci., 2021, 12, 12098–12106 RSC.
- G. Meng, T. Guo, T. Ma, J. Zhang, Y. Shen, K. B. Sharpless and J. Dong, Nature, 2019, 574, 86–89 CrossRef CAS PubMed.
- D. M. Turner, C. T. M. B. Tom and A. R. Renslo, ACS Comb. Sci., 2014, 16, 661–664 CrossRef CAS PubMed.
- S. Kitamura, Q. Zheng, J. L. Woehl, A. Solania, E. Chen, N. Dillon, M. V. Hull, M. Kotaniguchi, J. R. Cappiello, S. Kitamura, V. Nizet, K. B. Sharpless and D. W. Wolan, J. Am. Chem. Soc., 2020, 142, 10899–10904 CrossRef CAS PubMed.
- Z. Liu, J. Li, S. Li, G. Li, K. B. Sharpless and P. Wu, J. Am. Chem. Soc., 2018, 140, 2919–2925 CrossRef CAS PubMed.
- M. Congreve, R. Carr, C. Murray and H. Jhoti, Drug Discovery Today, 2003, 8, 876–877 CrossRef PubMed.
- H. Jhoti, G. Williams, D. C. Rees and C. W. Murray, Nat. Rev. Drug Discovery, 2013, 12, 644 CrossRef CAS PubMed.
- On most occasions, the aryl alcohol plates were found to be stable for longer than 6 months but were reprepared to align with good practices.
- Click chemistry stands as a transformative approach in synthesis and discovery. Inherently designed for precision, click reactions are notably reliable and selective-often producing predictable and consistent outcomes. For such reactions, monitoring using thin layer chromatography (TLC) proves sufficient for array-based synthesis.
- J. Zhang, X. Zhao, J. R. Cappiello, Y. Yang, Y. Cheng, G. Liu, W. Fang, Y. Luo, Y. Zhang, J. Dong, L. Zhang and K. B. Sharpless, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2103513118 CrossRef CAS PubMed.
- H. K. Patel and T. Bihani, Pharmacol. Ther., 2018, 186, 1–24 CrossRef CAS PubMed.
- A. E.-G. E. Amr, M. El-Naggar, M. A. Al-Omar, E. A. Elsayed and M. M. Abdalla, Molecules, 2018, 23, 1572 CrossRef PubMed.
- D. Sharma, S. Kumar and B. Narasimhan, Chem. Cent. J., 2018, 12, 107 CrossRef CAS PubMed.
- J. A. Aka, M. Zerradi, F. Houle, J. Huot and S.-X. Lin, Breast Cancer Res., 2012, 14, R92 CrossRef CAS PubMed.
- J. M. Day, H. J. Tutill, A. Purohit and M. J. Reed, Endocr.-Relat. Cancer, 2008, 15, 665–692 CrossRef CAS PubMed.
- F. E. F. Pardillo, T. T. Fajardo, R. M. Abalos, D. Scollard and R. H. Gelber, Clin. Infect. Dis., 2007, 44, 1096–1099 CrossRef PubMed.
- X. Y. Han, K. C. Sizer, E. J. Thompson, J. Kabanja, J. Li, P. Hu, L. Gómez-Valero and F. J. Silva, J. Bacteriol., 2009, 191, 6067–6074 CrossRef CAS PubMed.
- M. D. Coleman, Br. J. Dermatol., 1993, 129, 507–513 CrossRef CAS PubMed.
- D. L. Williams, L. Spring, E. Harris, P. Roche and T. P. Gillis, Antimicrob. Agents Chemother., 2000, 44, 1530–1537 CrossRef CAS PubMed.
- E. Cambau, L. Carthagena, A. Chauffour, B. Ji and V. Jarlier, Clin. Infect. Dis., 2006, 42, 238–241 CrossRef CAS PubMed.
- N. P. Mahajan, M. Lavania, I. Singh, S. Nashi, V. Preethish-Kumar, S. Vengalil, K. Polavarapu, C. Pradeep-Chandra-Reddy, M. Keerthipriya, A. Mahadevan, T. C. Yasha, B. N. Nandeesh, K. Gnanakumar, G. J. Parry, U. Sengupta and A. Nalini, Am. J. Trop. Med. Hyg., 2020, 102, 547–552 CrossRef CAS PubMed.
- Compound 9 was first prepared by Dr Grant Bare of the Sharpless Laboratory (unpublished) and was synthesized accordioning to a modified version of that method. Refer to the ESI† for full details.
- J. M. Thiede, S. L. Kordus, B. J. Turman, J. A. Buonomo, C. C. Aldrich, Y. Minato and A. D. Baughn, Sci. Rep., 2016, 6, 38083 CrossRef CAS PubMed.
- J. M. Thiede, S. L. Kordus, B. J. Turman, J. A. Buonomo, C. C. Aldrich, Y. Minato and A. D. Baughn, Sci. Rep., 2016, 6, 38083 CrossRef CAS PubMed.
- E. C. McLoughlin and N. M. O'Boyle, Pharmaceuticals, 2020, 13, 8 CrossRef CAS PubMed.
- M. O. Steinmetz and A. E. Prota, Trends Cell Biol., 2018, 28, 776–792 CrossRef CAS PubMed.
- T. Mühlethaler, D. Gioia, A. E. Prota, M. E. Sharpe, A. Cavalli and M. O. Steinmetz, Angew. Chem., Int. Ed., 2021, 60, 13331–13342 CrossRef PubMed.
- Y. Lu, J. Chen, M. Xiao, W. Li and D. D. Miller, Pharm. Res., 2012, 29, 2943–2971 CrossRef CAS PubMed.
- M. Dong, F. Liu, H. Zhou, S. Zhai and B. Yan, Molecules, 2016, 21, 1375 CrossRef PubMed.
- K. Sun, Z. Sun, F. Zhao, G. Shan and Q. Meng, Future Med. Chem., 2021, 13, 839–858 CrossRef CAS PubMed.
- G. C. Tron, T. Pirali, G. Sorba, F. Pagliai, S. Busacca and A. A. Genazzani, J. Med. Chem., 2006, 49, 3033–3044 CrossRef CAS PubMed.
- S. L. Gwaltney, H. M. Imade, K. J. Barr, Q. Li, L. Gehrke, R. B. Credo, R. B. Warner, J. Y. Lee, P. Kovar, J. Wang, M. A. Nukkala, N. A. Zielinski, D. Frost, S.-C. Ng and H. L. Sham, Bioorg. Med. Chem. Lett., 2001, 11, 871–874 CrossRef CAS PubMed.
- D. B. MacQueen and K. S. Schanze, J. Am. Chem. Soc., 1991, 113, 7470–7479 CrossRef CAS.
- V. Pophristic, L. Goodman and N. Guchhait, J. Phys. Chem. A, 1997, 101, 4290–4297 CrossRef CAS.
- S. Fortin, L. Wei, E. Moreau, J. Lacroix, M.-F. Côté, É. Petitclerc, L. P. Kotra and R. C. Gaudreault, J. Med. Chem., 2011, 54, 4559–4580 CrossRef CAS PubMed.
- R. O. Iakovenko, D. Chrenko, J. Kristek, E. Desmedt, F. Zálešák, F. D. Vleeschouwer and J. Pospíšil, Org. Biomol. Chem., 2022, 20, 3154–3159 RSC.
- F. Toulgoat, B. R. Langlois, M. Médebielle and J.-Y. Sanchez, J. Org. Chem., 2007, 72, 9046–9052 CrossRef CAS PubMed.
- The original SuFEx manifesto presented by Sharpless and co-workers extensively compares and contrasts the reactivity and stability of sulfonyl fluorides and sulfonyl chlorides; see ref. 5.
- T. Watanabe, T. Tsuruo, M. Naito and N. Kokubu, J. Natl. Cancer Inst., 1997, 89, 512–518 CrossRef CAS PubMed.
- L. Wang, K. W. Woods, Q. Li, K. J. Barr, R. W. McCroskey, S. M. Hannick, L. Gherke, R. B. Credo, Y.-H. Hui, K. Marsh, R. Warner, J. Y. Lee, N. Zielinski-Mozng, D. Frost, S. H. Rosenberg and H. L. Sham, J. Med. Chem., 2002, 45, 1697–1711 CrossRef CAS PubMed.
- C. Bingel, E. Koeneke, J. Ridinger, A. Bittmann, M. Sill, H. Peterziel, J. K. Wrobel, I. Rettig, T. Milde, U. Fernekorn, F. Weise, A. Schober, O. Witt and I. Oehme, Cell Death Dis., 2017, 8, e3013 CrossRef CAS PubMed.
- A. E. Prota, K. Bargsten, D. Zurwerra, J. J. Field, J. F. Díaz, K.-H. Altmann and M. O. Steinmetz, Science, 2013, 339, 587–590 CrossRef CAS PubMed.
- A. E. Prota, M. M. Magiera, M. Kuijpers, K. Bargsten, D. Frey, M. Wieser, R. Jaussi, C. C. Hoogenraad, R. A. Kammerer, C. Janke and M. O. Steinmetz, J. Cell Biol., 2013, 200, 259–270 CrossRef CAS PubMed.
- J. M. Andreu, B. Perez-Ramirez, M. J. Gorbunoff, D. Ayala and S. N. Timasheff, Biochemistry, 1998, 37, 8356–8368 CrossRef CAS PubMed.
- T. J. Fltzgerald, Biochem. Pharmacol., 1976, 25, 1383–1387 CrossRef PubMed.
- Y. Wang, H. Zhang, B. Gigant, Y. Yu, Y. Wu, X. Chen, Q. Lai, Z. Yang, Q. Chen and J. Yang, FEBS J., 2016, 283, 102–111 CrossRef CAS PubMed.
- M.-D. Canela, M.-J. Pérez-Pérez, S. Noppen, G. Sáez-Calvo, J. F. Díaz, M.-J. Camarasa, S. Liekens and E.-M. Priego, J. Med. Chem., 2014, 57, 3924–3938 CrossRef CAS PubMed.
- T. Chung, L. Garcia, M. M. Swamynathan, F. E. M. Froeling, L. C. Trotman, D. A. Tuveson and S. K. Lyons, Anal. Chem., 2023, 95, 5661–5670 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05729a |
| This journal is © The Royal Society of Chemistry 2024 |