
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Switching the hydrogenation selectivity of urea derivatives via subtly tuning the amount and type of additive in the catalyst system†
Jun
Zhu
a,
Yongtao
Wang
 a,
Jia
Yao
a and
Haoran
Li
*ab
a,
Jia
Yao
a and
Haoran
Li
*ab
aDepartment of Chemistry, ZJU-NHU United R&D Center, Zhejiang University, Hangzhou 310027, China. E-mail: lihr@zju.edu.cn
bState Key Laboratory of Chemical Engineering, College of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, China
First published on 23rd December 2023
Abstract
Catalytic hydrogenation of urea derivatives is considered to be one of the most feasible methods for indirect reduction functionalization of CO2 and synthesis of valuable chemicals and fuels. Among value-added products, methylamines, formamides and methanol are highly attractive as important industrial raw materials. Herein, we report the highly selective catalytic hydrogenation of urea derivatives to N-monomethylamines for the first time. More importantly, two- and six-electron reduction products can be switched on/off by subtly tuning 0.5 mol% KOtBu (2% to 1.5%): when the molar ratio of KOtBu/(PPh3)3RuCl2 exceeds 2.0, it is favorable for the formation of two-electron reduction products (N-formamides), while when it is below 2.0, the two-electron reduction products are further hydrogenated to six-electron reduction products (N-monomethylamines and methanol). Furthermore, changing the type of additive can also regulate this interesting selectivity. Control experiments showed that this selectivity is achieved by regulating the acid-base environment of the reaction to control the fate of the common hemiaminal intermediate. A feasible mechanism is proposed based on mechanistic experiments and characterization. This method has the advantages of being simple, universal and highly efficient, and opens up a new synthesis strategy for the utilization of renewable carbon sources.
Introduction
In recent years, carbon dioxide (CO2) reduction and functionalization have provided new insights into its chemical conversion and utilization.1–4 However, due to thermodynamic and kinetic limitations, to date, only a few processes using CO2 as a C1 source have been industrialized and these have been mainly used for the production of urea and its derivatives.5–8 Against this backdrop, catalytic hydrogenation of urea derivatives to value-added chemicals is a promising way to expand the resource utilization of CO2 (Scheme 1).9–13 | ||
| Scheme 1 Indirect conversion of CO2 and amines to value-added chemicals via hydrogenation of urea derivatives. | ||
N-Methylamine compounds are important solvents and play an important role in platform chemicals such as medicine, perfumes, synthetic resins, agricultural chemicals and dyes.14,15 Traditionally, producing N-methylamines mainly requires formaldehyde or toxic methylation reagents such as methyl iodide, methyl triflate, dimethyl sulfate, or diazomethane.16–19 Notably, in the process of producing N-monomethylamines, a mixture of N-monomethylamine and N,N-dimethylamine is often obtained because the formation of N,N-dimethylamine is a thermodynamically favorable reaction.20 Thus, highly selective synthesis of N-monomethylamine is a challenge. In this perspective, highly selective catalytic hydrogenation of CO2-derived urea derivatives to methylamines and the corresponding recyclable amines is a novel and environmentally friendly approach. In addition, although direct hydrogenation of CO2 to methanol has been extensively studied, it involves relatively harsh reaction conditions.21–26 In industry, formamides are produced by the reaction of an amine with toxic and flammable CO in methanol using an NaOCH3 catalyst,27–29 and methylamines are obtained by the reaction of an amine with methanol,30,31 both of which require methanol as the raw material. Thus, hydrogenation of urea derivatives may be an alternative strategy for the synthesis of valuable chemicals and fuels from CO2 under mild reaction conditions, and the resulting amine byproducts can be recycled.
In 2011, Milstein and colleagues reported the first example for hydrogenation of urea derivatives to methanol.10 Subsequently, several catalytic systems, including Ru and Ir, have been used for the hydrogenation of CO2-derived urea derivatives to formamide products.32–34 Notably, formamides can be further hydrogenated to yield the six-electron reduction products, such as methylamines and methanol.35–38 At present, methanol is still the main 6-electron reduction product reported by hydrogenation of urea derivatives, and the research on obtaining methylamine compounds is still limited. To date, only Leitner has reported the formation of methylaniline (24% yield) from diphenylurea using Ru/triphos in the presence of acid additives, accompanied by the formation of dimethylamine (8%) and methanol byproducts.32 Inspired by this result, we attempted to achieve the highly selective hydrogenation of urea derivatives to the monomethylamine as the main product. More importantly, an example of good selectivity for all three types of products using a single well-defined catalyst system by adjusting the process conditions is even more lacking.
Obviously, this step from formamides to methylamines and methanol is the key to controlling the selective reduction of urea derivatives to two-electron (N-formamides) and six-electron reduction products (N-monomethylamines and methanol). In view of this, further controlled reduction of formamide products to methylamine and methanol products is a feasible path to expand the utilization of urea and its derivatives. In addition, with formamides it is difficult to achieve selectivity between deoxidation hydrogenation (C–O bond cleavage) and deammonia hydrogenation (C–N bond cleavage).37,39,40 In this context, it is of great significance to develop an efficient catalyst system for hydrogenation of urea derivatives, which can adjust the chemoselectivity to obtain two-electron (N-formamides) and six-electron reduction products (N-monomethylamines and methanol) with high selectivity.
Herein, in order to achieve hierarchical control of hydrogenated urea derivatives to two- and six-electron reduction products, we attempt to further reduce formamide products to N-monomethylamines and CH3OH by tuning the reaction parameters on the basis of a Ru-triphos catalyst system for semi-hydrogenation of urea derivatives to formamides.
Results and discussion
In our previous study on the preparation of formamide from hydrogenated urea derivatives, we observed the formation of the 4-chloro-N-methylaniline by-product in addition to the N-(4-chlorophenyl)formamide main product using the Ru-triphos catalyst system in the absence of KOtBu.33 However, no significant 4-chloro-N-methylaniline product was detected in the presence of 2.5 mol% KOtBu. Based on these results, we wondered if further hydrogenation of formamide to 4-chloro-N-methylaniline could be facilitated by regulating the amount of KOtBu used. It can be clearly seen in Table 1 that when the amount of KOtBu added is 2 mol%, the 7% yield of N-methylamine (maximum yield is 100% based on the number of moles of urea, i.e., the maximum total yields of amine and methylamine is 200%) can be observed (Table 1, entry 1). Lowering the KOtBu loadings to 1.5 mol%, N-formamide was completely converted to N-methylamine (Table 1, entry 2). The results are surprising that just by changing 0.5 mol% KOtBu (2% to 1.5%), the selectivity switches from 84% formamide to 80% N-methylated amine. As the KOtBu loadings gradually decreased, the yield of methanol also decreased (entries 2–6). When the KOtBu loadings decreased to as low as 0.125 mol%, the reaction rate decreased significantly and even the intermediate formamide could be detected (Table 1, entries 6–8). By comparison, it was found that the selectivity of N-monomethylamine was the highest in the presence of 0.25 mol% KOtBu (Table 1, entry 5), but there was still a high content of 4-chloro-N,N-dimethylaniline product (8%). In order to improve the selectivity of N-monomethylamine, we continued to optimize the reaction conditions to reduce the amount of this byproduct. On the basis of the above optimization, the selectivity of N-monomethylamine was not improved with a catalyst loading of 2 mol% (Table 1, entry 9). Subsequently, we changed the reaction temperature and found that the yield of the N,N-dimethylamine by-product decreased with the increase of reaction temperature (Table 1, entries 10 and 11). When the reaction temperature was increased to 160 °C, only traces of N,N-dimethylamine could be detected, and the yield of 4-chloro-N-methylaniline was up to 98% (Table 1, entry 11). Interestingly, the selectivity of N-monomethylamine decreased when the hydrogen pressure was lowered, but the conversion rate did not decrease (Table 1, entries 12 and 13). Moreover, 89% and 81% yields of 4-chloro-N-methylaniline were achieved respectively when using NaHBEt3 or NaOH as additives instead of KOtBu (Table 1, entries 14 and 15).|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | KOtBu (mol%) | H2 (bar) | Temperature (°C) | Time (h) | Yield (%) of 1a | Yield (%) of 1b | Yield (%) of 1c | Yield (%) of 1d |
| a Reaction conditions: substrate (2 mmol), (PPh3)3RuCl2 (1 mol%), triphos (1.5 mol%), KOtBu (0–2.5 mol%), H2 (10–50 bar), THF (4 mL), 140–160 °C (bath temperature). Determined by GC using biphenyl as an internal standard. Identification of the products was also confirmed by GC-MS and 1H NMR; isolated yield in parentheses. b (PPh3)3RuCl2 (2 mol%) and triphos (3 mol%) were used. c NaHBEt3 was used instead of KOtBu. d NaOH was used instead of KOtBu. | ||||||||
| 1 | 2 | 50 | 140 | 12 | 2 | 84 | 7 | <1 |
| 2 | 1.5 | 50 | 140 | 12 | 9 | 0 | 80 | 6 |
| 3 | 1 | 50 | 140 | 12 | 7 | 0 | 79 | 8 |
| 4 | 0.5 | 50 | 140 | 12 | 2 | 0 | 83 | 7 |
| 5 | 0.25 | 50 | 140 | 12 | 1 | 0 | 87 | 8 |
| 6 | 0.125 | 50 | 140 | 12 | <1 | 4 | 80 | 5 |
| 7 | 0.125 | 50 | 140 | 18 | <1 | 0 | 85 | 6 |
| 8 | 0 | 50 | 140 | 24 | <1 | 7 | 74 | 6 |
| 9b | 0.25 | 50 | 140 | 8 | 2 | 0 | 81 | 8 |
| 10 | 0.25 | 50 | 150 | 10 | 1 | 0 | 96 | 2 |
| 11 | 0.25 | 50 | 160 | 8 | <1 | 0 | 98 (93) | <1 |
| 12 | 0.25 | 30 | 160 | 8 | 5 | 0 | 91 | 1 |
| 13 | 0.25 | 10 | 160 | 8 | 19 | 0 | 73 | 1 |
| 14c | 1 | 50 | 140 | 12 | 2 | 0 | 89 | 5 |
| 15d | 1 | 50 | 140 | 12 | 3 | 0 | 81 | 7 |

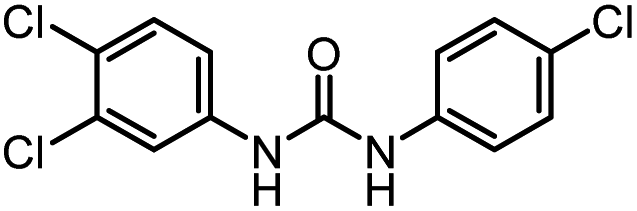
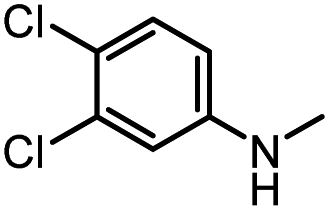
After investigating the optimum reaction conditions, the substrate range of the ruthenium-catalyzed selective preparation of N-monomethylamines from urea derivatives was discussed (Table 2). We initially chose substrates with electron-withdrawing groups on the aniline ring, with yields of N-monomethylamines ranging from 79 to 98% (entries 2–5). For 1,3-diphenylurea without substituents on the aromatic ring, the yield of N-methylaniline was 98% using NaHBEt3 as an additive instead of KOtBu. For 1,3-di(pyridin-2-yl)urea, which replaced the aromatic ring with pyridine, a moderate yield of 2-(methylamino)pyridine (61%) was obtained (entry 6). Surprisingly, substrates with strong electron-donating substituents on the aryl ring of aniline were also well tolerated, with 82–93% yields of the corresponding products (entries 7–9). As expected, asymmetric urea (1-(4-chlorophenyl)-3-(3,4-dichlorophenyl)urea) was also well converted to 4-chloro-N-methylaniline (65%) and 3,4-dichloro-N-methylaniline (22%) products (entry 10). The different yields may be related to the electronic and steric properties of substituents on the N-aromatic ring.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Time (h) | Product | Yieldb (%) of N-monomethylamine | Yieldb (%) of amine |
| a Reaction conditions: substrate (2 mmol), (PPh3)3RuCl2 (1 mol%), triphos (1.5 mol%), KOtBu (0.25 mol%), H2 (50 bar), THF (4 mL), 160 °C (bath temperature). b Yield determined by GC using biphenyl as an internal standard. Identification of the products was also confirmed by GC-MS and 1H NMR; isolated yield in parentheses. c NaHBEt3 was used instead of KOtBu. d 140 °C (bath temperature). | |||||
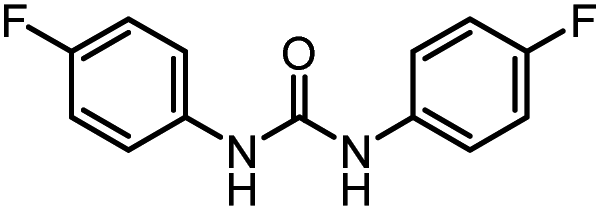
| 1 |
|
12 |

|
98 (94) | 100 |
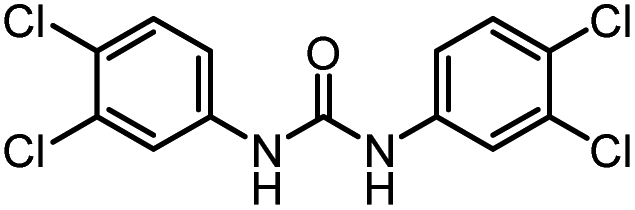
| 2 |
|
8 |
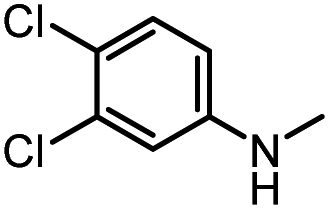
|
86 | 103 |
| 3 |

|
12 |

|
91 | 104 |
| 4 |

|
12 |
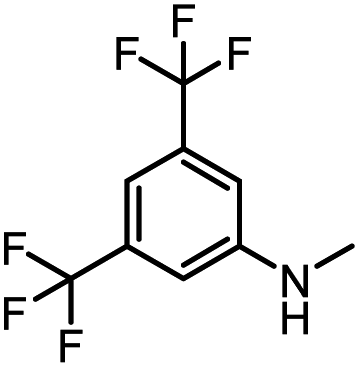
|
79 | 119 |
| 5 |

|
12c |

|
98 (91) | 100 |
| 6 |

|
12 |
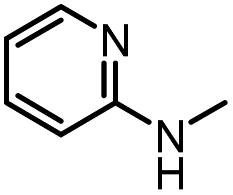
|
61 | 98 |
| 7 |
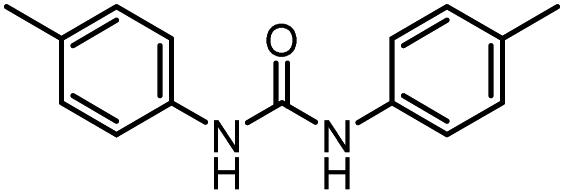
|
12 |
|
92 (89) | 102 |
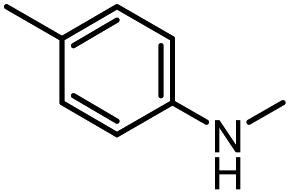
| 8 |

|
12 |
|
82 | 107 |
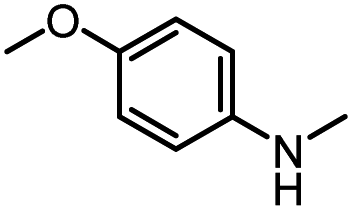
| 9 |

|
12d |

|
93 | 99 |
| 10 |
|
8 |

|
65 | 34 |
|
22 | 74 | |||
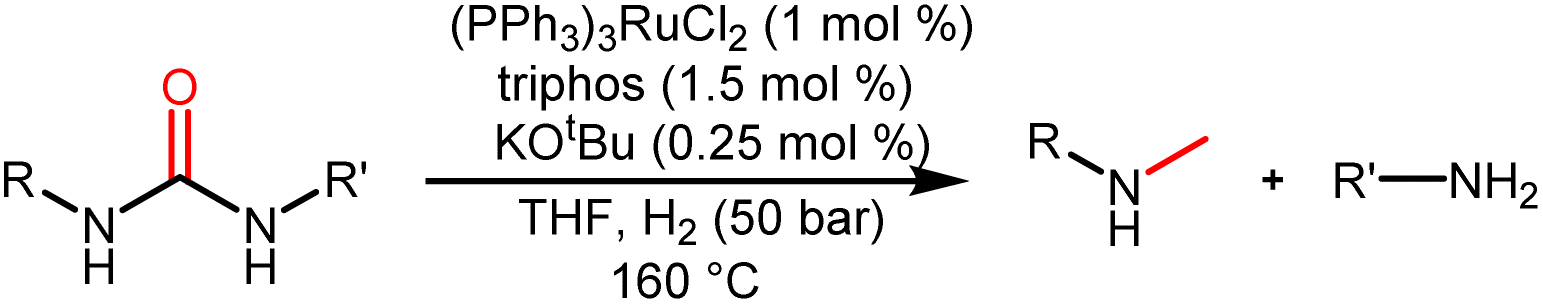
To understand the reaction path for hydrogenation of urea derivatives to methylamines, some control experiments were carried out. Remarkably, performing the catalytic hydrogenation reaction using (PPh3)3RuCl2 (1 mol%) and triphos (1.5 mol%) in the absence of KOtBu under H2 (50 bar) at 140 °C for 24 h in anhydrous tetrahydrofuran (THF), isocyanate and formamide were detected in addition to fully hydrogenated products (monomethylamine, dimethylamine and trace amounts of methanol) (Table 1, entry 8). In our previous studies,33 it was demonstrated that isocyanates were derived from the thermal decomposition of urea derivatives and were intermediates that form formamides. On this basis, 4-chlorophenyl isocyanate and N-(4-chlorophenyl)formamide were respectively used as substrates for hydrogenation experiments under identical reaction conditions, and these substrates were easily reduced to 4-chloro-N-methylaniline (Schemes 2a and b). Thus, according to preliminary experimental results, this reaction may be a sequential reaction of stepwise hydrogenation (Scheme 2d), in which urea derivatives underwent catalytic pyrolysis (step 1) and then the generated isocyanates were hydrogenated to formamides (step 2). They were eventually fully hydrogenated to form six-electron reduction products (step 3). In addition, the optimization experiment results showed that the conversion efficiency of the catalytic system with KOtBu was significantly higher than that of the catalytic system without KOtBu (Table 1). According to the results of control catalytic experiments in Schemes 2a (a Ru loading of 1% with 0.25 mol% KOtBu) and 2c (a low Ru loading of 0.1 mol% with 0.25 mol% KOtBu), it is speculated that the addition of KOtBu will accelerate the reaction rate for hydrogenation of ureas to formamides, while the catalytic system in the absence of KOtBu will promote the further hydrogenation of formamides to methylamine products.
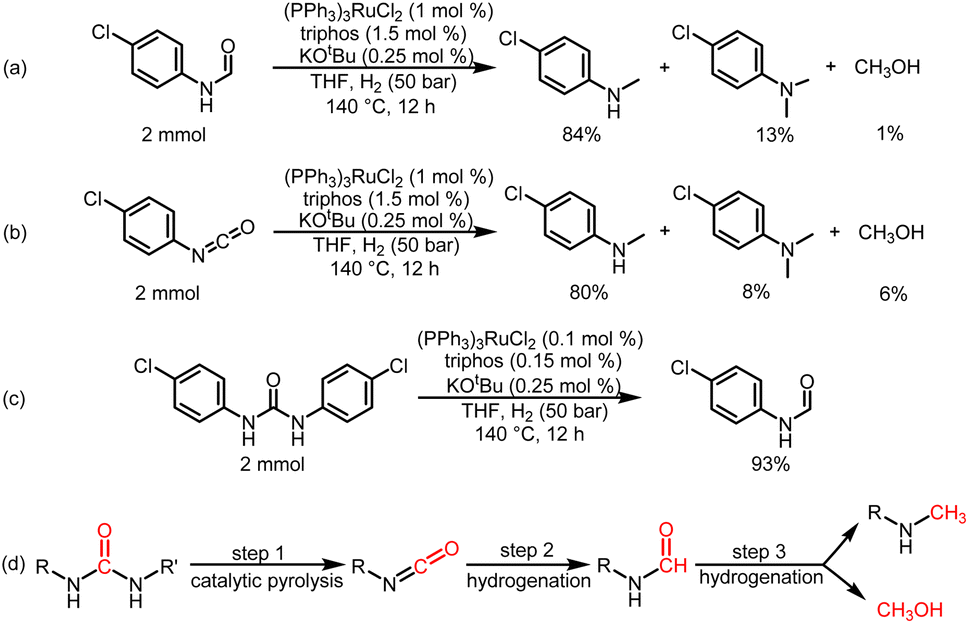 | ||
| Scheme 2 Control catalytic experiments. (a) Catalytic hydrogenation of N-(4-chlorophenyl)formamide in the presence of a Ru loading of 1% with 0.25 mol% KOtBu. (b) Catalytic hydrogenation of 4-chlorphenylisocyanate in the presence of a Ru loading of 1% with 0.25 mol% KOtBu. (c) Catalytic hydrogenation of 1,3-bis(4-chlorophenyl)urea in the presence of a low Ru loading of 0.1 mol% with 0.25 mol % KOtBu. (d) Possible reaction pathway for hydrogenation of urea derivatives to six-electron reduction products. | ||
We then attempted to gain insight into this interesting mechanism for switching the hydrogenation selectivity of urea derivatives by adjusting the amount of KOtBu. Multiple control experiments were conducted via tuning the amount of KOtBu to investigate its role. Increasing the KOtBu loadings from 0 to 2 mol% while keeping the remaining conditions the same results in a gradual increase in the conversion of 1,3-bis(4-chlorophenyl)urea (Fig. 1a). In this reaction, when the KOtBu loadings exceed 2.0 mol%, the hydrogenation efficiency of 1,3-bis(4-chlorophenyl)urea decreases slightly. Similarly, increasing the reaction temperature causes the conversion of 1,3-bis(4-chlorophenyl)urea to rise dramatically (Fig. S4†). It can be seen from Fig. 1b that the hydrogenation of isocyanate to formamide is a rapid hydrogenation process. With the increase of KOtBu loadings, the selectivity of formamide decreases obviously and the formamidine byproduct was mainly formed (see the ESI† for details). Fig. 1a and b show that when the molar ratio of KOtBu/(PPh3)3RuCl2 increases from 0 to 2.0, KOtBu significantly improves the catalytic activity of the Ru catalyst in the thermal decomposition of urea into isocyanate, and the formation of isocyanate is rapidly converted to formamide. In the experiments for hydrogenation of formamide (Fig. 1c), the selectivity of methanol gradually increases as the molar ratio of KOtBu/(PPh3)3RuCl2 increases from 0 to 2.0. When KOtBu/(PPh3)3RuCl2 = 1.0, the catalytic activity for hydrogenation of formamide to methylamine and methanol is the best. However, when the molar ratio of KOtBu/(PPh3)3RuCl2 is higher than 2.0, low hydrogenation activity of the catalyst system is observed, and only the strongest electrophilic reagent (isocyanate) can be reduced, while the weaker electrophilic reagent (formamide) will not be reduced. It is worth noting that isocyanate is difficult to convert in the absence of a Ru catalyst system (Fig. S1†). Thus, when the KOtBu loadings are higher than 2 (2 equiv. relative to (PPh3)3RuCl2), it is conducive to the hydrogenation of the urea derivative to produce formamide and inhibits further hydrogenation of formamide. When the molar ratio of KOtBu/(PPh3)3RuCl2 is lower than 2.0, high KOtBu loadings are conducive to breaking the C–N bond to produce methanol, while low KOtBu loadings tend to produce methylamine.
 | ||
| Fig. 1 Reaction conditions: substrate (2 mmol), (PPh3)3RuCl2 (0.5 mol%), triphos (0.75 mol%), H2 (50 bar), THF (4 mL). (a) Substrate (1,3-bis(4-chlorophenyl)urea), 120 °C, 1 h; (b) substrate (4-chlorophenyl isocyanate), 120 °C, 1 h; (c) substrate (N-(4-chlorophenyl)formamide), 140 °C, 1 h; (d) substrate (N-(4-chlorophenyl)formamide), KOtBu (1.75 equiv. relative to (PPh3)3RuCl2), 4 h. | ||
In addition, from the perspective of kinetics, the proportion of products can be changed by adjusting the temperature. To further improve the selectivity for hydrogenation of formamide to methanol, we tried to reduce the reaction temperature to achieve this goal. Fig. 1d shows that with the decrease of temperature, the reaction rate slows down, while the selectivity of methanol gradually increases. This makes it possible to obtain highly selective methanol from urea derivatives by a two-step process (urea derivatives are first semi-hydrogenated to formamides, followed by hydrogenation of formamides to produce the highly selective methanol product). Moreover, it has been reported that methanol may be the source of methyl groups.14,31,41 Based on this, we conducted relevant experiments and found that the reaction occurred with difficulty when keeping the reaction conditions the same (Schemes 3a and b). Thus, the possibility of monomethylamine/dimethylamine formation by methanol and amine/methylamine can be ruled out. Furthermore, in combination with the hydrogenation experiments of formamide in Scheme 2a and Fig. 1, obvious dimethylamine byproducts can also be detected, thus dimethylamine may be caused by further reaction between monomethylamine and formamide (Scheme 4c).
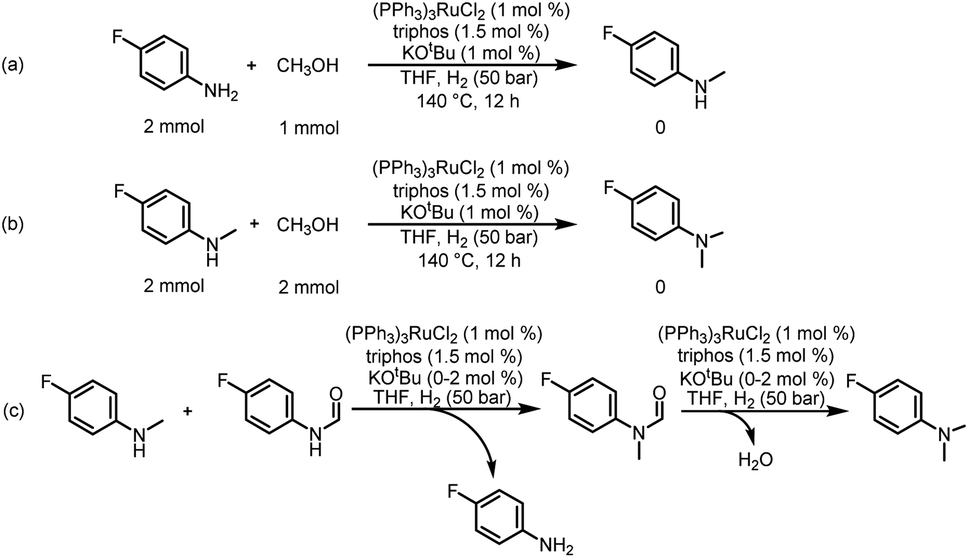 | ||
| Scheme 3 Possible pathway for the source of methyl groups. (a) Catalytic coupling of 4-fluoroaniline and methanol. (b) Catalytic coupling of 4-fluoro-N-methylaniline and methanol. (c) Possible reaction pathway for hydrogenation of urea derivatives to N,N-dimethylanilines. | ||
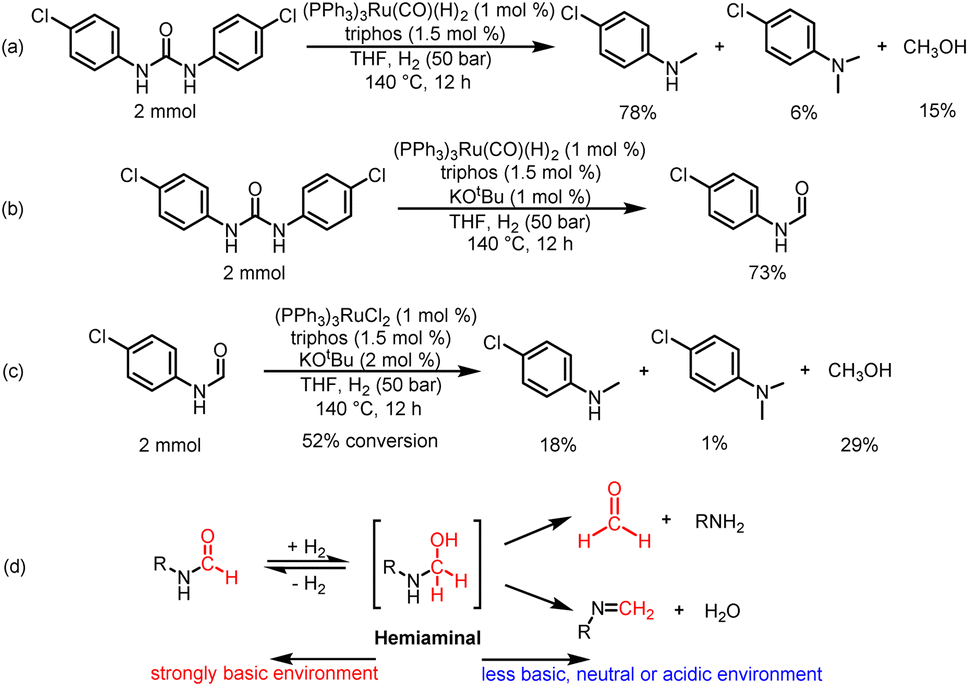 | ||
| Scheme 4 Mechanistic reactions. (a) Catalytic hydrogenation of 1,3-bis(4-chlorophenyl)urea with (PPh3)3Ru(CO)(H)2 as a metal precursor in the absence of KOtBu. (b) Catalytic hydrogenation of 1,3-bis(4-chlorophenyl)urea with (PPh3)3Ru(CO)(H)2 as a metal precursor in the presence of 1 mol% KOtBu. (c) Catalytic hydrogenation of N-(4-chlorophenyl)formamide in the presence of a Ru loading of 1 mol% with 2 mol% KOtBu. (d) Transformation pathways of hemiaminal intermediate in acid–base environments. | ||
In order to further understand the mechanism of this reaction, we first addressed the effect of base on the catalytic activity of the Ru-based catalyst system. According to the experimental results obtained, excess base may inhibit the catalytic activity of the catalyst system for the hydrogenation of formamide. This hypothesis is further supported by the results of an acid-base neutralization experiment: the excess HNTf2 neutralized the remaining base, leading to a significant change in the product from formamide to methylamine in the hydrogenation of 1,3-bis(4-chlorophenyl)urea (Fig. S5†). Unfortunately, HNTf2 may remain in the reaction solution. Lewis acids are known to activate amide bonds and promote nucleophilic addition to amides.41–46 Thus, it cannot be ruled out that residual HNTf2 promotes the hydrogenation of formamide.
Then, we discussed the identification and basic reactivity of Ru species for hydrogenation of urea derivatives. When a mixture of (PPh3)3RuCl2/triphos/2.5% KOtBu/1,3-bis(4-chlorophenyl)urea was submitted to the catalytic conditions, the ESI-MS and NMR spectra of the reaction mixture indicated the formation of (triphos)Ru(CO)(H)2 complexes (see the ESI† for details). Importantly, the (triphos)Ru(CO)(H)2 species were also formed under a KOtBu loading of 1%, which indicated that the catalytic species for catalytic hydrogenation of urea derivatives to two- and six-electron reduction products were associated with (triphos)Ru(CO)(H)2.41,47 When the (PPh3)3RuCl2/triphos/KOtBu mixture was heated under catalytic conditions, the ESI-MS data of the obtained crude reaction solution indicated that (triphos)Ru(PPh3)(H)2 may be the precatalyst formed in situ (see the ESI† for details). After the addition of the substrate, the dissociative replacement of PPh3 occurred on the (triphos)Ru(PPh3)(H)2 complex by CO (it may be derived from decarbonylation of various compounds containing C–O bonds in the reaction solution) to form the (triphos)Ru(CO)(H)2 species in situ.32,48,49 Moreover, the formation of dimeric Ru-containing species [(triphos)Ru(μ-H)]2 was inhibited effectively. Based on these results, (PPh3)3Ru(CO)(H)2 was used to replace (PPh3)3RuCl2 as a metal precursor for the hydrogenation of 1,3-bis(4-chlorophenyl)urea in the absence of KOtBu (Scheme 4a). Notably, the yield of the 4-chloro-N-methylaniline product was 78%, suggesting that (triphos)Ru(CO)(H)2 may be a catalytic intermediate for hydrogenation of urea derivatives to methylamines, and the coordination of the reaction substance is mainly through the transient loss of PPh3/CO. In addition, 1,3-bis(4-chlorophenyl)urea was successfully hydrogenated with (PPh3)3Ru(CO)(H)2 as a metal precursor in the presence of 1% KOtBu. Significantly, the major product was N-(4-chlorophenyl)formamide with a 73% yield, and no significant six-electron reduction products were detected (Scheme 4b). Similarly, the addition of base inhibited the further transformation of formamide. This suggests that the base may have a dual role: (a) to abstract the chloride ligand from the metal precursor to produce a ruthenium complex of dihydride structure, and (b) to inhibit the hydrogenation of formamide to methanol and methylamine.
Although we have some preliminary understanding of the dramatic switching selectivity from formamide to N-methylamine, it has not been rationalised appropriately. If the KOtBu loading was less than 2 mol%, its main role was to abstract the Cl− ligand on the metal precursor, which meant that the KOtBu loading determines the content of catalytic active components. That is, the lower the KOtBu loading, the poorer the catalytic efficiency. In fact, based on our experimental results, the outcome from the magic base loadings of 2 mol% and 1.5 mol% were very drastic (Table 1, entries 1 and 2). Further decrease in the base loading from 1.5 mol% to 0 mol% had no significant effect on the catalytic outcome (Table 1, entries 2–8). Unexpectedly, 1,3-bis(4-chlorophenyl)urea was able to convert well even in the absence of a base (Table 1, entry 8). By comparing the ESI-MS spectrum of the catalyst system lacking KOtBu under a N2 atmosphere, it is obvious that additional catalytic intermediate (triphos)Ru(CO)(H)2 was detected in the reaction solution of the catalyst system in the absence of KOtBu under a H2 atmosphere (Fig. S10 and S16†). Thus, the Cl− ligand on the metal precursor may not be abstracted only through KOtBu. H− ligands on Ru metals can also be generated by H2 dissociation exchange (Fig. S17†). At a relatively high reaction temperature, the stable 6-coordinated 18-electron catalyst precursor opens the vacant site of the 5-coordinated 16-electron reactive intermediate, followed by H2 coordination. Subsequently, the hydrogen ligands undergo heterolytic dissociation, dissociating into H+ and H−, where H− becomes a ligand, while H+ and Cl− become HCl (Scheme 5),50,51 resulting in an acidic reaction solution. This conclusion is supported by the acid-base test results of the reaction solution (the reaction solution without KOtBu is acidic, and the reaction solution in the presence of 2.5% KOtBu is basic).
 | ||
| Scheme 5 Proposed reaction mechanism for ruthenium-catalyzed hydrogenation of urea derivatives to formamides, methanol and monomethylamines. | ||
In theory, the complete exchange of the Cl− ligand on 1 mol% Ru metal by H− produces 2 mol% HCl, which explains why the selectivity switches from formamide to N-methylated amine just by a change of 0.5 mol% KOtBu (2 mol% to 1.5 mol%). The major role of base additive (KOtBu) is to reduce the content of HCl in the reaction system and increase the pH value of the reaction solution (Scheme 5). When the KOtBu loading is 2.5 mol%, the reaction solution is basic (in addition to neutralizing 2 mol% HCl, there is still a residual base), and the product is mainly formamide, a two-electron reduction product. When the KOtBu loading is 2 mol%, the reaction solution approaches neutral and the product begins to be converted into six-electron reduction products (Scheme 4d), which is in line with the phenomenon reported by Verpoort and co-workers in the dehydrogenative amidation of alcohols and amines.52 In the absence of Cs2CO3, no amide formation was observed (C–O bond cleavage). Use of the base amount ranging from 1 to 3 mol% (a catalyst loading of 1 mol%) resulted in amide as a main product. When the KOtBu loading is 1.5 mol%, the reaction solution becomes acidic and the products are dramatically and rapidly converted to six-electron reduction products. Previously, Cole–Hamilton and Leitner et al. also reported the dehydration of amides to form amines using the triphos/Ru catalyst system in the absence of additional additives and in the presence of an acid additive (MSA/HNTf2).32,44,45 Particularly, Leitner reported the formation of methyl aniline from diphenyl urea using Ru/triphos in the presence of an acid additive.32 In addition, we performed 1 mol% (PPh3)3RuHCl and (PPh3)3Ru(CO)HCl (metal Ru with one Cl− ligand and one H− ligand) substitution of (PPh3)3RuCl2 (metal Ru with two Cl− ligands) as metal precursors for hydrogenation of urea derivatives, respectively. The experimental results were consistent with our expectations (Fig. S2†). When the KOtBu loading is 1.5 mol% (higher than 1 mol%), the reaction product stays in the formamide stage. In the absence of KOtBu, formamide continued to hydrogenate to yield six-electron reduction products (Fig. 2). Although there is some literature reporting that the formation of N-methylamine involves a dehydration step which is usually promoted by KOtBu,53,54 this is a significant pathway in the hydrogen transfer reactions. In these catalytic reactions, the transfer of two hydrogen atoms may occur through the inner-sphere mechanism with deprotonation of the coordinated alcohol and β-H elimination of the resulting alkoxy group.55 Because the inner-sphere mechanism involves the coordination of the substrate to the metal fragment and then deprotonation with a Lewis base, additional bases are needed to facilitate the reaction to occur. The direct synthesis of imines from alcohols and amines reported by Milstein et al. does not require the addition of bases to achieve the dehydration step.56 Moreover, the formal deoxyhydrogenation of lactam reported by Kayaki can produce dehydrated products in the absence of bases.57
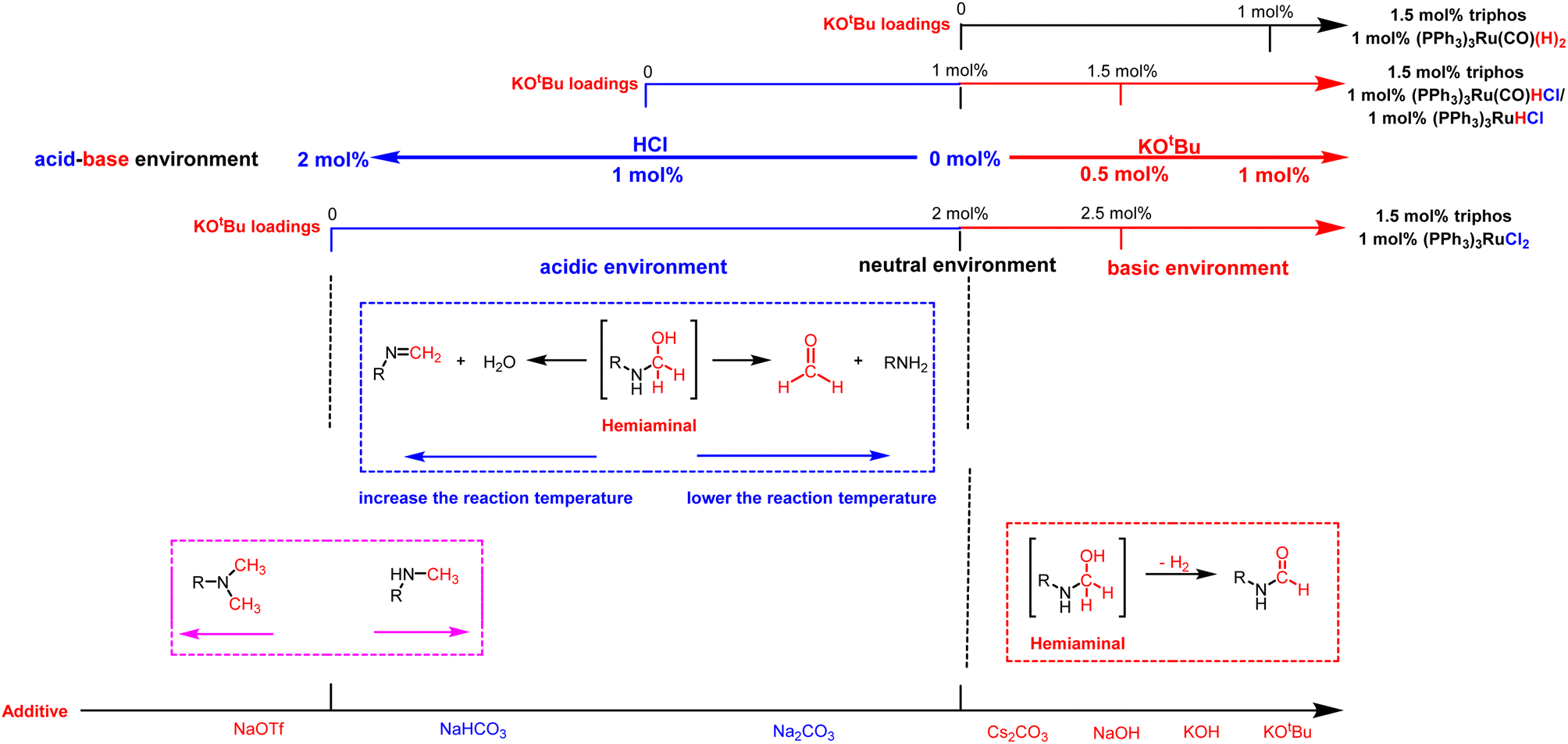 | ||
| Fig. 2 The effect of adding different amounts of bases and selecting different additives on the acidity–alkalinity of the reaction solution and selectivity of products. | ||
Based on these results, we examined the effect of base type in the presence of (PPh3)3RuCl2 (1 mol%), triphos (1.5 mol%), and base (2.5 mol%). 1,3-Bis(4-chlorophenyl)urea was hydrogenated under H2 (50 bar) at 140 °C for 12 h in THF as the benchmark reaction (Table 3). It was found that N-formamide was mainly formed with a strong base, and no significant six-electron reduction products were observed (Table 3, entries 1–4). Obviously, the yield of formamide with a Cs2CO3 loading of 2.5 mol% (Table 3, entry 3) was much lower than that of formamide with a low Cs2CO3 loading of 1.25 mol% (Table 3, entry 4). This may be caused by the excessive amount of base added, because neutralizing 2 mol% HCl requires about 1 mol% Cs2CO3 in theory, which is consistent with the result of relatively high KOH loadings (Table 3, entry 2). Importantly, the use of the weak bases Na2CO3 and NaHCO3 resulted in a substantial increase in the production of methylamine (Table 3, entries 5 and 7). When Na2CO3 and NaHCO3 loadings were increased, N-formamide yields were as high as 92% and 93%, and only a small amount of methylamine and methanol could be detected (Table 3, entries 6 and 8). It is worth noting that the formation of methylamine was also observed with sodium trifluoromethanesulfonate (NaOTf; an acid) as an additive, but the reaction rate was significantly slower (Table 3, entry 9). It is similar to the experimental results reported by Huynh et al., which can selectively catalyze both dehydrogenative amidation and N-alkylation through coupling of alcohols with amines.58 Thus, the interesting selectivity for hydrogenation of urea derivatives to two- and six-electron reduction products may also be achieved by controlling the fate of a common hemiaminal intermediate (Scheme 4c).55,59–64 In a basic environment, the process of dehydrogenation of hemiaminal is promoted by the base, as this is a reversible process.65 The dehydrogenation rate (from hemiaminal to formamide) is greater than the hydrogenation rate (from formamide to hemiaminal), which is conducive to the formation of the formamide product.52,58 In contrast, in a neutral or acidic environment, hemiaminal further breaks the C–N bond to form formaldehyde or breaks the C–O bond to form an imine.59
| Entry | Additive | Yield (%) of 1a | Yield (%) of 1b | Yield (%) of 1c | Yield (%) of 1d |
|---|---|---|---|---|---|
| a Reaction conditions: 1,3-bis(4-chlorophenyl)urea (2 mmol), (PPh3)3RuCl2 (1 mol%), triphos (1.5 mol%), additive (2.5 mol%), H2 (50 bar), THF (4 mL), 140 °C (bath temperature). b KOH (4 mol%). c Cs2CO3 (1.25 mol%). d Na2CO3 (1.25 mol%). e NaHCO3 (4 mol%). | |||||
| 1 | KOH | 0 | 94 | 0 | 0 |
| 2b | KOH | 0 | 42 | 0 | 0 |
| 3 | Cs2CO3 | 0 | 33 | <1 | 0 |
| 4c | Cs2CO3 | 0 | 87 | <1 | 0 |
| 5d | Na2CO3 | 0 | 0 | 91 | 7 |
| 6 | Na2CO3 | 2 | 92 | 3 | 0 |
| 7 | NaHCO3 | 7 | 1 | 82 | 5 |
| 8e | NaHCO3 | 2 | 93 | <1 | 0 |
| 9 | NaOTf | 0 | 22 | 4 | 46 |
Overall, we believe that selectivity in the formamide versus six-electron reduction products can be achieved by controlling the acidity and basicity of the reaction solution by adding different amounts of bases and choosing different additives (Fig. 2). In a basic environment (Scheme 4b), hemiaminal intermediates are more likely to release H2 to form formamide,65,66 while in a neutral environment (Scheme 4a) and acidic environment (Table 3, entry 9 and Fig. S5†), they eliminate H2O/amine to produce the corresponding imine/formaldehyde. When the acidity of the reaction solution was weakened or nearly neutral, the selectivity of formaldehyde was improved by eliminating amines with hemiaminal (Table 1 and Fig. 1c).38,67 Hemiaminal, on the other hand, helps eliminate the imine produced by H2O.32,44,45 It is worth noting that excessive acidity and alkalinity of the reaction environment can significantly reduce the efficiency of hydrogenation of urea derivatives (Fig. 1a and Table 3, entry 9). In addition, changing the reaction temperature can further improve the selectivity of the hemiaminal intermediates to eliminate H2O/amine to imine/formaldehyde (Fig. 1a). Within a certain reaction temperature range, increasing the reaction temperature is conducive to hemiaminal dehydration to methylamine, while lowering the reaction temperature can improve the selectivity of hemiaminal deamination to methanol.
Based on our experimental results and previous reports on the ruthenium-catalyzed hydrogenation of urea derivatives to formamides,33 we propose a plausible three competing catalytic cycles for hydrogenation of urea derivatives to N-formamides, N-monomethylamines, and methanol (Scheme 5). The precatalyst (triphos)Ru(PPh3)(H)2 (2) was prepared in situ by releasing HCl under H2 pressure using triphos and ruthenium precursor as raw materials. Then the PPh3 is dissociated to form the 16-electron dihydrogen complex 3, which is a reversible process. Catalytic thermal decomposition of the urea derivative occurs to eliminate R'NH3 to produce an isocyanate. Complex 3 is coordinated with the isocyanate to form substrate complex 4, which was then formed into hydride complex 5 by the classical migratory insertion step. The hydrogenolysis of the Ru–O bond is caused by the H2 molecular coordination to form complex 6, and the proton transfer to the oxygen through the adjacent H2 ligand leads to the formation of dihydride structure 7. Compound 8 is removed from 7 to regenerate active complex 3, and the released compound 8 undergoes tautomerism to form N-formamide as the product. N-formamide reacts with complex 3 to form complex 9, and neighboring H is transplanted and inserted to form 10. Complex 10 is coordinated with H2 to form 11, and protons are transferred to oxygen via the neighboring H2 ligand to form compound 12. In a basic environment, complex 12 terminates the subsequent reaction and dehydrogenates to formamide products via a reverse process.65,66 In contrast, the reaction continues, and the hemiaminal intermediate immediately loses water or the amine proceeds to hydrogenation. For formamide to methylamine conversion, complex 12 produces complex 13 by eliminating H2O.56,68 Then, complex 14 is formed by migration insertion, which is coordinated with hydrogen to form 15.69 The proton transfer to nitrogen via the adjacent H2 ligand (16) leads to the regeneration of the catalytically active species 3, releasing N-monomethylamine as a product. Similarly, for formamide to methanol conversion, complex 12 produces formaldehyde complex 17 at first by eliminating RNH2,11,64,70 then via migration insertion (18), coordination of an H2 molecule (19), and proton transfer (20), the catalytically active species 3 is finally regenerated and the methanol product is released.
Conclusions
In summary, we developed an approach to control the product selectivity for hydrogenation of urea derivatives by changing the amount and type of additive in the in situ triphos-Ru catalyst system. The addition of different amounts and different types of additives is mainly used to change the acid–base environment of the reaction. In a basic environment, the urea derivatives undergo highly selective catalytic hydrogenation to formamides. However, if the added base is too high, it will seriously reduce the yield of formamides. In a neutral or an acidic environment, further hydrogenation of formamides was promoted to produce six-electron reduction products. Meantime, the selectivity of methanol in a less acidic or nearly neutral environment is higher than that in an acidic environment. Furthermore, lowering the reaction temperature can further improve the selectivity of methanol for hydrogenation of formamides, and increasing the temperature is conducive to the formation of methylamines. Our mechanism study shows that the active species for catalytic hydrogenation of urea derivatives to two- and six-electron reduction products are dihydrogen compound (triphos)Ru(H)2. It is due mainly to the addition of excess base that inhibited the further conversion of formamides. This study provides a green and mild synthetic route for the highly selective production of N-monomethylamine compounds, and develops an effective Ru-based catalyst system for the controlled synthesis of methylamines, formamides and methanol. Also, it may provide new insights into the formylation and alkylation of amines using CO2 and H2.Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the ESI.† And all data can be obtained from the authors.Author contributions
H. L. conceived the project and supervised the work with Y. W. and J. Y. J. Z. performed the catalysis experiments and wrote the manuscript. All authors discussed the results and contributed to the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Key R&D Program of China (2022YFA1503200), Zhejiang Provincial Natural Science Foundation of China under Grant No. LQ22B030011, the Fundamental Research Funds for the Central Universities, and the National Natural Science Foundation of China (No. 22073081). We thank Prof. Ping Lu (Zhejiang University) for inspiring discussion on the mechanism of reaction, Mr Jiankai Zou (Zhejiang University) for GC-MS detection, Mrs Ling He (Zhejiang University) for NMR detection and Prof. Qiaohong He (Zhejiang University) for ESI-MS analysis.Notes and references
- R. P. Ye, J. Ding, W. Gong, M. D. Argyle, Q. Zhong, Y. Wang, C. K. Russell, Z. Xu, A. G. Russell, Q. Li, M. Fan and Y. G. Yao, Nat. Commun., 2019, 10, 5698 CrossRef CAS PubMed.
- A. Ramirez, X. Gong, M. Caglayan, S. F. Nastase, E. Abou-Hamad, L. Gevers, L. Cavallo, A. Dutta Chowdhury and J. Gascon, Nat. Commun., 2021, 12, 5914 CrossRef CAS PubMed.
- X. Zhang, Y. Liu, M. Zhang, T. Yu, B. Chen, Y. Xu, M. Crocker, X. Zhu, Y. Zhu, R. Wang, D. Xiao, M. Bi, D. Ma and C. Shi, Chem, 2020, 6, 3312–3328 CAS.
- C. Vogt, M. Monai, G. J. Kramer and B. M. Weckhuysen, Nat. Catal., 2019, 2, 188–197 CrossRef CAS.
- A. Tlili, E. Blondiaux, X. Frogneux and T. Cantat, Green Chem., 2015, 17, 157–168 RSC.
- Z. Tao, C. L. Rooney, Y. Liang and H. Wang, J. Am. Chem. Soc., 2021, 143, 19630–19642 CrossRef CAS PubMed.
- B.-X. Leong, Y.-C. Teo, C. Condamines, M.-C. Yang, M.-D. Su and C.-W. So, ACS Catal., 2020, 10, 14824–14833 CrossRef CAS.
- G. Ji, Y. Zhao and Z. Liu, Green Chem. Eng., 2022, 3, 96–110 CrossRef.
- J. Pouessel, O. Jacquet and T. Cantat, ChemCatChem, 2013, 5, 3552–3556 CrossRef CAS.
- E. Balaraman, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2011, 50, 11702–11705 CrossRef CAS PubMed.
- U. K. Das, A. Kumar, Y. Ben-David, M. A. Iron and D. Milstein, J. Am. Chem. Soc., 2019, 141, 12962–12966 CrossRef CAS PubMed.
- A. Kumar and J. Luk, Eur. J. Org Chem., 2021, 2021, 4546–4550 CrossRef CAS.
- X. Liu, J. G. de Vries and T. Werner, Green Chem., 2019, 21, 5248–5255 RSC.
- Y. Li, I. Sorribes, T. Yan, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 12156–12160 CrossRef CAS PubMed.
- K. Beydoun, T. vom Stein, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2013, 52, 9554–9557 CrossRef CAS PubMed.
- I. Sorribes, K. Junge and M. Beller, Chem.–Eur. J., 2014, 20, 7878–7883 CrossRef CAS.
- X.-D. Li, S.-M. Xia, K.-H. Chen, X.-F. Liu, H.-R. Li and L.-N. He, Green Chem., 2018, 20, 4853–4858 RSC.
- Y. Li, X. Fang, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 9568–9571 CrossRef CAS PubMed.
- W. Liu, B. Sahoo, A. Spannenberg, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2018, 57, 11673–11677 CrossRef CAS.
- H. Wang, H. Yuan, B. Yang, X. Dai, S. Xu and F. Shi, ACS Catal., 2018, 8, 3943–3949 CrossRef CAS.
- J. Zhu, F. Cannizzaro, L. Liu, H. Zhang, N. Kosinov, I. A. W. Filot, J. Rabeah, A. Bruckner and E. J. M. Hensen, ACS Catal., 2021, 11, 11371–11384 CrossRef CAS PubMed.
- J. Wei, R. Yao, Y. Han, Q. Ge and J. Sun, Chem. Soc. Rev., 2021, 50, 10764–10805 RSC.
- J. Zhong, X. Yang, Z. Wu, B. Liang, Y. Huang and T. Zhang, Chem. Soc. Rev., 2020, 49, 1385–1413 RSC.
- S. T. Bai, G. De Smet, Y. Liao, R. Sun, C. Zhou, M. Beller, B. U. W. Maes and B. F. Sels, Chem. Soc. Rev., 2021, 50, 4259–4298 RSC.
- S. Kar, A. Goeppert, J. Kothandaraman and G. K. S. Prakash, ACS Catal., 2017, 7, 6347–6351 CrossRef CAS.
- N. M. Rezayee, C. A. Huff and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 1028–1031 CrossRef CAS PubMed.
- D. Cheng, M. Wang, L. Tang, Z. Gao, X. Qin, Y. Gao, D. Xiao, W. Zhou and D. Ma, Angew. Chem., Int. Ed., 2022, 61, e202202654 CrossRef CAS PubMed.
- P. Ju, J. Chen, A. Chen, L. Chen and Y. Yu, ACS Sustain. Chem. Eng., 2017, 5, 2516–2528 CrossRef CAS.
- L. Zhang, Z. Han, X. Zhao, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2015, 54, 6186–6189 CrossRef CAS PubMed.
- X. Cui, X. Dai, Y. Zhang, Y. Deng and F. Shi, Chem. Sci., 2014, 5, 649–655 RSC.
- O. Jacquet, X. Frogneux, C. Das Neves Gomes and T. Cantat, Chem. Sci., 2013, 4, 2127 RSC.
- T. vom Stein, M. Meuresch, D. Limper, M. Schmitz, M. Holscher, J. Coetzee, D. J. Cole-Hamilton, J. Klankermayer and W. Leitner, J. Am. Chem. Soc., 2014, 136, 13217–13225 CrossRef CAS PubMed.
- J. Zhu, Y. Zhang, Z. Wen, Q. Ma, Y. Wang, J. Yao and H. Li, Chem.–Eur. J., 2023, e202300106, DOI:10.1002/chem.202300106.
- T. Iwasaki, K. Tsuge, N. Naito and K. Nozaki, Nat. Commun., 2023, 14, 3279 CrossRef CAS PubMed.
- J. G. Uranga, A. Gopakumar, T. Pfister, G. Imanzade, L. Lombardo, G. Gastelu, A. Züttel and P. J. Dyson, Sustainable Energy Fuels, 2020, 4, 1773–1779 RSC.
- M. Subaramanian, G. Sivakumar, J. K. Babu and E. Balaraman, Chem. Commun., 2020, 56, 12411–12414 RSC.
- V. Papa, J. R. Cabrero-Antonino, A. Spannenberg, K. Junge and M. Beller, Catal. Sci. Technol., 2020, 10, 6116–6128 RSC.
- J. R. Cabrero-Antonino, R. Adam, V. Papa and M. Beller, Nat. Commun., 2020, 11, 3893 CrossRef CAS PubMed.
- W. Yao, J. Wang, A. Zhong, J. Li and J. Yang, Org. Lett., 2020, 22, 8086–8090 CrossRef CAS PubMed.
- S. Kar, M. Rauch, A. Kumar, G. Leitus, Y. Ben-David and D. Milstein, ACS Catal., 2020, 10, 5511–5515 CrossRef CAS PubMed.
- I. Sorribes, J. R. Cabrero-Antonino, C. Vicent, K. Junge and M. Beller, J. Am. Chem. Soc., 2015, 137, 13580–13587 CrossRef CAS PubMed.
- M.-L. Yuan, J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, ACS Catal., 2016, 6, 3665–3669 CrossRef CAS.
- Y. Q. Zou, S. Chakraborty, A. Nerush, D. Oren, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2018, 8, 8014–8019 CrossRef CAS PubMed.
- J. R. Cabrero-Antonino, E. Alberico, K. Junge, H. Junge and M. Beller, Chem. Sci., 2016, 7, 3432–3442 RSC.
- J. Coetzee, D. L. Dodds, J. Klankermayer, S. Brosinski, W. Leitner, A. M. Slawin and D. J. Cole-Hamilton, Chem.–Eur. J., 2013, 19, 11039–11050 CrossRef CAS PubMed.
- M.-L. Yuan, J.-H. Xie and Q.-L. Zhou, ChemCatChem, 2016, 8, 3036–3040 CrossRef CAS.
- F. M. Geilen, B. Engendahl, A. Harwardt, W. Marquardt, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2010, 49, 5510–5514 CrossRef CAS PubMed.
- F. M. Geilen, B. Engendahl, M. Holscher, J. Klankermayer and W. Leitner, J. Am. Chem. Soc., 2011, 133, 14349–14358 CrossRef CAS PubMed.
- N. Sieffert, R. Reocreux, P. Lorusso, D. J. Cole-Hamilton and M. Buhl, Chem.–Eur. J., 2014, 20, 4141–4155 CrossRef CAS PubMed.
- J. H. Dam, G. Osztrovszky, L. U. Nordstrøm and R. Madsen, Chem.–Eur. J., 2010, 16, 6820–6827 CrossRef CAS PubMed.
- M. H. S. A. Hamid, C. L. Allen, G. W. Allen, A. C. Allen, H. C. Allen, A. J. A. Watson and J. M. J. Williams, J. Am. Chem. Soc., 2009, 131, 1766–1774 CrossRef CAS.
- X.-J. Wu, H.-J. Wang, Z.-Q. Yang, X.-S. Tang, Y. Yuan, W. Su, C. Chen and F. Verpoort, Org. Chem. Front., 2019, 6, 563–570 RSC.
- O. Ogata, H. Nara, M. Fujiwhara, K. Matsumura and Y. Kayaki, Org. Lett., 2018, 20, 3866–3870 CrossRef CAS PubMed.
- S. Elangovan, J. Neumann, J.-B. Sortais, K. Junge, C. Darcel and M. Beller, Nat. Commun., 2016, 7, 12641 CrossRef PubMed.
- A. Nova, D. Balcells, N. D. Schley, G. E. Dobereiner, R. H. Crabtree and O. Eisenstein, Organometallics, 2010, 29, 6548–6558 CrossRef CAS.
- B. Gnanaprakasam, J. Zhang and D. Milstein, Angew. Chem., Int. Ed., 2010, 122, 1510–1513 CrossRef.
- O. Ogata, H. Nara, K. Matsumura and Y. Kayaki, Org. Lett., 2019, 21, 9954–9959 CrossRef CAS PubMed.
- X. Xie and H. V. Huynh, ACS Catal., 2015, 5, 4143–4151 CrossRef CAS.
- A. Mukherjee, A. Nerush, G. Leitus, L. J. Shimon, Y. Ben David, N. A. Espinosa Jalapa and D. Milstein, J. Am. Chem. Soc., 2016, 138, 4298–4301 CrossRef CAS PubMed.
- A. Joshi, S. Kumari and S. Kundu, Adv. Synth. Catal., 2022, 364, 4371–4383 CrossRef CAS.
- Z. Shao, Y. Li, C. Liu, W. Ai, S. P. Luo and Q. Liu, Nat. Commun., 2020, 11, 591 CrossRef CAS PubMed.
- H. Yu, Z. Wu, Z. Wei, Y. Zhai, S. Ru, Q. Zhao, J. Wang, S. Han and Y. Wei, Commun. Chem., 2019, 2, 15 CrossRef.
- C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790–792 CrossRef CAS PubMed.
- L. Artus Suarez, Z. Culakova, D. Balcells, W. H. Bernskoetter, O. Eisenstein, K. I. Goldberg, N. Hazari, M. Tilset and A. Nova, ACS Catal., 2018, 8, 8751–8762 CrossRef CAS.
- J. A. Luque-Urrutia, T. Pèlachs, M. Solà and A. Poater, ACS Catal., 2021, 11, 6155–6161 CrossRef CAS.
- I. S. Makarov, P. Fristrup and R. Madsen, Chem.–Eur. J., 2012, 18, 15683–15692 CrossRef CAS PubMed.
- J. A. Garg, S. Chakraborty, Y. Ben-David and D. Milstein, Chem. Commun., 2016, 52, 5285–5288 RSC.
- A. Maggi and R. Madsen, Organometallics, 2011, 31, 451–455 CrossRef.
- F. E. Fernández, M. C. Puerta and P. Valerga, Organometallics, 2012, 31, 6868–6879 CrossRef.
- X. Liu and T. Werner, Chem. Sci., 2021, 12, 10590–10597 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05674k |
| This journal is © The Royal Society of Chemistry 2024 |