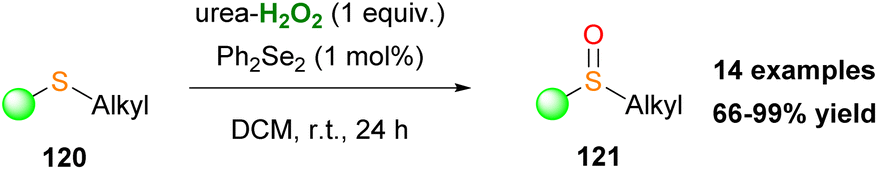DOI:
10.1039/D3SC05618J
(Perspective)
Chem. Sci., 2024,
15, 1177-1203
Organocatalytic activation of hydrogen peroxide: towards green and sustainable oxidations
Received
21st October 2023
, Accepted 15th December 2023
First published on 18th December 2023
Abstract
The advent of organocatalysis provided an additional option in every researcher's arsenal, towards the development of elegant and sustainable protocols for various organic transformations. Oxidation reactions are considered to be key in organic synthesis since oxygenated functionalities appear in many natural products. Hydrogen peroxide is categorized as a green oxidant, since its only by-product is water, offering novel opportunities for the development of green and sustainable protocols. In this review article, we intend to present recent developments in the field of the organocatalytic activation of hydrogen peroxide, providing useful insight into the applied oxidative protocols. At the same time, we will present some interesting mechanistic studies, providing information on the oxygen transfer processes.
 Efthymios T. Poursaitidis | Efthymios T. Poursaitidis is a PhD student at the Department of Chemistry, National and Kapodistrian University of Athens, under the supervision of Assoc. Professor Christoforos G. Kokotos. Efthymios studied Chemistry at the Department of Chemistry of the National and Kapodistrian University of Athens, graduating in 2021. He obtained his MSc degree in 2023 in the Kokotos group and his research interests focus on green organocatalytic oxidations. |
 Petros L. Gkizis | Dr Petros L. Gkizis was born in Athens, Greece. He obtained his BSc, MSc and PhD from the Aristotle University of Thessaloniki. In 2012, he was appointed as a postdoctoral fellow at Aristotle University of Thessaloniki in collaboration with the University of Crete (ERC-Grant). In 2014, he was appointed as a Technology Transfer Research Scientist at Pharmathen S.A. Pharmaceuticals, Thessaloniki, Greece. In 2020, he joined the group of Prof. Christoforos Kokotos at the National and Kapodistrian University of Athens as a postdoctoral researcher. His research interests focus on the development of photochemical reactions for the formation of C–C bonds and their application in the synthesis of pharmaceuticals and bioactive compounds. |
 Ierasia Triandafillidi | Dr Ierasia Triandafillidi was born in Athens, Greece. She obtained her BSc, MSc and PhD from the Department of Chemistry, National and Kapodistrian University of Athens, under the supervision of Assoc. Professor Christoforos G. Kokotos. In 2019, she spent 3 months as a visiting researcher at the University of Basel in the group of Professor Christof Sparr. In August 2019, she joined the group of Prof. George Kokotos at the National and Kapodistrian University of Athens as a postdoctoral researcher in medicinal chemistry. In 2020, she received a postdoctoral fellowship from the State Scholarships Foundation (IKY). Her research interests focus on the development of organocatalytic oxidation reactions and photochemical reactions for the formation of C–C bonds and their application in the synthesis of pharmaceuticals and bioactive compounds. |
 Christoforos G. Kokotos | Prof. Christoforos G. Kokotos was born in Athens, Greece (1981) and studied chemistry at the National and Kapodistrian University of Athens, Greece (2003). He completed his PhD studies at the University of Bristol, UK (2007) under the supervision of Prof. Varinder Aggarwal. He then carried out postdoctoral work at Princeton University, USA in the group of Prof. David W. C. MacMillan. He was elected as a Lecturer of Organic Chemistry in the Department of Chemistry of the National and Kapodistrian University of Athens, Greece and promoted to Assistant Professor (2016) and Associate Professor (2020). He was also elected a Chemistry Europe Fellow in 2022 (Class 2020–2021). His research interests lie in asymmetric organocatalysis, photocatalysis and green methodologies for oxidation reactions and their application in the synthesis of pharmaceuticals, bioactive molecules and high-added value chemicals. |
Introduction
The majority of organic molecules contain oxygen functionalities. These molecules find excellent applications in agrochemicals, materials science, petrochemistry and medicinal chemistry since many of these compounds exhibit pharmaceutical properties. For this reason, there is a high demand for synthetic protocols, targeting the oxidation of organic molecules. Alkanes, alkenes, alcohols, ketones, sulfides, etc. are considered as valuable precursors for the manufacture of a variety of functionalized derivatives, which serve as important building blocks in organic synthesis.1
Since 2000, organocatalysis, the use of small organic molecules as catalysts, has brought a revolution in modern organic synthetic chemistry. In 2021, the Nobel Prize in Chemistry was awarded to David W. C. MacMillan2 and Benjamin List,3 who pioneered the field of asymmetric organocatalysis. This revolution led many scientists, both in industry and academia, to explore eco-friendly procedures, replacing metal-based catalysts with small organic molecules that have a lower environmental impact. Thus, a plethora of organocatalytic protocols have been reported in the literature, aiming at organic transformations that were previously underexplored. Oxidative procedures were mostly investigated and many organocatalytic protocols were developed targeting the epoxidation of alkenes, the oxidation of sulfides into sulfoxides, the direct C–H functionalization of alkanes, etc. Aiming at the development of sustainable protocols that are characterized by lower environmental impact, reduced costs of reagents, as well as high applicability and broad scope of substrates, the use of the oxidant is key to fulfil the above-mentioned parameters. In the context of this review, we will discuss the use of the environmentally benign oxidant, hydrogen peroxide. Hydrogen peroxide is the simplest form of peroxides and was first identified and isolated by Luis Jaques Thenard, in 1818.4 Hydrogen peroxide (and its derivatives, such as the urea hydrogen peroxide complex) is considered as a green oxidant, since its only by-product is water.5 Hydrogen peroxide is mainly available in aqueous solutions, which are safer in storage, operation and transportation. In many cases, when the reagents are sensitive to moisture, ethereal hydrogen peroxide can be used, although it is not storable and must be used with great care. It is easy to handle, and this feature makes hydrogen peroxide the oxidant of choice in the pharmaceutical industry. However, its key limiting factor is the lack of reactivity to promote an oxidation reaction and thus an in situ or a beforehand activation is required. For many years, this was mainly achieved via metal catalysis.6 In many cases, its complex with urea appears to be more reactive, since urea activates hydrogen peroxide through hydrogen bonding. Nowadays, many efforts are targeting the activation of hydrogen peroxide using small organic molecules. In this review article, we intend to highlight the different approaches that have been followed to activate hydrogen peroxide towards racemic and asymmetric epoxidations, C–H hydroxylation, Baeyer–Villiger oxidation, etc. (Scheme 1). Also, some remarkable fundamental mechanistic studies will be mentioned in due course. The authors believe that such information will provide greater insights into the organocatalytic activation of hydrogen peroxide and this will be the stepping stone for the rational design of more efficient and sustainable protocols in the future.
 |
| | Scheme 1 Applications of hydrogen peroxide in oxidative organic transformations. | |
Organocatalytic activation of hydrogen peroxide by activated carbonyls
The uprise of organocatalysis prompted many researchers to search for small organic molecules that might act as potential catalysts to promote various organic transformations. The use of small organic molecules offers unlimited advantages in organic synthesis. Especially, in the pharmaceutical industry, the replacement of expensive metal catalysts by organic molecules that can be easily removed from the reaction mixture and in many cases may be reused, firstly offers a greener alternative and secondly reduces the overall cost of the process. Some extremely useful classes of molecules of great interest in organocatalysis are ketones and aldehydes. Pioneers in this field were the research groups of Denmark,7 Yang8 and Shi,9 who provided useful results for the basic application of ketones as catalysts in oxidation protocols.
Heteroatom oxidations with hydrogen peroxide
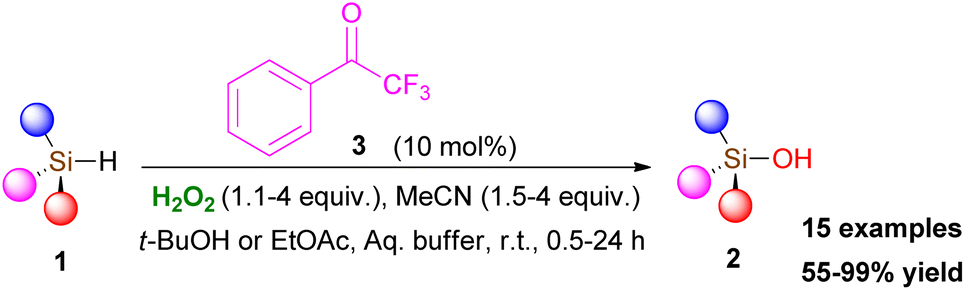
In 2013, Kokotos and coworkers, captivated by the above-mentioned elegant contributions in the field, envisaged that an activated ketone might activate hydrogen peroxide to form an intermediate, capable of inducing an oxidation reaction. Thus, they introduced the first organocatalytic oxidation protocol for the conversion of silanes 1 to silanols 2, an organic transformation with significant importance in the pharmaceutical industry, materials science and organic synthesis (Scheme 2).10 The authors tested a variety of commercially-available carbonyl derivatives and concluded that 2,2,2-trifluoroacetophenone (3) acts efficiently as the organocatalyst, activating hydrogen peroxide. It must be pointed out that acetonitrile plays a crucial role in the reaction output, since in its absence, the reaction does not take place. A series of optimization experiments illustrated that 10 mol% of 3, in the presence of 1.1 equiv. of hydrogen peroxide and 1.5 equiv. of acetonitrile led to quantitative formation of the desired product after 24 h. Increasing the equiv. of both hydrogen peroxide and acetonitrile (up to 4 equiv.) led to a significant reduction in the reaction time. A vast variety of symmetrical and non-symmetrical alkyl silanes was tested, leading to the corresponding silanols in good to excellent yields (Scheme 2). The oxidation of silanes bearing a double or a triple bond proved to be problematic. To circumvent this issue, the authors altered the reaction medium to ethyl acetate, leading to the desired products in good to excellent yields. Furthermore, the great advantage of this organocatalytic protocol appears to be its application to aryl silanes, where the Tamao–Fleming oxidation pathway does not take place.
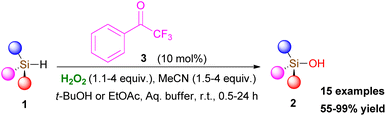 |
| | Scheme 2 Organocatalytic oxidation of silanes 1 to silanols 2. | |
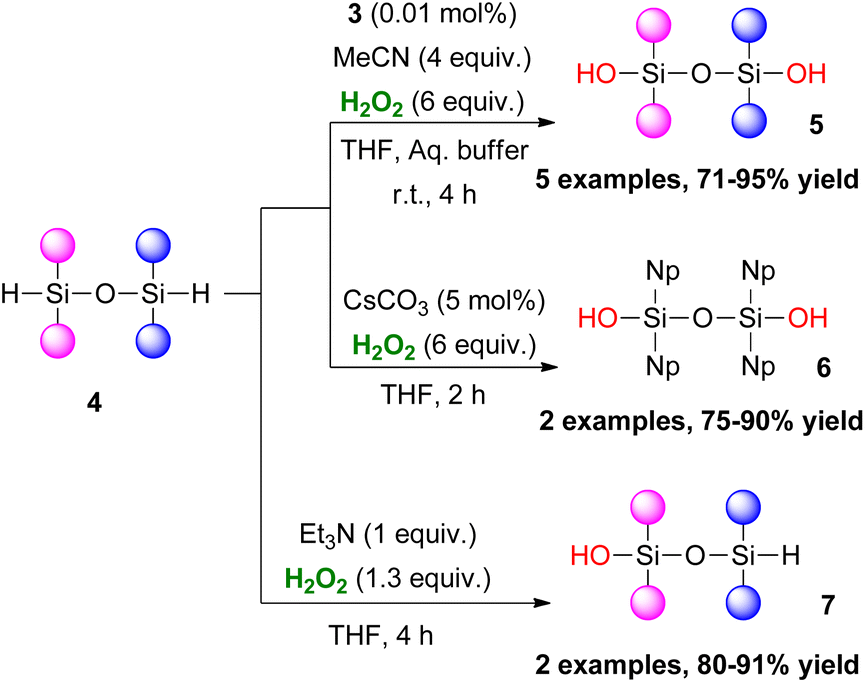
In 2019, Franz and coworkers studied the oxidative hydrolysis of hydrido-siloxanes 4 to siloxanols 5–7 using H2O2 (Scheme 3).11 They discovered that an extremely low catalyst loading of 3 (0.01 mol%) under basic conditions was able to promote the formation 1,3-disiloxanediols 5. The authors performed several control experiments, reporting that basic conditions in the absence of 3 also promoted the reaction. Tetra-aryl 1,3-dihydro-disiloxanes 6 showed excellent chemoselectivity when CsCO3 was used as the base. Interestingly, 1 equiv. of Et3N allowed an unprecedented chemoselective hydrolysis of 4 to 7. Regrettably, Tamao–Fleming oxidation persisted in all the above cases in phenyl-containing substrates. Further study on this matter, revealed that while ortho-substitution suppresses the effect, an electron-withdrawing group diminishes that suppression.
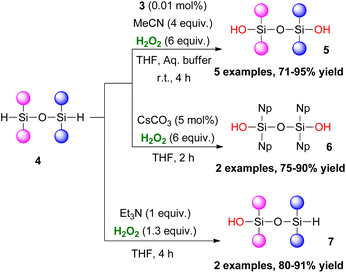 |
| | Scheme 3 Hydrolysis of hydrido-siloxanes to siloxanols. | |
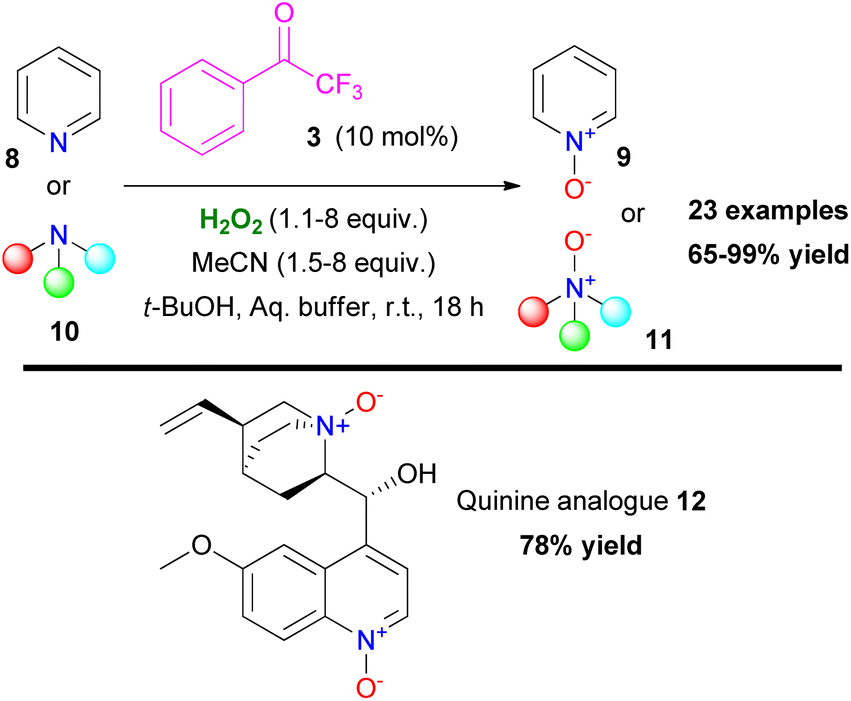
Based on their previous findings, Kokotos and coworkers expanded their work on heteroatom oxidation, utilizing azines or tertiary amines, providing access to N-oxides in excellent yields (Scheme 4).12 The developed protocol combines short reaction times and quantitative formation of the corresponding N-oxides (9 or 11) under mild reaction conditions. A variety of substrates was tested, proving the excellent functional group tolerance of the organocatalytic protocol. The authors employed the organocatalytic protocol for the synthesis of compound 12, derived from quinine, in high yield (78%) (Scheme 4). It must be mentioned though that secondary or primary amines were not tested, probably due to oxidative decomposition pathways that prevail on this occasion.
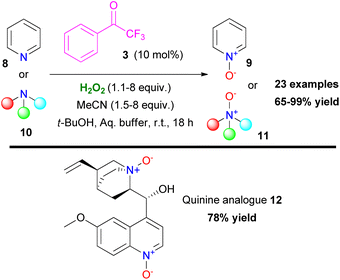 |
| | Scheme 4 Organocatalytic oxidation of tertiary amines or azines to the corresponding N-oxides. | |
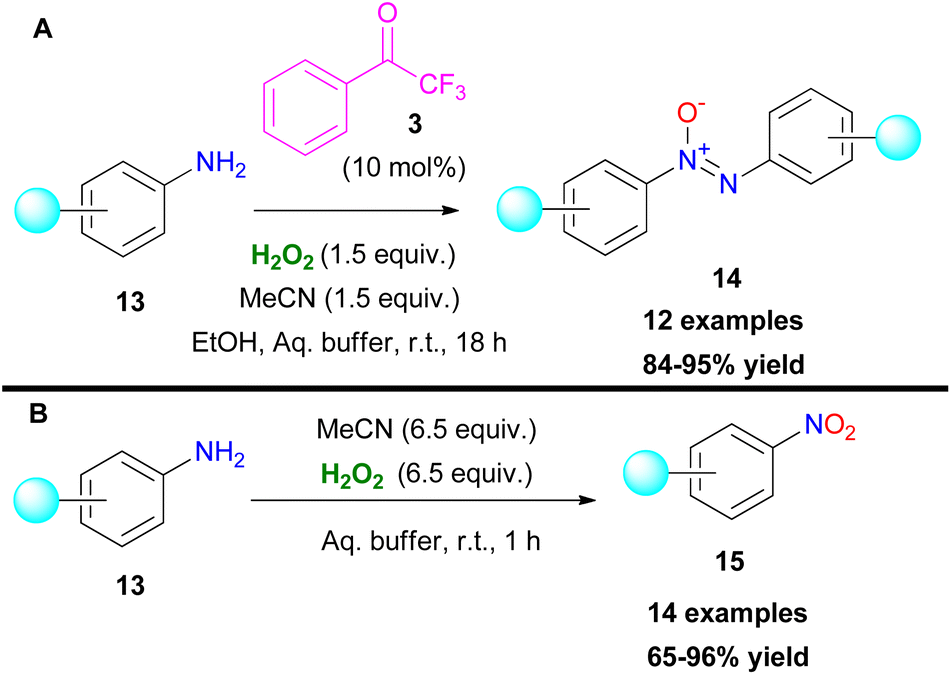
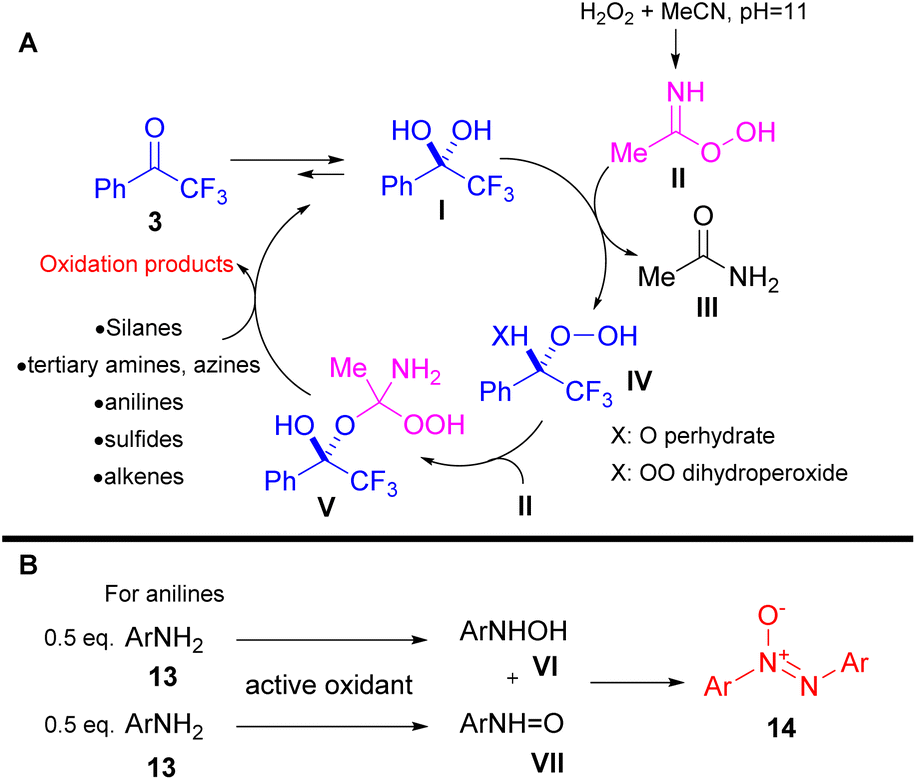
A few years later, the authors implemented their organocatalytic protocol towards the oxidation of anilines.13 In their attempt to probe the reaction mechanism, they noticed the formation of azoxy compounds 14, instead of the desired nitro derivatives 15 (Scheme 5). Even upon increasing the equiv. of hydrogen peroxide, the reaction rate for the synthesis of nitro compounds remained low.13 This can be attributed to the slow conversion of the formed nitroso compound to the nitro derivative. As a result, the hydroxylamine intermediate VI undergoes a condensation reaction with the nitroso intermediate VII, delivering the azoxy compound as depicted in Scheme 6B. In the absence of the organocatalyst, the reaction output was different.13 In this case, the use of 6.5 equiv. of hydrogen peroxide boosted the reaction towards the formation of the corresponding nitro derivative 15 (Scheme 5). Substituted anilines bearing either electron-withdrawing or electron-donating substituents were successfully applied to the organocatalytic protocol. On the other hand, 2-aminopyridine favoured the corresponding N-oxide. Under the catalyst-free conditions, the corresponding nitro compounds were formed in good to excellent yields. Interestingly, in this case, 2-aminopyridine was selectively converted to the nitro derivative in good yield. It is worth mentioning that naphthylamines and aliphatic amines were not viable under both reaction conditions. In order to elucidate the reaction mechanism, the authors performed a series of HRMS studies, identifying the possible intermediates that are involved in the oxidation procedure.
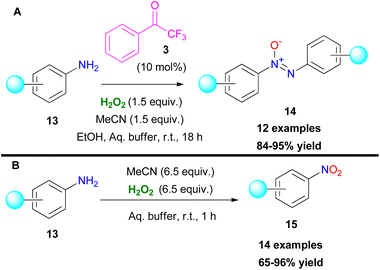 |
| | Scheme 5 Oxidation of anilines towards (A) azoxy compounds, (B) nitro compounds. | |
 |
| | Scheme 6 Proposed catalytic pathway of 2,2,2-trifluoroacetophenone towards novel oxidation transformations. (B) Proposed pathway for the formation of azoxy compounds. | |
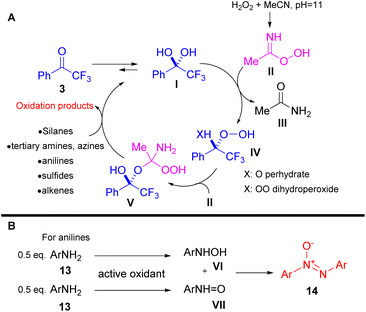
In their contributions, Kokotos and coworkers performed a series of mechanistic studies to elucidate the oxidation pathways and to identify the active oxidant species that are generated. The developed oxidative protocols are based on the equilibrium between the keto form (3) and the hydrate form I of the catalyst (Scheme 6A). In the presence of acetonitrile, Payne's intermediate II (ref. 14) is formed through acetonitrile's interaction with hydrogen peroxide at the appropriate pH (pH = 11). The latter interacts with hydrate form I, shifting the equilibrium to I, generating either the perhydrate IV (X = O) or dihydroperoxide IV (X = OO) intermediate (Scheme 6A). Based on HRMS studies performed by the authors, intermediate IV undergoes addition to Payne's intermediate, generating the active oxidant V, which is responsible for the oxidative organic transformations (Scheme 6A).10,12,13
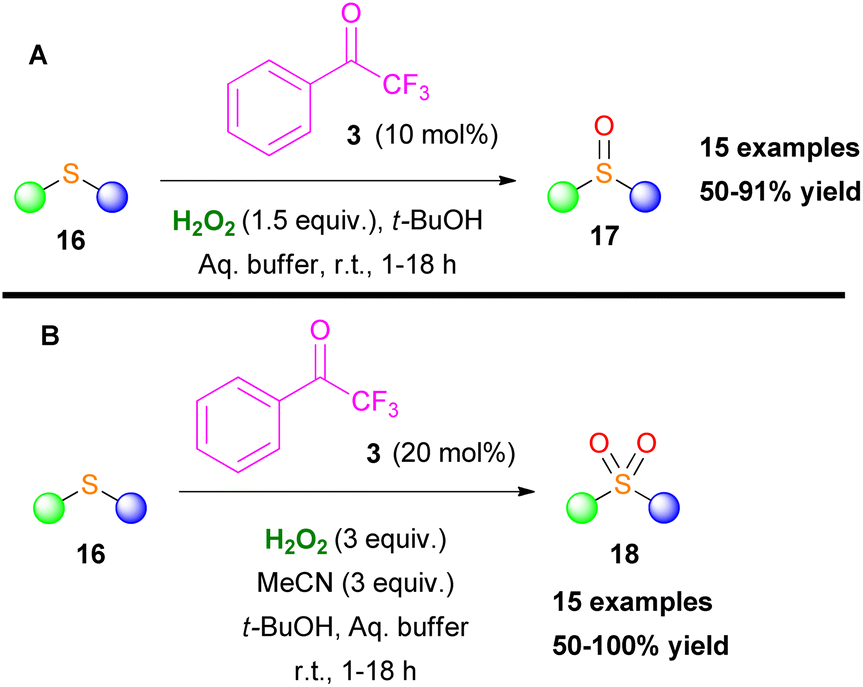
In the same vein, Kokotos and coworkers developed two oxidation protocols for the selective oxidation of sulfides either to sulfoxides or sulfones.15 Both families of compounds display a salient pharmaceutical profile and find many applications in medicinal chemistry and agriculture (Scheme 7).15 Acetonitrile plays a key role in the reaction output, since in its presence the overoxidized sulfone derivative is being formed. Based on mechanistic studies, the authors concluded that a perhydrate intermediate IV (X = O) from the perfluoroketone 3 is the active oxidant for the formation of sulfoxides, since Payne's intermediate14 was not detected (Scheme 6A).15
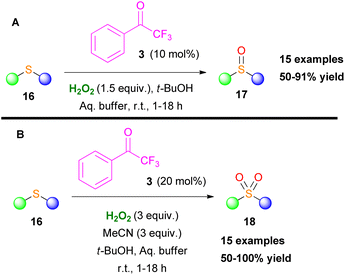 |
| | Scheme 7 Selective oxidation of sulfides to (A) sulfoxides and (B) sulfones. | |
Hydrogen peroxide-mediated epoxidation reactions
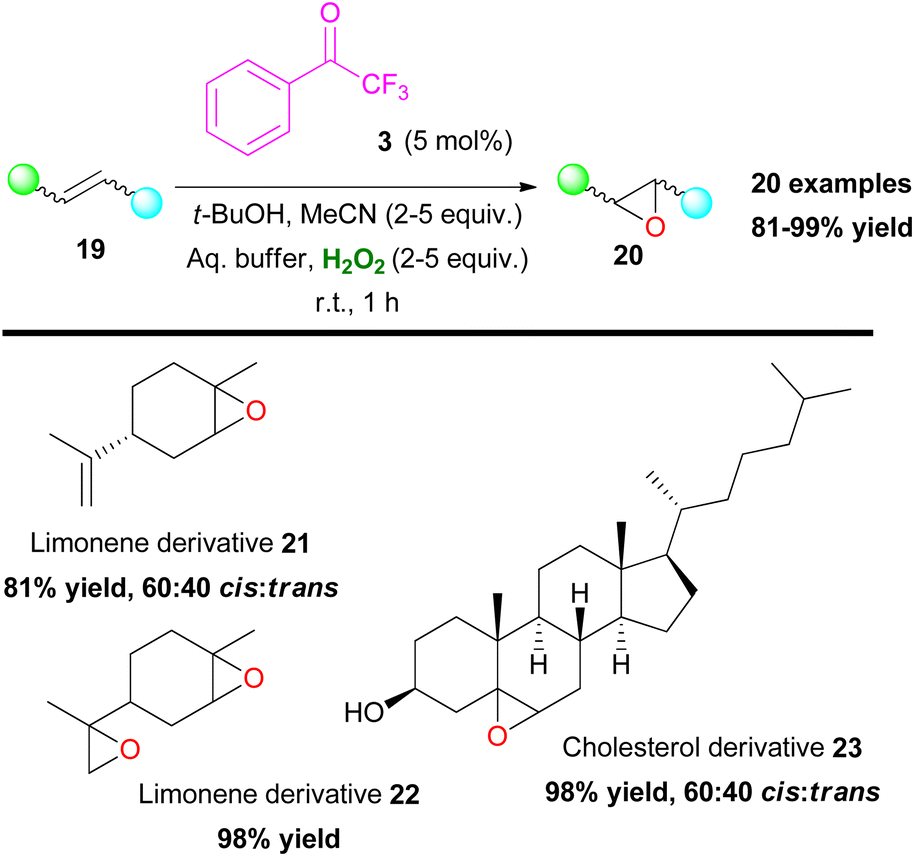
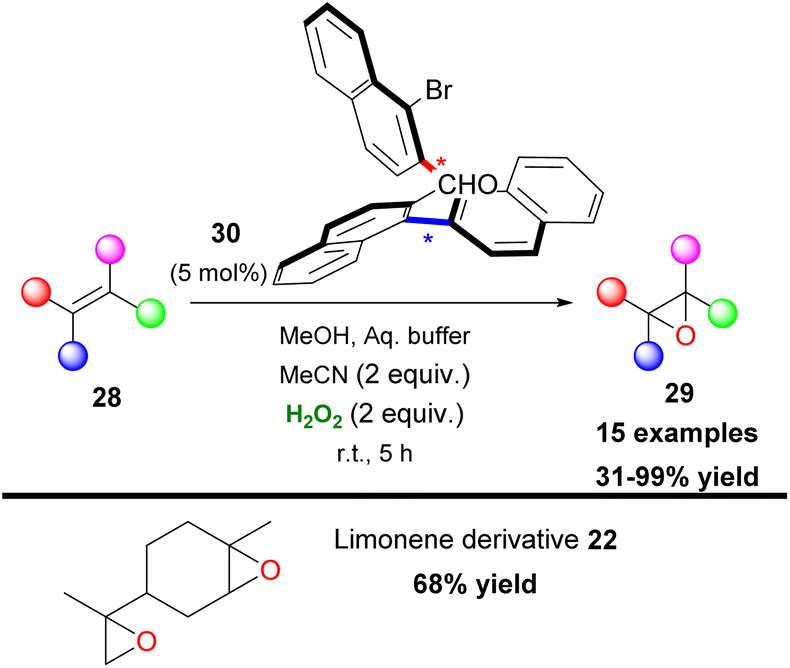
Another organic transformation with significant importance in organic synthesis is the formation of epoxides. The most common approach for the synthesis of epoxides is the epoxidation of unactivated olefins. This organic reaction has been well-studied by many research groups worldwide and there have been numerous contributions in the literature.16 Sharpless,17 Katsuki,18 Jacobsen19 and others pioneered the field, adding novel metal-based approaches towards oxirane synthesis. Furthermore, Denmark,7 Yang8 and Shi9 introduced dioxirane derivatives as the active oxidants in the first organocatalytic approaches in this area. Having established an organocatalytic approach in a series of organic transformations, Kokotos and coworkers expanded their strategy in investigating the use of 2,2,2-trifluoroacetophenone (3) towards the epoxidation reaction of unactivated alkenes (Scheme 8).20 The authors employed an extremely low catalyst loading (up to 5 mol%) of ketone 3, which was found to be capable of promoting with ease the selective epoxidation of various mono-, di- and trisubstituted alkenes and known natural compounds, such as limonene (21 and 22) or cholesterol (23) in excellent yields (up to 99%) combining short reaction times (Scheme 8). This was a major improvement, compared to the state of the art at that time. The authors based on NMR experiments evaluated a similar mechanistic pathway, to the one presented in Scheme 6A, where intermediate V is acknowledged as the active oxidant species.20
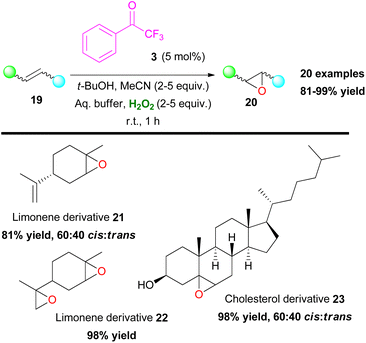 |
| | Scheme 8 Novel epoxidation protocol by Kokotos. | |
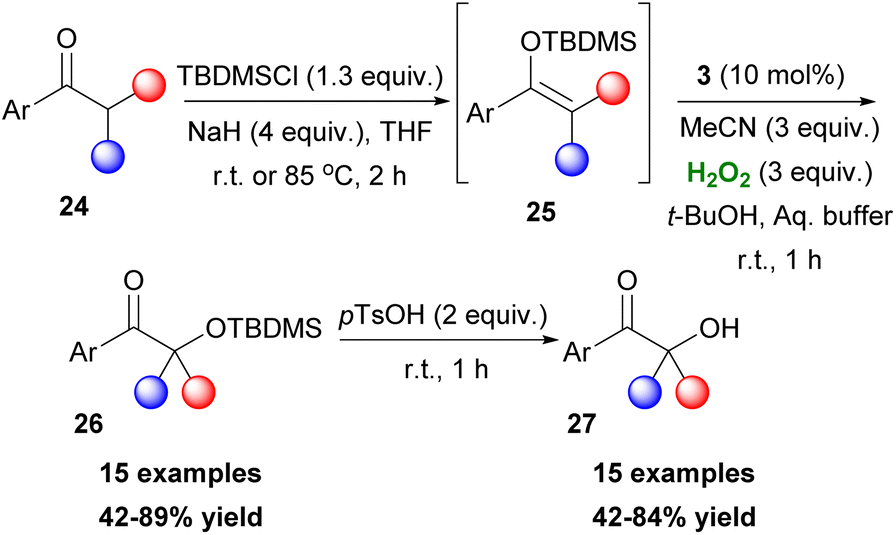
2,2,2-Trifluoroacetophenone 3 found an excellent application as the organocatalyst, by the Kokotos group, for the in situ oxidation of TBDMS-protected enols 25 to form TBDMS-protected 26 and free α-hydroxy ketones 27, upon treatment with an acid (Scheme 9).21 This method follows a similar concept proposed by Rubbotom.22 Aryl-methyl ketones, regardless of substitution in the aryl group, can yield both products successfully. Moreover, a substrate bearing a phenol scaffold showed a preference for the deprotection of the alcohol. This protocol can generate both secondary and tertiary alcohols at slightly reduced yields.
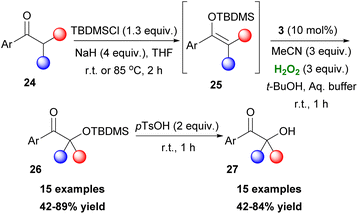 |
| | Scheme 9 Synthesis of TBDMS-protected and free α-hydroxy ketones. | |
Inspired by Kokotos' work on organocatalytic alkene epoxidation, Tyablikov and coworkers developed a heterogenous catalyst, where the 2,2,2-trifluoroacetophenone moiety of 3 was embedded in SBA-15 mesoporous silica, in their effort to prepare a catalyst that can be easily recycled.23 Furthermore, two different research groups24,25 sought to harness this protocol, for large-scale reactions. Employing continuous-flow reactor apparatus, Zheng and coworkers improved mass and heat transfer rates.24 After a thorough optimization of each reaction parameter, a fast and high yielding method was established. Chen and Xu25 utilized a microreaction continuous flow system that also provided high selectivity and yield of styrene oxide in shorter times, even when the amount of oxidant was reduced.
In an attempt to provide an asymmetric variant that failed and seeking other potential carbonyl-based molecules that might activate hydrogen peroxide, Kokotos, Sparr and coworkers, in 2021, developed an epoxidation protocol, introducing for the first time an aldehyde-catalysed epoxidation (Scheme 10).26 A good substrate scope was tested, providing an efficient sustainable protocol for the epoxidation of unactivated alkenes. Usually, in this type of reaction introduced by Mukaiyama27 in 1991, an overstoichiometric amount of aldehyde is required and the active intermediate is a matter under discussion.28 However, the authors performed a series of HRMS mechanistic studies and proved that atropoisomeric aldehyde 30, forms a dioxirane, which acts as the active oxidant. These findings are of great importance as it is indicated that aldehydes can serve as possible activators of hydrogen peroxide towards oxidative transformations even in sub-stoichiometric amounts. Unfortunately, the atropoisomeric aldehyde proved to have no impact on the enantioselectivity of the epoxidation protocol.
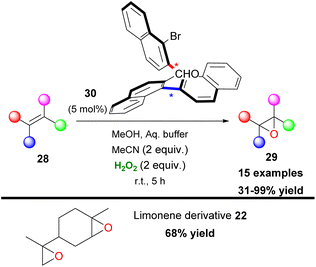 |
| | Scheme 10 First aldehyde-catalysed epoxidation protocol. | |
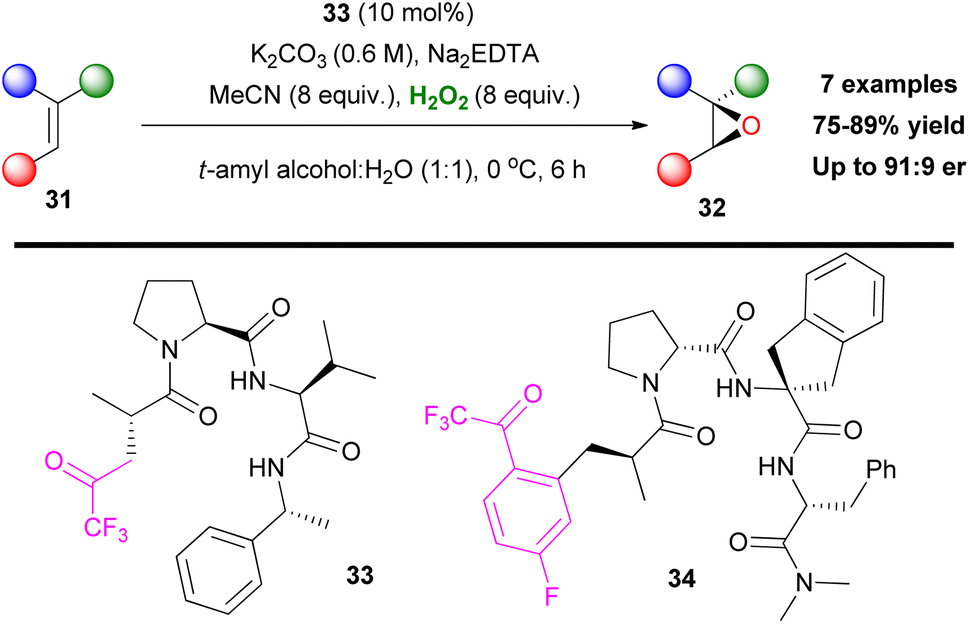
Asymmetric epoxidation has always been of great interest for organic synthesis, since its introduction. Inspired by Shi's contributions in this field,9 Miller and coworkers envisaged that a ketone moiety, embedded in a peptide, might induce enantioselectivity towards alkene epoxidation.29 For this reason, they prepared a series of peptides, decorated by the 2,2,2-trifluoromethylcarbonyl moiety (Scheme 11). Based on their initial idea, in the presence of hydrogen peroxide, a dioxirane should be formed. The peptide core can form an enantioselective transition state inducing asymmetry. Several alkenes were tested, affording the corresponding epoxides in good to excellent enantioselectivity.29 Under the optimized reaction conditions, trisubstituted epoxides were prepared in high enantioselectivity, while in disubstituted substrates selectivity dropped to modest (Scheme 11). Because of their innate chirality, peptide-based catalysts induce asymmetric induction. Exploiting this feature, Miller and coworkers, two years later, developed phenylalanine-based trifluoromethyl ketones as potential oxidation catalysts.30 In the case of catalyst 34, under the same reaction conditions, even though it was able to epoxidize less reactive olefins, such as N-(3-(prop-1-en-2-yl)phenyl)acetamide, the observed enantioselectivity was poor. This verifies indeed the fact that, even though high yielding epoxidation procedures are within reach, asymmetric induction is always a challenge.
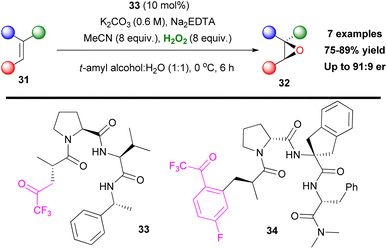 |
| | Scheme 11 Miller's perfluoroketone-embedded peptide catalysts, enantioselective epoxidation reaction with peptide catalyst 33. | |
One-pot transformations
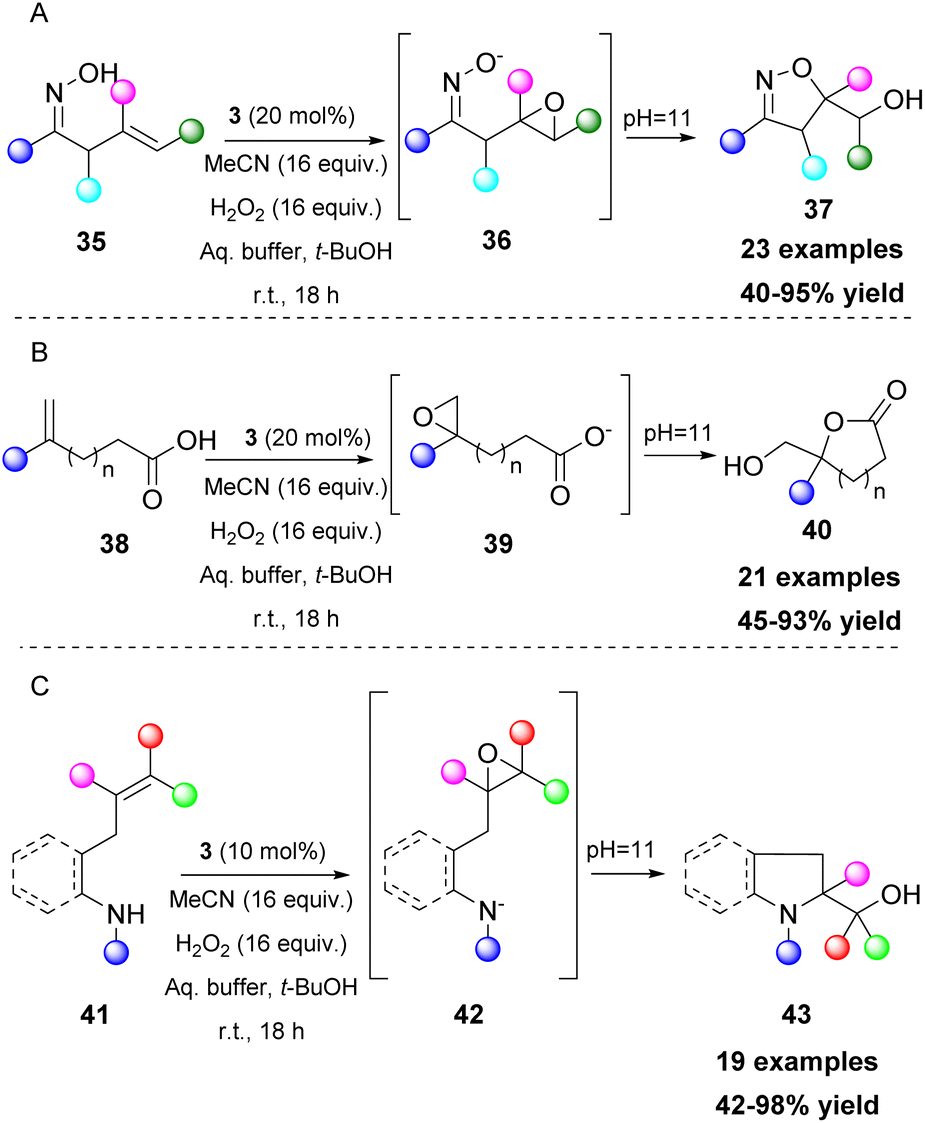
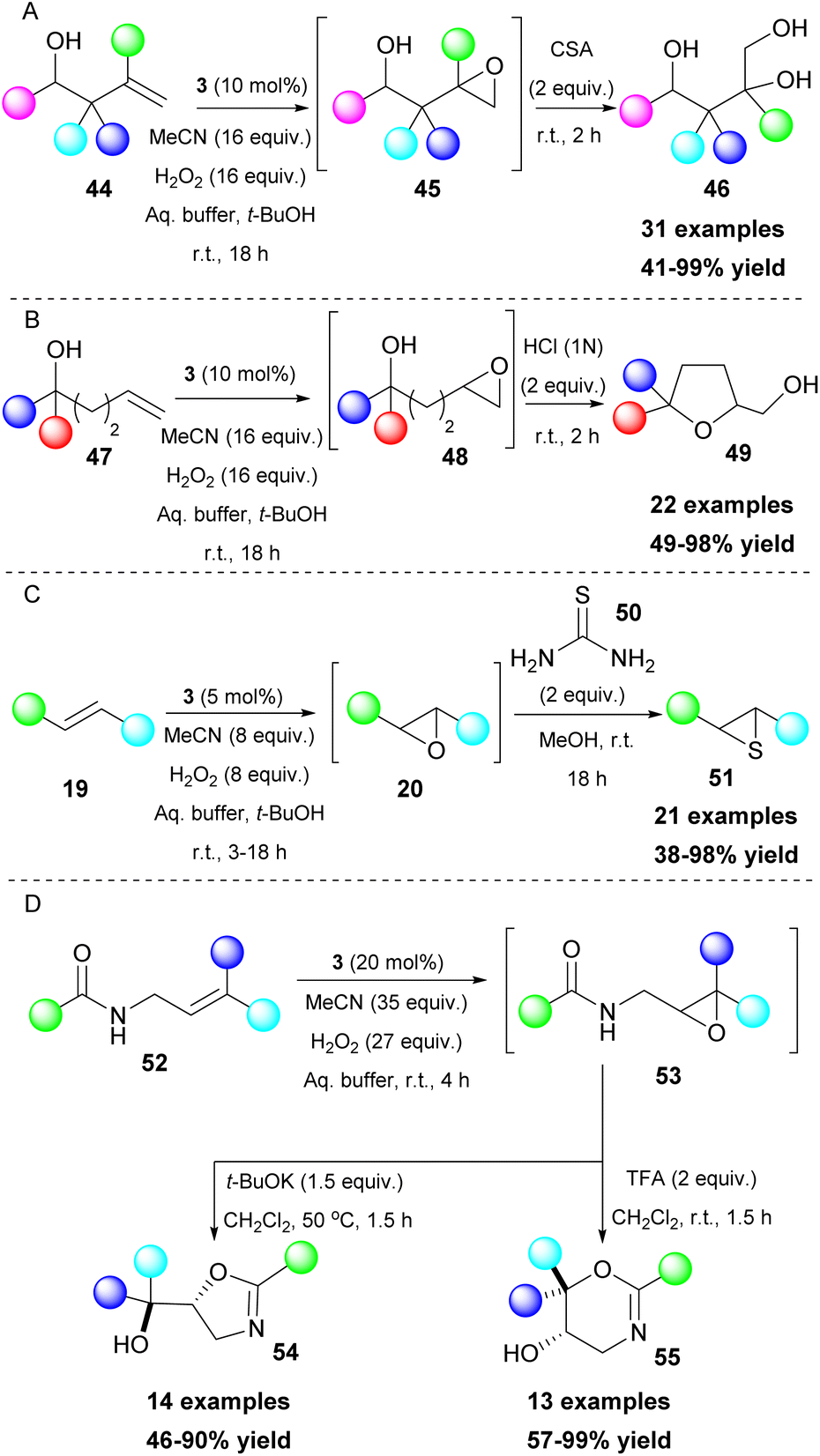
Epoxides are versatile intermediates and due to their ring strain, they are susceptible to ring opening reactions and other transformations. Kokotos and coworkers, having developed a series of organic transformations using 2,2,2-trifluoroacetophenone (3) as a potent catalyst, expanded its use towards one-pot transformations. A strategically placed functional group next to the formed epoxide was able to perform an intramolecular cyclization reaction, leading to the formation of various substrates, such as dihydrobenzofurans,31 isoxazolines,32 lactones,33 indolines and pyrrolidines (Scheme 12).34 In these cases, the aqueous buffer solution also provides the appropriate pH for the in situ deprotonation of the OH, N–OH, COOH and NH groups, enhancing their nucleophilicity and facilitating the ring opening of the epoxide (Scheme 12). To achieve the synthesis of pyrrolidines and indolines, protection of the nitrogen atom was vital to enhance the acidity of the free proton. Among all the substituents on the nitrogen atom, the mesyl group afforded the best results. Nevertheless, in some cases, a strong base (i.e. t-BuOK) was required (Scheme 12C).34
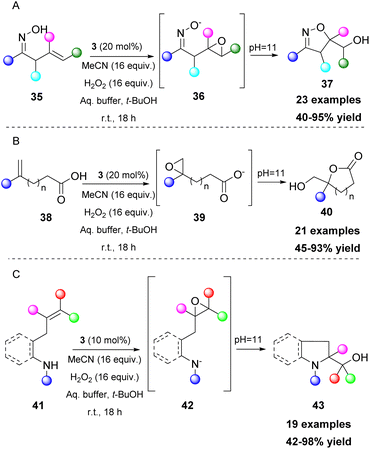 |
| | Scheme 12 Versatile one-pot transformations from functionalized epoxides to (A) isoxazolines, (B) lactones, (C) indolines and pyroolidines. | |
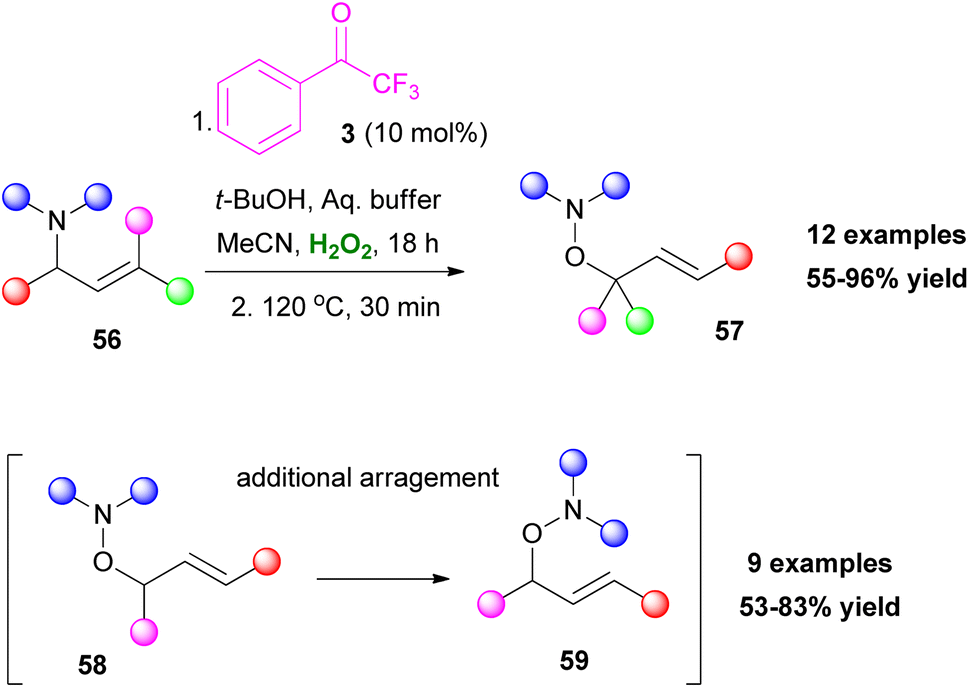
Tuning the reaction conditions and applying a variety of reagents provide access to various classes of molecules. Polyalcohols35 or polysubstituted tetrahydrofurans36 can be accessed by the addition of a strong acid (Scheme 13A and B), while in a more recent development, thiiranes can also be obtained, using thiourea 50 as the thiolating reagent (Scheme 13C).37 Interestingly, when substituted allylamides 52 were employed, the resulting epoxide 53 led to different cyclization products, depending on the additive that was used. In the presence of t-BuOK, a 5-exo-tet cyclization occurred, leading to the formation of oxazolines 54. On the contrary, in an acidic environment (TFA), a 6-endo-trig cyclization afforded dihydrooxazines 55 (Scheme 13D).38 An interesting feature is that allylamines show excellent chemoselectivity under the reaction conditions.38 This unique feature enabled the application of a Meisenheimer rearrangement step towards O-allylhydroxylamines 58 (Scheme 14). It needs to be highlighted that aromatic or conjugated aromatic allylic amines 56 allowed an additional rearrangement step (Scheme 14).39
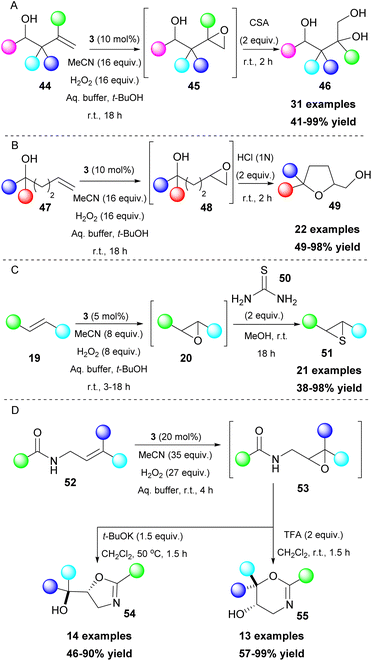 |
| | Scheme 13 One-pot transformations employing an additional step leading to (A) polyalcohols, (B) substituted tetrahydrofurans, (C) thiiranes, (D) oxazolines and dihydrooxazines. | |
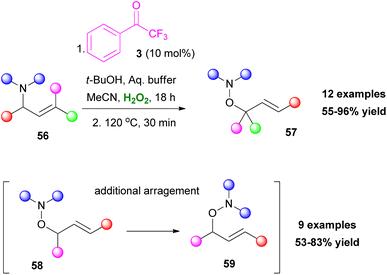 |
| | Scheme 14 Oxidation of tertiary allylic amines followed by a [2,3]-Meisenheimer rearrangement (additional arrangement for aromatic or conjugated aromatic allylic amines). | |
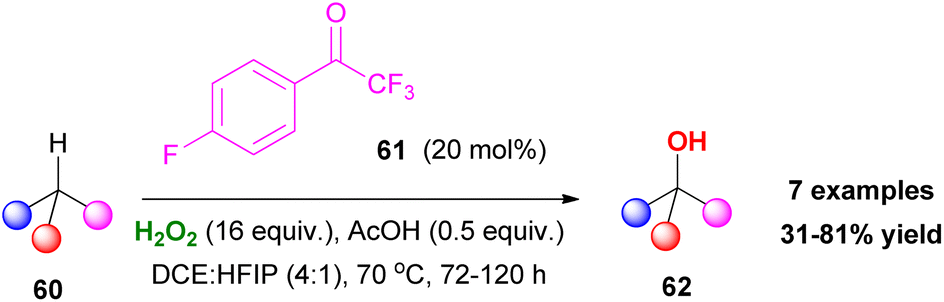
In this field, in 2014, Hilinski and coworkers proposed an elegant protocol for the C–H hydroxylation of alkenes, using hydrogen peroxide and a catalytic amount of ketone.40 In the same line with Kokotos' observations, the authors reported an organocatalytic protocol providing access to a variety of alcohols 62 (Scheme 15). Despite this elegant approach, the over-stoichiometric use of hydrogen peroxide posed significant difficulties, such as the formation of overoxidized ketone or ester by-products.
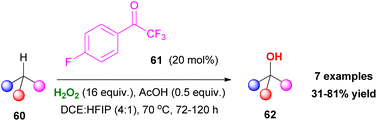 |
| | Scheme 15 Hilinski's chemoselective C–H hydroxylation. | |
Organocatalytic activation of hydrogen peroxide with peptide-embedded carboxylic acids for asymmetric oxidation
Asymmetric epoxidation derived from catalyst–substrate interactions
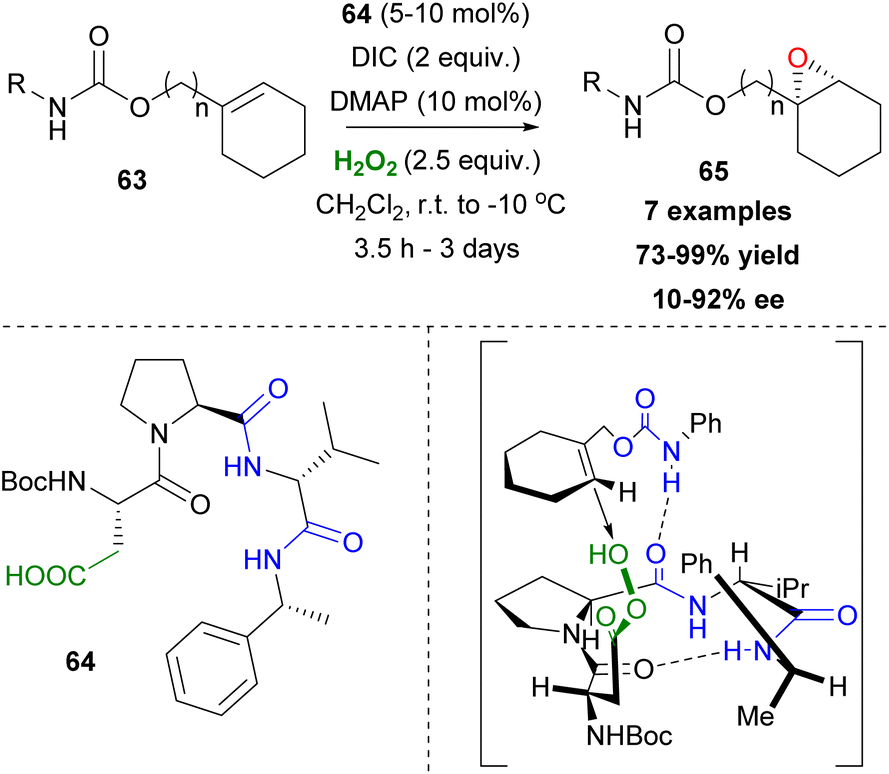
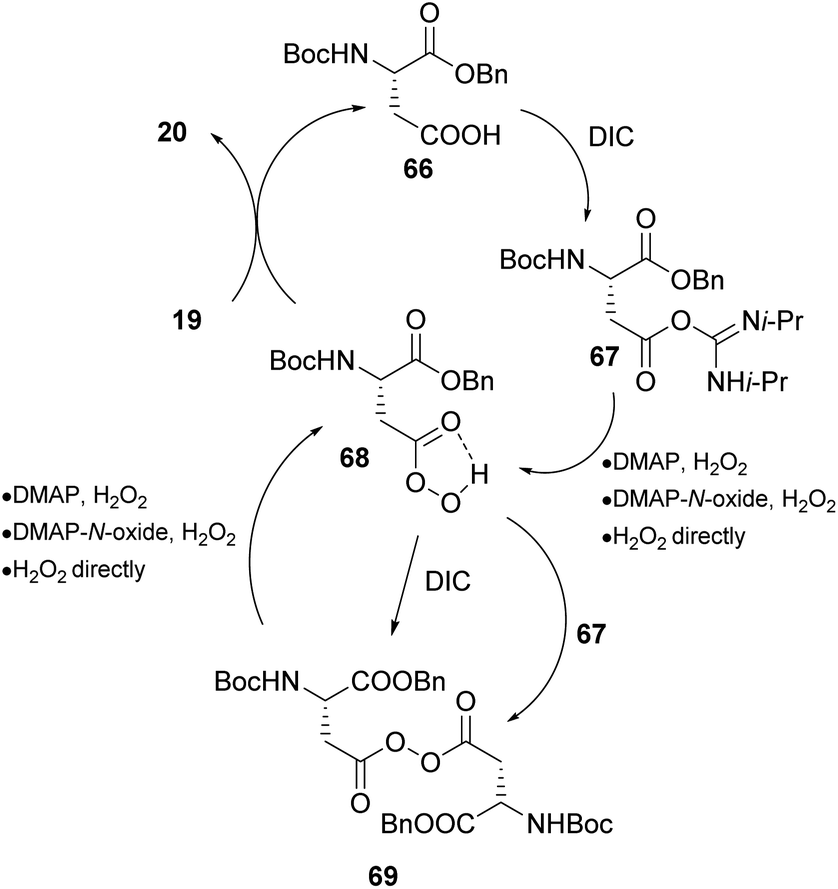
As mentioned previously, the asymmetric epoxidation of alkenes is considered as a valuable tool in organic synthesis. Miller and coworkers reported a series of contributions utilizing peptide-based carboxylic acids for the activation of hydrogen peroxide.41 Previous work in this field,42 led the authors to investigate a similar approach, utilizing the carboxylic residues in amino acids and peptides as potential catalysts.43 Knowing that peracids are famous epoxidizing agents, the authors envisioned that Asp- or Glu-based peptides that have free carboxylic groups in their structure could serve as catalysts in asymmetric epoxidation through the formation of the peracid intermediate (Scheme 16). Eventually, the Asp-based catalyst 64 in the presence of hydrogen peroxide, following a peracid activation pathway, afforded the desired epoxide 65 (R = Bn) in good yield and enantioselectivity (Scheme 16). The substitution of the nitrogen atom of the carbamate moiety and its position regarding the double bond appears to be key factors to the resulting enantioselectivity. This is reasonable, considering the appropriate transition state, based mainly on the formation of hydrogen bonding that is capable of inducing an enantioselective approach of hydrogen peroxide onto the double bond of the alkene (Scheme 16). Miller's group exploited the carbodiimide strategy that is usually used in peptide synthesis to activate hydrogen peroxide.43 Diisopropylcarbodiimide (DIC) enhances the electrophilicity of the catalyst, forming intermediate 67 and DMAP accelerates the oxygen atom transfer process, eventually generating the active electrophilic peracid intermediate 68 of the peptide catalyst (Scheme 17). Apart from its role in forming intermediate 68, DMAP also facilitates the perhydrolysis of intermediate 69, promoting the productivity of the catalytic cycle (Scheme 17). To gain greater insight into the interactions that govern the enantioselectivity of the epoxidation reaction, the authors proceeded with various changes on the catalyst's backbone. Their experiments showcased that H-bonding interactions in the transition state induce enantioselectivity in the corresponding epoxides.44
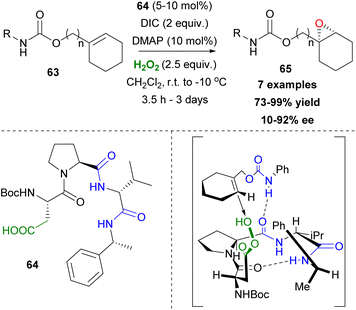 |
| | Scheme 16 Miller's asymmetric epoxidation. | |
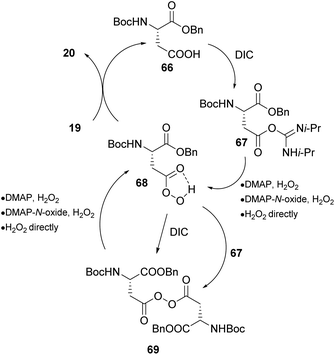 |
| | Scheme 17 Proposed mechanism using an Asp catalyst 66 following a peracid moiety 68 generation. | |
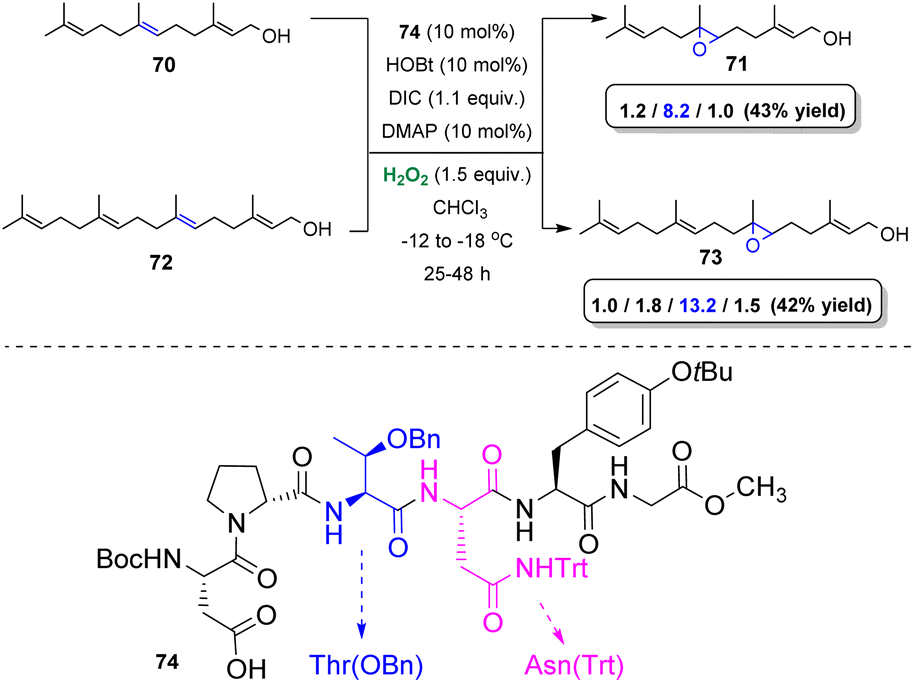
In order to identify the appropriate peptide-based catalyst, Miller and coworkers developed a high-throughput screening strategy. The authors combined split-and-pool synthesis and one-bead-one-compound (OBOC) type libraries45 to opt for the best catalyst towards the epoxidation of alkenes.46 Based on this strategy, a number of resin-bound peptide catalysts bearing the Boc–Asp–Pro motif were employed for the selective epoxidation of polyenes, such as farnesol. The selective oxidation of such kind of substrates is considered an extremely challenging task in organic chemistry and it is mainly performed employing enzymatic catalysis.47 After testing an enormous number of bead-bound peptides, an increased selectivity towards 2,3-epoxyfarnesol and towards 6,7-epoxyfarnesol was reported.47,48 Ultimately, β-turns along with NH directing groups of Thr(OBn) and Asn(Trt) of catalyst 74 proved to be the directing factors of the observed 6,7-selectivity (Scheme 18). Substituting the hydroxyl-directing group of farnesol (methyl ether) altered the reported selectivity. Interestingly, replacing the bulkier Thr(OBn) amino acid by Ile in the structure of the catalyst 74, according to the authors makes space for ether-directed epoxidation, providing promising regioselectivities. A few years later, Miller and coworkers based on the same strategy successfully assessed the basis of the 2,3-selectivity.49 (Z)-Allylic alcohols proved to be the best candidates, in terms of yields and enantioselectivity, while the addition of Et3N in the reaction mixture, in some cases enhanced the outcome.
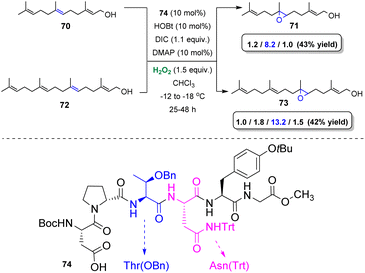 |
| | Scheme 18 Site selectivity of catalyst 74 towards 6,7-monoepoxides of farnesol and geranylgeraniol. | |
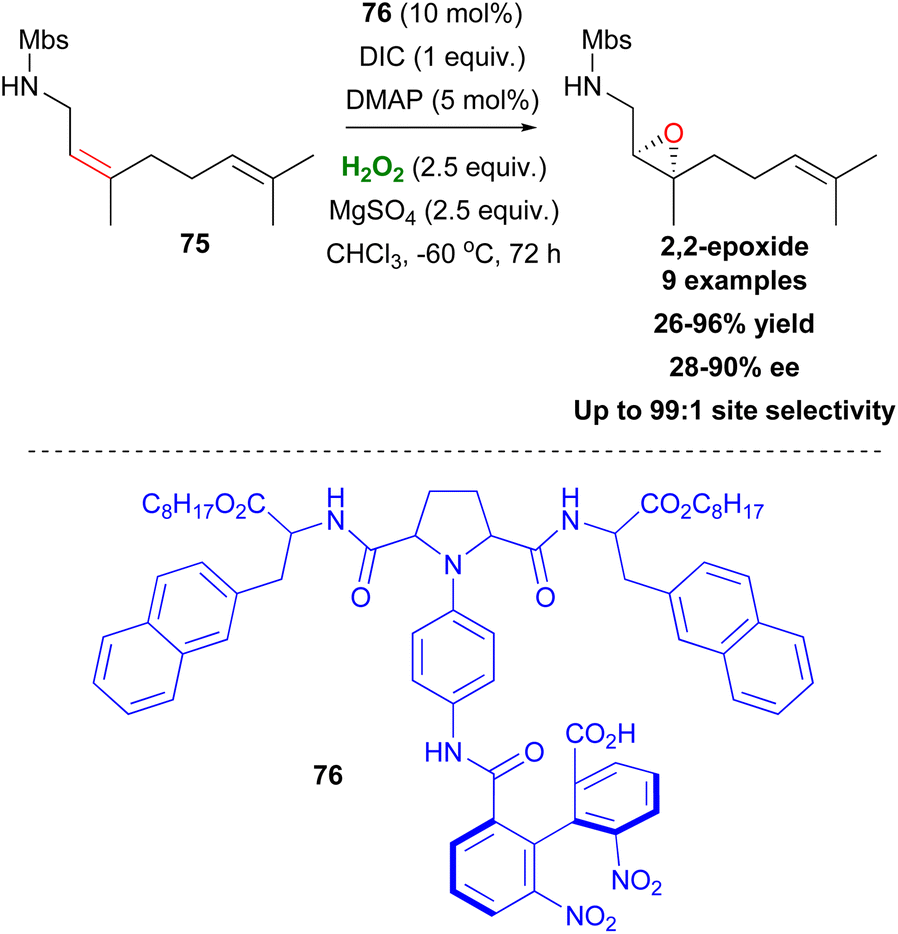
Inspired by Miller's studies, Kawabata proposed catalyst 76, capable of site-selective asymmetric epoxidation of nerylamine derivatives, favouring the formation of 2,3-epoxides (Scheme 19).50 According to the authors, site selectivity is controlled by the biphenyl moiety with an amide group of 76 and the pyrrolidine moiety being responsible for the enantioselectivity. Interestingly, substitution of the NHMbs group by NMeMbs leads to total inversion of site selectivity, favouring the 6,7-epoxides, instead. Using epoxidation conditions with m-CBPA as the oxidant, also led to 6,7-epoxidation. The presence of MgSO4 as a drying agent was also important, as it removed the excess water produced by H2O2, which freezes at very low temperatures, leading to reduced yields (Scheme 19).
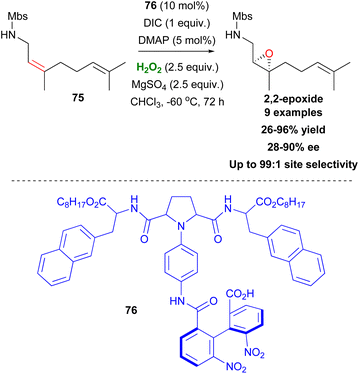 |
| | Scheme 19 Site selective asymmetric epoxidation of nerylamine derivatives. | |
Baeyer–Villiger oxidation
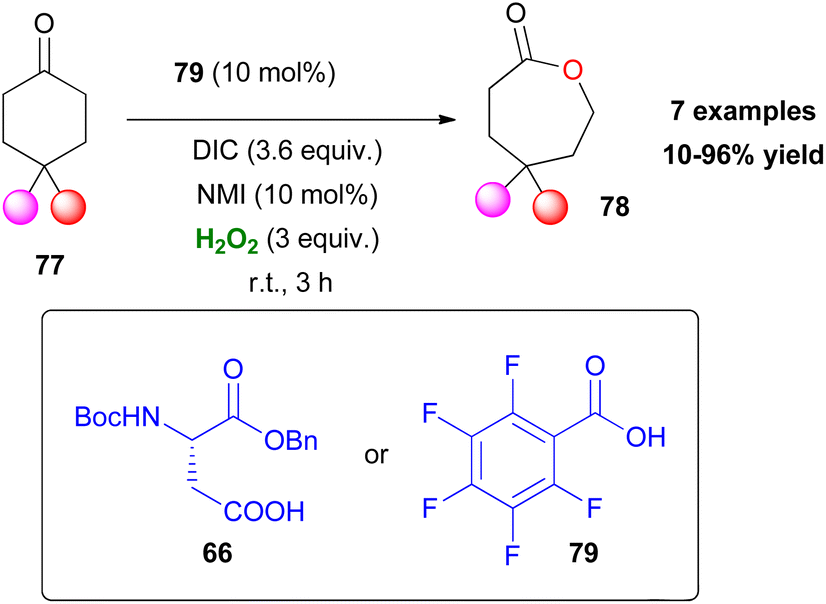
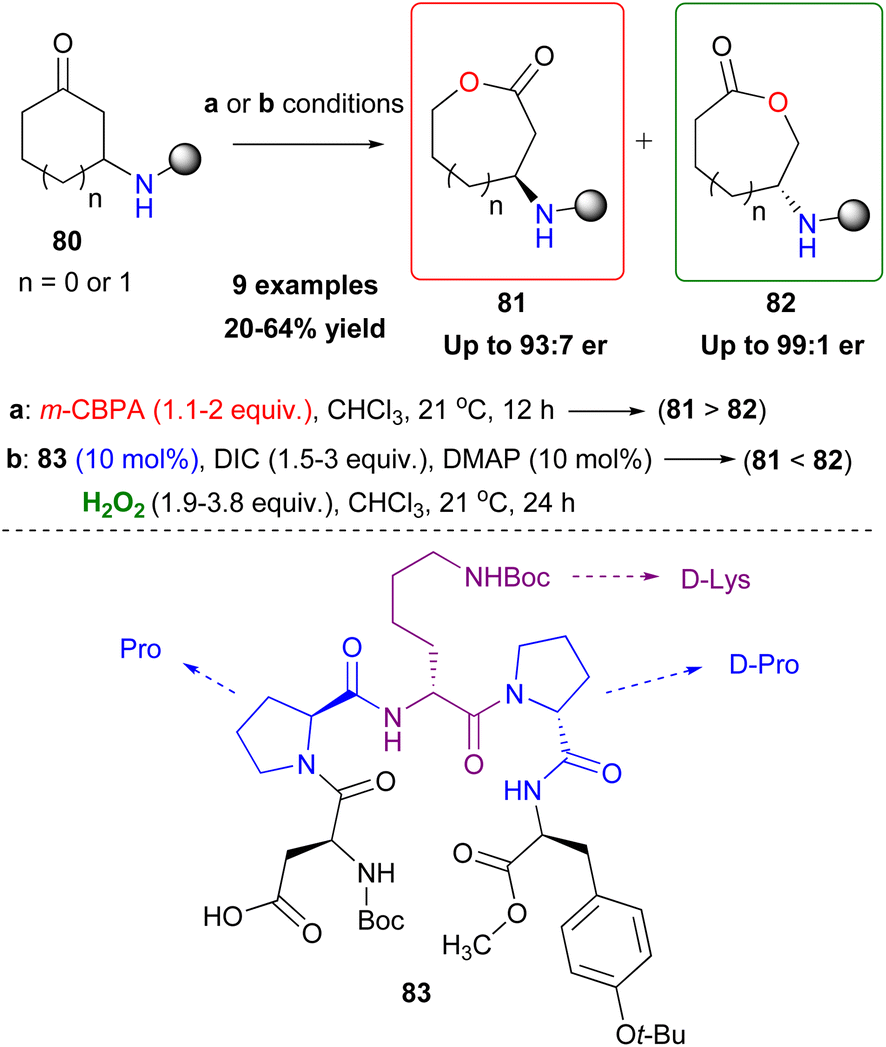
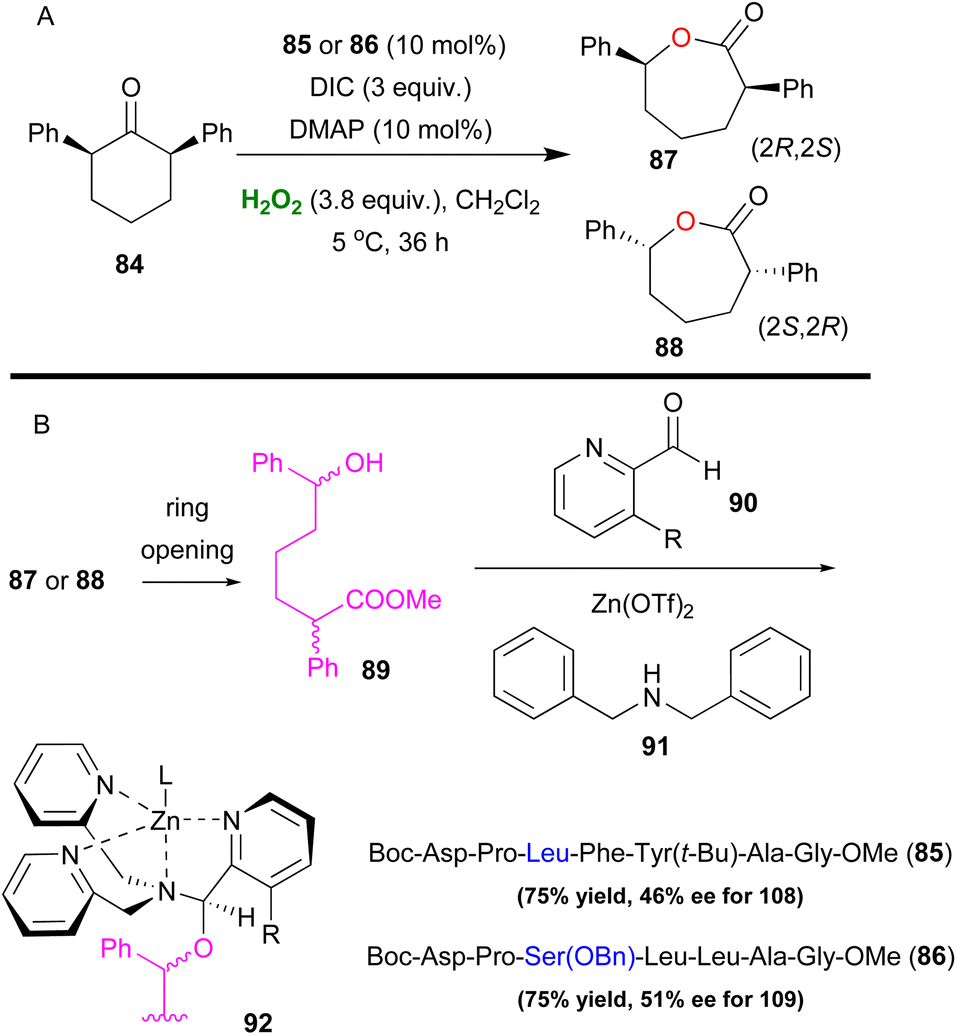
The Baeyer–Villiger oxidation is a very useful oxidative organic transformation,51 which has attracted many research groups worldwide to develop more sustainable and easy-to-execute protocols. In 2008, Miller and coworkers envisaged that the same peracid catalytic route, which was previously reported, could be applied for a Baeyer–Villiger oxidation of ketones using hydrogen peroxide as the oxidant.52 However, the major drawback in the process is the amount of the inactive diacyl peroxide 69 (Scheme 17), that is formed in the presence of DIC. This was successfully encountered in the epoxidation protocol, utilizing DMAP as the additive to promote its perhydrolysis. In the Baeyer–Villiger oxidation process, this drawback was circumvented by the slow addition of DIC, decreasing the formation of 69. To avoid this, the authors envisaged that a more acidic catalyst such as pentafluorobenzoic acid 79 (pKa = 1.74 compared to catalyst 66 pKa = 4.4) will solve this issue as the formation of the lactone was attained in higher yields (Scheme 20). Even though asymmetric induction was not achieved at the time, the authors implied its possibility by testing catalyst 64. Following their research, Miller and coworkers investigated the use of a peptide-based catalyst to achieve a more rigid transition state, inducing regio- and enantioselectivity. Applying their high throughput screening strategy, the authors turned their attention to catalyst 83 (Scheme 21).53 Compared to m-CBPA, the selected peptide catalyst 83 showed increased affinity, towards the disfavoured reverse oxidation product (81vs.82) as depicted in Scheme 21. Even though the measured total yield was mostly mediocre in most substrates, the regioselectivity and enantioselectivity of reverse product 82 were favourable. According to the authors, these can be attributed mainly to the Pro groups in i + 1 and i + 3 positions of the catalyst and of course to the NH functional group attached to the substrate's structure, likely leading to H-bonding directing interactions. Further analysis regarding the conformations that catalyst 83 can exhibit in solution on its own or in the presence of the substrate or its respective favoured lactone product revealed a much more complex reason behind its catalytic behaviour. The importance of D-Lys in the i + 2 position, regarding this behaviour, was also highlighted.54 Substrates deprived of these directing groups are a special challenge in asymmetric induction, therefore this “uncharted” area was further explored by Miller, Anslyn and coworkers.55 Screening a library of catalysts, which was composed solely of L-amino acids, the authors concluded that the use of two catalysts, one with a Leu in the i + 2 position (85) and one with a Ser(OBn) in the i + 2 position (86) favoured a different enantiomer formation, which was validated by HPLC and circular dichroism (CD) assays (Scheme 22A).56 The latter method, which was on par with HPLC results, allowed not only for the rapid catalyst evaluation but also the analysis of the enantiomeric excess of alcohols, derived from the ring opening of the parent lactone products, which serve as ligands to zinc complex 92, exhibiting characteristic Cotton effects56 (Scheme 22B). The two peptide-based catalysts were found to induce an enantioselective Baeyer–Villiger oxidation. Catalysts containing Leu in the i + 2 position favoured the 2R,6S-lactone derivative (87), while the 2S,6R-lactone derivative (88) was favoured in the presence of the Ser(OBn) catalyst. In both cases, it is believed thus, to proceed via a peptide-Criegee intermediate (Scheme 22B).
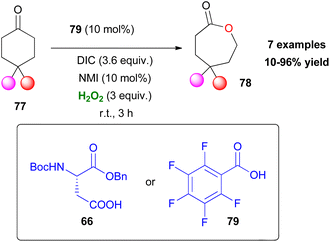 |
| | Scheme 20 Initial Bayer–Villiger oxidation by Miller. | |
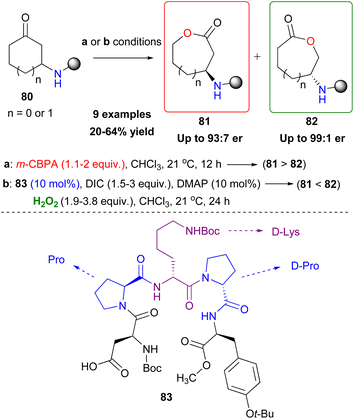 |
| | Scheme 21 Enantioselective and regioselective Bayer–Villiger oxidation of functionalized ketones. | |
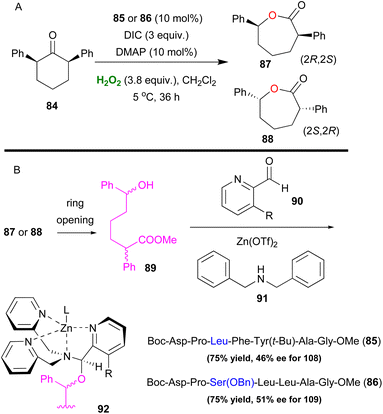 |
| | Scheme 22 (A) Unfunctionalized ketone enantioselective Baeyer–Villiger oxidation. (B) Analysis of absolute stereochemistry and enantioselectivity from the Zn-complex with CD arrays. | |
Baeyer–Villiger oxidation vs. epoxidation reaction
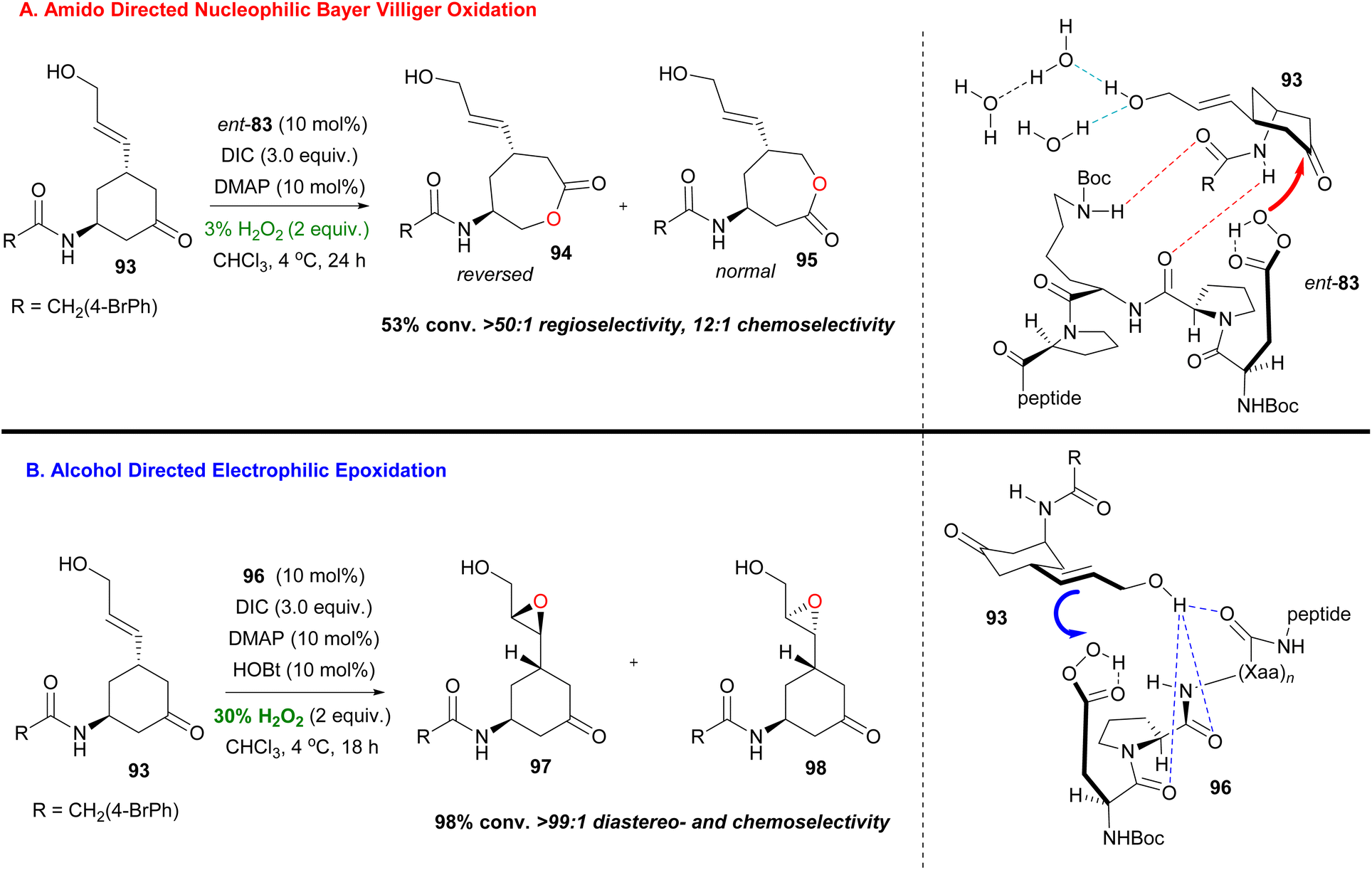
In general, the most difficult task in organic chemistry is chemoselectivity. Biocatalysis, mainly, offers the opportunity to tune the chemical reactivity of the functional groups that appear in a molecule. In 2016, Miller and coworkers in a ground-breaking contribution synthesised bifunctional substrates that are susceptible to both Baeyer–Villiger and epoxidation and can exhibit orthogonal reactivity, depending on the peptide catalyst used.57 The embedded aspartyl peracid moiety can serve as a nucleophilic or electrophilic oxidant and peptides 96 and 83 already showed their catalytic activity in directed epoxidation of allylic alcohols47 and Baeyer–Villiger oxidation of functionalized ketones,53 respectively (Scheme 23). When 96 was used in the presence of a free hydroxyl group, excellent regio- and diastereoselectivity can be obtained for the corresponding epoxide, with only traces of other undesired oxidation products (Scheme 23). While using catalyst 83, the epoxidation product prevailed over the Baeyer–Viliger oxidation product and this was attributed to the stereochemical mismatch between 83 and substrate 93. This speculation proved to be right since the use of ent-83 afforded the desired chemoselectivity (Scheme 23). Further investigation of the reaction conditions led the authors to the conclusion that increasing the ratio of water, compared to H2O2, enhanced the Baeyer–Villiger oxidation pathway on the free hydroxyl group substrate. Indeed, at the cost of reducing the yield (50–58%), water anchors the hydroxyl group with competing H-bonding, allowing the desired outcome (Scheme 23).
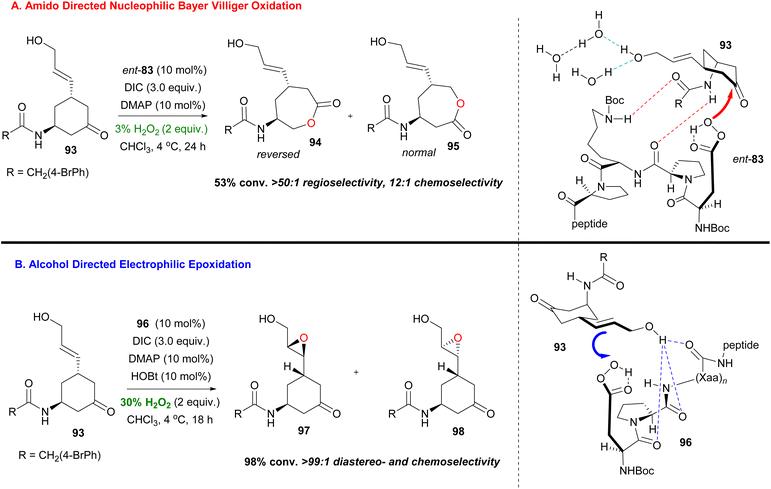 |
| | Scheme 23 Catalyst dependent chemoseletive oxidations of bifunctional substrates using (A) ent-83 or (B) 96. | |
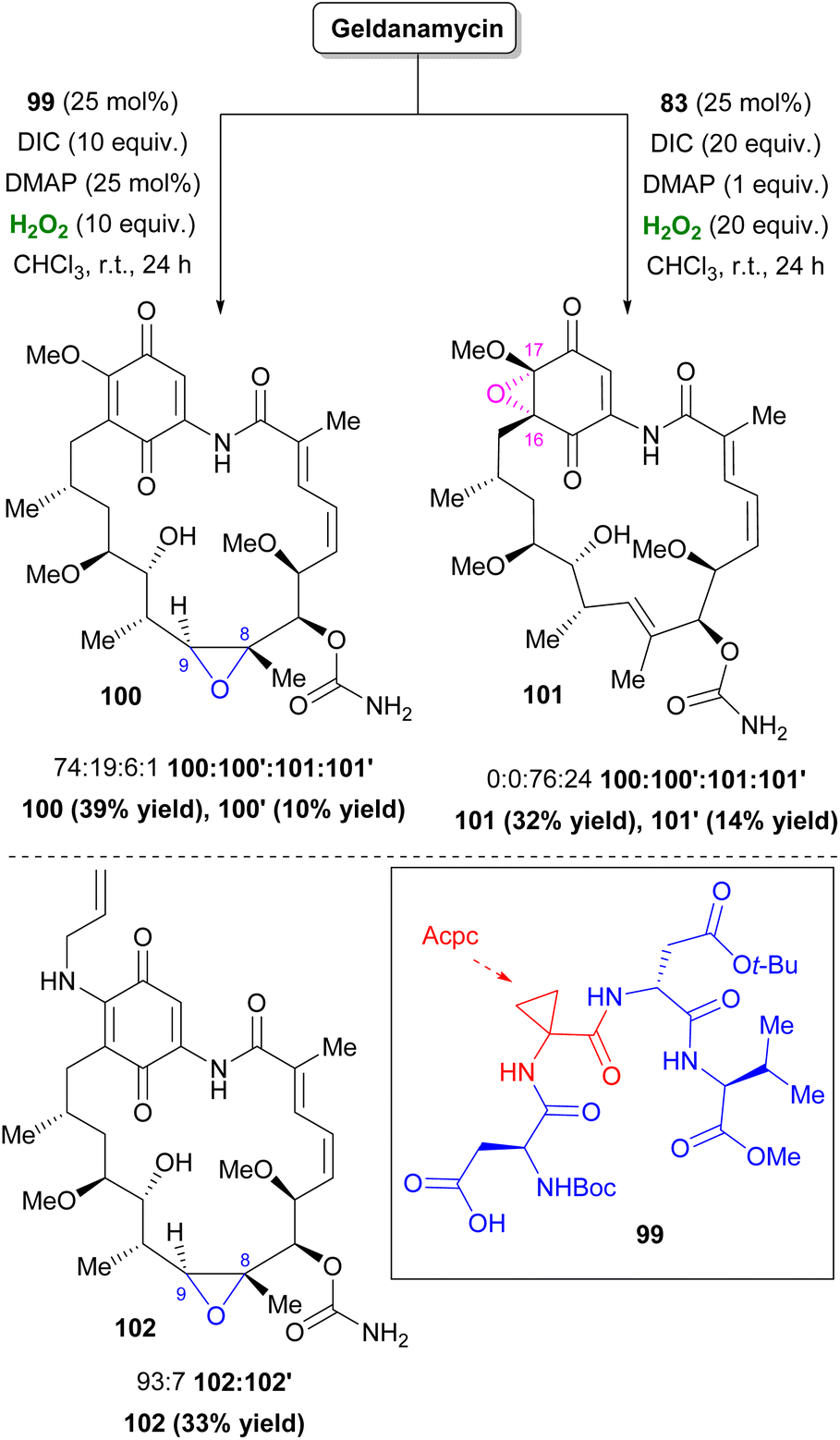
In 2020, Miller and co-workers incorporating their Asp-based peptide method, found two catalysts for the selective oxidation of Geldanamycin, a compound that inhibits the function of Hsp90 protein.58 Catalyst 99 was selective towards 8,9-epoxide 100, suggesting that β-turn conformation allowed interaction of the carbamate in C7 with the i + 1 Acpc group of 99. On the other hand, catalyst 83, which previously showed potential in nucleophilic Bayer–Villiger oxidation, favored an unprecedented quinone epoxidation in the 16,17-position (101) (Scheme 24).59 A truncated study revealed that, in this case, Asp–Pro is mainly responsible for this outcome. In contrast, this was not repeated with simpler quinones. Tanespimycin or 17-AAG, an improved Geldanamycin analogue, exhibited similar behavior using catalyst 99, providing 102 with elevated selectivity (Scheme 24).
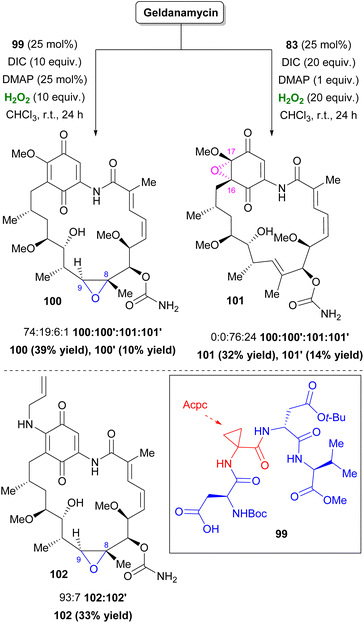 |
| | Scheme 24 Geldanamycin's chemoselective epoxidation. | |
Applications of the Asp–peracid oxidation
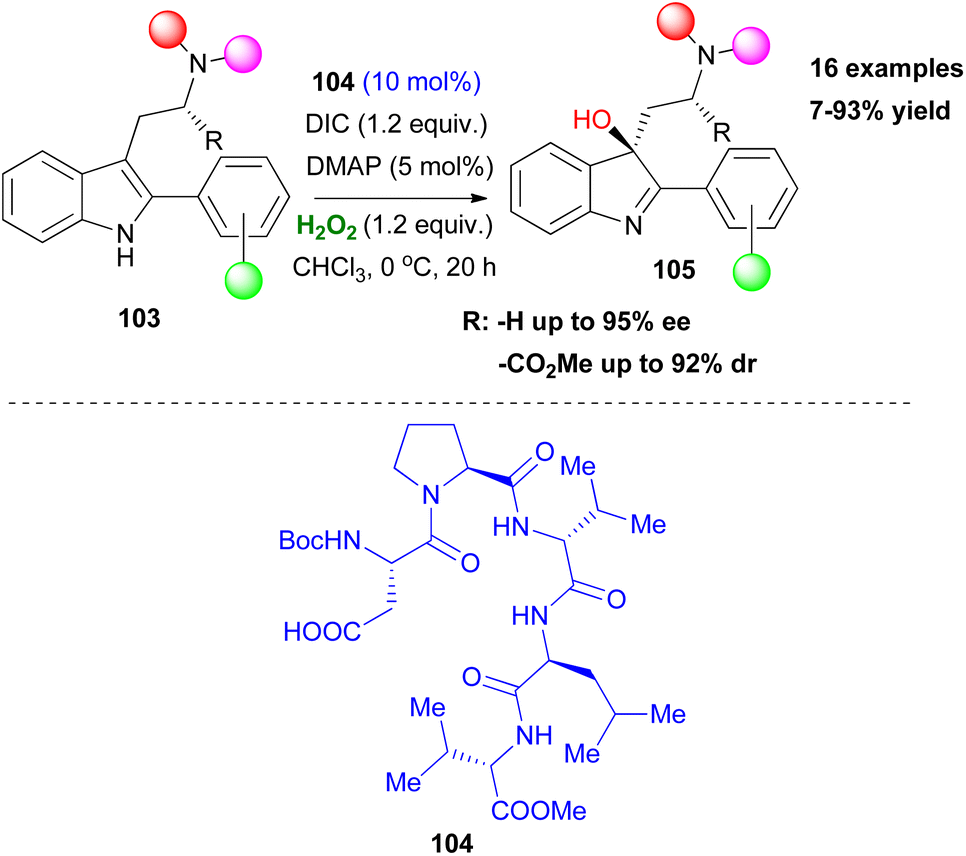
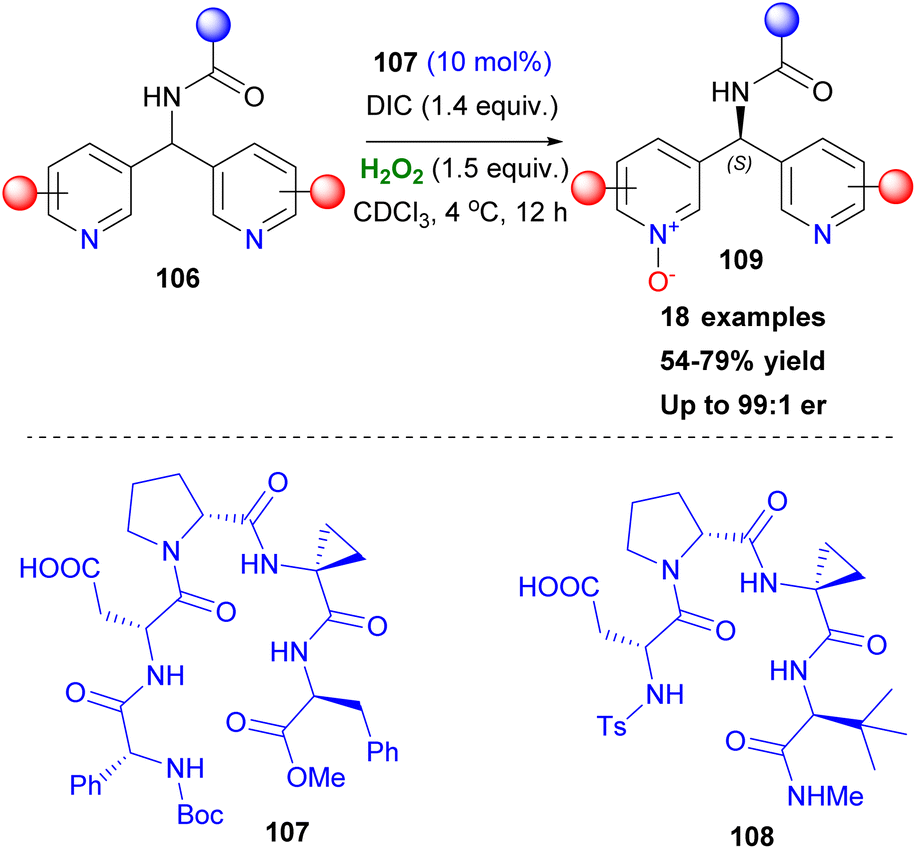
After the successful application of Asp peptide-based catalysts in oxidation protocols, such as the asymmetric epoxidation and Baeyer Villiger oxidation, Miller and coworkers attempted to expand the general affinity of the aspartyl peracid route in other oxidative transformations. Their first attempt was focused on the selective oxidation of indoles, which is considered important, due to the existence of the indole moiety in a variety of natural products. In 2011, in collaboration with Movassaghi, they investigated indole oxidation under the already known reaction conditions.60 After screening the peptide catalyst used, not only diastereoselective oxidation of tryptophan-based indoles 103 was at hand, but also enantiomeric induction of achiral scaffolds was introduced (Scheme 25). Interestingly, the presence of other free indole or NH-groups in the structure did not dampen the pre-established chemoselectivity, excluding the need for protecting groups. Seeking more potential candidates that might undergo an oxidative transformation, Miller and coworkers studied the oxidation of a series of substituted pyridine scaffolds.61 Desymmetrisation of bis(pyridines), with a H-bonding group strategically placed in a pro-stereogenic center was achieved by peptide-based catalyst 107, after catalyst screening employing the OBOC method (Scheme 26).61 It needs to be clarified that these molecules can function as co-catalysts as depicted in Scheme 26, rendering the use of DMAP unnecessary. Substitution at the 6,6′-position of the substrates proved vital for inducing excellent enantioselectivity. To showcase the advantages offered by this protocol, the authors performed several derivatization experiments, highlighting the utility of the formed N-oxides 109. Furthermore, application in scaffolds resembling medicines, like Loratadine or Varenicline, exhibited promising results in terms of generality of the current method. In the same vein, Miller and coworkers expand their protocol proposing a chemoselective N-oxidation that leads to asymmetric induction in pyridyl sulfoximines employing catalyst 108.62
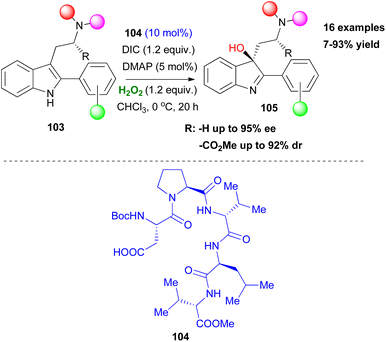 |
| | Scheme 25 Asymmetric oxidation of indoles to 3-hydroxy-indolenines. | |
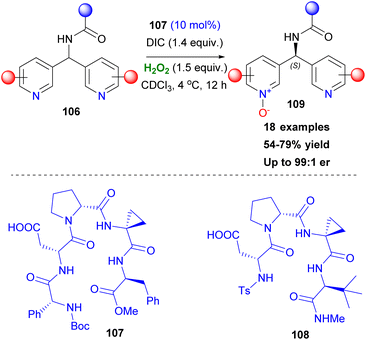 |
| | Scheme 26 Desymmetrisation of bis(pyridines) by N-oxidation. | |
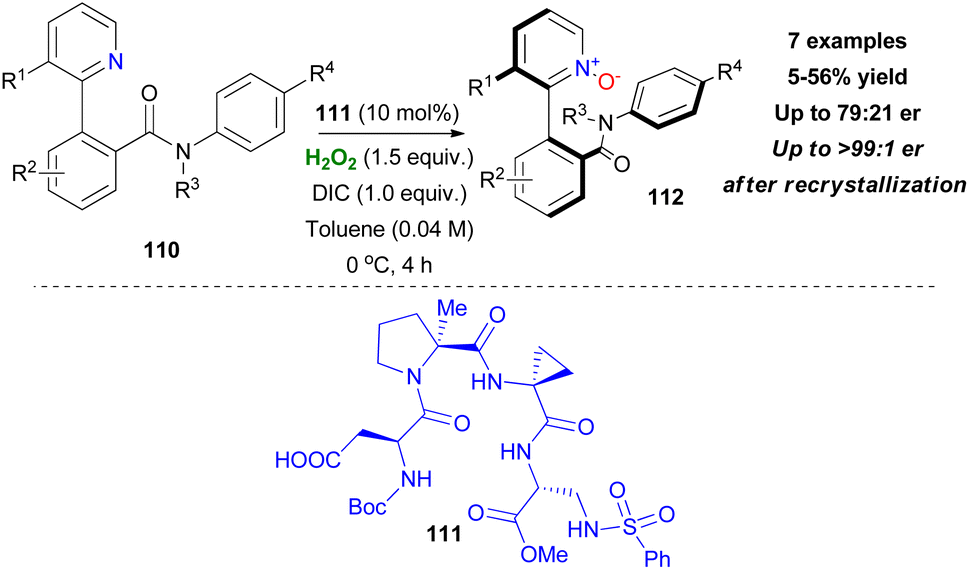
The most recent work of Miller's group focuses on stabilisation of stereogenic axis centres via N-oxidation with catalyst 111 (Scheme 27).63 Trisubstituted biaryl scaffolds with substitution in the 3-position of the pyridine moiety prevented the unwanted racemization, which occurs by rotation of the axis, to some extent.
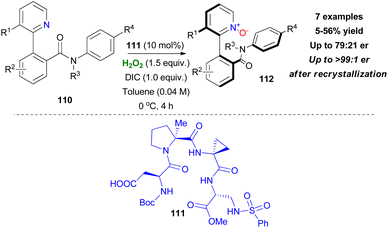 |
| | Scheme 27 Stabilisation of the atropoisomeric axis by N-oxidation. | |
The proton of the amide group was necessary in terms of selectivity for catalyst–substrate interactions, however at the risk of racemization. Even though the reaction conditions were optimized for enantiomeric excess and atropoisomeric stability, an inevitable protonation of the pyridine moiety by various potential proton sources in the reaction mixture seemed to influence the reaction outcome eventually. However, recrystallization of the final product provided enantiomeric enrichment (Scheme 27).63
Organocatalytic activation of hydrogen peroxide with iminium salts
Applications of iminium salts in organic oxidations
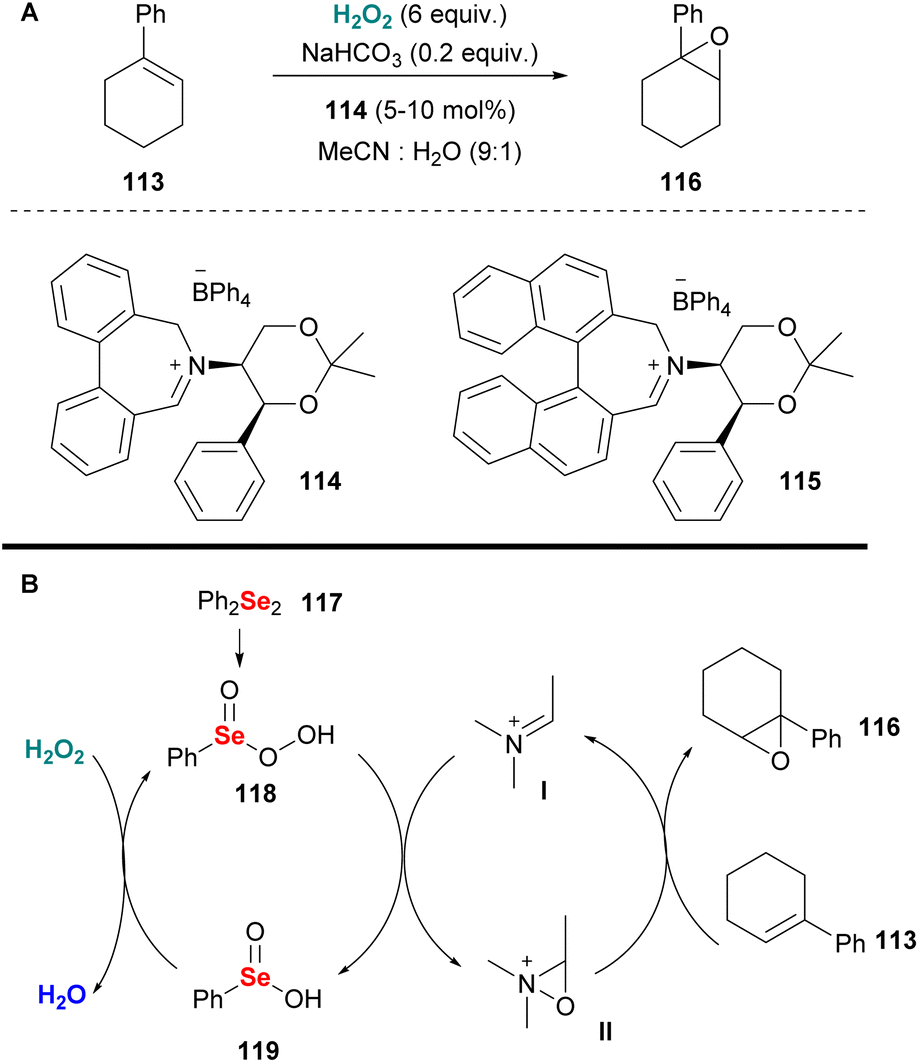
Since 1976, when Lusinchi first reported the use of iminium salts as potential organocatalysts in asymmetric epoxidations,64 many researchers introduced a plethora of iminium salt catalysts. Page and coworkers were among the first to introduce a series of iminium salt catalysts, based on the tetrahydroisoquinoline scaffold decorated with a chiral substituent on the nitrogen atom.65 Having successfully employed these novel organocatalysts in alkene epoxidation, employing Oxone as the oxidant, the authors investigated the use of other potential oxidants, aiming at high enantioselectivity. Therefore, hydrogen peroxide was employed as an alternative eco-friendly oxidant. The authors opted for sodium bicarbonate as the optimum base to promote the epoxidation of 1-phenylacetylene, minimizing the background reaction (Scheme 28A). Additionally, they examined the effect of the reaction parameters (temperature, co-solvent, base etc.) and reported a plausible double mechanistic scenario, where the base might act either as a base or a peroxidophore, providing a useful guide for future attempts. Unfortunately, this elegant organocatalytic protocol suffers from low enantioselectivity and only 1-phenylcyclohexene was tested. To address this drawback and increase the enantioselectivity of the epoxidation process the authors, inspired by Santoro's and Du Bois' work66 exploited diphenyl diselenide 117 as the co-catalyst.67 This novel oxidation system generates benzeneperseleninic acid 118, which oxidises iminium salt I, facilitating the enantioselective oxygen transfer as depicted in Scheme 28B. With these reaction conditions, Page and coworkers achieved higher enantioselectivities, increasing the reaction rate.67
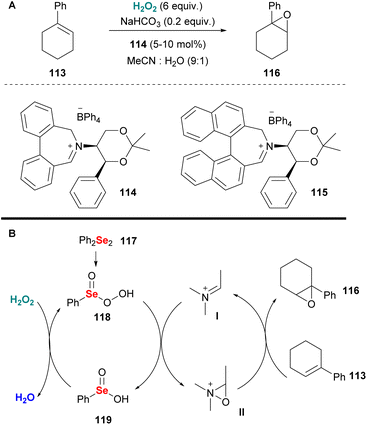 |
| | Scheme 28 (A) Olefin epoxidation employing catalyst 114. (B) Catalytic cycle with the use of diselenide 117 as the co-catalyst. | |
Following their research in oxidative protocols, employing hydrogen peroxide as the oxidant, Page and coworkers applied Du Bois' reaction conditions66b towards sulfide oxidation.68 This contribution provides easy access to sulfoxides 121, avoiding the formation of sulfone (Scheme 29). Nevertheless, this protocol cannot be used, when sulfide's substituents are decorated with a double bond.
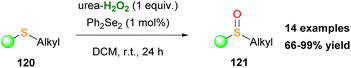 |
| | Scheme 29 Sulfide oxidation protocol by Page and coworkers. | |
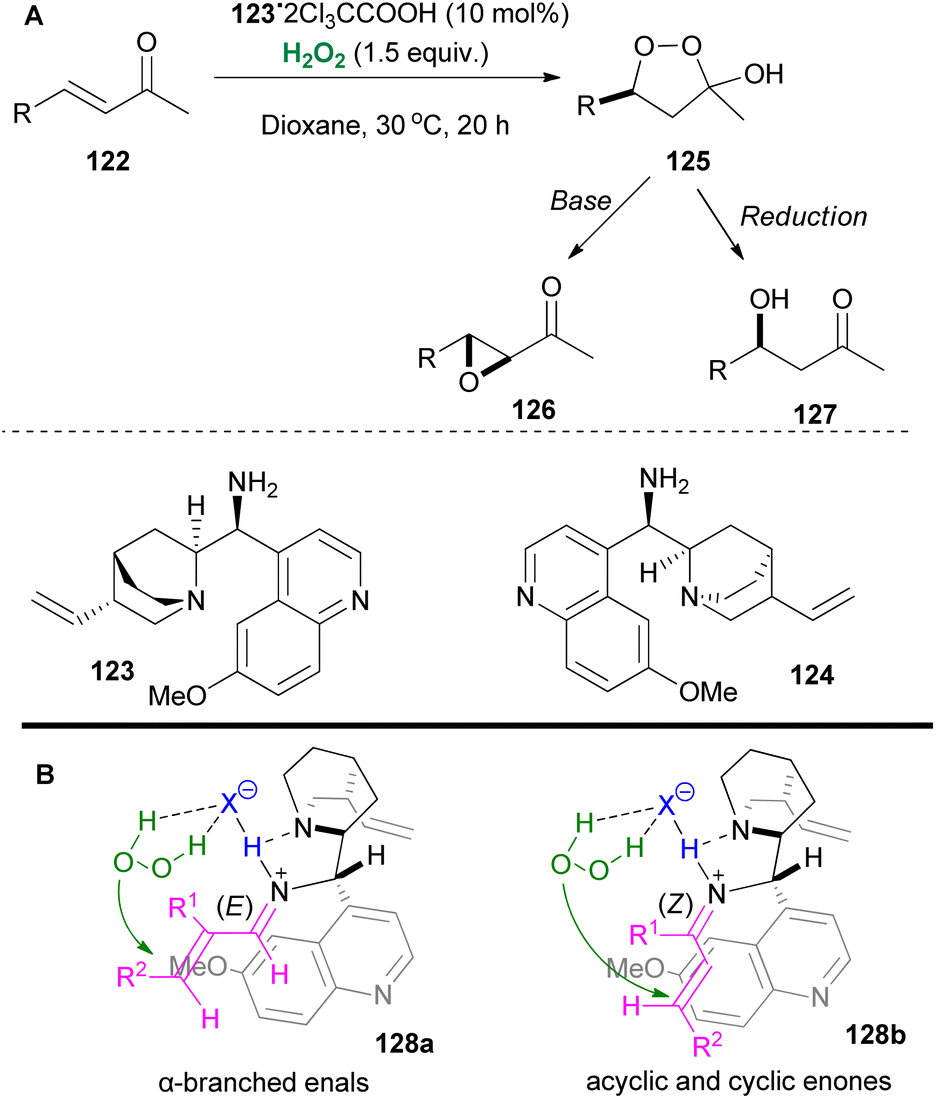
Iminium catalysis has been employed not only towards the hydrogen peroxide activation but also towards the substrate activation, inducing an oxidation reaction. Ground-breaking contributions by Jorgensen69 and MacMillan70 introduced this feature as a powerful strategy for the enantioselective epoxidation of α,β-unsaturated carbonyl compounds. Many researchers employed this strategy in oxidation reactions.71 In 2008, pioneering the field of enantioselective catalysis,72 List and coworkers developed an elegant asymmetric hydroperoxidation protocol of α,β-unsaturated ketones 122, employing Cinchona alkaloid derivatives 123 or 124 as the catalysts (Scheme 30).73 In this contribution, the authors reported easy access to enantiopure peroxyhemiacetals 125, intermediates of great importance in natural product synthesis, using hydrogen peroxide as the oxidant. These versatile intermediates provided easy access to enantiopure epoxides 126 or alcohols 127, upon workup. In this contribution, the main problem faced by the authors appeared to be the substrate scope, since aromatic and trisubstituted enones proved to be unreactive (Scheme 30).
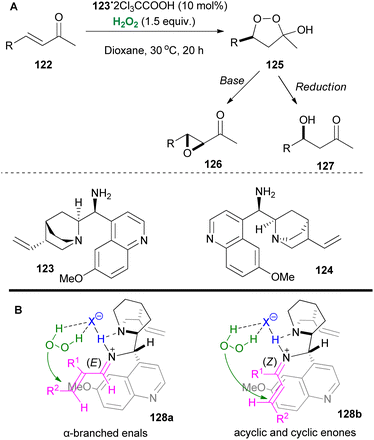 |
| | Scheme 30 (A) Peroxide formation and its applications. (B) Proposed transition states from List and coworkers. | |
In a later study, the same group managed to expand the substrate scope even more with some alterations in the reaction conditions, thus including acyclic enones, cyclic enones and α-branched enals. Through extensive research, the authors also proposed possible transition states (128a and 128b) that explain the observed stereoselectivities (Scheme 30B).74
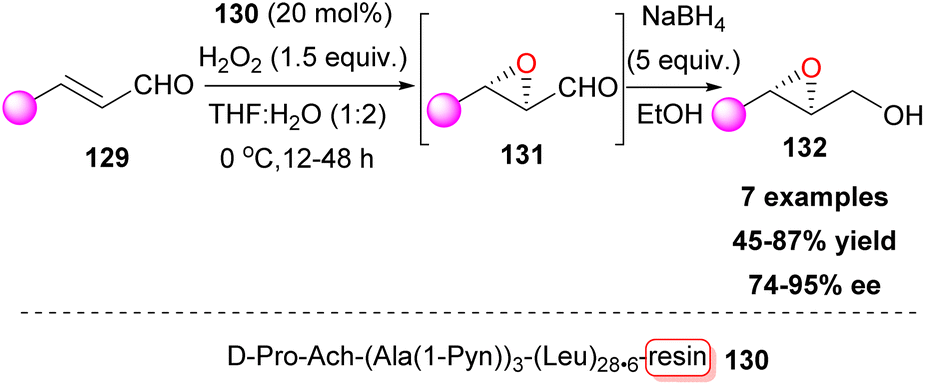
Knowing that asymmetric induction in aqueous media is more challenging compared to organic solvents, Kudo and co-workers envisaged a resin-bound peptide that provided a hydrophobic microenvironment.75 To this end, peptide catalysts with a resin-supported poly-Leu chain, that adopt an α-helix structure, were tested in search of novel catalytic sequences for the enantioselective epoxidation of α,β-unsaturated cinnamaldehydes 129, which were subjected to reduction afterwards. After a thorough screening, peptide catalyst 130 emerged, capable of three reuses without loss of enantioselectivity (Scheme 31). Other cinnamaldehydes and some aliphatic aldehydes also yielded high stereo- and enantioselectivities.
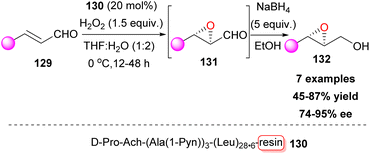 |
| | Scheme 31 Resin-bound peptide catalyst for asymmetric epoxidation of α,β-unsaturated cinnamaldehydes. | |
After a novel study proposed by List,76 combining iminium and Brønsted acid catalysis for the nucleophilic epoxidation of α,β-unsaturated ketones, Schröder and coworkers77 tested a series of chiral diamines and flexible phosphoric acids in search of a new counteranion pair for this reaction (Scheme 32). Diamine 134 and phosphoric acid 135 proved to be an excellent pair in ether-like solvents, with dioxane being the optimum, since its low dielectric constant and its miscibility with H2O2 facilitated high enantioselectivity and yield, respectively. More in-depth investigation revealed that these ion-paired catalysts behave as one stereoisomer making their antipode counterpart accessible, and therefore employing one “stereoisomer” each time led to stereochemical control of the final product 136 (Scheme 32). The substrate scope was extended to cyclic and some acyclic enones with excellent enantioselectivities. Eventually, this robust catalytic system was successfully expanded to other asymmetric transformations in the same work.
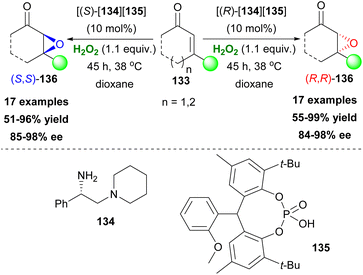 |
| | Scheme 32 Phosphoric acid and diamine synergistic effect in the asymmetric epoxidation of enones. | |
Applications of iminium salts in the C–H hydroxylation reaction
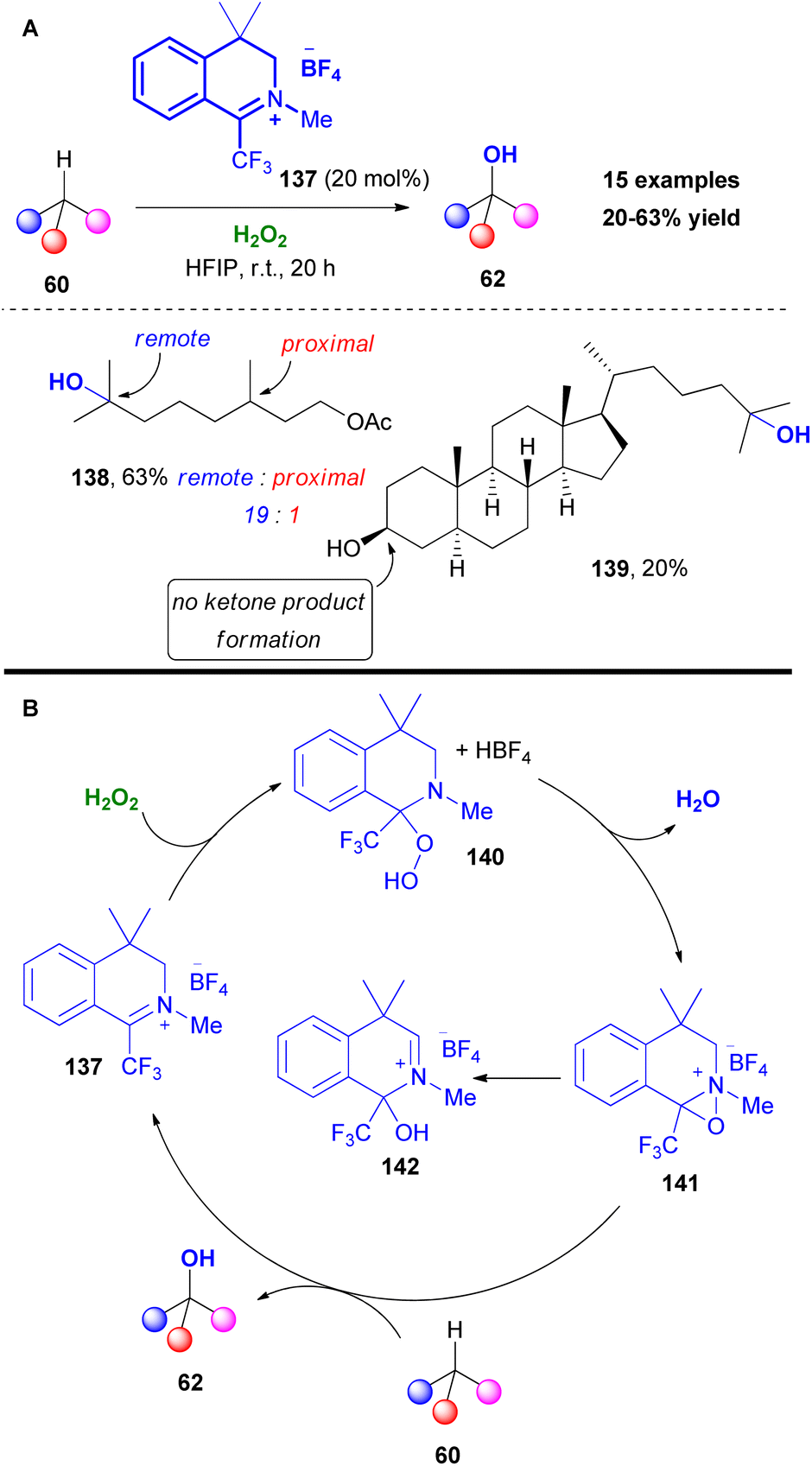
Another challenging task, which attracted the researchers' interest, is the chemoselective C–H bond hydroxylation. The increased number of contributions in the literature, including numerous late-stage functionalizations of complex molecules, pointed out the importance of this useful transformation. Based on the reaction output and inspired by Du Bois' work,66b Hilinski and coworkers, investigated the use of oxaziridinium salts in C–H hydroxylation reactions.78 Initially, the authors unsuccessfully used Page's catalyst,79 in the presence of hydrogen peroxide as the oxidant (Scheme 33). Slight modification in the catalyst's core afforded promising results. It must be mentioned that the catalytic activity of 137 was enhanced in the presence of a gem-dimethyl substitution at the benzylic position, which prevented the possibility of aromatization and N-methyl substitution. Furthermore, the use of HFIP was essential, since it is known to activate hydrogen peroxide. The authors reported higher selectivity for hydroxylation of 3° C–H bonds that are remote to an electron-withdrawing group, compared to what is known in the literature. This effect can be attributed to the solvent effect, since HFIP has the ability to deactivate potential reactive sites via hydrogen bonding. Testing their protocol in a series of substrates, the authors noticed that C–H hydroxylation took place in the presence of a primary or secondary alcohol.
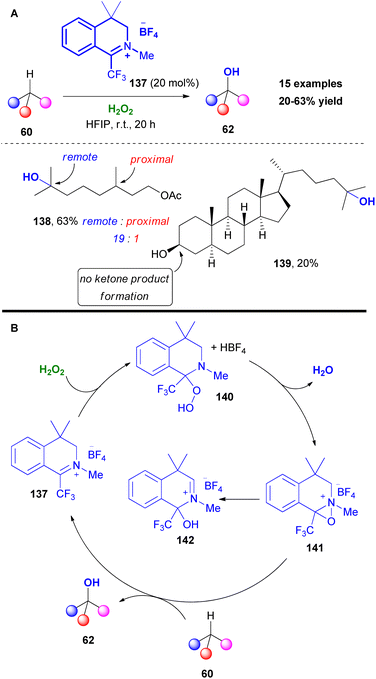 |
| | Scheme 33 (A) C–H hydroxylation by Hilinski and coworkers. (B) Catalytic cycle of C–H hydroxylation. | |
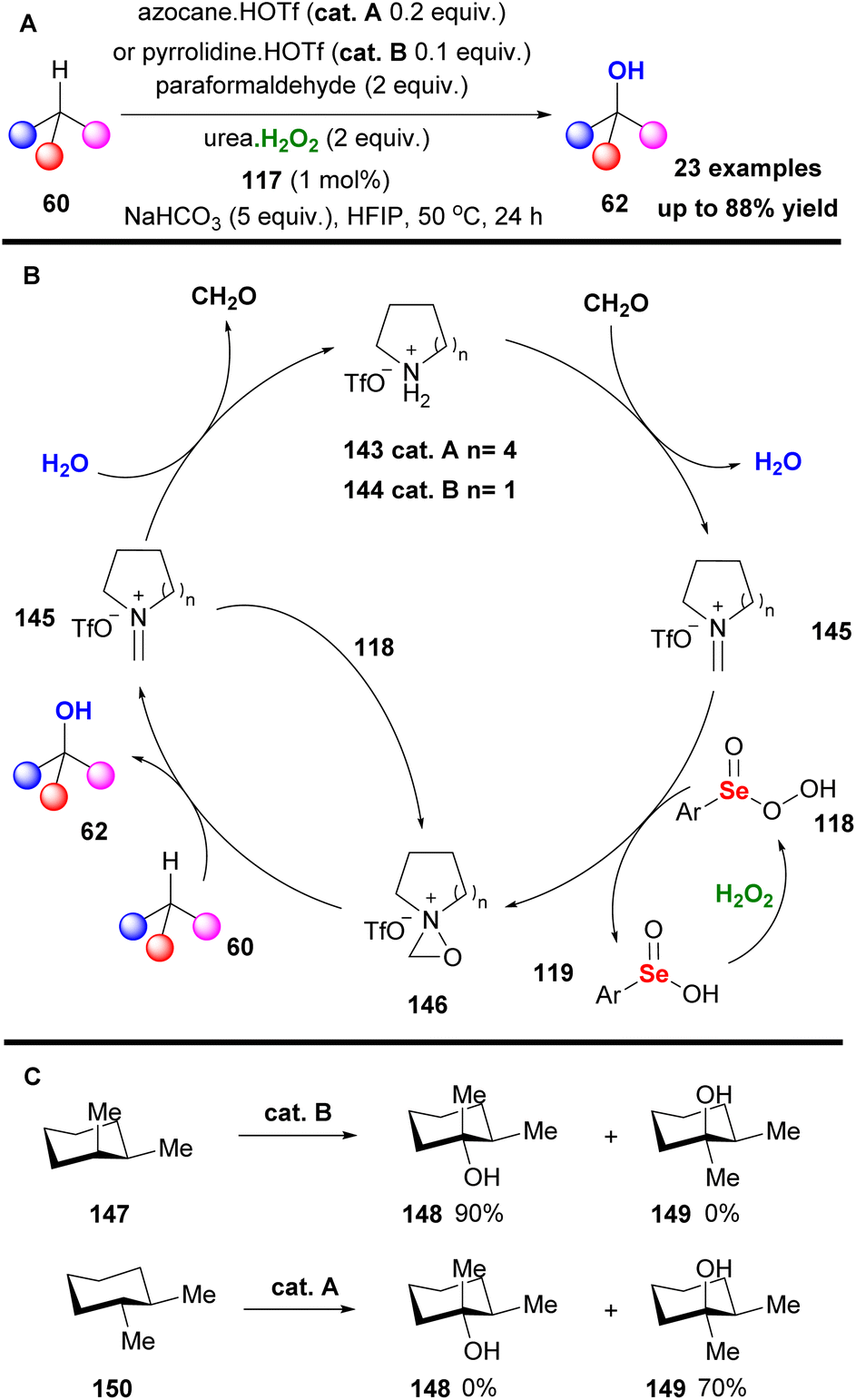
Contrary to what is stated in the literature, this protocol promoted the hydroxylation of unactivated secondary C–H bonds, minimizing the overoxidation pathway (Scheme 33A). To probe the reaction mechanism, Hilinski and coworkers conducted NMR studies, verifying the formation of oxaziridinium salt 141, proposing the mechanism depicted in Scheme 33B. Initially, addition of hydrogen peroxide onto catalyst 137 liberates fluoboric acid, forming intermediate 140. The acidic environment promotes the formation of the oxaziridinium salt 141, which reacted with substrate 60, delivering the desired product. Following their continuous interest in C–H hydroxylation reactions, Hilinski and coworkers acknowledging the advantages offered by iminium ion catalysis, envisaged the in situ generation of an oxaziridinium salt, combining a secondary amine, a ketone or aldehyde and an oxidant.80 This concept was elegantly employed by Yang and coworkers in epoxidation reactions.81 The authors performed a series of optimisation experiments identifying azocane triflate salt and paraformaldehyde as the starting material for the imine synthesis, 1,2-bis(3,5-bis(trifluoromethyl)phenyl)diselane 117 as the peroxide promoter and urea-H2O2 as the oxidant (Scheme 34A). With these conditions in hand, excellent chemoselectivity and site selectivity were acquired, along with high yields. In many cases, including rigid substrates, pyrrolidine triflate salt 144 afforded the highest yield. This protocol was successfully tested in a vast substrate scope, affording excellent results in remote chemoselectivity. Furthermore, it was found to be effective in late-stage functionalization and was successfully tested in a gram scale reaction. According to the proposed mechanism, the in situ formed iminium salt reacts with the perselinic acid 118 to deliver oxaziridinium salt 146 (Scheme 34B). Then, the active oxidant performs the C–H hydroxylation. Since the hydroxylation reaction of 60 is governed by the formation rate of intermediate 145, the size of the secondary amine is believed to play a prominent role in the reaction mechanism (Scheme 34B). Furthermore, the authors investigated the stereospecificity of the reaction. For this reason, cis- and trans-1,2-dimethylcyclohexane were employed, showing complete retention of stereochemistry (Scheme 34C).
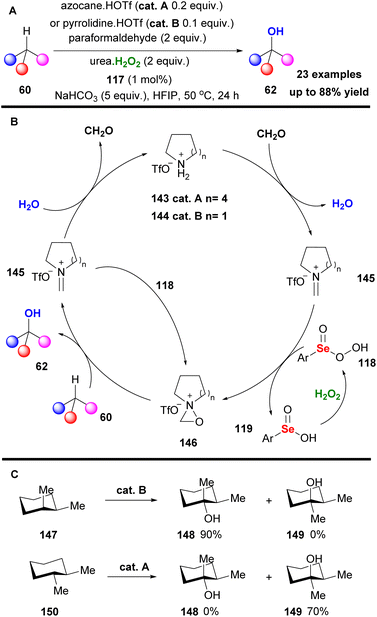 |
| | Scheme 34 (A) C–H hydroxylation employing the in situ generated iminium salt 145. (B) Studies on the retention of stereochemistry. (C) Hydroxylation of cyclohexanes. | |
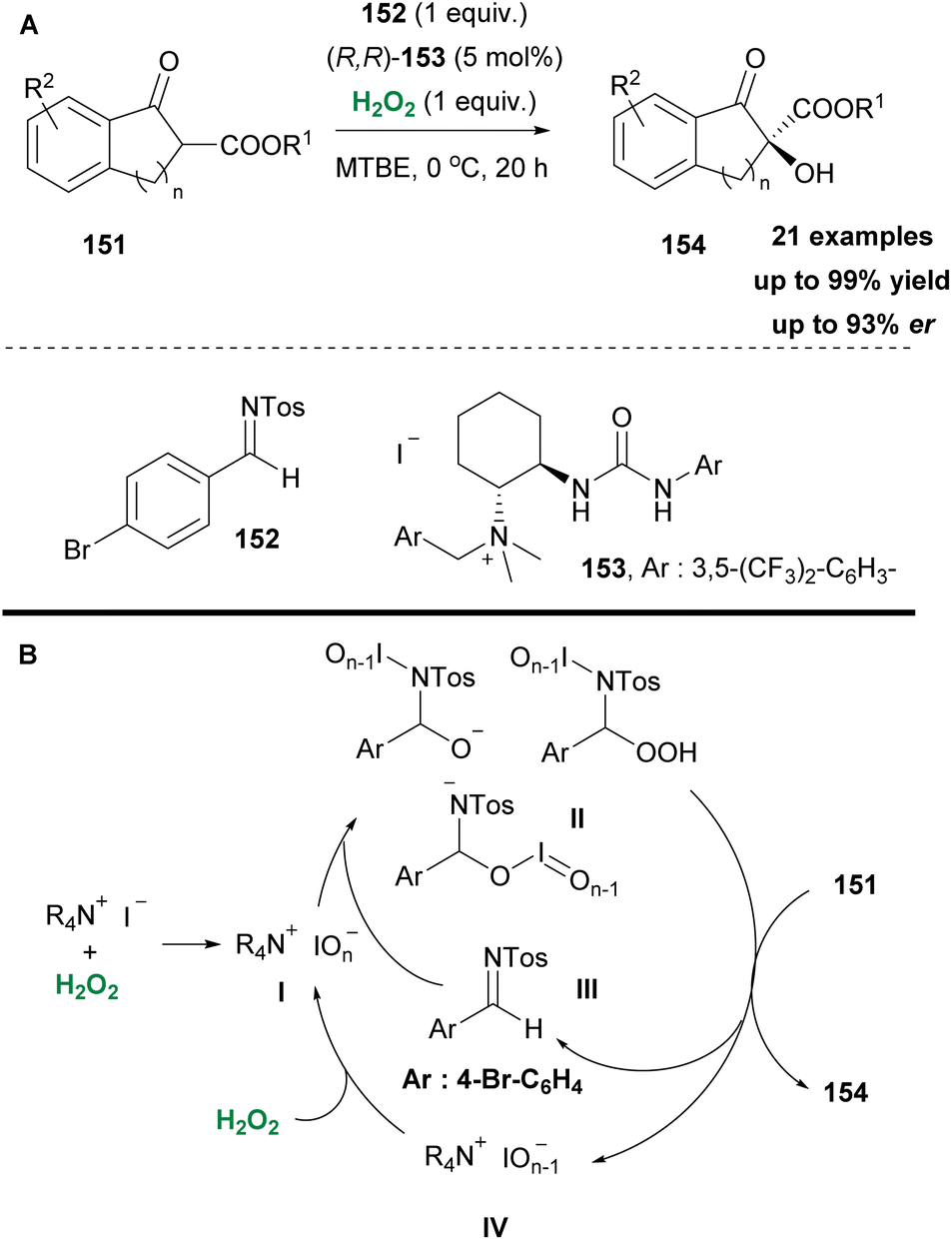
In the same vein, Waser and coworkers few years earlier based on their previous work,82 enriched by Ooi's contributions to asymmetric oxaziridine synthesis,83 envisioned a straightforward asymmetric α-hydroxylation of β-ketoesters via an in situ generation of oxaziridine (Scheme 35A).84 A series of investigations to obtain the optimum reaction conditions revealed the crucial role of the counter anion, which proved to be responsible for the hydrogen peroxide activation, instead of the formation of an oxaziridine intermediate. This was elegantly reported in the past by Ishihara and coworkers.85 Based on previous reports in the literature, in their mechanistic scenario the authors supported the formation of iodine hypervalent species I, upon interaction of hydrogen peroxide with ammonium iodide (Scheme 35B). This species reacts with imine 152 to form an unknown oxidant partner II, which actually promotes asymmetric hydroxylation (Scheme 35). It is supported by the authors that hypervalent species II, apart from their role in reacting with the imine 152, also behave as the base to deprotonate β-ketoesters 151. Based on their finding, iodide is necessary as the oxidizable counter-anion, since bromide afforded mediocre results. A good substrate scope was evaluated with excellent results. Nevertheless, regarding the realistic mechanistic approach, more experiments should be performed.
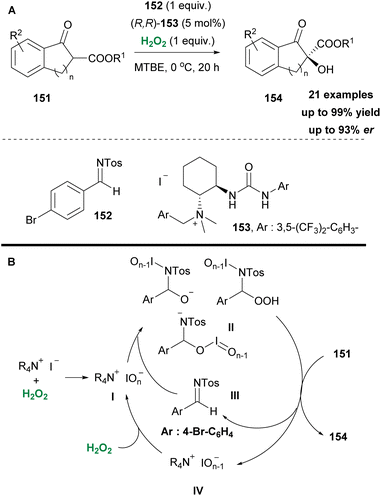 |
| | Scheme 35 (A) C–H hydroxylation protocol by Waser and co-workers. (B) Catalytic cycle. | |
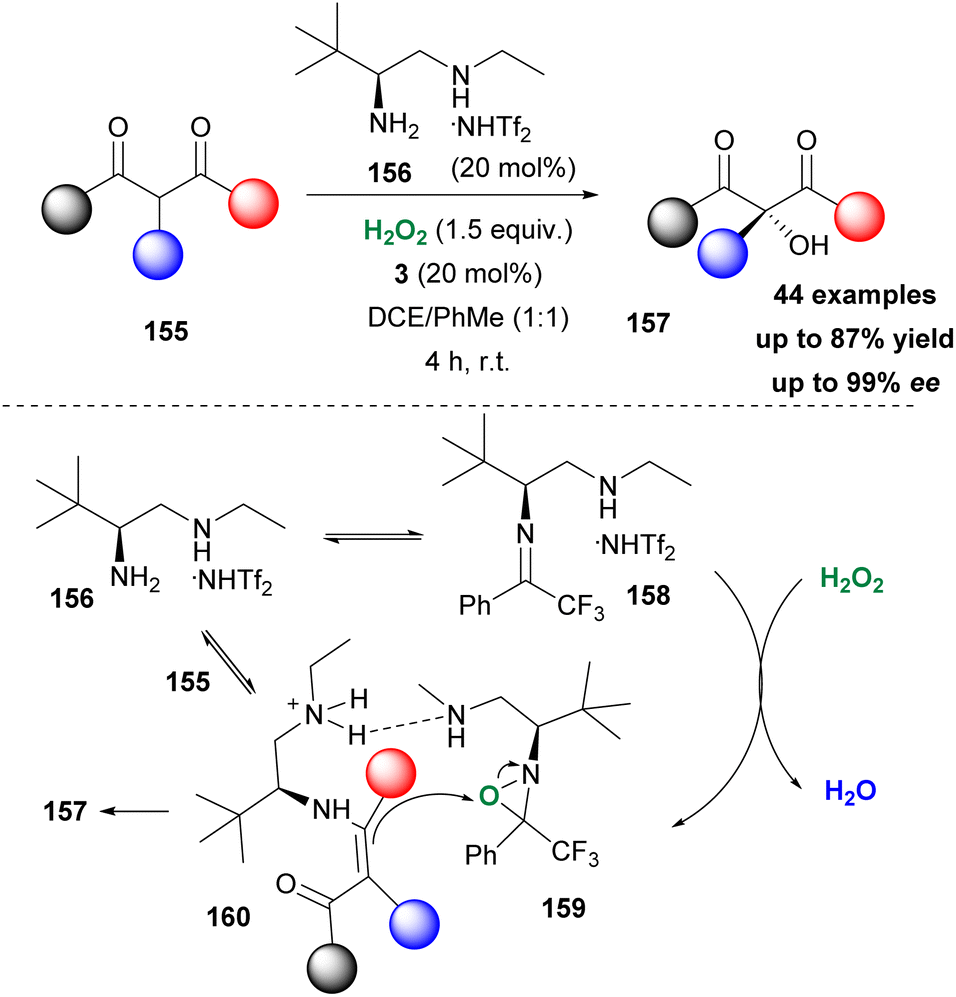
Recently, Zhang, Luo and coworkers reported a dual amine-ketone cooperative catalysis for the asymmetric α-hydroxylation of keto-compounds (Scheme 36).86 Screening a number of primary-secondary diamines, the authors opted for amine 156 as the optimum partner for ketone 3 to afford an excellent organocatalytic system. Hydrogen peroxide was identified as the oxidant of choice. A vast variety of substrates was tested affording the desired hydroxylated derivatives in good to excellent yields with high enantioselectivities. Not only β-keto-esters but also β-keto-amides afforded excellent results. The authors performed several mechanistic studies (DFT calculations and HRMS experiments) to probe a plausible mechanistic pathway. In their proposal, amine reacts in the acidic environment (induced by NHTf2) to form iminium salt 158. The latter in the presence of hydrogen peroxide forms oxaziridinium salt 159, which transfers an oxygen atom to enamine 160 to deliver the desired product. As depicted in Scheme 36, amine 156 has a dual role, as it forms the active oxidant and at the same time forms enamine 160, upon reaction with 155, facilitating the oxidation reaction.
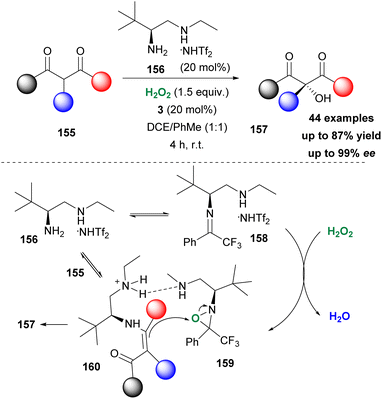 |
| | Scheme 36 Dual amine/ketone organocatalytic activation of hydrogen peroxide. | |
Use of hydrogen peroxide to form endoperoxides and their applications in oxidation reactions
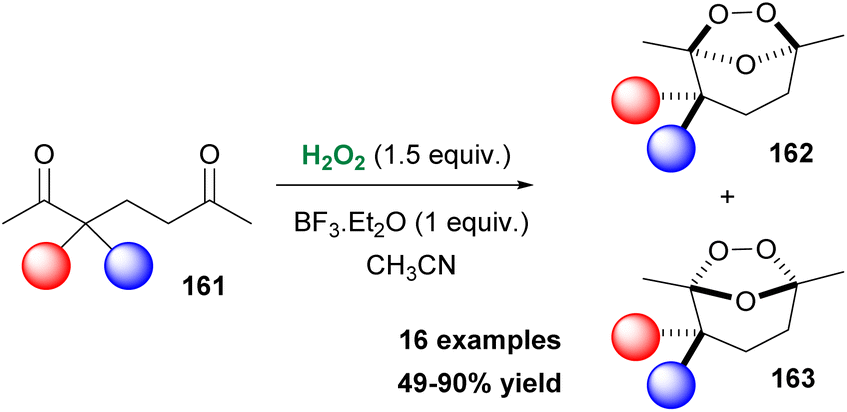
Over the past decade, many researchers acknowledged the great importance of endoperoxides in modern synthetic chemistry, as well as in medicinal chemistry. Organic peroxides are found in many natural products and compounds of medicinal interest, exhibiting anticancer, antimalarial and antiviral activity. In 2015, a naturally-occurring product, Artemisinin, was recognized by the Nobel Prize in Medicine as an antimalarial agent.87 Furthermore, peroxides derived from ketones find excellent application as radical initiators in polymer production.88 As a consequence, many synthetic approaches have been reported, providing access to various classes of peroxides. Additionally, in the past years, many efforts have been made, targeting also the synthesis of azaperoxides. In 2018, Alabugin, Terent'ev and coworkers reported an excellent study on ozonide synthesis from 1,5-diketones, employing hydrogen peroxide as the oxidant.89 The major advantage of this study, compared to what already existed in the literature, is the one-step formation of ozonides without the formation of other peroxides or even oxidized by-products. To probe the optimal reaction conditions, the authors employed diketone 161 as the model substrate (Scheme 37). Hydrogen peroxide was found to be the reagent of choice, while BF3·Et2O as the catalyst (0.5 equiv.) provided the highest yield of products (95%), along with a modest selectivity towards 162. This oxidative protocol exhibited high functional group tolerance. A variety of 1,5-diketones decorated with electron-withdrawing or electron-donating groups afforded ozonides in good to excellent yields (Scheme 37). It must be mentioned that compounds bearing a double or a triple bond remained intact and the authors did not mention the formation of by-products, derived from epoxidation reactions or other oxidative pathways. The authors performed a series of computational studies to probe the thermodynamic stability of the product. The efficient stability of the formed ozonides under several reaction conditions might find excellent application as potential protecting groups.89
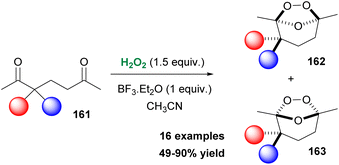 |
| | Scheme 37 Ozonide synthesis. | |
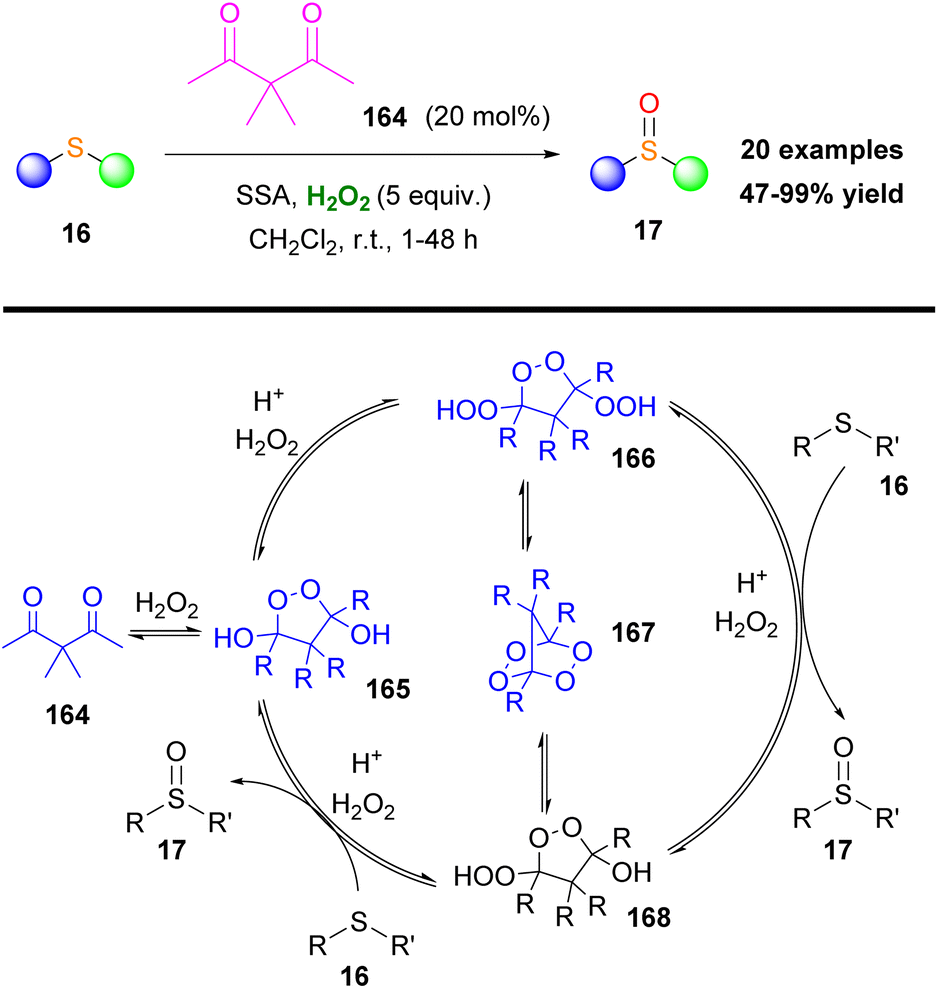
In a more recent study, Tomkinson and coworkers employed peroxides to achieve organocatalytic sulfoxidation.90 Specifically, the authors examined a series of diketones, like 164, as catalysts, along with silica-sulfuric acid (SSA) as an effective recyclable co-catalyst (Scheme 38). The presence of α-hydrogens, between the two carbonyls, reduces their electrophilicity and therefore renders the catalyst less effective. A variety of sulfides 16 were applied to this protocol. Electron-rich sulfides required faster reaction times, while sulfides with bulky groups required longer reaction times. Oxidizable and acid sensitive groups remained intact. The proposed catalytic cycle in Scheme 38 involves the formation of three important peroxides (165, 166 and 167) in equilibrium, confirmed by single X-ray analysis. X-ray analysis demonstrated the trans-relationship of the exocyclic oxygen substituents in the intermediates 165 and 166. The active species is peroxide 166, which can oxidize two different molecules of sulfide, leading to peroxide 168 and eventually returning to 165 (Scheme 38).
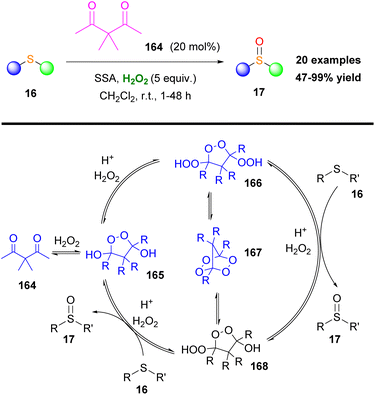 |
| | Scheme 38 Tomkinson's sulfoxidation and proposed mechanistic scenario. | |
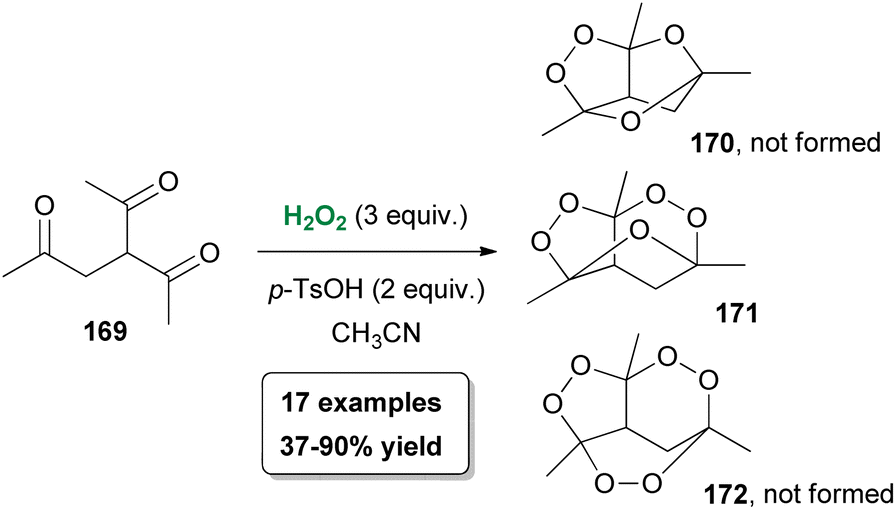
A few years later, following their interest in peroxide chemistry, Alabugin, Terent'ev and coworkers reported the use of hydrogen peroxide towards the synthesis of oxygen-rich tricyclic heterocycles.91 Based on their previous findings, the authors exploited triketones 169, which are considered flexible precursors, as the starting materials providing theoretical access to oxygen-rich structures 170–172 (Scheme 39). For this purpose, β,γ′-triketones 169 were used, due to their capability of forming more easily a peroxide bridge. A series of optimization experiments provided the optimum conditions, where p-TsOH·H2O was identified as the appropriate Lewis acid. Under the optimal reaction conditions, bis-peroxide derivative 171 was obtained in high yield as the sole product, while neither the monoperoxide 170 nor the tris-peroxide analogue 172 was formed. In the presence of a substituent at the α-position, along with the formation of the bis-peroxide derivative, the formation of the monoperoxide 170 also took place. In their effort to broaden the scope of the peroxidation protocol, the authors proved that in the presence of excess hydrogen peroxide, monoperoxides can be converted into bis-peroxide analogues. Despite the complexity of the structure, the bis-peroxide derivatives were found to be stable. Computational and mechanistic studies provided a plausible mechanism for this oxidative process.91
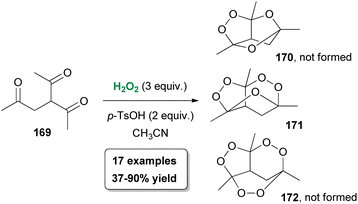 |
| | Scheme 39 Oxidation of substituted triketones 169. | |
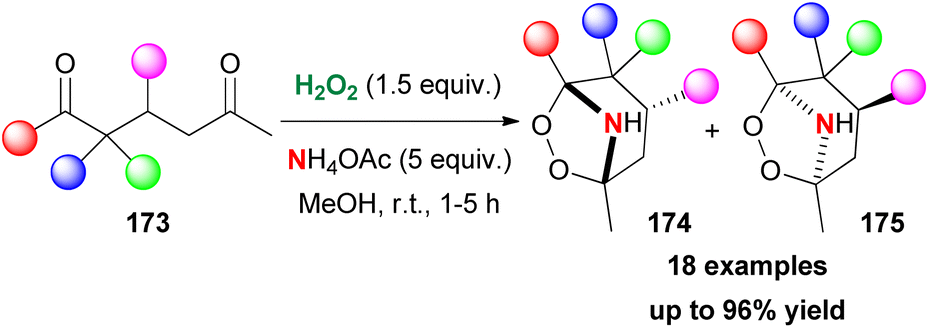
Having developed a series of contributions towards the synthesis of organic peroxides, Alabugin and Terent'ev turned their attention to implementing their protocols in the synthesis of azaperoxides, a family of compounds whose synthesis was unexplored at the time.92 For this purpose, 1,5-diketones as the starting materials, hydrogen peroxide and a nitrogen group source were employed. Contrary to their previous studies, in this case, the presence of a Lewis acid was not necessary. 1,5-Diketone 173, in the presence of hydrogen peroxide and 22% aqueous NH3 in MeOH, led to the formation of two azaozonides 174 (kinetically controlled product) and 175 (thermodynamically controlled product) as shown in Scheme 40. Ammonium acetate was opted as the nitrogen source. A variety of substrates was tested with excellent results. But, the presence of an ester group led to the formation of a dihydropyridine ring, due to the enolization that takes place initially. The authors performed experiments, excluding the possible formation of azaozonides from the reaction of the ammonium salt with the ozonides. Based on computational studies, the authors proposed a mechanistic scenario involving initial addition of the NH2 group to the carbonyl group. A series of condensation steps led to the formation of the desired product. With these results in hand, they also investigated the access to more complex molecules. Employing β,β′-triketones as the starting materials, the authors reported a protocol en route to bridged tricyclic azaperoxides.93 Despite the structural complexity, the formed products showed excellent stability. The main mechanistic proposal involves the addition of hydrogen peroxide and the formation of peroxy-hemiacetals or peroxy-hemiaminals.
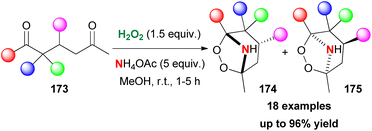 |
| | Scheme 40 Synthesis of azaozonides. | |
Activation of hydrogen peroxide by various organic molecules
Since hydrogen peroxide is considered as a green oxidant, many researchers turned their attention to alternative ways for its activation. For this purpose, a vast variety of organic molecules has been employed, targeting the activation of hydrogen peroxide. Julia, Colonna and coworkers pioneered the field reporting a series of publications employing poly-amino acids, activating chalcones via hydrogen bond formation promoting asymmetric epoxidation.94 In the context of this review, we will not discuss the activation of hydrogen peroxide employing pyrazinium flavinium salts or artificial flavin systems, since these two categories are already discussed in an elegant review article by Cibulka.95
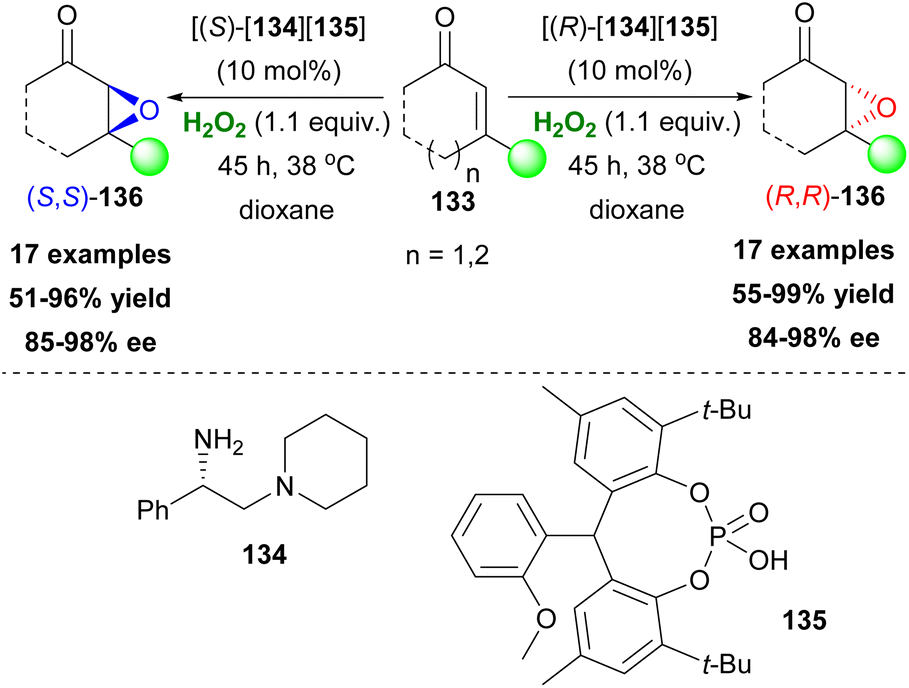
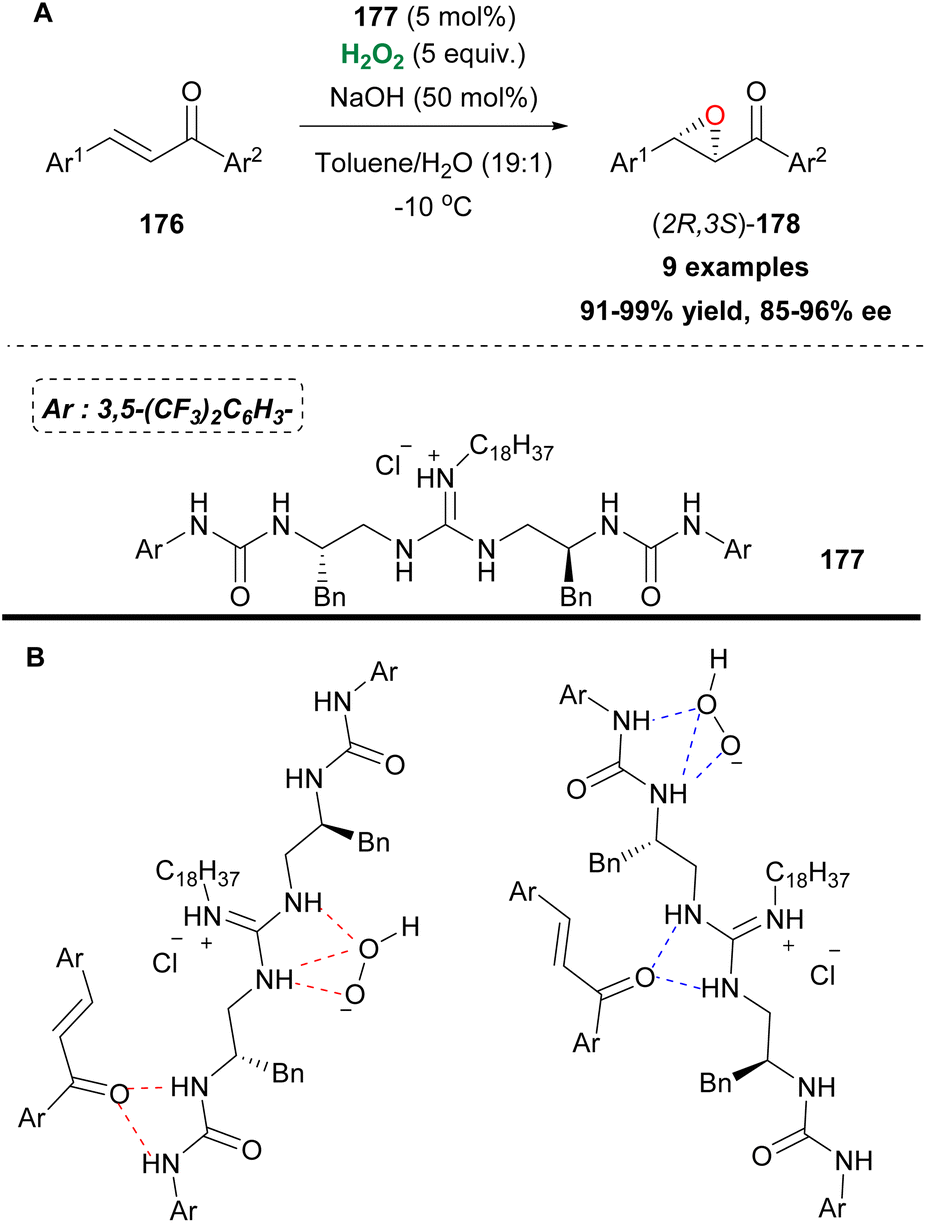
Small organic molecules can activate hydrogen peroxide via the formation of hydrogen bonds, enhancing its electrophilic nature. Guanidine-based catalysts have been explored in asymmetric epoxidations presenting mediocre results. Nagasawa and coworkers designed a bifunctional catalyst, combining the guanidine motif with a urea functional group (Scheme 41). Since urea is known to activate hydrogen peroxide and based on the ability of both functional groups, attached to the catalyst, to interact with either hydrogen peroxide or chalcones via hydrogen bonding, they developed an elegant protocol for the asymmetric epoxidation of 1,3-diarylenones 176 (Scheme 41A and B).96 The authors identified compound 177, decorated with a long alkyl chain on the guanidine group, to improve the catalytic activity in a biphasic system, as the catalyst of choice (Scheme 41A). The 3,5-bistrifluoromethyl groups attached to the phenyl group proved to have a positive impact, regarding the enantioselectivity of the oxidation process. Additionally, the catalyst can be easily reused without any significant loss of the catalytic activity. A variety of substituted chalcones were tested, affording excellent yields and high enantioselectivities. Recently, another bifunctional urea catalyst was introduced for the synthesis of chiral glycidic ester derivatives, with a quinidine scaffold, providing high yields and enantioselectivities, due to its bulky t-butyl group.97
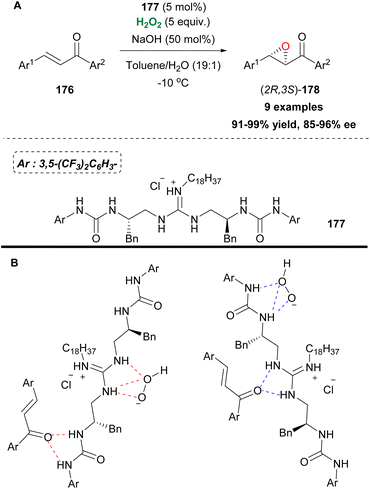 |
| | Scheme 41 (A) Epoxidation employing bifunctional catalyst 177. (B) Hydrogen bond formation leading to the desired product. | |
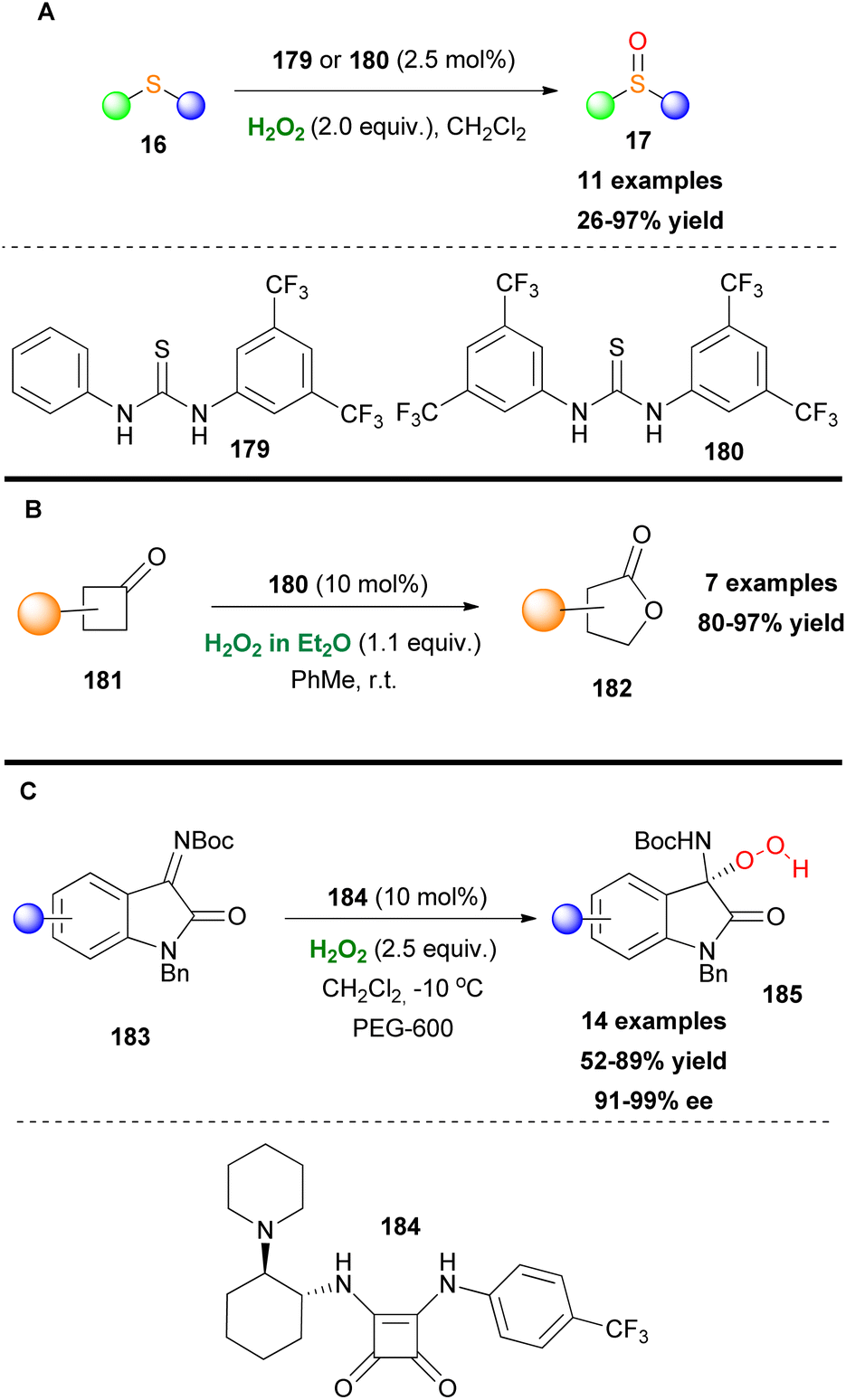
In the same line with Nagasawa, Cai and coworkers designed recyclable thiourea organocatalysts, decorated with 3,5-bisfluoromethyl groups, and employed them towards the chemoselective oxidation of sulfides (Scheme 42A).98 The novel thiourea-based catalysts demonstrated excellent functional group tolerance and were reused up to five times without any significant loss of reactivity. Another interesting use of thiourea-based catalysts was reported by Kotsuki and coworkers, in 2012.99 Based on hydrogen bond formation between the thiourea-based catalyst, hydrogen peroxide and the cyclobutanone scaffold, the authors developed a novel efficient Baeyer–Villiger oxidation protocol of cyclobutanones 181. Despite this elegant procedure which is taking place under mild reaction conditions the authors did not study the possible enantioselective pathway (Scheme 42B). Another well-known class of compounds with excellent catalytic activities are squaramides. The nitrogen-substituted cyclobutenodione core resembles the active center of the urea catalyst. Liu and Li used an acid–base bifunctional chiral squaramide 184 as the chiral organocatalyst to enhance the nucleophilicity of H2O2 (Scheme 42C).100 Initially, these hydroperoxide products were unstable in the reaction mixture and prone to dimerization. The authors speculated that a crowding environment of hydrogen bonding provided by an additive could stabilize and facilitate their isolation. Polyethylene glycols (PEGs) specifically PEG-600 served that purpose. A study of the substrate scope showed great tolerance of this method when various functional groups were present on the aryl ring and replacement of the Boc group with the 1-adamantyloxycarbonyl group (Adoc). Noteworthy is the first enantioselective 1,2-hydroperoxidation of isatin-derived ketimines 183.
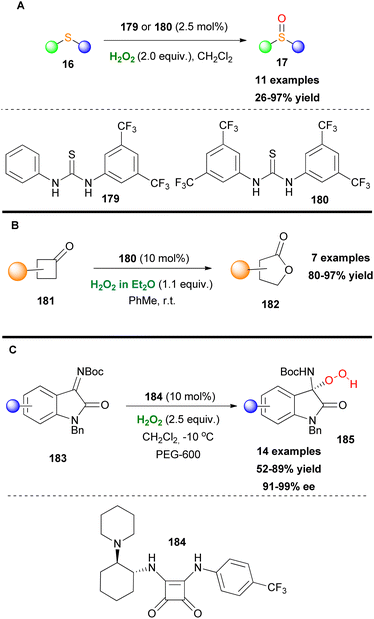 |
| | Scheme 42 (A) Sulfide oxidation induced by thiourea-based catalysts. (B) Baeyer–Villiger oxidation of cyclobutanones. (C) Organocatalytic activation of hydrogen peroxide by squaramides. | |
Another category of organic molecules that can easily activate hydrogen peroxides through hydrogen bond formation is perfluorinated alcohols. The strong electron-withdrawing properties of the fluorine atom, combined with the ability of the hydroxyl group to form hydrogen bonds lead to an electrophilic activation of hydrogen peroxide. Based on this idea, Neumann and coworkers exploited 2,2,2-trifluoroethanol (TFE) and 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) to promote the epoxidation of alkenes.101 A series of cycloalkenes and terminal alkenes were tested, affording the corresponding epoxides in good to excellent yields. In the case of a tetrasubstituted alkene, the epoxidation proceeded sufficiently, but due to the acidic conditions, ring opening followed by pinacol metathesis led to the formation of a ketone by-product. This protocol was furthermore applied successfully, towards the sulfide oxidation. 1,1,1,3,3,3-Hexafluoroisopropanol (HFIP) was also used by Bonnet-Delpon and coworkers to activate urea-hydrogen peroxide via hydrogen bonding in a catalytic epoxidation reaction.102 The major concern derived from the use of perfluorinated solvents is their high toxicity. This problem led many scientists to find ways to reduce their use. Towards this direction, Berkessel and coworkers designed a series of dendritic fluorinated catalysts, aiming at reducing the amount of the solvent, and maintaining the local concentration of the fluoroalcohol in order to promote the oxidation reaction.103 The authors prepared two hyperbranched polyglycerol-based HFIP derivatives, where the active fluoroalcohol group was determined to be 2.9 and 3.0 mmol per gram of catalyst. Both catalysts promoted the epoxidation reaction sufficiently using only 20 mol%. Open chain alkenes afforded moderate yields. The main advantage, derived from the use of those catalysts, appeared to be their potential recovery for multiple uses. Additionally, the authors employed the catalyst in the oxidation of thioanisole to the corresponding sulfoxide without the formation of sulfone. Despite this interesting finding, the scope of sulfides remained unexplored.
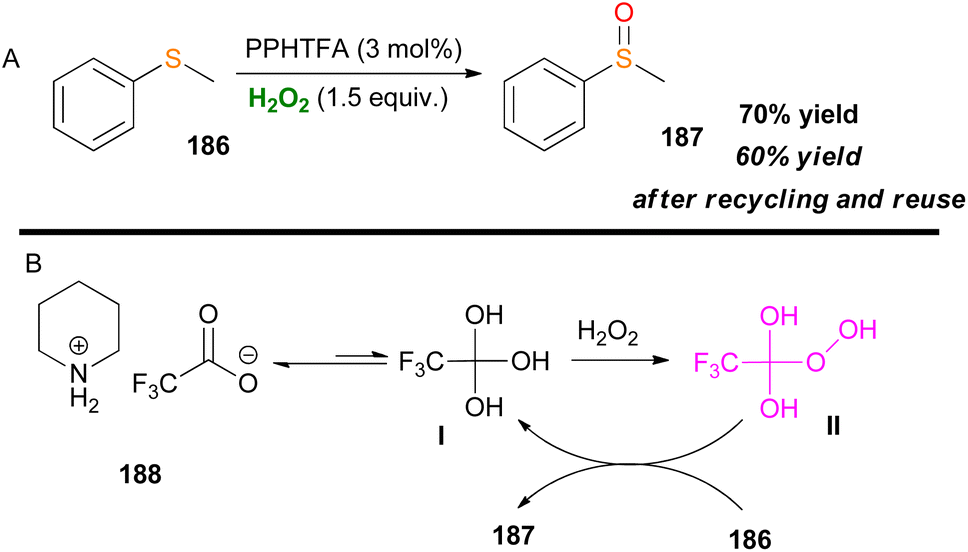
An interesting approach is the use of piperidinium trifluoroacetate (PPHTFA) ionic liquid as a catalytic activator of H2O2 for the selective oxidation of thioanisole 186, from Alinezhad and coworkers (Scheme 43).104 After a series of mechanistic experiments, the proposed active species was a trifluoro hydroperoxy species II (Scheme 43B). Recycling and reuse of the ionic liquid gave a desirable outcome with total chemoselectivity to sulfoxide 187. Higher temperatures gave room for overoxidation, which was also verified by DFT calculations.
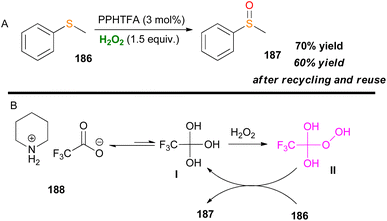 |
| | Scheme 43 (A) Activation of H2O2 by an ionic liquid catalyst. (B) Proposed reaction mechanism. | |
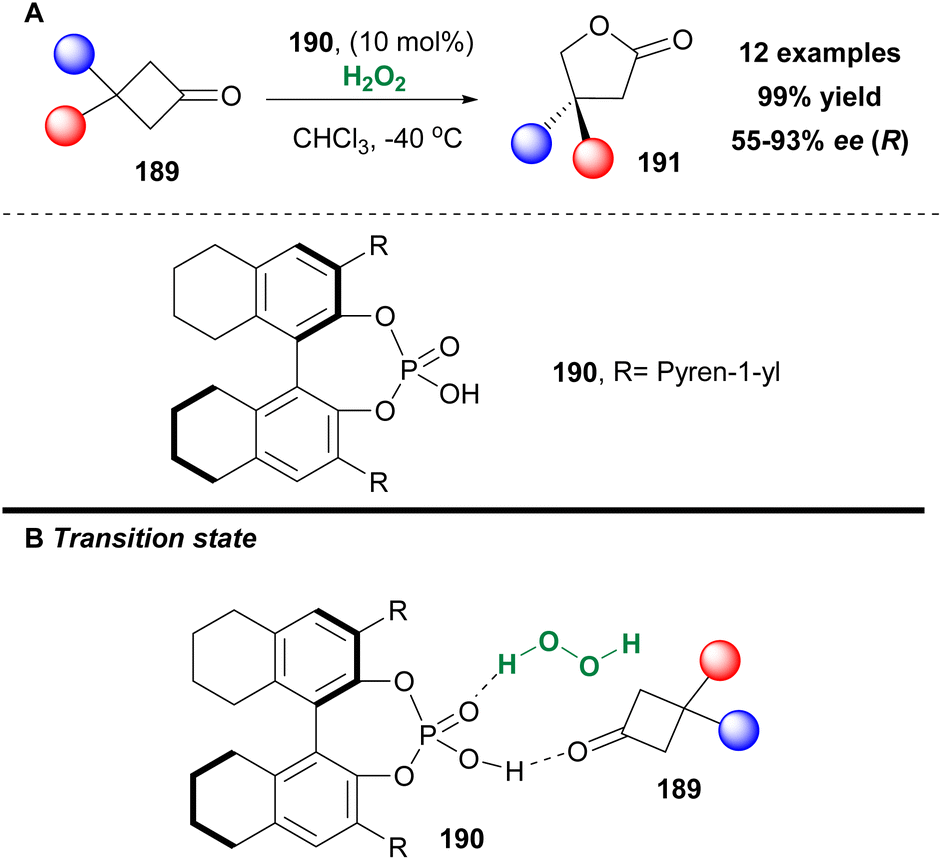
A well-known family of compounds that are able to form hydrogen bonds and likewise might serve as potential activators of hydrogen peroxide are chiral phosphoric acids. Ding and coworkers were the first to introduce their use in an enantioselective Baeyer–Villiger oxidation in excellent yield and high enantioselectivity contrary to what was reported in the literature.105 In their effort to optimize their preliminary results, they tested a variety of BINOL- and octahydroBINOL-derivatives to achieve higher enantioselectivities (Scheme 44). Chiral phosphoric acid 190 afforded the highest enantioselectivity, along with the highest yield. It must be mentioned that when the catalyst was washed with 4 N HCl and water prior to use, its catalytic activity was enhanced. The authors examined the good scope of 3,3-bissubstituted cyclobutanones 189, which afforded the corresponding lactones 191 in excellent yields and up to 93% ee (Scheme 44).106 The authors managed to showcase the capability of Brønsted acids to induce an asymmetric Baeyer–Villiger oxidation, though without providing any mechanistic details at the time. Two years later, the authors revisited the mechanism of the Baeyer–Villiger oxidation of 3-substituted cyclobutanones 189 and performed a series of mechanistic studies.107 Their major conclusion derived from these studies was that the chiral phosphoric acid is capable of activating both the substrate, by increasing its electrophilicity, and hydrogen peroxide, by increasing its nucleophilicity, through the formation of hydrogen bonds (Scheme 44B).
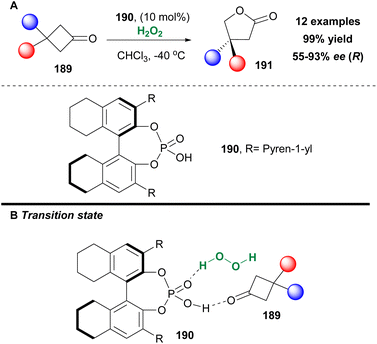 |
| | Scheme 44 (A) Baeyer–Villiger oxidation employing catalyst 190 by Ding and coworkers. (B) Proposed transition state. | |
Having established an elegant oxidation protocol, they expanded its use in a variety of bicyclic and tricyclic cyclobutanone derivatives, providing access to the synthesis of complex ester derivatives.108 These ground-breaking contributions by Ding and coworkers found excellent application in the synthesis of prostaglandins. Inspired by Ding's work, Chen, Peng and coworkers employed the asymmetric organocatalytic Baeyer–Villiger oxidation to synthesize a generic lactone intermediate, which served as a versatile building block for the synthesis of prostaglandin derivatives in high enantiomeric purity.109
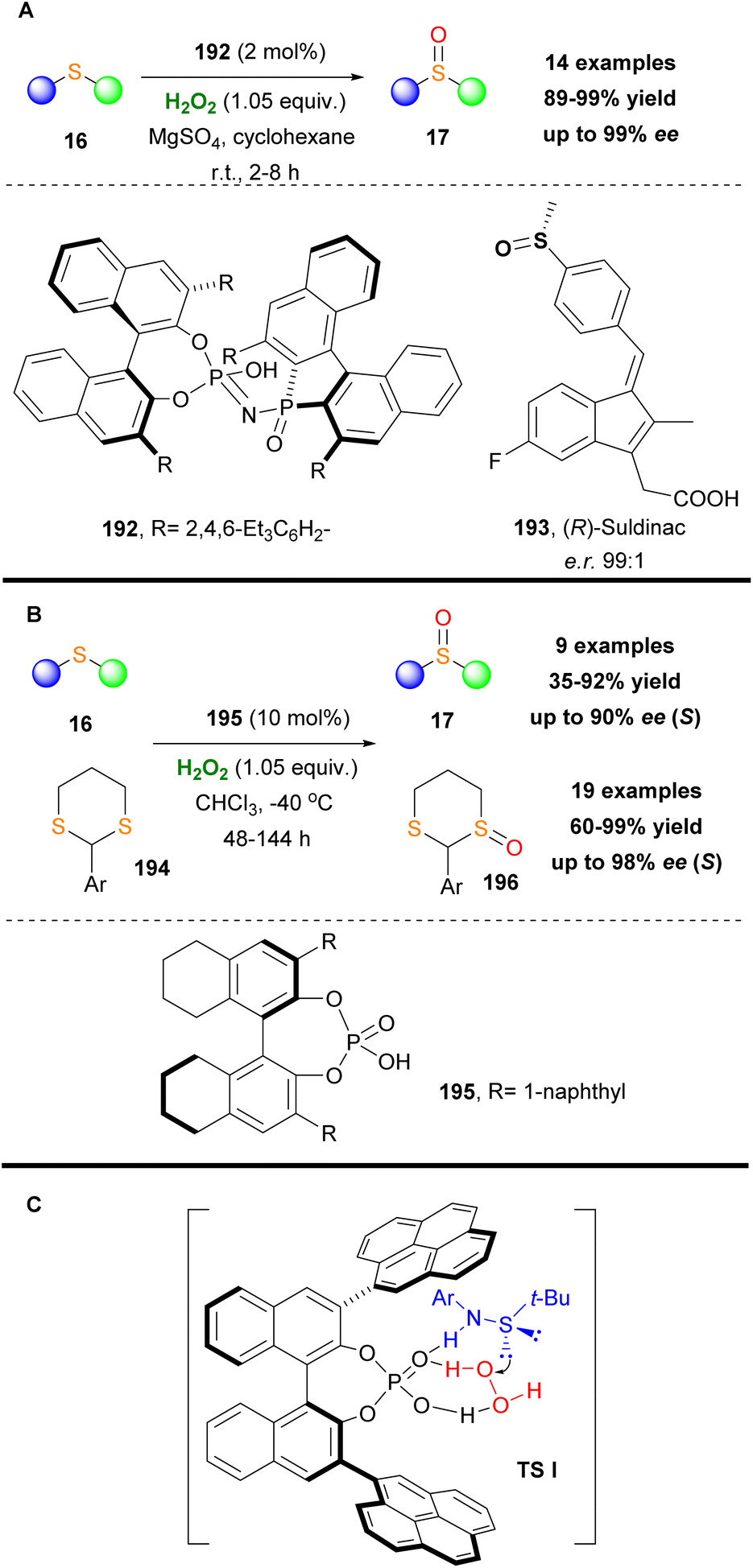
Another useful application of chiral phosphoric acids was reported by List and coworkers.110 Acknowledging the great importance of chiral sulfoxides, they successfully utilize chiral phosphoric acid 192 in a highly enantioselective oxidative protocol, converting sulfides 16 into the corresponding sulfoxides 17 (Scheme 45A).110 It is worth mentioning that the use of MgSO4 to remove water from the reaction mixture led to significantly shortened reaction times. The enantioselective protocol was successfully applied to a variety of sulfides displaying excellent functional group tolerance. Additionally, it must be mentioned that the overoxidized sulfone byproduct was detected in low yields. The authors tested their enantioselective protocol in real-life applications synthesizing successfully, on a 4 g scale, in high yields and enantioselectivity the nonsteroidal anti-inflammatory drug Suldinac 193 (Scheme 45A).
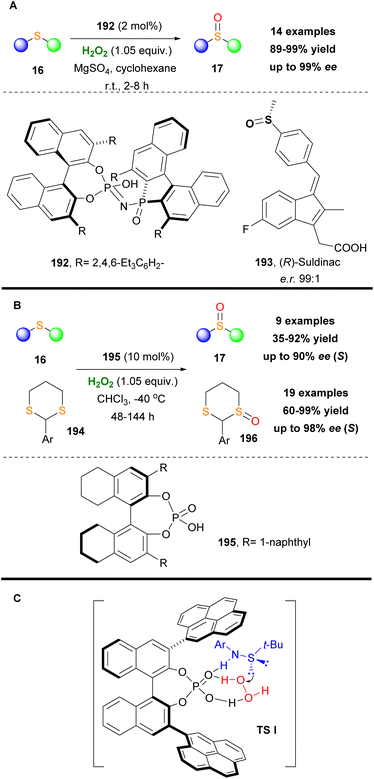 |
| | Scheme 45 (A) Sulfide oxidation by List and coworkers. (B) Sulfide and dithiane oxidation using catalyst 195. (C) Oxidation transition state. | |
Almost at the same period, Wang and coworkers reported a similar protocol, where chiral phosphoric acid 195 was found to be the optimum catalyst. In their contribution, the authors enriched the substrate scope, where apart from the use of sulfides, they applied their methodology towards the oxidation of 1,3-dithianes. Despite the fact that the oxidative protocol suffered from prolonged reaction times (up to 144 h), it was characterized by high yields and high enantioselectivities (Scheme 45B).111
Seeking more sulfur-containing molecules that can be easily oxidized, Tang and coworkers, in 2019, reported the asymmetric oxidation of sulfenamides, employing hydrogen peroxide as the oxidant.112 The corresponding chiral sulfonamides are useful intermediates, leading to the synthesis of chiral sulfoxides and sulfinamides in high yields. The mechanistic scenario involves the formation of the transition state as depicted in Scheme 45C, where based on the formation of hydrogen bonding, the oxidation proceeds in high enantioselectivity. Similar to List's approach, also in this study, the use of MgSO4 was an essential feature, since its dehydration activity does not allow water to destroy hydrogen bonding that is formed in the transition state.
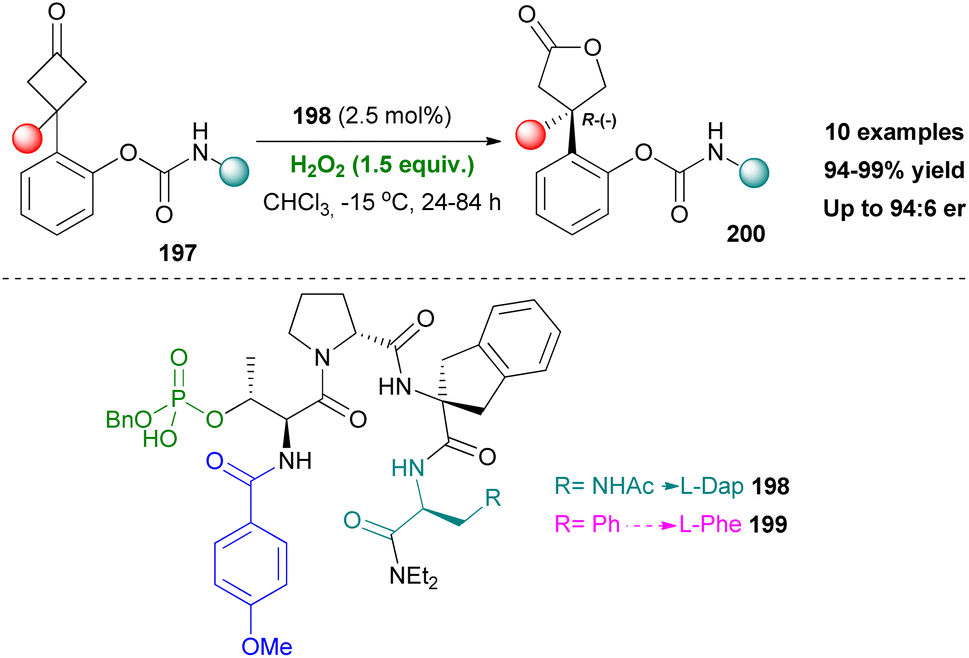
In 2019, Miller's group established a new asymmetric Baeyer–Villiger oxidation method.113 This time, however, the active group of the proposed peptide catalyst would be a pThr moiety instead of an Asp, introducing a different functionalisation for peptide catalysts. The authors began their investigation using unfunctionalized cyclobutanones, while screening various pThr-embedded peptides adopting β-turns. Initially, enantiomeric excess was not observed. Believing that the absence of an H-directing group in the substrate was behind this problem, carbamate functionality was once again added. This turned out to be half the solution to the problem, since the decision to incorporate L-Dap, as a side chain director in the i + 3 position, had a dramatic effect. Moreover, meddling on the N-terminus part, led to introduction of the electron-rich p-MeO-benzamide group, thus creating the optimal peptide catalyst 198 (Scheme 46). Catalyst 198 provided high enantioselectivity and yields, on ortho-carbamate substrates 197. An interesting stereodivergence was observed when the position of the carbamate group of the substrate changed to meta- and para-. While the ortho-position provided the (R)-(−) absolute configuration, the other two favored the (S)-(+) configuration, which was validated by X-ray analysis. Noteworthy is the fact that changing the i + 3 position to Phe led to inversion of the absolute configuration in meta- and para-analogues ((R)-(−)). The authors concluded that while both 198 and 199 have the same conformation, 198 permits H-bonding interaction and 199 engages in steric repulsive interactions. This can very well mean that this protocol is mainly substrate-oriented, rather than catalyst-oriented.
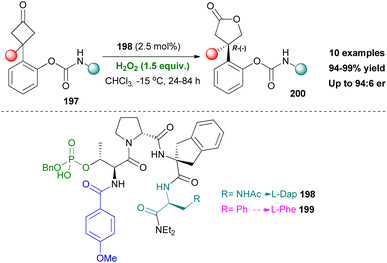 |
| | Scheme 46 pThr-embedded peptide catalysts for asymmetric Bayer–Villiger oxidation. | |
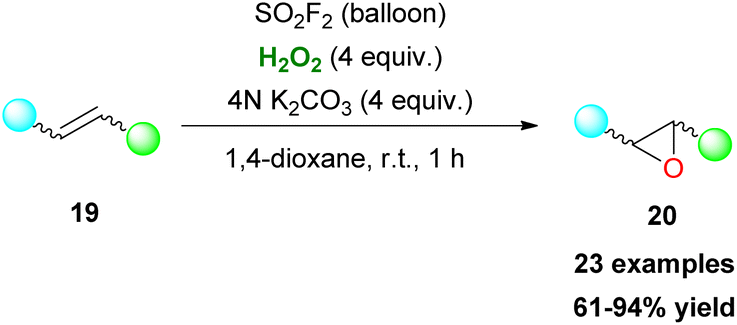
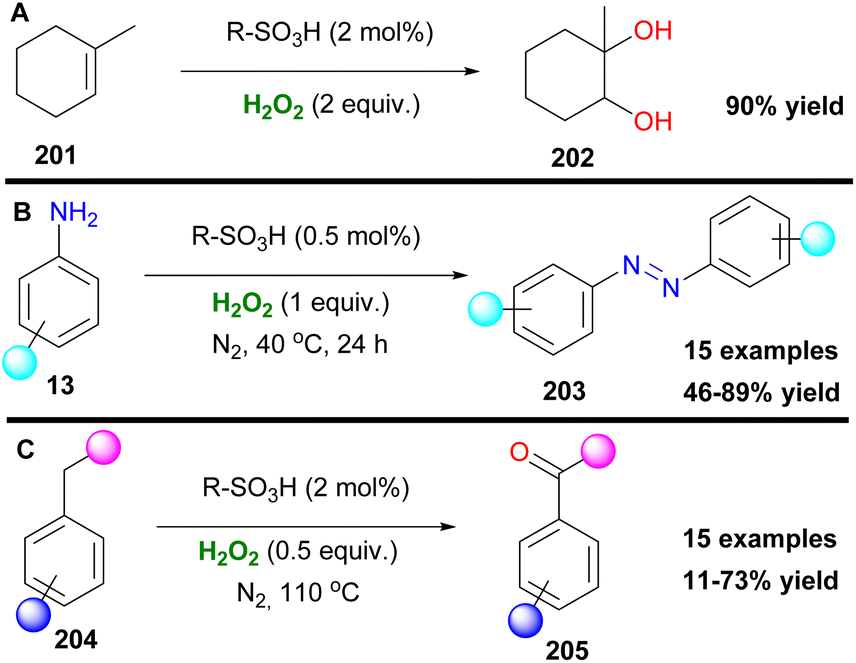
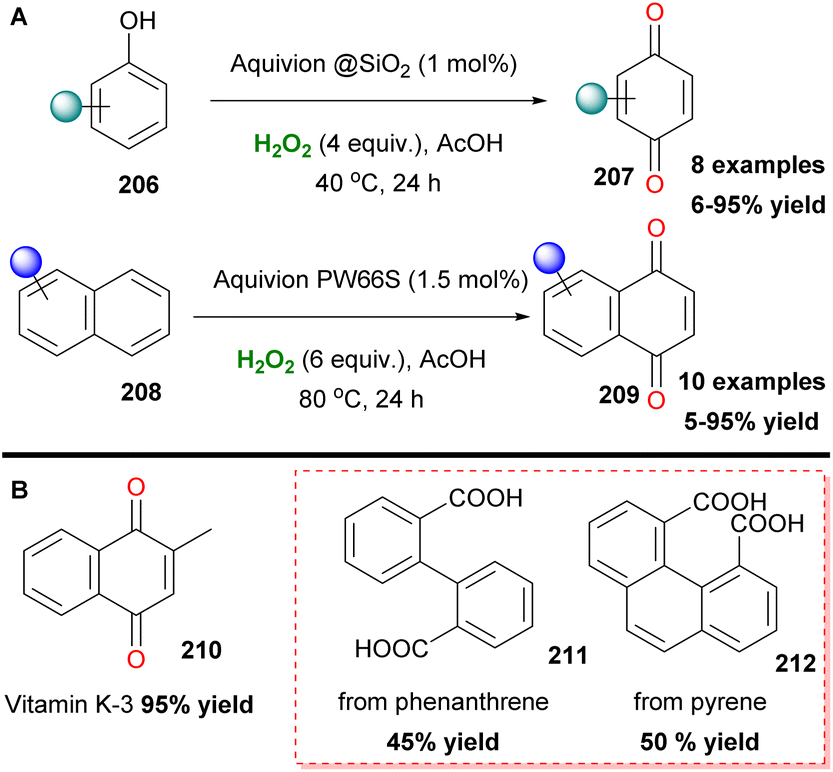
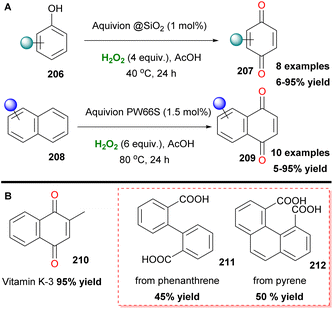
Organosulfonic peracids, a family of unstable but potent oxidants, can be formed in situ from H2O2 as depicted by Schulz and Kim for epoxidation reactions.114 However, harsh reaction conditions and expensive or explosive reagents were required in these cases. In an effort to address these issues, Yan, Lin and coworkers proposed the use of the SO2F2/K2CO3/H2O2 system for a cheap and mild activation of hydrogen peroxide.115 This method works well in a variety of double bonds and has the potential for upscaling (Scheme 47). In the past few years, Maggi and coworkers have been interested in the use of various silica-supported sulfonic acid catalysts for mild oxidative transformations (Scheme 48) such as dihydroxylation of 1-methylcyclohexene 201 to diol 202 (Scheme 48A),116 dimerization of anilines 13 to azobenzenes 203 (Scheme 48B)117 and selective Csp3 oxidation of benzylic methylenes 204 to aryl carbonyls 205 (Scheme 48C),118 under neat reaction conditions, thus minimizing waste production. In their latest work, however, the authors employed a catalyst derived from Aquivinion, a commercially available polymer with sulfonic groups in side chains of a perfluorinated scaffold. The presence of a polar, water-soluble solvent was imperative for the effective dearomatisation of phenols 206 to quinones 207 (Scheme 49A).119 In order to incorporate naphthalenes 208 and some polycyclic arenes in this method, re-optimisation of the reaction conditions led to an unsupported polymeric sulfonic acid as the catalyst and an increase of the oxidant and temperature (Scheme 49A). Vitamin K3210 was successfully retrieved almost quantitively from 2-methylnaphthalene (Scheme 49B). Moreover, electron-poor substrates were not suitable in any case, while phenanthrene and pyrene were oxidatively cleaved giving 211 and 212, respectively (Scheme 49B).
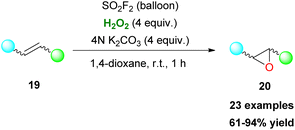 |
| | Scheme 47 SO2F2-mediated epoxidation of double bonds. | |
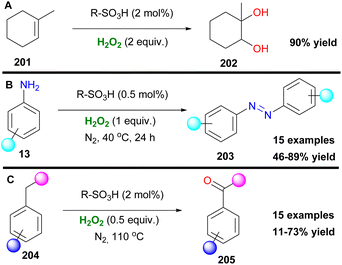 |
| | Scheme 48 Various oxidative transformations with sulfonic acids. (A) Dihydroxylation. (B) Synthesis of azobenzenes. (C) Oxidation to ketones. | |
 |
| | Scheme 49 (A) Dearomatisation of phenols and polycylic arenes and (B) selected examples. | |
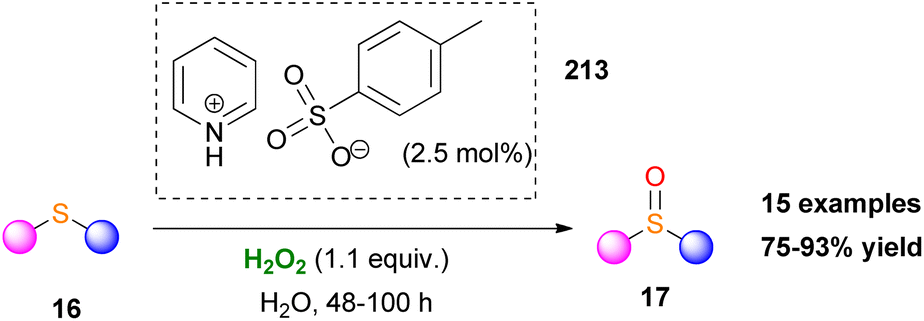
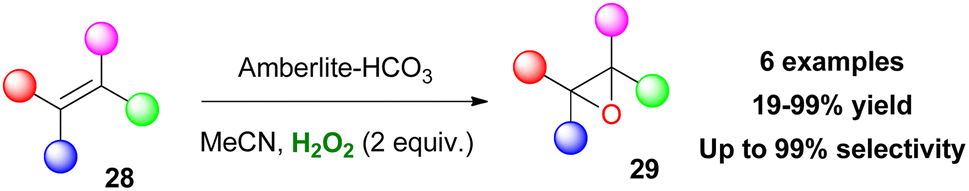
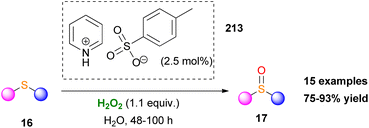
In another study, sulfonic acids have been applied as counteranions in metal-free ammonium and pyridinium salts.120 Employing catalyst 213, various aromatic and aliphatic sulfides were selectively oxidized to sulfoxides in high yields. However, long reaction times were needed and any attempt to induce enantioselectivity was fruitless (Scheme 50). Quaternary ammonium bicarbonate catalysts have also been utilised for epoxidation reactions. Mielby and Kegnæs applied an easily accessible and recyclable heterogenous Amberlite-bicarbonate catalyst that provided promising epoxidation yields and selectivities (Scheme 51).121 It was suggested that epoxidation possibly occurs by a peroxycarboximidic acid intermediate, since acetamide was detected as a by-product, along with a bicarbonate-activation of H2O2 as Yao and Richardson proposed.122
 |
| | Scheme 50 Air stable pyridinium salt catalyst for sulfoxidation. | |
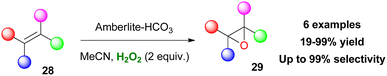 |
| | Scheme 51 Application of quaternary ammonium bicarbonate catalysts in olefin epoxidation. | |
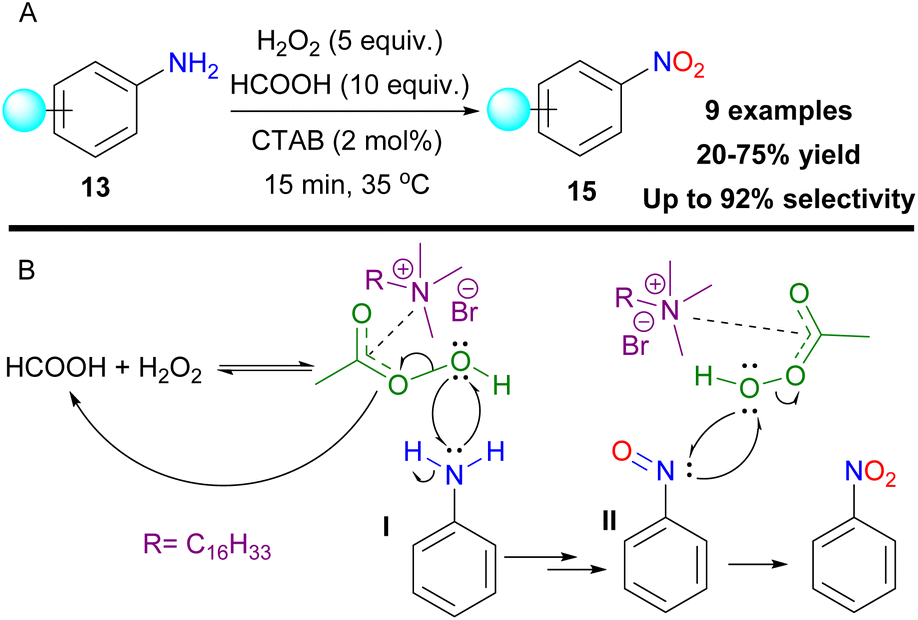
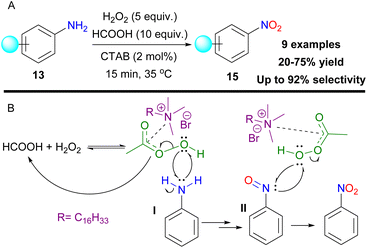
In an effort to selectively oxidize anilines 13 to nitro compounds 15, Birada and colleagues used formic acid and hydrogen peroxide to generate performic acid as the active oxidant (Scheme 52A).123 To maintain the desired selectivity, the authors used cetyltrimethylammonium bromide (CTAB), a cationic surfactant, that stabilizes aniline I and the nitroso intermediate II, by inducing radical pathways (Scheme 52B) and short reaction times. 4-Nitro and halogen-substituted anilines could not maintain desirable selectivity. The authors also concluded that the aromatic ring is mandatory to activate the oxidation.
 |
| | Scheme 52 (A) Aniline oxidation by in situ generated performic acid and (B) proposed reaction mechanism. | |
Conclusions
The progress in the field of organocatalysis achieved from 2000 until today has been one of the major landmarks in organic chemistry and offered unique opportunities to researchers in academia and industry. The use of small organic molecules that can activate either the substrate or the reagents, promoting fundamental organic transformations was highly appreciated. Organocatalysis offers an alternative and more sustainable approach, replacing metal-based catalysts, developing environmentally benign procedures, and reducing the overall cost of the synthetic route. Since hydrogen peroxide and its derivatives are considered as green oxidants, organocatalysis found excellent application in oxidative organic transformation. From the literature illustrated in this review, asymmetric epoxidation, Baeyer–Villiger oxidation, oxidation of sulfides, amines, silanes etc. were thoroughly investigated by the use of a variety of organocatalysts, which were able to activate hydrogen peroxide. In particular the field of peptide-based catalysts provides numerous different approaches in the area of asymmetric oxidation and will continue to do so in the future. Few examples of C–H activation–oxidation have been reported, but this field remained unexplored. We strongly believe that this field is still promising and future efforts will target the induction of enantioselectivity. The rational design of the catalysts offered the possibility of asymmetric induction. The oxidation of double bonds was thoroughly investigated, applying a variety of organocatalysts. Despite the plethora of contributions in the literature, there is still space for novel organocatalytic (asymmetric) epoxidation of unfunctionalized alkenes. Furthermore, there are applications, where the organocatalysts are embedded in a solid support, providing the opportunity of its reuse or recycling. Overall, the area of organocatalysed oxidation with green oxidants, like hydrogen peroxide, is demonstrating rapid progress in both quantitative and qualitative respects. The future for the development of organocatalytic protocols activating hydrogen peroxide seems to be bright.
Author contributions
E. T. P., P. L. G. and I. T. prepared the first draft of the manuscript and C. G. K. performed the final editing. The manuscript was written through contributions of all authors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge the Hellenic Foundation for Research and Innovation (HFRI) for financial support through a grant, which is financed by 1st Call for H.F.R.I. Research Projects to Support Faculty Members & Researchers and the procurement of high-cost research equipment grant (grant number 655).
Notes and references
- S. Stebbins, The World's 15 Top Selling Drugs, http://247wallst.com/special-report/2016/04/26/top-selling-drugs-in-the-world/2/, accessed July 29, 2017.
- K. A. Ahrendt, C. J. Borths and D. W. C. MacMillan, New strategies for organic catalysis: the first highly enantioselective organocatalytic Diels-Alder reaction, J. Am. Chem. Soc., 2000, 122, 4243–4244 CrossRef CAS.
- B. List, R. A. Lerner and C. F. Barbas, Proline-catalyzed direct asymmetric aldol reaction, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS.
- L. J. Thenard, Observations sur des combinaisons nouvelles entre l'oxigene et divers acides, Ann. Chim. Phys., 1818, 8, 306–313 Search PubMed.
- I. Triandafillidi, D. I. Tzaras and C. G. Kokotos, Green organocatalytic oxidative methods using activated ketones, ChemCatChem, 2018, 10, 2521–2535 CrossRef CAS.
-
R. A. Sheldon and J. K. Kochi, Metal-catalyzed oxidations of organic compounds, Academic Press, New York, 1981 Search PubMed.
-
(a) S. E. Denmark, Z. Wu, C. M. Crudden and H. Matsuhashi, Catalytic epoxidation of alkenes with oxone. 2. Fluoro ketones, J. Org. Chem., 1997, 62, 8288–8289 CrossRef CAS PubMed;
(b) S. E. Denmark and H. Matsuhashi, Chiral fluoro ketones for catalytic asymmetric epoxidation of alkenes with oxone, J. Org. Chem., 2002, 67, 3479–3486 CrossRef CAS PubMed.
- D. Yang, Ketone-catalyzed asymmetric epoxidation reactions, Acc. Chem. Res., 2004, 37, 497–505 CrossRef CAS.
-
(a) Z.-X. Wang, Y. Tu, M. Frohn, J.-R. Zhang and Y. Shi, An efficient catalytic asymmetric epoxidation method, J. Am. Chem. Soc., 1997, 119, 11224–11235 CrossRef CAS;
(b) H. Tian, X. She, L. Shu, H. Yu and Y. Shi, Highly enantioselective epoxidation of cis-olefins by chiral dioxirane, J. Am. Chem. Soc., 2000, 122, 11551–11552 CrossRef CAS.
- D. Limnios and C. G. Kokotos, Organocatalytic oxidation of organosilanes to silanols, ACS Catal., 2013, 3, 2239–2243 CrossRef CAS.
- A. T. Kelly and A. K. Franz, Metal-free synthesis of 1,3-disiloxanediols and aryl siloxanols, ACS Omega, 2019, 4, 6295–6300 CrossRef CAS PubMed.
- D. Limnios and C. G. Kokotos, 2,2,2-Trifluoroacetophenone as an organocatalyst for the oxidation of tertiary amines and azines to N-oxides, Chem.–Eur. J., 2014, 20, 559–563 CrossRef CAS PubMed.
- E. Voutyritsa, A. Theodorou, M. G. Kokotou and C. G. Kokotos, Organocatalytic oxidation of substituted anilines to azoxybenzenes and nitro compounds: mechanistic studies excluding the involvement of a dioxirane intermediate, Green Chem., 2017, 19, 1291–1298 RSC.
- G. B. Payne, P. H. Deming and P. H. Williams, Reactions of hydrogen peroxide. VII. Alkali-catalyzed epoxidation and oxidation using a nitrile as co-reactant, J. Org. Chem., 1961, 26, 659–663 CrossRef CAS.
- E. Voutyritsa, I. Triandafillidi and C. G. Kokotos, Green organocatalytic oxidation of sulfides to sulfoxides and sulfones, Synthesis, 2017, 49, 917–924 CAS.
- For a review, see: Y. Shi, Organocatalytic asymmetric epoxidation of olefins by chiral ketones, Acc. Chem. Res., 2004, 37, 488–496 CrossRef CAS PubMed.
- T. Katsuki and K. B. Sharpless, The first practical method for asymmetric epoxidation, J. Am. Chem. Soc., 1980, 102, 5974–5976 CrossRef CAS.
- R. Irie, K. Noda, Y. Ito, N. Matsumoto and T. Katsuki, Catalytic asymmetric epoxidation of unfunctionalized olefins, Tetrahedron Lett., 1990, 31, 7345–7348 CrossRef CAS.
- W. Zhang, J. L. Loebach, S. R. Wilson and E. N. Jacobsen, Enantioselective epoxidation of unfunctionalized olefins catalyzed by sale manganese complexes, J. Am. Chem. Soc., 1990, 112, 2801–2803 CrossRef CAS.
- D. Limnios and C. G. Kokotos, 2,2,2-Trifluoroacetophenone: an organocatalyst for an environmentally friendly epoxidation of alkenes, J. Org. Chem., 2014, 79, 4270–4276 CrossRef CAS.
- E. Voutyritsa, A. Theodorou and C. G. Kokotos, Green organocatalytic α-hydroxylation of ketones, Org. Biomol. Chem., 2016, 14, 5708–5713 RSC.
-
(a) G. M. Rubottom, M. A. Vazquez and D. R. Pelegrina, Peracid oxidation of trimethylsilyl enol ethers: a facile α-hydroxylation procedure, Tetrahedron Lett., 1974, 15, 4319–4322 CrossRef;
(b) G. M. Rubottom and J. M. Gruber,
m-Chloroperbenzoic acid oxidation of 2-trimethylsilyloxy-1,3-dienes. Synthesis of α-hydroxy and α-acetoxy enones, J. Org. Chem., 1978, 43, 1599–1602 CrossRef CAS.
- I. Tyablikov and B. Romanovsky, A heterogeneous organocatalyst for olefin epoxidation, Catal. Today, 2016, 278, 40–44 CrossRef CAS.
- W.-Q. Yuan, S.-Q. Zhou, Y.-Y. Jiang, H.-H. Li and H.-D. Zheng, Organocatalyzed styrene epoxidation accelerated by continuous-flow reactor, J. Flow Chem., 2020, 10, 227–234 CrossRef CAS.
- Y. Wu, Z. Chen, F. Wang, J. Xu and Y. Wang, Efficient organocatalytic synthesis of styrene oxide from styrene and its kinetic study in a continuous-flow microreaction system, Chem. Eng. Sci., 2022, 247, 117045 CrossRef CAS.
- I. Triandafillidi, M. G. Kokotou, D. Lotter, C. Sparr and C. G. Kokotos, Aldehyde-catalyzed epoxidation of unactivated alkenes with aqueous hydrogen peroxide, Chem. Sci., 2021, 12, 10191–10196 RSC.
- T. Yamada, T. Takai, O. Rhode and T. Mukaiyama, Highly efficient method for epoxidation of olefins with molecular oxygen and aldehydes catalyzed by nickel(II) complexes, Chem. Lett., 1991, 20, 1–4 CrossRef.
- For mechanistic studies on Mukaiyama epoxidation, see:
(a) K. R. Lassila, F. J. Waller, S. E. Werkheiser and A. L. Wressell, Aldehyde/olefin cooxidations: parallel epoxidation pathways and concerted decomposition of the peroxyacyl-olefin adduct, Tetrahedron Lett., 1994, 35, 8077–8080 CrossRef CAS;
(b) W. Nam, H. J. Kim, S. H. Kim, R. Y. N. Ho and J. S. Valentine, Metal complex-catalyzed epoxidation of olefins by dioxygen with co-oxidation of aldehydes. A mechanistic study, Inorg. Chem., 1996, 35, 1045–1049 CrossRef CAS PubMed;
(c) B. B. Wentzel, P. A. Gosling, M. C. Feiters and R. J. M. Nolte, Mechanistic studies on the epoxidation of alkenes with molecular oxygen and aldehydes catalysed by transition metal-β-diketonate complexes, J. Chem. Soc., Dalton Trans., 1998, 13, 2241–2246 RSC;
(d) B. B. Wentzel, P. L. Alsters, M. C. Feiters and R. J. M. Nolte, Mechanistic studies on the Mukaiyama epoxidation, J. Org. Chem., 2004, 69, 3453–3464 CrossRef CAS PubMed.
- D. K. Romney and S. J. Miller, A Peptide-embedded trifluoromethyl ketone catalyst for enantioselective epoxidation, Org. Lett., 2012, 14, 1138–1141 CrossRef CAS.
- A. L. Featherston and S. J. Miller, Synthesis and evaluation of phenylalanine-derived trifluoromethylketones for peptide-based oxidation catalysis, Bioorg. Med. Chem., 2016, 24, 4871–4874 CrossRef PubMed.
- I. Triandafillidi, I. K. Sideri, D. I. Tzaras, N. Spiliopoulou and C. G. Kokotos, Green organocatalytic synthesis of dihydrobenzofurans by oxidation-cyclization of allylphenols, Synthesis, 2017, 49, 4254–4260 CrossRef CAS.
- I. Triandafillidi and C. G. Kokotos, Green organocatalytic synthesis of isoxazolines via a one-pot oxidation of allyloximes, Org. Lett., 2017, 19, 106–109 CrossRef CAS PubMed.
- I. Triandafillidi, M. Raftopoulou, A. Savvidou and C. G. Kokotos, Organocatalytic synthesis of lactones via an oxidation of alkenoic-acids, ChemCatChem, 2017, 9, 4120–4124 CrossRef CAS.
- A. Theodorou and C. G. Kokotos, Green organocatalytic synthesis of indolines and pyrrolidines from alkenes, Adv. Synth. Catal., 2017, 359, 1577–1581 CrossRef CAS.
- A. Theodorou, I. Triandafillidi and C. G. Kokotos, Green organocatalytic dihydroxylation of alkenes, Eur. J. Org Chem., 2017, 11, 1502–1509 CrossRef.
- A. Theodorou and C. G. Kokotos, Organocatalytic synthesis of polysubstituted tetrahydrofurans from alkenes, Green Chem., 2017, 19, 670–674 RSC.
- A. Tsoukaki, E. Skolia, I. Triandafillidi and C. G. Kokotos, Organocatalytic synthesis of thiiranes from alkenes, Eur. J. Org Chem., 2022, e202200463 CrossRef CAS.
- A. Theodorou, I. Triandafillidi and C. G. Kokotos, Organocatalytic synthesis of oxazolines and dihydrooxazines from allyl-amides: bypassing the inherent regioselectivity of the cyclization, Adv. Synth. Catal., 2018, 360, 951–957 CrossRef CAS.
- A. Theodorou, D. Limnios and C. G. Kokotos, One-pot synthesis of O-allylhydroxylamines through the organocatalytic oxidation of tertiary allylic amines followed by a [2,3]-Meisenheimer rearrangement, Chem.–Eur. J., 2015, 21, 5238–5241 CrossRef CAS.
- C. J. Pierce and M. K. Hilinski, Chemoselective hydroxylation of aliphatic sp3 C-H bonds using a ketone catalyst and aqueous H2O2, Org. Lett., 2014, 16, 6504–6507 CrossRef CAS PubMed.
- For a recent review, see: A. J. Metrano, A. J. Chinn, C. R. Shugrue, E. A. Stone, B. Kim and S. J. Miller, Asymmetric catalysis mediated by synthetic peptides, version 2.0: expansion of scope and mechanisms, Chem. Rev., 2020, 120, 11479–11615 CrossRef CAS PubMed.
- A. Berkessel, B. Koch, C. Toniolo, M. Rainaldi, Q. B. Broxterman and B. Kaptein, Asymmetric enone epoxidation by short solid-phase bound peptides: further evidence for catalyst helicity and catalytic activity of individual peptides stands, Biopolymers, 2006, 84, 90–96 CrossRef CAS PubMed.
- G. Peris, C. E. Jakobsche and S. J. Miller, Aspartate-catalyzed asymmetric epoxidation reactions, J. Am. Chem. Soc., 2007, 129, 8710–8711 CrossRef CAS PubMed.
- C. E. Jakobsche, G. Peris and S. J. Miller, Functional analysis of an aspartate-based epoxidation catalyst with amide-to-alkene peptidomimetic catalyst analogues, Angew. Chem., Int. Ed., 2008, 47, 6707 CrossRef CAS PubMed.
- For initial introduction on split and pool synthesis and OBOC applications, see:
(a) A. Furka, F. Sebestyen, M. Asgedom and G. Dibo, General method for rapid synthesis of multicomponent peptide mixtures, Int. J. Pept. Protein Res., 1991, 37, 487–493 CrossRef CAS PubMed;
(b) K. S. Lam, S. E. Salmon, E. M. Hersh, V. J. Hruby, W. M. Kazmierski and R. J. Knapp, A new type of synthetic peptide library for identifying ligand-binding activity, Nature, 1991, 354, 82–84 CrossRef CAS;
(c) K. S. Lam, M. Lebl and V. Krchnák, The “one-bead-one-compound” combinatorial library method, Chem. Rev., 1997, 97, 411–448 CrossRef CAS PubMed.
- P. A. Lichtor and S. J. Miller, One-bead-one-catalyst approach to aspartic acid-based oxidation catalyst discovery, ACS Comb. Sci., 2011, 13, 321–326 CrossRef CAS PubMed.
- P. A. Lichtor and S. J. Miller, Combinatorial evolution of site- and enantioselective catalysts for polyene epoxidation, Nat. Chem., 2012, 4, 990–995 CrossRef CAS PubMed.
- P. A. Lichtor and S. J. Miller, Experimental lineage and functional analysis of a remotely directed peptide epoxidation catalyst, J. Am. Chem. Soc., 2014, 136, 5301–5308 CrossRef CAS PubMed.
- N. C. Abascal, P. A. Lichtor, M. W. Giuliano and S. J. Miller, Function-oriented investigations of a peptide-based catalyst that mediates enantioselective allylic alcohol epoxidation, Chem. Sci., 2014, 5, 4504–4511 RSC.
- T. Nobuta and T. Kawabata, Catalyst-controlled site-selective asymmetric epoxidation of nerylamine and geranylamine derivatives, Chem. Commun., 2017, 53, 9320–9323 RSC.
- For a review, see: M. Uyanik and K. Ishihara, Baeyer-Villiger oxidation using hydrogen peroxide, ACS Catal., 2013, 3, 513–520 CrossRef CAS.
- G. Peris and S. J. Miller, A nonenzymatic acid/peracid catalytic cycle for the Baeyer-Villiger oxidation, Org. Lett., 2008, 10, 3049–3052 CrossRef CAS PubMed.
- D. K. Romney, S. M. Colvin and S. J. Miller, Catalyst control over regio- and enantioselectivity in Baeyer-Villiger oxidations of functionalized ketones, J. Am. Chem. Soc., 2014, 136, 14019–14022 CrossRef CAS PubMed.
- N. C. Abascal and S. J. Miller, Solution structures and molecular associations of a peptide-based catalyst for the stereoselective Baeyer-Villiger oxidation, Org. Lett., 2016, 18, 4646–4649 CrossRef CAS PubMed.
- M. W. Giuliano, C.-Y. Lin, D. K. Romney, S. J. Miller and E. V. Anslyn, A synergistic combinatorial and chiroptical study of peptide catalysts for asymmetric Baeyer-Villiger oxidation, Adv. Synth. Catal., 2015, 357, 2301–2309 CrossRef CAS PubMed.
-
(a) L. You, J. S. Berman and E. V. Anslyn, Dynamic multi-component covalent assembly for the reversible binding of secondary alcohols and chirality sensing, Nat. Chem., 2011, 3, 943–948 CrossRef CAS PubMed;
(b) L. You, G. Pescitelli, E. V. Anslyn and L. Di Bari, An exciton-coupled circular dichroism protocol for the determination of identity, chirality, and enantiomeric excess of chiral secondary alcohols, J. Am. Chem. Soc., 2012, 134, 7117–7125 CrossRef CAS PubMed;
(c) L. You, J. S. Berman, A. Lucksanawichien and E. V. Anslyn, Correlating sterics parameters and diastereomeric ratio values for a multicomponent assembly to predict exciton-coupled circular dichroism intensity and thereby enantiomeric excess of chiral secondary alcohols, J. Am. Chem. Soc., 2012, 134, 7126–7134 CrossRef CAS PubMed.
- J. S. Alford, N. C. Abascal, C. R. Shugrue, S. M. Colvin, D. K. Romney and S. J. Miller, Aspartyl oxidation catalysts that dial in functional group selectivity, along with regio- and stereoselectivity, ACS Cent. Sci., 2016, 2, 733–739 CrossRef CAS PubMed.
-
(a) L. Whitesell, E. G. Mimnaugh, B. De Costa, C. E. Myers and L. M. Neckers, Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 8324–8328 CrossRef CAS PubMed;
(b) L. Li, L. Wang, Q.-D. You and X.-L. Xu, Heat shock protein 90 inhibitors: an update on achievements, challenges, and future directions, J. Med. Chem., 2020, 63, 1798–1822 CrossRef CAS PubMed;
(c) J. G. Supko, R. L. Hickman, M. R. Grever and L. Malspeis, Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent, Cancer Chemother. Pharmacol., 1995, 36, 305–315 CrossRef CAS PubMed.
- M. J. Hilton, C. M. Brackett, B. Q. Mercado, B. S. J. Blagg and S. J. Miller, Catalysis-enabled access to cryptic geldanamycin oxides, ACS Cent. Sci., 2020, 6, 426–435 CrossRef CAS PubMed.
- F. Kolundzic, M. N. Noshi, M. Tjandra, M. Movassaghi and S. J. Miller, Chemoselective and enantioselective oxidation of indoles employing aspartyl peptide catalysts, J. Am. Chem. Soc., 2011, 133, 9104–9111 CrossRef CAS PubMed.
- S.-Y. Hsieh, Y. Tang, S. Crotti, E. A. Stone and S. J. Miller, Catalytic enantioselective pyridine N-oxidation, J. Am. Chem. Soc., 2019, 141, 18624–18629 CrossRef CAS.
- Y. Tang and S. J. Miller, Catalytic enantioselective synthesis of pyridyl sulfoximines, J. Am. Chem. Soc., 2021, 143, 9230–9235 CrossRef CAS.
- S. E. Huth, E. A. Stone, S. Crotti and S. J. Miller, On the ability of the N-O bond to support a stable stereogenic axis: peptide-catalyzed atroposelective N-oxidation, J. Org. Chem., 2023, 88, 12857–12862 CrossRef CAS PubMed.
- P. Milliet, A. Picot and X. Lusinchi, Formation d'un sel d'oxaziridinium quaternaire par methylation d’un oxaziranne-mise en evidence de ses proprieties oxydantes, Tetrahedron Lett., 1976, 17, 1573–1576 CrossRef.
- P. C. B. Page, P. Parker, G. A. Rassias, B. R. Buckley and D. Bethell, Iminium salt-catalysed asymmetric epoxidation using hydrogen peroxide as stoichiometric oxidant, Adv. Synth. Catal., 2008, 350, 1867–1874 CrossRef CAS.
- For contributions on the use of diselenide as the co-catalyst, see:
(a) S. Santoro, C. Santi, M. Sabatini, L. Testaferri and M. Tiecco, Eco-friendly olefin dihydroxylation catalyzed by diphenyl diselenide, Adv. Synth. Catal., 2008, 350, 2881–2884 CrossRef CAS;
(b) B. H. Brodsky and J. Du Bois, Oxaziridine-mediated catalytic hydroxylation of unactivated 3° C-H bonds using hydrogen peroxide, J. Am. Chem. Soc., 2005, 127, 15391–15393 CrossRef CAS PubMed.
- B. R. Buckley, C. E. Elliott, Y. Chan, N. Dreyfus and P. C. B. Page, Activation of hydrogen peroxide by diphenyl diselenide for highly enantioselective oxaziridinium salt mediated catalytic asymmetric epoxidation, Synlett, 2013, 24, 2266 CrossRef CAS.
- P. C. B. Page, B. R. Buckley, C. Elliott, Y. Chana, N. Dreyfus and F. Marken, Chemoselective oxidation of sulfides to sulfoxides with urea-hydrogen peroxide complex catalysed by diselenide, Synlett, 2016, 27, 80–82 CrossRef.
- M. Marigo, J. Franzen, T. B. Poulsen, W. Zhuang and K. A. Jorgensen, Asymmetric organocatalytic epoxidation of α,β-unsaturated aldehydes with hydrogen peroxide, J. Am. Chem. Soc., 2005, 127, 6964–6965 CrossRef CAS.
- S. Lee and D. W. C. MacMillan, Enantioselective organocatalytic epoxidation using hypervalent iodine reagents, Tetrahedron, 2006, 62, 11413–11424 CrossRef CAS.
- For selected examples, see:
(a) G.-L. Zhao, I. Ibrahem, H. Sunden and A. Cordova, Amine-catalyzed asymmetric epoxidation of α,β-unsaturated aldehydes, Adv. Synth. Catal., 2007, 349, 1210–1224 CrossRef CAS;
(b) B. P. Bondzic, T. Urushima, H. Ishikawa and Y. Hayashi, Asymmetric epoxidation of α-substituted acroleins catalyzed by diphenylprolinol silyl ether, Org. Lett., 2010, 12, 5434–5437 CrossRef CAS PubMed.
- X. Wang and B. List, Asymmetric counteranion-directed catalysis for the epoxidation of enals, Angew. Chem., Int. Ed., 2008, 47, 1119–1121 CrossRef CAS PubMed.
- C. M. Reisinger, X. Wang and B. List, Catalytic asymmetric hydroperoxidation of α,β-unsaturated ketones: an approach to enantiopure peroxyhemiketals, epoxides, and aldols, Angew. Chem., Int. Ed., 2008, 47, 8112–8115 CrossRef CAS.
- O. Lifchits, M. Mahlau, C. M. Reisinger, A. Lee, C. Farès, I. Polyak, G. Gopakumar, W. Thiel and B. List, The Cinchona primary amine-catalyzed asymmetric epoxidation and hydroperoxidation of α,β-unsaturated carbonyl compounds with hydrogen peroxide, J. Am. Chem. Soc., 2013, 135, 6677–6693 CrossRef CAS.
- K. Akagawa and K. Kudo, Asymmetric epoxidation of α,β-unsaturated aldehydes in aqueous media catalyzed by resin-supported peptide-containing unnatural amino acids, Adv. Synth. Catal., 2011, 353, 843–847 CrossRef CAS.
- X. Wang, C. M. Reisinger and B. List, Catalytic asymmetric epoxidation of cyclic enones, J. Am. Chem. Soc., 2008, 130, 6070–6071 CrossRef CAS.
- F. Scharinger, Á. M. Pálvölgyi, M. Weisz, M. Weil, C. Stanetty, M. Schnürch and K. Bica-Schröder, Sterically demanding flexible phosphoric acids for constructing efficient and multi-purpose asymmetric organocatalysts, Angew. Chem., Int. Ed., 2022, 61, e202202189 CrossRef CAS PubMed.
- D. Wang, W. G. Shuler, C. J. Pierce and M. K. Hilinski, An iminium salt organocatalyst for selective aliphatic C-H hydroxylation, Org. Lett., 2016, 18, 3826–3829 CrossRef CAS PubMed.
- P. C. B. Page, M. M. Farah, B. R. Buckley and A. J. Blacker, New chiral binaphthalene-derived iminium salt organocatalysts for asymmetric epoxidation of alkenes, J. Org. Chem., 2007, 72, 4424–4430 CrossRef CAS PubMed.
- P. L. Hahn, J. M. Lowe, Y. Xu, K. L. Burns and M. K. Hilinski, Amine organocatalysis of remote, chemoselective C(sp3)-H hydroxylation, ACS Catal., 2022, 12, 4302–4309 CrossRef CAS.
- M.-K. Wong, L.-M. Ho, Y.-S. Zheng, C.-Y. Ho and D. Yang, Asymmetric epoxidation of olefins catalyzed by chiral iminium salts generated in situ from amines and aldehydes, Org. Lett., 2001, 3, 2587–2590 CrossRef CAS PubMed.
- K. Gratzer and M. Waser, Investigations concerning the syntheses of TADDOL-derived secondary amines and their use to access novel chiral organocatalysts, Synthesis, 2012, 44, 3661–3670 CrossRef CAS PubMed.
- D. Uraguchi, R. Tsutsumi and T. Ooi, Catalystic asymmetric oxidation of N-sulfonyl imines with hydrogen peroxide – trichloroacetonitrile system, J. Am. Chem. Soc., 2013, 135, 8161–8164 CrossRef CAS.
- C. Mairhofer, J. Novacek and M. Waser, Synergistic ammonium (hypo)iodite/imine catalysis for the asymmetric α-hydroxylation of β-ketoesters, Org. Lett., 2020, 22, 6138–6142 CrossRef CAS PubMed.
- M. Uyanik, N. Sasakura, E. Kaneko, K. Ohori and K. Ishihara, Chiral ammonium hypoiodite-catalysed enantioselective oxidative dearomatization of 1-naphthols using hydrogen peroxide, Chem. Lett., 2015, 44, 179–181 CrossRef.
- M. Cai, K. Xu, Y. Li, Z. Nie, L. Zhang and S. Luo, Chiral primary amine/ketone cooperative catalysis for asymmetric α-hydroxylation with hydrogen peroxide, J. Am. Chem. Soc., 2021, 143, 1078–1087 CrossRef CAS PubMed.
-
https://www.nobelprize.org/nobel_prizes/medicine/laureates/2015/tu-facts.html, accessed October 30, 2023.
-
E. T. Denisov, T. G. Denisova and T. S. Pokidova, Handbook of free radical initiators, John Wiley and Sons, 2003 Search PubMed.
- I. A. Yaremenko, G. P. Gomes, P. S. Radulov, Y. Y. Belyakova, A. E. Vilikotskiy, V. A. Vil', A. A. Korlyukov, G. I. Nikishin, I. V. Alabugin and A. O. Terent'ev, Ozone-free synthesis of ozonides: assembling bicyclic structures from 1,5-diketones and hydrogen peroxide, J. Org. Chem., 2018, 83, 4402–4426 CrossRef CAS PubMed.
- S. C. Davidson, G. P. Gomes, L. R. Kuhn, I. V. Alabugin, A. R. Kennedy and N. C. O. Tomkinson, Organocatalytic sulfoxidation, Tetrahedron, 2021, 78, 131784–131794 CrossRef CAS.
- I. A. Yaremenko, P. S. Radulov, M. G. Medvedev, N. V. Krivoshchapov, Y. Y. Belyakova, A. A. Korlyukov, A. I. Ilovaisky, A. O. Terent'ev and I. V. Alabugin, How to build rigid oxygen-rich tricyclic heterocycles from triketones and hydrogen peroxide: control of dynamic covalent chemistry with inverse α-effect, J. Am. Chem. Soc., 2020, 142, 14588–14607 CrossRef CAS PubMed.
- I. A. Yaremenko, Y. Y. Belyakova, P. S. Radulov, R. A. Novikov, M. G. Medvedev, N. V. Krivoshchapov, A. A. Korlyukov, I. V. Alabugin and A. O. Terent'ev, Marriage of peroxides and nitrogen heterocycles: selective three-component assembly, peroxide-preserving rearrangement, and stereoelectronic source of unusual stability of bridged azaozonides, J. Am. Chem. Soc., 2021, 143, 6634–6648 CrossRef CAS PubMed.
- I. A. Yaremenko, Y. Y. Belyakova, P. S. Radulov, R. A. Novikov, M. G. Medvedev, N. V. Krivoshchapov, A. A. Korlyukov, I. V. Alabugin and A. O. Terent'ev, Inverse α-effect as the Ariadne's thread on the way to tricyclic aminoperoxides: avoiding thermodynamic traps in the labyrinth of possibilities, J. Am. Chem. Soc., 2022, 144, 7264–7282 CrossRef CAS PubMed.
- S. Julia, J. Guixer, J. Masana, J. Rocas, S. Colonna, R. Annuziata and H. Molinari, Synthetic enzymes. Part 2. Catalytic asymmetric epoxidation by means pf polyaminoacids in a triphase system, J. Chem. Soc., Perkin Trans. 1, 1982, 1317–1324 RSC.
- R. Cibulka, Artificial flavin systems for chemoselective and stereoselective oxidations, Eur. J. Org Chem., 2015, 915–932 CrossRef CAS.
- S. Tanaka and K. Nagasawa, Guanidine-urea bifunctional organocatalyst for asymmetric epoxidation of 1,3-diarylenones with hydrogen peroxide, Synlett, 2009, 4, 667–670 Search PubMed.
- N. Ji, Q. Tian, Q. Yang, M. Li and W. He, Chiral urea-catalyzed enantioselective epoxidation of α,β-unsaturated esters, Tetrahedron Lett., 2021, 68, 152909 CrossRef CAS.
- Y.-B. Huang, W.-B. Yi and C. Cai, A recyclable fluorous thiourea organocatalyst for the chemoselective oxidation of sulfides, J. Fluorine Chem., 2011, 132, 554–557 CrossRef CAS.
- N. Sasakura, K. Nakano, Y. Ichikawa and H. Kotsuki, A new environmentally friendly method for the Baeyer-Villiger oxidation of cyclobutanones catalyzed by thioureas using H2O2 as an oxidant, RSC Adv., 2012, 2, 6135–6139 RSC.
- W. Guo, Y. Liu and C. Li, Asymmetric catalytic 1,2-hydroperoxidation of isatin-derived ketimine with hydrogen peroxide in the crowding environment of PEGs, Org. Lett., 2017, 19, 1044–1047 CrossRef CAS PubMed.
- K. Neimann and R. Neumann, Electrophilic activation of hydrogen peroxide: selective oxidation reactions in perfluorinated alcohol solvents, Org. Lett., 2000, 2, 2861–2863 CrossRef CAS PubMed.
- J. Legros, B. Crousse, D. Bonnet-Delpon and J. P. Begue, Urea-hydrogen peroxide/hexafluoro-2-propanol: an efficient system for a catalytic epoxidation reaction without a metal, Eur. J. Org Chem., 2002, 3290–3293 CrossRef CAS.
- A. Berkessel, J. Krämer, F. Mummy, J.-M. Neudörfl and R. Haag, Dendritic fluoroalcohols as catalysts for alkene epoxidation with hydrogen peroxide, Angew. Chem., Int. Ed., 2013, 52, 739–743 CrossRef CAS PubMed.
- H. Alinezhad, M. Vafaeezadeh and M. M. Hashemi, Formation of a catalytically active intermediate for oxidation in H2O2/ionic liquid system: experimental and theoretical investigations, Res. Chem. Intermed., 2017, 43, 2615–2625 CrossRef CAS.
- A. Watanabe, T. Uchida, K. Ito and T. Katsuki, Highly enantioselective Baeyer-Villiger oxidation using Zr(salen) complex as catalyst, Tetrahedron Lett., 2002, 43, 4481–4485 CrossRef CAS.
- S. Xu, Z. Wang, X. Zhang, X. Zhang and K. Ding, Chiral Brønsted acid catalyzed asymmetric Baeyer-Villiger reaction of 3-substituted cyclobutanones by using aqueous H2O2, Angew. Chem., Int. Ed., 2008, 47, 2840–2843 CrossRef CAS PubMed.
- S. Xu, Z. Wang, Y. Li, X. Zhang, H. Wang and K. Ding, Mechanistic investigation of chiral phosphoric acid catalyzed asymmetric Baeyer-Villiger reaction of 3-substituted cyclobutanones with H2O2 as the oxidant, Chem.–Eur. J., 2010, 16, 3021–3035 CrossRef CAS.
- S. Xu, Z. Wang, X. Zhang and K. Ding, Asymmetric Baeyer-Villiger oxidation of 2,3- and 2,3,4-substituted cyclobutanones catalyzed by chiral phosphoric acids with aqueous H2O2 as the oxidant, Eur. J. Org Chem., 2011, 110–116 CrossRef CAS.
- K. Zhu, S. Hu, M. Liu, H. Peng and F.-E. Chen, Access to a key building block for the prostaglandin family via stereocontrolled organocatalytic Baeyer-Villiger oxidation, Angew. Chem., Int. Ed., 2019, 58, 9923–9927 CrossRef CAS PubMed.
- S. Liao, I. Čorić, Q. Wang and B. List, Activation of H2O2 by chiral confined Brønsted acids: a highly enantioselective catalytic sulfoxidation, J. Am. Chem. Soc., 2012, 134, 10765–10768 CrossRef CAS PubMed.
- Z.-M. Liu, H. Zhao, M.-Q. Li, Y.-B. Lan, Q.-B. Yao, J.-C. Tao and X.-W. Wang, Chiral phosphoric acid-catalyzed asymmetric oxidation of aryl alkyl sulfides and aldehyde-derived 1,3-dithianes: using aqueous hydrogen peroxide as the terminal oxidant, Adv. Synth. Catal., 2012, 354, 1012–1022 CrossRef CAS.
- L.-J. Ma, S.-S. Chen, G.-X. Li, J. Zhu, Q.-W. Wang and Z. Tang, Chiral Brønsted-acid-catalyzed asymmetric oxidation of sulfenamide by using H2O2: a versatile access to sulfinamide and sulfoxide with high enantioselectivity, ACS Catal., 2019, 9, 1525–1530 CrossRef CAS.
- A. L. Featherston, C. R. Shugrue, B. Q. Mercado and S. J. Miller, Phosphothreonine (pThr)-based multifunctional peptide catalysis for asymmetric Baeyer-Villiger oxidations of cyclobutanones, ACS Catal., 2019, 9, 242–252 CrossRef CAS PubMed.
-
(a) M. Schulz, R. Kluge and M. Lipke, Substitution of arylsulfonyl imidazolides by hydrogen peroxide: aryl sulfonic peracids as oxidants for olefins, Synlett, 1993, 915–918 CrossRef CAS;
(b) S. G. Yang, J. P. Hwang, M. Y. Park, K. Lee and Y. H. Kim, Highly efficient epoxidation of electron-deficient olefins with tetrabutylammonium peroxydisulfate, Tetrahedron, 2007, 63, 5184–5188 CrossRef CAS.
- C. Ai, F. Zhu, Y. Wang, Z. Yan and S. Lin, SO2F2-Mediated epoxidation of olefins with hydrogen peroxide, J. Org. Chem., 2019, 84, 11928–11934 CrossRef CAS PubMed.
- R. Maggi, G. Martra, C. G. Piscopo, G. Alberto and G. Sartori, Oxidation of alkenes to 1,2-diols: FT-IR and UV studies of silica-supported sulfonic acid catalysts and their interaction with H2O and H2O2, J. Catal., 2012, 294, 19–28 CrossRef CAS.
- E. Paris, F. Bigi, D. Cauzzi, R. Maggi and G. Maestri, Oxidative dimerization of anilines with heterogeneous sulfonic acid catalysts, Green Chem., 2018, 20, 382–386 RSC.
- E. Paris, C. Oldani, A. S. Aricò, C. D'Urso, F. Bigi, G. Maestri, F. Pancrazzi and R. Maggi, Silica nanoparticles decorated with polymeric sulfonic acids trigger selective oxidation of benzylic methylenes to aldehydic and ketonic carbonyls, ACS Sustain. Chem. Eng., 2019, 7, 5886–5891 CrossRef CAS.
- F. Pancrazzi, G. Maestri, R. Maggi and R. Viscardi, Oxidative dearomatization of phenols and polycyclic aromatics with hydrogen peroxide triggered by heterogeneous sulfonic acids, Eur. J. Org Chem., 2021, 5407–5414 CrossRef CAS.
- F. Secci, A. Frongia and P. P. Piras, Ammonium salt catalyzed oxidation of organosulfides to organosulfoxydes, Tetrahedron Lett., 2014, 55, 603–605 CrossRef CAS.
- J. Mielby and S. Kegnæs, Epoxidation of alkenes with aqueous hydrogen peroxide and quaternary ammonium bicarbonate catalysts, Catal. Lett., 2013, 143, 1162–1165 CrossRef CAS.
- H. Yao and D. E. Richardson, Epoxidation of alkenes with bicarbonate-activated hydrogen peroxide, J. Am. Chem. Soc., 2000, 122, 3220–3221 CrossRef CAS.
- K. Ravi, B. D. Bankar, S. Jindani and A. V. Biradar, Surfactant-assisted selective oxidation of aromatic amines to nitro compounds by in situ-formed performic acid, ACS Omega, 2019, 4, 9453–9457 CrossRef CAS PubMed.
Footnote |
| † Equal contribution. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Petros L.
Gkizis†
,
Petros L.
Gkizis†