
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceResponsive macrocyclic and supramolecular structures powered by platinum
Miguel A.
Soto
 *a and
Mark J.
MacLachlan
*abc
*a and
Mark J.
MacLachlan
*abc
aDepartment of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, British Columbia V6T 1Z1, Canada. E-mail: msoto@chem.ubc.ca; mmaclach@chem.ubc.ca
bQuantum Matter Institute, University of British Columbia, 2355 East Mall, Vancouver, British Columbia V6T 1Z4, Canada
cWPI Nano Life Science Institute, Kanazawa University, Kanazawa, 920-1192, Japan
First published on 11th December 2023
Abstract
Humankind's manipulation of platinum dates back more than two millennia to burial objects. Since then, its use has evolved from purely decorative purposes in jewelry to more functional applications such as in catalysts, pharmaceuticals, and bioimaging agents. Platinum offers a range of properties arguably unmatched by any other metal, including electroactivity, photoluminescence, chromic behaviour, catalysis, redox reactivity, photoreactivity, and stimuli-controlled intermetallic interactions. The vast body of knowledge generated by the exploration of these and other properties of platinum has recently merged with other areas of chemistry such as supramolecular and host–guest chemistry. This has shown us that platinum can incorporate its responsive character into supramolecular assemblies (e.g., macrocycles and polymers) to produce materials with tailorable functions and responses. In this Perspective Article, we cover some platinum-powered supramolecular structures reported by us and others, hoping to inspire new and exciting discoveries in the field.
Introduction
The potential of non-covalent interactions and reversible covalent bonds has been gradually uncovered since the seminal work of 1987 Nobel laureates Cram, Lehn, and Pedersen.1–4 Today, literature abounds with examples of craftily designed supramolecular structures and materials, in which dynamic bonds are key to enabling responsiveness.5–7 These systems undergo unique electronic, stereochemical, and constitutional changes upon physical and chemical stimulation. This is attractive to various fields of chemistry and biochemistry, as it can lead to systems with tailor-made functions.8–11Metal ions have become an important part of the available toolkit for designing these responsive supramolecular assemblies.12–14 The ever-increasing number of metal–organic cages, macrocycles, and polymers attests to this — not to mention the fascinating work of Sauvage (2016 Nobel Laureate)15 on the use of copper coordination complexes to power up molecular machines. Metal centers have become an essential component for supramolecular design, as they unlock a whole range of properties that are unrivaled by organic analogues. These include tunable luminescence,16 redox activity,17 magnetic susceptibility,18 anti-aromaticity,19 catalytic activity,20 and multi-responsiveness.21
In recent years, our group has focused on incorporating platinum into small molecules using cyclometalating ligands to elicit responsive behavior. This research contributes to the growing body of knowledge on Pt-containing supramolecular systems, alongside the foundational work of other groups. In most of the reported examples, the stimuli-responsiveness is harnessed either from the ligands (e.g., via photoisomerization) or from the metal center through metallophilic interactions.22–31 In this Perspective Article, we aim to go beyond these explored approaches, and discuss how, in our view, other platinum-centered responses may become crucial in the design and realization of new supramolecular assemblies. These include classic reactivity of platinum complexes such as redox and ligand exchange processes that may lead to changes in, for example, ligand distribution, oxidation state of the metal center, nuclearity of the complex, and stereochemistry (Fig. 1).
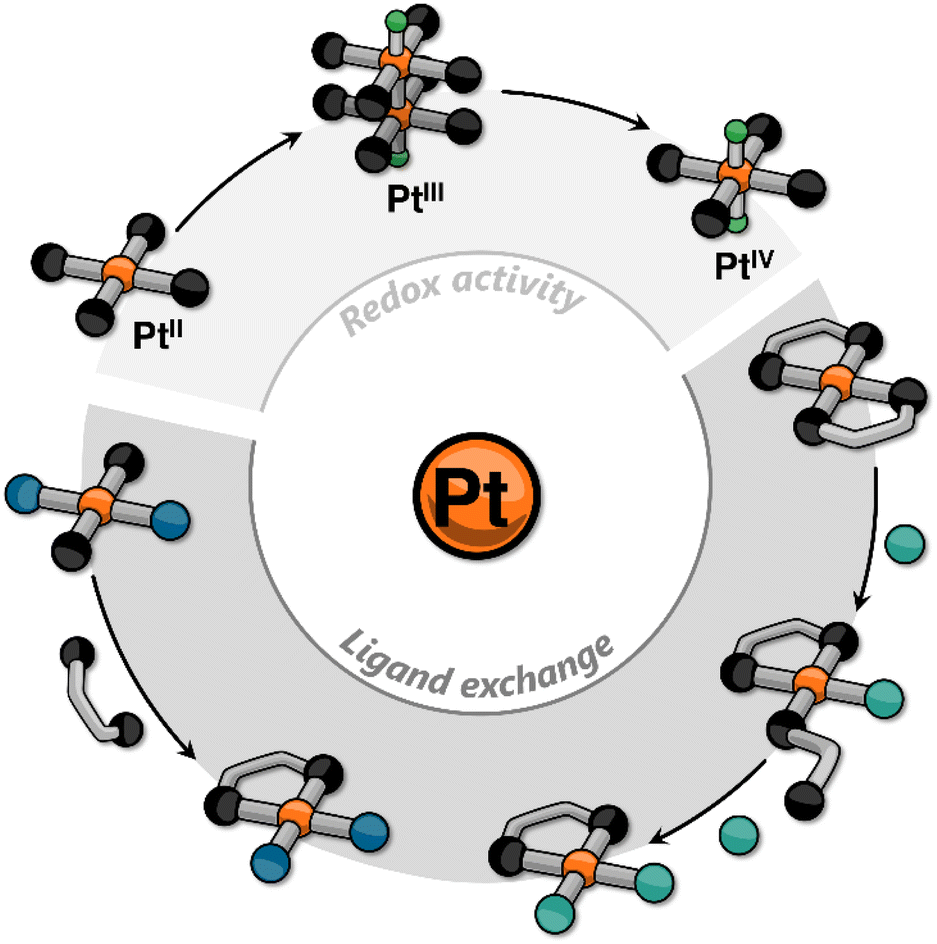 | ||
| Fig. 1 Cartoon representation of some platinum-centered responses that have been incorporated into supramolecular assemblies. | ||
Platinum in brief
Platinum (from the Spanish platina, meaning 'little silver') is a transition metal with a wide range of chemical properties, and its coordination chemistry has been extensively studied because of its importance in biological, medical, and industrial applications. Platinum-based materials are widely used in electronics and jewelry because of their high electrical conductivity and lustre.32–34 In the field of anti-cancer research, platinum complexes have revolutionized cancer treatment by providing a new class of drugs that act by binding to DNA, ultimately leading to cell death.35 Amazingly, more than 50% of the platinum metal we mine today goes into the catalytic converters in our cars.In the field of catalysis, platinum complexes have been extensively used as highly active and selective catalysts in various industrial processes.36 Current research efforts are focused on the design of novel platinum complexes with greater catalytic performance and stability, while also improving their recyclability. Platinum also finds applications in renewable energy, particularly in fuel cells, where it serves as a catalyst for hydrogen oxidation.37 Furthermore, platinum has applications in the field of light-emitting diodes, where it is critical to improving their efficiency and performance through advanced material and device design strategies.38
Platinum coordination chemistry
In coordination complexes, platinum is commonly found in +2 and +4 oxidation states. Platinum(II) complexes generally have square planar geometry, whilst platinum(IV) complexes are usually octahedral. The oxidation state of the Pt centre is crucial to the reactivity and electronic properties of the complexes. In addition to the most commonly observed PtII and PtIV, PtIII has attracted considerable attention in the past few decades as new methods have emerged to access PtIII complexes. In the +3 oxidation state, platinum shows diverse coordination geometries and unique reactivity. Platinum(III) complexes often have distorted octahedral or square planar structures, depending on the ligands involved. The ability to control the oxidation state of platinum allows for the design of materials with tailored properties and functions, expanding their use across various applications.39,40There are many approaches to assemble platinum coordination complexes, depending on the desired features. The use of monodentate ligands, for example, may lead to cis or trans arrangements, which can influence the electronic and steric properties of the resulting complex. Bidentate or polydentate ligands, on the other hand, produce a single (non-isomeric) complex that is stabilized by the chelate effect. Examples of such ligands include diamines, bis(phosphines), and 2,2′-bipyridine derivatives. Another approach is to use bridging ligands that can connect two or more platinum centres, leading to the formation of polynuclear complexes. Molecules equipped with carboxylic acids, imines, and pyridyl moieties are examples of the commonly used multidentate ligands. The obtained polynuclear complexes can show unique properties, such as cooperative binding, catalytic activity, and redox communication between the platinum centres.41
In addition to these approaches, recent studies have explored the use of supramolecular self-assembly to construct platinum-based materials with unique properties. For example, platinum ions can be incorporated into self-assembled organic nanostructures, such as micelles and vesicles, to provide additional functionality, including catalytic activity or drug delivery. The use of self-assembly allows for the construction of highly ordered and tunable materials with precise control over the size, shape, and composition of the resulting structures.42
The assembly of platinum coordination complexes is a versatile and highly customizable process that allows for the design of materials with bespoke architectures. By exploring different ligands, bridging groups, and self-assembly strategies, one can create platinum-based materials with intriguing properties for a wide range of applications.
Responsiveness powered by platinum
While fundamental studies of platinum have been a subject of research for many decades, the focus is increasingly shifting to exploiting the properties of platinum to construct responsive systems. Platinum is being used as a key element to impart responsiveness and advanced functionality to molecular architectures, creating opportunities for innovative applications in areas such as sensing, drug delivery, and smart materials. In the following sections, we discuss the advances made by our group and others in which responsiveness to competitive species (ligand exchange and insertion), and redox inputs are advantageous for the design and realization of dynamic supramolecular assemblies.Responsiveness to competitive species
In the early 2000s, the Lusby group recognized the importance of achieving switchable supramolecular assemblies via ligand exchange to advance the design of molecular ratchets and artificial molecular machines. They relied on platinum since greater thermodynamic biases can be introduced between two switchable states in comparison to their palladium counterparts.47–49Fig. 2a shows one of the systems studied by the Lusby group.50
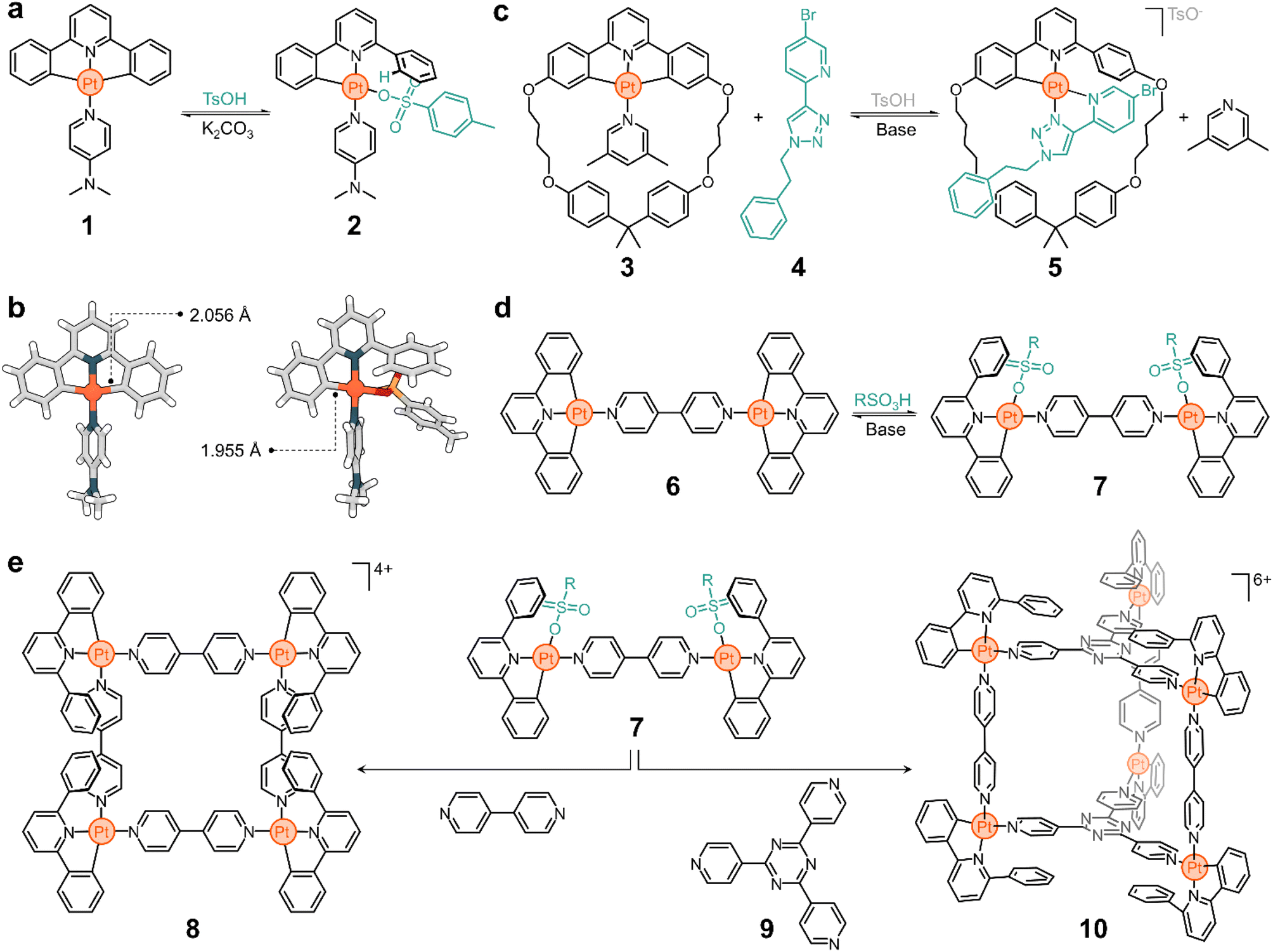 | ||
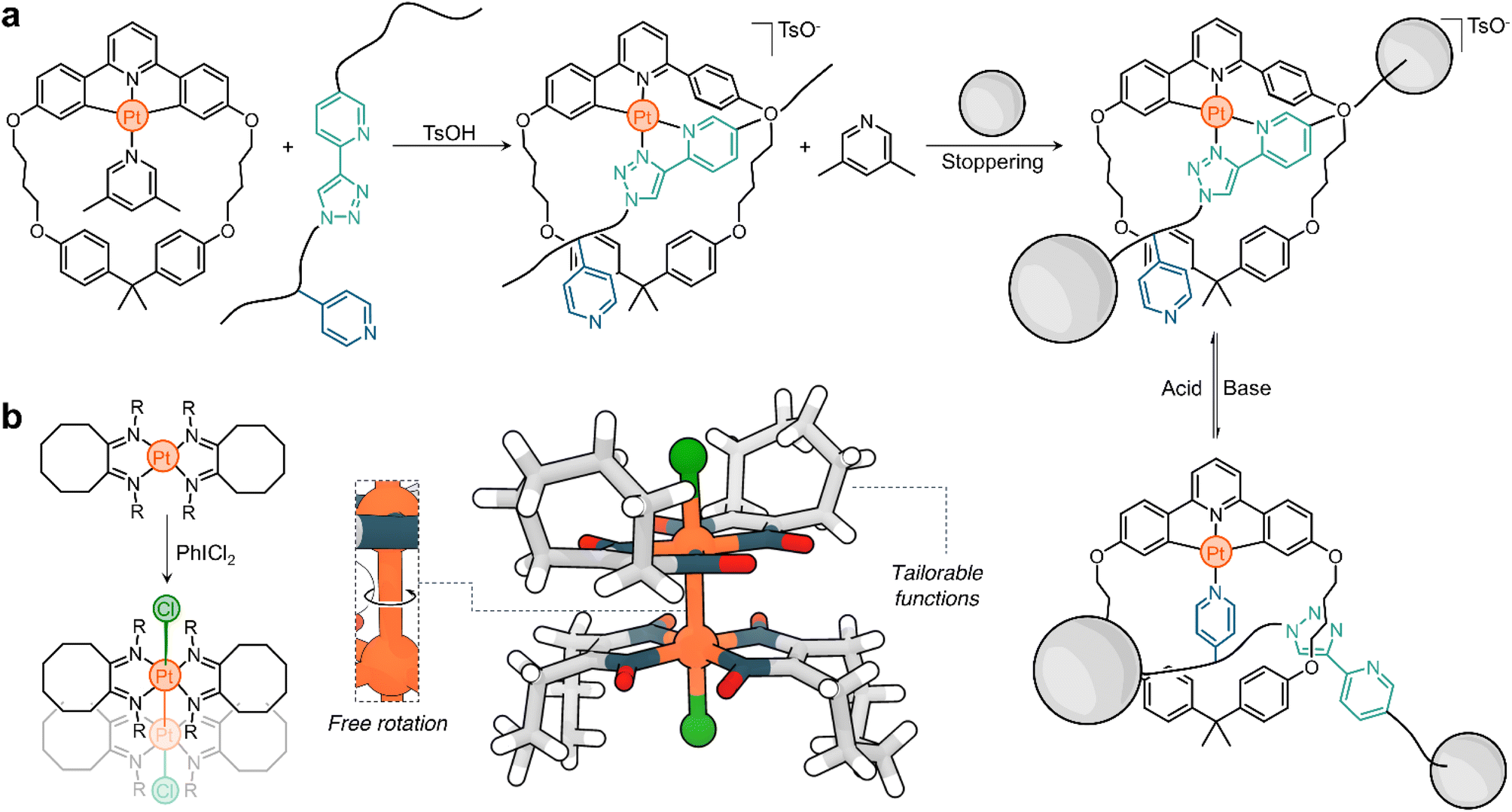
| Fig. 2 Lusby's responsive platform. (a) Reversible transformation of 1 into 2 through acid–base stimuli. (b) Solid-state structures of 1 and 2 as determined by SCXRD. (c) Synthesis of threaded complex 5 by reversible ligand exchange. (d) pH-Responsive behaviour of complex 6 to yield 7. (e) Assembly of metal–organic structures 8 and 10 using the exchange-active precursor 7. TsOH = toluenesulfonic acid; R = p-C6H4CH3. | ||
Complex 1 consists of a tridentate ligand (2,6-diphenylpyridine), a PtII center, and an ancillary 4-dimethylaminopyridine (DMAP) ligand. Compound 1 is stable in solution but undergoes rapid transformation upon adding a Brønsted-Lowry acid. Addition of p-toluenesulfonic acid selectively cleaves a C–Pt bond, protonating and releasing a phenyl ring from the metal complex. The resulting vacant coordination site at the Pt center is rapidly filled by p-toluenesulfonate to yield compound 2 (Fig. 2a). Based on the solid-state structures of 1 and 2 (Fig. 2b), it is suggested that this process is driven by an increase in the binding energy of the C–Pt bond upon acidification, which correlates well with a decrease in the dC–Pt, from 2.056 Å to 1.955 Å for 1 and 2, respectively. This process is reversible and the addition of base (e.g., K2CO3) to a solution of 2 quickly regenerates 1. The excellent conversion ratios of the forward (>70%) and backward (>80%) reactions make this a promising platform from which to build more complex structures.
The transformation of 1 to 2 is an example of partial ligand displacement, in which neither the tridentate nor the ancillary ligands are exchanged. In fact, both the addition of acid (which activates the compound for exchange) and the presence of a competitive ligand are required to observe exchange. For example, complex 3 is stable in the presence of 4 (a bidentate ligand), but rapidly converts to 5 when acid is added (Fig. 2c).51 Acid leads to partial displacement of the cyclic tridentate ligand, followed by exchange (2,6-lutidine to 4) driven by the chelate effect of the competing ligand. The resulting compound has a threaded geometry in which both the ring and linear moieties are linked by a PtII centre. Furthermore, this transformation is accompanied by (i) a change in the charge of the complex, with 3 being neutral and 5 being positively charged, and (ii) a change in the binding mode: 3 has one monodentate and one tridentate ligand (3 + 1), while 4 has two bidentate ligands (2 + 2). This process is reversible and 3 can be recovered from 5 by addition of base.
The complexity of the system is easily increased by changing the ancillary ligands. For example, if 4,4′-bipyridine (4,4′-bipy) is used instead of DMAP in complex 1, the dinuclear structure 6 is obtained (Fig. 2d). Addition of acid to 6 generates 7 (Fig. 2d), which is activated for exchange.50 The subsequent addition of 4,4′-bipy displaces the coordinated sulfonate ligand to give a metal–organic macrocycle (8, Fig. 2e). This structure has four 4,4′-bipy pillars and four PtII corners, all supporting a multicharged, square-shaped molecule. Similarly, when the activated structure 7 is treated with the tritopic ligand 9, cage 10 spontaneously forms (Fig. 2e). As expected, all these processes are reversible. The addition of base triggers the dissociation of the superstructures and yields the starting material 6 (inactive for exchange).
This platform illustrates the potential of responsive Pt complexes to reversibly generate metal–organic structures through hierarchical subcomponent assembly. This also opens other interesting avenues of exploration. For example, (i) the transformation of 6 into 8 and 10 occurs with a change in electrostatic charge, which may be advantageous to tune the solubility of the superstructures and their host performance; and (ii) the obtained cyclometalated assemblies are emissive, and pairing this with the neutralizing anions may lead to intriguing applications in aggregation-induced emission (AIE),52 circularly polarized luminescence (CPL), and optoelectronics.
Ligand exchange has also been explored to switch between macrocycles of different nuclearity using acetylide-based molecules. Trans-PtII acetylide complexes with triphenylphosphine ancillary ligands have a linear structure, whereas their cis analogues have a 90° bent conformation.53 These isomeric forms can be interchanged by switching the phosphine ligands (Scheme 1a). The Yang group took advantage of this property to construct a structurally switchable assembly.54 They designed a pyridine-containing trans-PtII acetylide ligand (11) that reacts with 12 (Scheme 1b) to form macrocycle 13.
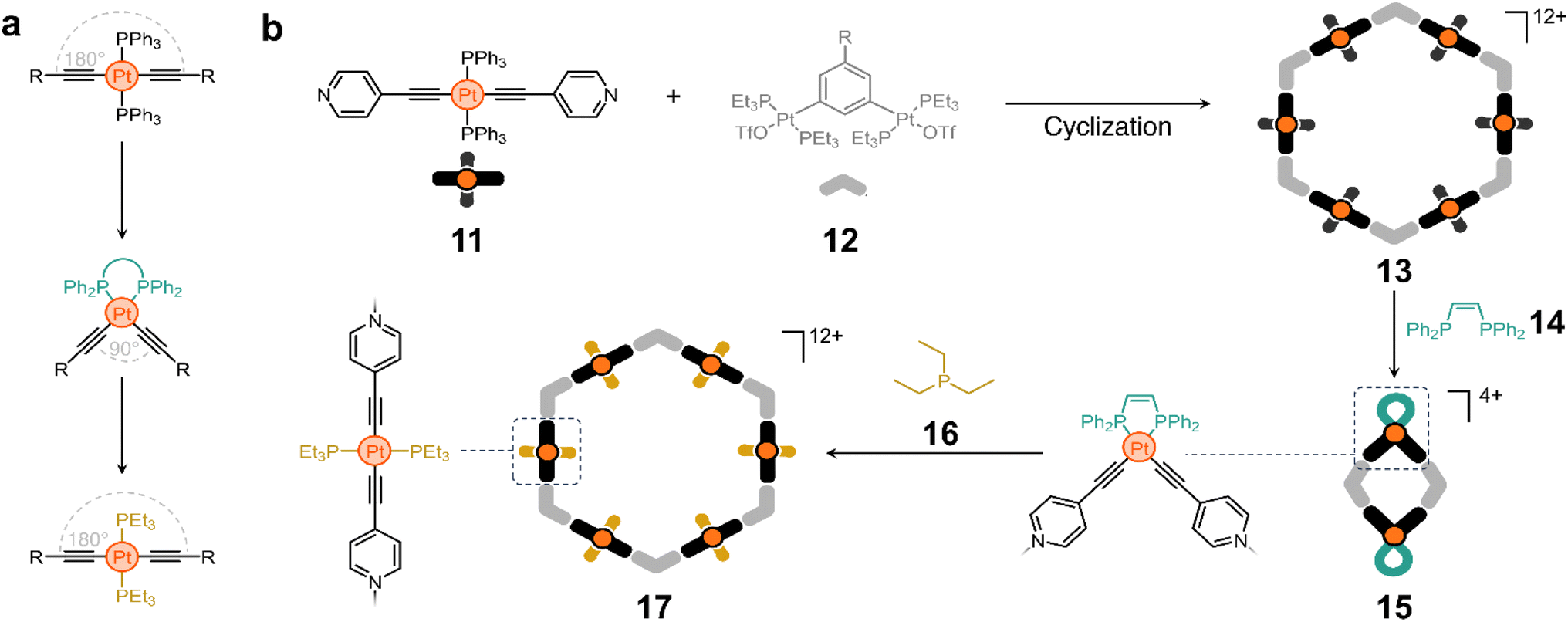 | ||
| Scheme 1 Switchable cyclic structures through ligand exchange. (a) Exchange of ancillary ligands in Pt acetylide complexes. (b) Precursors used for the assembly of a hexagonal macrocycle and reshaping of macrocycle 13 into 15 and 17via ligand exchange. | ||
Interestingly, treatment of compound 13 with cis-1,2-bis(diphenylphosphino)ethylene (14) causes ligand exchange from triphenylphosphine to 14, bending all pillars from 180° to nearly 90°. This in turn collapses 13 to form a new rhomboidal macrocycle (15, Scheme 1b). Considering that triethylphosphine (16) is a stronger electron-donating ligand, its addition to the medium displaces 14 from the rhombus and reshapes the macrocycle back into a hexagon (17).54
This approach illustrates an elegant strategy for controlling the shape of cyclic molecules, which has proven useful for the creation of polymer-anchored structures while maintaining good reactivity and processability. For example, it is possible to switch between macrocycles with four and six polymer chains covalently anchored at their vertices, which affects the physicochemical properties of the materials and their on-surface self-assembly. Similarly, polymer anchoring with this class of metallomacrocycles has been achieved by host–guest interactions alone, with the added advantage of controlling polymer chain unanchoring by degradation of the Pt-containing macrocycles.55
Exchange on PtII acetylide complexes has also been used to make other classes of supramolecular structures, including helicates and linear polymers, enabling dynamic behaviour and reversible structural changes.56–59 In principle, this strategy could also be useful to control the size and shape of nano/micrometre-sized particles while maintaining the intrinsic porosity of the materials.
In a different example, the Mirkin lab used the weak-link approach60 to modulate the cavity size of a Pt containing cavitand. Weak-link approach complexes are typically composed of hemilabile phosphinoalkyl-chalcoether61 (or amine,62 or carbene63) ligands and d8 transition metals (e.g., RhI, PdII or PtII). These molecules can be switched between rigid, condensed species and flexible, open complexes via the abstraction and introduction of a small molecule (or elemental anion) that displaces the metal–X (weak-link) interaction, while leaving the metal–P (strong-link) interaction intact. This is illustrated in Scheme 2a, where complex 18 (condensed, rigid form) transforms into a partially open analogue (19) and finally into 20, the fully open and flexible form of the system. The process is reversible, so 20 reverts to 18 upon chloride abstraction.64
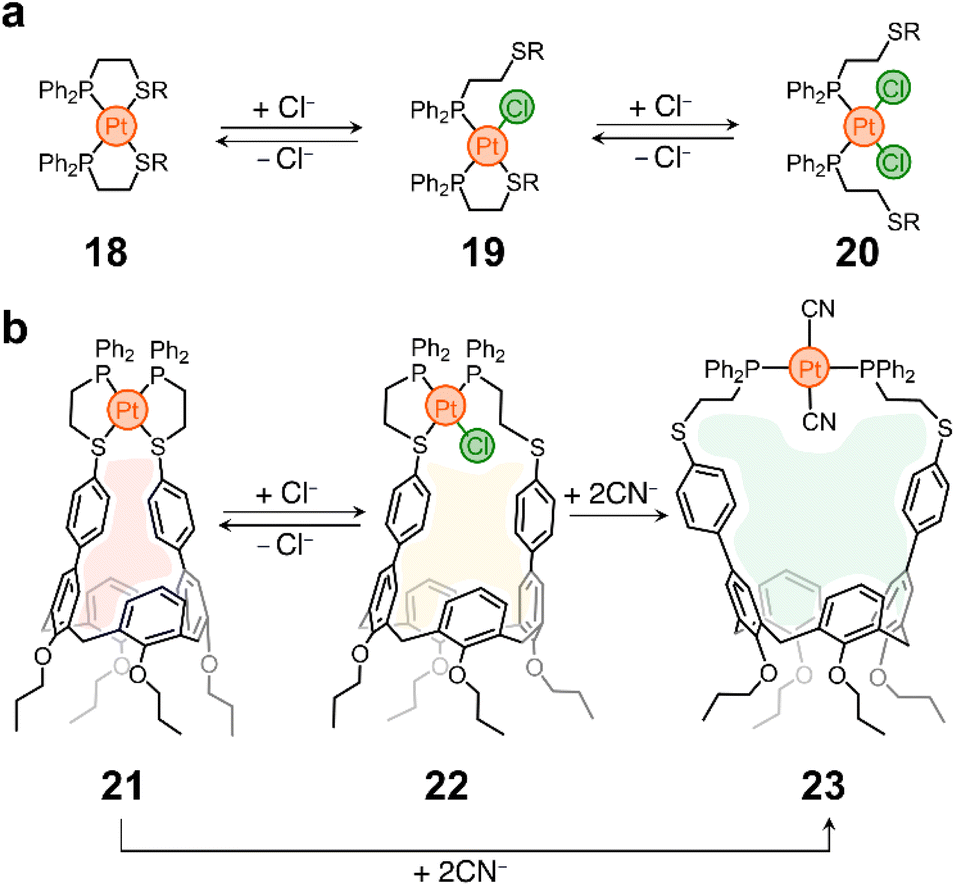 | ||
| Scheme 2 Weak-link approach. (a) Prototypical example of Mirkin group's weak-link approach.60 (b) Weak-link approach applied to the opening/closing of a host's pocket. | ||
This chemistry was cleverly incorporated in the design of a responsive calixarene compound.65 This compound (21) contains a Pt node that blocks the opening of the host, making it ineffective for guest recognition (Scheme 2b). However, the addition of chloride eliminates the weak-link and generates a new species (22) with a larger opening (Scheme 2b). It was shown that 21 is inactive as a host, while 22 can incorporate pyridine N-oxide into its pocket with a tight fit. It is noteworthy that Cl− abstraction from 22 reforms 21 and causes guest ejection. Conversion of 21 to 22 and back to 21 can be repeated several times with fidelity of guest uptake/ejection. Given that 21 has two weak-links, it was also shown that both can be eliminated by introducing strong electron-donating ligands such as CN−, yielding compound 23 (Scheme 2b), which has the largest opening of the three species and is thus able to accommodate a larger guest, such as N-methylpyridinium, in its cavity. Compounds 21 and 22 show no binding to this guest.
The weak-link approach is a powerful tool not only for reversibly controlling the shape of molecular containers, but also for producing responsive heteroleptic complexes, metallopolymers, redox-responsive structures, metallomacrocycles and more.66–69 The accessible synthesis and response to simple ions also has important advantages over other strategies, making it promising for incorporation into biologically compatible systems and nanostructured materials. Coupling this platform with supramolecular synthons that rely on non-covalent interactions (e.g., H-bonding) could open interesting new avenues for chemical discovery.
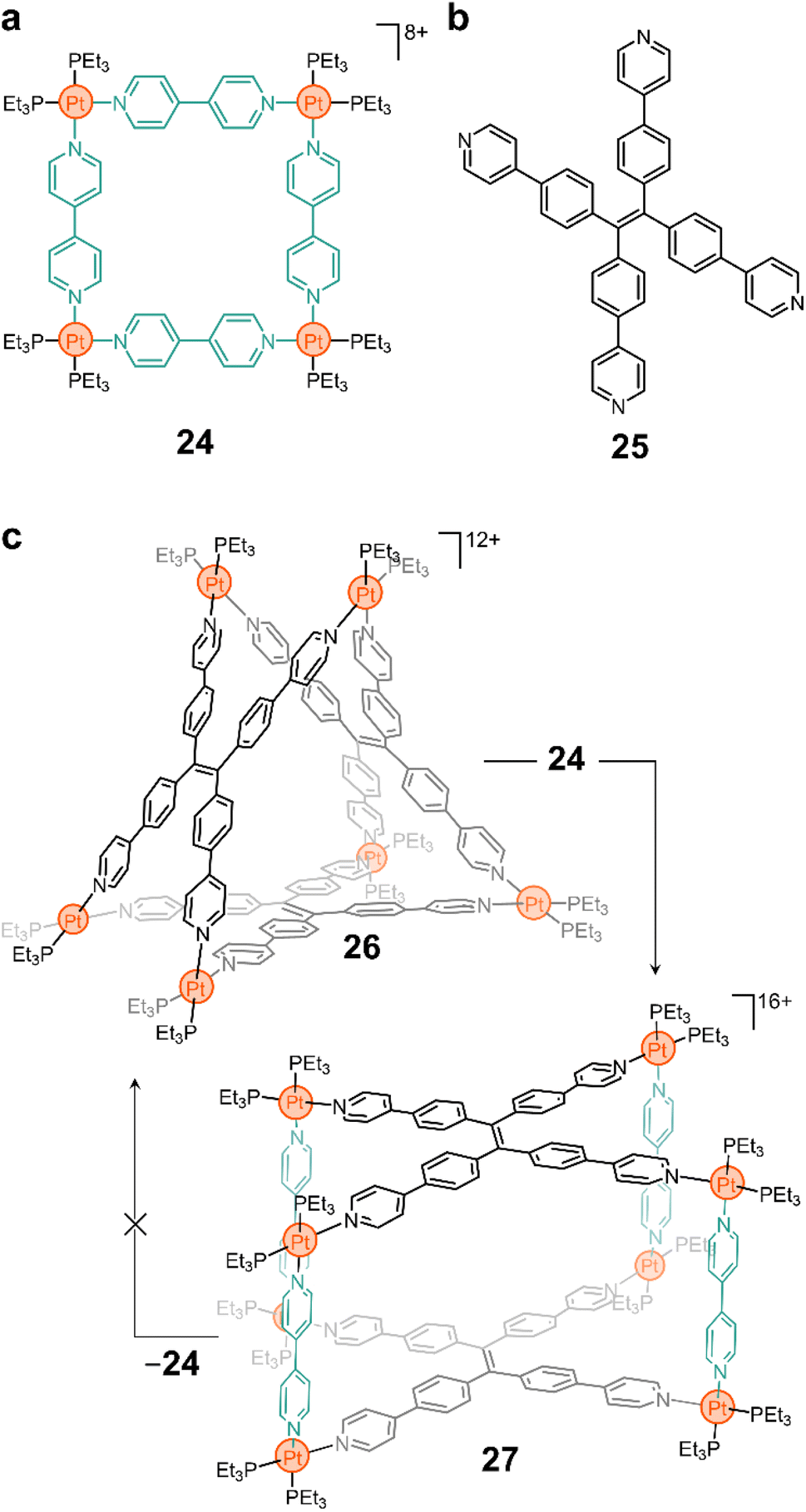 | ||
| Fig. 3 Ligand insertion for cage-to-cage transformation. (a) Classical bipyridine-based square with PtII corners (24). (b) Tetrapyridyl ligand 25 used in the construction of triangular prism 26 and cuboid cage 27. (c) Fusion of cage 26 and macrocycle 24 to yield cage 27. | ||
More complex 3-D constructs can be assembled by using non-linear linkers. For example, the Yan group showed that ligand 25 (Fig. 3b) yields the triangular prism 26 when complexed with a PtII node (Fig. 3c).80 As in many of the examples shown above, the N–Pt bond in this compound is labile and, thus, the ligands can be displaced when competitive species are introduced into the system. More interestingly, Yan and coworkers showed that this is still true when preformed macrocycles are mixed together, resulting in reorganization of the ligands to form a product in solution.80 For example, the addition of 24 to a solution of 26 results in the fusion of the two cyclic structures to form a new molecule (27, Fig. 3c) that contains both ligands 4,4′-bipy and 25 in its backbone. Although 24 is luminescent, the fused product 27 is not, providing a macroscopic handle from which to probe molecular rearrangement.
More sophisticated designs (e.g., using porphyrinoid linkers) and fluorescent sensors with rapid response to amino acids have also been constructed using this type of ligand insertion and fusion of preassembled structures.81,82 Exploiting the lability of the N–Pt bond is an interesting approach to induce topological changes with precise molecular control and atom economy. While challenges such as reversibility and limited experimental conditions will continue to plague supramolecular chemists, it is important to recognize that there are a vast number of possibilities that can be exploited from this approach. And this can lead to potential applications in cargo delivery, sensing, bioimaging, and more.
Redox-active assemblies
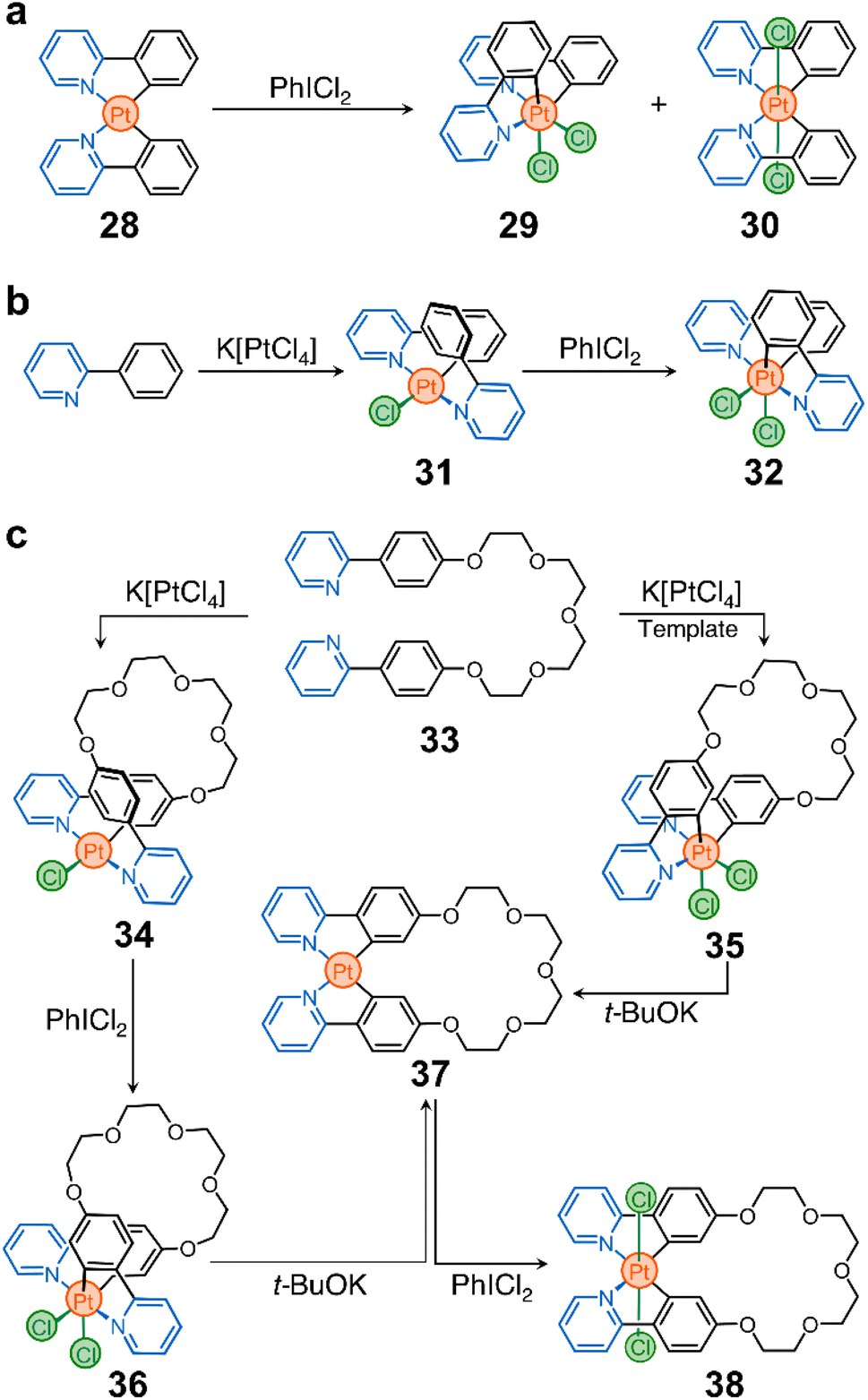 | ||
| Scheme 3 Redox-active unbridged phenylpyridine complexes for ligand reorganization. (a) First homoleptic, phenylpyridine-based cyclometalated PtII complex (28). (b) Products (29 and 30) of the oxidation of 28. (c) Partially cyclometalated complex 31 and its oxidation product (32). (d) Ring-closing reactions of proligand 33 into 34 and 35 and their postprocessing to generate rings 36–38. | ||
These advances inspired us to use cyclometalated PtII complexes as nodes in ring-shaped molecules.90 In this context, two important discoveries were crucial for the realization of our system. First, Sanford et al. showed that oxidation of 28 with iodobenzene dichloride (PhICl2) yields the PtIV stereoisomers 29 and 30 (Scheme 3a).91 Second, Rourke et al. found that phenylpyridines can lead to the formation of partially cyclometalated complexes, such as 31, which can also be oxidized to form 32 (Scheme 3b) – the trans-N,N stereoisomer of 29 and 30.92 This rich stereochemistry and redox activity led our team to wonder whether these species (e.g., 28) could be used to ring close flexible ligands while retaining their redox activity.
As a prototypical example, we synthesized proligand 33 (Scheme 3c), containing two phenylpyridine units bridged by a tetraethylene glycol chain. This proligand can be ring-closed to selectively generate macrocycles 34 and 35 (Scheme 3c). They differ in oxidation state (PtIIvs. PtIV) and stereochemistry (cis vs. trans).90 Importantly, these two compounds can be further processed. For example, treatment of 34 with PhICl2 yields a new macrocycle (36, Scheme 3c) with PtIV and trans-N,N stereochemistry. In addition, both 35 and 36 can be reduced to produce complex 37, which can finally be oxidized into PtIV complex 38 (Scheme 3c).
It is noteworthy that while toggling between oxidation states and geometries of the Pt nodes in macrocycles 34–38, the ligand distribution around the metal center also changes, subtly reshaping the cavity of the macrocycles. This alters the host performance of the rings and modulates their binding affinity towards a specific guest. Importantly, when the cavity of a host, e.g.37, is occupied by a guest, its redox reactivity is altered, either by the introduced steric bulk, by the modification of the electronic properties of the host, or by both.
This platform offers new possibilities for the construction of other classes of macrocycles (with different sizes, shapes, and chemistries) for applications in confinement reactivity, enzymatic mimicry, and molecular machines. However, it is important to recognize that there are challenges that may need to be addressed for the more diverse and faster growth of these cyclometalated Pt nodes. These include exploring milder reduction conditions for the transformation of, for example, 35 and 36 to 37, and investigating the reverse transformation of 36 to 34.
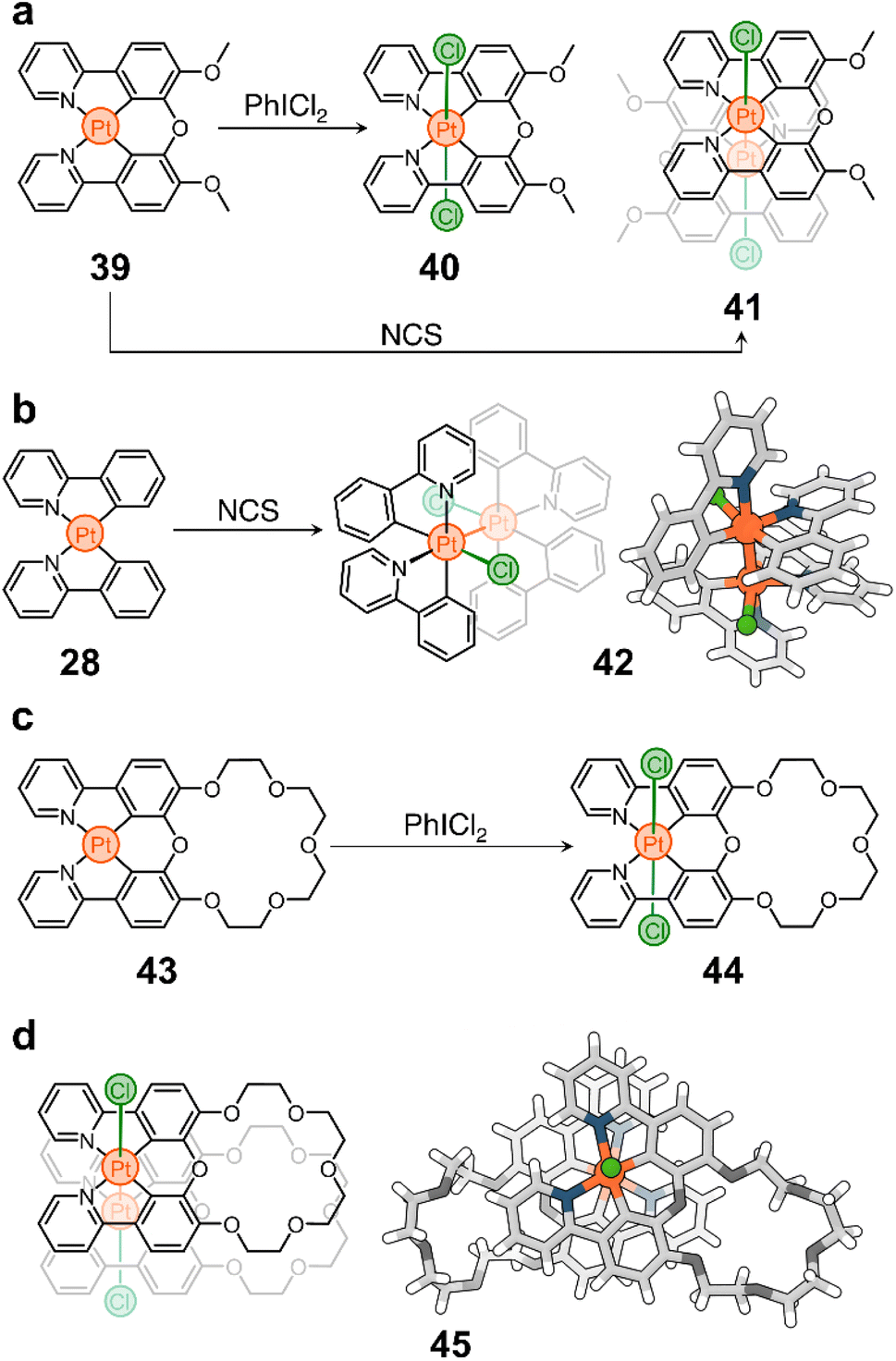 | ||
| Scheme 4 Redox-active oxygen-bridged phenylpyridine complexes for oxidation-dimerization. (a) Prototypical PtII cyclometalated complex 39 and its oxidation products 40 and 41. (b) Synthesis of PtIII dimer 42 from 28. (c) PtIV product 44 resulting from the oxidation of 43. (d) Molecular container 45 resulting from the oxidation-dimerization of complex 43. | ||
Although little studied, unbridged phenylpyridine PtII complexes can be oxidized to PtIII species to form dimeric structures held together by intermetallic bonds.91,102 For example, the oxidation of 28 with N-chlorosuccinimide (NCS) yields compound 42 in high conversion (Scheme 4b). Based on this observation, we tested the oxidation of 39 with NCS and found that complex 41 is the only dimeric PtIII species formed in solution (Scheme 4a), which is remarkable considering that the synthesis of PtIII complexes typically requires bridging ligands to bring two Pt centers together and direct the oxidation and metal–metal bond formation.103,104 The stability of 41 and its ease of synthesis led us to believe that this redox-controlled dimerization could be useful for designing new supramolecular structures. Thus, following our research on complex 39, we targeted compound 43 (Scheme 4c), a PtII complex containing a crown ether blended to its chelating backbone.105
As expected, 43 shows interesting photoluminescent properties in response to guests occupying its cavity. This complex is also redox active and can be converted to a PtIV analog (44, Scheme 4c) by oxidation with PhICl2. More importantly, we realized that the oxidation of 43 into a PtIII dimer could generate a new form of molecular container (45, Scheme 4d) consisting of two crown ethers linked by a Pt–Pt bond. Indeed, treatment of 43 with NCS produces compound 45 almost quantitatively. It is noteworthy that this host can trap large cations such as Rb+ and Cs+ using its two crown ether moieties, which is helpful to remove these cations, and potentially toxic ions, from solution.106
It is interesting to note that the oxidation of 43 can also be achieved photochemically. Irradiation of the compound with blue or white light, in the presence of dichloromethane (CH2Cl2), triggers the conversion of the PtII structure to its PtIII parent. This is caused by the photolysis of the solvent, which generates reactive chlorine that is ultimately responsible for the oxidation of the metal center. Prolonged illumination facilitates the further conversion of 45 to its PtIV counterpart (44). This whole system can function as a trap-release mechanism, allowing the capture and precipitation of ions from solution by successive assembly and disassembly of the container through PtII to PtIII to PtIV photooxidation. Notably, compound 44 can be reduced back to its precursor (43).
We believe that intermetallic bonding, which can be accessed through the reversible oxidation of PtII complexes, has great potential for exploration and could open a variety of avenues for scientific investigation. In particular, we are interested in studying the electrochemical formation and reversibility of intermetallic bonds. We believe that this platform could provide access to materials with high performance guest sequestration and recovery. In addition, the reversible nature of the dimerization could provide a new way to glue components together in polymeric structures and nanostructured materials.
Future directions
Platinum chemistry has seen significant research interest for many decades, with goals mainly revolving around novel synthetic methodologies and applications of the resulting complexes. With the advent of supramolecular chemistry, platinum has emerged as a unique element from which function and responsive character can be harnessed. This is evidenced in part by the examples described in this Perspective, which only scratch the surface of the many possibilities that can be uncovered when decades of Pt research intertwine with concepts from supramolecular chemistry, materials chemistry, systems chemistry, and nanotechnology.Platinum's electronic properties, electroactivity, chromic behaviour, redox reactivity, photoreactivity, and intermetallic interactions make it a unique element from which to program responsiveness into more functional and intricate molecules and materials. The platforms described here can already serve as inspiration for the design and fabrication of new platinum-based supramolecular assemblies. For example, the Lusby group's system could be used to synthesize quasirotaxanes (interlocked molecules in which a threading linear molecule is covalently linked to a ring) and molecular shuttles (Scheme 5a).51 The design of complementary threads and macrocycles could allow the assembly of metal–organic interlocked molecules with switchable states that could differ in luminescence or catalytic activity.107
 | ||
| Scheme 5 Prototypical examples of the use of Pt responsive systems for the generation of (a) switchable quasirotaxanes and (b) molecular rotors. | ||
Given the unique properties of platinum and its potential in supramolecular chemistry, it would also be plausible to use it in the construction of molecular machines. One specific example worth exploring is the case of PtIII dimers. Research has shown that these structures have free rotation about the Pt–Pt bond; see for example one of the first PtIII dimers reported by Willis in the 1990s108 (Scheme 5b). This rotation could potentially be controlled or modulated by the introduction of specifically designed ligands. Several factors support the idea of using these Pt dimers in the design of molecular rotors/motors. First, the synthesis of these complexes is relatively straightforward. Second, they exhibit a balance of stability and dynamism, ensuring they remain intact under various conditions, but still allowing for their functional mobility. These characteristics make them promising candidates as hinges to be incorporated into molecular rotors (as depicted in Scheme 5b). An additional advantage of these PtIII dimers is the potential to form them using photochemical methods. Using light as a stimulus has advantages, particularly due to its non-invasive nature. This means it can be used to trigger specific reactions without causing undesired side effects or damage to the surrounding environment. Moreover, the ability to disassemble these structures on demand provides an added layer of control, further highlighting their potential as building blocks for molecular machinery.
Richard J. Puddephatt once made an important observation: “Supramolecular chemistry and the chemistry of alkyl derivatives of the transition metals [platinum] are both topics of considerable current interest, but the combination of the two fields is still underdeveloped. The challenges are, in large part, experimental in nature”.109 We can further extend this to metalated platinum systems in general. In light of this, there is a pressing need to draw on the skills and innovation of synthetic chemists. By doing so, we could pave the way for the development of sophisticated supramolecular structures that effectively exploit the unique properties of platinum. New possibilities and applications in molecular science could be unlocked by this fusion.
Author contributions
M. A. S. and M. J. M. wrote the manuscript.Conflicts of interest
The authors declare no conflict of interest to declare.Acknowledgements
The authors are grateful to their co-workers who have explored supramolecular Pt chemistry in the MacLachlan group. MJM acknowledges funding from NSERC (Discovery Grant) and thanks the Canada Research Chairs Program. We also thank WPI NanoLSI (Kanazawa, Japan) for support.Notes and references
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, Wiley, 3rd edn, 2022 Search PubMed.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS.
- F. Huang and E. V. Anslyn, Chem. Rev., 2015, 115, 6999–7000 CrossRef CAS.
- A. J. Savyasachi, O. Kotova, S. Shanmugaraju, S. J. Bradberry, G. M. Ó'Máille and T. Gunnlaugsson, Chem, 2017, 3, 764–811 CAS.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4098 CrossRef CAS PubMed.
- D. B. Amabilino, D. K. Smith and J. W. Steed, Chem. Soc. Rev., 2017, 46, 2404–2420 RSC.
- X. Chen, M. A. Würbser and J. Boekhoven, Acc. Mater. Res., 2023, 4, 416–426 CrossRef CAS PubMed.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- M. Wei, Y. Gao, X. Li and M. J. Serpe, Polym. Chem., 2017, 8, 127–143 RSC.
- M. Mrinalini and S. Prasanthkumar, ChemPlusChem, 2019, 84, 1103–1121 CrossRef CAS PubMed.
- D. Yan, Z. Wang and Z. Zhang, Acc. Chem. Res., 2022, 55, 1047–1058 CrossRef CAS.
- Transition Metals in Supramolecular Chemistry, ed. J. P. Sauvage, Wiley, 2008 Search PubMed.
- T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS.
- M. H.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2022, 144, 22805–22825 CrossRef CAS.
- J.-P. Sauvage, Angew. Chem., Int. Ed., 2017, 56, 11080–11093 CrossRef CAS PubMed.
- Z. Meng, F. Yang, X. Wang, W.-L. Shan, D. Liu, L. Zhang and G. Yuan, Inorg. Chem., 2023, 62, 1297–1305 CrossRef CAS PubMed.
- D. E. Herbert, J. B. Gilroy, W. Y. Chan, L. Chabanne, A. Staubitz, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2009, 131, 14958–14968 CrossRef CAS.
- V. Brega, M. Zeller, Y. He, H. P. Lu and J. K. Klosterman, Chem. Commun., 2015, 51, 5077–5080 RSC.
- M. Yamashina, Y. Tanaka, R. Lavendomme, T. K. Ronson, M. Pittelkow and J. R. Nitschke, Nature, 2019, 574, 511–515 CrossRef CAS PubMed.
- L. Li, L. Yang, X. Li, J. Wang, X. Liu and C. He, Inorg. Chem., 2021, 60, 8802–8810 CrossRef CAS.
- L. Li, Y. Cong, L. He, Y. Wang, J. Wang, F.-M. Zhang and W. Bu, Polym. Chem., 2016, 7, 6288–6292 RSC.
- O. S. Wenger, Chem. Rev., 2013, 113, 3686–3733 CrossRef CAS.
- X. Zhang, Z. Chi, Y. Zhang, S. Liu and J. Xu, J. Mater. Chem. C, 2013, 1, 3376–3390 RSC.
- A. Kobayashi and M. Kato, Eur. J. Inorg. Chem., 2014, 2014, 4469–4483 CrossRef CAS.
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS.
- P. Xue, J. Ding, P. Wang and R. Lu, J. Mater. Chem. C, 2016, 4, 6688–6706 RSC.
- M. Yoshida and M. Kato, Coord. Chem. Rev., 2018, 355, 101–115 CrossRef CAS.
- B. Li, H.-T. Fan, S.-Q. Zang, H.-Y. Li and L.-Y. Wang, Coord. Chem. Rev., 2018, 377, 307–329 CrossRef CAS.
- M. Yoshida and M. Kato, Coord. Chem. Rev., 2020, 408, 213194 CrossRef CAS.
- B. Li, Z. Liang, H. Yan and Y. Li, Mol. Syst. Des. Eng., 2020, 5, 1578–1605 RSC.
- M. A. Soto, R. Kandel and M. J. MacLachlan, Eur. J. Inorg. Chem., 2021, 2021, 894–906 CrossRef CAS.
- J. Emsley, Nature's Building Blocks: An A–Z Guide to The Elements, Oxford University Press, 2001 Search PubMed.
- J. Emsley, The Elements, Clarendon Press, 1998 Search PubMed.
- T. Bell, The Properties and Applications of Platinum, https://www.thoughtco.com/metal-profile-platinum-2340149, accessed 26 September 2023 Search PubMed.
- R. Sharma, V. J. Singh and P. A. Chawla, Anti-Cancer Agents Med. Chem., 2022, 22, 821–835 CrossRef CAS.
- T. K. Meister, K. Riener, P. Gigler, J. Stohrer, W. A. Herrmann and F. E. Kühn, ACS Catal., 2016, 6, 1274–1284 CrossRef CAS.
- R. J. Spiegel, Transp. Res. D: Transp. Environ., 2004, 9, 357–371 CrossRef.
- C. Cebrián and M. Mauro, Beilstein J. Org. Chem., 2018, 14, 1459–1481 CrossRef PubMed.
- C. E. Holloway and M. Melnik, Rev. Inorg. Chem., 2005, 25, 93–270 CAS.
- E. Lu and S. T. Liddle, in Molecular Metal–Metal Bonds: Compounds, Synthesis, Properties, ed. S. T. Liddle, Wiley, 2015, ch. 10, pp. 352–395 Search PubMed.
- R. H. Crabtree and H. Torrens, Palladium: Inorganic & Coordination Chemistry, in Encyclopedia of Inorganic Chemistry, Wiley, 2006 Search PubMed.
- B. Z. Momeni and A. S. Abd-El-Aziz, Coord. Chem. Rev., 2023, 486, 215113 CrossRef CAS.
- A. C. Albéniz and P. Espinet, Palladium: Inorganic & Coordination Chemistry, in Encyclopedia of Inorganic Chemistry, Wiley, 2006 Search PubMed.
- F. Ibukuro, T. Kusukawa and M. Fujita, J. Am. Chem. Soc., 1998, 120, 8561–8562 CrossRef CAS.
- M. Yoshizawa, K. Ono, K. Kumazawa, T. Kato and M. Fujita, J. Am. Chem. Soc., 2005, 127, 10800–10801 CrossRef CAS.
- H. Takezawa, K. Shitozawa and M. Fujita, Nat. Chem., 2020, 12, 574–578 CrossRef CAS.
- S. J. Pike and P. J. Lusby, Chem. Commun., 2010, 46, 8338–8340 RSC.
- O. Chepelin, J. Ujma, P. E. Barran and P. J. Lusby, Angew. Chem., Int. Ed., 2012, 51, 4194–4197 CrossRef CAS PubMed.
- J. Ujma, M. De Cecco, O. Chepelin, H. Levene, C. Moffat, S. J. Pike, P. J. Lusby and P. E. Barran, Chem. Commun., 2012, 48, 4423–4425 RSC.
- P. J. Lusby, P. Müller, S. J. Pike and A. M. Z. Slawin, J. Am. Chem. Soc., 2009, 131, 16398–16400 CrossRef CAS.
- D. Sooksawat, S. J. Pike, A. M. Z. Slawin and P. J. Lusby, Chem. Commun., 2013, 49, 11077–11079 RSC.
- N. Liu, T. Lin, M. Wu, H.-K. Luo, S.-L. Huang and T. S. A. Hor, J. Am. Chem. Soc., 2019, 141, 9448–9452 CrossRef CAS.
- K. Campbell, R. McDonald, M. J. Ferguson and R. R. Tykwinski, J. Organomet. Chem., 2003, 683, 379–387 CrossRef CAS.
- W. Zheng, W. Wang, S.-T. Jiang, G. Yang, Z. Li, X.-Q. Wang, G.-Q. Yin, Y. Zhang, H. Tan, X. Li, H. Ding, G. Chen and H.-B. Yang, J. Am. Chem. Soc., 2019, 141, 583–591 CrossRef CAS PubMed.
- Z. Zhang, J. Zhao, Z. Guo, H. Zhang, H. Pan, Q. Wu, W. You, W. Yu and X. Yan, Nat. Commun., 2022, 13, 1393 CrossRef CAS PubMed.
- Y. Furusho, Y. Tanaka and E. Yashima, Org. Lett., 2006, 8, 2583–2586 CrossRef CAS PubMed.
- M. Horie, N. Ousaka, D. Taura and E. Yashima, Chem. Sci., 2015, 6, 714–723 RSC.
- Y. Miyagi, T. Ishida, M. Marumoto, N. Sano, T. Yajima and F. Sanda, Macromolecules, 2018, 51, 815–824 CrossRef CAS.
- T. Ishida, T. Sotani, N. Sano, H. Sogawa and F. Sanda, Macromolecules, 2022, 55, 309–321 CrossRef CAS.
- J. R. Farrell, C. A. Mirkin, I. A. Guzei, L. M. Liable-Sands and A. L. Rheingold, Angew. Chem., Int. Ed., 1998, 37, 465–467 CrossRef CAS.
- M. S. Rosen, A. M. Spokoyny, C. W. Machan, C. Stern, A. Sarjeant and C. A. Mirkin, Inorg. Chem., 2011, 50, 1411–1419 CrossRef CAS.
- H. J. Yoon, J. Kuwabara, J.-H. Kim and C. A. Mirkin, Science, 2010, 330, 66–69 CrossRef CAS.
- M. S. Rosen, C. L. Stern and C. A. Mirkin, Chem. Sci., 2013, 4, 4193–4198 RSC.
- R. D. Kennedy, C. W. Machan, C. M. McGuirk, M. S. Rosen, C. L. Stern, A. A. Sarjeant and C. A. Mirkin, Inorg. Chem., 2013, 52, 5876–5888 CrossRef CAS PubMed.
- J. Mendez-Arroyo, J. Barroso-Flores, A. M. Lifschitz, A. A. Sarjeant, C. L. Stern and C. A. Mirkin, J. Am. Chem. Soc., 2014, 136, 10340–10348 CrossRef CAS PubMed.
- A. M. Lifschitz, M. S. Rosen, C. M. McGuirk and C. A. Mirkin, J. Am. Chem. Soc., 2015, 137, 7252–7261 CrossRef CAS PubMed.
- A. I. d'Aquino, Z. S. Kean and C. A. Mirkin, Chem. Mater., 2017, 29, 10284–10288 CrossRef.
- H. F. Cheng, A. I. d'Aquino, J. Barroso-Flores and C. A. Mirkin, J. Am. Chem. Soc., 2018, 140, 14590–14594 CrossRef CAS PubMed.
- B. D. Coleman, A. I. d'Aquino, Z. Kean, Y. Wang, J. K. Hedlund Orbeck, C. L. Stern and C. A. Mirkin, Polyhedron, 2022, 227, 116116 CrossRef CAS.
- Y. Sun, C. Chen, J. Liu and P. J. Stang, Chem. Soc. Rev., 2020, 49, 3889–3919 RSC.
- P. J. Stang, D. H. Cao, S. Saito and A. M. Arif, J. Am. Chem. Soc., 1995, 117, 6273–6283 CrossRef CAS.
- P. S. Mukherjee, N. Das, Y. K. Kryschenko, A. M. Arif and P. J. Stang, J. Am. Chem. Soc., 2004, 126, 2464–2473 CrossRef CAS.
- S. Shanmugaraju, A. K. Bar, K.-W. Chi and P. S. Mukherjee, Organometallics, 2010, 29, 2971–2980 CrossRef CAS.
- J.-L. Zhu, L. Xu, Y.-Y. Ren, Y. Zhang, X. Liu, G.-Q. Yin, B. Sun, X. Cao, Z. Chen, X.-L. Zhao, H. Tan, J. Chen, X. Li and H.-B. Yang, Nat. Commun., 2019, 10, 4285 CrossRef.
- H. Zhu, Q. Li, B. Shi, H. Xing, Y. Sun, S. Lu, L. Shangguan, X. Li, F. Huang and P. J. Stang, J. Am. Chem. Soc., 2020, 142, 17340–17345 CrossRef CAS PubMed.
- S. Lv, Y. Miao, D. Zheng, X. Li, D. Liu and F. Song, Mol. Pharm., 2021, 18, 1229–1237 CrossRef CAS PubMed.
- Y. Yin, Z. Chen, R.-H. Li, C. Yuan, T.-Y. Shao, K. Wang, H. Tan and Y. Sun, Inorg. Chem., 2021, 60, 9387–9393 CrossRef CAS PubMed.
- N. Wang, J. Zhao, Q. Xu, P. Xing, S. Feng, X.-D. Xu and H.-B. Yang, J. Mater. Chem. C, 2022, 10, 13860–13870 RSC.
- G. Durán-Sampedro, E. Y. Xue, M. Moreno-Simoni, C. Paramio, T. Torres, D. K. P. Ng and G. de la Torre, J. Med. Chem., 2023, 66, 3448–3459 Search PubMed.
- G. Li, Z. Zhou, C. Yuan, Z. Guo, Y. Liu, D. Zhao, K. Liu, J. Zhao, H. Tan and X. Yan, Angew. Chem., Int. Ed., 2020, 59, 10013–10017 CrossRef CAS PubMed.
- Y. Shi, I. Sánchez-Molina, C. Cao, T. R. Cook and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9390–9395 CrossRef CAS PubMed.
- M. Zhang, M. L. Saha, M. Wang, Z. Zhou, B. Song, C. Lu, X. Yan, X. Li, F. Huang, S. Yin and P. J. Stang, J. Am. Chem. Soc., 2017, 139, 5067–5074 CrossRef CAS.
- L. Chassot, E. Mueller and A. von Zelewsky, Inorg. Chem., 1984, 23, 4249–4253 CrossRef CAS.
- D. S. Black, G. B. Deacon and G. L. Edwards, Aust. J. Chem., 1994, 47, 217–227 CrossRef CAS.
- M. M. Mdleleni, J. S. Bridgewater, R. J. Watts and P. C. Ford, Inorg. Chem., 1995, 34, 2334–2342 CrossRef CAS.
- J.-Y. Cho, K. Y. Suponitsky, J. Li, T. V. Timofeeva, S. Barlow and S. R. Marder, J. Organomet. Chem., 2005, 690, 4090–4093 CrossRef CAS.
- C. P. Newman, K. Casey-Green, G. J. Clarkson, G. W. V. Cave, W. Errington and J. P. Rourke, Dalton Trans., 2007, 3170–3182 RSC.
- W. Jiang and C. A. Schalley, Beilstein J. Org. Chem., 2010, 6, 14 Search PubMed.
- K. R. Neff, K. Basemann, N. Romano, J. J. Becker and M. R. Gagné, Organometallics, 2023, 42, 2177–2184 CrossRef CAS.
- M. A. Soto, V. Carta, I. Suzana, B. O. Patrick, F. Lelj and M. J. MacLachlan, Angew. Chem., Int. Ed., 2023, 62, e202216029 CrossRef CAS PubMed.
- S. R. Whitfield and M. S. Sanford, Organometallics, 2008, 27, 1683–1689 CrossRef CAS.
- J. Mamtora, S. H. Crosby, C. P. Newman, G. J. Clarkson and J. P. Rourke, Organometallics, 2008, 27, 5559–5565 CrossRef CAS.
- D. A. K. Vezzu, J. C. Deaton, J. S. Jones, L. Bartolotti, C. F. Harris, A. P. Marchetti, M. Kondakova, R. D. Pike and S. Huo, Inorg. Chem., 2010, 49, 5107–5119 CrossRef CAS PubMed.
- H. Fukagawa, T. Shimizu, H. Hanashima, Y. Osada, M. Suzuki and H. Fujikake, Adv. Mater., 2012, 24, 5099–5103 CrossRef CAS PubMed.
- E. Turner, N. Bakken and J. Li, Inorg. Chem., 2013, 52, 7344–7351 CrossRef CAS PubMed.
- S. Huo, Chem. Rec., 2018, 18, 1583–1595 CrossRef CAS PubMed.
- I. Allison, H. Lim, A. Shukla, V. Ahmad, M. Hasan, K. Deshmukh, R. Wawrzinek, S. K. M. McGregor, J. K. Clegg, V. V. Divya, C. Govind, C. H. Suresh, V. Karunakaran, K. N. Narayanan Unni, A. Ajayaghosh, E. B. Namdas and S.-C. Lo, ACS Appl. Electron. Mater., 2019, 1, 1304–1313 CrossRef CAS.
- T. Theiss, S. Buss, I. Maisuls, R. López-Arteaga, D. Brünink, J. Kösters, A. Hepp, N. L. Doltsinis, E. A. Weiss and C. A. Strassert, J. Am. Chem. Soc., 2023, 145, 3937–3951 CrossRef CAS PubMed.
- S. Buss, M. V. Cappellari, A. Hepp, J. Kösters and C. A. Strassert, Chemistry, 2023, 5, 1243–1255 CrossRef CAS.
- J.-M. Kim, K. Cheong, J. Jiang, S. O. Jeon, W. P. Hong and J. Y. Lee, Trends Chem., 2023, 5, 267–278 CrossRef CAS.
- M. A. Soto, V. Carta, R. J. Andrews, M. T. Chaudhry and M. J. MacLachlan, Angew. Chem., Int. Ed., 2020, 59, 10348–10352 CrossRef CAS.
- T. Yamaguchi, O. Kubota and T. Ito, Chem. Lett., 2004, 33, 190–191 CrossRef CAS.
- J. J. Wilson and S. J. Lippard, Inorg. Chem., 2012, 51, 9852–9864 CrossRef CAS PubMed.
- X. Wu, D.-G. Chen, D. Liu, S.-H. Liu, S.-W. Shen, C.-I. Wu, G. Xie, J. Zhou, Z.-X. Huang, C.-Y. Huang, S.-J. Su, W. Zhu and P.-T. Chou, J. Am. Chem. Soc., 2020, 142, 7469–7479 CrossRef CAS PubMed.
- M. A. Soto, V. Carta, M. T. Cano, R. J. Andrews, B. O. Patrick and M. J. MacLachlan, Inorg. Chem., 2022, 61, 2999–3006 CrossRef CAS PubMed.
- M. A. Soto, M. T. Chaudhry, G. K. Matharu, F. Lelj and M. J. MacLachlan, Angew. Chem., Int. Ed., 2023, 62, e202305525 CrossRef CAS PubMed.
- D. A. Leigh, V. Marcos and M. R. Wilson, ACS Catal., 2014, 4, 4490–4497 CrossRef CAS.
- L. A. M. Baxter, G. A. Heath, R. G. Raptis and A. C. Willis, J. Am. Chem. Soc., 1992, 114, 6944–6946 CrossRef CAS.
- R. J. Puddephatt, Dalton Trans., 2022, 51, 7011–7024 RSC.
| This journal is © The Royal Society of Chemistry 2024 |