
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic acceptorless dehydrogenative borylation of styrenes enabled by a molecularly defined manganese complex†
Kuhali
Das‡
a,
Abhishek
Kundu‡
b,
Koushik
Sarkar
a,
Debashis
Adhikari§
 *b and
Biplab
Maji§
*a
*b and
Biplab
Maji§
*a
aDepartment of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur 741246, India. E-mail: bm@iiserkol.ac.in
bDepartment of Chemical Sciences, Indian Institute of Science Education and Research Mohali, SAS Nagar 140306, India. E-mail: adhikari@iisermohali.ac.in
First published on 11th December 2023
Abstract
In this study, we employed a 3d metal complex as a catalyst to synthesize alkenyl boronate esters through the dehydrogenative coupling of styrenes and pinacolborane. The process generates hydrogen gas as the sole byproduct without requiring an acceptor, rendering it environmentally friendly and atom-efficient. This methodology demonstrated exceptional selectivity for dehydrogenative borylation over direct hydroboration. Additionally, it exhibited a preference for borylating aromatic alkenes over aliphatic ones. Notably, derivatives of natural products and bioactive molecules successfully underwent diversification using this approach. The alkenyl boronate esters served as precursors for the synthesis of various pharmaceuticals and potential anticancer agents. Our research involved comprehensive experimental and computational studies to elucidate the reaction pathway, highlighting the B–H bond cleavage as the rate-determining step. The catalyst's success was attributed to the hemilability and metal–ligand bifunctionality of the ligand backbone.
Introduction
Fostering a greener, more efficient, and cost-effective chemical synthesis entails comprehensively redesigning established catalytic methodologies to minimize the reliance on Earth's critical resources.1 The more accessibility, lower toxicity level, and potential for unveiling novel catalytic properties make the Earth-abundant transition metals attractive candidates to substitute their heavier congeners for these purposes.2 Within this context, the molecularly defined complexes of manganese have garnered substantial interest in sustainable hydrogen transfer and hydroelementation reactions.3 In particular, the hydrosilylation and hydroboration reactions of alkenes leading to alkyl silanes and boranes have been explored with multidentate manganese-based metal complexes by the groups of Trovitch, Zheng, Thomas, Kirchner, Wang, Xie, and others (Fig. 1a).4 Their effort leads to linear or branched alkyl products depending upon the chosen catalyst and the microenvironment of the reaction. In comparison, the dehydrogenative functionalization of alkenes leading to alkenyl silanes is a relatively new area of research (Fig. 1b).4b,f,5 Xi and Wang independently utilized Mn2(CO)10 and di-phosphine/Mn2(CO)10 complexes as catalysts to synthesize alkenyl silanes in an acceptorless pathway via a manganese-centered radical.4b,f Kirchner's (PP)Mn(I)-alkyl complex catalyzes the same reaction by forming an Mn-silyl species, consuming one-third of the alkene as an H2 acceptor.5 However, similar ligand backbones lead to hydroboration products, and manganese-catalyzed synthesis of alkenyl boronate has remained elusive. | ||
| Fig. 1 [a] Mn-catalyzed hydroelementation reactions, [b] Mn-catalyzed acceptorless dehydrogenative silylation of olefinic C–H bonds, [c] this work: Mn-catalyzed acceptorless dehydrogenative borylation of alkenes. | ||
The reports on the borylation of readily available alkenes surfaced employing Pd, Rh, Ir, Pt, Fe, Co, and Cu complexes with HBpin or B2pin2 as borylating agents.6 However, the state-of-the-art borylation of alkenes requires a stoichiometric, sacrificial alkene, ketone, or alcohol to scavenge the produced byproducts, H2 or HBpin. The chemo- and regioselectivity are also compromised due to the formation of a competitive 1,2-diborylation adduct.6j,p,7 Zr-catalyzed dehydrogenative borylation of alkenes in an acceptorless pathway has been recently reported.8 The first-row transition metal-complex catalyzed acceptorless dehydrogenative borylation (ADB) of alkenes without needing an acceptor for the release of hydrogen remained unknown. Besides, mechanistically driven catalyst design is still underdeveloped in this area, leaving significant room for further exploration.
Ligand design is crucial in enabling the 3d metal catalysts towards a selective bond activation and subsequent transformation. In this vein, we recently developed phosphine-free Mn(I)-complexes derived from multifunctional ligands that, apart from the conventional metal–ligand cooperativity, utilized the hemilability of the soft sidearm for bond activation.9 We hypothesized that Mn(I)-complexes with similar ligand backbones would be tenable to achieve the dehydrogenative borylation of alkenes (Fig. 1c). Notably, the catalyst had to be highly selective to minimize the competitive hydroboration reactions.4 Herein, we report the first example of a bench stable Mn(I)-complex catalyzed ADB of alkenes with exclusive regio- and stereoselectivity and high chemoselectivity. The reaction operated under mild conditions without needing a hydrogen acceptor and delivered products in high yields.
Results and discussion
The dehydrogenative borylation was initially conducted on a model substrate styrene 1 (Table 1). Pleasingly, the Mn1, possessing a thiomethoxy arm, catalyzed the reaction of 1 (1 equiv.) with HBpin (1.5 equiv.) and delivered trans-2-phenylvinylboronic acid pinacol ester 2 in 30% yield at 65 °C. The reaction was performed in THF in the presence of a catalytic amount of KOtBu (entry 1). While the anti-Markovnikov hydroboration and 1,2-diborylation adducts were not detected, alkyl boronic acid pinacol ester 3 was noticed in 5% yield. Gratifyingly, ethylbenzene 3′, the complete hydrogenated product of styrene was undetected. Besides, the headspace analysis of the reaction tube detected hydrogen gas, proving the acceptorless nature of the reaction (Section S8.1†). The manganese(I) complex Mn2 with a thiophene sidearm displayed slightly better reactivity, yielding 37% of 2 (entry 2).|
|
|||
|---|---|---|---|
| Entry | Variation from the standard conditions | % Yield of 2 | Selectivity 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:3′ :3:3′ |
| a Reaction conditions: 1 (0.1 mmol), HBpin (0.15 mmol), Mn3 (6 mol%), KHMDS (10 mol%), THF (33 μL), 65 °C, 24 h, Ar. Yields were determined by 1H NMR using mesitylene as the internal standard. Isolated yields are in the parenthesis. b KOtBu instead of KHMDS. | |||
| 1b | Mn1 | 30 | 86:14:0 |
| 2b | Mn2 | 37 | 83:17:0 |
| 3b | Mn3 | 46 | 88:12:0 |
| 4 | Mn3 without KHMDS | <5 | — |
| 5 | None | 76 (72) | 93:7:0 |
| 6 | KH instead of KHMDS | 33 | 82:18:0 |
| 7 | KOMe instead of KHMDS | 23 | 74:26:0 |
| 8 | LiEt3BH instead of KHMDS | 29 | 60:40:0 |
| 9 | 1,4-Dioxane as solvent | 40 | 80:20:0 |
| 10 | CH3CN as solvent | 41 | 80:20:0 |
| 11 | CH2Cl2, PhMe and benzene | <10 | — |
| 12 | Without Mn3 | 0 | — |
Under this situation, we hypothesized that incorporating a more π-accepting ligand framework around the metal would further improve the reactivity and selectivity by reducing the hydricity of the intermediate Mn–H. We have synthesized the complex Mn3 with a quinoline framework (Fig. 2 and S1†). The CO bands in the infrared spectra in 2018, 1936, and 1907 cm−1 are consistent with the formulation. The 1H and 13C NMR data also confirmed its formation. The single crystal X-ray diffraction shows an octahedral geometry around manganese with a bidentate coordination of the NN-ligand. Trans to the nitrogen atoms are placed two carbonyl groups forming a quasi-meridional plane, and the other carbonyl is positioned trans to the bromide in its perpendicular plane.
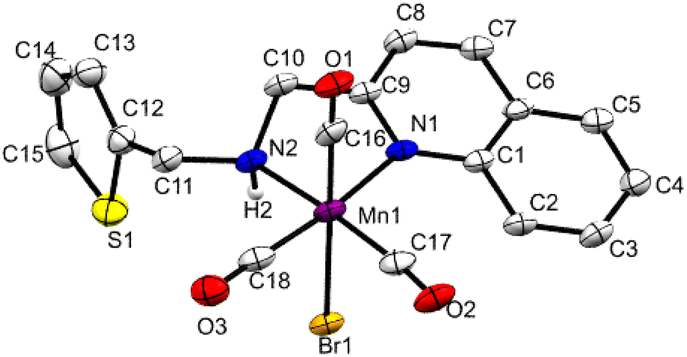 | ||
| Fig. 2 Molecular structure of Mn3 (50% probability ellipsoids).10 | ||
As hypothesized, Mn3 displayed improved catalytic activities, delivering 46% yield of 2 (Table 1, entry 3). Further optimization revealed that the reaction was susceptible to the nature of the catalyst activator used. While the reaction stops without an activator (entry 4), using KHMDS remarkably improved the outcome, delivering 76% of 2 at high chemoselectivity; 2:3:3′ = 93:7:0 (entry 5). In comparison, LiEt3BH, KH, and KOMe gave inferior results (entry 6–8). Employing solvents other than THF also leads to poor yields or selectivity (entry 9–11). As anticipated, the absence of Mn3 does not yield any product (entry 12) as proved by a control reaction.
We then probed the generality of the Mn3-catalyzed ADB reaction (Table 2). Gratifyingly, a range of styrene derivatives containing steric and electronically biased substituents underwent smooth reaction under these conditions, yielding trans-vinyl boronic esters in high yields and selectivities. The presence of electron-donating groups at the para-position leads up to 78% yields of the products 4–7 with high chemoselectivity (up to 96:4), favoring ADB over hydroborated products. An electronically neutral p-phenyl substituent 8 also furnished comparable results. The product yield and chemoselectivity reduced a little as we moved from slightly electron-donating F to a highly electron-withdrawing trifluoromethyl group (9–12). However, the electronic effect on the reaction rate was negligible (vide infra). Notably, the halogens were retained, leaving scope for further functionalization. A similar trend is observed after changing the substituents from para to meta (13–16). The reaction was minimally affected by sterics; ortho-substituted styrenes 2-methyl, and 2-bromostyrene (17 and 18) furnished 58 and 51% yields, respectively. However, α-, and β-methyl styrene fails to give a product (Section S6.2†). The 3,5-dimethoxy and 3,4,5-trimethoxy styrenes furnished the alkenyl boronate esters (19 and 20) with good yields and excellent selectivities, which could be further employed to synthesize some bioactive compounds (vide infra). Notably, 2-vinylnaphthalene (21) and 3-(trifluoromethyl)-4′-vinyl-1,1′-biphenyl (22) reacted smoothly, and the desired products were isolated in 50 and 71% yields, respectively. Styrenes containing thiomethoxy (23) and heterocyclic moieties such as furan (24) and pyridine (25) were also tolerated, furnishing the alkenyl boronates in moderate to good yields. Noteworthily, 2-vinyl ferrocene also delivered the product (26), maintaining the high chemoselectivity. The aliphatic alkene 1-octene provided small quantities of hydroboration and dehydrogenative borylation product mixture under these conditions.
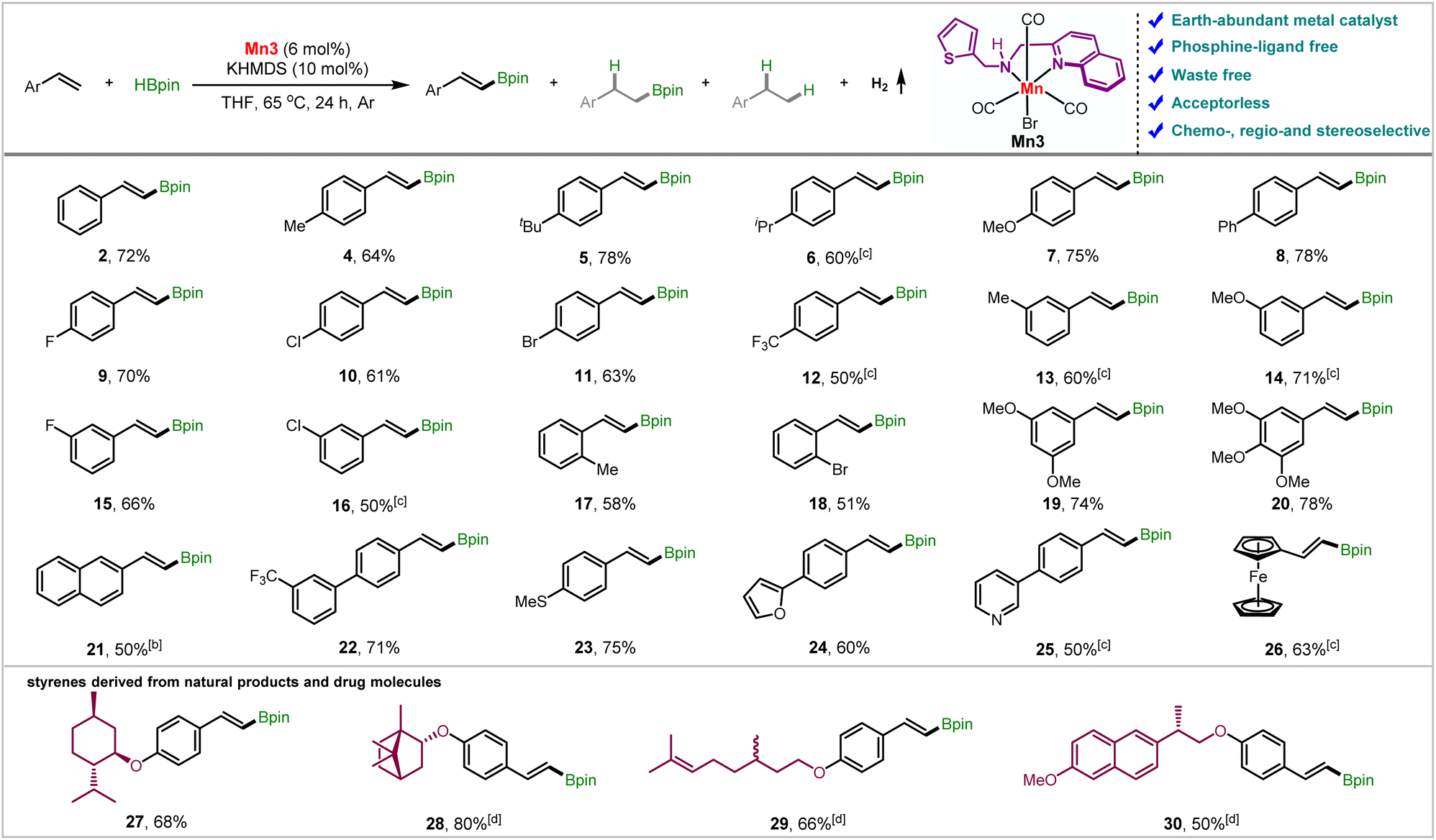
We have further borylated the derivatives of some natural products and drug molecules to demonstrate the synthetic applicability of our developed protocol. The styrenes prepared from (−)-menthol, (−)-borneol, and (+/−)-citronellol could quickly be diversified to the respective alkenyl boronates (27–29) in 66–80% yields while maintaining high selectivity for ADB. The aliphatic alkene counterpart in citronellol derivative (29) was utterly untouched, featuring the chemoselectivity towards aromatic alkenes over aliphatic ones. Additionally, the styrenyl ether of the alcohol obtained from naproxen underwent smooth borylation, furnishing 50% yield of 30.
The utility of the alkenyl boronate ester products was then showcased by carrying out an array of transformations (Fig. 3). The oxidation of alkenyl boronate ester 20 with Oxone® led to the aldehyde 31 in 62% isolated yield. The vinyl halides are crucial coupling partners in different transition metal-catalyzed syntheses. The alkenyl boronate ester group could easily be converted to various vinyl halides (Cl, Br, I, 32–34) up to 88% yield in a highly stereoselective trans-fashion. The enol ether 35 could be synthesized in good yields in a Cu-catalyzed pathway in the presence of allyl alcohol. The [2 + 2]-cycloaddition of 2 with 4-methoxy styrene under photochemical irradiation of 456 nm blue LED with fac-Ir(ppy)3 as a photosensitizer and subsequent oxidation with NaBO3·4H2O led to cyclobutanol derivative 36 in 67% yields and 4:1 diastereomeric ratio.11
 | ||
| Fig. 3 Diversification of alkenyl boronate ester. Reaction conditions: [a] 20, Oxone®, acetone/H2O, 0 °C, 2 h; [b] 20, CuCl2, THF/MeOH/H2O, 100 °C, 36 h, N2; [c] 20, CuSO4·5H2O, NaBr, MeOH, 50 °C, 12 h, N2; [d] 20, I2, NaOH, THF, rt, 1 h; [e] 20, allyl alcohol, Cu(OAc)2, NEt3, 4 Å molecular sieves, neat, rt, 16 h; [f] 2, 4-methoxystyrene, fac-Ir(ppy)3, CH2Cl2, 456 nm blue LED, 25 h, Ar then NaBO3·4H2O, THF/H2O, rt, 10 h; [g] 20, 4-iodoanisole, Pd(OAc)2, PPh3, Na2CO3, PhMe/EtOH/H2O, 100 °C, 24 h, N2; [h] 20, 2-iodobenzothiophene, Pd(PPh3)4, NaOH, 1,4-dioxane, 100 °C, 24 h, N2; [i] 20, 2-chlorobenzothiazole, Pd(PPh3)4, NaOH, dioxane, 100 °C, 24 h, N2; [j] 19, 4-bromophenol, Pd(OAc)2, SPhos, Na2CO3, PhMe/EtOH/H2O, 100 °C, 24 h, N2; [k] 40, BBr3, CH2Cl2, -78 °C to rt, 12 h, N2. | ||
The methodology was then employed to synthesize several bioactive compounds. We utilized 20 to synthesize stilbene derivatives 37–39. The compound 37 (DMU-212) exhibited superior activity in colon cancer than resveratrol.12 The stilbenes 38, 39 have showcased potent inhibition to the growth of human cancer cells.13 The antioxidant and anti-carcinogenic pterostilbene 4014 was synthesized in 64% yield via the Suzuki–Miyaura coupling of 19 and 4-bromophenol. The methyl deprotection of pterostilbene 40 furnished anti-carcinogenic drug resveratrol 41.15
Sets of kinetic, isotope labeling, control experiments, and high-level computation studies were then performed to gain detailed insight into the reaction mechanism. A proposed mechanism is depicted in Fig. 4a, and corresponding energies for the viable intermediate and transition states are shown in Fig. 4b. Upon treatment with KHMDS, Mn3 underwent thermodynamically downhill dehydrohalogenation, followed by the loss of one CO ligand, resulting in a pentacoordinate Mn(I)-complex I. The thiophene arm is bound to the Mn-center in the computed structure, resulting in a distorted trigonal pyramidal geometry across manganese (Y-shaped TBP), where the CO–Mn–CO bond angle is 94.38°. Such deviation (from the ideal 120°) is attributed to a strong π-donor amido ligand trans to the CO–Mn–CO that shortens the Mn–N bond (1.86 Å). As an outcome of this geometrical influence, all six electrons on manganese are paired, and the ground state remains on the singlet spin surface. Experimentally, the formation of this complex was proved by two new CO stretching frequencies (1921 and 1793 cm−1) in the IR spectra of Mn3 upon treatment of KHMDS (Section S8.9†). We have encountered similar manganese intermediate in our previous studies.9a,b
 | ||
| Fig. 4 (a) Proposed pathway, and (b) potential energy surface for Mn-catalyzed acceptorless dehydrogenative borylation of olefins. | ||
Then HBpin activation occurs across the Mn-amido bond through TS-I, having a transition barrier of 11.39 kcal mol−1 from intermediate I. The crucial part of this activation is the pivotal role of the Mn–N bond that switches back to predominantly σ-donor mode, reflected in the increased Mn–N bond length (2.05 Å) in intermediate II. During this step, a manganese hydride forms, whose formation is also traced by 1H NMR spectroscopy, showing a resonance at −14.07 ppm (Section S8.10†).
The proposed N–H metal–ligand bifunctionality in activating the B–H bond is then experimentally probed. We have prepared the Mn(I)-complex Mn4, having an N–Me substitution instead of N–H as in Mn3 (Section S2.1–2.2†).10 When the reaction was performed using Mn4, a poor 20% isolated yield of 2 was obtained (Fig. 5a). The lower yet unignorable amount of product formation hints at a relatively high-energy reaction channel different from our proposed mechanistic sketch. Plausibly, the deprotonation of the quinolinyl CH2 group would lead to (de)aromatization of the quinolyl motif, enabling bond activation, as demonstrated earlier by Milstein.16
 | ||
| Fig. 5 Mechanistic studies. [a] Probing metal–ligand bifunctionality and hemilability, [b] Eyring analysis, [c] kinetic isotope effect analysis, [d] Hammet analysis, [e] deuterium labeling studies, [f] control experiment. | ||
In the next stage of the catalytic cycle, the hemilabile thiophene arm detaches from the metal center to vacate a site for incoming substrate. It leads to intermediate III (Fig. 4). Recently, we delineated the hemilability of the sulfur-containing arm in hydrogen transfer reactions.9 To probe whether the weak (de)coordination of sulfur-arm is operative in this catalysis, we have prepared the Mn(I)-complex Mn5, where the sulfur atom in Mn3 has been replaced with weakly polarizable oxygen (Fig. 5a, Section S2.1–2.2†). Mn5 as a catalyst significantly reduces the yield of 2. The hemilabile behavior is further probed by applying exogenous strong Lewis basic ligand PCy3, which resulted in a poor yield of 2 with an increasing amount of the external ligand (Section S8.3†).
Then, adding HBPin to intermediate III leads to the formation of IV, featuring a η1-HBPin ligand (Fig. 4). This HBPin coordination step is energetically uphill by 6.15 kcal mol−1. The follow-up step overcomes a transition barrier of 23.55 kcal mol−1viaTS-II to afford intermediate V, featuring η2-dihydrogen coordinated to Mn. This step possesses the highest computational barrier of potential energy surface, indicative of the rate-determining step. We have explored the temperature dependence on the rate constant (Fig. 5b). The Eyring analysis provided ΔG(298) = 23.3 ± 2.9 kcal mol−1, which is in excellent agreement with the computed value.
We have probed it further by the kinetic isotope effect analysis (Fig. 5c, eqn (i)). The reaction with DBpin (kD = 3.82 × 10−3 M min−1) is slower than HBpin (kH = 9.13 × 10−3 M min−1). The KIE (kH/kD) of 2.39 suggests that the B–H bond session is likely to be involved in the rate-determining, i.e., the slowest step of the reaction. The theoretically calculated KIE (kH/kD) value of 2.7, also aligned with the experimental value.17 For the KIE consideration, contribution from changes in the isotopic partition function was ignored and changes in zero-point energy was included solely.
Since the substrate alkene is not involved in this step, its electronic nature would have little influence on this step. In strong support of this assertion, the Hammett correlation studies disclose the reaction to be slightly biased towards the electron-donating substrates regarding yields and chemoselectivity. At the same time, the electronic effect was minimal (ρ = −0.60) on the reaction rate (Fig. 5d).
Furthermore, we performed deuterium labeling studies with 42-d and HBpin (Fig. 5e, eqn (i)). It resulted in a little <5% deuterium scrambling, implying that the Mn–H addition across the alkene double bond is not taking place and the formation of 8 is irreversible. The same conclusion could be derived by employing DBpin as the reagent, which does not result in deuterium incorporation in product 8 (Fig. 5e, eqn (ii)). The addition of alkyl boronic ester 3 under the standard reaction conditions with or without HBpin does not yield alkenyl boronic ester 2 (Fig. 5f). It concludes that 3 is not an intermediate for the reaction and that 2 does not form via the dehydrogenation of 3.
We note that, unlike dehydrogenative silylation by the Xie group,4b the possibility of radical-promoted activation of HBPin does not exist here. In the previous case, the starting manganese species is Mn(0), which, being a metalloradical, facilitates a hydrogen atom transfer from substrate silane to generate Mn(I)-hydride and silyl radical. In contrast, the present scenario represents a Mn(I) species with an explicit exhibition of M–L bifunctional motif,18 nullifying the probability of radical-mediated bond activation.
Additionally, the possibility of in situ conversion of Mn(I) to Mn(0) was discarded by employing Mn2(CO)10 in combination with L3. This led to 30% yield of the saturated hydroboration product with a trace of alkenyl boronate ester (Section S8.2†).
In the next stage, H2 is released from V, which leads to intermediate VI (Fig. 4). This H2 release is energetically downhill from η2-dihydrogen bound intermediate by 9.64 kcal mol−1. Encouragingly, GC analysis of the reaction tube's headspace confirmed the liberation of H2 gas (Section S8.1†).
The thiophene arm then detaches to give intermediate VII, and styrene binds to the vacant manganese site in an η2-fashion, leading to intermediate VIII. Boryl migration to the terminal carbon site of olefin results in a 16e− Mn complex IX, which further reorganizes and gives a comparatively low energy intermediate X, stabilized by C–H agostic interaction. A close look at the optimized intermediate X reveals a stretched C–H bond (1.15 Å), validating such agostic interaction. Also, in X, the distance between Mn-center and that alkyl hydrogen is 1.93 Å, and the M–H–C bond angle is 97.77°. This C–H agostic interaction provides a proper platform for the subsequent β-hydride elimination step, resulting in the desired ADB product traversing through TS-IV, with an energy barrier of 11.65 kcal mol−1. Liberation of the product returns the catalyst's resting state, and further binding of the second molecule of HBpin ensues to keep the process catalytic.
We further determined the kinetic isotope effect in the process where the styrene was deuterium labeled (Fig. 5c, eqn (ii)). The KIE (kH/kD) of 1.74 with 42 (kH = 9.13 × 10−3 M min−1) and deuterated substrate 42-d (kD = 5.26 × 10−3 M min−1) indicates that the C–H bond cleavage of the alkene substrate might not be involved in the RDS. This experimental data further corroborates well with the computational insight where β-hydride elimination is less energy-demanding and not part of a rate-determining step. Although it is not part of the rate-determining step, the β-hydride elimination step determines the E-selectivity (Section S9.1†). Intermediate X is the most stable intermediate compared to intermediate XA or XB, which leads to the Z- and geminal-isomer, respectively. Also, TS-IV possesses a lower transition barrier than the other two (TS-IVA and TS-IVB) corresponding to other regioisomers. An alternative pathway has been discussed in detail in the ESI (Section S9.2†).
Conclusion
In summary, we have disclosed a synthetic protocol for alkenyl boronate esters via the dehydrogenative coupling of alkenes with pinacolborane. The chemo-, stereo-, and regioselective reaction was catalyzed by Mn(I)-complex possessing a multifunctional phosphine-free ligand architecture. Hydrogen gas is produced as the only byproduct and is liberated without an acceptor. Following the established protocol, derivatives of different natural products and drug molecules were fabricated in good yields. Furthermore, alkenyl boronate esters were employed as precursors to drugs like resveratrol, pterostilbene, and other potential anticancer agents. The functionalization of boronate esters via oxidation, halogenation, etherification, and photochemical [2 + 2] cycloaddition led to the corresponding products in good yields and excellent stereocontrol. Detailed experimental and high-level computation studies were performed to delineate the reaction pathway. The metal–ligand bifunctionality and hemilability of the thiophene arm are crucial to the catalyst's success. The deuterium labeling studies and other kinetic experiments aligned with the computational analyses and suggest that the B–H bond cleavage of pinacolborane is involved in the rate-determining step.Data availability
The ESI† includes all experimental details, including optimization of the synthetic method, synthesis, and characterization of all starting materials and products reported in this study, and mechanistic studies. NMR spectra of all products, crystallography, and computation details are included as well.Author contributions
KD and BM conceived the project. KD and KS performed experiments, analyzed products, and performed experimental mechanistic studies with input from BM. AK performed the DFT calculations with input from DA. KD and AK wrote the initial draft with input from BM and DA. BM and DA edited the manuscript. BM and DA acquired financial support for the development of this project.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
BM thanks IISER Kolkata and CSIR 02(0405)/21/EMR-II for financial support. DA is thankful for the financial support from MoE-STARS (STARS2/2023-0474). KD, AK and KS thank CSIR for the PhD fellowship.References
- P. Chirik and R. Morris, Acc. Chem. Res., 2015, 48, 2495–2495 CrossRef CAS PubMed.
- (a) P. T. Anastas and J. B. Zimmerman, Green Chem., 2019, 21, 6545–6566 RSC; (b) R. M. Bullock, Catalysis without Precious Metals, Wiley-VCH, Weinheim, 2010 CrossRef.
- K. Das, S. Waiba, A. Jana and B. Maji, Chem. Soc. Rev., 2022, 51, 4386–4464 RSC.
- (a) S. Weber, D. Zobernig, B. Stöger, L. F. Veiros and K. Kirchner, Angew. Chem., Int. Ed., 2021, 60, 24488–24492 CrossRef CAS; (b) J. Dong, X.-A. Yuan, Z. Yan, L. Mu, J. Ma, C. Zhu and J. Xie, Nat. Chem., 2021, 13, 182–190 CrossRef CAS; (c) G. Zhang, H. Zeng, J. Wu, Z. Yin, S. Zheng and J. C. Fettinger, Angew. Chem., Int. Ed., 2016, 55, 14369–14372 CrossRef CAS PubMed; (d) J. R. Carney, B. R. Dillon, L. Campbell and S. P. Thomas, Angew. Chem., Int. Ed., 2018, 57, 10620–10624 CrossRef CAS PubMed; (e) T. K. Mukhopadhyay, M. Flores, T. L. Groy and R. J. Trovitch, Chem. Sci., 2018, 9, 7673–7680 RSC; (f) X. Yang and C. Wang, Chin. J. Chem., 2018, 36, 1047–1051 CrossRef CAS.
- S. Weber, M. Glavic, B. Stöger, E. Pittenauer, M. Podewitz, L. F. Veiros and K. Kirchner, J. Am. Chem. Soc., 2021, 143, 17825–17832 CrossRef CAS PubMed.
- (a) T. Davan, E. W. Corcoran and L. G. Sneddon, Organometallics, 1983, 2, 1693–1694 CrossRef CAS; (b) J. M. Brown and G. C. Lloyd-Jones, J. Chem. Soc. Chem. Commun., 1992, 710–712 RSC; (c) S. A. Westcott, T. B. Marder and R. T. Baker, Organometallics, 1993, 12, 975–979 CrossRef CAS; (d) M. Murata, S. Watanabe and Y. Masuda, Tetrahedron Lett., 1999, 40, 2585–2588 CrossRef CAS; (e) R. B. Coapes, F. E. S. Souza, R. L. Thomas, J. J. Hall and T. B. Marder, Chem. Commun., 2003, 614–615 RSC; (f) A. Caballero and S. Sabo-Etienne, Organometallics, 2007, 26, 1191–1195 CrossRef CAS; (g) V. J. Olsson and K. J. Szabó, Angew. Chem., Int. Ed., 2007, 46, 6891–6893 CrossRef CAS PubMed; (h) T. Ohmura, Y. Takasaki, H. Furukawa and M. Suginome, Angew. Chem., Int. Ed., 2009, 48, 2372–2375 CrossRef CAS; (i) A. Kondoh and T. F. Jamison, Chem. Commun., 2010, 46, 907–909 RSC; (j) J. Takaya, N. Kirai and N. Iwasawa, J. Am. Chem. Soc., 2011, 133, 12980–12983 CrossRef CAS PubMed; (k) I. Sasaki, H. Doi, T. Hashimoto, T. Kikuchi, H. Ito and T. Ishiyama, Chem. Commun., 2013, 49, 7546–7548 RSC; (l) A. N. Brown, L. N. Zakharov, T. Mikulas, D. A. Dixon and S.-Y. Liu, Org. Lett., 2014, 16, 3340–3343 CrossRef CAS; (m) M. Morimoto, T. Miura and M. Murakami, Angew. Chem., Int. Ed., 2015, 54, 12659–12663 CrossRef CAS; (n) C. Wang, C. Wu and S. Ge, ACS Catal., 2016, 6, 7585–7589 CrossRef CAS; (o) T.-J. Hu, G. Zhang, Y.-H. Chen, C.-G. Feng and G.-Q. Lin, J. Am. Chem. Soc., 2016, 138, 2897–2900 CrossRef CAS; (p) H. Wen, L. Zhang, S. Zhu, G. Liu and Z. Huang, ACS Catal., 2017, 7, 6419–6425 CrossRef CAS; (q) T. J. Mazzacano and N. P. Mankad, ACS Catal., 2017, 7, 146–149 CrossRef CAS; (r) D. Yoshii, X. Jin, N. Mizuno and K. Yamaguchi, ACS Catal., 2019, 9, 3011–3016 CrossRef CAS.
- N. Kirai, S. Iguchi, T. Ito, J. Takaya and N. Iwasawa, Bull. Chem. Soc. Jpn., 2013, 86, 784–799 CrossRef CAS.
- (a) X. Shi, S. Li and L. Wu, Angew. Chem., Int. Ed., 2019, 58, 16167–16171 CrossRef CAS; (b) Q. Zhao, X.-F. Wu, X. Xiao, Z.-Y. Wang, J. Zhao, B.-W. Wang and H. Lei, Organometallics, 2022, 41, 1488–1500 CrossRef CAS.
- (a) A. Jana, K. Das, A. Kundu, P. R. Thorve, D. Adhikari and B. Maji, ACS Catal., 2020, 10, 2615–2626 CrossRef CAS; (b) K. Sarkar, K. Das, A. Kundu, D. Adhikari and B. Maji, ACS Catal., 2021, 11, 2786–2794 CrossRef CAS; (c) K. Das, K. Sarkar and B. Maji, ACS Catal., 2021, 11, 7060–7069 CrossRef CAS; (d) S. Waiba, S. K. Jana, A. Jati, A. Jana and B. Maji, Chem. Commun., 2020, 56, 8376–8379 RSC.
- CCDC-2192558 (Mn3), 2297411 (Mn4) Contains the supplementary crystallographic data.
- Y. Liu, D. Ni, B. G. Stevenson, V. Tripathy, S. E. Braley, K. Raghavachari, J. R. Swierk and M. K. Brown, Angew. Chem., Int. Ed., 2022, 61, e202200725 CrossRef CAS.
- S. Sale, R. G. Tunstall, K. C. Ruparelia, G. A. Potter, W. P. Steward and A. J. Gescher, Int. J. Cancer, 2005, 115, 194–201 CrossRef CAS.
- N. R. Penthala, S. Thakkar and P. A. Crooks, Bioorg. Med. Chem. Lett., 2015, 25, 2763–2767 CrossRef CAS.
- D. McCormack and D. McFadden, Oxid. Med. Cell. Longevity, 2013, 2013, 575482 Search PubMed.
- S. Fulda, Drug Discovery Today, 2010, 15, 757–765 CrossRef CAS.
- U. K. Das, Y. Ben-David, Y. Diskin-Posner and D. Milstein, Angew. Chem., Int. Ed., 2018, 57, 2179–2182 CrossRef CAS PubMed.
- R. A. M. O'Ferrall, J. Phys. Org. Chem., 2010, 23, 572–579 CrossRef.
- C. Erken, A. Kaithal, S. Sen, T. Weyhermüller, M. Hölscher, C. Werlé and W. Leitner, Nat. Commun., 2018, 9, 4521 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2192558 and 2297411. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05523j |
| ‡ These authors contributed equally. |
| § Lead contact. |
| This journal is © The Royal Society of Chemistry 2024 |