
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect conversion of carboxylic acids to free thiols via radical relay acridine photocatalysis enabled by N–O bond cleavage†
Dmitry L.
Lipilin
,
Mikhail O.
Zubkov
 ,
Mikhail D.
Kosobokov
and
Alexander D.
Dilman
*
,
Mikhail D.
Kosobokov
and
Alexander D.
Dilman
*
N. D. Zelinsky Institute of Organic Chemistry, Leninsky Prosp. 47, 119991 Moscow, Russian Federation. E-mail: adil25@mail.ru
First published on 5th December 2023
Abstract
Carboxylic acids and thiols are basic chemical compounds with diverse utility and widespread reactivity. However, the direct conversion of unprotected acids to thiols is hampered due to a fundamental problem – free thiols are incompatible with the alkyl radicals formed on decarboxylation of carboxylic acids. Herein, we describe a concept for the direct photocatalytic thiolation of unprotected acids allowing unprotected thiols and their derivatives to be obtained. The method is based on the application of a thionocarbonate reagent featuring the N–O bond. The reagent serves both for the rapid trapping of alkyl radicals and for the facile regeneration of the acridine-type photocatalyst.
Introduction
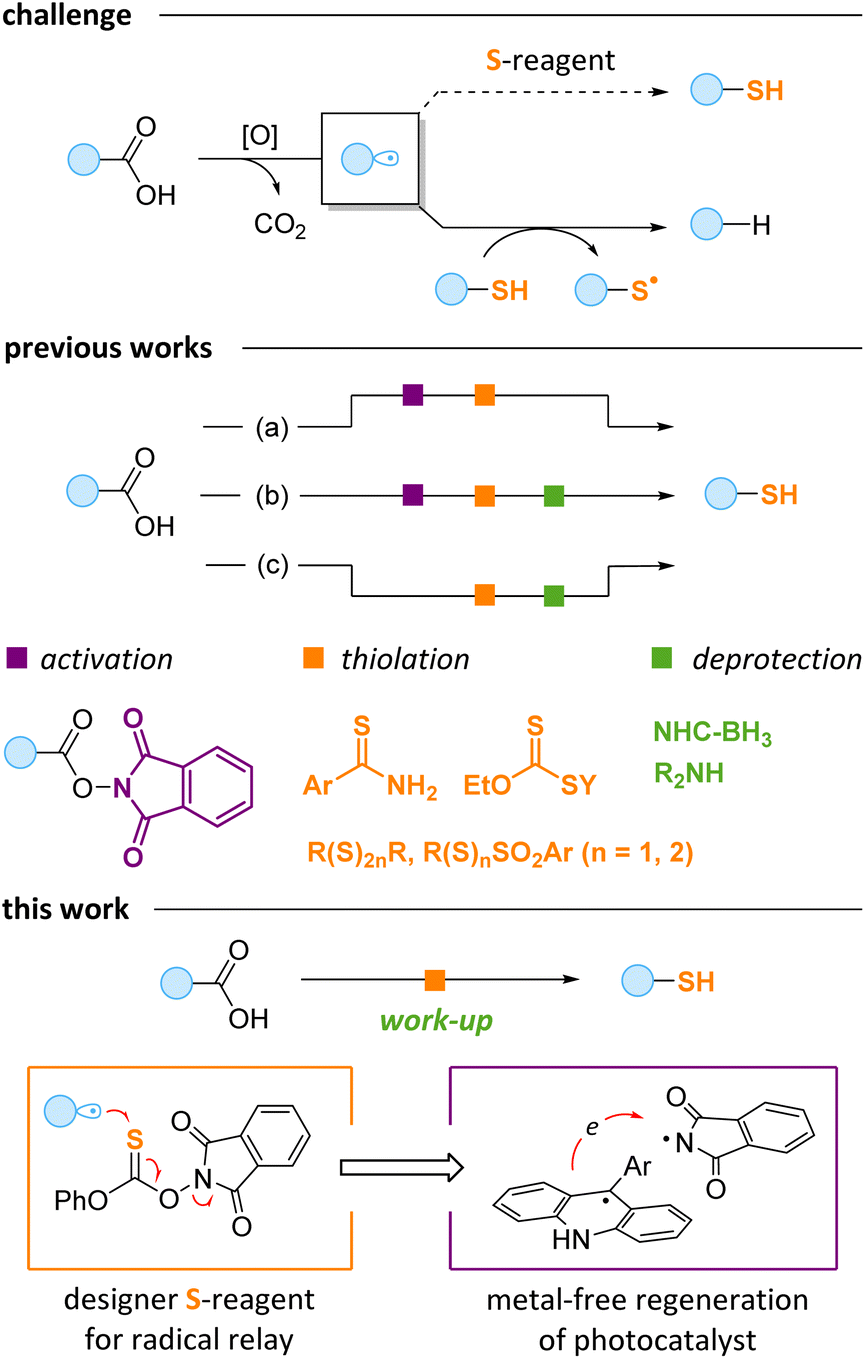
The development of mild and efficient protocols for carbon–sulfur bond formation is a long-standing goal in organic chemistry. Several methodologies have been introduced to address the growing demand for this type of transformation in the pharmaceutical industry and materials science.1–8 Modern organocatalytic methods,1 transition metal catalysis2 and currently rising photoredox catalysis3 were employed in various C–S bond forming reactions. However, the majority of the established methods deliver disubstituted sulfides. The production of sensitive free thiols usually requires additional steps or auxiliary groups.4 At the same time, thiols have found significant application in polymerization and 3D printing5 and nanoparticle stabilization6 and are extremely important for biocompatible thiol–ene/yne “click” chemistry7 – one of the general methodologies for polypeptide structure modification.8The carboxylic group is ubiquitous in natural products, peptides and drugs.9 Therefore, a mild transformation of the carboxylic moiety to free thiol would be a valuable tool for drug design and late-stage modification of bioactive compounds. At the recent surge of visible light driven chemistry,10 multiple preparative methods based on photoinduced radical decarboxylation have been developed.11 Radicals generated in these protocols were successfully used for carbon–carbon and carbon–heteroatom bond formation. However, the photoredox promoted thiolation of free carboxylic acids is challenging due to several reasons. A key problem is that the resulting thiols efficiently quench alkyl radicals to form alkanes, which disrupt the thiolation process (Scheme 1, top).12 Moreover, some low-valent sulfur sources are unstable under strongly oxidative conditions required for the oxidation of the carboxylate group.13
 | ||
| Scheme 1 Conversion of carboxylic acids to thiols. | ||
To avoid the alkyl radical quenching, the thiol should be produced in a protected form, which would liberate the free thiol only after the radical process is completed. Therefore, a delicate balance is needed: the protecting group on the sulfur should be effective in preventing premature release of the thiol, and, at the same time, it should be easily removed, preferably without an additional synthetic step. As a result, known approaches to the decarboxylative preparation of free thiols require at least one additional derivatization, activation or deprotection step (Scheme 1). For instance, benzothioamides were recently reported as useful sulfur sources for decarboxylative thiolation of redox-active esters (RAEs).14a In this case, free thiols were liberated under basic conditions (path a). RAEs were also coupled with tetrasulfides14b and thioacids14c to provide masked thiols, which require a separate deprotection step (path b). In these processes, preliminary synthesis of RAEs11b,c is needed with the overall protocol being less practical. Alternatively, the thiolation of free carboxylic acids can be achieved by using specific sulfur-containing reagents (path c). Thus, Alexanian discovered that acids can be subjected to direct O–H hydrogen atom abstraction by the electron-deficient amidyl radical from amidoxanthates, and the thiol group is then released in reaction with primary amines or alkoxides.15 Recently, we reported another thiolation method exploiting the unique properties of 4-perfluoropyridine disulfide,12c but the free thiols were later obtained in a separate photochemical protocol upon treatment with NHC-borane.16 Finally, Pratt and co-workers suggested trisulfur dioxides as suitable sulfur donors providing unsymmetric disulfides, which give mixtures of two thiols as a result of reduction.17
Overall, the existing protocols for the conversion of acids to thiols are tedious and become inconvenient if further derivatization is needed. Herein, we report an efficient one-pot protocol for converting acids to thiols. A key idea of our method is to use a thionocarbonate reagent allowing for the radical relay process by virtue of facile cleavage of the N–O bond formally serving as an “internal oxidant” (Scheme 1, bottom). For the activation of carboxylic acids, we employed acridine-type photocatalysis,18 which in our case is facilitated by the reagent-determined catalyst recycling. Thus, the reaction is based on the interplay between the reagent and the photocatalyst, with the thiol being released on mildly basic work-up.
Results and discussion
Isobutyric acid 1a was selected as a model substrate, and its thiolation was evaluated using various reagents 2 in the presence of acridine photocatalysts (Scheme 2). Thionocarbonate 2a, which can be easily accessed from common reagents – thiophosgene, N-hydroxyphthalimide and phenol,19 was found optimal. The replacement of the phthalimide fragment by smaller succinimide (2b) or diacetimide (2c) led to a drop in the yield. The reagent with the dimethylamino moiety (2d) was completely ineffective. Surprisingly, the performance of di-Boc-substituted thionocarbonate 2e was similar to that of 2a with 86% isolated yield, but it was more expensive and difficult to prepare. The replacement of the phenyl group with methyl (2f) also resulted in a low yield. Acridine A-1 under irradiation of violet light (400 nm) in methylene chloride was optimal. The progressive addition of acridine photocatalyst A-1 with a syringe pump and the use of 1.5 equivalents of 2a significantly improved the reaction outcome; the product 3a was isolated in 85% yield. Bulkier catalysts A-2 and A-320 gave slightly diminished yields in comparison to A-1, while unsubstituted acridine A-4 gave almost no product. The reaction did not proceed without catalyst or light, and blue irradiation (450 nm) was not enough to initiate the process. The addition of catalyst in one portion also gave reduced yields. Importantly, reactions performed under an argon or air atmosphere gave identical yields and no traces of radical scavenging by oxygen were detected. Correspondingly, all further experiments were carried out under air.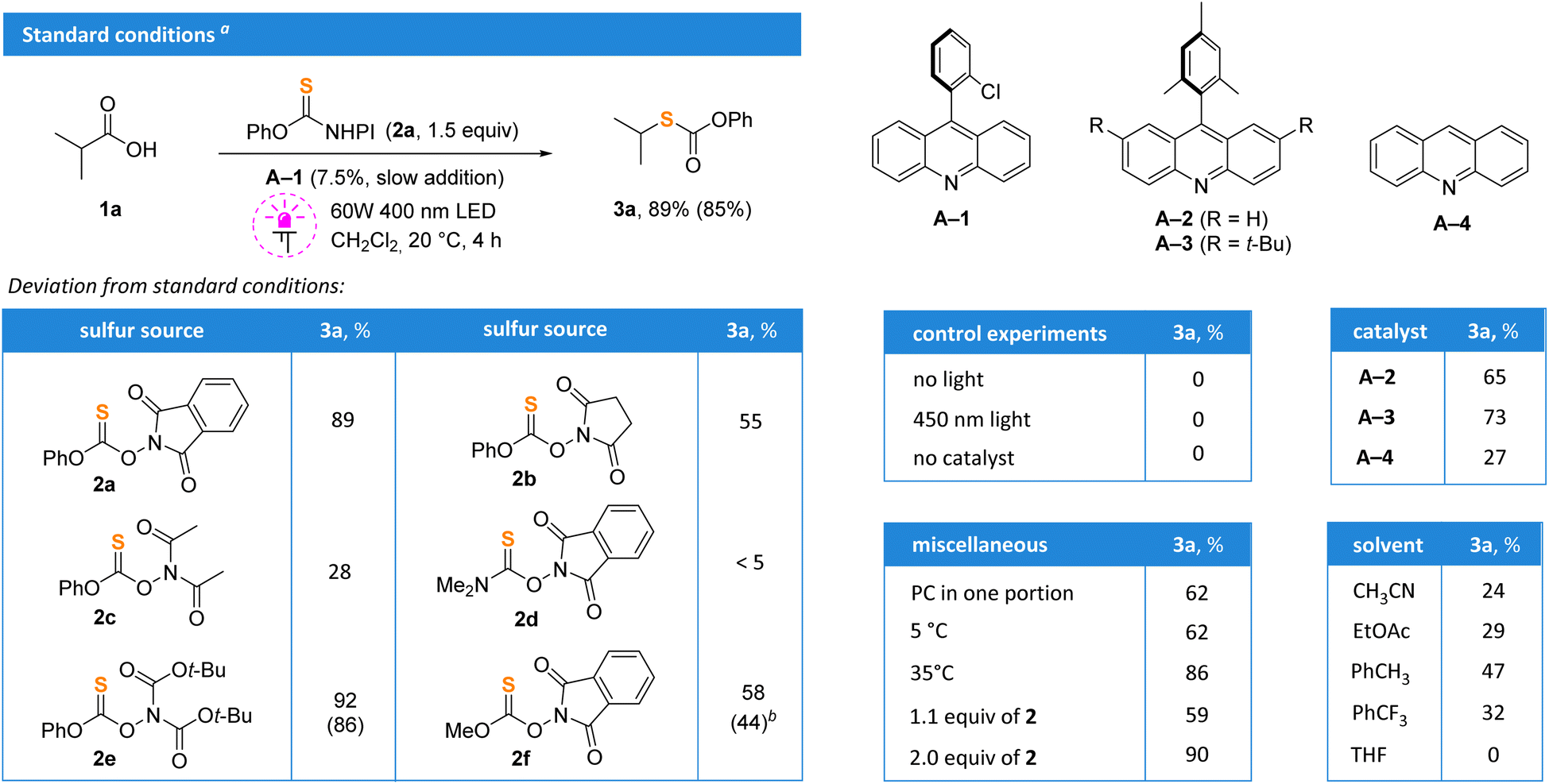 | ||
| Scheme 2 Optimization studies. a Yields were determined by 1H NMR using CH2Br2 as an internal standard. Isolated yields are shown in parentheses. b Methyl-substituted thiocarbonate 3a-Me was isolated. | ||
With the optimized conditions in hand, we investigated the scope of the decarboxylation–thiolation reaction (Scheme 3). Initially formed thiocarbonates 3 can be smoothly converted to corresponding thiols after work-up with aqueous potassium carbonate solution, and free thiols 4a–g were isolated. However, since free thiols are smelly and prone to oxidation, it is more convenient to isolate them in the protected form. Thus, compounds 3 derived from acids generating primary, secondary and tertiary radicals were prepared in good yields. Some reactions proceeding through the formation of primary radicals, gave products in decreased yields (products 3i–k). The reaction worked nicely with substrates having O-, S- and N-substituents adjacent to the carboxylic group (products 3k–u). Due to the instability of α-heteroatom-substituted alkylthiols, the corresponding derivatives were isolated only in the form of thiocarbonates. The reaction tolerates a range of functionalities such as free hydroxyl (3w), amide (3e) and halide (3n, q, ad, af) groups. Pyridine (3p,q), pyrazole (3t), indole (3j), piperidine (3e,f), oxetane (3v), benzodioxane (3o) and tetrahydrofuran (3k) heterocyclic cores were compatible with the reaction conditions. N-Protected amino acids were also successfully converted into the corresponding thiocarbonates (products 3z, aa, ae). Late-stage functionalization of lipid regulators gemfibrozil and clofibrate was successfully performed (products 3ab and 3ad). Carboxylic groups of valproic acid (3ac), picamilon (3ag) and herbicide 2,4-D (3af) were also replaced with the thiocarbonate in good yields (Scheme 3, bottom).
 | ||
| Scheme 3 Scope of the reaction. Isolated yields are shown. Reaction conditions: 1 (0.5 mmol), 2a (0.75 mmol), A-1 (37.5 μmol, slow addition), CH2Cl2 (2 mL), 60 W 400 nm LEDs, 20 °C, 4 h. Workup conditions (for the synthesis of thiols 4): K2CO3 (5 mmol), MeOH (10 mL), H2O (2 mL), 20 °C, 4 h. a 2 mmol scale reaction. b 10% of A-1 was used. c MeCN as solvent. d DMSO as solvent. | ||
Unfortunately, ibuprofen (product 3ah) and other benzylic acids cannot be thiolated, with almost no acid conversion observed.
To demonstrate the synthetic utility of the thiolation method, we performed several one-pot transformations starting from carboxylic acids 1 (Scheme 4). Importantly, thiocarbonates 3 can be further functionalized at the sulfur atom without deprotection to free thiols. For example, treatment of regular reaction mixtures containing 3 with chlorine and acetic acid gave sulfonyl chlorides, which were directly combined with amines affording sulfonamides 6a–c in good overall yields. Carbonates 3 formed in situ can also be alkylated with various electrophilic halides including butyl (5a) and allyl (5b) bromides, ethyl bromoacetate (5c) and pteridine-derived bromide (5d). The arylation reaction was performed by aromatic halogen substitution of electron deficient heterocycles leading to oxadiazole (5e) and pyridine (5f, g) sulfides. Finally, under the action of Selectfluor, the intermediate thiocarbonates 3 were smoothly converted into sulfonyl fluorides (7a–c) valuable for the SuFEx click chemistry.21
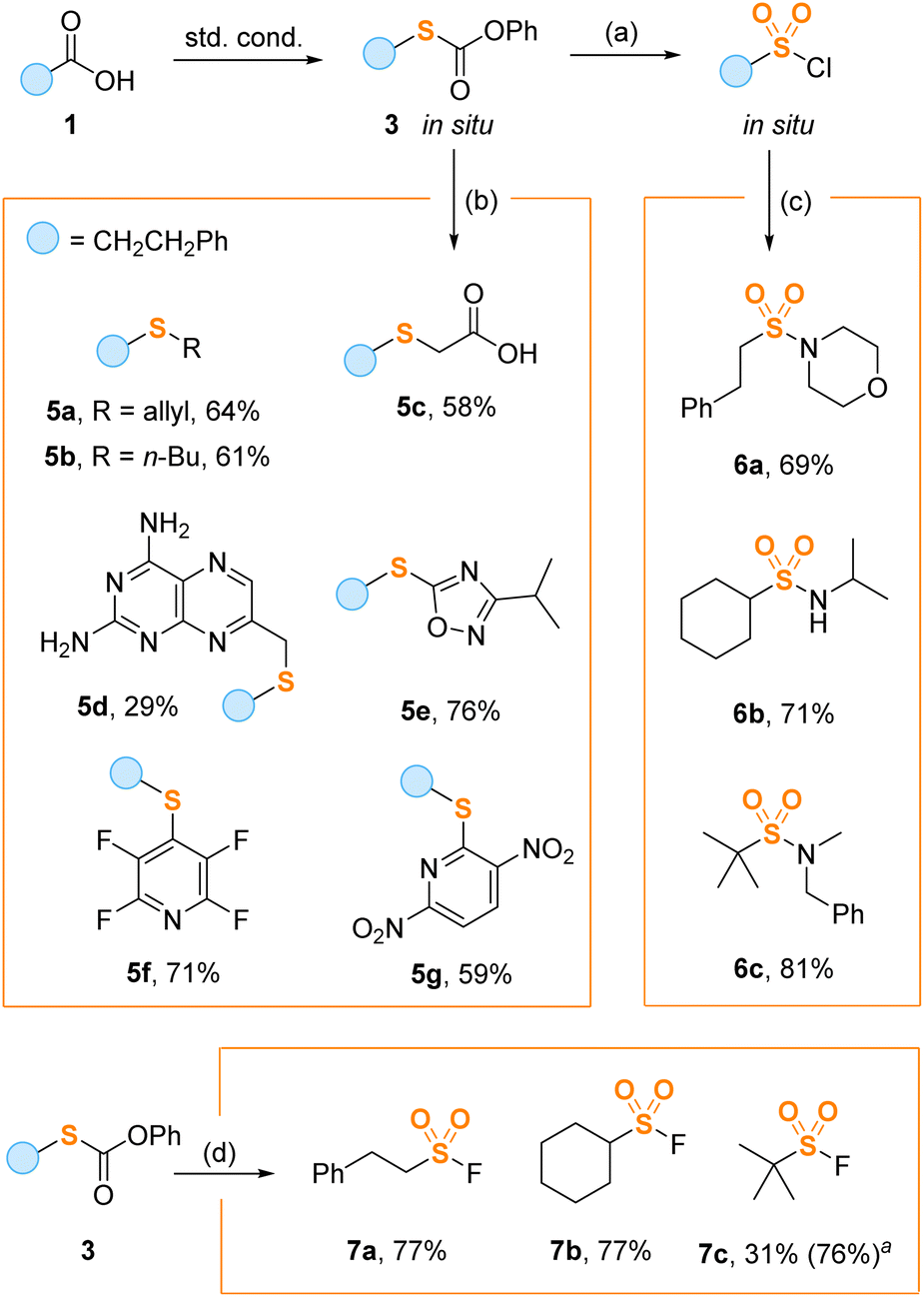 | ||
| Scheme 4 Derivatization reactions. Isolated yields based on carboxylic acids 1 are shown. Reaction conditions: (a) Cl2, AcOH, rt., 15 min; (b) R-Hal (5 equiv.), K2CO3 (5 equiv.), reflux, 4 h; (c) amine (5 equiv.), MeCN, 0 °C, 15 min; (d) Selectfluor, MeCN/H2O 10/1, 85 °C, 2 h. aYield was determined by 1H NMR using CH2Br2 as an internal standard. | ||
The proposed reaction mechanism involving the photocatalytic decarboxylative cycle is shown in Scheme 5. In the first step, photoexcited acridine reacts with carboxylic acid via a proton-coupled electron transfer (PCET) followed by decarboxylation with the formation of an alkyl radical, as previously reported by the Larionov group.18 This radical attacks the carbon–sulfur double bond of 2a followed by N–O bond cleavage and generation of the phthalimide radical. We believe that the latter species is responsible for the regeneration of the acridine catalyst through consecutive electron (ET) and proton (PT) transfer steps. To the best of our knowledge, this is the first case of acridine regeneration by nitrogen-centered radicals. Previously, this possibility was shown only for sulfur-centered radicals12c,18f,g and metal complexes.18a–e
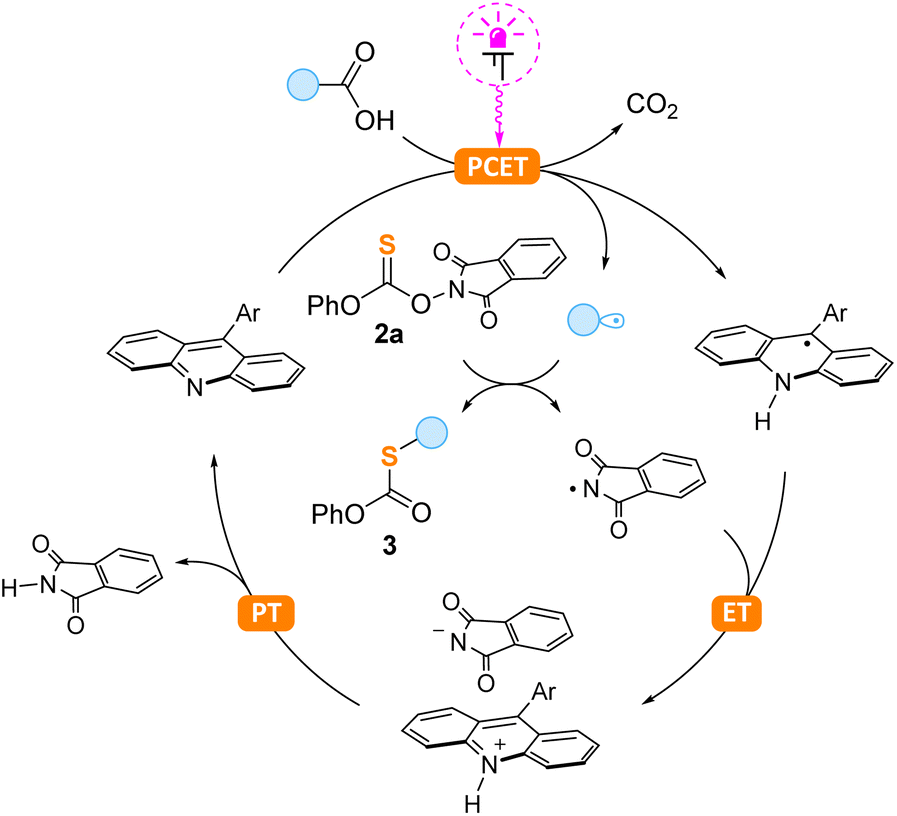 | ||
| Scheme 5 Proposed mechanism. | ||
To support the reaction mechanism, computational studies of the thiolation of isobutyric acid were performed using density functional theory. In Scheme 6, the calculated Gibbs free energy profile is shown (blue path). The formation of alkyl radicals on decarboxylation is a well-known exergonic process, which was previously analyzed in detail.22 The interaction of the isopropyl radical with thionocarbonate 2a is expected to proceed through the addition intermediate Int-1, which was located on the total electronic energy surface (orange path). However, due to entropic contribution, this species may not be a minimum on the reaction coordinate. Subsequent cleavage of the N–O bond is highly exergonic, and from Int-1 it requires only 4.4 kcal mol−1 (for TS-2) suggesting this to be extremely fast. Interestingly, in intermediate Int-1, the spin density is fully located on the phthalimide fragment (which corresponds to the canonical structure with separated charges), thereby making it predisposed for the formation of the phthalimide radical.
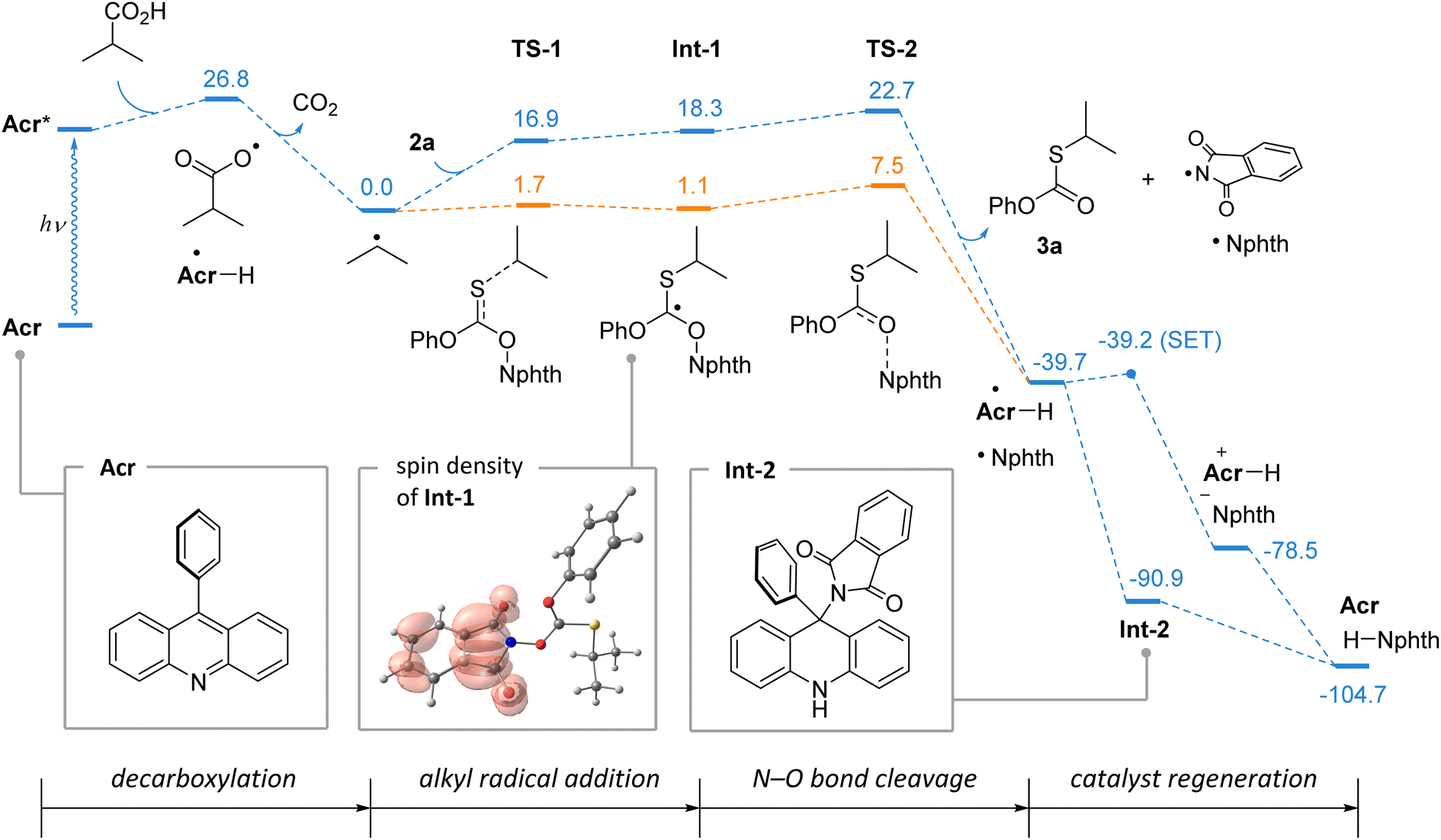 | ||
| Scheme 6 DFT calculations of the reaction mechanism. Performed at the ωB97XD/def2-TZVP/PCM(CH2Cl2) level of theory. Gibbs free energy profile (blue); for selected intermediates, total electronic energy is given (orange). Energies are given in kcal mol−1. | ||
The catalyst regeneration step involves interaction of phthalimide and acridinyl radicals resulting in two stable closed-shell molecules. This step is highly favorable with a driving force of 65 kcal mol−1, and is likely to be very efficient. A single electron transfer can generate the acridinium cation and phthalimide anion, and its barrier was estimated employing the Marcus theory to be 0.5 kcal mol−1, meaning that it is virtually diffusion controlled. Direct hydrogen atom transfer (HAT) between phthalimide and acridinyl radicals cannot be ruled out, which may further be alleviated by contribution of the electron transfer. Alternatively, two radicals may recombine leading to intermediate Int-2, and a similar process for the trapping of C-centered radicals was previously reported as the pathway of catalyst degradation.20 However, in the case of Int-2, its fragmentation is driven by aromatization of the acridine system, and is favored by 13.8 kcal mol−1.
To evaluate the importance of the nature of the alkyl radical in its addition to thionocarbonates, we studied the energetics of this step with various radicals and thione reagents (Scheme 7). Increase of stability of the alkyl radical decreases the thermodynamic efficiency of the addition (left part). For the benzyl radical, the reaction is most unfavorable with the Gibbs free energy of 27.6 kcal mol−1, while the enthalpy of benzyl radical addition is about six times higher than that of the ethyl radical. These values are consistent with the fact that the thiolation does not proceed with benzylic acids. Besides, we have shown that the phthalimide fragment makes a significant contribution to the destabilization of the intermediate Int-1 compared to derivatives of more conventional thionocarbonates and xanthates (right part). Indeed, replacement of the phthalimide fragment of 2a with a methoxy group leads to a drop in the Gibbs free energy by 9.5 kcal mol−1. In contrast, for conventional thionocarbonates (Y = OMe), the phenoxy substituent may favor the formation of the addition intermediate by 3.8 kcal mol−1. However, for phthalimide-substituted reagents (Y = ONphth), this effect virtually disappears, apparently, because of the localization of the spin density on the phthalimide fragment. This illustrates profile of the reaction by changing the nature of the addition that the phthalimide not only serves for the formation of the nitrogen-centered radical, but also may change the energetics of the intermediate.
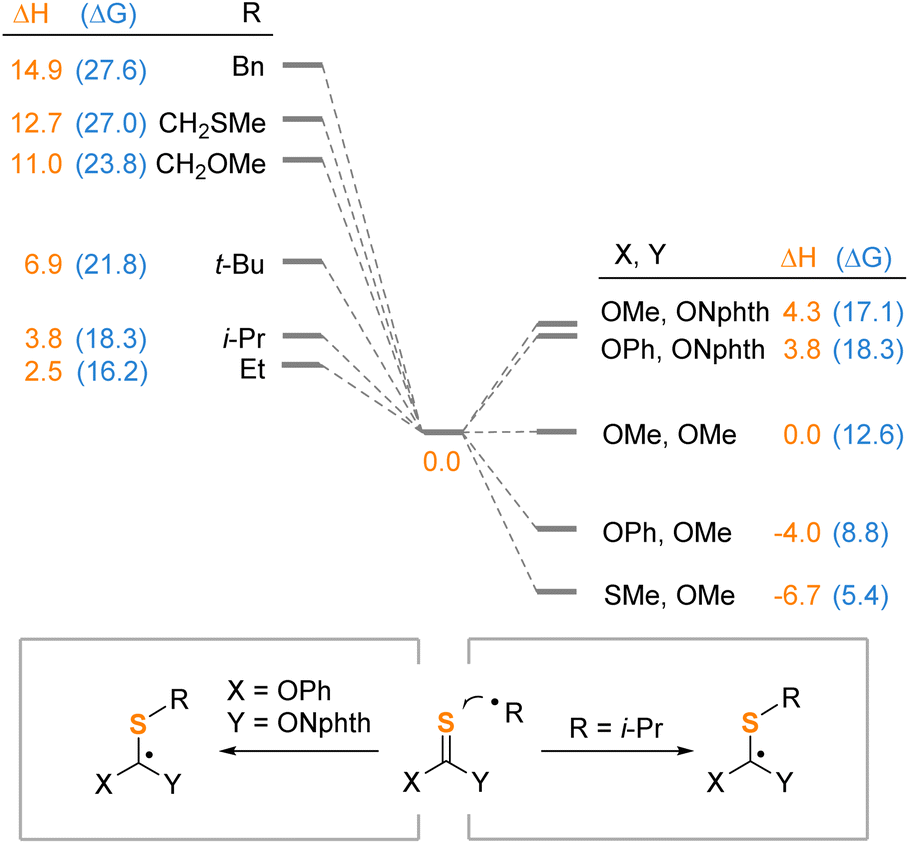 | ||
| Scheme 7 Substituent effects on the radical addition. Performed at the ωB97XD/def2-TZVP/PCM(CH2Cl2) level of theory. Energies are given in kcal mol−1. | ||
To validate the radical character of the thiolation protocol, we conducted the model reaction in the presence of common radical inhibitors (Scheme 8A). Thus, stable radical TEMPO completely inhibited the reaction by absorbing the intermediate isopropyl radical almost quantitatively forming product 8. The addition of butylated hydroxytoluene (BHT) serving as a hydrogen donor also considerably inhibited the process affording only 39% of 3a along with 94% conversion of the starting acid. Finally, presence of 1,1-diphenylethylene had almost no deleterious effect on the yield of 3a. The radical clock experiment with α-cyclopropyl substituted acid 10 afforded S-alkenyl thiocarbonate 11 exclusively (Scheme 8B).
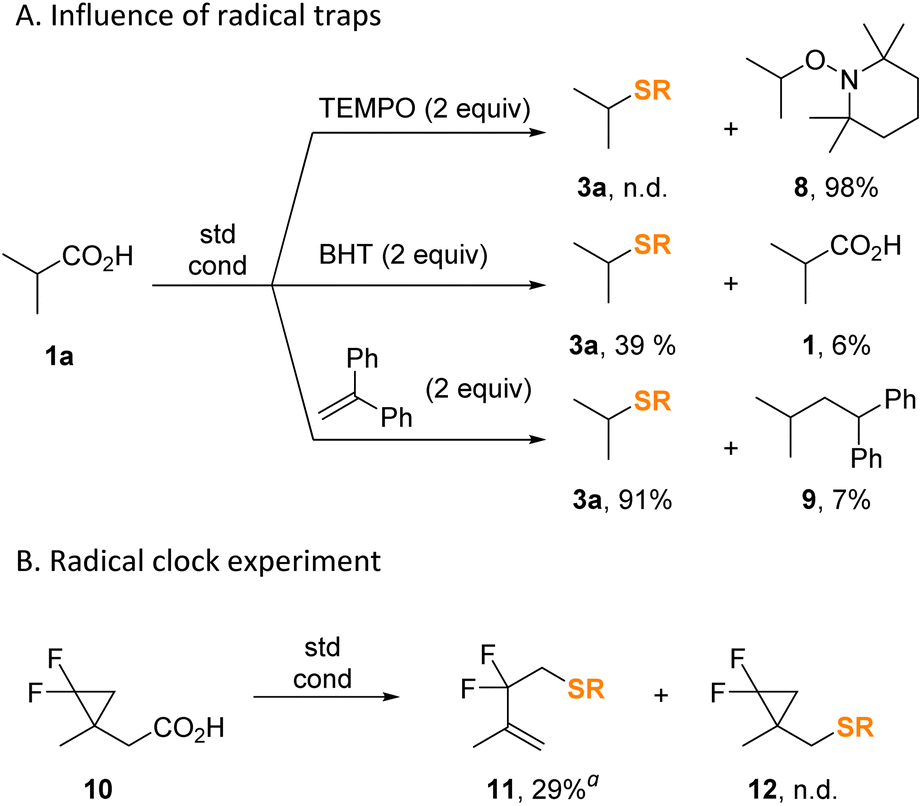 | ||
| Scheme 8 Mechanistic experiments. Yields were determined by 1H NMR using CH2Br2 as internal standard. Abbreviations: TEMPO, (2,2,6,6-tetramethylpiperidin-1-yl)oxyl; BHT, 2,6-di-tert-butyl-4-methylphenol. a Isolated yield. | ||
Conclusions
In summary, a practical method for the direct transformation of carboxylic acids to thiols was developed. The use of acridine photocatalysis in combination with a predesigned thionocarbonate reagent as a sulfur source is a key feature determining the reaction efficiency. The process is based on a favorable balance between several elementary steps including radical addition, N–O bond cleavage, and photocatalyst regeneration. The thiolation protocol allowed convenient one-pot conversion of unprotected acids into a variety of valuable sulfur compounds.Data availability
Procedures, characterization data, NMR spectra, and details of DFT calculations are available in the ESI.†Author contributions
Investigation, D. L. L. and M. O. Z.; conceptualization, M. O. Z.; data Curation, M. D. K.; writing – original draft, M. D. K.; writing – review & editing, M. O. Z. and A. D. D.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Russian Science Foundation (project 21-73-10129).References
- P. Chauhan, S. Mahajan and D. Enders, Chem. Rev., 2014, 114, 8807–8864 CrossRef CAS PubMed.
- (a) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2022, 122, 16110–16293 CrossRef CAS PubMed; (b) T. Kondo and T.-a. Mitsudo, Chem. Rev., 2000, 100, 3205–3220 CrossRef CAS PubMed; (c) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596–1636 CrossRef CAS PubMed.
- (a) Z. Wu and D. A. Pratt, Nat. Rev. Chem, 2023, 7, 573–589 CrossRef; (b) A. Wimmer and B. König, Beilstein J. Org. Chem., 2018, 14, 54–83 CrossRef CAS PubMed; (c) D. Yang, Q. Yan, E. Zhu, J. Lv and W.-M. He, Chin. Chem. Lett., 2022, 33, 1798–1816 CrossRef CAS; (d) R. Cannalire, S. Pelliccia, L. Sancineto, E. Novellino, G. C. Tron and M. Giustiniano, Chem. Soc. Rev., 2021, 50, 766–897 RSC.
- (a) Y. Liu, S. Liu and Y. Xiao, Beilstein J. Org. Chem., 2017, 13, 589–611 CrossRef CAS PubMed; (b) G. Mercey, V. Reboul, M. Gulea, J. Levillain and A.-C. Gaumont, Eur. J. Org Chem., 2012, 2012, 5423–5434 CrossRef CAS; (c) J. Clayden and P. MacLellan, Beilstein J. Org. Chem., 2011, 7, 582–595 CrossRef CAS PubMed; (d) I. V. Koval, Russ. J. Org. Chem., 2005, 41, 631–648 CrossRef CAS; (e) C. M. Rayner, Contemp. Org. Synth., 1996, 3, 499–533 RSC.
- (a) D. Sticker, R. Geczy, U. O. Häfeli and J. P. Kutter, ACS Appl. Mater. Interfaces, 2020, 12, 10080–10095 CrossRef CAS; (b) A. B. Lowe, Polym. Chem., 2014, 5, 4820–4870 RSC; (c) A. C. Weems, K. R. Delle Chiaie, R. Yee and A. P. Dove, Biomacromolecules, 2020, 21, 163–170 CrossRef CAS PubMed; (d) A. C. Weems, K. R. Delle Chiaie, J. C. Worch, C. J. Stubbs and A. P. Dove, Polym. Chem., 2019, 10, 5959–5966 RSC; (e) A. Oesterreicher, J. Wiener, M. Roth, A. Moser, R. Gmeiner, M. Edler, G. Pinter and T. Griesser, Polym. Chem., 2016, 7, 5169–5180 RSC.
- (a) V. Montes-García, M. A. Squillaci, M. Diez-Castellnou, Q. K. Ong, F. Stellacci and P. Samorì, Chem. Soc. Rev., 2021, 50, 1269–1304 RSC; (b) K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- (a) Q. Xiao, Q. X. Tong and J. J. Zhong, Molecules, 2022, 27, 619 CrossRef CAS PubMed; (b) C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed; (c) A. K. Sinha and D. Equbal, Asian J. Org. Chem., 2019, 8, 32–47 CrossRef CAS; (d) J. V. Burykina, A. D. Kobelev, N. S. Shlapakov, A. Y. Kostyukovich, A. N. Fakhrutdinov, B. Konig and V. P. Ananikov, Angew Chem. Int. Ed. Engl., 2022, 61, e202116888 CrossRef CAS PubMed; (e) V. V. Levin and A. D. Dilman, J. Org. Chem., 2019, 84, 8337–8343 CrossRef CAS PubMed.
- (a) M. Ahangarpour, I. Kavianinia, P. W. R. Harris and M. A. Brimble, Chem. Soc. Rev., 2021, 50, 898–944 RSC; (b) M. D. Nolan and E. M. Scanlan, Front. Chem., 2020, 8 Search PubMed; (c) K. S. Lim, J. H. Galarraga, X. Cui, G. C. J. Lindberg, J. A. Burdick and T. B. F. Woodfield, Chem. Rev., 2020, 120, 10662–10694 CrossRef CAS PubMed; (d) T. O. Machado, C. Sayer and P. H. H. Araujo, Eur. Polym. J., 2017, 86, 200–215 CrossRef CAS.
- (a) J. Iglesias, I. Martínez-Salazar, P. Maireles-Torres, D. Martin Alonso, R. Mariscal and M. López Granados, Chem. Soc. Rev., 2020, 49, 5704–5771 RSC; (b) Q. Wu, X. Bao, W. Guo, B. Wang, Y. Li, H. Luo, H. Wang and N. Ren, Biotechnol. Adv., 2019, 37, 599–615 CrossRef CAS; (c) P. Ertl and T. Schuhmann, J. Nat. Prod., 2019, 82, 1258–1263 CrossRef CAS PubMed; (d) T. Henkel, R. M. Brunne, H. Müller and F. Reichel, Angew. Chem., Int. Ed., 1999, 38, 643–647 CrossRef CAS.
- (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS; (b) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS; (c) F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC; (d) S. Crespi and M. Fagnoni, Chem. Rev., 2020, 120, 9790–9833 CrossRef CAS PubMed; (e) L. Candish, K. D. Collins, G. C. Cook, J. J. Douglas, A. Gómez-Suárez, A. Jolit and S. Keess, Chem. Rev., 2022, 122, 2907–2980 CrossRef CAS PubMed; (f) H. Wang, Z. Liu, A. Das, P. Bellotti, S. Megow, F. Temps, X. Qi and F. Glorius, Nat. Synth., 2023, 2, 1116–1126 CrossRef.
- (a) S. B. Beil, T. Q. Chen, N. E. Intermaggio and D. W. C. MacMillan, Acc. Chem. Res., 2022, 55, 3481–3494 CrossRef CAS PubMed; (b) S. K. Parida, T. Mandal, S. Das, S. K. Hota, S. De Sarkar and S. Murarka, ACS Catal., 2021, 11, 1640–1683 CrossRef CAS; (c) S. Karmakar, A. Silamkoti, N. A. Meanwell, A. Mathur and A. K. Gupta, Adv. Synth. Catal., 2021, 363, 3693–3736 CrossRef CAS; (d) J. Schwarz and B. König, Green Chem., 2018, 20, 323–361 RSC; (e) J. Xuan, Z.-G. Zhang and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632–15641 CrossRef CAS; (f) G. A. Lutovsky, S. N. Gockel, M. W. Bundesmann, S. W. Bagley and T. P. Yoon, Chem, 2023, 9, 1610–1621 CrossRef CAS PubMed.
- (a) F. Dénès, M. Pichowicz, G. Povie and P. Renaud, Chem. Rev., 2014, 114, 2587–2693 CrossRef PubMed; (b) V. I. Supranovich, V. V. Levin, V. A. Kokorekin and A. D. Dilman, Adv. Synth. Catal., 2021, 363, 2888–2892 CrossRef CAS; (c) M. O. Zubkov, M. D. Kosobokov, V. V. Levin and A. D. Dilman, Org. Lett., 2022, 24, 2354–2358 CrossRef CAS PubMed; (d) L. I. Panferova, M. O. Zubkov, V. A. Kokorekin, V. V. Levin and A. D. Dilman, Angew. Chem., Int. Ed., 2021, 60, 2849–2854 CrossRef CAS; (e) J. D. Griffin, M. A. Zeller and D. A. Nicewicz, J. Am. Chem. Soc., 2015, 137, 11340–11348 CrossRef CAS PubMed; (f) J.-P. R. Chauvin, M. Griesser and D. A. Pratt, J. Am. Chem. Soc., 2017, 139, 6484–6493 CrossRef CAS PubMed; (g) M. Newcomb, A. G. Glenn and M. B. Manek, J. Org. Chem., 1989, 54, 4603–4606 CrossRef CAS.
- M. Galicia and F. J. González, J. Electrochem. Soc., 2002, 149, D46 CrossRef CAS.
- (a) T. Cao, T. Xu, R. Xu, X. Shu and S. Liao, Nat. Commun., 2020, 11, 5340 CrossRef CAS; (b) Z. Wu and D. A. Pratt, J. Am. Chem. Soc., 2020, 142, 10284–10290 CrossRef CAS; (c) T. Xu, T. Cao, M. Yang, R. Xu, X. Nie and S. Liao, Org. Lett., 2020, 22, 3692–3696 CrossRef CAS.
- (a) C. G. Na, D. Ravelli and E. J. Alexanian, J. Am. Chem. Soc., 2020, 142, 44–49 CrossRef CAS; (b) R. Mao, S. Bera, A. C. Turla and X. Hu, J. Am. Chem. Soc., 2021, 143, 14667–14675 CrossRef CAS PubMed.
- L. I. Panferova, M. O. Zubkov, M. D. Kosobokov and A. D. Dilman, Org. Lett., 2022, 24, 8559–8563 CrossRef CAS PubMed.
- Z. Wu and D. A. Pratt, Angew Chem. Int. Ed. Engl., 2021, 60, 15598–15605 CrossRef CAS PubMed.
- (a) H. T. Dang, G. C. Haug, V. T. Nguyen, N. T. H. Vuong, V. D. Nguyen, H. D. Arman and O. V. Larionov, ACS Catal., 2020, 10, 11448–11457 CrossRef CAS PubMed; (b) V. T. Nguyen, V. D. Nguyen, G. C. Haug, H. T. Dang, S. Jin, Z. Li, C. Flores-Hansen, B. S. Benavides, H. D. Arman and O. V. Larionov, ACS Catal., 2019, 9, 9485–9498 CrossRef CAS PubMed; (c) V. D. Nguyen, R. Trevino, S. G. Greco, H. D. Arman and O. V. Larionov, ACS Catal., 2022, 12, 8729–8739 CrossRef CAS PubMed; (d) V. T. Nguyen, G. C. Haug, V. D. Nguyen, N. T. H. Vuong, H. D. Arman and O. V. Larionov, Chem. Sci., 2021, 12, 6429–6436 RSC; (e) V. T. Nguyen, V. D. Nguyen, G. C. Haug, N. T. H. Vuong, H. T. Dang, H. D. Arman and O. V. Larionov, Angew. Chem., Int. Ed., 2020, 59, 7921–7927 CrossRef CAS PubMed; (f) V. T. Nguyen, G. C. Haug, V. D. Nguyen, N. T. H. Vuong, G. B. Karki, H. D. Arman and O. V. Larionov, Chem. Sci., 2022, 13, 4170–4179 RSC; (g) V. D. Nguyen, G. C. Haug, S. G. Greco, R. Trevino, G. B. Karki, H. D. Arman and O. V. Larionov, Angew. Chem., Int. Ed., 2022, 61, e202210525 CrossRef CAS PubMed; (h) H. T. Dang, A. Porey, S. Nand, R. Trevino, P. Manning-Lorino, W. B. Hughes, S. O. Fremin, W. T. Thompson, S. K. Dhakal, H. D. Arman and O. V. Larionov, Chem. Sci., 2023, 14, 13384–13391 RSC; (i) J. A. Andrews, J. Kalepu, C. F. Palmer, D. L. Poole, K. E. Christensen and M. C. Willis, J. Am. Chem. Soc., 2023, 145, 21623–21629 CrossRef CAS PubMed.
- S. Klaus, W. Richard and G. Ferdinand, US Pat., US2872450, 1959 Search PubMed.
- K. A. Zhilyaev, D. L. Lipilin, M. D. Kosobokov, A. I. Samigullina and A. D. Dilman, Adv. Synth. Catal., 2022, 364, 3295–3301 CrossRef CAS.
- (a) J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430–9448 CrossRef CAS PubMed; (b) C. J. Smedley, J. A. Homer, T. L. Gialelis, A. S. Barrow, R. A. Koelln and J. E. Moses, Angew. Chem., Int. Ed., 2022, 61, e202112375 CrossRef CAS PubMed.
- (a) G. dos Passos Gomes, A. Wimmer, J. M. Smith, B. König and I. V. Alabugin, J. Org. Chem., 2019, 84, 6232–6243 CrossRef CAS PubMed; (b) L. Kuhn, V. A. Vil’, Y. A. Barsegyan, A. O. Terent’ev and I. V. Alabugin, Org. Lett., 2022, 24, 3817–3822 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2271075. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05513b |
| This journal is © The Royal Society of Chemistry 2024 |