
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControl of the fluorescence lifetime in dye based nanoparticles†
Stine G.
Stenspil
 a,
Junsheng
Chen
a,
Mikkel B.
Liisberg
a,
Amar H.
Flood
b and
Bo W.
Laursen
*a
a,
Junsheng
Chen
a,
Mikkel B.
Liisberg
a,
Amar H.
Flood
b and
Bo W.
Laursen
*a
aNano-Science Center & Department of Chemistry, University of Copenhagen, Universitetsparken 5, 2100 København Ø, Denmark. E-mail: bwl@chem.ku.dk
bDepartment of Chemistry, Indiana University, 800 East Kirkwood Avenue, Bloomington, Indiana 47405, USA
First published on 29th February 2024
Abstract
Fluorescent dye based nanoparticles (NPs) have received increased interest due to their high brightness and stability. In fluorescence microscopy and assays, high signal to background ratios and multiple channels of detection are highly coveted. To this end, time-resolved imaging offers suppression of background and temporal separation of spectrally overlapping signals. Although dye based NPs and time-resolved imaging are widely used individually, the combination of the two is uncommon. This is likely due to that dye based NPs in general display shortened and non-mono-exponential lifetimes. The lower quality of the lifetime signal from dyes in NPs is caused by aggregation caused quenching (ACQ) and energy migration to dark states in NPs. Here, we report a solution to this problem by the use of the small-molecule ionic isolation lattices (SMILES) concept to prevent ACQ. Additionally, incorporation of FRET pairs of dyes locks the exciton on the FRET acceptor providing control of the fluorescence lifetime. We demonstrate how SMILES NPs with a few percent rhodamine and diazaoxatriangulenium FRET acceptors imbedded with a cyanine donor dye give identical emission spectra and high quantum yields but very different fluorescence lifetimes of 3 ns and 26 ns, respectively. The two spectrally identical NPs are easily distinguished at the single particle level in fluorescence lifetime imaging. The doping approach for dye based NPs provides predictable fluorescence lifetimes and allows for these bright imaging reagents to be used in time-resolved imaging detection modalities.
Introduction
Nanoparticles (NPs) based on organic fluorophores and building blocks have recently attracted growing interest as they display a series of advantages compared to organic single molecule fluorophores and fluorescent proteins.1,2 For instance, NPs can offer very bright and stable emission compared to their molecular counterparts. To achieve these attractive properties in a NP small enough for bioimaging a high density of dyes needs to be realized in the NP. While packing hundreds of fluorophores in a small NP offers great advantages in terms of brightness,3 it also poses new challenges such as aggregation caused quenching (ACQ) and altered photophysical properties of the dyes due to dye–dye interactions.4,5 These challenges have limited the applications of dye based NPs, and only a small subset of the diverse photophysical properties applied in molecular probes and labels has been transferred to bright NPs with high dye density. One application not widely explored for dye based NPs is fluorescence lifetime imaging (FLIM) and time-resolved imaging. These techniques are gaining traction as they offer efficient suppression of background and auto-fluorescence, as well as temporal separation of spectrally similar signals and multiplexing.6–11 In particular, lanthanide-based emitters have shown large potential for a wide range of time-resolved applications with their μs to ms lifetimes (e.g., for time gating or using energy transfer to other emitting species).12–15 Compared to lanthanide-based emitters, organic fluorophores with ns lifetimes allow for a higher photon flux (photons per time). The low abundance of organic dye based NPs for lifetime imaging is likely due to their unpredictable fluorescence lifetimes. The close dye–dye distances in bright NPs with high dye density result in unwanted excited state processes, producing fluorescence lifetimes very different from that in solution.16 The potential of lifetime imaging using dye based NPs has been shown by Hoffmann et al. for polystyrene NPs with low dye loading.17,18 Despite low dye loading the fluorescence lifetime readout is still strongly affected by dye–dye interactions.Fluorophores with long fluorescence lifetimes (>10 ns) are particularly interesting to complement abundant dye classes with short ns lifetimes and because of their easy separation from background autofluorescence.19,20 However, dyes with long fluorescence lifetimes intrinsically have low molar absorption coefficients, as dictated by the Strickler–Berg relation,21 and thus low brightness. This limitation is difficult to bypass via synthetic design of the fluorophore itself.22 It is therefore favourable to combine molecular fluorophore properties in supramolecular fluorophore assemblies. One way to design such a supramolecular “hybrid” fluorophore was shown with the dyad design by Kacenauskaite et al. where the covalent combination of the highly absorbing perylene (donor) with a triangulenium (acceptor) dye generated a bright supramolecular dyad with a long fluorescence lifetime, enabling single molecule time-gated detection.23 We envision that the same supramolecular principle of hybrid properties generated by combination of molecular dyes can be realized in dye based NPs offering super-bright labels24–27 and markers with a broad range of predictable fluorescence lifetimes for lifetime based imaging and detection.
Previously, we introduced small-molecule ionic isolation lattices (SMILES) as an approach for maintaining solution state properties of cationic fluorophores in solid state materials such as crystals, thin films, and nanoparticles.28–30 By introducing the cyanostar (CS) anion receptor31 (Fig. 1a) the dyes become separated from each other by the large anion receptor complex (Fig. 1d), thus minimizing dye–dye interactions and ACQ, while still maintaining a very high dye density in the resulting materials. Although spectral properties, quantum yields, and brightness are greatly improved in SMILES NPs compared to their non-SMILES counterparts, fluorescence lifetimes are still inhomogeneous (non-mono-exponential) and often shortened compared to solution. This is a common trend for all dye based NPs with high dye density.
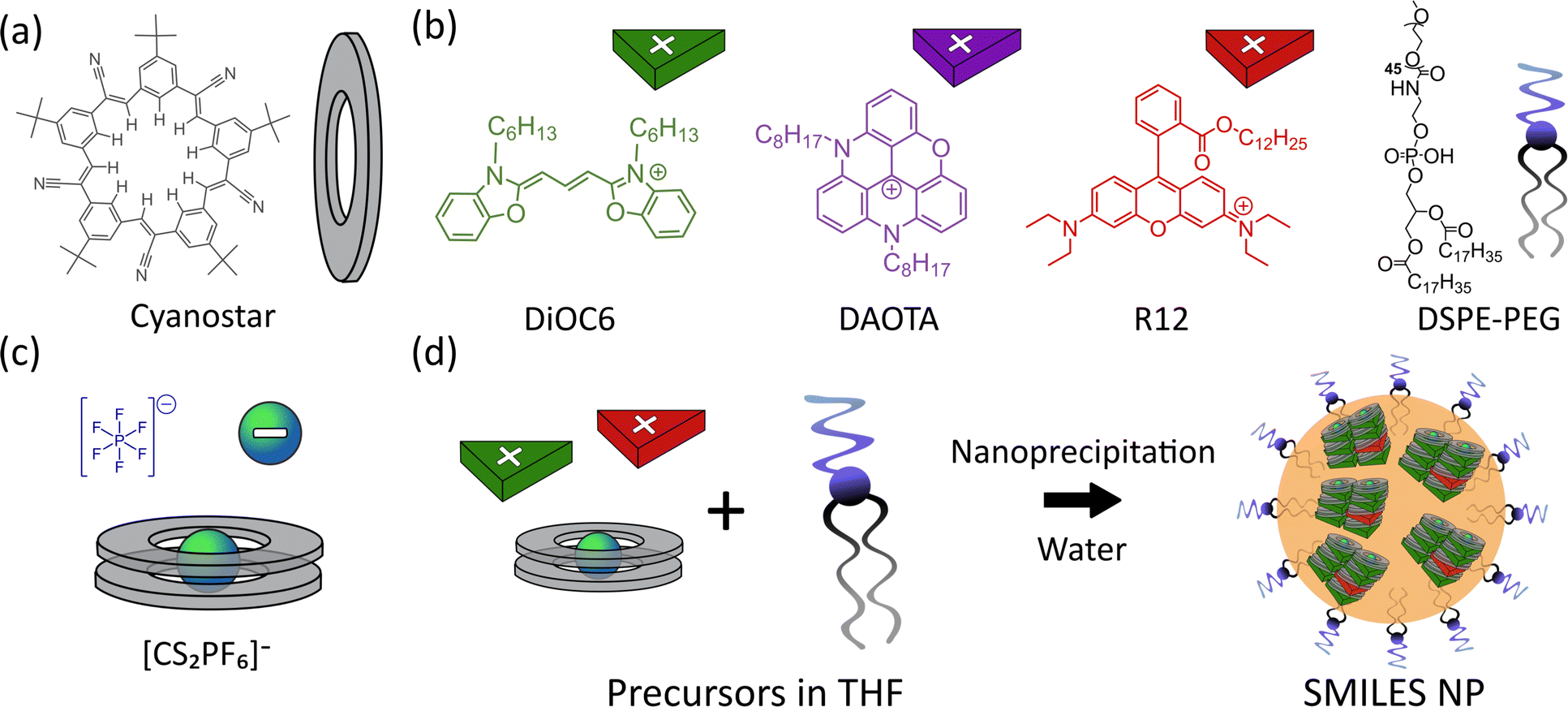 | ||
| Fig. 1 (a) Structure of the cyanostar anion receptor. (b) Structures of the cationic dyes (DiOC6, DAOTA, and R12) and DSPE-PEG used in the formation of SMILES NPs. (c) Schematic of the anion (PF6−) and anionic complex formed by 2 equivalents of cyanostar to 1 anion. (d) Schematic of the formation of doped SMILES NPs by nanoprecipitation of THF precursor solution in a large excess of water. | ||
The poor lifetime properties stem from exciton migration (homo-FRET) between the dyes in the NPs with high density, which increases the risk of fluorescence quenching by trap states in the NP material. The result of this quenching is both reduced fluorescence intensity and shortened multi-exponential fluorescence lifetimes. We recently studied these effects in crystals and thin films and showed that the introduction of a dopant fluorophore (FRET acceptor) into SMILES materials solved these issues and reinstated the mono-exponential lifetime of the acceptor (dopant) dye.16
In this work, we for the first time introduce FRET pairs in SMILES NPs. The use of specifically chosen FRET pairs increases the absorption and gives the possibility of modulating the emission properties very precisely. This allows for increasing the brightness many fold with the use of up to 50 donor dyes acting as antennas for each acceptor dye. By selecting acceptor dyes (R12 and DAOTA, Fig. 1b) previously shown to form SMILES with nearly identical emission spectra but very different fluorescence lifetime,16 we can engineer the fluorescence lifetime and obtain super bright NPs with lifetimes of ∼4 ns and 26 ns that are easily differentiable at the single particle level in FLIM and time-gated imaging.
Results and discussion
Preparation and characterization of NPs
Five NP compositions were prepared, with three neat SMILES NPs consisting of only the cyanine donor dye (DiOC6), or the acceptor dyes (rhodamine, R12, and diazaoxatriangulenium, DAOTA). These were prepared to establish the intrinsic properties of each dye in the SMILES NPs. Two doped NPs consisting of the FRET pairs DiOC6:R12 and DiOC6:DAOTA were prepared and optimized to achieve the best possible emission properties.All SMILES NPs were produced following a previously reported nanoprecipitation procedure.29 Briefly, a precursor solution in tetrahydrofuran (THF) was prepared with all the components needed to form SMILES NPs, including: the cationic dye(s) (DiOC6, R12 and DAOTA) as either PF6− or BF4− salts, the cyanostar macrocycle (2.5 mol eq.), and a 56 wt% amphiphilic surface capping agent (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-poly(ethylene glycol-2000) (DSPE-PEG)). The precursor solution was injected into a large excess of water under sonication to form the NPs. The doped NPs were prepared with varying mol% of the dopant (see the ESI† for details).
Dynamic light scattering of the NPs was carried out for both neat and doped NPs, which showed average sizes of ≈17 nm irrespective of the dye constituents. The hydrodynamic sizes are displayed in Tables S1, S2, Fig. S1 and S2.†
Neat SMILES NPs
SMILES NPs consisting of only one type of dye have been reported previously using either DiOC6 or R12.29,30 The emission for all neat SMILES NPs displays a slight red-shift ∼450 cm−1 compared to DCM solution, also observed previously. This shift is likely due to the difference in the dielectric environment. In the case of DiOC6, both the fluorescence quantum yield and lifetime are increased in SMILES NPs compared to DCM solution as shown in Fig. 2a and Table S4.† This is due to the rigid environment in the SMILES lattice compared to solution, which limits the non-radiative deactivation of this flexible cyanine dye.16 However, for R12 SMILES NPs (Fig. 2b), the emission intensity is lower and the fluorescence lifetime shows a shortened multi-exponential decay of 1.42 ns compared to 3.77 ns in DCM. Similar trends have been observed previously in crystalline SMILES materials16 and have been assigned to trap states. Even with a very low abundance of trap states, they can have a substantial impact on the photophysical properties due to very efficient energy migration in NPs with high dye density. In the case of DAOTA SMILES NPs the lifetime is longer (25 ns) than that in DCM (21 ns), but the fluorescence intensity is halved (Fig. 2c and Table S4†). The decay of the NP is fitted bi-exponentially, where one of the components is significantly shorter (12 ns) than the DAOTA lifetime in solution. This indicates that, while there may be less efficient vibrational relaxation, the fluorescence is still quenched by trap states in the NP structure. While each of the three dyes displays different changes in decay rates between solution and SMILES NPs, it is clear that the decays are non-mono-exponential and display short lifetime components, in particular for DiOC6 and R12 that have strong transition moments, thus the most efficient energy migration.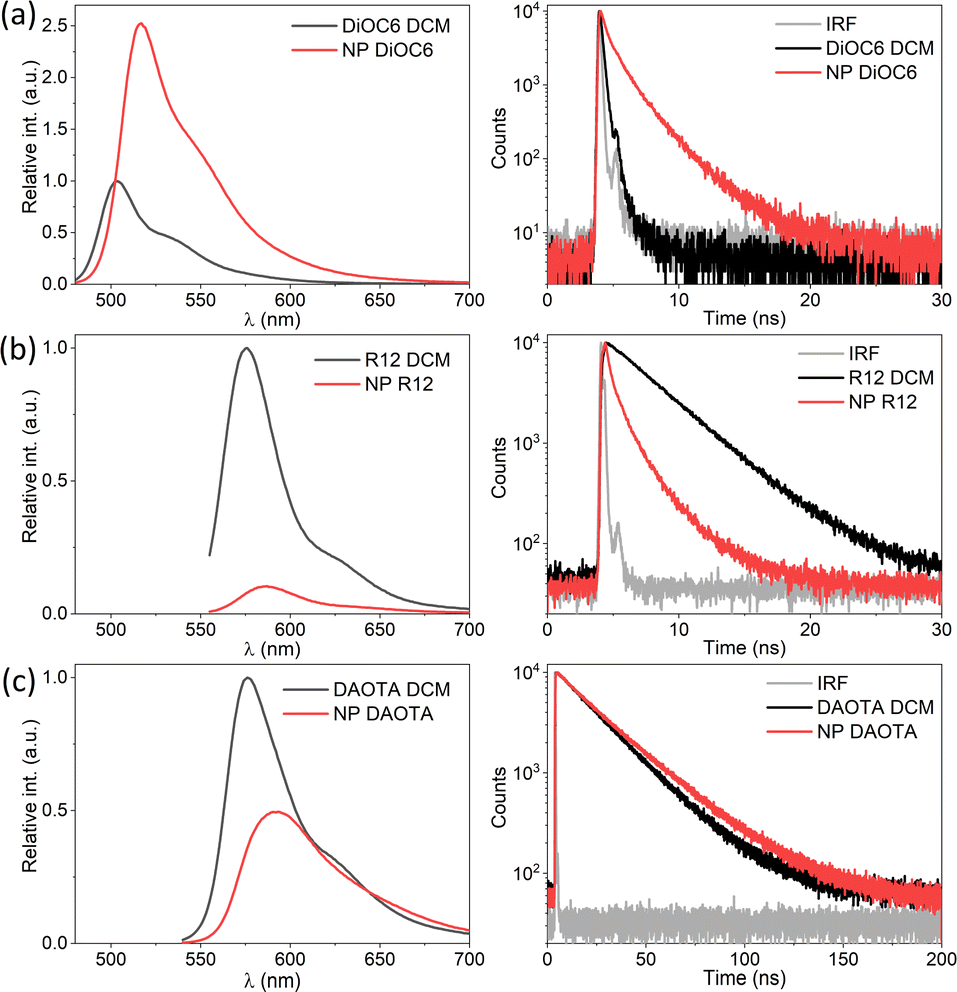 | ||
| Fig. 2 Fluorescence spectra and decays of neat NPs (red) compared to DCM solutions (black) for (a) DiOC6 (λex = 470 nm and λem det = 550 nm), (b) R12 (λex = 550 nm, λex = 530 nm and λem det = 550 nm) and (c) DAOTA (λex = 530 nm and λem det = 650 nm). | ||
FRET SMILES NPs
The neat SMILES NPs containing only one type of dye are vulnerable to trap sites (as seen in the data in Fig. 2). The traps can be of different origins, either as synthetic impurities in the fluorophore stock, impurities present at the nanoparticle formation or defects in the lattice order due to the rapid formation of NPs. No matter the origin, a single trap can quench a large number of dyes due to energy migration, as illustrated in the state diagram in Fig. 3a. This can be seen for both neat R12 and DAOTA NPs where their fluorescence intensity is drastically lowered and a new short fluorescence lifetime component appears. For the R12 NPs, this is particularly significant as shown above (Fig. 2b). To suppress quenching from trap states, we introduce dopant fluorophores (FRET acceptors) to the nanoparticles. We expect that most of the excitations will be transferred to the dopants (Fig. 3b). Since the S1 states of the dopants are lower in energy and are on average far apart, the exciton will be transferred to and locked on a single dopant and decay with the intrinsic radiative properties of that dopant.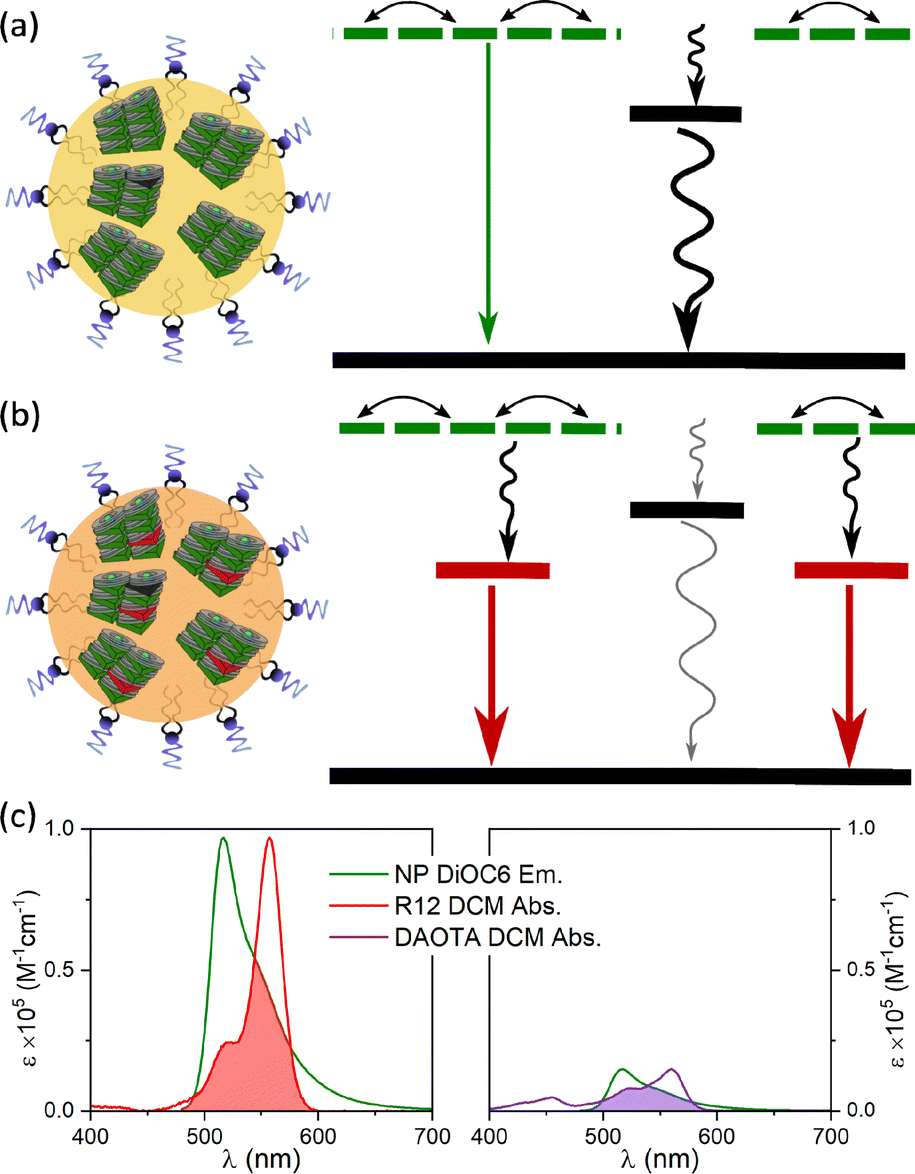 | ||
| Fig. 3 State diagram depicting (a) NPs with trap states quenching the fluorescence and (b) doped NPs where the excitation energy is primarily funnelled to emissive dopant sites. (c) NP DiOC6 emission normalised to the acceptor dye molar absorption spectra R12 (left) and DAOTA (right). The spectral overlaps are shaded in red and purple respectively. | ||
To improve the brightness of the SMILES NPs we use the highly absorbing DiOC6 fluorophore as the antenna (FRET donor, ε ≈ 143![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1) and introduce a redshifted dye (R12 or DAOTA) as the dopant (absorption and emission spectra are compared in Fig. S4†). Fig. 3C shows the large spectral overlap between DiOC6 NP emission and the absorption of either R12 or DAOTA in DCM, normalised to the acceptor absorption coefficient. This illustrates the different spectral overlap integrals, which are crucial for the efficiency of energy transfer. As the absorption coefficient of R12 is approximately 5 times larger than that of DAOTA (Table S5†), rhodamine is expected to be a more efficient acceptor at lower doping percentages than DAOTA. Previous work on crystals, powders, and thin films has shown that a doping degree of 1–12 mol% will yield materials with highly efficient energy transfer.16
000 M−1 cm−1) and introduce a redshifted dye (R12 or DAOTA) as the dopant (absorption and emission spectra are compared in Fig. S4†). Fig. 3C shows the large spectral overlap between DiOC6 NP emission and the absorption of either R12 or DAOTA in DCM, normalised to the acceptor absorption coefficient. This illustrates the different spectral overlap integrals, which are crucial for the efficiency of energy transfer. As the absorption coefficient of R12 is approximately 5 times larger than that of DAOTA (Table S5†), rhodamine is expected to be a more efficient acceptor at lower doping percentages than DAOTA. Previous work on crystals, powders, and thin films has shown that a doping degree of 1–12 mol% will yield materials with highly efficient energy transfer.16
Doped NPs also allow easy tuning of the spectral properties to fit specific needs and the combination of the most valuable properties of several fluorophores. We chose these three dyes specifically as a proof of concept as R12 and DAOTA have strikingly similar emission spectra but significantly different fluorescence lifetimes.
Optimizing doping of nanoparticles
Fig. 4a shows the absorption of SMILES DiOC6 NPs with increasing mol% R12. The main absorption bands that can be seen are from the cyanostar–anion complex [CS2PF6]− at ∼310 nm and DiOC6 at ∼490 nm. The absorption from R12 at 560 nm is only visible at higher doping degrees (Fig. S5a†) due to the low concentration compared to DiOC6 and CS. The fluorescence from R12, on the contrary, is very pronounced, even at low doping degrees; this is clearly seen when comparing neat DiOC6 NPs to NPs doped with R12 (Table 1).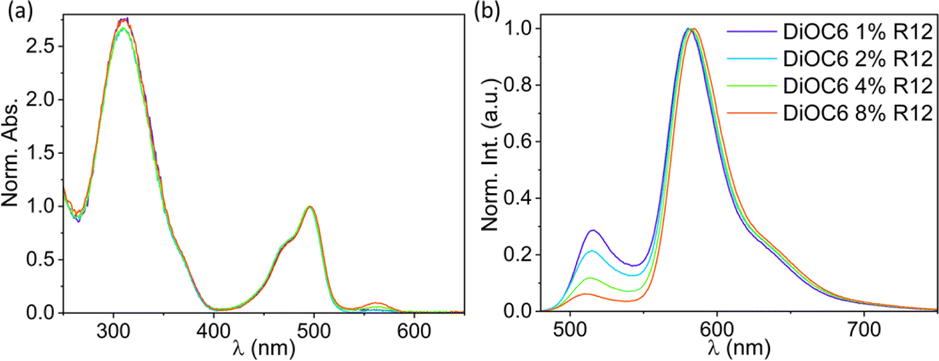 | ||
| Fig. 4 Normalized (a) absorption and (b) emission spectra (λex = 470 nm) of DiOC6 NPs doped with R12 ranging from 1% to 8%. The same colour coding is used for the absorption spectra as for the emission spectra. | ||
| R12% mol | τ int,520nm (ns) | τ int,650nm (ns) | QY | QY for emission in the R12 banda |
|---|---|---|---|---|
| a QY was calculated for emission in the R12 band beyond 545 nm. b Relative QY from Table S4. | ||||
| 0 | 1.41 | n.a. | 0.13 | n.a. |
| 1 | 1.25 | 3.19 | 0.32 | 0.27 |
| 2 | 1.53 | 3.19 | 0.27 | 0.24 |
| 4 | 1.58 | 2.59 | 0.27 | 0.24 |
| 8 | 1.41 | 1.89 | 0.20 | 0.19 |
| 100 | n.a. | 1.42 | n.a. | 0.06b |
Already after introduction of 1% R12, the fluorescence quantum yield is more than doubled and the emission from the R12 dopant displays a lifetime of 〈τ〉 = 3.2 ns (detected at 650 nm). Both observations agree with the dopant model (Fig. 3b), predicting an increase in quantum yield as excitons get locked on R12 instead of on dark trap states.
As the doping degree is increased, the emission from DiOC6 ∼510 nm decreases compared to emission from the R12 dopant at ∼580 nm, as shown in Fig. 4b. This indicates that more excitation energy is transferred to the R12 dopant dyes. This interpretation is also reflected in the quantum yield calculated only based on the R12 band (Table 1). The fluorescence decays detected in the DiOC6 band do not change significantly with the doping level (Table 1 and Fig. S6C†). This suggests that residual emission from the donor dye likely stems from particles or regions with no acceptor dyes present, while in other particles/regions, where there are acceptor dyes, the energy transfer is so fast that the donor emission is not detectable within the time-resolution of the instrument. This leads us to conclude that the energy transfer is faster than our time-resolution (∼50 ps) and that the efficiency is not limited by the rate but by the inhomogeneity of the NPs.
As the doping degree increases the energy transfer will be more complete and a larger fraction of the emission will stem from the dopant dye. However, the quantum yield and fluorescence lifetime start to decrease beyond 2 mol% of the dopant (Table 1) indicating that new pathways of quenching are present. These could be the generation of new trap states by R12 and/or that the R12 dopants are now close enough to facilitate energy migration between R12 sites and thus also to dark trap states in the structure. It would be possible to achieve 100% energy transfer from the donor to the acceptor, but this will come at the cost of increased energy migration (homo-FRET) between the acceptor dyes and thereby a reduced fluorescence lifetime. The optimal quantum yield and lifetime are found using 1–2 mol% with ϕ ≈ 0.3 and 〈τ〉 = 3.2 ns, which, compared to the neat R12 NPs (Table S4,†ϕ ≈ 0.06 and 〈τ〉〉 = 1.4 ns), is a substantial improvement. Due to the ∼6 times lower absorption coefficient of DAOTA (Table S5†), a higher doping concentration is needed to achieve efficient energy transfer to this dopant. Still, similar quantum yield and fluorescence lifetime trends are observed for the DiOC6:DAOTA NPs, as shown in Fig. S6† and summarized in Table 2. In this case, the optimal doping level is found to be 4 mol% DAOTA resulting in a NP with ϕ = 0.43 and τ = 26 ns. For both doped NPs, a completely mono-exponential decay is not achieved. Yet, the fluorescence lifetime of the R12 doped NPs (∼3.2 ns) is significantly increased compared to the neat (100% mol) R12 NPs (∼1.42 ns, Table 1 and Fig. S7†) and closer to the lifetime of R12 in organic solution (3.7 ns). Such a large improvement is not seen for NPs doped with DAOTA (∼26 ns compared to neat DAOTA NPs, 24.8 ns, Table 2). This is most likely due to the moderate oscillator strength of DAOTA that reduces the rate of homo-FRET and energy migration in neat DAOTA SMILES NPs, and therefore also quenching by trap states. In comparison to FRET SMILES microcrystals, the lifetimes measured for R12 doped NPs display a slightly shorter fluorescence lifetime (3.6 ns in crystals to 3.2 ns in NPs). For NPs with DAOTA as an acceptor, the fluorescence lifetime is longer than that observed in microcrystalline SMILES (23.3 ns to 25.6 ns in NPs). This lifetime is also close to the radiative lifetime of DAOTA, 27.9 ns, suggesting that there is very low non-radiative relaxation when the exciton is on the DAOTA dye. The two optimized FRET doped SMILES NPs (DiOC6:R12 1 mol% and DiOC6:DAOTA 4 mol%) display remarkably similar absorption and emission profiles but very different fluorescence decays (Fig. 5). In this regard, the indistinguishable emission spectra of the two NPs are ideal to demonstrate applications of dye based NPs for time-resolved imaging.
| DAOTA% mol | τ int,520nm (ns) | τ int,650nm (ns) | QY | QY for emission in the DAOTA banda |
|---|---|---|---|---|
| a QY was calculated for emission in the DAOTA band beyond 545 nm. b Relative QY from Table S4. | ||||
| 0 | 1.41 | n.a. | 0.13 | n.a. |
| 1 | 2.30 | 25.6 | 0.41 | 0.35 |
| 2 | 2.31 | 26.2 | 0.43 | 0.39 |
| 4 | 2.92 | 25.6 | 0.43 | 0.41 |
| 8 | 2.03 | 23.3 | 0.40 | 0.38 |
| 100 | n.a | 24.8 | n.a. | 0.34b |
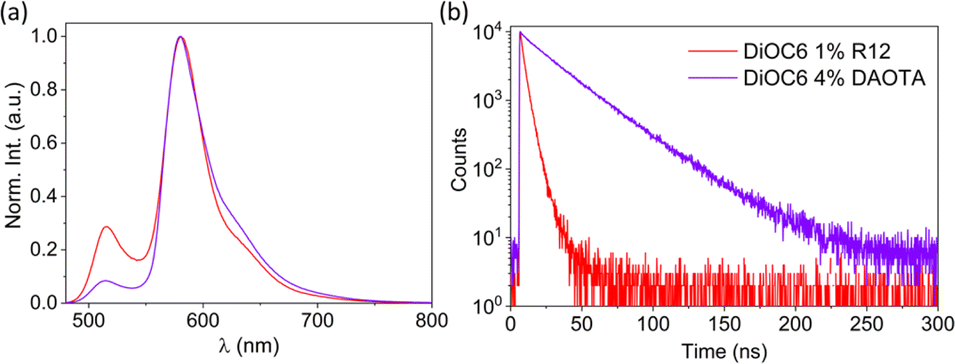 | ||
| Fig. 5 (a) Normalized emission spectra (λex = 470 nm) and (b) fluorescence decays (λex = 470 nm and λem det = 650 nm) of DiOC6 NPs doped with 1% R12 or 4% DAOTA. | ||
Fluorescence lifetime imaging
Fig. 6 displays FLIM images of single doped NPs without DSPE-PEG spincoated on a glass coverslip recorded using a 561 nm longpass optical filter to selectively detect the acceptor dye emission. For these measurements of NPs on dry substrates, the DSPE-PEG coating was omitted from the NP preparation. This was to avoid the detergent mixing with (dissolving) the NP SMILES structure as water evaporates and leaves the PEG chains less polar and mixable with the lipophilic dye components. While the NP sizes without coating are slightly smaller ∼14 nm (Fig. S3 and Table S3†), their optical properties remain largely unaffected (Fig. S8†), as previously reported.29 FLIM images of the doped NPs with either R12 or DAOTA show good lifetime homogeneity either being short ∼4 ns (Fig. 6a) or long ∼25 ns (Fig. 6b). We note that the DAOTA NPs apparently show a broader lifetime distribution than the R12 NPs (Fig. S12†). The broad distribution could indicate a larger particle-to-particle variation; however the percentage deviations from the mean lifetimes of the two NPs are not that different. Nonetheless, the DAOTA NPs exhibit significantly longer lifetimes than the R12 NPs, and we proceeded by preparing a mixture of the two types of NPs, as shown in Fig. 6c. In the mixed sample, both characteristic short- and long-lived NPs show up as red and blue, respectively.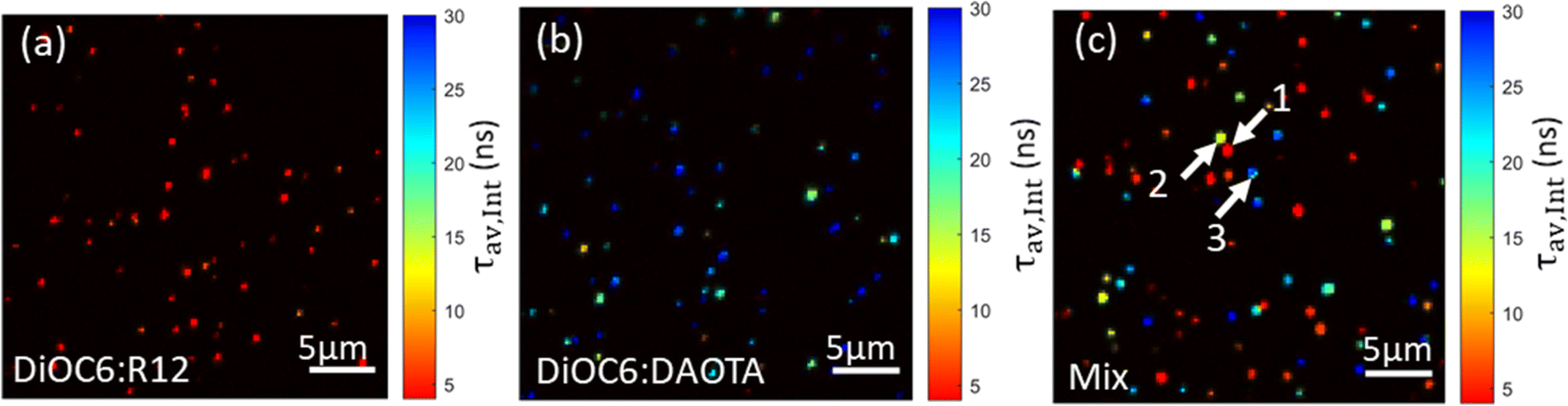 | ||
| Fig. 6 Pseudocolored average intensity weighted lifetime images of doped SMILES NPs; (a) DiOC6 1% R12, (b) DiOC6 4% DAOTA, and (c) a mixture of both NPs. | ||
However, some NPs with an average lifetime of between 4 and 25 ns show up in yellow/green. Even though highly dilute solutions were used to ensure single NP imaging some NPs will likely by chance be in close vicinity to each other and result in overlapping signals. The particles that display ∼12 ns lifetime might be due to two NPs with a short and long lifetime being too close to be resolved spatially. Analysis of the fluorescence decays of three different areas/spots in the image (marked by arrows in Fig. 6c) seems to support this hypothesis. Particle 1 and particle 3 reveal good mono-exponential decays of 3 ns and 28 ns in agreement with cuvette measurements of R12 and DAOTA doped NPs (Fig. S14†). For particle 2, a bi-exponential fit describes the decay best yielding two lifetimes of 8 and 24 ns (Fig. S14B†). The deviation of the lifetimes from cuvette measurements is likely due to the low photon count acquired for each pixel in the image.
Another approach, where different lifetimes can be utilized, is time-gating. In this case, a two-coloured image is generated by depicting pixels with photons arriving in specific time intervals after the excitation pulse in red and blue, respectively, as shown in Fig. 7a and c (re-analysis of the FLIM image shown in Fig. 6c). In this case, we have set the gate boundaries to achieve the same number of photons. The short gate accumulates all photons from 0 ns to 13 ns and the long gate accumulates from 58 ns to 193 ns after excitation. The short time-gate depicts all NPs, as both short- and long-lived particles contribute with photons at fast timescales (zoom-in shown in Fig. 7b, while full field of views are shown in Fig. S15†). In the short time-gate, there are 4 bright particles, and the less bright long-lived NPs (Y shape) are difficult to discern from the background. Instead, with the long time-gate, the long fluorescence lifetime NPs can be easily observed exclusively. We note that particle number 2 (Fig. 6c and 7b, arrow) is apparent in both time gates; again, this indicates that the spot consists of two NPs with short and long lifetimes in close proximity and appears purple in the pseudocoloured time-gate image (Fig. 7c). This form of time-gating can be a strong tool to gain an even larger imaging contrast, for example, by removing the short-lived auto-fluorescence in cells.
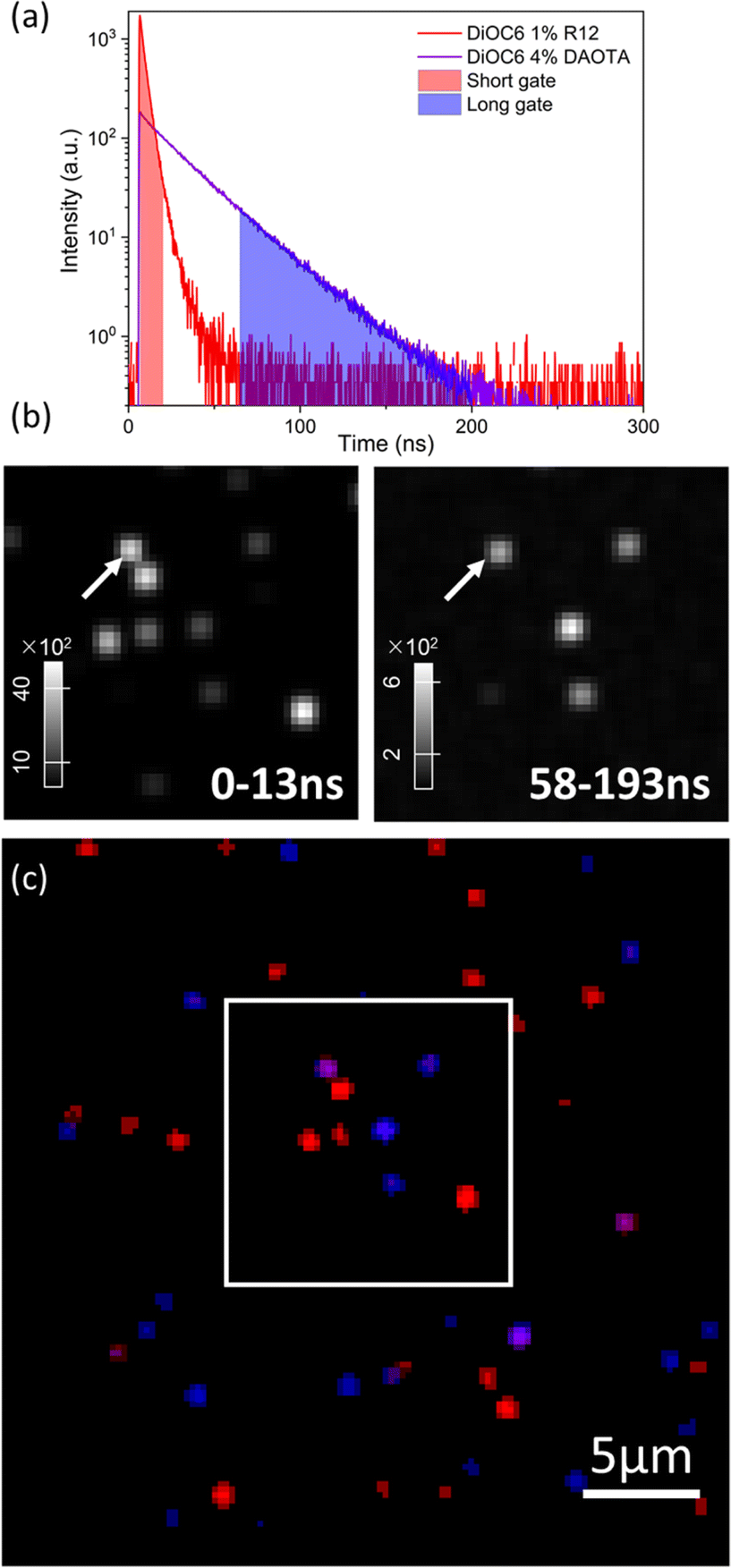 | ||
| Fig. 7 Time-gated imaging. (a) Normalized fluorescence decays of doped NPs according to their QY. (b) Short and long time gated images in time intervals 0–13 ns (left) and 58–193 ns (right) after excitation. The arrow indicates particle 2. (c) Pseudocolored image of mixed particles, where red and blue indicate photons that arrived a short or long time after excitation, respectively. | ||
Conclusions
We report the first high density dye based fluorescent NPs designed for fluorescence lifetime imaging. By doping SMILES NPs with FRET acceptors, we outcompete trap states in the NP lattice and gain increased fluorescence quantum yields and control of the emission lifetime. These bright lifetime-tuneable NPs are very easily assembled from commercial dyes with the desired properties using the SMILES approach. Compared to covalently synthesized dyads and other multi-chromophores with tailored photophysical properties both the SMILES NPs have the advantage of simple synthesis and modification as well as the large brightness stemming from the hundreds of dyes in each NP. We expected the new FRET SMILES NPs to be a useful tool for adding more channels in multiplexed fluorescence imaging, thereby allowing for increasing the amount of information gained in a single image. For applications in bioimaging, development of reactive surface coatings for targeted imaging is necessary, which would also enable immobilization of the NPs and further work on single particle imaging. The long-lived NPs are also an excellent candidate for time-gated imaging, where short-lived fluorescence, that can occur from cell auto-fluorescence, can be removed, which will increase the signal-to-noise ratio drastically.Data availability
Experimental data are available from the corresponding author upon reasonable request.Author contributions
SGS, JC, AHF and BWL conceived the study. SGS carried out the experiments and data analysis. MBL assisted with FLIM and data analysis. SGS and BWL wrote the original manuscript. All authors commented and reviewed the manuscript and figures.Conflicts of interest
There are no conflict to declare.Acknowledgements
This work was supported by the Danish Council of Independent Research (DFF-0136-00122B). A. H. F. acknowledges support from the US National Science Foundation (DMR-2118423). J. C. acknowledges funding support from the Novo Nordisk Foundation (NNF22OC0073582).Notes and references
- A. Reisch and A. S. Klymchenko, Fluorescent Polymer Nanoparticles Based on Dyes: Seeking Brighter Tools for Bioimaging, Small, 2016, 12, 1968–1992 CrossRef CAS PubMed
.
- W. R. Algar, M. Massey, K. Rees, R. Higgins, K. D. Krause, G. H. Darwish, W. J. Peveler, Z. Xiao, H.-Y. Tsai, R. Gupta, K. Lix, M. V. Tran and H. Kim, Photoluminescent Nanoparticles for Chemical and Biological Analysis and Imaging, Chem. Rev., 2021, 121, 9243–9358 CrossRef CAS PubMed
- A. H. Ashoka, I. O. Aparin, A. Reisch and A. S. Klymchenko, Brightness of fluorescent organic nanomaterials, Chem. Soc. Rev., 2023, 52, 4525–4548 RSC
- J. Gierschner, J. Shi, B. Milián-Medina, D. Roca-Sanjuán, S. Varghese and S. Park, Luminescence in Crystalline Organic Materials: From Molecules to Molecular Solids, Adv. Opt. Mater., 2021, 9, 2002251 CrossRef CAS
- D. Bialas, E. Kirchner, M. I. S. Röhr and F. Würthner, Perspectives in Dye Chemistry: A Rational Approach toward Functional Materials by Understanding the Aggregate State, J. Am. Chem. Soc., 2021, 143, 4500–4518 CrossRef CAS PubMed
- T. Niehörster, A. Löschberger, I. Gregor, B. Krämer, H.-J. Rahn, M. Patting, F. Koberling, J. Enderlein and M. Sauer, Multi-target spectrally resolved fluorescence lifetime imaging microscopy, Nat. Methods, 2016, 13, 257–262 CrossRef PubMed
- N. Oleksiievets, J. C. Thiele, A. Weber, I. Gregor, O. Nevskyi, S. Isbaner, R. Tsukanov and J. Enderlein, Wide-Field Fluorescence Lifetime Imaging of Single Molecules, J. Phys. Chem. A, 2020, 124, 3494–3500 CrossRef CAS PubMed
- H. S. Afsari, M. Cardoso Dos Santos, S. Lindén, T. Chen, X. Qiu, P. M. P. van Bergen en Henegouwen, T. L. Jennings, K. Susumu, I. L. Medintz, N. Hildebrandt and L. W. Miller, Time-gated FRET nanoassemblies for rapid and sensitive intra- and extracellular fluorescence imaging, Sci. Adv., 2016, 2, e1600265 CrossRef PubMed
- M. B. Liisberg and T. Vosch, Time gated Fourier transform spectroscopy as a technique for disentangling short- and long-lived luminescence, Commun. Mater., 2023, 4, 57 CrossRef
- Y. Xie, M. C. Arno, J. T. Husband, M. Torrent-Sucarrat and R. K. O'Reilly, Manipulating the fluorescence lifetime at the sub-cellular scale via photo-switchable barcoding, Nat. Commun., 2020, 11, 2460 CrossRef CAS PubMed
- D. Kage, K. Hoffmann, M. Wittkamp, J. Ameskamp, W. Göhde and U. Resch-Genger, Luminescence lifetime encoding in time-domain flow cytometry, Sci. Rep., 2018, 8, 16715 CrossRef PubMed
- M. Cardoso Dos Santos, A. Runser, H. Bartenlian, A. M. Nonat, L. J. Charbonnière, A. S. Klymchenko, N. Hildebrandt and A. Reisch, Lanthanide-Complex-Loaded Polymer Nanoparticles for Background-Free Single-Particle and Live-Cell Imaging, Chem. Mater., 2019, 31, 4034–4041 CrossRef CAS
- X. Qiu, J. Xu, M. Cardoso Dos Santos and N. Hildebrandt, Multiplexed Biosensing and Bioimaging Using Lanthanide-Based Time-Gated Förster Resonance Energy Transfer, Acc. Chem. Res., 2022, 55, 551–564 CrossRef CAS PubMed
- C. Chen, B. Corry, L. Huang and N. Hildebrandt, FRET-Modulated Multihybrid Nanoparticles for Brightness-Equalized Single-Wavelength Barcoding, J. Am. Chem. Soc., 2019, 141, 11123–11141 CrossRef CAS PubMed
- L. Haye, N. Fayad, R. C. Knighton, A. Combes, O. Jeannin, A. Klymchenko, T. Gallavardin, N. Hildebrandt, L. J. Charbonnière and A. Reisch, NIR FRET Luminescence in Rhenium Complex and Dye Co-Loaded Polymer Nanoparticles, Adv. Mater. Technol., 2023, 8, 2301016 CrossRef CAS
- L. Kacenauskaite, S. G. Stenspil, A. H. Olsson, A. H. Flood and B. W. Laursen, Universal Concept for Bright, Organic, Solid-State Emitters–Doping of Small-Molecule Ionic Isolation Lattices with FRET Acceptors, J. Am. Chem. Soc., 2022, 144, 19981–19989 CrossRef CAS PubMed
- K. Hoffmann, T. Behnke, D. Drescher, J. Kneipp and U. Resch-Genger, Near-Infrared-Emitting Nanoparticles for Lifetime-Based Multiplexed Analysis and Imaging of Living Cells, ACS Nano, 2013, 7, 6674–6684 CrossRef CAS PubMed
- K. Hoffmann, T. Behnke, M. Grabolle and U. Resch-Genger, Nanoparticle-encapsulated vis- and NIR-emissive fluorophores with different fluorescence decay kinetics for lifetime multiplexing, Anal. Bioanal. Chem., 2014, 406, 3315–3322 CrossRef CAS PubMed
- M. Rosenberg, K. R. Rostgaard, Z. Liao, A. O. Madsen, K. L. Martinez, T. Vosch and B. W. Laursen, Design, synthesis, and time-gated cell imaging of carbon-bridged triangulenium dyes with long fluorescence lifetime and red emission, Chem. Sci., 2018, 9, 3122–3130 RSC
- R. M. Rich, D. L. Stankowska, B. P. Maliwal, T. J. Sørensen, B. W. Laursen, R. R. Krishnamoorthy, Z. Gryczynski, J. Borejdo, I. Gryczynski and R. Fudala, Elimination of autofluorescence background from fluorescence tissue images by use of time-gated detection and the AzaDiOxaTriAngulenium (ADOTA) fluorophore, Anal. Bioanal. Chem., 2013, 405, 2065–2075 CrossRef CAS PubMed
- S. J. Strickler and R. A. Berg, Relationship between Absorption Intensity and Fluorescence Lifetime of Molecules, J. Chem. Phys., 1962, 37, 814–822 CrossRef CAS
- H. Osaki, C.-M. Chou, M. Taki, K. Welke, D. Yokogawa, S. Irle, Y. Sato, T. Higashiyama, S. Saito, A. Fukazawa and S. Yamaguchi, A Macrocyclic Fluorophore Dimer with Flexible Linkers: Bright Excimer Emission with a Long Fluorescence Lifetime, Angew. Chem., Int. Ed., 2016, 55, 7131–7135 CrossRef CAS PubMed
- L. Kacenauskaite, N. Bisballe, R. Mucci, M. Santella, T. Pullerits, J. Chen, T. Vosch and B. W. Laursen, Rational Design of Bright Long Fluorescence Lifetime Dyad Fluorophores for Single Molecule Imaging and Detection, J. Am. Chem. Soc., 2021, 143, 1377–1385 CrossRef CAS PubMed
- K. Trofymchuk, A. Reisch, P. Didier, F. Fras, P. Gilliot, Y. Mely and A. Klymchenko, Giant light-harvesting nanoantenna for single-molecule detection in ambient light, Nat. Photonics, 2017, 11, 657–663 CrossRef CAS PubMed
- C. Wu, B. Bull, C. Szymanski, K. Christensen and J. McNeill, Multicolor Conjugated Polymer Dots for Biological Fluorescence Imaging, ACS Nano, 2008, 2, 2415–2423 CrossRef CAS PubMed
- C. Wu, Y. Zheng, C. Szymanski and J. McNeill, Energy Transfer in a Nanoscale Multichromophoric System: Fluorescent Dye-Doped Conjugated Polymer Nanoparticles, J. Phys. Chem. C, 2008, 112, 1772–1781 CrossRef CAS PubMed
- D. Genovese, E. Rampazzo, S. Bonacchi, M. Montalti, N. Zaccheroni and L. Prodi, Energy transfer processes in dye-doped nanostructures yield cooperative and versatile fluorescent probes, Nanoscale, 2014, 6, 3022–3036 RSC
- C. R. Benson, L. Kacenauskaite, K. L. VanDenburgh, W. Zhao, B. Qiao, T. Sadhukhan, M. Pink, J. Chen, S. Borgi, C.-H. Chen, B. J. Davis, Y. C. Simon, K. Raghavachari, B. W. Laursen and A. H. Flood, Plug-and-Play Optical Materials from Fluorescent Dyes and Macrocycles, Chem, 2020, 6, 1978–1997 CAS
- J. Chen, S. M. A. Fateminia, L. Kacenauskaite, N. Bærentsen, S. G. Stenspil, J. Bredehoeft, K. L. Martinez, A. H. Flood and B. W. Laursen, Ultra-Bright Fluorescent Organic Nanoparticles Based on Small-Molecule Ionic Isolation Lattices, Angew. Chem., Int. Ed., 2021, 60, 9450–9458 CrossRef CAS PubMed
- J. Chen, S. G. Stenspil, S. Kaziannis, L. Kacenauskaite, N. Lenngren, M. Kloz, A. H. Flood and B. W. Laursen, Quantitative Energy Transfer in Organic Nanoparticles Based on Small-Molecule Ionic Isolation Lattices for UV Light Harvesting, ACS Appl. Nano Mater., 2022, 5, 13887–13893 CrossRef CAS
- S. Lee, C.-H. Chen and A. H. Flood, A pentagonal cyanostar macrocycle with cyanostilbene CH donors binds anions and forms dialkylphosphate [3]rotaxanes, Nat. Chem., 2013, 5, 704–710 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05496a |
| This journal is © The Royal Society of Chemistry 2024 |