
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOpen shell (4n + 2)π and closed shell 4nπ planar core-modified decaphyrins†‡
Pragati
Shukla
 ,
Madan D.
Ambhore
and
Venkataramanarao G.
Anand
*
,
Madan D.
Ambhore
and
Venkataramanarao G.
Anand
*
Department of Chemistry, Indian Institute of Science Education and Research (IISER), Pune 411008, Maharashtra, India. E-mail: vg.anand@iiserpune.ac.in
First published on 11th March 2024
Abstract
Planar 44π and 46π core-modified decaphyrins with ten thiophene units have been synthesized from short thiophene oligomers. They have been structurally characterized by single crystal X-ray diffraction with further support from spectroscopic analysis and quantum chemical calculations. Our analysis revealed diradicaloid characteristics for 46π species in contrast to the closed shell property of the 44π congener. Further, 44π and 46π undergo reversible two-electron chemical oxidation, as observed by spectro-electrochemical measurements.
Introduction
Synthetic expanded porphyrinoids are excellent model systems to study Hückel, Möbius and Baird aromaticity.1–4 Sustaining planarity in porphyrinoids containing forty or more π-electrons has remained a formidable challenge for synthetic chemists. Thermodynamic stability invariably favours non-planar topology at the expense of (anti)aromatic characteristics in π-expanded porphyrinoids. Sessler's tetracationic 40π deca-pyrrole, turcasarin, 1, was the first giant porphyrinoid to be characterized with a figure-of-eight topology.5 Subsequently, decaphyrins have regularly featured as non-planar macrocycles. For example, a decaphyrin, 2, with ten pyrroles and ten bridging carbons also adopted a twisted topology.6a,b It is pertinent to note that a 40π octafuran and a 38π octaphyrins are the only large planar porphyrinoids known to display ring current effects.7,8a,b A couple of synthetic protocols have been explored to avoid a twisted topology by covalent/non-covalent support in the cavity of a decaphyrin. Osuka and co-workers attempted to induce a planar topology by covalently linking a phenylene ring in the centre of a redox-active 46π decaphyrin.9 Later, Okujima and Kobayashi employed an anion template to synthesize a 38π dicationic cyclo[10]pyrrole through oxidative coupling of densely π-substituted bipyrrole.10 Redox-active pyrrolic porphyrinoids with an identical number of heterocyclic units can be synthesized and isolated with different numbers of π-electrons in the macrocyclic network by varying: (a) the ratio between pyrrole units and bridging carbon atoms and (b) the number of imine/amine-like pyrrolic nitrogens.11 Hence, (4n + 2)π pyrrolic decaphyrins are known to adopt between 46 and 38π electrons, while partially core-modified decaphyrins12 vary between 48π and 40π electrons. Non-pyrrolic planar porphyrinoids display significantly altered electronic and redox properties compared to their corresponding pyrrole analogues, as reflected in their structural and electronic properties Fig. 1.13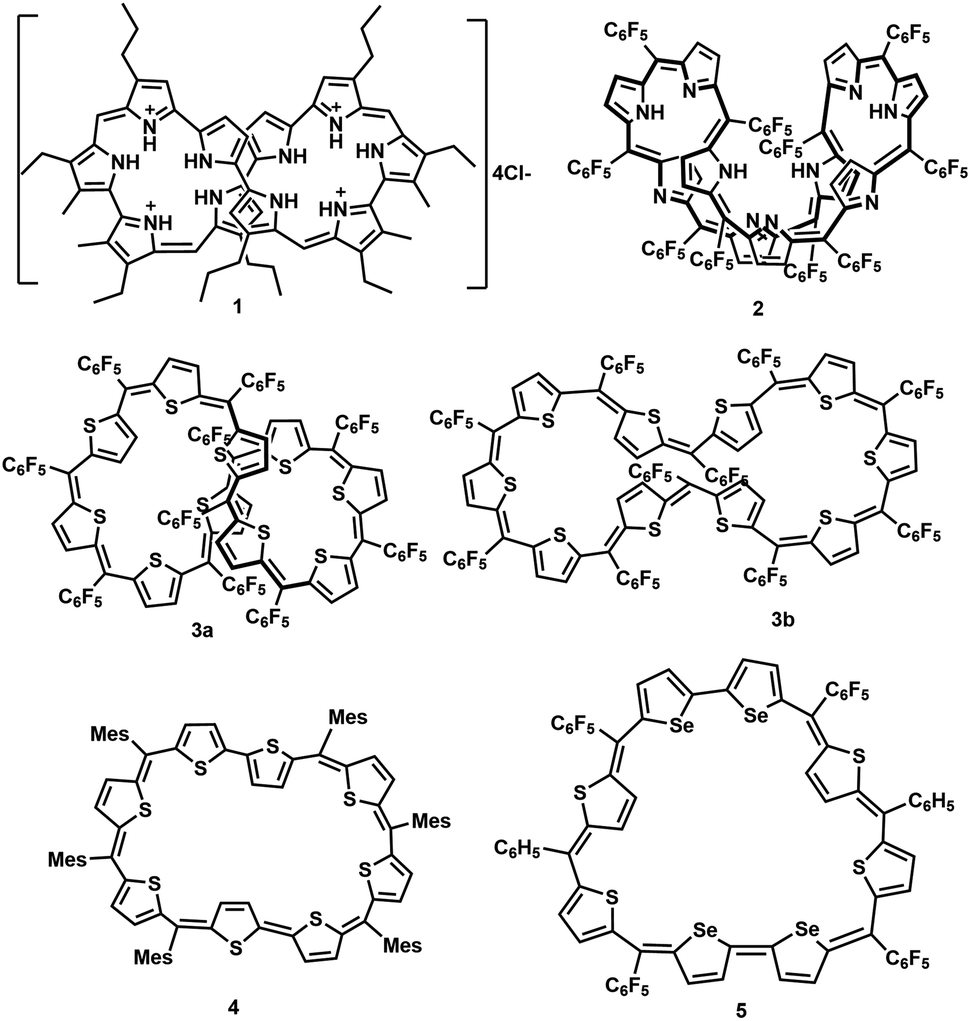 | ||
| Fig. 1 Selected examples of expanded porphyrinoids with ten and eight heterocyclic units. | ||
A completely core-modified deca-thia decaphyrin, 3, with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of thiophene and bridging carbons contains a maximum of 50π electrons and can be ring-oxidized to 48π dicationic species.14 In the solid state, 3 can be tuned to adopt either a twisted (3a) or near-planar (3b) topology depending on the solvent of crystallization. Planar non-pyrrolic (4n + 2)π octaphyrins, 4 and 5, with bithiophene/biselenophene units display unique open shell configurations depending on the macrocyclic topology.15,16a In most known diradical species, such as linear oligothiophene or bisphenalenyls, an increase in the length of the spacer is known to increase the diradical character due to extra resonance energy attained by the diradicals.16b In this regard, it can be envisaged that a planar (4n + 2)π decathiophene macrocycle could display pronounced diradicaloid features. Therefore, all the synthesized core-modified planar decaphyrins with varying number of π-electrons can be tested for their diradical properties. Herein, we report the first examples of planar 44π and 46π deca-thia porphyrinoids which display closed shell and open shell characteristics, respectively. Moreover, 42π dications of 44π macrocycles displayed moderate diradicaloid features.
:1 ratio of thiophene and bridging carbons contains a maximum of 50π electrons and can be ring-oxidized to 48π dicationic species.14 In the solid state, 3 can be tuned to adopt either a twisted (3a) or near-planar (3b) topology depending on the solvent of crystallization. Planar non-pyrrolic (4n + 2)π octaphyrins, 4 and 5, with bithiophene/biselenophene units display unique open shell configurations depending on the macrocyclic topology.15,16a In most known diradical species, such as linear oligothiophene or bisphenalenyls, an increase in the length of the spacer is known to increase the diradical character due to extra resonance energy attained by the diradicals.16b In this regard, it can be envisaged that a planar (4n + 2)π decathiophene macrocycle could display pronounced diradicaloid features. Therefore, all the synthesized core-modified planar decaphyrins with varying number of π-electrons can be tested for their diradical properties. Herein, we report the first examples of planar 44π and 46π deca-thia porphyrinoids which display closed shell and open shell characteristics, respectively. Moreover, 42π dications of 44π macrocycles displayed moderate diradicaloid features.
Results and discussion
A synthetic strategy to induce a rigid planar structure for 50π decathiophene, 2, can be envisaged by decreasing the number of bridging carbons in the macrocyclic framework. An attempt to synthesize a 44π decaphyrin was designed from short thiophene oligomers (See ESI,‡ for details of the synthesis). Acid-assisted condensation of terthiophene diol, 6, with bithiophene, 7, under dark and inert conditions in dichloromethane was followed by DDQ oxidation open to air (Scheme 1a). The MALDI-TOF/TOF mass spectrum of the reaction mixture revealed the formation of the expected 44π decathiophene, 10 (ESI S1‡). Under similar reaction conditions, the condensation of terthiophene diol 6 with either biselenophene, 8, or bifuran, 9, yielded 44π decaphyrins 11 and 12, respectively (ESI S3 and S5‡). Encouraged by this success, we designed modified strategy (Scheme 1b) to synthesize a 46π decaphyrin, with six bridging carbons, by condensing terthiophene, 13, with the diol of thia dipyrrane, 14, under conditions similar to the synthesis of 10. MALDI TOF/TOF mass spectral analysis of the reaction mixture (ESI S8‡) confirmed the identity of the target 46π decaphyrin, 15.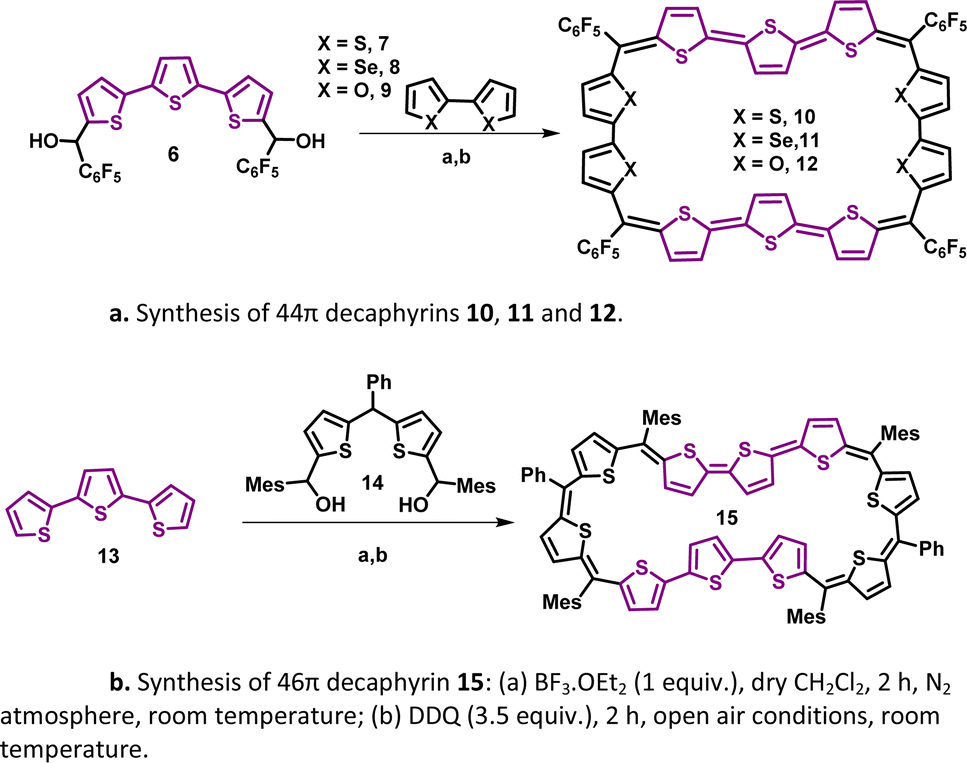 | ||
| Scheme 1 (a) Synthesis of 44π decaphyrins 10, 11 and 12. (b) Synthesis of 46π decaphyrin 15: (a) BF3 OEt2 (1 equiv.), dry CH2Cl2, 2 h, N2 atmosphere, room temperature; (b) DDQ (3.5 equiv.), 2 h, open air conditions, room temperature. | ||
All the decaphyrins were isolated in 2.2 to 7.2% yields by column chromatography with dichloromethane and n-hexane as eluents. The absolute structure of these macrocycles was determined by 1H NMR spectroscopy and single crystal X-ray diffraction studies. The ESI-TOF high resolution mass spectrum displayed an m/z value of 1535.8490 (calcd. for C68H20F20S20; [M]+ 1535.8453) for 10 (ESI S2a‡), 1727.6244 (calcd. for C68H20F20S6Se4; [M]+ 1727.6231) for 11 (ESI S4‡), m/z value of 1471.9408 and 735.9665 (calcd. for C68H20F20O4S6; [M]+ 1471.9366 and [M]2+ 735.9683) for 12 (ESI S6‡) and m/z value of 761.1470 (calcd. for C94H74S10; [M]+ 1522.2998 and [M]2+ 761.1499) for 15 (ESI S9‡). The proton NMR spectrum of decathiophene 10 recorded in CDCl3 displayed well-resolved signals at room temperature. A sharp singlet at δ 8.69 ppm and four distinct doublets at δ 6.58, 6.55, 6.09 and 5.87 ppm corresponding to four protons each (ESI S11‡) were observed. The 1H–1H two-dimensional NMR spectrum revealed two sets of correlations for doublets (δ 6.58, 6.09 ppm and δ 6.55, 5.87 ppm) in support of the coupling of β-CH protons of thiophene rings (ESI S12‡). A lower number of signals suggested higher symmetry for the macrocycle in the solution state. A similar 1H NMR spectral pattern at room temperature in CDCl3 was observed for tetra-selena derivative, 11, with five signals in the region between δ 6.04 and 8.41 ppm (ESI S13‡). Tetraoxa derivative, 12, also displayed a similar well-resolved spectrum with relatively downfield chemical shift values between δ 6.80 and 7.53 ppm (ESI S14‡).
The absolute molecular structure of 10 was determined by X-ray diffraction of single crystals obtained by vapor diffusion of n-hexane into its solution in chloroform. 10 adopted a squarish arrangement with all the thiophene rings in same plane and meso-pentafluoro substituents oriented almost perpendicular to the macrocyclic plane defined by four bridging carbons. The central thiophene in both the terthiophene units was inverted with its sulfur atom facing away from the macrocyclic core (Fig. 2a and b). Such a symmetrical molecular structure is in good agreement with the observed 1H NMR spectrum of 10. Further, the value +5.93 ppm for the estimated nucleus-independent chemical shift (NICS)17 calculated at the B3LYP/6-31G(d,p) level of theory suggested poor antiaromatic character for the 44π macrocycle. Similar to 10, the tetra-oxa derivative 12, also displayed a near-planar structure in the solid state (Fig. 2c and d). Unexpectedly, it revealed inversion of two adjacent thiophene rings in each of the terthiophene units with symmetry much lower than that of decaphyrin 10. This conformation is perhaps more pronounced in the solid state and may not correspond to the solution state structure as analyzed from its 1H NMR spectral features. Earlier reports revealed that it is not uncommon for expanded porphyrinoids to adopt different structures in solid and liquid states.9,16a The molecular structure of 46π decaphyrin 15 determined from X-ray diffraction studies revealed a planar and rectangular topology. Two adjacent thiophene rings were inverted in each terthiophene, while the sulfur atoms of the other six thiophene rings are oriented towards the macrocyclic core (Fig. 2e and f). To the best of our knowledge, these are the largest planar porphyrinoids known to date. Being a planar and globally π-conjugated structure comprising 46 π-electrons, it adheres to Hückel's aromatic structure. An estimated NICS value of −17.94 ppm at the macrocyclic centre further substantiated Hückel aromaticity for the 46π macrocycle (ESI S33b‡). A clockwise direction of ring current in 15 obtained from the plot of computed anisotropy of induced current density (ACID)18 further supports the aromatic character in this (4n + 2)π macrocycle. However, the ACID plot for 10 did not reveal an exclusive direction of the ring current in tune with the estimated NICS value (ESI S31 and S32‡). In addition, the computationally calculated harmonic oscillator model of aromaticity (HOMA)19a values of 0.74 and 0.88 for 10 and 15 corroborate respective aromatic characteristics for these π circuits. The estimated values for the electronic aromaticity indices AV1245 and AVmin19b,c (for 10) are 1.68 and 1.00, respectively, and relatively higher values of 2.84 and 1.78 were obtained for 15. Thus, comparable increases in the index values for 15 suggest the aromatic characteristics of the 46π decaphyrin.
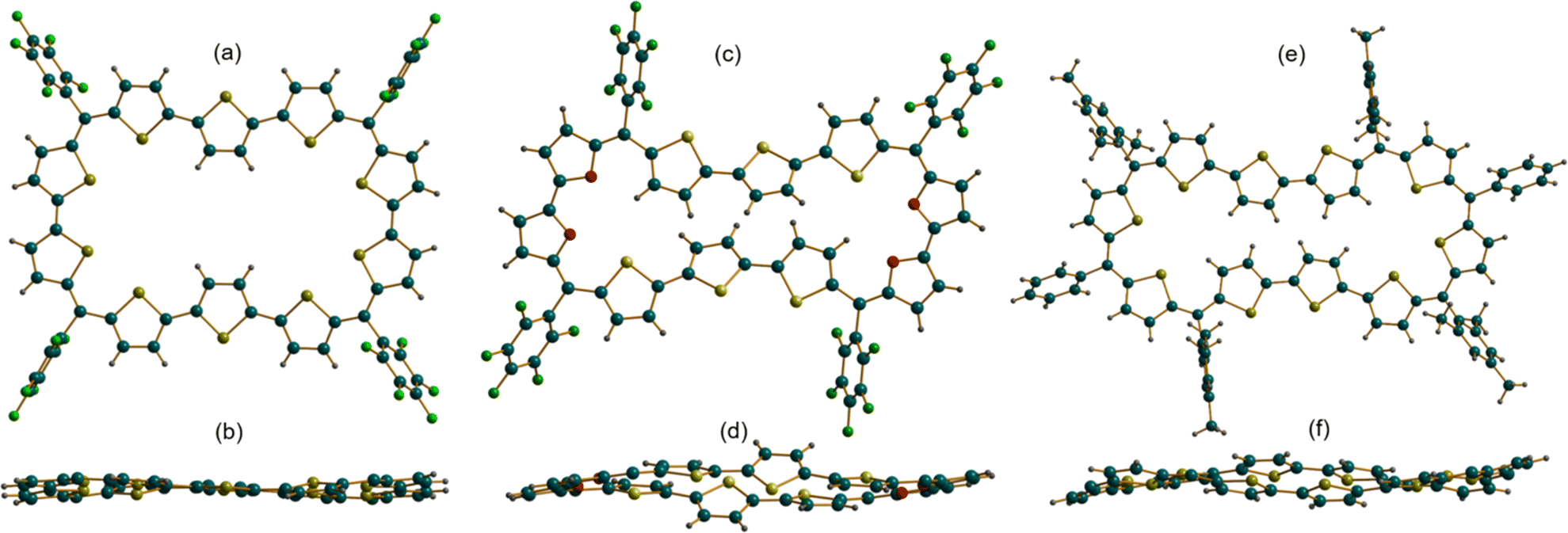 | ||
| Fig. 2 Molecular structure of decaphyrins 10 (a and b), 12 (c and d) and 15 (e and f) with side view and top view. Images a, c and e represent the top view while images b, d and f represent the side view, where meso-substituents are omitted for clarity. | ||
In stark contrast to structural and computational studies, 46π decaphyrin 15 did not display well resolved signals in its 1H NMR at room temperature. Suspecting its fluxional nature, we recorded 1H NMR spectra at lower temperatures up to 198 K (ESI S18‡), which revealed only broad signals in the region between δ 6.00 and 9.00 ppm supporting diradicaloid characteristics for the planar 46π macrocycle. Our earlier reports suggest that such (4n + 2)π systems with relatively broad 1H NMR signals signify the diradicaloid character of the macrocycle.15,16a To further confirm the diradicaloid properties of 15, variable-temperature EPR was recorded, and it displayed a broad signal with a g value of 2.0054. The EPR signal intensity decreases with a decrease in temperature, suggesting a singlet ground state (Fig. 3a). To support experimental findings, quantum chemical calculations using the UCAM-B3LYP/6-31G(d,p) method revealed a singlet–triplet energy gap ΔES–T of −2.19 kcal mol−1. Moreover, it yielded an occupation number NOON (natural orbital occupation number)20 of 0.84 for the LUMO and a y0 value of 0.69, suggesting a diradicaloid character (ESI Table S1a‡). The obtained NOON value for decathiophene is much higher than that reported for 38π octathiophene,15,16a which is close to 0.40% with an occupation number of 0.67 for the LUMO. The frontier molecular orbital diagram (FMO) shows the electron density of two electrons localised over two halves of conjugated macrocycle 15 (Fig. 4). It can be envisaged that the quinoidal form 15a is fully conjugated and is in resonance with the benzenoidal form 15b of the macrocycle corresponding to the open shell conformation (Fig. 3b). Its additional stability can be attributed to the increased number of aromatic thiophene rings of the terthiophene moiety in the planar topology. However, antiaromatic 44π decathiophene, 10, did not display any diradicaloid features, as analyzed in experimental and computational studies (ESI Table S2‡). Various basis sets have been employed for determining the aromatic characteristics of poly-aromatic hydrocarbons and porphyrin-based systems.21 However, our calculations with B3LYP/6-31G(d,p) level of theory matched the observed experimental results. The diradicaloid character was estimated with the UCAM B3LYP/6-31G(d,p) basis set.
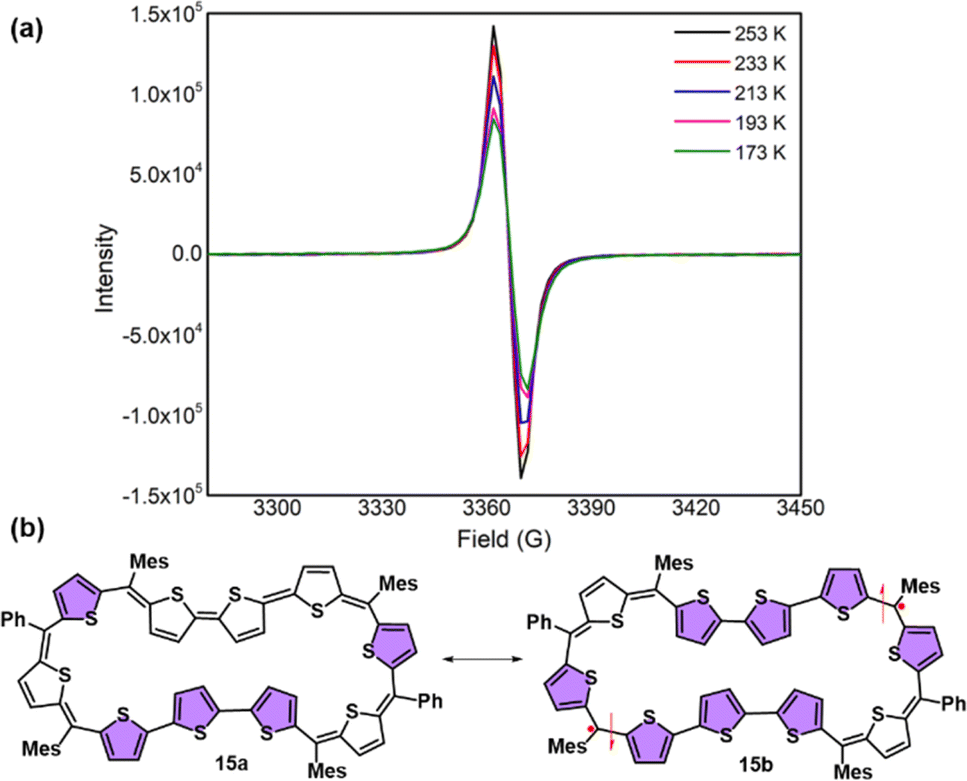 | ||
| Fig. 3 (a) Variable-temperature EPR spectra of 15. (b) Fully conjugated form 15a and diradicaloid form 15b of decaphyrin 15. | ||
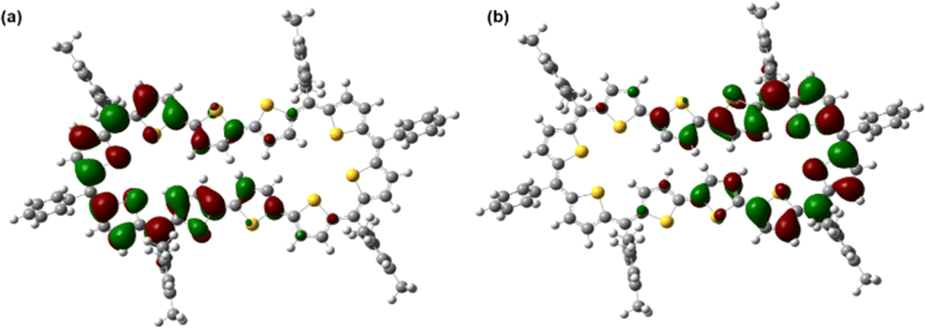 | ||
| Fig. 4 Frontier SOMO profiles, calculated using spin-unrestricted UCAM-B3LYP/6-31G(d,p) for 15: (a) SOMO α spin and (b) SOMO β spin. | ||
By virtue of extended π-conjugation, 44π decaphyrin, 10, displayed strong absorptions at 507 (136500) and 602 (185000) nm (ESI S23‡). Tetra-selena derivative 11 also displayed similar absorptions, while the tetra-oxa derivative 12 displayed strong absorption (Fig. 5a) at 494 (187000) nm and relatively weak bands at 384 (112400), 583 (41200) and 624 (52900) nm. In contrast, 46π decaphyrin, 15, displayed multiple absorptions (Fig. 5b) at 432 (117800), 546 (sh), 605 (153800), 664 (sh) and 725 (128700) nm. A relatively intense and broad absorption band at 966 (112900) nm extending to NIR supports the diradicaloid nature of 15 (Table 1). Such wide-ranging absorptions in the visible part of the electromagnetic spectrum are uncommon in giant porphyrinoids.
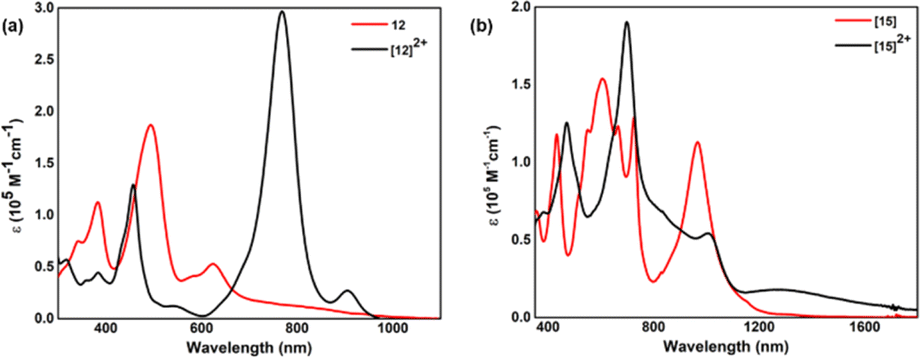 | ||
| Fig. 5 UV-visible absorption spectra of (a) 12 and [12]2+, (b) 15 and [15]2+. | ||
| Decaphyrin | No. of πe− | λ max (nm) ε (M−1 cm−1) |
|---|---|---|
| 10 | 44 | 507 (136500), 559 (80500) and 602 (185000) |
| 11 | 44 | 505 (94900), 547 (63700) and 589 (124700) |
| 12 | 44 | 384 (112400), 494 (187000), 583 (41200) and 624(52900) |
| 15 | 46 | 432 (117800), 546 (sh), 605 (153800), 664 (sh), 725 (128700) and 966 (112900) |
Since 4nπ porphyrinoids are susceptible to reversible two-electron ring oxidation,13 the redox property of 44π decaphyrins was studied by chemical and spectro-electrochemical methods. The 44π decaphyrins 10–12 could be reversibly oxidized to their respective 42π dicationic species, by various reagents such as trifluoroacetic acid, [Et3O]+[SbCl6]−, NOBF4, NOSbF6 or triflic acid (Scheme 2). Upon oxidation, a subtle change from a purple to bluish color along with intense red-shifted absorptions at 772 nm (450800) nm were observed for [10]2+ (ESI S23‡). Unfortunately, dications were less stable in common organic solvents over a period of time and hence eluded their complete characterization. Hence, we could not successfully grow single crystals to determine their absolute structure. However, dicationic [10]2+ (m/2) species could be identified from the HR-MS spectrum to confirm the ring oxidation of the macrocycle (ESI S2b‡). Since dication [10]2+ is a Hückel (4n + 2)π aromatic system, we suspected it to exhibit a diradicaloid feature in its planar conformation. Hence, computational studies for [10]2+ were performed with similar parameters to those employed for 15. As anticipated, it displayed a singlet triplet energy gap ΔES–T of −2.61 kcal mol−1. These values suggested a thermally accessible triplet state, and it also had a NOON value of y0 = 0.50 displaying a moderately diradicaloid character (ESI Table S2‡). These results are in further support of our findings of a significant increase in diradicaloid character with increasing length of π-conjugation in aromatic expanded porphyrinoids. Analogous color changes and red-shifted absorptions were observed upon oxidation of the tetra-selena and tetra-oxa 44π decaphyrins 11 and 12. [12]2+ displayed intense red-shifted absorptions at 768 (296000) and 457 (129400) nm, which were associated with a color change from brownish-green to yellowish-green upon oxidation.
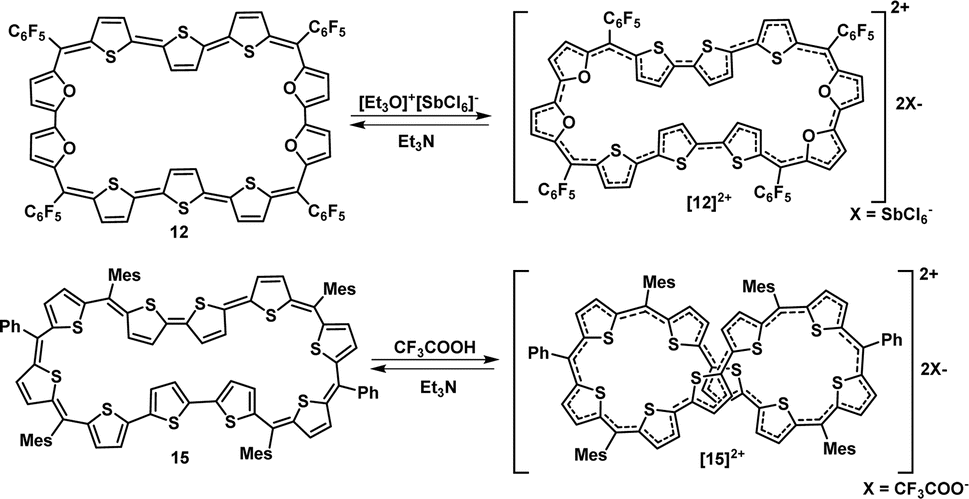 | ||
| Scheme 2 Oxidation of 12 and 15 to their corresponding dications [12]2+ and [15]2+. | ||
Cyclic voltammetry studies revealed four oxidations at 0.40, 0.75, 1.08 and 1.35 V and one reduction at −0.88 V for the 44π macrocycle 12 (ESI S27‡). Spectro-electrochemical measurements (ESI S29‡) also revealed an absorption at 768 nm at an applied potential of 0.87 V validating dicationic species derived upon chemical oxidation by a Meerwein salt.22 Fortunately, the tetra-oxa decaphyrin [12]2+ could be isolated and characterized by 1H NMR spectroscopy. It displayed a well-resolved spectrum with ten doublets in the region between δ 4.0 and 9.00 ppm (ESI S16‡) at room temperature. The number of resonances was twice the number observed in its freebase form. The 1H–1H COSY spectrum (ESI S17‡) revealed five correlations, suggesting inversion of two adjacent thiophene rings in both the terthiophene units, as observed from X-ray diffraction studies of the parent macrocycle. Its estimated NICS value of −17.82 ppm further supported the aromaticity of 42π dication [12]2+. All our efforts to crystallize (4n + 2)π dicationic species [12]2+ were futile.
Similar two-electron ring oxidation was also observed for 46π diradicaloid decaphyrin, 15 (Scheme 2). Upon addition of a Meerwein salt or TFA, intense red-shifted absorptions were observed at 470 (125200), 698 (190100) and 1008 (54000) nm for 44π dicationic species, [15]2+. Two-electron ring oxidation for the (4n + 2)π species was further supported by the observation of the m/2 value from high resolution mass spectrometry (ESI S10‡) and from spectro-electrochemical studies (ESI S30‡). The 1H NMR spectrum of [15]2+ revealed only broad signals at room temperature. However, in contrast to 46π species, the 44π dication species [15]2+ displayed sharp resonances upon lowering the temperature to 203 K (ESI S21‡). Ten resonances were observed within a small region between δ 6.75 and 8.12 ppm, resulting in the overlap of many signals. Methyl protons of mesityl rings were found to resonate between δ 1.41 and 2.99 ppm. These chemical shift values did not reflect the paratropic ring current effect expected of planar 4nπ species.
Crystallization of [15]2+ was attempted with various solvent systems, and the best possible crystals were obtained by vapor diffusion of heptane into a solution of [15]2+ in dichloromethane. The molecular structure of 44π dication [15]2+·2(CF3COO)− obtained from X-ray diffraction analysis revealed a twisted conformation, resulting in the loss of the anticipated antiaromatic character. The dicationic nature of the figure-of-eight conformation was confirmed by two associated CF3COO− counter-anions for the dipositive charge on the macrocycle (Fig. 6). This structure also represents an unusual criss-crossing of the two terthiophene units in the centre of the macrocycle forming two five-membered pockets. Structure-induced loss of antiaromaticity led to a closed shell 44π species, which could be reduced back to the parent planar 46π macrocycle with appropriate reducing agents such as triethylamine or Zn dust. Even the 42π dication of decaphyrins 12 could be reduced back to the parent 44π macrocycle with similar reducing agents.
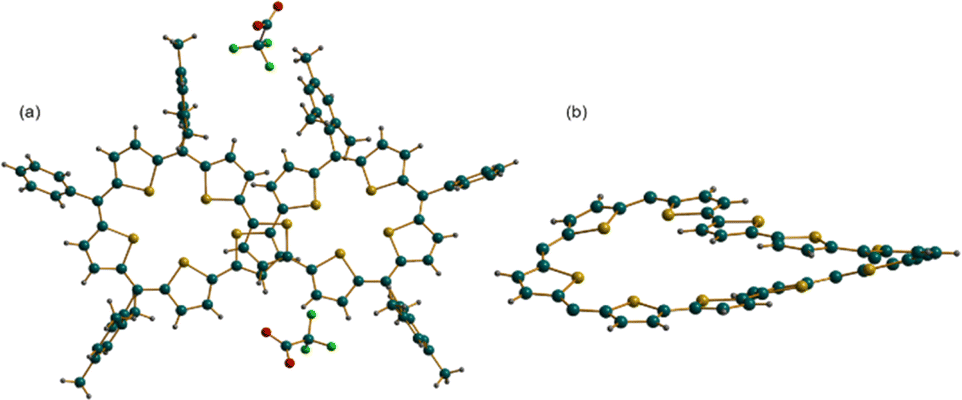 | ||
| Fig. 6 Molecular structure obtained from an X-ray diffraction study of decaphyrin dication [15]2+·2(CF3COO)−: (a) side view and (b) top view. | ||
Conclusions
We have described novel examples of planar decaphyrins bearing 44π and 46π electrons. Their structural characterization was accomplished by X-ray diffraction studies and spectroscopic investigations. Our analyses reveal that a planar (4n + 2)π species is stabilized as a diradicaloid, and such macrocyclic character can be enhanced by extending the length of conjugation, while the 44π species adopts a closed shell configuration with a flat topology. Both species undergo reversible two-electron ring oxidation to yield the corresponding 42π and 44π dicationic species. Electrochemical studies suggest multiple oxidation states which have yet to be accessed by appropriate chemical reagents. Significantly, these studies suggest that accommodating short thiophene oligomers in planar (4n + 2)π electron giant porphyrinoids may have a greater tendency to adopt radicaloid characteristics to sustain aromaticity. The synthesis of such giant porphyrinoids is underway in this laboratory.Data availability
Details of synthesis, spectroscopic characterization, co-ordinates obtained from computational calculations and CCDC numbers for the molecular structures are made available in ESI.‡Author contributions
P. S. contributed to syntheses, spectroscopic characterizations, X-ray data, computational calculations and assisted in writing the manuscript. M. D. A. was involved in synthetic design and computational calculations. V. G. A. conceptualized and managed the whole project and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
VGA thanks Department of Science and Technology (DST), New Delhi, India, for the Swarnajayanti Fellowship and SERB (Grant no: CRG/2019/005046), New Delhi, India for the financial support. PS and MDA thank Indian Institute of Science Education and Research (IISER), Pune, India, for their respective research fellowships. Authors acknowledge DST-FIST, New Delhi, India, grant (SR/FST/CS-II/2019/105) for the VT EPR facility at IISER Pune, India. Authors acknowledge IISER Pune, India, for the high performance computating cluster facility for the quantum chemical calculations.References
- D. Seidel and J. L. Sessler, Angew. Chem., Int. Ed., 2003, 42, 5134–5175 CrossRef PubMed.
- T. Tanaka and A. Osuka, Chem. Rev., 2017, 117, 2584–2640 CrossRef CAS PubMed.
- M. Stepien, N. Sprutta, P. Chwalisz, L. Szterenberg and L. L. Grazynski, Angew. Chem., Int. Ed., 2007, 46, 7869–7873 CrossRef CAS PubMed.
- S. Venkatraman and T. K. Chandrashekar, Acc. Chem. Res., 2003, 36, 676–691 CrossRef PubMed.
- J. L. Sessler, S. J. Weghorn, V. Lynch and M. R. Johnson, Angew Chem. Int. Ed. Engl., 1994, 33, 1509–1511 CrossRef.
- (a) S. Shimizu, N. Aratani and A. Osuka, Chem.–Eur. J., 2006, 12, 4909–4918 CrossRef CAS; (b) Y. Tanaka, J.-Y. Shin and A. Osuka, Eur. J. Org Chem., 2008, 1341–1349 CrossRef CAS.
- J. S. Reddy, S. Mandal and V. G. Anand, Org. Lett., 2006, 8(24), 5541–5543 CrossRef CAS.
- (a) W. Y. Cha, T. Soya, T. Tanaka, H. Mori, Y. Hong, S. Lee, K. H. Park, A. Osuka and D. Kim, Chem. Commun., 2016, 52, 6076–6078 RSC; (b) M. D. Ambhore, A. Basavarajappa and V. G. Anand, Chem. Commun., 2019, 55, 6763–6766 RSC.
- V. G. Anand, S. Saito, S. Shimizu and A. Osuka, Angew. Chem., Int. Ed., 2005, 44, 7244–7248 CrossRef CAS PubMed.
- T. Okujima, C. Ando, S. Agrawal, H. Matsumoto, S. Mori, K. Ohara, I. Hisaki, T. Nakae, M. Takase, H. Uno and N. Kobayashi, J. Am. Chem. Soc., 2016, 138, 7540–7543 CrossRef CAS PubMed.
- M. Broring, J. Jendrny, L. Zander, H. Schmickler, J. Lex, Y. D. Wu, M. Nendel, J. Chen, D. A. Plattner, K. N. Houk and E. Vogel, Angew Chem. Int. Ed. Engl., 1995, 34, 2515–2516 CrossRef.
- A. Ghosh, S. Dash, A. Srinivasan, C. H. Suresh, S. Peruncheralathan and T. K. Chandrashekar, Org. Chem. Front., 2019, 6, 3746–3753 RSC.
- B. K. Reddy, A. Basavarajappa, M. D. Ambhore and V. G. Anand, Chem. Rev., 2017, 117, 3420–3443 CrossRef CAS PubMed.
- H. S. Udaya, A. K. Basavarajappa, T. Y. Gopalakrishna and V. G. Anand, Chem. Commun., 2022, 58, 13931–13934 RSC.
- Y. Ni, T. Y. Gopalakrishna, S. Wu and J. Wu, Angew. Chem., Int. Ed., 2020, 59, 7414–7418 CrossRef CAS PubMed.
- (a) M. D. Ambhore, P. Shukla, R. G. Gonnade and V. G. Anand, Chem. Commun., 2022, 58, 8946–8949 RSC; (b) Z. Zeng, X. Shi, C. Chi, J. T. Lopez Navarrete, J. Casado and J. Wu, Chem. Soc. Rev., 2015, 44, 6578–6596 RSC.
- P. V. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
- D. Geuenich, K. Hess, F. Kohler and R. Herges, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS PubMed.
- (a) T. M. Krygowski and M. K. Cyranski, Chem. Rev., 2001, 101, 1385–1420 CrossRef CAS PubMed; (b) E. Matito, Phys. Chem. Chem. Phys., 2016, 18, 11839–11846 RSC; (c) I. C. Reig, T. Woller, J. C. Garcıa, M. Alonso, M. T. Sucarrat and E. Matito, Phys. Chem. Chem. Phys., 2018, 20, 2787–2796 RSC.
- D. Dohnert and J. Koutecky, J. Am. Chem. Soc., 1980, 102, 1789–1796 CrossRef.
- (a) I. C. Reig, E. R. Cordoba, M. T. Sucarrat and E. Matito, Molecules, 2020, 25, 711 CrossRef; (b) I. C. Reig, R. G. Aviles, E. R. Cordoba, M. T. Sucarrat and E. Matito, Angew. Chem., Int. Ed., 2021, 60, 24080–24088 CrossRef PubMed; (c) J. Wu, Diradicaloids, Jenny Stanford Publishing, 2022 CrossRef.
- R. Rathore, A. S. Kumar, S. V. Lindeman and J. K. Kochi, J. Org. Chem., 1998, 63, 5847–6682 CrossRef CAS.
Footnotes |
| † Dedicated to Prof. Vadapalli Chandrasekhar on the occasion of his 65th birthday |
| ‡ Electronic supplementary information (ESI) available. CCDC 2293502, 2294107, 2294108 and 2294163. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05251f |
| This journal is © The Royal Society of Chemistry 2024 |