
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery and bioinspired total syntheses of unprecedented sesquiterpenoid dimers unveiled bifurcating [4 + 2] cycloaddition and target differentiation of enantiomers†
Tao-Bin
He‡
a,
Bing-Chao
Yan‡
 a,
Yuan-Fei
Zhou‡
c,
Yue-Qian
Sang‡
b,
Xiao-Nian
Li
a,
Han-Dong
Sun
a,
Chu
Wang
c,
Xiao-Song
Xue
*b and
Pema-Tenzin
Puno
*a
a,
Yuan-Fei
Zhou‡
c,
Yue-Qian
Sang‡
b,
Xiao-Nian
Li
a,
Han-Dong
Sun
a,
Chu
Wang
c,
Xiao-Song
Xue
*b and
Pema-Tenzin
Puno
*a
aState Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, Yunnan Key Laboratory of Natural Medicinal Chemistry, Kunming 650201, China. E-mail: punopematenzin@mail.kib.ac.cn
bKey Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Shanghai200032, China. E-mail: xuexs@sioc.ac.cn
cSynthetic and Functional Biomolecules Center, Beijing National Laboratory for Molecular Sciences, Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China
First published on 29th November 2023
Abstract
[4 + 2] cycloaddition has led to diverse polycyclic chiral architectures, serving as novel sources for organic synthesis and biological exploration. Here, an unprecedented class of cadinane sesquiterpene [4 + 2] dimers, henryinins A–E (1–5), with a unique 6/6/6/6/6-fused pentacyclic system, were isolated from Schisandra henryi. The divergent total syntheses of compounds 1–5 and their enantiomers (6–10) were concisely accomplished in eight linear steps using a protection-free approach. Mechanistic studies illustrated the origin of selectivity in the key [4 + 2] cycloaddition as well as the inhibition of reaction pathway bifurcation via desymmetrization. The chemical proteomics results showed that a pair of enantiomers shared common targets (PRDX5 C100 and BLMH C73) and had unique targets (USP45 C588 for 4 and COG7 C419 for 9). This work provides experimental evidence for the discovery of unprecedented cadinane dimers from selective Diels–Alder reaction and a powerful strategy to explore the biological properties of natural products.
Introduction
Natural products arising from [4 + 2] cycloaddition account for a large number of substances in nature, with varying degrees of structural complexity and pharmacological activities.1–3 The inherent chiral states of polycyclic natural products produced from selective cycloadditions exert diverse biological effects. Given the important relationship between stereochemistry and biological activity, the syntheses of natural products and their enantiomers are expected to increase the potential of screening new frameworks for drug discovery.4 As one of the most powerful and widely used transformations for carbon–carbon bond formation, Diels–Alder reactions enable the expedient syntheses of structurally diverse natural products.5–7 Sesquiterpene dimers, which are naturally occurring metabolites having 30 skeleton carbons, originate biogenetically from two identical or different sesquiterpenoid units.8 Many bioactive dimeric sesquiterpenoids, particularly guaiane and lindenane dimers, are formed via key Diels–Alder reactions (gochnatiolide,9,10 artemisianin A,11 schizukaol A,12–14 and japonicone A15 in Fig. 1A). The total syntheses of these dimeric sesquiterpenoids remain a great challenge. Furthermore, it is still unclear how the selectivity of cycloaddition is achieved during the formation of these natural enantiomeric dimers.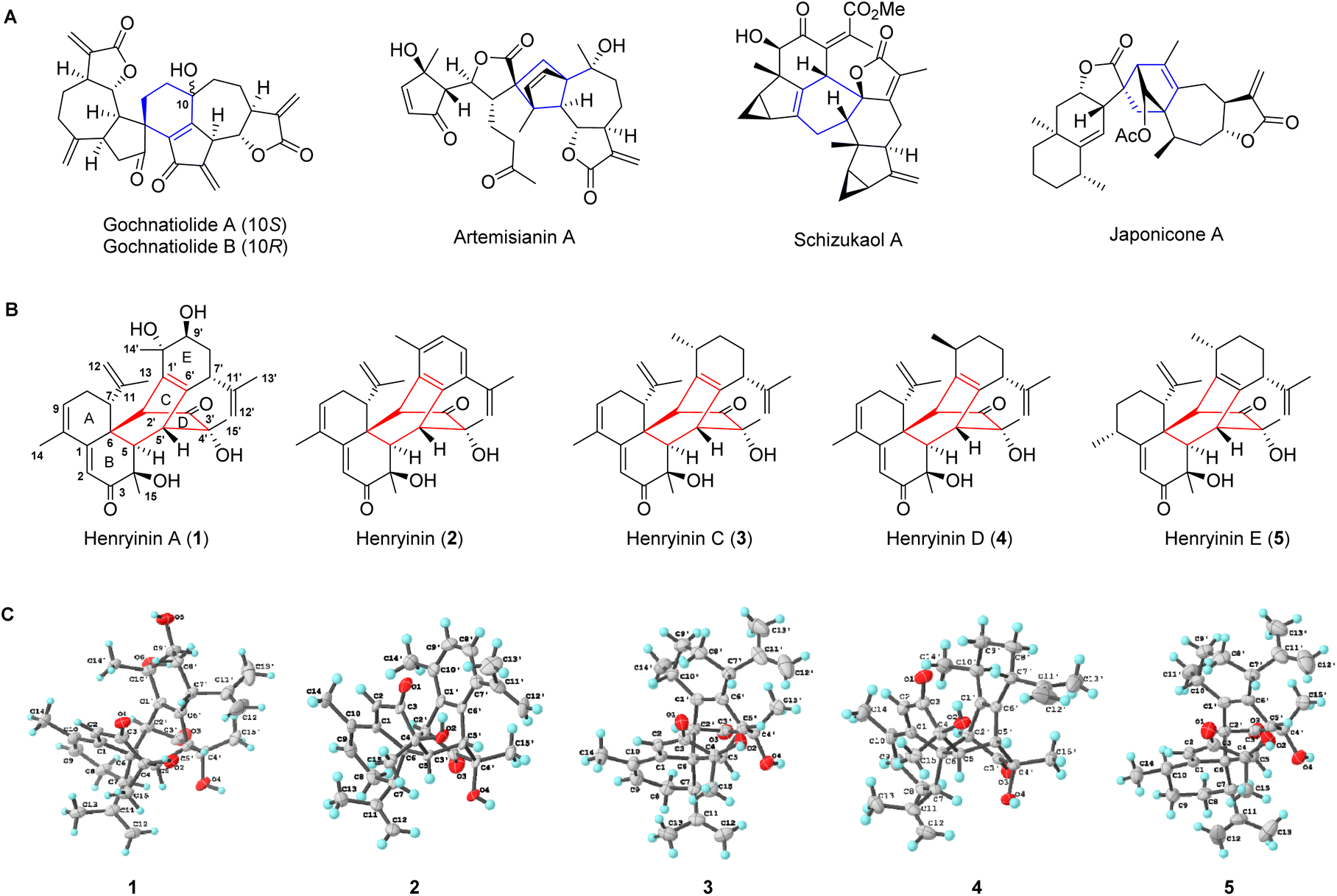 | ||
| Fig. 1 The structure of dimeric sesquiterpenoids. (A) Selected examples of dimeric sesquiterpenoids. (B and C) Chemical and X-ray structures of henryinins A–E (1–5) bearing complex stereogenic centers (ten, seven, nine, nine, and ten chiral carbons for 1–5, respectively). | ||
Cadinane sesquiterpene dimers represent a unique class of sesquiterpene dimers with a wide array of biological activities, including antitumor, anti-inflammation, and anti-HIV properties.8 Herein, a new class of cadinane sesquiterpene [4 + 2] dimers, henryinins A–E (1–5) featuring a unique 6/6/6/6/6-fused pentacyclic skeleton, were isolated from the stems and leaves of Schisandra henryi (Fig. 1B). They possess complex stereogenic centers (ten, seven, nine, nine, and ten chiral carbons for 1–5, respectively) as single enantiomers, demonstrating the peri-, regio-, and stereo-selective features of their biogenetic pathways.
Both enantiomers can be found in the majority of natural products. Additionally, they may exhibit markedly distinct biological and pharmacological behavior in chiral living systems because of their different spatial orientations.16,17 Thus, to investigate the biological activities and provide evidence for the biogenetic pathways of these isolated dimers, the bioinspired and protection-group-free total syntheses of 1–5 and their enantiomers (6–10) were achieved in eight linear steps. The steps mainly included oxidative aromatization via Saegusa–Ito oxidation, oxidative dearomatization, and bifurcating Diels–Alder reactions with high peri-, regio-, and stereo-selectivity. Computational modelling demonstrated the origin of selectivity and the modulation of ambimodal cycloadditions. Given that the enantiomers could bind to different targets, their potential targets were detected using chemical proteomics. The results showed that a pair of enantiomers could share common targets (PRDX5 C100 and BLMH C73) and have their own unique targets (USP45 C588 for 4 and COG7 C419 for 9), providing a powerful strategy to explore the biological properties of natural products.
Results and discussion
Structural elucidation and plausible biogenetic pathways for 1–5
Henryinin A (1), a colorless crystal, possesses a molecular formula of C30H38O6, as determined using high-resolution electrospray ionization mass spectroscopy (HRESIMS) ([M + K]+m/z 533.2304, calculated 533.2300), indicating 12 degrees of unsaturation. The carbon-13 (13C) nuclear magnetic resonance (NMR), distortionless enhancement by polarization transfer (DEPT), and heteronuclear single quantum coherence (HSQC) spectra of 1 (Table S1, ESI†) revealed 30 carbon signals attributable to six methyl, four methylene (two olefinic), eight methine (two olefinic and one oxygenated), and 12 quaternary carbon (two carbonyl, three oxygenated and six olefinic) groups. The presence of an α, β, γ, δ-unsaturated ketone was indicated by the deshielded chemical shift of C-1 (δC 153.4) and the shielded chemical shifts of C-2 (δC 123.2) and C-3 carbonyl (δC 203.1) together with olefinic C-9 (δC 133.4) and C-10 (δC 131.9). The above evidence indicated that 1 might be a sesquiterpene dimer fused with a pentacyclic scaffold.The planar structure of 1 was assigned using two-dimensional (2D) NMR data (Fig. S1, ESI†). The key 1H–1H correlation spectroscopy (COSY) correlations of H-7/H2-8/H-9 and the heteronuclear multiple bond correlation (HMBC) results of H-7 with C-1, C-11, C-12, and C-13, H2-8 with C-6 and C-10, and Me-14 with C-1, C-9, and C-10 suggested the presence of a six-membered ring A substituted with C-10 methyl and C-7 isopropenyl groups. The HMBC correlations from H-2 to C-1, C-4, C-6, and C-10, H-5 to C-3, C-4, and C-7, H3-15 to C-3, C-4, and C-5 suggested that a six-membered ring B was fused with ring A. Similarly, a bicyclo[2,2,2]octane-bridged ring system was established using the HMBC correlations of H-2′ with C-5, C-7, and C-10′, H-5′ with C-1′, C-3′, and C-6′, and H3-15 with C-3′, C-4′ and C-5′, together with the 1H–1H COSY correlations of H-5/H-5′. The key 1H–1H COSY correlations of H-7′/H2-8′/H-9′ along with the HMBC correlations of H-7′ with C-11′, C-12′, and C-13′; H-14′ with C-9′ and C-10′; H-2′ with C-10′; and H-5′ with C-7′ showed the presence of a six-membered ring E substituted with C-10′ methyl and C-7′ isopropenyl groups. In rotating frame Overhauser effect spectroscopy (ROESY) (Fig. S1†), the cross-peaks of H-5/Me-15 indicated that H-5 and Me-15 were cofacial and adopted α-orientations. The ROESY correlations of H-2′/H-7 suggested that H-2′ and H-7 were vicinally disposed. Considering the biogenetic pathway involving the intermolecular Diels–Alder reaction, H-7 and H-2′ were deduced to be the opposite orientations of H-5 and H-5′, respectively. Thus, H-7 and H-5′ were tentatively assigned as β-orientations, whereas H-2′ was α-oriented. The orientation of Me-15′ was deduced to be β-oriented based on the ROESY correlations of Me-15′/H-5′.
To confirm the structure of 1, high-quality crystals were obtained via recrystallization in methanol. An X-ray diffraction study for 1 was successfully performed using anomalous scattering of CuKα radiation providing conclusive evidence for its absolute configuration (4R,5R,6R,7R,2′S,4′R,5′R,7′R,9′S,10′S) with a Flack parameter of 0.09(4) (Fig. 1C). Compounds 2–5 were determined to be similar to 1, of which the structural elucidation was detailed in the ESI.† Their structures and absolute configurations were confirmed by single-crystal X-ray diffraction analysis (Fig. 1C).
A plausible biogenetic pathway for 1–5 was proposed (Fig. 2A). Based on the reported hypothesis, the formation of sesquiterpenes was considered to originate from farnesyl pyrophosphate (FPP).18,19 Therefore, ring-forming reactions involving the ionization and cyclization of FPP are proposed to be the initial step in the formation of these compounds. The cationic intermediate 11 would further be transformed into intermediate 12via quenching of the positive charge and loss of a proton or capture of an external nucleophile such as water or the original pyrophosphate anion. Intermediate 12 would further be transformed into intermediate 13via oxidation, dehydration, and cyclization. The corresponding precursors (14a, 15a, and 16a) of these compounds could be derived from key intermediate 13via hydrogenation and oxidation. Finally, the precursors (14a, 15a, and 16a) would be transformed into sesquiterpene dimers with a new carbon skeleton via a key Diels–Alder cycloaddition.
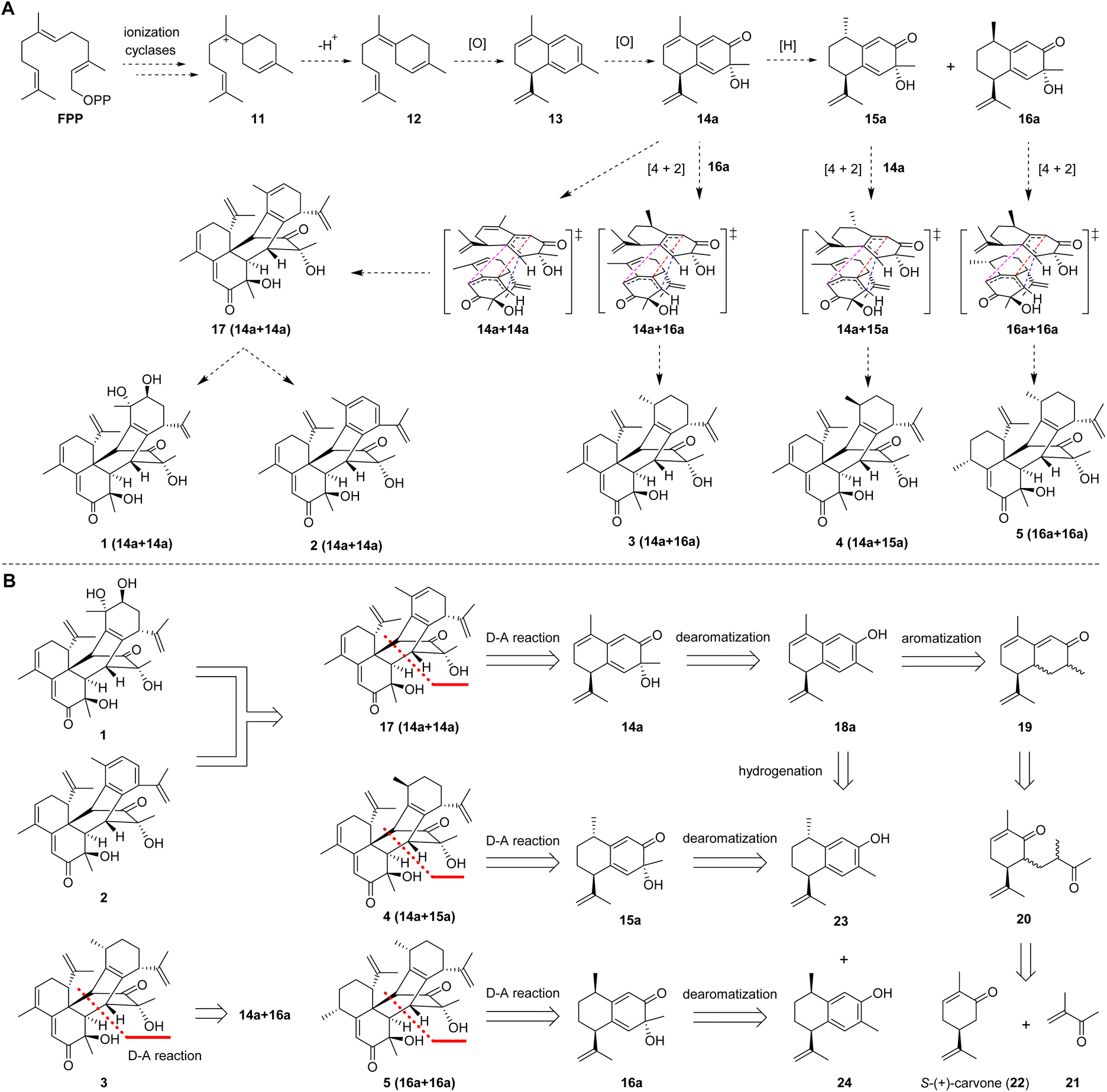 | ||
| Fig. 2 Design of the total syntheses of henryinins A–E (1–5). (A) Plausible biogenetic pathways of henryinins A–E. (B) Retrosynthetic analysis of henryinins A–E (1–5) in a divergent and protection-free approach. | ||
Based on putative biosynthetic pathways, the retrosynthetic analysis of henryinins A–E (1–5) is outlined in Fig. 2B. Considering the structural characteristics of the targets, we envisioned that 1 and 2 could be derived from 17via aromatization20–23 and dihydroxylation,24 respectively. Compound 17 could be constructed from intermediate 14a through an intermolecular Diels–Alder reaction.25,26 Intermediate 14a could be generated via dearomatization of 18a,27 which could be created by aromatization of 19.23,28 Finally, 19 could be prepared from the aldol condensation of 20, which could be accessed from 21 and S-(+)-carvone (22) through a Michael reaction.29–31 Using a similar synthetic route, 3–5 could be synthesized from intermediates 23 and 24, respectively (Fig. 2B).
Bioinspired total syntheses of 1–5 and their enantiomers 6–10
Based on our retrosynthetic analysis, we commenced with the bioinspired and protection group-free syntheses of 1–5 starting from building compound 19, which was prepared from S-(+)-carvone (22) according to a reported procedure (Fig. 3A).32 The Mukaiyama–Michael reaction of 21 with the kinetic silyl enol ether was accomplished using 5% tritylium hexachloroantimonate as the catalyst to give 20 as a single diastereomer in an overall yield of 53%.31 The cyclization and methyl isomerization of diketone 20 in the presence of potassium hydroxide (KOH) in methanol at room temperature afforded cadinene sesquiterpenoid 19 in 72% yield.31 The selective reduction of the γ,δ-double bond of 19 was screened under several conditions, including Pd/C in benzyl alcohol,33 Pd/C in the presence of hydrogen in hexane,34 and NaBH4 and NiCl2·6H2O in methanol (MeOH) (see Table S6† for details).35 Finally, the treatment of 19 with palladium on calcium carbonate (CaCO3) produced enone epimers 25a and 25b (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) in an isolated yield of 91%.34 The absolute configuration of 25a was confirmed by single-crystal X-ray diffraction analysis.
:3) in an isolated yield of 91%.34 The absolute configuration of 25a was confirmed by single-crystal X-ray diffraction analysis.
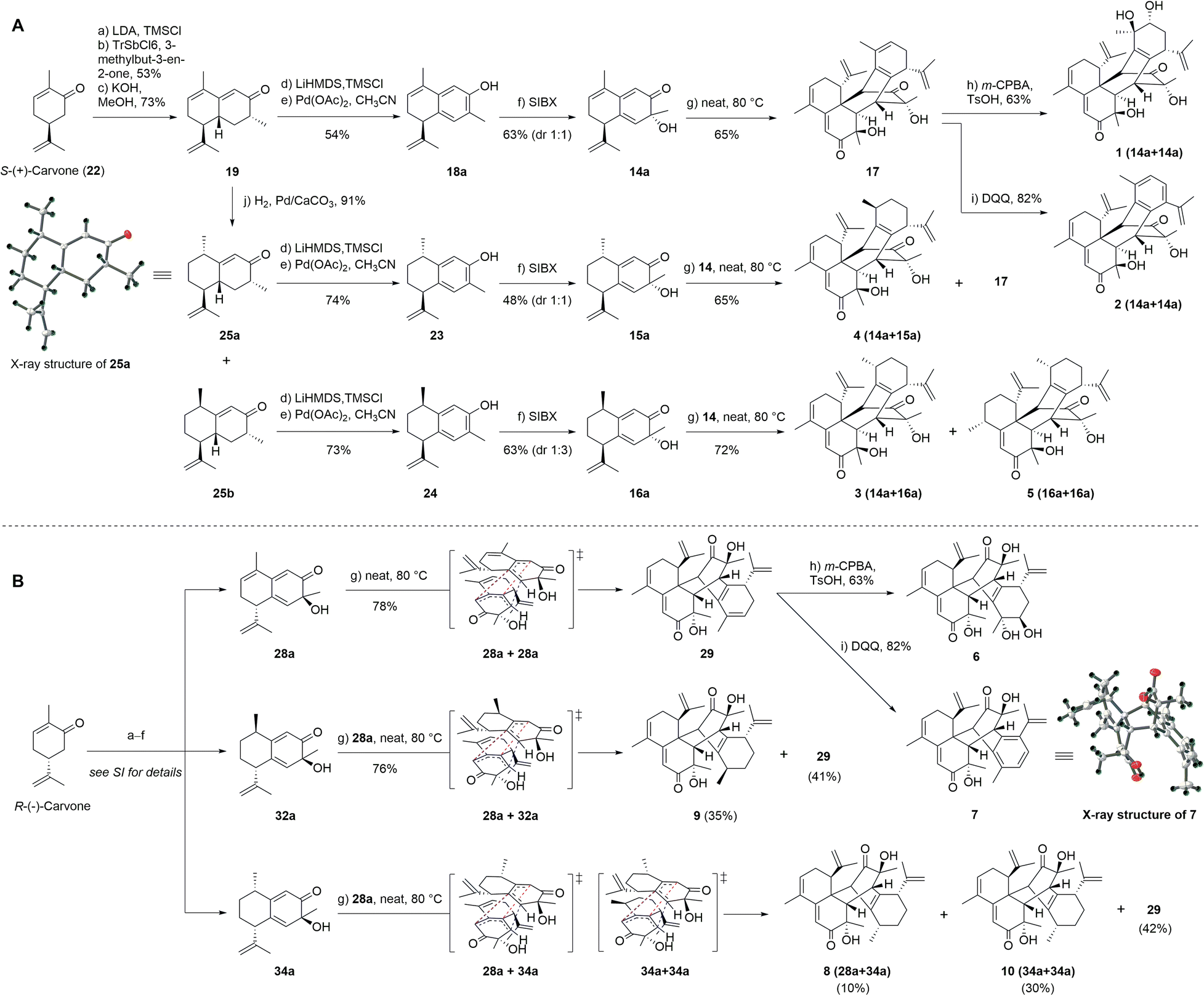 | ||
| Fig. 3 Bioinspired total syntheses. (A) Henryinins A–E (1–5). (B) Enantiomers 6–10 based on the peri-, regio-, and stereo-selectivity of Diels–Alder reaction. | ||
To achieve the key intermediate phenols 18a, 23, and 24, aromatization reactions were investigated under a variety of conditions.20,22,23,28,36–39 Initially, palladium(II) trifluoroacetate [Pd(TFA)2] and the corresponding additives [pyridine, and toluenesulfonic acid (TsOH)] were used as catalysts based on the report of Stahl et al.20 However, the aromatization of compound 19 was not observed in our investigation (Table 1, entry 1). Then, we examined the conversion of 19, 25a, and 25b to phenol using lithium diisopropylamide (LDA) and phenylselenyl chloride (PhSeCl) in tetrahydrofuran (THF) followed by oxidation of selenide with 30% hydrogen peroxide (H2O2).37,38 These conditions were suitable for converting 25a and 25b; however, the undesired product 18b was observed for 19 (Table 1, entry 2). We subjected 19 to 2,3-dichloro-5,6-dicyano-benzoquinone (DDQ); however, the aromatization of 19 did not occur (Table 1, entry 3).39 When 19 and DDQ were reacted in the presence of 5 mol% tert-butyldimethylsilyl chloride (TBSCl) in dioxane at room temperature, the product 18c was obtained (Table 1, entry 4).22 However, we could not achieve the desired product via the dearomatization of 18c when we attempted to synthesize the target product 2 by the dearomatization and Diels–Alder reaction of 18c. Given these challenges, we sequentially sought to explore the dehydrogenative aromatization of 19via metal-free I2 as the catalyst and dimethylsulfoxide (DMSO) as the oxidant.23 Likewise, undesired product 18c was generated (Table 1, entry 5). Gratifyingly, inspired by Saegusa–Ito oxidation for enone synthesis,40 phenols 18a, 23, and 24 were afforded via oxidative dehydrogenation of 19, 25a, and 25b, respectively (Table 1, entry 6). Although diverse synthetic methods for the dehydrogenation of cyclohexanone have been developed for the preparation of phenols in recent decades, such as metal-catalyzed20,28,36 or metal-free23,37,39 oxidative dehydrogenation, to our knowledge, this was the first example of the total synthesis of phenols via Saegusa–Ito oxidation. To improve the efficiency, we further explored the conditions of aromatization using this method (see Table S7† for details).
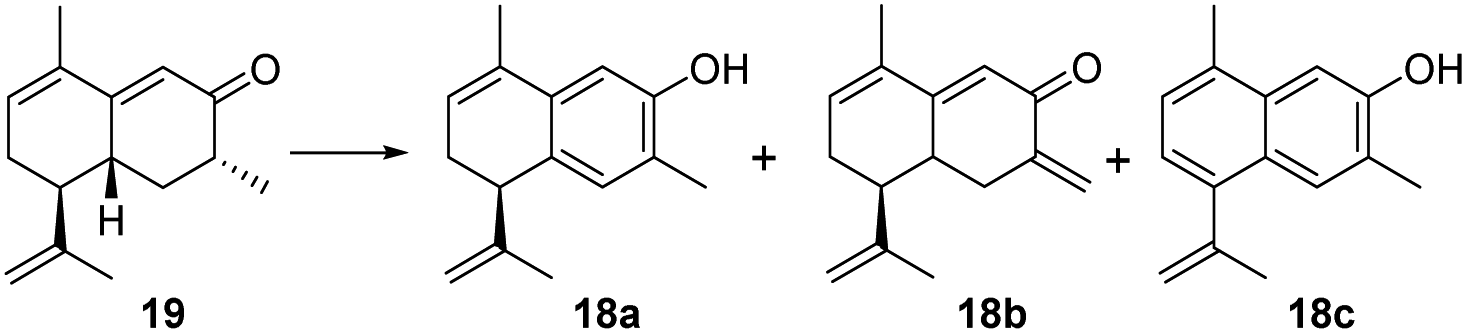
With ample quantities of phenols 18a, 23, and 24 in hand, the key dearomatizing transformation of these phenols was performed using different methods (the detailed optimization procedures are shown in Table S8†). These phenols underwent oxidative dearomatization after treatment with lead(IV) acetate [Pb(OAc)4] to furnish the acetylated product (Table S8,† entry 5). However, the obtained products failed to undergo Diels–Alder cycloaddition and were broken via deacetylation with various bases. Finally, the dearomatizing transformation was efficiently accomplished via stabilized l5-iodane 2-iodoxybenzoic acid (SIBX)-mediated hydroxylative phenol.41,42 The oxidation of phenol 18a with a suspension of SIBX at room temperature produced 14a and 14b (1:1) in an overall yield of 63%. With 14a and 14b in hand, we focused on the formation of a bicyclo[2,2,2]octane-bridged system. Interestingly, we only found the formation of an endo diastereomeric adduct (17) of 14a, which showed the peri-, regio-, and stereo-specific natures of the Diels–Alder reaction. The treatment of key intermediate 17 with meta-chloroperoxybenzoic acid (m-CPBA) in the presence of p-TsOH provided compound 1 in an isolated yield of 67%.43 The aromatization of 17 proceeded smoothly using DDQ to deliver compound 2.37 The treatment of 25a with a suspension of SIBX at room temperature provided epimers 15a and 15b (1:1) in 48% yield. Unexpectedly, neither 15a nor 15b underwent the Diels–Alder reaction, which may have been a result of the Me-14 configuration.
Fortunately, 15a and 14a (1:3) were heated at 80 °C to produce compounds 4 and 9. Compound 25b underwent dearomatization with a suspension of SIBX at room temperature to generate epimers 16a and 16b (1:3), resulting from the propensity of spontaneous [4 + 2] cyclodimerizations of 16a. Compounds 16a and 14a were heated at 80 °C to produce compounds 3, 5, and 9 (1:1:2). Thus, the enantioselective preparation of natural products (1–5) was effectively accomplished based on the chiral starting material.
In recent years, many enantiomeric natural products have been discovered with distinct biological activities.44 Furthermore, the high level of regioselectivity and periselectivity of the Diels–Alder reaction has also been reported recently.45 To further explore whether the isopropenyl group located at C-7 affected the peri-, regio-, and stereo-selectivity of the Diels–Alder reaction of our intermediates and the biological activities of enantiomeric natural products, we performed the total syntheses of their enantiomers using R-(−)-carvone as the starting material. As expected, the total syntheses of enantiomers of natural products (6–10) was effectively completed and the Diels–Alder reaction also demonstrated peri-, regio-, and stereo-selectivity (Fig. 3B). The fine synthetic route is outlined in Scheme S1 (ESI†).
Computational studies on the reactivity and origin of selectivity
Density functional theory calculations were conducted to explore the influence of structural differences on the reactivity and selectivity of the dimerization reaction. We chose 14a and its stereoisomer 14b for the initial computations. The homodimerization of 14a is both thermodynamically and kinetically favored over that of 14b (Fig. 4A). The transition state of 14a (TS14a+14a) is more stable than that of TS14b+14b (14b) by 15.1 kcal mol−1, which was consistent with the absence of the cycloaddition product for 14b in the experiments. Moreover, the formation of 17 is more exergonic (by 10.5 kcal mol−1) and is favored over the dimerization product of 14b (by 5.4 kcal mol−1). The two distorted reactants in TS14a+14a form a double-layer shape staggered in C2 symmetry, as the dihedral C6–C5–C5′−C6′ angle is 56.1°. TS14a+14a is an ambimodal transition state that led to two identical adducts, in which the formed C5–C5′ bond is shared, and the competing C6–C2′ or C2–C6′ bond formation afford two equivalent products denoted as [2 + 4] or [4 + 2], respectively.46–48 Because of the shorter C5–C5′ and longer C6–C2′ or C2–C6′ distances, TS14a+14a is concerted but asynchronous.49 The significant free energy difference of 15.1 kcal mol−1 between TS14a+14a and TS14b+14b is merely caused by the inversion of the C4 configuration. As shown in Fig. 4B, a glimpse of the TS14b+14b structure may readily lead to the conclusion that this energy difference is caused by the steric repulsion between the equatorial methyl groups attached to C4 and the neighboring C–H bonds of the other reactant. However, substituting the Me group with H in both TS14a+14a and TS14b+14b to release the steric repulsion of Me only decreased the free energy difference to 10.9 kcal mol−1 (Fig. S11A†), illustrating this steric effect is not the major reason for the energy difference.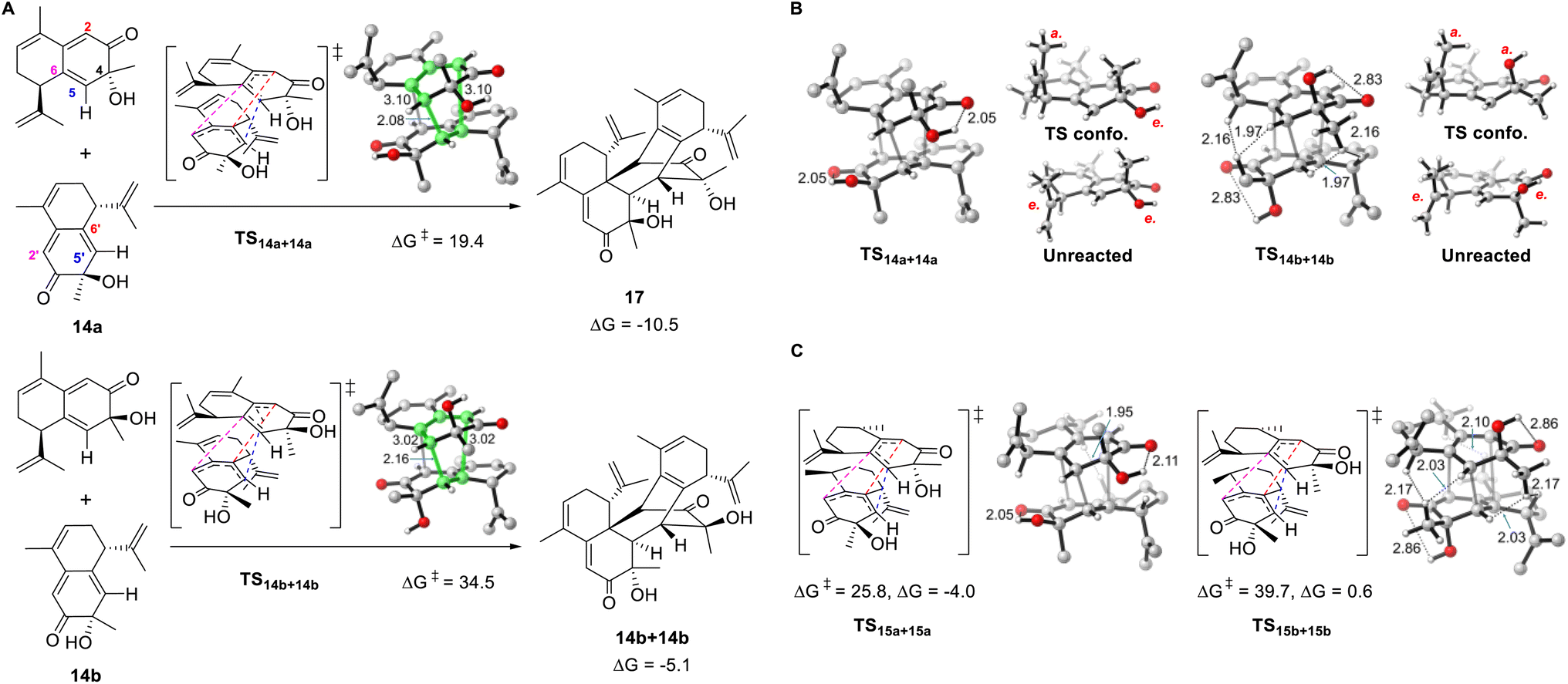 | ||
| Fig. 4 Computational results. (A) Free energy barriers and reaction energies (kcal mol−1) for the homo-dimerization reaction of 14a and 14b. (B) Comparison between TS14a+14a and TS14b+14b illustrates the origin of stereoselectivity. (C) Homo-dimerization reaction of 15a and 15b. | ||
Distortion/interaction analysis was then conducted for the transition states and the derived intrinsic reaction coordinates.50 Plotting the energies against the length of the forming bond, we noticed that the distortion energies of 14a and 14b maintain a constant difference of ∼10 kcal mol−1, and the interaction energy difference was approximately 5 kcal mol−1 (Fig. S12†). To maximize the deserved interactions in the transition state, both reactants change conformation as the OH/Me group leaves the equatorial for the axial position of 14b/14a, respectively. The intramolecular hydrogen bond between OH and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in 14a is kept in TS14a+14a, but disappeared in TS14b+14b, implying that its absence caused the high distortion energy of TS14b+14b. Indeed, if OH is substituted by H, the energy difference of the corresponding transition states decreases to 3.3 kcal mol−1 (Fig. S11B†). Therefore, the strong preference for TS14a+14a over TS14b+14b is ascribed to the destruction of intramolecular hydrogen bonds and the steric repulsion caused by the Me group in TS14b+14b, with the former playing a major role. Notably, this origin of stereoselectivity not only reflects the effect of the C4 configuration, but is also suitable for 28b with an inverted C7 configuration because the corresponding transition state is the mirror image of TS14b+14b.
O in 14a is kept in TS14a+14a, but disappeared in TS14b+14b, implying that its absence caused the high distortion energy of TS14b+14b. Indeed, if OH is substituted by H, the energy difference of the corresponding transition states decreases to 3.3 kcal mol−1 (Fig. S11B†). Therefore, the strong preference for TS14a+14a over TS14b+14b is ascribed to the destruction of intramolecular hydrogen bonds and the steric repulsion caused by the Me group in TS14b+14b, with the former playing a major role. Notably, this origin of stereoselectivity not only reflects the effect of the C4 configuration, but is also suitable for 28b with an inverted C7 configuration because the corresponding transition state is the mirror image of TS14b+14b.
In addition to the aforementioned pericyclic reaction of 14a to produce 17, the pathways leading to other stereoisomers and regiomers were calculated to be inaccessible (Fig. S13 and S14†), as the energy barriers are >36.0 kcal mol−1. Both intra- and inter-molecular steric repulsions were observed in these transition states. Similar to the unmodified 14a and 14b, the homodimerization of 16a is both thermodynamically and kinetically favored over that of 16b (Fig. S15†). Unlike 14a, the homodimerization product of 15a was not observed in the experiments. Correspondingly, the reaction barrier was calculated to be 25.8 kcal mol−1, in which steric repulsion exists between the Me groups at C10 and C10′ (Fig. 4C and S16†).
The cross-dimerization between 14a and 16a demonstrated that subtle structural modifications could inhibit the reaction pathway bifurcation and produce a single product. In theory, both 14a and 16a could act as dienophiles to react with the other reactant; however, only the former adduct 3 was observed. Accordingly, our computation located TS14a+16a for the former reaction pathway; however, attempts to find the transition state for the latter reaction failed and led to TS14a+16a. The merged structure indicates that TS14a+16a is ambimodal as well (Fig. 5). Compared with the equivalent C6–C2′ or C2–C6′ distance of 3.10 Å in TS14a+14a, the larger steric hindrance of the saturated C9–C10 of 16a than that of the CC of 14a elongates the C6–C2′ distance to 3.40 Å and increases the dihedral C6–C5–C5′−C6′ to 62.6°. Concomitantly, the C2–C6′ distance decreases to 2.96 Å because of the seesaw motion of 16a. Weaker/stronger secondary orbital interactions between C6–C2' and C2–C6′ suggest a lower/higher yield of the corresponding product.51 According to Houk−Yang's empirical equation,51 the distribution of 3/16a+14a was predicted to be 64:1. A quasi-classical direct molecular dynamics simulation was conducted; however, none of the trajectories initiated from TS14a+16a terminates at 16a+14a (Fig. S17†).52 Moreover, the Cope rearrangement of 3 to produce 16a+14a has an inaccessible energy barrier of 31.2 kcal mol−1, thus rationalizing the absence of the adduct with 16a as a dienophile. These results illustrate that the steric hindrance created by structural modification breaks the C2 symmetry and inhibits reaction pathway bifurcation.51,53–56 Although the homodimerization of 15a is accessible, its transformation was enabled by cross-dimerization with 14a, which has a barrier of 22.5 kcal mol−1viaTS14a+15a (Fig. S18†). The energy difference of 3.3 kcal mol−1 between TS15a+15a and TS14a+15a excludes the homodimerization reaction. Moreover, the computed barriers increase in an order from TS14a+14a to TS15a+15a, which reproduced the experimental product distribution. Similar to 14a and 16a, the cross-dimerization of 14a and 15a failed to generate the adduct with 15a as a dienophile because bifurcation is inhibited (Fig. S19†).
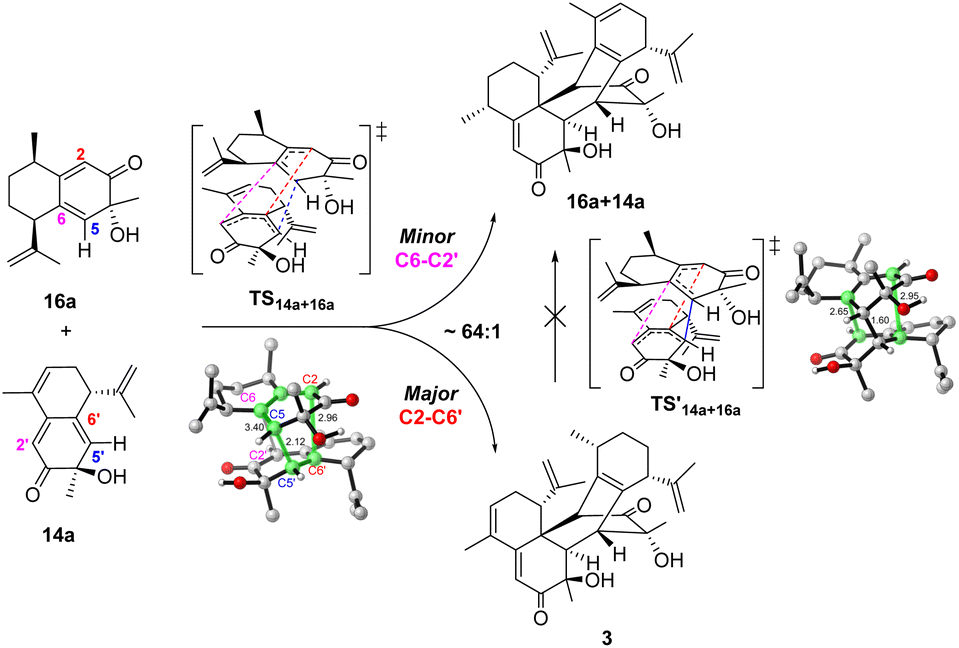 | ||
| Fig. 5 Reaction pathway bifurcation and the ambimodal TS14a+16a of the cross-dimerization reaction between 14a and 16a. | ||
Chemoproteomic profiling of a pair of enantiomers by the competitive rdTOP-ABPP method
We reasoned that the α,β,γ,δ-unsaturated ketone groups of compounds 1–10 would capture thiol groups and form C–S covalent bonds with the cysteine residues of the target proteins. Therefore, we could explore whether there are active cysteines in the proteome that can covalently bind to the compounds, thereby identifying the potential target of the compound. The isotopic tandem orthogonal proteolysis-activity-based protein profiling (isoTOP-ABPP) method, an advanced derivative of Activity-Based Protein Profiling (ABPP),57 employs a cysteine-reactive iodoacetamide probe functionalized with a bioorthogonal alkyne handle [iodoacetamide (IA)-alkyne] and a pair of isotopically encoded cleavable azide-biotin tags to enable quantification of intrinsic cysteine reactivity.58 Additionally, it can be used for mapping druggable hotspots targeted by covalently acting metabolites.59 Recently, the rdTOP-ABPP method was introduced to enhance the pipeline by incorporating reductive dimethylation for triplex quantitation to broadly functional cysteinomes.60 The above methods were designed to determine if the compounds can covalently bind to cysteines and compete with IA-alkyne. Briefly, the α,β,γ,δ-unsaturated ketone groups in 1–10 likely reacted with cysteine residues of target proteins because the sulfur atoms of cysteine residues will have strong nucleophilic reactivity, when the sulfhydryl groups are deprotonated. When cysteine residues form covalent bonds with compounds, fewer cysteine residues undergo nucleophilic substitution reactions with IA-alkyne compared with the DMSO group. Subsequent visualization of IA-alkyne using azide-rhodamine will produce less fluorescence. Therefore, we first evaluated whether these compounds could compete with IA-alkyne in cell lysates using in-gel fluorescence. HT1080 cell lysates were treated with 100 μM compounds 1–10, labelled with IA-alkyne and then visualized using azide-rhodamine via copper-catalyzed azide–alkyne cycloaddition (CuAAC) (Fig. S19†).Compounds 3, 4 and 9 showed weaker fluorescence than the vehicle group (vehicle group was added DMSO as a control), indicating that the three compounds likely have stronger reactivity with cysteine residues than other groups and showed competition with IA-alkyne (Fig. 6A). Given that 4 and 9 are a pair of enantiomers, the two compounds were also studied. The fluorescence of 4 and 9 decreased with increasing compound concentrations, suggesting that competition against IA-alkyne was concentration-dependent (Fig. 6B). Although cytotoxic experiments on HT1080 cells demonstrated that 4 showed similar inhibition rates to 9 at three concentrations (Fig. 6C), whether the targets of the natural product (4) differed from its enantiomer (9) remains unclear. Therefore, we performed comprehensive profiling of 4 and 9 in HT1080 cell lysates using rdTOP-ABPP. The HT1080 whole cell lysates were first treated with DMSO, 4 or 9 (10 μM and 100 μM), and then labeled separately with the IA-alkyne probe. After enrichment with streptavidin and on-bead trypsin digestion, the probe-adducted peptides from DMSO, 4 or 9 (10 μM and 100 μM), were isotopically labeled with light, medium, and heavy dimethylation reagents, respectively (Fig. 6D). The samples were subjected to acid cleavage, and the released peptides were analyzed using liquid chromatography with tandem mass spectrometry (LC-MS/MS) and quantified using CIMAGE 2.0 software.61 We performed three biological replicates for both experiments, and the peptides quantified in at least two out of the three replicates were considered for further analysis. The light/medium [DMSO/4 or 9 (10 μM)] ratio and the light/heavy [DMSO/4 or 9 (100 μM)] ratio reflected the competitiveness of the given compounds with IA-alkyne (Fig. 6D and S20†). Finally, 1901 targets were quantified under the 10 μM compound treatment, and 1660 targets were quantified under the 100 μM compound treatment (Fig. 6E). Among the candidate targets (Fig. 6F), we chose these targets for two reasons. One is that the spectrum has a high degree of confidence, requiring a good fit (R2) value between the light/medium/heavy groups and stable performance across three replicates. The other is its concentration-dependent features, meaning that the ratio of the high-concentration group (100 μM) was higher than that of the low-concentration group (10 μM). In the candidates, PRDX5 C100 and BLMH C73 both met the above criteria in the two groups and were considered as common targets of compounds 4 and 9 (Fig. 6G, for more see the ESI†). USP45 C588 was detected only in the compound 4 group but not in the compound 9 group and was thought to be a specific target for 4. The dose–effect relationship of USP45 C588 was not shown at high and low concentrations in the compound 4 group, mainly because the competition reached saturation at low concentrations (the highest ratio value was 15) (Fig. 6G, for more see ESI†). COG7 C419 was detected in both the compound 4 group and compound 9 group. However, COG7 C419 showed a concentration-dependent feature only in the compound 9 group and was considered to be a specific target for 9.
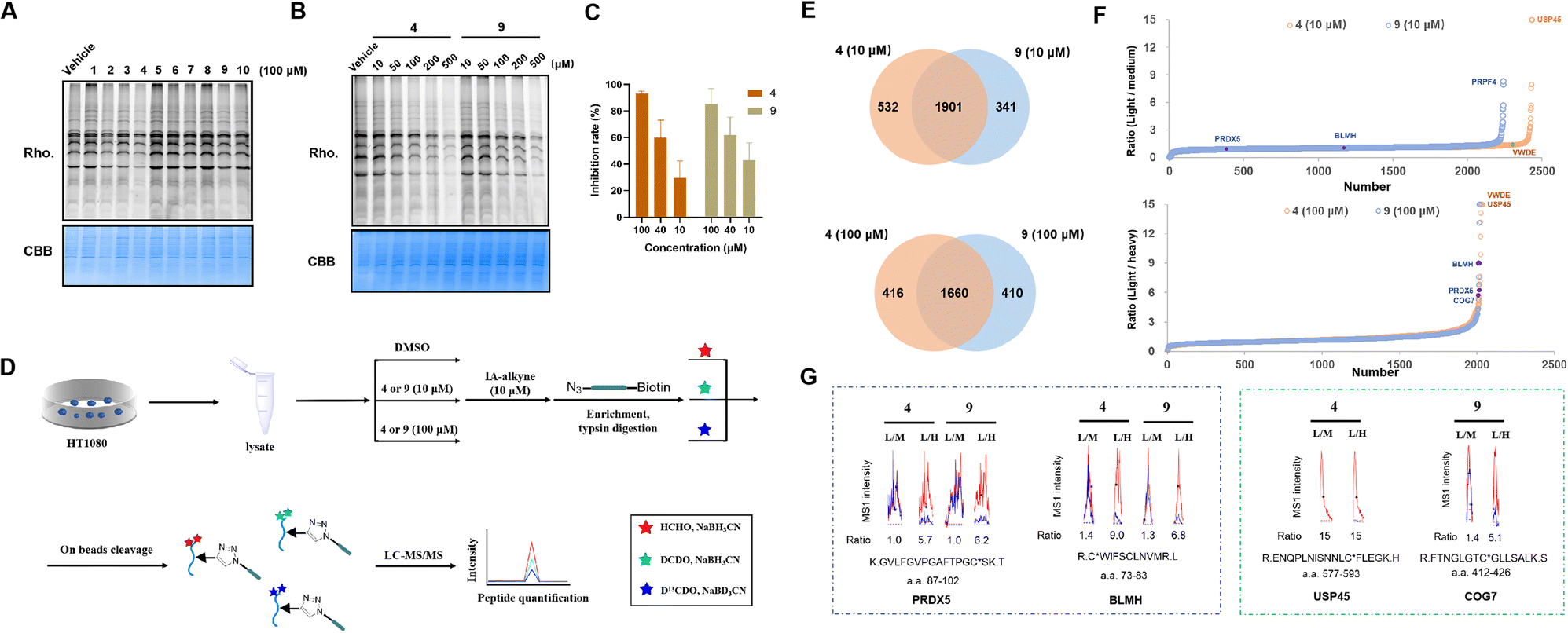 | ||
| Fig. 6 Chemical proteomic profiling by competitive rdTOP-ABPP. (A) The HT1080 cell lysates were incubated with compounds 1–10 and visualized by in-gel fluorescence; (B) the fluorescence decreased as the concentrations of 4 and 9 increased; (C) cytotoxic experiments demonstrated that 4 showed similar inhibition rates to 9 at three concentrations; (D) the scheme of quantitative profiling of reactive cysteines by rdTOP-ABPP; (E) a Venn diagram showing the number of re-active cysteine peptides quantified from 4 and 9 at a 10 or 100 μM level; (F) the rdTOP-ABPP ratios for each cysteine site quantified at a 10 or 100 μM level; (G) representative MS1 profiles for peptides that show common targets (PRDX5 and BLMH) for 4 and 9, and unique targets for 4 (USP45) and 9 (COG7). | ||
Data availability
All experimental and characterization data, as well as NMR spectra, are available in the ESI. Crystallographic data for the structures reported in this paper have been deposited at the Cambridge Crystallographic Data Centre “https://www.ccdc.cam.ac.uk/”, under the accession numbers CCDC: 2212935, 2212936, 2212937, 2212938, 2212942, 2212943, 2212947, 2212950 for 1–5, 7, 25a, and 30a, respectively.†Author contributions
T.-B. H., B.-C. Y., Y.-F. Z., and Y.-Q. S. contributed equally to this work. T.-B. H.: conceptualization, data curation, investigation, writing original draft, and visualization, B.-C. Y.: conceptualization, data curation, visualization, validation, and writing – review & editing, Y.-F. Z.: conceptualization, data curation, investigation, writing original draft, and visualization, Y.-Q. S.: conceptualization, data curation, investigation, writing original draft, and visualization. X.-N. L.: data curation and investigation, H.-D. S.: supervision, C. W.: supervision, writing – review & editing, conceptualization, and validation, X.-S. X.: supervision, writing – review & editing, conceptualization, and validation, P.-T. P.: supervision, funding acquisition, writing – review & editing, conceptualization, resources, and validation.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We are grateful to Prof. Jun Deng for helpful discussion. This project was supported financially by the National Science Fund for Distinguished Young Scholars (82325047), NSFC-Joint Foundation of Yunnan Province (U2002221), Second Tibetan Plateau Scientific Expedition and Research (STEP) program (2019QZKK0502), Major Projects for Fundamental Research of Yunnan Province (202201BC070002), CAS “Light of West China” Program, CAS Interdisciplinary Innovation Team (Pema-Tenzin Puno), National Natural Science Foundation of China (22007089), Natural Science Foundation of Yunnan Province (202001AT070074), Youth Innovation Promotion Association CAS (B.-C. Yan), China Postdoctoral Science Foundation (2023M730106) and Yunnan Province Science and Technology Department (202305AH340005).Notes and references
- J. Clardy and C. Walsh, Lessons from natural molecules, Nature, 2004, 432, 829–837 CrossRef CAS PubMed.
- H. Oikawa and T. Tokiwano, Enzymatic catalysis of the Diels–Alder reaction in the biosynthesis of natural products, Nat. Prod. Rep., 2004, 21, 321–352 RSC.
- C. Li, T. Dong, L. Dian, W. Zhang and X. Lei, Biomimetic syntheses and structural elucidation of the apoptosis-inducing sesquiterpenoid trimers: (−)-ainsliatrimers A and B, Chem. Sci., 2013, 4, 1163–1167 RSC.
- Y. Wang, M. M. Dix, G. Bianco, J. R. Remsberg, H.-Y. Lee, M. Kalocsay, S. P. Gygi, S. Forli, G. Vite, R. M. Lawrence, C. G. Parker and B. F. Cravatt, Expedited mapping of the ligandable proteome using fully functionalized enantiomeric probe pairs, Nat. Chem., 2019, 11, 1113–1123 CrossRef CAS PubMed.
- R.-B. Wang, S.-G. Ma, C. S. Jamieson, R.-M. Gao, Y.-B. Liu, Y. Li, X.-J. Wang, Y.-H. Li, K. N. Houk, J. Qu and S.-S. Yu, Library construction of stereochemically diverse isomers of spirooliganin: their total synthesis and antiviral activity, Chem. Sci., 2021, 12, 7003–7011 RSC.
- K.-I. Takao, R. Munakata and K.-I. Tadano, Recent advances in natural product synthesis by using intramolecular Diels−Alder reactions, Chem. Rev., 2005, 105, 4779–4807 CrossRef CAS PubMed.
- J.-L. Li, S.-L. Zhou, P.-Q. Chen, L. Dong, T.-Y. Liu and Y.-C. Chen, Asymmetric Diels–Alder reaction of β,β-disubstituted enals and chromone-fused dienes: construction of collections with high molecular complexity and skeletal diversity, Chem. Sci., 2012, 3, 1879–1882 RSC.
- L.-F. Ma, Y.-L. Chen, W.-G. Shan and Z.-J. Zhan, Natural disesquiterpenoids: an update, Nat. Prod. Rep., 2020, 37, 999–1030 RSC.
- Y. Chen, W. Li, Z. Zeng and Y. Tang, (−)-Gochnatiolide B, synthesized from dehydrocostuslactone, exhibits potent anti-bladder cancer activity in vitro and in vivo, Sci. Rep., 2018, 8, 8807 CrossRef PubMed.
- C. Li, L. Dian, W. Zhang and X. Lei, Biomimetic syntheses of (−)-gochnatiolides A–C and (−)-ainsliadimer B, J. Am. Chem. Soc., 2012, 134, 12414–12417 CrossRef CAS PubMed.
- G.-M. Xue, D.-R. Zhu, C. Han, X.-B. Wang, J.-G. Luo and L.-Y. Kong, Artemisianins A–D, new stereoisomers of seco-guaianolide involved heterodimeric [4+2] adducts from Artemisia argyi induce apoptosis via enhancement of endoplasmic reticulum stress, Bioorg. Chem., 2019, 84, 295–301 CrossRef CAS PubMed.
- B. Du, Z. Huang, X. Wang, T. Chen, G. Shen, S. Fu and B. Liu, A unified strategy toward total syntheses of lindenane sesquiterpenoid [4 + 2] dimers, Nat. Commun., 2019, 10, 1892 CrossRef PubMed.
- J.-L. Wu, Y.-S. Lu, B. Tang and X.-S. Peng, Total syntheses of shizukaols A and E, Nat. Commun., 2018, 9, 4040 CrossRef PubMed.
- C. Yuan, B. Du, H. Deng, Y. Man and B. Liu, Total syntheses of sarcandrolide J and shizukaol D: lindenane sesquiterpenoid [4+2] dimers, Angew. Chem., Int. Ed., 2017, 56, 637–640 CrossRef CAS PubMed.
- J. J. Qin, H. Z. Jin, J. J. Fu, X. J. Hu, Y. Wang, S. K. Yan and W. D. Zhang, Japonicones A–D, bioactive dimeric sesquiterpenes from Inula japonica thunb, Bioorg. Med. Chem. Lett., 2009, 19, 710–713 CrossRef CAS PubMed.
- X. Kang, E. R. Stephens, B. M. Spector-Watts, Z. Li, Y. Liu, L. Liu and Y. Cui, Challenges and opportunities for chiral covalent organic frameworks, Chem. Sci., 2022, 13, 9811–9832 RSC.
- M. Quack, G. Seyfang and G. Wichmann, Perspectives on parity violation in chiral molecules: theory, spectroscopic experiment and biomolecular homochirality, Chem. Sci., 2022, 13, 10598–10643 RSC.
- D. E. Cane, Enzymic formation of sesquiterpenes, Chem. Rev., 1990, 90, 1089–1103 CrossRef CAS.
- Y. J. Hong and D. J. Tantillo, Consequences of conformational preorganization in sesquiterpene biosynthesis: theoretical studies on the formation of the bisabolene, curcumene, acoradiene, zizaene, cedrene, duprezianene, and sesquithuriferol sesquiterpenes, J. Am. Chem. Soc., 2009, 131, 7999–8015 CrossRef CAS PubMed.
- D. Pun, T. Diao and S. S. Stahl, Aerobic dehydrogenation of cyclohexanone to phenol catalyzed by Pd(TFA)2/2-dimethylaminopyridine: evidence for the role of Pd nanoparticles, J. Am. Chem. Soc., 2013, 135, 8213–8221 CrossRef CAS PubMed.
- K. J. Shea, L. D. Burke and R. J. Doedens, A novel synthesis of metacyclophanes. Thermal and DDQ induced aromatization of bridgehead dienes, J. Am. Chem. Soc., 1985, 107, 5305–5306 CrossRef CAS.
- K. Chen, C. Liu, L. Deng and G. Xu, A practical Δ1-dehydrogenation of Δ4-3-keto-steroids with DDQ in the presence of TBDMSCl at room temperature, Steroids, 2010, 75, 513–516 CrossRef CAS PubMed.
- Y.-F. Liang, S. Song, L. Ai, X. Li and N. Jiao, A highly efficient metal-free approach to meta- and multiple-substituted phenols via a simple oxidation of cyclohexenones, Green Chem., 2016, 18, 6462–6467 RSC.
- C. Bonini and G. Righi, A critical outlook and comparison of enantioselective oxidation methodologies of olefins, Tetrahedron, 2002, 58, 4981–5021 CrossRef CAS.
- A. Sib and T. A. M. Gulder, Stereoselective total synthesis of bisorbicillinoid natural products by enzymatic oxidative dearomatization/dimerization, Angew. Chem., Int. Ed., 2017, 56, 12888–12891 CrossRef CAS PubMed.
- S. Dong, K. J. Cahill, M.-I. Kang, N. H. Colburn, C. J. Henrich, J. A. Wilson, J. A. Beutler, R. P. Johnson and J. A. Porco Jr, Microwave-based reaction screening: tandem retro-Diels–Alder/Diels–Alder cycloadditions of o-quinol dimers, J. Org. Chem., 2011, 76, 8944–8954 CrossRef CAS PubMed.
- K. C. Nicolaou, K. B. Simonsen, G. Vassilikogiannakis, P. S. Baran, V. P. Vidali, E. N. Pitsinos and E. A. Couladouros, Biomimetic explorations towards the bisorbicillinoids: total synthesis of bisorbicillinol, bisorbibutenolide, and trichodimerol, Angew. Chem., Int. Ed., 1999, 38, 3555–3559 CrossRef CAS PubMed.
- I. Y. El-Deeb, M. Tian, T. Funakoshi, R. Matsubara and M. Hayashi, Conversion of cyclohexanones to alkyl aryl ethers by using a Pd/C–ethylene system, Eur. J. Org Chem., 2017, 2017, 409–413 CrossRef CAS.
- K.-S. Ngo and G. D. Brown, Synthesis of amorphane and cadinane sesquiterpenes from fabiana imbricata, Tetrahedron, 1999, 55, 15099–15108 CrossRef CAS.
- J. Beauhaire, P.-H. Ducrot and I. Simon, Synthesis of diaprepal A2 cadinane analogs; efficient access to polyoxygenated cadinanes, Synth. Commun., 1995, 25, 3015–3025 CrossRef CAS.
- A. V. Baranovsky, B. J. M. Jansen, T. M. Meulemans and A. de Groot, Enantioselective synthesis of cadinanes starting from R-(−)- or S-(+)-carvone, Tetrahedron, 1998, 54, 5623–5634 CrossRef CAS.
- E. F. Landau and E. P. Irany, Preparation of methyl isopropenyl ketone from methyl ethyl ketone and formaldehyde, J. Org. Chem., 1947, 12, 422–425 CrossRef CAS PubMed.
- R. Vitali, G. Caccia and R. Gardi, Benzyl alcohol as hydrogen donor in selective transfer hydrogenation of unsaturated steroids, J. Org. Chem., 1972, 37, 3745–3746 CrossRef CAS.
- P. Kraft, S. Jordi, N. Denizot and I. Felker, On the dienone motif of musks: synthesis and olfactory properties of partially and fully hydrogenated dienone musks, Eur. J. Org Chem., 2014, 2014, 554–563 CrossRef CAS.
- J. Liu, J. Wu, J.-H. Fan, X. Yan, G. Mei and C.-C. Li, Asymmetric total synthesis of cyclocitrinol, J. Am. Chem. Soc., 2018, 140, 5365–5369 CrossRef CAS PubMed.
- J. Zhang, Q. Jiang, D. Yang, X. Zhao, Y. Dong and R. Liu, Reaction-activated palladium catalyst for dehydrogenation of substituted cyclohexanones to phenols and H2 without oxidants and hydrogen acceptors, Chem. Sci., 2015, 6, 4674–4680 RSC.
- C. X. Zhang, F. Q. Bi and Y. L. Li, The first total synthesis of ligudentatin A, Chin. Chem. Lett., 2008, 19, 805–806 CrossRef CAS.
- M. I. Al-hassan, Conversion of 19-nortestosterone to β-estradiol, Synth. Commun., 1989, 19, 453–461 CrossRef CAS.
- J. A. Robl, A new and versatile route for the synthesis of highly substituted benzenoids, Tetrahedron Lett., 1990, 31, 3421–3424 CrossRef CAS.
- Y. Ito, T. Hirao and T. Saegusa, Synthesis of α,β-unsaturated carbonyl compounds by palladium(II)-catalyzed dehydrosilylation of silyl enol ethers, J. Org. Chem., 1978, 43, 1011–1013 CrossRef CAS.
- N. Lebrasseur, J. Gagnepain, A. Ozanne-Beaudenon, J.-M. Léger and S. Quideau, Efficient access to orthoquinols and their [4 + 2] cyclodimers via SIBX-mediated hydroxylative phenol dearomatization, J. Org. Chem., 2007, 72, 6280–6283 CrossRef CAS PubMed.
- J. Gagnepain, F. Castet and S. Quideau, Total synthesis of (+)-aquaticol by biomimetic phenol dearomatization: double diastereofacial differentiation in the Diels–Alder dimerization of orthoquinols with a C2-symmetric transition state, Angew. Chem., Int. Ed., 2007, 46, 1533–1535 CrossRef CAS PubMed.
- T. W. Bingham, L. W. Hernandez, D. G. Olson, R. L. Svec, P. J. Hergenrother and D. Sarlah, Enantioselective synthesis of isocarbostyril alkaloids and analogs using catalytic dearomative functionalization of benzene, J. Am. Chem. Soc., 2019, 141, 657–670 CrossRef CAS PubMed.
- Y. Chen, G. B. Craven, R. A. Kamber, A. Cuesta, S. Zhersh, Y. S. Moroz, M. C. Bassik and J. Taunton, Direct mapping of ligandable tyrosines and lysines in cells with chiral sulfonyl fluoride probes, Nat. Chem., 2023, 15, 1616–1625 CrossRef CAS PubMed.
- M. Ohashi, C. S. Jamieson, Y. Cai, D. Tan, D. Kanayama, M.-C. Tang, S. M. Anthony, J. V. Chari, J. S. Barber, E. Picazo, T. B. Kakule, S. Cao, N. K. Garg, J. Zhou, K. N. Houk and Y. Tang, An enzymatic Alder-ene reaction, Nature, 2020, 586, 64–69 CrossRef CAS PubMed.
- P. Caramella, P. Quadrelli and L. Toma, An unexpected bispericyclic transition structure leading to 4+2 and 2+4 cycloadducts in the endo dimerization of cyclopentadiene, J. Am. Chem. Soc., 2002, 124, 1130–1131 CrossRef CAS PubMed.
- D. H. Ess, S. E. Wheeler, R. G. Iafe, L. Xu, N. Çelebi-Ölçüm and K. N. Houk, Bifurcations on potential energy surfaces of organic reactions, Angew. Chem., Int. Ed., 2008, 47, 7592–7601 CrossRef CAS PubMed.
- D. J. Tantillo, Dynamic effects on organic reactivity—Pathways to (and from) discomfort, J. Phys. Org. Chem., 2021, 34, e4202 CrossRef CAS.
- Z. Yang, C. S. Jamieson, X.-S. Xue, M. Garcia-Borràs, T. Benton, X. Dong, F. Liu and K. N. Houk, Mechanisms and dynamics of reactions involving entropic intermediates, Trends Chem., 2019, 1, 22–34 CrossRef CAS.
- F. M. Bickelhaupt and K. N. Houk, Analyzing reaction rates with the distortion/interaction-activation strain model, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed.
- S. Chen, P. Yu and K. N. Houk, Ambimodal dipolar/Diels–Alder cycloaddition transition states involving proton transfers, J. Am. Chem. Soc., 2018, 140, 18124–18131 CrossRef CAS PubMed.
- S. Pratihar, X. Ma, Z. Homayoon, G. L. Barnes and W. L. Hase, Direct chemical dynamics simulations, J. Am. Chem. Soc., 2017, 139, 3570–3590 CrossRef CAS PubMed.
- H. Yamataka, M. Sato, H. Hasegawa and S. C. Ammal, Dynamic path bifurcation for the Beckmann reaction: observation and implication, Faraday Discuss., 2010, 145, 327–340 RSC.
- J. B. Thomas, J. R. Waas, M. Harmata and D. A. Singleton, Control elements in dynamically determined selectivity on a bifurcating surface, J. Am. Chem. Soc., 2008, 130, 14544–14555 CrossRef CAS PubMed.
- R. B. Campos and D. J. Tantillo, Designing reactions with post-transition-state bifurcations: asynchronous nitrene insertions into C–C σ bonds, Chem, 2019, 5, 227–236 CAS.
- S. R. Hare, R. P. Pemberton and D. J. Tantillo, Navigating past a fork in the road: carbocation−π interactions can manipulate dynamic behavior of reactions facing post-transition-state bifurcations, J. Am. Chem. Soc., 2017, 139, 7485–7493 CrossRef CAS PubMed.
- Y. Liu, M. P. Patricelli and B. F. Cravatt, Activity-based protein profiling: the serine hydrolases, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 14694–14699 CrossRef CAS PubMed.
- E. Weerapana, C. Wang, G. M. Simon, F. Richter, S. Khare, M. B. D. Dillon, D. A. Bachovchin, K. Mowen, D. Baker and B. F. Cravatt, Quantitative reactivity profiling predicts functional cysteines in proteomes, Nature, 2010, 468, 790–795 CrossRef CAS PubMed.
- C. Wang, E. Weerapana, M. M. Blewett and B. F. Cravatt, A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles, Nat. Methods, 2014, 11, 79–85 CrossRef CAS PubMed.
- F. Yang, J. Gao, J. Che, G. Jia and C. Wang, A dimethyl-labeling-based strategy for site-specifically quantitative chemical proteomics, Anal. Chem., 2018, 90, 9576–9582 CrossRef CAS PubMed.
- J. Gao, Y. Liu, F. Yang, X. Chen, B. F. Cravatt and C. Wang, CIMAGE2.0: an expanded tool for quantitative analysis of activity-based protein profiling (ABPP) Data, J. Proteome Res., 2021, 20, 4893–4900 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2212935–2212938, 2212942, 2212943, 2212947 and 2212950. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05233h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |