
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective chiral dimerization and folding driven by arene–perfluoroarene force†
Qiuhong
Cheng
,
Aiyou
Hao
 * and
Pengyao
Xing
*
* and
Pengyao
Xing
*
Key Laboratory of Colloid and Interface Chemistry of Ministry of Education, School of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, People's Republic of China. E-mail: haoay@sdu.edu.cn; xingpengyao@sdu.edu.cn
First published on 29th November 2023
Abstract
Oligomerization and folding of chiral compounds afford diversified chiral molecular architectures with interesting chiroptical properties, but their rational and precise control remain poorly understood. In this work, we employed arene–perfluoroarene (AP) interaction to manipulate the folding and dimerization of alanine derivatives bearing pyrene and a perfluoronaphthalene derivative. Based on X-ray crystallography and nuclear magnetic resonance, the compound with a smaller tether and high skeleton rigidity self-assembled into double helical dimers by duplex hydrogen bonding and AP forces in a less polar solvent. Reversible disassociation occurred upon switching to a dipolar solvent or applying heating–cooling cycles. In comparison, the compound with increased skeleton flexibility folds into chiral molecular clamps in a less polar solvent, and is transformed into planar dimers upon switching to a polar solvent. The dynamic geometrical transformation between dimerization and folding was accompanied by chiroptical switching. Beyond the molecular and supramolecular level, we showed hierarchy control in the self-assembled nanoarchitectures and columnar and lamellar arrangements of their molecular packing. This work utilized AP forces to prepare and manipulate the chiral architectures at different hierarchical levels, enriching methodologies in precise chiral synthetic chemistry.
Introduction
Supramolecular self-assembly from chiral units widely occurs in natural products.1,2 Point chirality in amino acids or nucleic acid transfer on the supramolecular or macroscopic scale through noncovalent force-driven self-assembly provide functions of bio-information storage and selective chiral recognition.3–8 In addition to self-assembly, intrinsic folding by noncovalent forces also creates complicated helical structures, such as the secondary structures of peptides, proteins and polysaccharides.9–13 The preference towards chiral folding and self-assembly dramatically influences the resulting structure and functions, while the structural basis still remains a mystery.14,15 For simpler synthetic chiral building units, rational control of the balance between intramolecular folding and intermolecular oligomerization is crucial for the precise synthesis of chiral self-assembled materials.16Small chiral compounds bearing two or multiple aromatic entities are effective building units in constructing functional soft materials, which is ascribed to the directional growth aided by diverse aromatic forces.17–21 Peptides from aromatic amino acids, and N-terminal aryl peptides and amino acids fall into this scope.22–25 Their potentials in self-assembly have been intensively explored, while the intramolecular folding by π-forces between aryl segments is rarely referred to ref. 26–28. Identification of the folding behaviors and their structural basis facilitates the manipulation and fabrication of novel functional chiral materials for asymmetric catalysis, recognition and sensing applications. For example, recently, Alcázar and our group unveiled folded molecular tweezers in ditryptophan peptide derivatives and explored their functions in the recognition of electron-deficient guests and chiroptical properties.29,30 Possible interactions between aryl entities include π–π stacking (ionic-π and cation-π), charge-transfer interaction and arene–perfluoroarene (AP) interaction.22,24,31–33 AP interaction occurs between planar fused aromatic rings and perfluorinated aromatic compounds.34 Fluorination produces an inverted electron distribution compared to hydrocarbons, so that perfluoroarene with π-holes would generate multiple F⋯H hydrogen bonds and π⋯π-hole interactions. Thus, the AP interaction has stronger binding affinity than the π–π interaction.35 For example, in the gas phase, the binding energy of benzene–hexafluorobenzene is about twice as high as that of benzene–benzene complexes.36 AP interaction has been used in tuning supramolecular chirality, structure and properties in multiple constituent systems, yet its application in tuning folding and self-assembly behaviors has not been reported so far.37,38
In this work, naphthalene-F7 was conjugated to pyrene appended alanine. The resulted building units, namely PMA (N-(1-oxo-1-((3-((perfluoronaphthalen-2-yl)amino)propyl)amino)propan-2-yl)pyrene-1-carboxamide) and PBA (N-(1-oxo-1-((3-((perfluoronaphthalen-2-yl)amino)propyl)amino)propan-2-yl)-4-(pyren-1-yl)butanamide), bear both the AP donor and an acceptor with either a rigid or flexible tether. In the solid state, X-ray crystallography indicates that LPMA and LPBA adopt a double helical dimer and planar dimer structure, respectively. In contrast, D/LPMA in the racemic state afforded a planar dimer, while D/LPBA folded into chiral foldamers. In the homochiral and heterochiral modality, AP interaction and hydrogen bonds are dominating. In the solution phase, solvent polarity varied the preference towards folding and oligomerization. In dimethyl sulfoxide (DMSO), PMA remains in a monomer state, and in less polar chloroform (CHCl3), dimers are generated, and the monomer–dimer transformation is reversibly controlled by a heating–cooling process. In contrast, PBA with a flexible tether forms a dimer in CHCl3, and folds in dipolar solvent DMSO. Further heating of the DMSO solution would break down the AP and hydrogen bonds into a monomeric state. Finally, we explored the self-assembly into nanoarchitectures, where double helical and planar dimers of LPMA and LPBA packed into columnar and lamellar structures, respectively, showed a hierarchical impact from molecular to supramolecular to nanoscale. This work introduces an AP interaction to manipulate folding and hierarchical self-assembly with controlled supramolecular chirality that may enrich the rational and precise synthesis of chiral functional materials (Scheme 1).
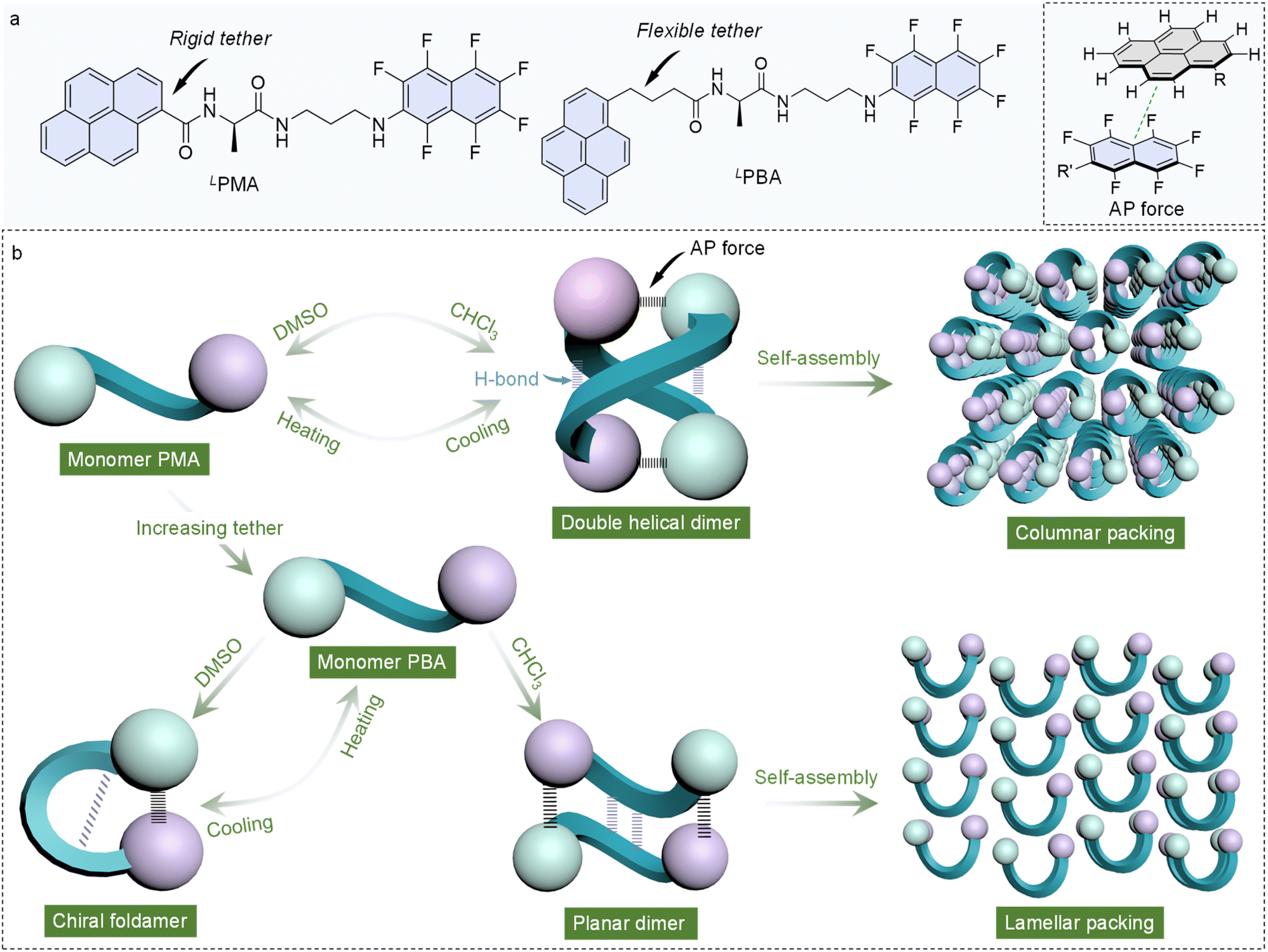 | ||
| Scheme 1 (a) Molecular structure of building units and the presentation of the AP force; (b) structural evolution of the two building units controlled by tether flexibility, solvent and heating/cooling process. | ||
Results and discussion
The synthesis of PMA and PBA was through several steps including aromatic substitution and amide condensation reactions (Schemes S1 and S2†). 1H, 13C and 19F nuclear magnetic resonance (NMR) and mass spectra were used to fully characterize the enantiomerically pure compounds (Fig. S1–S24†). Most AP complexation was realized in perfluorinated compounds such as naphthalene-F8, as the substitution by electron-rich nitrogen would degenerate the electron density of π-holes and the corresponding AP forces.36 In order to probe the occurrence of AP-initiated self-assembly, single crystals were carefully cultivated via solvent evaporation and the corresponding enantiopure X-ray structures are shown in Fig. 1 (Tables S1–S4†). LPMA with a rigid skeleton features a dimeric geometry (Fig. 1a), where pyrene and naphthalene-F7 segments are closely packed in a head-to-tail manner. Two PMA molecules are packed into a double helical screw sense driven structure and stabilized by multiple noncovalent forces. Between the amide groups, two hydrogen bonds are formed, and the nearly parallel (d = 3.43 Å) pyrene/naphthalene-F7 arrays imply the occurrence of the AP force. Non-covalent interaction (NCI)/reduced density gradient (RDG) analysis on the dimers suggests the presence of aromatic forces between the pyrene and naphthalene-F7 moieties (green regions in Fig. 1b) as well as H-bond interaction (blue regions in Fig. 1b and S25†).39,40 Hirshfeld surfaces (bottom of Fig. 1b) also confirmed the crucial role of hydrogen bonds and AP forces in dimerization.41 Decomposed intermolecular contact distributions based on the Hirshfeld surfaces of the dimers are highlighted on the fingerprint graphics (Fig. 1c). F⋯H occupied the largest fraction of up to 35.4%. Other related forces including C⋯C and F⋯C show fractions of 12.1% and 7.5%, respectively. In contrast, O⋯H only shows a fraction of 8.5%. Due to AP interaction primarily consisting of F⋯H and π⋯π-hole interactions, the above results suggest that AP forces mainly drive the dimerization, and are also aided by hydrogen bonds (Fig. S26†). In comparison, PBA possesses an elongated alkyl spacer with enhanced structural flexibility. A dimeric structure was found in LPBA as well (Fig. 1d). LPBA adopts a face-to-face dimer orientation supported by duplex hydrogen bonds and AP interaction (d = 3.51 Å). NCI analysis suggests the crucial role of hydrogen bonds and AP forces as well (Fig. 1e). The calculated fingerprint diagram shows an F⋯H fraction of 22.2%, lower than that of LPMA (35.4%). Also, the fraction of C⋯C (8%, assigned as the π⋯π hole short contacts) is lower than that of LPMA (12.1%) (Fig. S27†). The contribution of AP forces in the dimerization is reduced, along with increased spacer length and structural flexibility.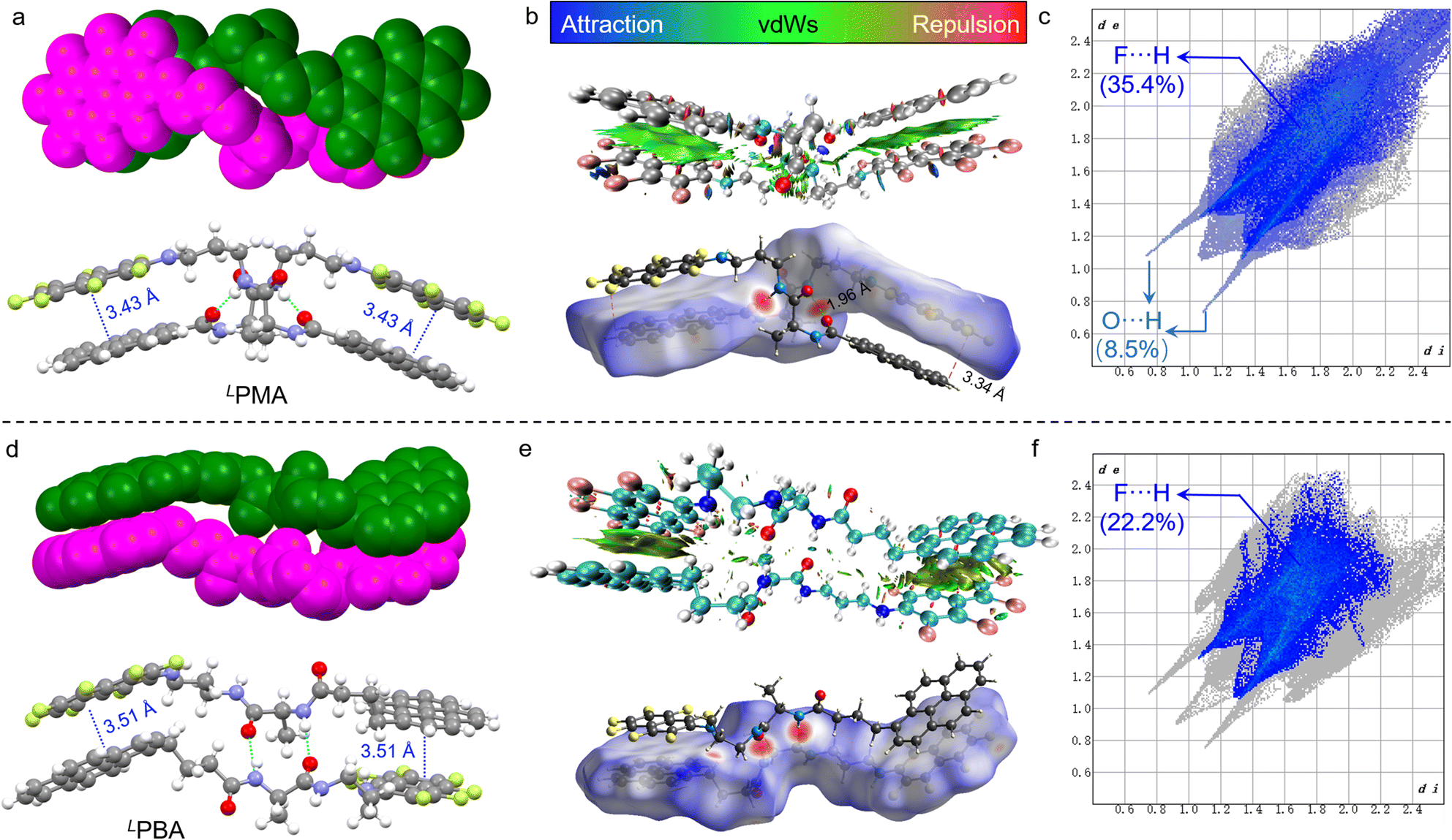 | ||
| Fig. 1 (a) X-ray structures of LPMA dimers. (b) Noncovalent interaction analysis of LPMA dimers. The green regions represent vdWs interactions. (c) Short contact fraction distributions in LPMA dimers. (d–f) X-ray structures and the corresponding noncovalent interaction analysis of LPBA dimers. | ||
Wallach's rule tells us that the single crystal of a racemic mixture is always denser than that of its enantiomers.42,43 In addition to Wallach's rule, we speculated that, in the racemic form, an alternative self-assembly modality might be adopted. Single crystals of the racemic mixtures were cultivated, as shown in Fig. 2. D/LPMA surprisingly afforded a dimeric structure, consistent with its enantiomeric counterparts. Duplex hydrogen bonds and AP interactions are present as well. NCI analysis of the D/LPMA dimer and LPBA folder also suggest the presence of AP and hydrogen bond interactions (Fig. S28 and S29†). However, we noticed that the overlapping of pyrene/naphthalene-F7 regions is deficient compared to LPMA. The F⋯H fraction (27.7%) is significantly lower than LPMA (35.4%) (Fig. S30†). Instead of screw sense, face-to-face dimerization emerged. Our intuitive understanding is that D/LPMA dimerization is less energetically favored, in contrast to the enantiopure form. Based on the X-ray structures, the dimerization interaction energies were calculated at the theory level of b3lyp/def2SVP-D3(BJ) (left diagram of Fig. 2c). In the enantiopure form, the mean dimerization energy is determined as −35.3 kcal mol−1. This value is much higher than that for most noncovalent forces such as hydrogen bonds and π–π stacking, due to the presence of multiple interactions. In the racemic form, the dimerization binding energy is 27.0 kcal mol−1, much lower than that of the enantiopure state, which is in good agreement with the X-ray structure analysis and assumptions. Wallach's rule emphasizes the high space utilization in the racemic form, yet in the present case, the binding affinity is lower than for the enantiopure counterpart. Nevertheless, chiral self-resolution was not observed in the crystallization of PMA racemates.
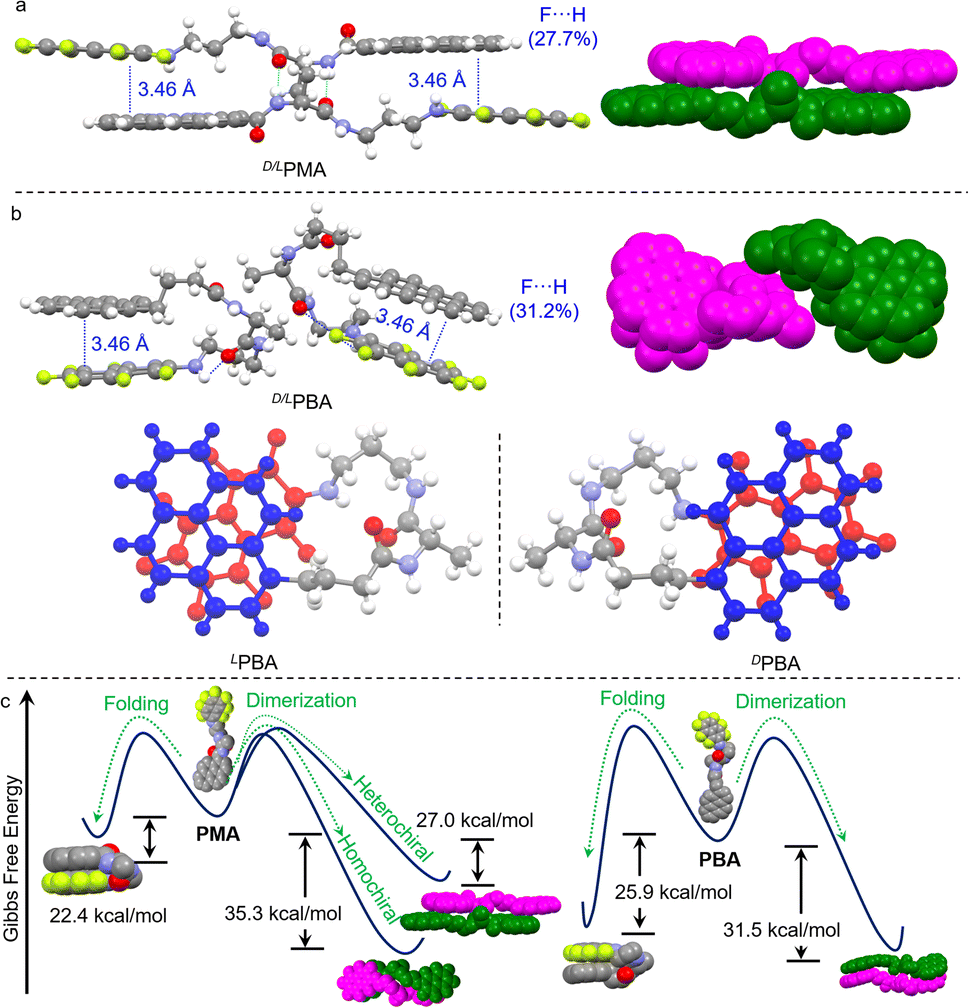 | ||
| Fig. 2 (a) X-ray structures of D/LPMA. (b) X-ray structures of D/LPBA, and an illustration of the folded enantiomers. (c) Energy landscape of the self-assemblies in the homochiral and heterochiral complexation (left: PMA, right: PBA). | ||
Next, the self-assembly of PBA in the racemic form was probed using X-ray crystal structures. Surprisingly, D/LPBA does not show dimers. Instead, intramolecular folding occurs (Fig. 2b). DPMA and LPMA individually folded into enantioselective mirror conformations. Folding is facilitated by the hydrogen bonds and AP interaction. For PBA, hydrogen bonding occurred between the amine NH adjacent to the naphthalene-F7 and amide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. The intramolecular folding allows for sufficient overlap between pyrene and naphthalene-F7, which gives rise to a high F⋯H fraction (30.6%) (Fig. S31†). This implies that PBA, compared to PMA, has the potential to form folded structures, which might be due to the low binding affinity to afford heterochiral dimers or other self-assemblies, while the flexibility of alkyl chains conformationally favors the folding topology. The energy landscapes give insights into the pathway complexity (Fig. 2c). PMA folding shows lower interaction energy (22.4 kcal mol−1) compared to that of PBA (25.9 kcal mol−1), and the energy gaps (Δ) between homochiral dimerization and the folding of PMA and PBA are 12.9 kcal mol−1 and 5.6 kcal mol−1, respectively. The small Δ value of PBA between dimerization and folding implies that, in the enantiopure form, PBA may realize transformation between the two states, while the transformation for PMA is energetically unfavorable.
O. The intramolecular folding allows for sufficient overlap between pyrene and naphthalene-F7, which gives rise to a high F⋯H fraction (30.6%) (Fig. S31†). This implies that PBA, compared to PMA, has the potential to form folded structures, which might be due to the low binding affinity to afford heterochiral dimers or other self-assemblies, while the flexibility of alkyl chains conformationally favors the folding topology. The energy landscapes give insights into the pathway complexity (Fig. 2c). PMA folding shows lower interaction energy (22.4 kcal mol−1) compared to that of PBA (25.9 kcal mol−1), and the energy gaps (Δ) between homochiral dimerization and the folding of PMA and PBA are 12.9 kcal mol−1 and 5.6 kcal mol−1, respectively. The small Δ value of PBA between dimerization and folding implies that, in the enantiopure form, PBA may realize transformation between the two states, while the transformation for PMA is energetically unfavorable.
To further demonstrate the importance of AP interaction in forming a folded structure, DFT-based computational studies were employed. A model compound was constructed without AP interaction through modifying the single crystal structure of the PBA dimer. We replaced naphthalene-F7 with a methyl group to eliminate AP interaction (Fig. S32†). After structural optimization, more hydrogen bonds appear and the methyl group is away from the pyrene ring, indicating that the molecule does not prefer to form a folded structure without AP interaction. As shown in Fig. S33a,† the main hydrogen bond to drive the folding is N1–H⋯O1. To completely avoid the influence of hydrogen bonding, N1 and N2 were both replaced with O (Fig. S33c and d†). After optimization, no hydrogen bond was found, while a folded structure was retained, driven by AP interaction. This suggests the vital role of AP interaction in driving the formation of the folder (Fig. S33e and S34†). Molecular dynamics (MD) simulation was also used to monitor the transformation process from a non-folding to a folding conformation of a model molecule. A non-folding conformation was used as the initial structure and placed in a box with a volume of 5 × 5 × 5 nm3 to observe the conformation transition over time. The MD simulation was conducted using the GROMACS 2020 program, with a total simulation time of 30 ns. As shown in Fig. S35,† the simulation system reached equilibrium in less than 1 ns. The molecular structure changes dramatically in the early stage of the simulation. After reaching equilibrium, the molecule existed in a folded conformation induced by the AP interaction. The distance between C1 and C2 obviously decreased and eventually stabilized around 4 Å, demonstrating the conformation transformation from a non-folded to a folded state. Moreover, hydrogen bonds were not observed in this simulation system.
Noncovalent forces including AP and hydrogen bonds are sensitive to solvent environments. Protic or polar solvents such as DMSO would normally destroy noncovalent forces via solvation, while less polar solvents such as CHCl3, dichloroethane (DCE) or methyl cyclohexane (MCH) may prefer the occurrence of noncovalent forces. Additionally, the volume ratio between DMSO and CHCl3 was altered to probe the structural evolution in solution. With increasing volume fraction of DMSO (fDMSO) against CHCl3, absorbance at around 350 nm was enhanced with slight hypochromic shifts (Fig. 3a). This change is reminiscent of the disassociation of self-assemblies driven by aromatic stacking forces. The disassembly process of PMA and PBA leads to the variations in the fluorescence.44–46 For PMA, the emission intensity was enhanced initially, and then declined in high fDMSO regions (Fig. 3a). The major emission peak at 407 nm hypsochromically shifted to 402 nm once DMSO was added, which is in accordance with the disappearance of AP interaction. LPBA shares a similar structure with LPMA, while with increasing fDMSO, the absorbance decreased slightly, in sharp contrast to LPMA (Fig. 3b). The emission intensity at around 400 nm decreased with increasing intensity at a bathochromic-shifted wavelength of around 500 nm. The spectroscopic changes with increasing fDMSO suggest structural evolution beyond the disassociation. We speculated that first, DMSO destroys the hydrogen bonds leading to the disassembly of the PBA dimer. Then, due to the flexible tether of PBA compared with PMA, AP interaction drives the formation of a foldamer. Further heating of the DMSO solution would destroy hydrogen bonds and intramolecular AP interaction to afford monomers. AP complexation lowers the energy gap of frontier orbitals. For example, LPBA in the free monomer state gives a gap of 3.91 eV between the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO), while this is slightly reduced to 3.78 eV after AP-driven folding. Due to the more sufficient AP overlap in foldamers (F⋯H fraction: 30.6%) than dimers (F⋯H fraction: 22.2%), more bathochromic shifts would be generated. This thus supports the assumption that self-assemblies and folded structures are formed in CHCl3 and DMSO, respectively. Temperature-variable emission spectroscopy was performed using high boiling point DCE to replace CHCl3 (Fig. 3c–e). Upon heating LPMA from 293 to 343 K, LPMA shows a decreased emission intensity ascribed to the disassociation of self-assemblies with enhanced molecular motion, and the subsequent cooling results in full recovery to the initial intensity (Fig. 3e). However, the emission intensity of LPBA was slightly enhanced (Fig. 3d) upon heating, and upon subsequent cooling it could hardly be recovered. This difference verifies that LPMA may adopt an assembly/disassociation cycle while LPBA is under structural transformation from dimerization to folding in the heating–cooling process. In the subsequent cooling, foldamer formation resulted in hysteresis that delays the emission recovery.
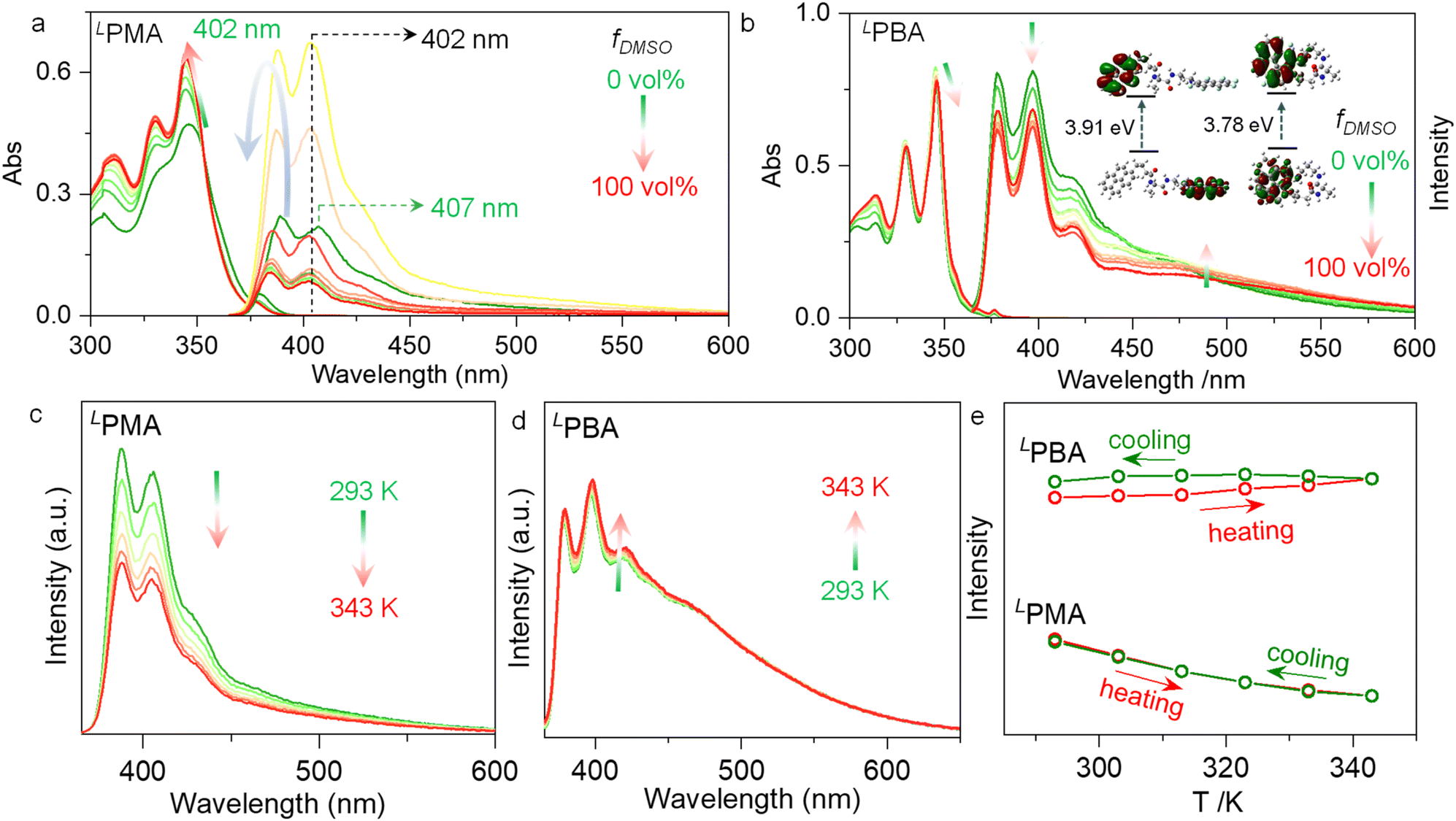 | ||
| Fig. 3 (a) Absorption and emission spectra of LPMA (0.2 mM) with different volume fractions of DMSO (fDMSO) against CHCl3. (b) Absorption and emission spectra of LPBA (0.2 mM) with different volume fractions of DMSO (fDMSO) against CHCl3. The inset shows the HOMO–LUMO gap of the free monomer and folded LPBA. Temperature-variable emission spectra of (c) LPMA and (d) LPBA in DCE (0.2 mM), as well as (e) the summarized max. intensity as a function of temperature. | ||
To correlate the spectral variations in the solutions, we performed 1H NMR spectroscopy in CDCl3 and DMSO-d6. Concentration dependent 1H NMR spectra from 0.05 mM to 2.0 mM in CDCl3 were collected (Fig. 4a, b, S36 and S37†). The active protons of Ha, Hb and Hc are located between 5.0 and 7.0 ppm. With increasing concentration, amide protons Ha and Hb shifted to lower fields, with self-assembly contributing to the shift. Based on the X-ray structures (Fig. 1), the dimerization involves the amide complementary hydrogen bonds, while Hc does not participate. Thus, in the low polarity CDCl3, dimerization is expected. In contrast, concentration-dependent 1H NMR spectra in DMSO-d6 showed no apparent shifts, supporting the absence of self-assembly (Fig. S38 and S39†). Partial 1H NMR spectra of Hc (Fig. S40†) indicate the different locations of Hc. Hc of LPMA and Hc of LPBA are located at 6.13 and 6.30 ppm, respectively, while in CDCl3 they share an identical location (Fig. 4b). This phenomenon suggests that Hc is in different chemical environments for LPMA and LPBA. Also, we observed that when increasing the concentration from 0.1 to 10 mM in DMSO-d6, Hc of LPBA shows larger shifts to higher fields, which may be caused by the intramolecular folding. Based on the X-ray structure, Hc is involved in hydrogen bonds in the LPBA foldamers (Fig. S41 and S42†). Then, temperature-variable 1H NMR spectroscopy was performed (Fig. 4c, S43 and S44†). From 298 K to 343 K, the aromatic protons of LPBA are barely shifted, indicating that the conformations are retained. In comparison, the aromatic protons (pyrene domain) of LPMA shift to lower fields upon heating. Meanwhile, Hf and He of the alkyl spacers at low temperatures are split, caused by the chiral conformations, which are merged upon heating. These changes are possibly due to the transformation from a folded to unfolded structure. Two-dimensional (2D) nuclear Overhauser effect spectroscopy (NOESY) is a powerful tool for precise assignment of protons and proton–proton correlations. For LPMA, we found correlations including Hd–Hg and Hd–Hi in CDCl3. The long distances between the protons evidence the formation of dimers with structures similar to those in the solid phase X-ray analysis (Fig. 4d). However, in DMSO-d6 (c = 10 mM), no proton correlations were found, disregarding the protons with intrinsic close contact (Fig. 4e). Therefore, it is confirmed that LPMA adopts dimers and free monomers in CHCl3 and DMSO, respectively. For LPBA, due to the low solubility in CDCl3, the splitting of hydrogen in 2D NOESY is not obvious where Hi/Hh/Hk and He/Hf overlap. However, long distance proton correlations such as Hj–Hi, Hi–Hk and Hg–He/Hf probably exist, in good agreement with the dimerization conformation found in solid structure X-ray analysis (Fig. S45 and S46†). In contrast, in DMSO-d6, strong proton–proton correlations exist, such as Ha–Hi, Hj–Hi, He–Hi, He–Hk and Hg–Hk. This verifies the formation of foldamers in the dipolar DMSO phase. Based on the 1D and 2D 1H NMR, it can be concluded that LPBA gives dimers and foldamers in CHCl3 and DMSO, respectively.
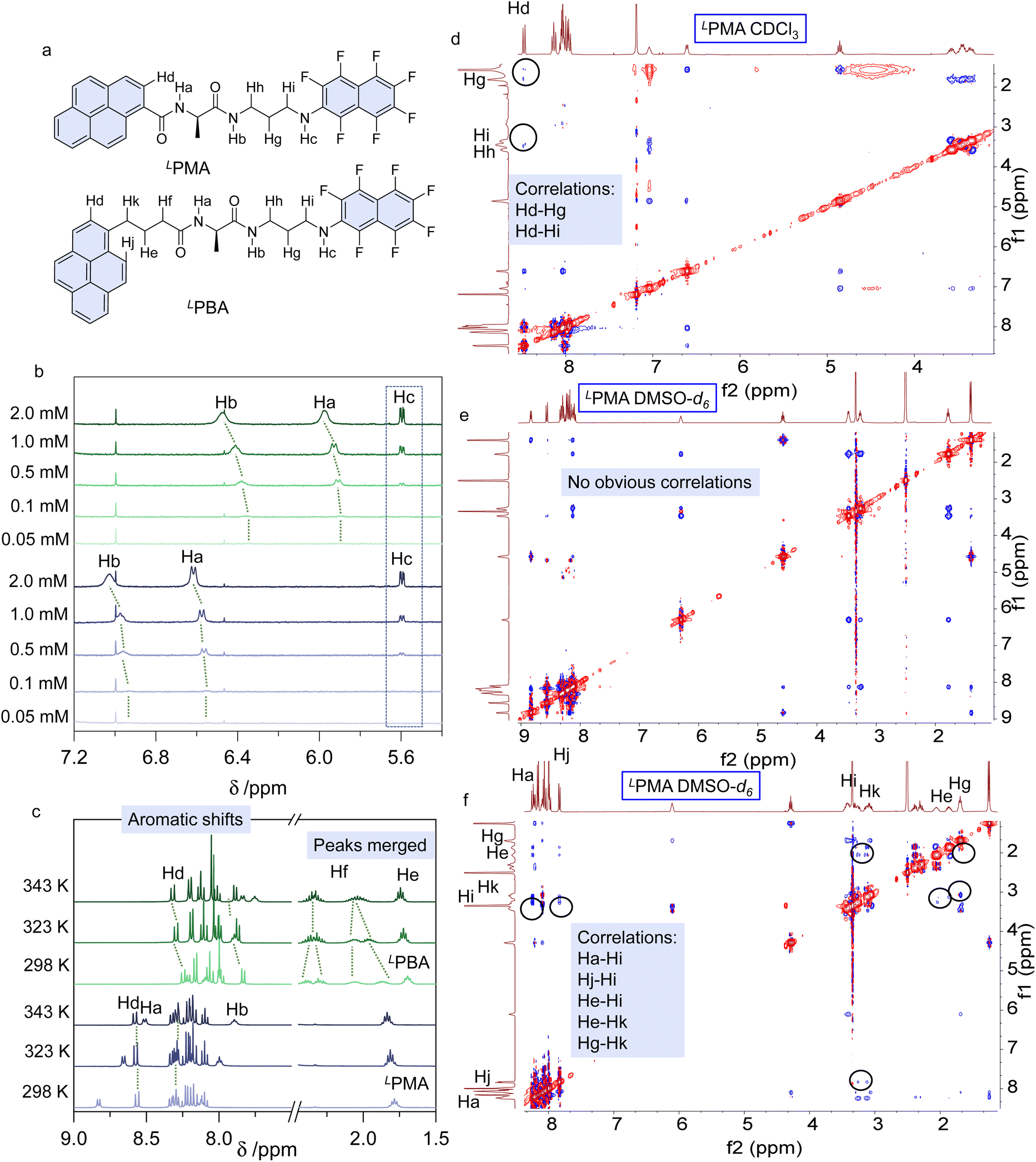 | ||
| Fig. 4 1H NMR studies. (a) Proton assignments of LPMA and LPBA. (b) Concentration-dependent 1H NMR spectra of LPMA and LPBA in CDCl3. (c) Temperature-variable 1H NMR spectra of LPMA and LPBA in DMSO-d6 (10 mM). (d–f) 2D NOESY spectra of LPMA and LPBA in CDCl3 (saturated) and DMSO-d6 (10 mM). | ||
Two-dimensional diffusion ordered NMR spectroscopy (DOSY) is a powerful technique to study molecular geometries in solution. According to the Stokes–Einstein equation, the diffusion coefficient D is associated with the hydrodynamic radius, and thus the two-dimensional geometry can be predicted.47–49 The viscosities of solvents at various temperature were recorded using a fitting curve (Fig. S47 and S48†). DOSY spectra of LPMA and LPBA in CDCl3 and DMSO-d6 at different temperatures were collected (Fig. S49–S56†), and the diffusion coefficients as well as the hydrodynamic radii are summarized in Table 1 (Fig. S57–S64†). Using an oblate spheroid model (length b > width a), the size parameters were predicted based on the Stokes–Einstein equation. The r values, defined as the ratios of the hydrodynamic radii of the oblate model to the equivalent radii from the volume of the spheroid, are close to 1.0, indicating the validity of the prediction. The p values calculated using b/a represent the aspect ratio. In CDCl3, upon increasing the temperature from 25 °C to 45 °C, the p value of LPMA decreases from 4.2 to 2.3, and LPBA decreases from 3.2 to 1.7. The decreased aspect ratio agrees with the disassociation of dimers by heating. In DMSO-d6, the p value of LPMA is not changed by heating. This means that LPMA retains a monomeric free state independent of thermovariations. However, the p value of LPBA shows a significant increase from 1.7 to 3.0 upon heating. This increasing aspect ratio is consistent with the unfolding of the foldamers, which also verifies the assumption based on the 1D 1H NMR results (Fig. 4).
| Entry | T [°C] | D [μm2 s−1] | Radius [Å] | b [Å] | a [Å] | p | r |
|---|---|---|---|---|---|---|---|
| a T and D represent the temperature and diffusion coefficients; b and a are the length and width of the oblate spheroid model, p is the aspect ratio, and r is a parameter close to 1.0 to reflect the reliability. | |||||||
| LPMA (CDCl3) | 25 | 1040 | 3.9 | 5.5 | 1.3 | 4.2 | 1.02 |
| 45 | 1050 | 4.5 | 7.0 | 3.0 | 2.3 | 1.02 | |
| LPBA (CDCl3) | 25 | 691 | 5.9 | 8.0 | 2.5 | 3.2 | 1.03 |
| 45 | 974 | 4.8 | 5.7 | 3.3 | 1.7 | 1.00 | |
| LPMA (DMSO-d6) | 25 | 161 | 6.9 | 8.9 | 3.0 | 3.0 | 0.99 |
| 50 | 310 | 6.0 | 8.5 | 2.8 | 3.0 | 1.08 | |
| LPBA (DMSO-d6) | 25 | 157 | 7.1 | 8.2 | 4.9 | 1.7 | 0.99 |
| 50 | 284 | 6.6 | 8.7 | 2.9 | 3.0 | 1.01 | |
Based on the above results, a brief summary of the structures in solution are shown in Fig. 5. For PMA, in the low polar solvent (CHCl3), dimeric self-assembly is generated by AP force and hydrogen bonds. Upon replacing CHCl3 with the dipolar solvent DMSO, disassociation and transformation into monomers occur. Heating also breaks down the noncovalent forces, generating monomers in low polarity solvents. PMA failed to afford folded structures due to the rigid skeleton and energy barrier. Elongating alkyl spacers with enhanced structural flexibility result in the possibility of forming folded structures. PBA with enhanced flexibility also features dimerization in CHCl3. At a high temperature in CHCl3, PBA forms foldamers. Switching the solvent to polar DMSO transforms PBA into folded structures. Further heating the DMSO solution would destroy hydrogen bonds and intramolecular AP interaction to afford monomers. The structural evolution provides a rational and precise control strategy for molecular folding and supramolecular complexation.
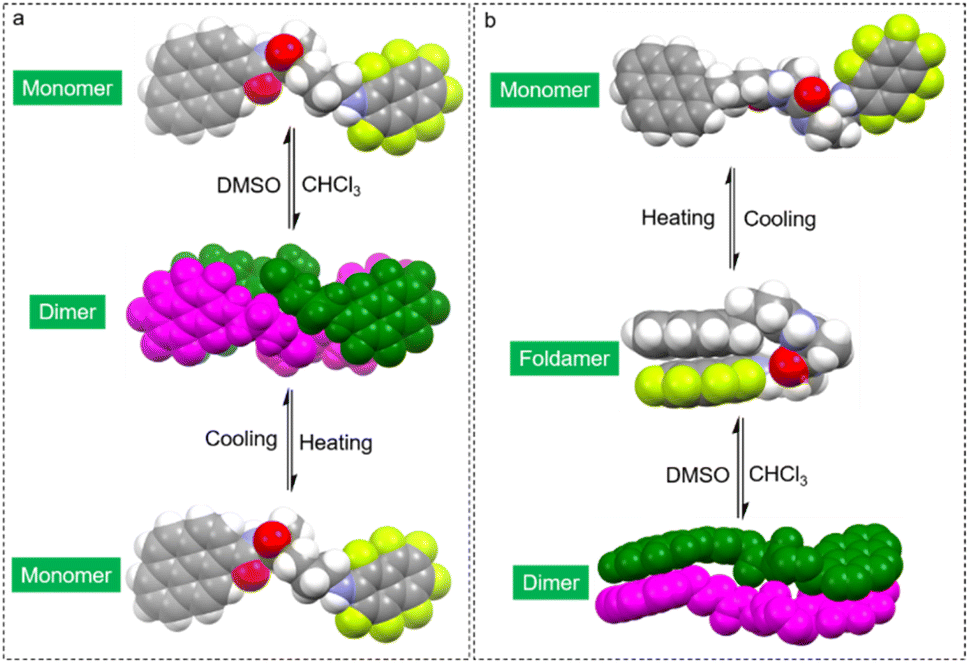 | ||
| Fig. 5 Space-filling models to demonstrate the structural evolution by solvent and thermal treatments. (a) LPMA, (b) LPBA. | ||
The building units are intrinsically chiral due to the alanine segments. In CHCl3, LPMA and DPMA feature negative and positive signals in the pyrene absorption region (300 to 350 nm), indicating the chirality transfer from alanine segments to pyrene pendants (Fig. 6a). The dimeric form possesses better shape resistance compared to the monomers that afford enhanced chiroptical signals. In DMSO, the monomers show reduced CD signals, agreeing with our speculations (Fig. S65†). The tendency is inverted for PBA (Fig. 6b). The DMSO solution gives more intense CD signals than CHCl3 (Fig. S66†). Based on the NMR studies, PBA in DMSO and CHCl3 preferably adopts folded and dimer geometries, respectively. In the foldamer form, the structures are regarded as more rigid due to the clamp topology, and thus enhanced Cotton effects are expected. Next, we evaluated the thermal effects on the chiroptical properties. In DMSO, LPMA remains in a monomeric state, and increasing the temperature from 293 K to 358 K barely changes the CD signals. The negative changes agree with the monomer state, which shows no structural variation upon heating–cooling cycling. However, in CHCl3, an increase from 293 K to 328 K results in a CD signal reduction, which can be fully recovered after cooling (Fig. S67†). The phenomenon is consistent with the dimer to monomer structural transformation. LPBA exhibited more profound chiroptical changes with the variation of temperature (Fig. 6c, d and S68†). Increasing the temperature of LPBA in DMSO from room temperature to 363 K resulted in continuously decreasing Cotton effects. This process is accompanied by the unfolding behavior, and complete recovery occurs during the cooling process without any hysteresis. In DCE (replacing CHCl3 considering the boiling point), a similar propensity was observed.
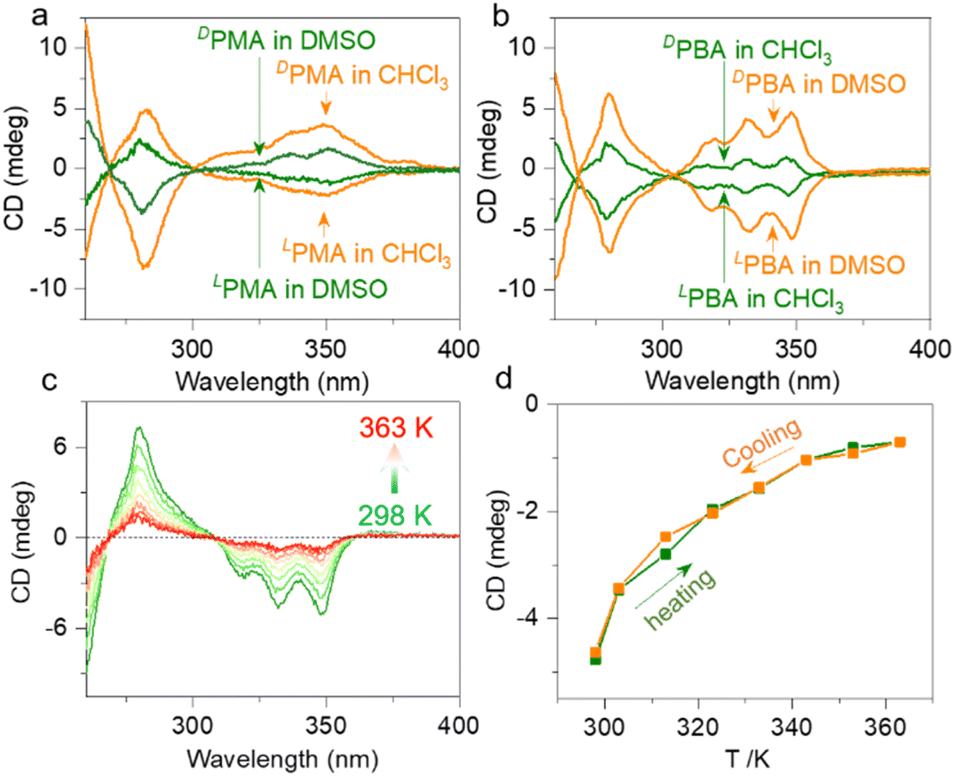 | ||
| Fig. 6 Chiroptical properties. (a) CD spectra of PMA enantiomers in CHCl3 and DMSO (0.3 mM). (b) CD spectra of PBA enantiomers in CHCl3 and DMSO (0.3 mM). (c) Temperature-variable CD spectra of LPBA (0.3 mM) in DMSO. (d) CD signal at 350 nm in DMSO of LPBA (0.3 mM) during heating and cooling cycles. | ||
Spontaneous self-assembly by solvent processing can generate ordered nanoarchitectures. This structural evolution was realized using a nanoprecipitation protocol.50 By dispersing building units from CHCl3 into MCH, self-assembly was triggered through a good/poor solvent exchange. Before testing, the samples were aged for 12 h. The final concentration of the samples is 1 mM and the volume ratio of CHCl3 and MCH is 2/8. The dispersion of LPMA under transmission electron microscopy (TEM) shows planar nanosheets with a rectangular shape, indicating a crystallization-induced aggregation pathway (Fig. 7a). In contrast, LPBA generated a nanorod architecture with lengths up to several micrometers and widths of about 200 nm (Fig. 7b). The screw sense and helical nanoarchitectures were not found, possibly because of the dimerization effect that hinders 1D extension to amplify the macroscopic chirality. Atomic force microscopy (AFM) was employed to examine the thickness (Fig. 7c and d). A high aspect ratio was found in the nanosheet morphology of LPMA. A thickness of 35 nm was observed from the cross-section profile, indicating its 2D planar nature. Rod structures with thicknesses of about 60 nm were observed in the LPBA self-assemblies (Fig. 7d). The CD spectra of the self-assemblies were recorded (Fig. 7e, f and S69†). The spectra are different to those measured in solution phase, due to aggregation introduced asymmetric packing of building units to afford supramolecular chirality, in addition to the chirality in the dimers or foldamers. Additionally, the relatively intense CD signals originate from the more closely packed nanosheets or nanorods, which showed similar peaks to those of the crystalline powder, indicating that the nanostructures have the same molecular packing model (Fig. S70†). Consequently, self-assembly behaves as a valid protocol to manipulate chiroptical properties in addition to the above discussed control methods. The aggregates were collected and subjected to X-ray diffraction (XRD) to explore the molecular arrangements (Fig. 7g). Compared with the simulated powder patterns from X-ray single crystallography, similar patterns were found, suggesting that these nanostructures share similar molecular packing arrays. However, there was clear specific plane selective exposure or growth present in the aggregation thanks to the relative kinetic control of the aggregation. In addition, the experimental XRD of LPMA consists of the first and second order diffraction peaks with a distance ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1/√3, and is assigned as hexagonal packing. LPBA features a different packing mode, with a distance ratio of 1:1/2:1/3, which is consistent with a typical lamellar structure. In the X-ray structures of LPMA (Fig. 7h), the dimers (intertwined magenta/green structure) in the top view pack into a hexagonal structure, between which hydrogen bonds behave as linkers. This packing is in good agreement with the XRD patterns. For LPBA, the dimers pack into layers via hydrogen bonds as well, and the layers further pack into lamellar structures, which verifies the observation from the XRD patterns. For the self-assembly of LPMA and LPBA in a polar solvent environment (DMSO/H2O), various nanostructures are shown, compared with the aggregations in a nonpolar solvent (CHCl3/MCH). Both LPMA and LPBA assembled into fused micelles, as shown by the TEM and AFM results, of about 100 nm in diameter, which also showed in dynamic light scattering (DLS) patterns (Fig. S71–S73†). The CD spectra of the self-assemblies are different to those measured in nonpolar environments, due to the fact that the polar solvent could form hydrogen bonds with LPMA and LPBA, resulting in competition, which does not benefit the growth of 2D and 3D nanostructures. Moreover, the direction and shape of the CD signals were similar to those in the solution phase (Fig. S74†). Thus, the assembled morphology and corresponding chiroptical properties could also be controlled by adjusting the solvent polarity.
:1/√3, and is assigned as hexagonal packing. LPBA features a different packing mode, with a distance ratio of 1:1/2:1/3, which is consistent with a typical lamellar structure. In the X-ray structures of LPMA (Fig. 7h), the dimers (intertwined magenta/green structure) in the top view pack into a hexagonal structure, between which hydrogen bonds behave as linkers. This packing is in good agreement with the XRD patterns. For LPBA, the dimers pack into layers via hydrogen bonds as well, and the layers further pack into lamellar structures, which verifies the observation from the XRD patterns. For the self-assembly of LPMA and LPBA in a polar solvent environment (DMSO/H2O), various nanostructures are shown, compared with the aggregations in a nonpolar solvent (CHCl3/MCH). Both LPMA and LPBA assembled into fused micelles, as shown by the TEM and AFM results, of about 100 nm in diameter, which also showed in dynamic light scattering (DLS) patterns (Fig. S71–S73†). The CD spectra of the self-assemblies are different to those measured in nonpolar environments, due to the fact that the polar solvent could form hydrogen bonds with LPMA and LPBA, resulting in competition, which does not benefit the growth of 2D and 3D nanostructures. Moreover, the direction and shape of the CD signals were similar to those in the solution phase (Fig. S74†). Thus, the assembled morphology and corresponding chiroptical properties could also be controlled by adjusting the solvent polarity.
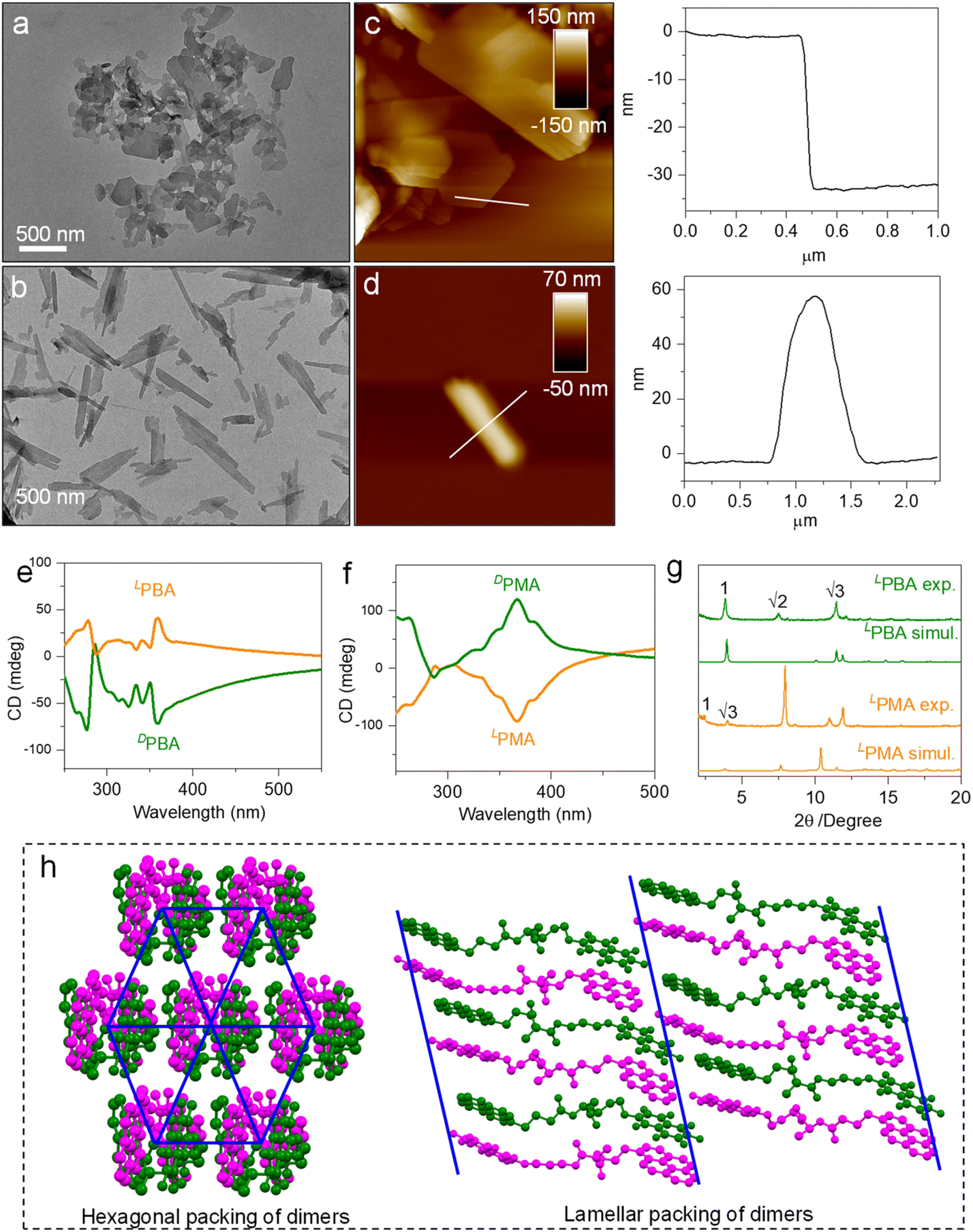 | ||
| Fig. 7 Self-assemblies. (a and b) TEM images of self-assembled LPMA and LPBA in a mixture of CHCl3/MCH (2/8 by volume, 1 mM); (c and d) the corresponding AFM images and cross-section profiles (3 × 3 μm and 5 × 5 μm, respectively); (e and f) CD spectra of self-assemblies. (g) XRD pattern comparison between simulated powder patterns and experimental results; (h) hexagonal and lamellar packing of dimers of LPMA and LPBA, respectively. | ||
Conclusions
In this work, amino acid derivatives PMA and PBA bearing pyrene and naphthalene-F7 segments were designed and synthesized. Using X-ray crystallography, diverse self-assembly modalities were unveiled. PMA self-assembled into double helical dimers in the enantiopure state, while the racemates generated planar achiral dimers. PBA self-assembled into planar dimers in its enantiopure form, while the racemic form gave foldamers. All the folding and dimerized structures featured hydrogen bonding and AP interaction. In the solution phase, we used 1D and 2D 1H NMR to evidence the structural evolution. This demonstrated that the PMA monomer in DMSO is transformed into dimers when the solvent is changed to a less polar solvent such as CHCl3. Additionally, PBA adopts dimer and foldamer structures in CHCl3 and DMSO, respectively. Both of the self-assemblies and foldamers show reversible responses to heating–cooling cycles. By differential self-assembly behavior, PMA and PBA displayed distinct chiroptical activities in response to the solvent polarity. Finally, we showed control of the self-assembly at the nanoscale, and PBA and PMA adopted hexagonal and lamellar packing, respectively. This work introduces a multi-control protocol to tune the preference towards chiral folding and self-assembly at different hierarchical levels, which may facilitate the rational fabrication of functional chiral materials.Experimental
Materials, experimental details, and additional CD, 1H NMR, and MS spectra can be found in the ESI.†Data availability
Crystallographic data in CIF format has been deposited with the Cambridge Structural Database. Date supporting this manuscript has been provided in the ESI.†Author contributions
Q. Cheng carried out the main experiments and data analyzing. P. Xing and A. Hao proposed the assumption and wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 21901145, 22171165) and Natural Science Foundation of Shandong Province (No. ZR2022MB080). We also acknowledge the financial support from Youth Cross-Scientific Innovation Group of Shandong University (2020QNQT003). We thank Prof. Di Sun at Shandong University for assistance with the data collection of X-ray crystal structures.Notes and references
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS.
- L. E. Mackenzie and P. Stachelek, Nat. Chem., 2021, 13, 521–522 CrossRef CAS.
- H. Sakaino, D. J. Broer, S. C. J. Meskers, E. W. Meijer and G. Vantomme, Angew. Chem., Int. Ed., 2022, 61, e202200 CrossRef.
- C. Kulkarni, A. K. Mondal, T. K. Das, G. Grinbom, F. Tassinari, F. J. Mathijs, M. E. W. Meijer and R. Naaman, Adv. Mater., 2020, 32, 1904965 CrossRef CAS.
- Z. Cao, H. Gao, M. Qiu, W. Jin, S. Deng, K.-Y. Wong and D. Lei, Adv. Mater., 2020, 32, 1907151 CrossRef CAS.
- S. Huang, H. Yu and Q. Li, Adv. Sci., 2021, 8, 2002132 CrossRef.
- Y. Li, K. Liu, X. Li, Y. Quan and Y. Cheng, Chem. Commun., 2020, 56, 1117–1120 RSC.
- J. Liu, F. Yuan, X. Ma, D. Y. Auphedeous, C. Zhao, C. Liu, C. Shen and C. Feng, Angew. Chem., Int. Ed., 2018, 57, 6475–6479 CrossRef CAS PubMed.
- J. L. Greenfield, E. W. Evans, D. D. Nuzzo, M. D. Antonio, R. H. Friend and J. R. Nitschke, J. Am. Chem. Soc., 2018, 140, 10344–10353 CrossRef.
- L. Milanesi, J. P. Waltho, C. A. Hunter and M. Volk, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19563–19568 CrossRef CAS.
- H. Li, L. Kou, L. Liang, B. Li, W. Zhao, X. Yang and B. Wu, Chem. Sci., 2022, 13, 4915 RSC.
- D. Mondal, M. Ahmad, B. Dey, A. Mondal and P. Talukdar, Nat. Commun., 2022, 13, 6507 CrossRef CAS PubMed.
- K. R. Strom and J. W. Szostak, J. Am. Chem. Soc., 2022, 144, 18350–18358 CrossRef CAS PubMed.
- I. Okamoto, M. Nabeta, Y. Hayakawa, N. Morita, T. Takeya, H. Masu, I. Azumaya and O. Tamura, J. Am. Chem. Soc., 2007, 129, 1892–1893 CrossRef CAS.
- V. Berl, I. Huc, R. G. Khoury, M. J. Krische and J. M. Lehn, Nature, 2000, 407, 720–723 CrossRef CAS.
- D. Nuńez-Villanueva, G. Iadevaia, A. E. Stross, M. A. Jinks, J. Swain and C. A. Hunter, J. Am. Chem. Soc., 2017, 139, 6654–6662 CrossRef.
- S. Shen, G. V. Baryshnikov, Q. Xie, B. Wu, M. Lv, H. Sun, Z. Li, H. Ågren, J. Chen and L. Zhu, Chem. Sci., 2023, 14, 970 RSC.
- J. Li, P. Li, M. Fan, X. Zheng, J. Guan and M. Yin, Angew. Chem., Int. Ed., 2022, e202202532 CAS.
- S. Datta and D. Chaudhuri, Angew. Chem., Int. Ed., 2022, e202201956 CAS.
- M. Dharmarwardana, S. Pakhira, R. P. Welch, C. Caicedo-Narvaez, M. A. Luzuriaga, B. S. Arimilli, G. T. McCandless, B. Fahimi, J. L. Mendoza-Cortes and J. J. Gassensmith, J. Am. Chem. Soc., 2021, 143, 5951–5957 CrossRef CAS PubMed.
- M. Mahl, M. A. Niyas, K. Shoyama and F. Würthner, Nat. Chem., 2022, 14, 457–462 CrossRef CAS.
- Z. Wang, A. Hao and P. Xing, Angew. Chem., Int. Ed., 2020, 59, 11556–11565 CrossRef CAS.
- Z. Wang, A. Hao and P. Xing, Chin. Chem. Lett., 2021, 32, 1390–1396 CrossRef CAS.
- Z. Wang, Y. Li, A. Hao and P. Xing, Angew. Chem., Int. Ed., 2021, 60, 3138–3314 CrossRef CAS PubMed.
- W. Ji, Y. Tang, P. Makam, Y. Yao, R. Jiao, K. Cai, G. Wei and E. Gazit, J. Am. Chem. Soc., 2021, 143, 17633–17645 CrossRef CAS.
- M. Dergham, S. Lin and J. Geng, Angew. Chem., Int. Ed., 2022, 61, e2021142 CrossRef PubMed.
- J. Kim, J. Lee, W. Y. Kim, H. Kim, S. Lee, H. C. Lee, Y. S. Lee, M. Seo and S. Y. Kim, Nat. Commun., 2015, 6, 6959 CrossRef CAS.
- S. Bagatur and T. Fuhrmann-Lieker, J. Eur. Opt. Soc.: Rapid Publ., 2019, 15, 1530 Search PubMed.
- E. Sánchez-Santos, J. J. Garrido-González, L. F. Rodríguez-Sahagún, A. Habib, Á. L. F. de Arriba, F. Sanz, E. M. M. del Valle, J. R. Morán and V. Alcázar, Org. Biomol. Chem., 2022, 20, 7972–7980 RSC.
- Z. Wang, H. Ai, A. Hao and P. Xing, Chem. Mater., 2022, 34, 10162–10171 CrossRef CAS.
- W. Ji, B. Xue, Y. Yin, S. Guerin, Y. Wang, L. Zhang, Y. Cheng, L. J. W. Shimon, Y. Chen, D. Thompson, R. Yang, Y. Cao, W. Wang, K. Cai and E. Gazit, J. Am. Chem. Soc., 2022, 144, 18375–18386 CrossRef CAS.
- J. Xu, Q. Chen, S. Li, J. Shen, P. Keoingthong, L. Zhang, Z. Yin, X. Cai, Z. Chen and W. Tan, Angew. Chem., Int. Ed., 2022, e202202571 CAS.
- E. K. Roesner, D. Asheghali, A. Kirillova, M. J. Strauss, A. M. Evans, M. L. Becker and W. R. Dichtel, Chem. Sci., 2022, 13, 2475 RSC.
- T. Kim, J. Hong, J. Kim, J. Cho and Y. Kim, J. Am. Chem. Soc., 2023, 145, 1793–1802 CrossRef CAS.
- H. Lin, X. Chang, D. Yan, W.-H. Fanga and G. Cui, Chem. Sci., 2017, 8, 2086 RSC.
- G. Y. Lee, E. Hu, A. L. Rheingold, K. N. Houk and E. M. Sletten, J. Org. Chem., 2021, 86, 8425–8436 CrossRef CAS.
- H. Zhang, J. Han, X. Jin and P. Duan, Angew. Chem., Int. Ed., 2021, 60, 4575–4580 CrossRef CAS PubMed.
- J. Zhao, B. Wang, A. Hao and P. Xing, Nanoscale, 2022, 14, 1779 RSC.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. J. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolf, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- O. Wallach, Liebigs Ann. Chem., 1895, 286, 90–143 CrossRef CAS.
- J. Marciniak, M. Andrzejewski, W. Cai and A. Katrusiak, J. Phys. Chem. C, 2014, 118, 4309–4313 CrossRef CAS.
- Y. Xu, C. Li, Q. Cao, B. Wang and Y. Xie, Dyes Pigm., 2017, 139, 681–687 CrossRef CAS.
- M. Elldrissi, S. J. Teat, P. F.-X. Corvini, M. J. Paterson and S. J. Dalgarno, Chem. Commun., 2017, 53, 1973–1976 RSC.
- Y. Zhang, B. He, J. Liu, S. Hu, L. Pan, Z. Zhao and B. Tang, Phys. Chem. Chem. Phys., 2018, 20, 9922 RSC.
- L. Avram and Y. Cohen, Chem. Soc. Rev., 2015, 44, 586–602 RSC.
- Z. Zhang, H. Wang, X. Wang, Y. Li, B. Song, O. Bolarinwa, R. A. Reese, T. Zhang, X. Wang, J. Cai, B. Xu, M. Wang, C. Liu, H.-B. Yang and X. Li, J. Am. Chem. Soc., 2017, 139, 8174–8185 CrossRef CAS PubMed.
- N. Giuseppone, J.-L. Schmitt, L. Allouche and J.-M. Lehn, Angew. Chem., Int. Ed., 2008, 47, 2235–2239 CrossRef CAS.
- S. Bobbala, S. D. Allen and E. A. Scott, Nanoscale, 2018, 10, 5078–5088 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2246067, 2246071, 2246072 and 2246095. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05212e |
| This journal is © The Royal Society of Chemistry 2024 |