
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePseudomorphic amorphization of three-dimensional superlattices through morphological transformation of nanocrystal building blocks†
Masaki
Saruyama
 *a,
Ryo
Takahata
a,
Ryota
Sato
a,
Kenshi
Matsumoto
a,
Lingkai
Zhu
b,
Yohei
Nakanishi
a,
Motoki
Shibata
cd,
Tomotaka
Nakatani
cd,
So
Fujinami
cd,
Tsukasa
Miyazaki
cd,
Mikihito
Takenaka
a and
Toshiharu
Teranishi
*ab
*a,
Ryo
Takahata
a,
Ryota
Sato
a,
Kenshi
Matsumoto
a,
Lingkai
Zhu
b,
Yohei
Nakanishi
a,
Motoki
Shibata
cd,
Tomotaka
Nakatani
cd,
So
Fujinami
cd,
Tsukasa
Miyazaki
cd,
Mikihito
Takenaka
a and
Toshiharu
Teranishi
*ab
aInstitute for Chemical Research, Kyoto University, Gokasho, Uji, Kyoto 611-0011, Japan. E-mail: saruyama@scl.kyoto-u.ac.jp; teranisi@scl.kyoto-u.ac.jp
bGraduate School of Science, Kyoto University, Gokasho, Uji, Kyoto 611-0011, Japan
cOffice of Society-Academia Collaboration for Innovation, Kyoto University, Yoshida-Honmachi, Kyoto 606-8501, Japan
dOffice of Society-Academia Collaboration for Innovation, Kyoto University, Gokasho, Uji, Kyoto 611-0011, Japan
First published on 5th January 2024
Abstract
Nanocrystal (NC) superlattices (SLs) have been widely studied as a new class of functional mesoscopic materials with collective physical properties. The arrangement of NCs in SLs governs the collective properties of SLs, and thus investigations of phenomena that can change the assembly of NC constituents are important. In this study, we investigated the dynamic evolution of NC arrangements in three-dimensional (3D) SLs, specifically the morphological transformation of NC constituents during the direct liquid-phase synthesis of 3D NC SLs. Electron microscopy and synchrotron-based in situ small angle X-ray scattering experiments revealed that the transformation of spherical Cu2S NCs in face-centred-cubic 3D NC SLs into anisotropic disk-shaped NCs collapsed the original ordered close-packed structure. The random crystallographic orientation of spherical Cu2S NCs in starting SLs also contributed to the complete disordering of the NC array via random-direction anisotropic growth of NCs. This work demonstrates that an understanding of the anisotropic growth kinetics of NCs in the post-synthesis modulation of NC SLs is important for tuning NC array structures.
Introduction
In recent years, a variety of nanocrystal (NC) superlattices (SLs) have been developed by assembling uniform-sized inorganic NCs as “artificial atoms”. When NCs are brought close together, we observe not only various physical interactions but also nanopores among NC components, which can be exploited to provide new collective electronic, magnetic, mechanical and catalytic properties.1,2 Close-packing of NCs enhances interparticle interactions, which should lead to emergence of concerted physical properties such as faster electronic transport,3 lower lasing threshold4 and superfluorescence.5 Non-ordered assembly of NCs supplies larger interparticle gaps and broader pore size distribution, which should accelerate molecular transport within SLs.6,7 Therefore, tuning the assembly manner of NCs is important to control the properties of SLs.The most basic SL is a regular array of spherical NCs with uniform size. Typically, long-range ordered NC SLs in two or three dimensions are fabricated by slow evaporation of a solution containing purified NCs.8 In the case of NCs capped with common ligands containing hydrophobic alkyl chains, the NCs tend to assemble into close-packed SLs with a face-centred-cubic (fcc) or hexagonal-close-packed (hcp) structure via interparticle interactions and/or entropic forces.1 Assembling non-spherical NCs produces SLs with spatial symmetries that differ from those of simple fcc and hcp structures. For example, nanocubes, nanoplates, and nanorods form SLs with simple cubic,5 columnar,9 and bundle10 structures, respectively. These anisotropic SLs are recognized as new types of mesoscopic materials because of their unique directional physical properties.10
Recently, spontaneous formation of three-dimensional (3D) SLs during NC synthesis has been reported,8–12 in which self-assembly is driven by the increase in van der Waals (vdW) attraction forces between NCs during their growth. Because vdW attraction is a universal force commonly found in matter, this in situ self-assembly process is potentially applicable to the rapid, scalable synthesis of SLs from various kinds of NCs, which can provide us with the opportunity to apply NC SLs to various fields. This self-assembly method can be adopted for not only spherical NCs but also nanorods11 and nanodisks12 to form anisotropic 3D SLs. One surprising phenomenon in this process is that NCs continue to grow even after the formation of SLs,8,9,11 which is presumably because NC precursors penetrate the interior of SLs through NC gaps. Previous studies have demonstrated that NCs inside SLs grow while maintaining their shape and size uniformity,8,9,11 which increases the overall lattice constant of SLs but does not change the arrangement of NCs.
Here, we show the dynamic evolution of NC arrangements in 3D SLs, specifically the morphological transformation of NC constituents during the direct liquid-phase synthesis of 3D NC SLs. Because low-chalcocite Cu2S NCs can transform from spheres to disks during the growth process, they were used as model building blocks. In addition to ex situ characterization, synchrotron-based in situ small-angle X-ray scattering (SAXS) experiments were conducted to monitor overall events, including the nucleation, growth and assembly of Cu2S NCs and the subsequent structural change of Cu2S NC SLs in the synthesis solution. These measurements revealed that the transformation of spherical Cu2S NCs into anisotropic disks in preformed fcc 3D SLs collapses the original ordered close-packed structure. Random crystallographic orientation of spherical Cu2S NCs in starting SLs further contributes to the complete disordering of the NC array via random-direction anisotropic growth of NCs. We also show that this process leads to the formation of unique, kinetically stabilized NC assemblies (faceted random aggregates) through pseudomorphic (templated) amorphization during post-assembly growth. These results indicate that an understanding of the anisotropic growth kinetics of NCs in the post-synthesis modulation of NC SLs is important for obtaining desired NC array structures.
Results and discussion
Copper sulphide (Cu2−xS) NCs have been widely studied as both unique hole-based plasmonic materials active in the near infrared (NIR) region and potential starting materials for cationic exchange reactions.13–15 Methods for the precise control of the size, morphology and crystal phase of Cu2−xS NCs are well established, and the formation mechanism of Cu2−xS NCs has been thoroughly investigated.16 The assembly of Cu2−xS NCs has been achieved mainly by solvent evaporation17 and controlled precipitation by adding poor solvents.18 The self-assembly of Cu2−xS NCs occurs during liquid-phase synthesis,12,19 and considerable interest has focused on the evolution of the Cu2−xS phase, while the formation mechanism of Cu2−xS SLs has not been comprehensively characterized. Our previous study demonstrated that spontaneous self-assembly of NCs can be induced during the reactions of general colloidal synthesis protocols.9 To achieve in situ formation of Cu2−xS NC SLs, we used a typical, simple synthesis protocol of Cu2−xS NCs. Specifically, we heated a mixture of CuCl2·2H2O, 1-dodecanethiol (DDT) and 1-dodecylamine (DDA) to 230 °C at 10 °C min−1, and we kept the mixture at that temperature. When DDT was injected into the deep-blue solution containing the Cu-DDA complex at 80 °C, the solution turned turbid light-yellow, indicative of the formation of large Cu-DDT complexes with a lamellar supermolecular structure (Fig. S1a†).20 The solution turned transparent at 130–140 °C due to the melting of Cu-DDT.20 At approximately 220 °C, the reaction solution gradually turned from orange to dark-brown, indicative of the nucleation of Cu2−xS NCs formed by the thermolysis of Cu-DDT precursors. Approximately 4 min after the reaction temperature reached 230 °C, the solution became cloudy due to the formation of large microparticles, indicative of the self-assembly of NCs. To characterize these large particles, we collected the product by purification without non-polar good solvents and centrifugation to avoid collapse of the NC-assembled structure.First, we characterized the product obtained with the reaction temperature held at 230 °C for 10 min. The X-ray diffraction (XRD) pattern of this product showed diffraction peaks assignable to low-chalcocite Cu2S (Fig. S2†). From the scanning electron microscopy (SEM) image in Fig. 1a and S3,† we observed Cu2S NC SLs with a micrometre-sized faceted structure together with the sheet-like unreacted Cu-DDT complex, which could not be removed owing to its insolubility in the solvent used for purification (Fig. S1b†). The magnified SEM image in Fig. 1e revealed that monodisperse spherical Cu2S NCs were closely packed into highly ordered 3D NC SLs. We further characterized the products formed at longer reaction times (20, 30 and 35 min at 230 °C) to observe the temporal evolution of Cu2S NC SLs. The XRD patterns of these products were also assignable to the low-chalcocite Cu2S phase, although the peak width narrowed with increasing reaction time, indicating that Cu2S NCs grew without changing their crystal phase (Fig. S2†). SEM observations showed that the faceted morphology of the products was roughly preserved regardless of the reaction time (Fig. 1b–d). The highly ordered array of monodisperse Cu2S NCs was still observed at 20 min (Fig. 1f); however, the regularity of the Cu2S NC array was totally collapsed at 30 min (Fig. 1g). Moreover, the product obtained after another 5 min of reaction (at 35 min) consisted of randomly oriented disk-shaped Cu2S NCs (Fig. 1h). The ex situ SAXS patterns of these Cu2S NC SLs clearly showed the temporal change in the ordering degree of the NC array in SLs (Fig. 1i). Cu2S NC SLs formed at 10 min and 20 min had diffraction peaks assignable to the fcc arrangement of spherical Cu2S NC components. The Bragg peaks shifted to a lower scattering vector (q) with increasing reaction time, and the lattice constant of fcc Cu2S NC SLs increased from 12.8 nm (10 min) to 14.2 nm (20 min). However, these Bragg peaks disappeared after 30 min, which was consistent with the disordered assembly of Cu2S NCs observed in the SEM images (Fig. 1g and h).
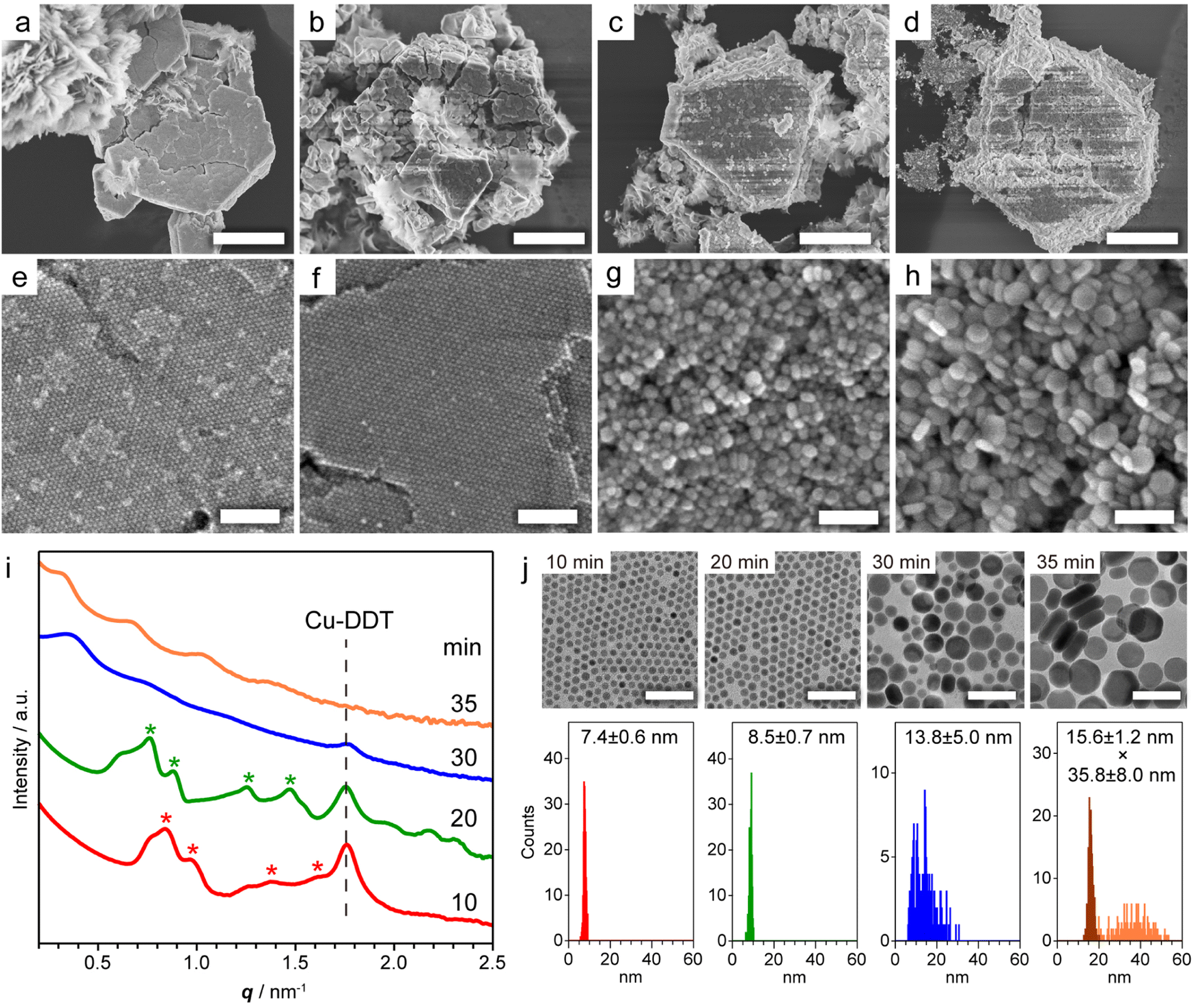 | ||
| Fig. 1 (a–d) Low and (e–h) high magnification SEM images of Cu2S NC SLs obtained by holding the reaction temperature at 230 °C for (a and e) 10, (b and f) 20, (c and g) 30, and (d and h) 35 min. (i) Ex situ SAXS patterns of the products obtained at various reaction times. Red and green asterisks indicate the Bragg peaks from fcc SLs with lattice constants of 12.8 and 14.2 nm, respectively. The peak from residual Cu-DDT was also detected (Fig. S1c†). (j) TEM images and size histograms of dispersed Cu2S NCs at various reaction times. The thickness (dark-orange) and width (light-orange) of disk-shaped NCs were determined in the sample at 35 min. Scale bars = 5 μm (a–d); 100 nm (e–h); 50 nm (j). | ||
To investigate how the size and shape evolution of Cu2S NC constituents in SLs affect the regularity of SLs, the morphological change of Cu2S NCs was determined using transmission electron microscopy (TEM) (Fig. 1j). Cu2S NCs were disassembled from SLs by sonication in chloroform and then drop-cast on a TEM grid. The TEM images of Cu2S NCs in SLs formed at 10 min and 20 min showed monodisperse spherical NCs with diameters of 7.4 ± 0.6 nm (σ = 8%) and 8.5 ± 0.7 nm (σ = 8%), respectively (Fig. 1j), indicating that isotropic growth of spherical Cu2S NC constituents expanded the fcc lattice of SLs. The diameters of Cu2S NC cores were smaller than the calculated diameters of spherical components in SLs using lattice constants (9.1 nm for 10 min and 10.0 nm for 20 min) because of the presence of DDT/DDA ligands. The surface-to-surface interparticle distance was estimated to be 1.7 nm for 10 min and 1.5 nm for 20 min. Considering the length of DDT/DDA with the all-trans conformation (1.8 nm), alkyl chains might be intercalated or folded between Cu2S NCs in SLs. The shorter interparticle distance for 20 min might be caused by the enhanced vdW attraction force between larger Cu2S cores. Moreover, Cu2S NCs obtained at 30 min were larger in size with a broader size distribution [13.8 ± 5.0 nm (σ = 36%)], as shown in Fig. 1j. After further reaction (35 min), Cu2S NCs evolved into larger disk-shaped NCs with a polydisperse width of 35.8 ± 8.0 nm (σ = 22%) and uniform thickness (15.6 ± 1.2 nm, σ = 8%) (Fig. 1j and S4†). This sphere-to-disk conversion due to the preferential surface stabilization of specific crystal planes with protecting ligands is often observed in the synthesis of Cu2−xS NCs.21 In our case, the high-resolution TEM (HRTEM) images of disk-shaped Cu2S NCs (35 min) showed that growth along [![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 02] was suppressed to induce anisotropic elongation from initially formed spherical Cu2S NCs (Fig. S5†). The above observations suggest that both broadening of the size distribution and anisotropic transformation of monodisperse spherical NCs disrupt the initially formed regular arrangement of NCs in SLs.
02] was suppressed to induce anisotropic elongation from initially formed spherical Cu2S NCs (Fig. S5†). The above observations suggest that both broadening of the size distribution and anisotropic transformation of monodisperse spherical NCs disrupt the initially formed regular arrangement of NCs in SLs.
Spontaneous assembly of NCs into SLs during NC synthesis has been reported for many materials (i.e., Pd,8 Au,22 Fe3O4,8 PbS,8 ZrO2,10 Co,11 NiPx9 and Cu2−xS12,19). For SLs formed from spherical Pd NCs or rod-shaped Co NCs, the lattice parameters of SLs change during the reaction,8,11 while the assembly structures of SLs are maintained because the shape and size-uniformity of NCs are retained. To the best of our knowledge, the drastic change from a crystalline to an amorphous structure observed in our Cu2S NC SLs has not been reported.
To elucidate the details of the overall process, including the nucleation and assembly of Cu2S NCs and the structural evolution of SLs, in situ SAXS measurements were conducted using a synchrotron beamline. The reaction was reproduced in a custom-made glass vessel with an X-ray window to detect X-rays scattered by Cu2S NCs and SLs formed in the solution (Fig. S6†).9 SAXS signals were acquired every 12 s as the temperature increased from 113 °C (Movie S1†). The heating rate was programmed at approximately 10 °C min−1 and the temperature reached 230 °C at 12.6 min. The contour plot of time-evolved in situ SAXS patterns clearly showed that various events took place during the reaction (Fig. 2a). The scattering peak at 1.75 nm−1 from layered Cu-DDT complexes disappeared at 139 °C because of the melting of the lamellar structure (Fig. 2a, S1c and d†).20 Obvious scattering was not observed until the reaction temperature reached ∼220 °C because small Cu-DDT molecules and [Cu–S] monomers (∼1.4 nm on average) were the dominant scattering species in the solution (Fig. S7†).12 When the reaction temperature exceeded 227 °C (12.2 min), the scattering intensity started to increase in the low q region (<2 nm−1), indicating initiation of the nucleation of Cu2S NCs (Fig. 2b). This diffuse scattering intensity from dispersed NCs became stronger with increasing reaction time because of the size-growth of Cu2S NCs (Fig. 2b). The emergence of oscillation peaks indicated the formation of monodisperse NCs. We applied data fitting to a series of scattering patterns with a spherical form factor to estimate the size and size-distribution of Cu2S NCs (Fig. 2c and d). The average size of Cu2S NCs grew from 2.0 nm (12.2 min) to 6.8 nm (16.8 min), while the corresponding relative standard deviation decreased from 29% to 7% (Fig. 2d). This size-focusing in the “heating-up” process can be explained by considering the generation of [Cu–S] monomer species by thermolysis of Cu-DDT precursors.23 Cu2S NCs start to nucleate when the [Cu–S] monomer concentration reaches supersaturation and grow by consuming [Cu–S] monomers in the solution. Because the slow thermolysis of stable Cu-DDT limits the supply rate of [Cu–S] monomers after nucleation in our case, the growth of Cu2S NCs is preferred to another nucleation,12 resulting in a narrow size-distribution of the system. During the growth of Cu2S NCs, sharp Bragg reflection peaks suddenly appeared at 16.8 min (4.2 min after reaching 230 °C, Fig. 2a), indicating that Cu2S NCs started to assemble at that time. The Bragg pattern was attributed to an fcc lattice, which indicated that monodisperse spherical Cu2S NCs were assembled into a cubic close-packed array. The high size uniformity at the assembly step owing to the size-focusing process contributes to the formation of highly ordered NC SLs.9 The surface-to-surface interparticle distance calculated from the lattice constant of SLs (13.2 nm) and the average diameter of Cu2S NCs (6.85 nm) was 2.48 nm, suggesting that DDT ligands are partially intercalated and/or folded between NCs in SLs. This distance is longer than that in the ex situ SAXS experiment, probably because the swelling of solvent in SLs and high temperature expand the lattice of SLs. This self-assembly is likely triggered by vdW attraction forces among Cu2S NCs grown in the solution. We calculated the interaction potential between two DDT-capped spherical Cu2S NCs as a function of interparticle distance, considering the vdW attractive force between Cu2S cores and the steric repulsion force between DDT ligands (Fig. S8†).24 The obtained Lennard–Jones type curve showed potential wells that deepened as the size of Cu2S cores increased, indicating that self-assembly proceeded readily for large NCs owing to the increased vdW force. This calculation qualitatively explains the experimental results, specifically the sudden commencement of self-assembly during the growth of Cu2S NCs. Note that the NCs experimentally obtained when the self-assembly started (6.85 nm) was not large enough to overcome the thermal energy (1 kBT) in this potential calculation (Fig. S8c†). In addition, the interparticle distance at the deepest point in the attraction well for 6.85 nm Cu2S NCs was calculated to be 2.8 nm, which is longer than the experimental value (2.48 nm). These differences suggest that the calculated attraction force is weaker than the experimental one, and the additional forces such as vdW attraction between intercalated DDT ligands also contributed to the stabilization of assemblies.
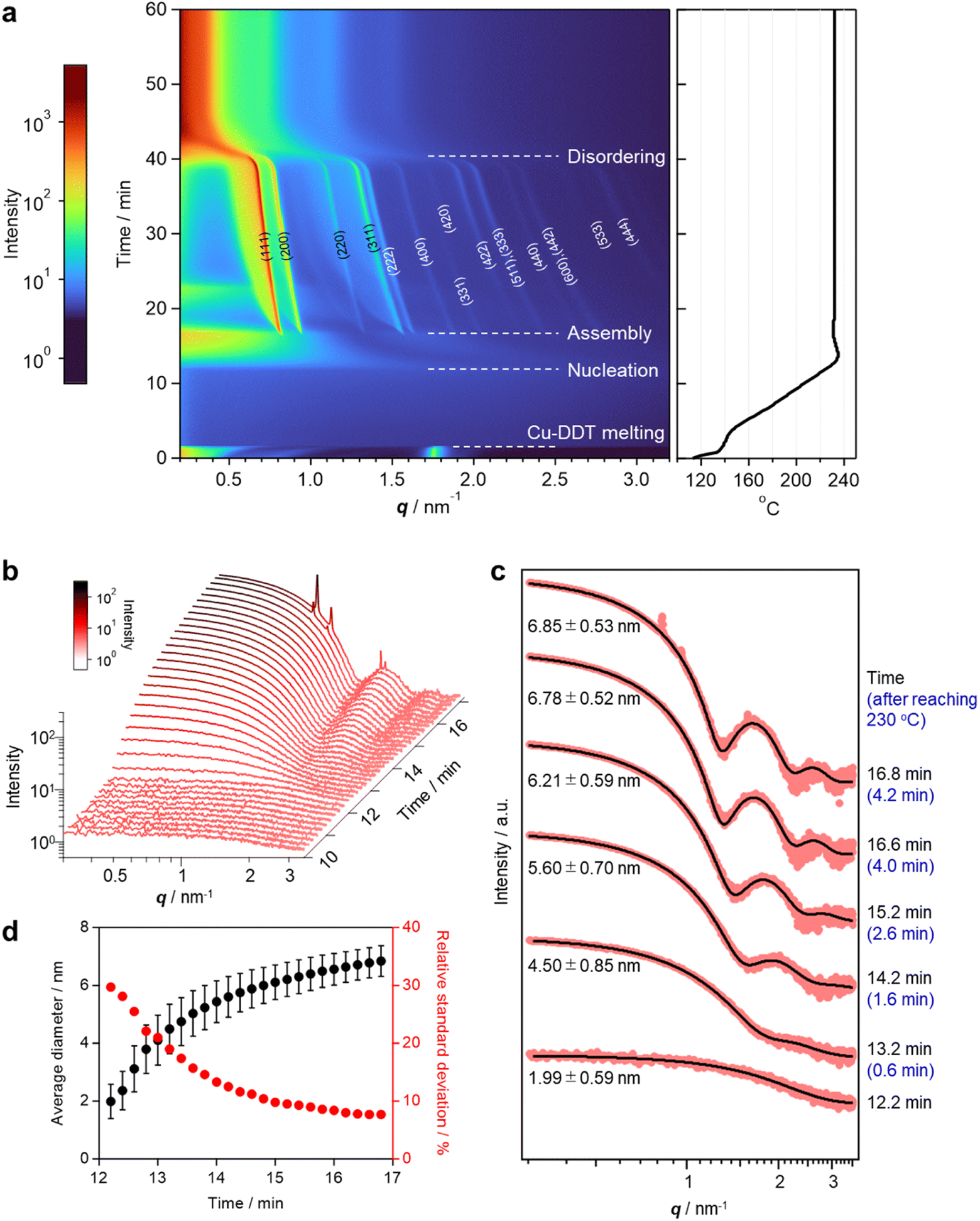 | ||
| Fig. 2 (a) (left) Contour plot of in situ SAXS patterns and (right) temperature profile over the reaction time. (b) Time evolution of background-subtracted in situ SAXS patterns before the self-assembly of Cu2S NCs. (c) Representative SAXS patterns from NC nucleation to SL formation. Pink dots and black solid lines represent the background-subtracted and fitted data, respectively. (d) Time-evolved average size and relative standard deviation of Cu2S NCs dispersed in the solution. | ||
After the self-assembly started, following the decrease in the number of dispersed Cu2S NCs, the Bragg peak intensity increased while the diffuse scattering intensity from dispersed Cu2S NCs rapidly decreased (Fig. S9†). Notably, a series of Bragg peaks gradually shifted to lower q during the reaction while the fcc arrangement of NCs was preserved (Fig. 2a and 3a). The lattice constant successively expanded from 13.2 nm to 17.9 nm, which was consistent with the change in ex situ SAXS patterns (Fig. 1i), suggesting that precursor molecules were incorporated into interparticle spaces for the growth of NC components inside SLs.8 The diameter of spherical units in the fcc lattice calculated from the lattice constant increased from 9.3 nm to 12.7 nm, suggesting that spherical Cu2S NCs grew to approximately 10 nm in SLs, considering the surface-to-surface interparticle distance of approximately 2.5 nm during the in situ SAXS experiment (Fig. S10†). Subsequently, sharp Bragg peaks suddenly disappeared at 41 min (28.4 min after reaching 230 °C, Fig. 3a), indicating loss of the long-range ordering of Cu2S NCs in SLs, as observed in Fig. 1. A closer look at the time-evolved in situ SAXS patterns revealed that a satellite shoulder peak close to the 111 diffraction peak appeared immediately after the self-assembly (Fig. 3a and S11†). The area of this shoulder peak increased with increasing reaction time, which reflects the broadening of the size distribution of Cu2S NCs. The shoulder peak finally merged with the sharp peak to form a broad pattern at 41.0 min (Fig. 3a). This suggested that SLs were partly disordered in the Cu2S NC array at an early stage of the self-assembly (approximately 1 min after self-assembly), and the disordered region gradually expanded.
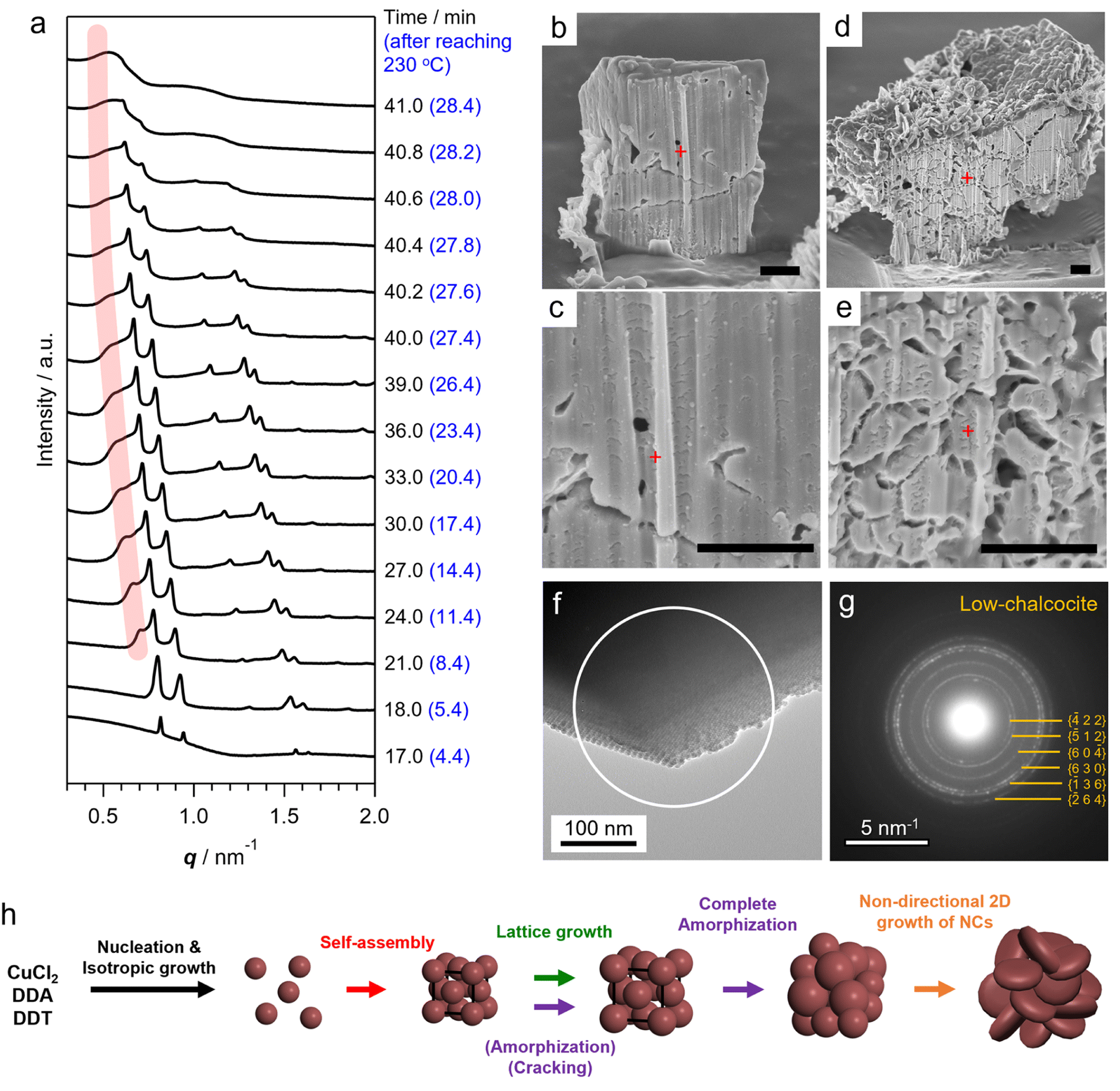 | ||
| Fig. 3 (a) Time-evolved in situ SAXS patterns after self-assembly. Pink line indicates the shoulder peaks close to 111 diffraction peaks. (b–e) Cross-sectional FIB-SEM images of SLs obtained by holding the reaction temperature at 230 °C for (b and c) 10 min and (d and e) 20 min, which correspond to 22.6 min and 32.6 min total reaction time of in situ SAXS measurement, respectively. Scale bars are 1 μm. Red crosses indicate the position for the SEM operation. (f) TEM image of an edge of Cu2S NC SLs (10 min after reaching 230 °C). (g) SAED pattern of Cu2S SLs within the white circle in (f). Ring profile indicates random crystallographic orientation of low-chalcocite spherical Cu2S NCs in SLs. (h) Schematic illustration of the overall process of the structural evolution of Cu2S NC SLs synthesized using DDT. | ||
The coexistence of ordered and disordered phases in a single SL would lead to stress at the interfaces of these phases, making the SL fragile. Indeed, the SEM image of SLs formed after holding the reaction temperature at 230 °C for 20 min (corresponding to 32.6 min in the in situ SAXS experiment) showed large cracks at the surface (Fig. 1b). Focused-ion-beam (FIB)-SEM observations clearly showed a larger number of cracks on the cross-section of the SL at the middle stage (Fig. 3d and e) than that at the early stage (10 min at 230 °C, Fig. 3b and c). These observations revealed that the disordered array inside the SL induced large stresses at the interfaces with ordered regions to generate cracks throughout the SL. Such cracks would accelerate the penetration of precursors into SLs, resulting in an increased growth rate of NCs within SLs. Indeed, the lattice expansion rate of ordered parts drastically increased before entire amorphization (Fig. S10†).
In addition, the crystallographic orientation of NCs within SLs also affects the NC arrangement after post-assembly anisotropic growth. Selected area electron diffraction (SAED) of the spherical Cu2S NC array in SLs showed a ring-like pattern of the Cu2S phase, indicating that the crystallographic orientation of low-chalcocite Cu2S NCs in SLs was random (Fig. 3f and g). Nearly spherical Cu2S cores (Fig. 1j and S5a†) and ligands in high-density coverage can interact with each other as soft spheres, which diminishes the anisotropic interaction potential between NCs.25 This indicates that the preferential lateral growth of Cu2S nanodisks along 〈210〉 and 〈010〉 leads to a random direction in SLs (Fig. S5h†), which further disrupts the original ordered array and promotes amorphization of the SLs. Fig. 3h summarizes the overall structural evolution of SL in this one-pot reaction.
Notably, the original polyhedral shape of ordered SLs is maintained even after amorphization of the NC array (Fig. 1c and d and S12†). The shape of 3D NC SLs is generally determined by the surface energy. Ordered SLs from monodisperse spherical NCs tend to have a faceted shape because close-packed facets are preferentially exposed to minimize the surface energy (i.e., (111) of fcc SLs).26 In contrast, polydisperse NCs tend to assemble into spherical aggregates because of the absence of specific stable surfaces.9 Therefore, amorphous polyhedral SLs in this study are thought to be kinetically stabilized structures resulting from pseudomorphic transformation of the preformed ordered SL template. This demonstrates that post-assembly growth of NC components can bring about additional structural diversity to NC SLs.
The above order-to-disorder transition of SLs was caused by the self-assembly of spherical Cu2S NCs prior to the anisotropic transformation of NC building blocks, owing to the slow growth of Cu2S NCs under the present conditions. Therefore, tuning the growth kinetics of Cu2S NCs before self-assembly would result in the different final structures of SLs.9 To accelerate the growth of Cu2S NCs, tert-dodecanethiol (t-DDT), instead of DDT, was used because the former is a more reactive sulphur precursor with a weaker C–S bond than the latter.16 As a result, the self-assembly started at approximately 200 °C, sooner than in the case of DDT, and the one-dimensional (1D) close-packed columnar structure was formed by stacking Cu2S nanoplates (Fig. S13a–c†). This stacking is achieved by maximizing the attractive force between nanoplates, as observed for various NCs with similar shapes.6 This suggested that the high reactivity of t-DDT induced the fast two-dimensional anisotropic growth of NCs prior to the self-assembly, which led to the formation of 1D columnar SLs. For 90 s after reaching 200 °C, the columnar-assembly was maintained while the width of nanoplates rapidly increased from 41 ± 2 nm (0 s sample) to 120 ± 8 nm (Fig. S13c†), owing to the high reactivity of t-DDT. The SAXS pattern of columnar SLs (90 s sample) showed strong peaks at multiples of the first-order peak position (q*, 2q* and 3q*) derived from the lamellar structure with a periodic thickness of 7.1 nm. This thickness is in good agreement with that of Cu2S nanoplates (6.1 ± 0.5 nm) with a surface ligand layer (Fig. S13d†).
To elucidate the formation process of the columnar structure, in situ SAXS measurement was performed for the reaction using t-DDT (Fig. S13e†). As the precursor solution was heated, the scattering intensity began to increase at 194 °C due to the nucleation of Cu2S NCs (Fig. S14a†). The nucleation temperature is significantly lower compared to the case using DDT, owing to the higher reactivity of t-DDT. Notably, a diffraction peak emerged at 196 °C, signifying that the self-assembly starts immediately after nucleation, driven by the rapid growth of the Cu2S nuclei (Fig. S14a†). Subsequently, high-order diffraction peaks at 2q*, 3q* and 4q* (q*: primary peak) appeared and the intensity of each peak gradually increased with the reaction time, indicating the formation of an ordered lamellar superstructure. During the growth stage, the periodic thickness expanded from 8.52 nm to 9.36 nm. These values are approximately 2 nm larger than the corresponding thickness of the powder sample in Fig. S13d† (7.1 nm). Since the average thickness of Cu2S nanoplates after the in situ SAXS experiment (6.4 nm, Fig. S14b†) was similar to that of the powdery sample, this difference can be attributed to the swelling of the ligand layer between the Cu2S nanoplates in the liquid phase.27,28 During the measurement, peak positions consistently appeared at integral multiples of q*, indicating that the columnar structure was preserved in the anisotropic growth after assembly. This preservation was attributed to the consistent preferential in-plane growth of Cu2S nanoplates within the initially formed columnar superstructures (Fig. S13f†). These results emphasize the importance of crystallographic orientation of NC components in dictating the arrangement of NCs within SLs subsequent to post-assembly growth.
Conclusions
In summary, the structural evolution of Cu2S NC SLs through the anisotropic transformation of NC components was investigated using electron microscopy and synchrotron-based in situ SAXS experiments. In the conventional liquid-phase synthesis of Cu2S NCs, highly ordered 3D Cu2S NC SLs were directly formed by spontaneous self-assembly of monodisperse spherical Cu2S NCs driven by strong vdW attractive forces. At the subsequent growth stage, non-directional anisotropic transformation of spherical Cu2S NC components with random crystallographic orientation within SLs completely disordered the preformed periodic array of Cu2S NCs. This study suggests that further growth of NCs inside SLs can be exploited to modulate the original array structure of NCs. It is necessary to understand the growth process of NCs to tune the crystallographic orientation of NCs within SLs for fine control of the lattice parameters of SLs. Future work will be directed at predictable modulation of the structural symmetry of NC SLs between distinct ordered phases (order-to-order phase transition) by means of the post-growth process to create functional mesoscopic materials with desired directional collective opto-electronic properties.Data availability
Additional experimental data supporting this article are included in the ESI.† Reasonable requests for additional information can be made to the corresponding authors.Author contributions
M. Saruyama designed the research, performed the synthesis and characterization and wrote the manuscript. M. Saruyama, R. T., K. M., L. Z., Y. N., M. Shibata, T. N., S. F., T. M. and M. T. performed SAXS using the synchrotron. T. T. supervised the research. All authors have given approval to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A part of this work was conducted at Nagoya University (Proposal No. JPMXP09A19NU0039) and Nagoya Institute of Technology (Proposal No. JPMXP09S20NI0020), supported by the Nanotechnology Platform program of the Ministry of Education, Culture, Sports, Science and Technology (MEXT). We thank Prof. S. Arai, Ms. M. Nakano and Ms. C. Yukita for the FIB-SEM observations. We thank Prof. T. Asaka for the HRTEM and SAED measurements. SAXS experiments were performed at SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2020A1577, 2021A1076, 2021B1662, 2021B7702, 2022A1614, 2022A7702, 2022B1389, 2022B7702 and 2023A1267). Our thanks also go to Dr N. Ohta for experimental support at SPring-8 and Prof. M. Tosaka and Prof. S. Yamago for the ex situ SAXS measurements at Kyoto University. We acknowledge the support of the Quantum Beam Analyses Alliance (QBAA). This work was supported by JST-CREST (Grant No. JPMJCR21B4) (T. T.), JST-FOREST (Grant No. JPMJFR213I) (M. Saruyama), JSPS KAKENHI for Scientific Research S (Grant No. JP19H05634) (T. T.), Scientific Research B (Grant No. JP23H01802) (M. Saruyama) and Challenging Research (Exploratory) (Grant No. JP20K21236) (M. Saruyama). We thank Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.References
- M. A. Boles, M. Engel and D. V. Talapin, Chem. Rev., 2016, 116, 11220–11289 CrossRef CAS PubMed.
- A. B. Grommet, M. Feller and R. Klajn, Nat. Nanotechnol., 2020, 15, 256–271 CrossRef CAS.
- X. Lan, M. Chen, M. H. Hudson, V. Kamysbayev, Y. Wang, P. Guyot-Sionnest and D. V. Talapin, Nat. Mater., 2020, 19, 323–329 CrossRef CAS.
- V. I. Klimov, A. A. Mikhailovsky, S. Xu, A. Malko, J. A. Hollingsworth, C. A. Leatherdale, H. J. Eisler and M. G. Bawendi, Science, 2000, 290, 314–317 CrossRef CAS.
- G. Rainò, M. A. Becker, M. I. Bodnarchuk, R. F. Mahrt, M. V. Kovalenko and T. Stöferle, Nature, 2018, 563, 671–675 CrossRef.
- F. Rechberger and M. Niederberger, Nanoscale Horiz., 2017, 2, 6–30 RSC.
- F. Nakagawa, M. Saruyama, R. Takahata, R. Sato, K. Matsumoto and T. Teranishi, J. Am. Chem. Soc., 2022, 144, 5871–5877 CrossRef CAS PubMed.
- N. Vogel, M. Retsch, C. A. Fustin, A. Del Campo and U. Jonas, Chem. Rev., 2015, 115, 6265–6311 CrossRef CAS PubMed.
- T. Paik, D. K. Ko, T. R. Gordon, V. Doan-Nguyen and C. B. Murray, ACS Nano, 2011, 5, 8322–8330 CrossRef CAS PubMed.
- T. Wang, J. Zhuang, J. Lynch, O. Chen, Z. Wang, X. Wang, D. LaMontagne, H. Wu, Z. Wang and Y. C. Cao, Science, 2012, 338, 358–363 CrossRef CAS PubMed.
- L. Wu, J. J. Willis, I. S. McKay, B. T. Diroll, J. Qin, M. Cargnello and C. J. Tassone, Nature, 2017, 548, 197–201 CrossRef CAS PubMed.
- A. Pucci, M. G. Willinger, F. Liu, X. Zeng, V. Rebuttini, G. Clavel, X. Bai, G. Ungar and N. Pinna, ACS Nano, 2012, 6, 4382–4391 CrossRef CAS PubMed.
- B. Cormary, T. Li, N. Liakakos, L. Peres, P. F. Fazzini, T. Blon, M. Respaud, A. J. Kropf, B. Chaudret, J. T. Miller, E. A. Mader and K. Soulantica, J. Am. Chem. Soc., 2016, 138, 8422–8431 CrossRef CAS.
- W. Van Der Stam, F. T. Rabouw, J. J. Geuchies, A. C. Berends, S. O. M. Hinterding, R. G. Geitenbeek, J. Van Der Lit, S. Prévost, A. V. Petukhov and C. De Mello Donega, Chem. Mater., 2016, 28, 6381–6389 CrossRef CAS.
- C. Coughlan, M. Ibáñez, O. Dobrozhan, A. Singh, A. Cabot and K. M. Ryan, Chem. Rev., 2017, 117, 5865–6109 CrossRef CAS PubMed.
- Y. Liu, M. Liu and M. T. Swihart, J. Phys. Chem. C, 2017, 121, 13435–13447 CrossRef CAS.
- M. Saruyama, R. Sato and T. Teranishi, Acc. Chem. Res., 2021, 54, 765–775 CrossRef CAS PubMed.
- Y. Zhai and M. Shim, Chem. Mater., 2017, 29, 2390–2397 CrossRef CAS.
- T. H. Larsen, M. Sigman, A. Ghezelbash, R. C. Doty and B. A. Korgel, J. Am. Chem. Soc., 2003, 125, 5638–5639 CrossRef CAS.
- Z. Zhuang, Q. Peng, B. Zhang and Y. Li, J. Am. Chem. Soc., 2008, 130, 10482–10483 CrossRef CAS.
- P. Nørby, S. Johnsen and B. B. Iversen, ACS Nano, 2014, 8, 4295–4303 CrossRef.
- P. Espinet, M. C. Lequerica and J. M. Martín-Alvarez, Chem.–Eur. J., 1999, 5, 1982–1986 CrossRef CAS.
- M. B. Sigman, A. Ghezelbash, T. Hanrath, A. E. Saunders, F. Lee and B. A. Korgel, J. Am. Chem. Soc., 2003, 125, 16050–16057 CrossRef CAS.
- B. Abécassis, F. Testard and O. Spalla, Phys. Rev. Lett., 2008, 100, 1–4 CrossRef.
- G. K. Soon, Y. Piao, J. Park, S. Angappane, Y. Jo, N. M. Hwang, J. G. Park and T. Hyeon, J. Am. Chem. Soc., 2007, 129, 12571–12584 CrossRef PubMed.
- J. N. Israelachvili, Intermolecular and Surface Forces, Academic Press, 3rd edn, 2011, ch. 13 Search PubMed.
- N. Olichwer, A. Meyer, M. Yesilmen and T. Vossmeyer, J. Mater. Chem. C, 2016, 4, 8214–8225 RSC.
- Y. Wan, N. Goubet, P. A. Albouy, N. Schaeffer and M. P. Pileni, Langmuir, 2013, 29, 13576–13581 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, additional characterization and results. See DOI: https://doi.org/10.1039/d3sc05085h |
| This journal is © The Royal Society of Chemistry 2024 |