
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNo-wash fluorogenic labeling of proteins for reversible photoswitching in live cells†
Kenji
Torii
a,
Sam
Benson
bc,
Yuichiro
Hori
*d,
Marc
Vendrell
 *bc and
Kazuya
Kikuchi
*ae
*bc and
Kazuya
Kikuchi
*ae
aGraduate School of Engineering, Osaka University, Suita, Osaka 565-0871, Japan. E-mail: kkikuchi@mls.eng.osaka-u.ac.jp
bCentre for Inflammation Research, The University of Edinburgh, Edinburgh EH16 4UU, UK. E-mail: Marc.Vendrell@ed.ac.uk
cIRR Chemistry Hub, Institute for Regeneration and Repair, The University of Edinburgh, Edinburgh EH16 4UU, UK
dFaculty of Science, Kyushu University, Fukuoka, Fukuoka 819-0395, Japan. E-mail: hori@chem.kyushu-univ.jp
eImmunology Frontier Research Center, Osaka University, Suita, Osaka 565-0871, Japan
First published on 18th December 2023
Abstract
Photoswitchable fluorescent molecules (PSFMs) are positioned as valuable tools for biomolecule localization tracking and super-resolution imaging technologies due to their unique ability to reversibly control fluorescence intensity upon light irradiation. Despite the high demand for PSFMs that are suitable for live-cell imaging, no general method has been reported that enables reversible fluorescence control on proteins of interest in living cells. Herein, we have established a platform to realize reversible fluorescence switching in living cells by adapting a protein labeling system. We have developed a new PSFM, named HTL–Trp–BODIPY–FF, which exhibits strong fluorogenicity upon recognition of Halo-tag protein and reversible fluorescence photoswitching in living cells. This is the first example of a PSFM that can be applicable to a general-purpose Halo-tag protein labeling system for no-wash live-cell imaging.
Introduction
The use of photoswitchable fluorescent molecules (PSFMs) for tracking cellular dynamics1 and visualizing cellular components with high spatial resolution using super-resolution imaging2–6 has greatly increased in recent years. While photoactivatable fluorophores irreversibly photoswitch from non-fluorescent to fluorescent states upon light irradiation,7–12 PSFMs exhibit reversible fluorescence switching upon light irradiation allowing each PSFM to be localized multiple times. This is useful for live-cell studies where molecular dynamics can be visualized over long periods. Notably, PSFMs are also used in RESOLFT (reversible saturable optical fluorescence transition) techniques which enable live-cell super-resolution imaging with lower-intensity light irradiation mitigating phototoxicity compared to conventional STED (stimulated emission depletion) nanoscopy.13 Despite the high demand for PSFMs for live-cell imaging, there have only been a few examples of reversible photoswitching in living cells, namely fluorescent proteins,13–15 fluorene-based chromophore–hCRBPII (human cellular retinol-binding protein II) complexes,16 and self-blinking fluorophores.7,17–19 In general, synthetic fluorophores have greater photostability than fluorescent proteins and are thus more favorable for live-cell imaging. Chromophore–hCRBPII complexes exhibit fast thermal isomerization and slow photoisomerization rates from fluorescence ON to OFF states. Self-blinking fluorophores have been used for single-molecule localization nanoscopy with high spatial resolution; however, the temporal resolution is still lower than other super-resolution techniques, including STED and RESOLFT imaging. Therefore, the establishment of a platform to perform reversible photoswitching using synthetic PSFMs in living cells is highly desirable for present live-cell imaging technologies.Cy3–Alexa647 heterodimer2 and fluorescent diarylethenes3,20 have both been developed as PSFMs for RESOLFT imaging. However, neither can be used for live-cell imaging because cyanine dyes require high concentrations of primary thiols (over 100 mM) and potassium iodide for reversible photoswitching, and diarylethenes contain multiple carboxylic acids to improve water solubility, resulting in poor membrane permeability. Recently, PSFMs based on photochromic molecules that undergo reversible photoisomerization upon photoirradiation have been described. In addition to the diarylethenes,3,20–23 spiropyran,4–6 arylazopyrazole,24 and fulgimide25,26-based PSFMs have been reported in the last few years. However, these photochromic PSFMs are highly hydrophobic and can form intermolecular aggregates in aqueous solution,4,6,25 and display reduced photostability. Our group has previously demonstrated that a PSFM modified on the protein surface enables persistent reversible photoswitching performance in biological media.25 The keystone of this mechanism is that chemical modification of PSFMs on the protein surface suppresses physical intermolecular aggregation among PSFMs resulting in a significant increase in their photostability. We have demonstrated the utility of this strategy in fixed cells by immunostaining;25 however, this approach is not suitable for live cell imaging. Therefore, no methodology has been established to use PSFMs for live-cell imaging without compromising their photostability.
To address the above-mentioned issue, we have implemented a protein-tag labeling system using a PSFM with fluorogenic capabilities. Some environmentally sensitive fluorophores have been used as fluorogenic probes for no-wash imaging due to their unique property of exhibiting strong fluorescence emission after binding to a protein tag while minimizing the fluorescence signal from unlabeled free fluorophores.27–29 No-wash imaging is highly desirable for live-cell imaging because it avoids extensive washing of cells and provides simplified experimental procedures. Therefore, it is greatly advantageous to develop PSFMs that allow no-wash fluorogenic labeling in living cells.
In this study, we have developed a novel PSFM named HTL–Trp–BODIPY–FF, which contains the Halo-tag ligand,30 the environmentally sensitive fluorophore Trp–BODIPY31–37 and furylfulgimide (FF)25,38,39 (Fig. 1a). To achieve site-specific protein labeling in living cells, we chose the Halo-tag system, which is versatile for live-cell imaging applications,30 and Trp–BODIPY, which exhibits brighter fluorescence emission in hydrophobic environment.31–37HTL–Trp–BODIPY–FF exhibits limited fluorescence emission in aqueous solutions predominantly due to the free rotation between the phenyl group and the BODIPY core which facilitates non-radiative decay rather than radiative decay as fluorescence (OFF state). On the other hand, HTL–Trp–BODIPY–FF showed a fluorescence intensity increase after binding Halo-tag (ON state), because it was adjacent to the hydrophobic Halo-tag protein surface and sterically inhibited the rotation of the phenyl groups (Fig. 1b). It helps to minimize the fluorescence signal derived from unlabeled HTL–Trp–BODIPY–FF and allow no-wash labeling. FF is a photochromic molecule that undergoes quantitative reversible photoisomerization between colorless open- and colored closed-ring forms. The open-ring form of HTL–Trp–BODIPY–FF shows fluorescence after labeling Halo-tag (ON state). On the other hand, after UV light irradiation induces photoisomerization to the closed-ring form of FF that works as a Förster resonance energy transfer (FRET) acceptor for Trp–BODIPY, resulting in fluorescence quenching (OFF state) (Fig. 1b). Although the use of UV light can cause phototoxicity in living samples, FF outperforms other photochromic compounds in the desired spectral change between two states, making it a suitable photoswitchable FRET quencher. The labeled HTL–Trp–BODIPY–FF exhibited reversible fluorescence switching upon light irradiation with higher photostability compared to the unlabeled probe, assisted by the Halo-tag protein surface that prevents intermolecular aggregation among probes. We used this feature to visualize the reversible fluorescence control of HTL–Trp–BODIPY–FF to Halo-tag expressed in live cells. To the best of our knowledge, this is the first example of a fluorogenic PSFM modified with a Halo-tag protein showing reversible fluorescence photoswitching and represents a valuable tool for the development of future PSFM structures.
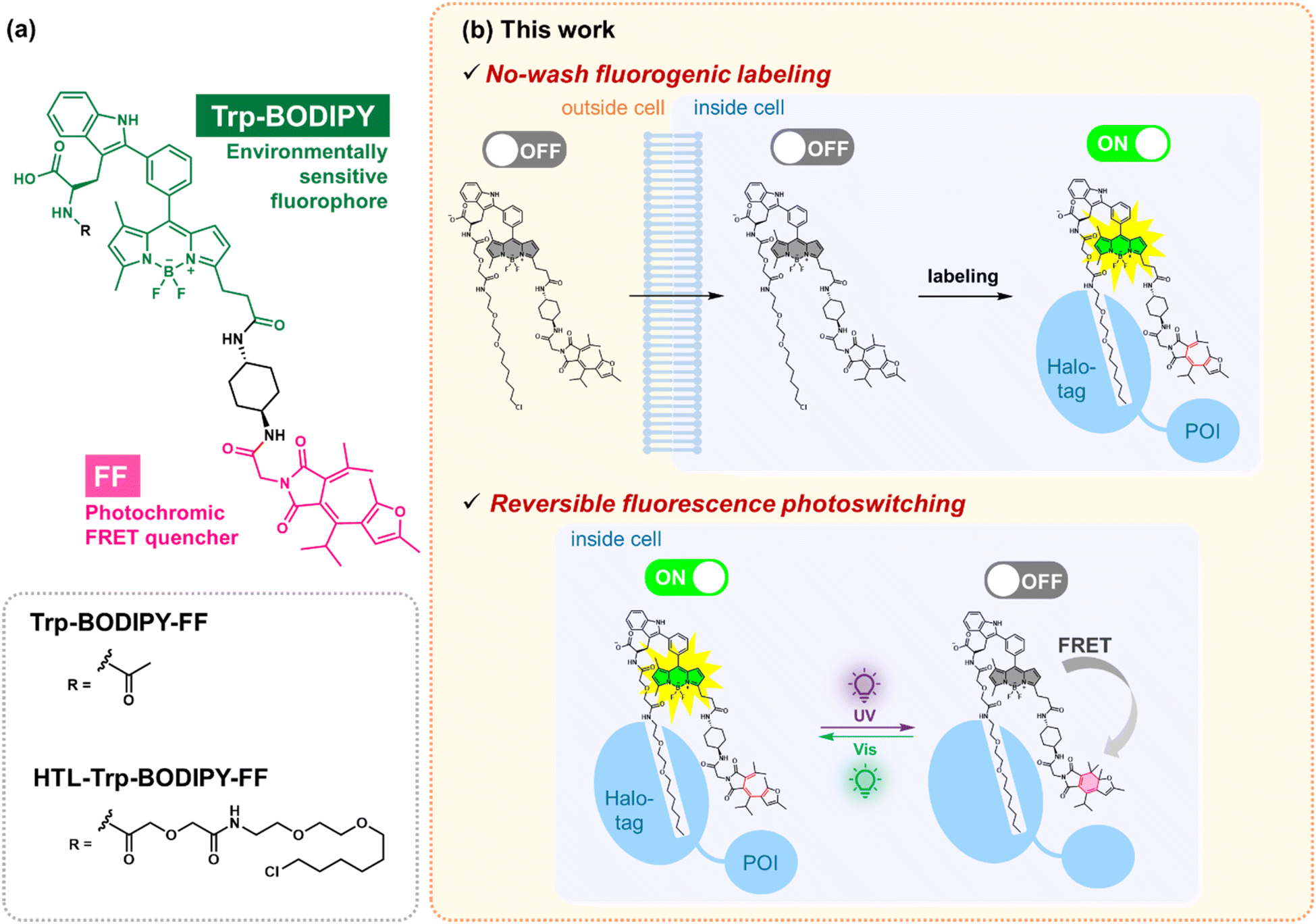 | ||
| Fig. 1 (a) Chemical structures of Trp–BODIPY–FF and HTL–Trp–BODIPY–FF. (b) Mechanism of reversible fluorescence switching in living cells using HTL–Trp–BODIPY–FF. The increase in fluorescence intensity was induced on the hydrophobic Halo-tag protein surface. After labeling, the fluorescence intensity was reversibly controlled by photoisomerization of furylfulgimide (FF) based on Förster energy transfer (FRET) efficiency. The persistent reversibility was assisted by Halo-tag protein surface. POI: protein of interest. | ||
Results and discussion
Molecular design and synthesis of Trp–BODIPY–FF
First, we designed the photoswitchable fluorogenic molecule Trp–BODIPY–FF to scrutinize its photophysical properties, including both environmental sensitivity and photoswitching performance (Fig. 1a). Trp–BODIPY is an environmentally sensitive fluorophore that emits green fluorescence in high viscosity or hydrophobic environments.31–37 In the previous study, we optimized the core structure of Trp–BODIPY, which has high environmental sensitivity and ease of synthesis.32 Trp–BODIPY has lower fluorescence quantum yields (ΦFL = 0.22 in phospholipid membranes31) compared to the conventional meso-phenyl BODIPY (ΦFL = 0.65 in methanol40) (chemical structures are shown in Fig. S1†). This may be due to photoinduced electron transfer from donor tryptophan to the acceptor BODIPY core.35 In addition, the introduction of tryptophan can lower the transition energy barrier to access non-radiative decay, which improves fluorogenicity.35 Considering this advantage of tryptophan, we speculate that Trp–BODIPY exhibits significant fluorogenic ability in hydrophobic environments, and it is suitable for our purpose of fluorogenic protein labeling. FF is known to exhibit quantitative photoisomerization to the closed-ring form upon irradiation at 365 nm and to the open-ring form upon irradiation at 530 nm.25 The open-ring form of FF has no visible absorption (Fig. S2 and Table S1†) and doesn't show any FRET with Trp–BODIPY; therefore, Trp–BODIPY is able to fluoresce in a hydrophobic environment (ON state). While the closed-ring form has a large absorption band in the visible region (Fig. S2 and Table S1†) and functions as a FRET-based fluorescence quencher for Trp–BODIPY (OFF state). In addition, a rigid cyclohexyl linker between Trp–BODIPY and FF was incorporated to prevent intramolecular quenching.25,41 Taken together, Trp–BODIPY–FF was designed to exhibit its fluorogenic nature after interacting with the protein surface, and the fluorescence intensity was regulated by light irradiation. The detailed synthetic scheme and full characterization data are described in the ESI (Fig. S3a and b).†Photophysical properties of Trp–BODIPY–FF
Next, we measured the photophysical properties of Trp–BODIPY–FF. The fluorescence intensity of Trp–BODIPY–FF is significantly higher with increasing glycerol concentration in phosphate buffer (PB), and displays its strongest fluorescence emission in high viscosity environments (Fig. 2a). This viscosity sensitivity is consistent with our previously reported Trp–BODIPY derivatives.31–37 The viscosity sensitivity is closely related to the C–C bond rotation between the BODIPY core and the phenyl group. In high viscosity environments, this rotation is restricted, while the fluorescence intensity is enhanced because the nonradiative decay is inactivated.42 The fluorescence intensity also increased upon its dissolution in low polarity alcohols with longer carbon numbers (n-octanol > n-hexanol > n-butanol > ethanol > PB), indicating that BODIPY–FF shows strong fluorescence in low polarity environments (Fig. 2b). The reason that higher polarity environments cause loss of fluorescence may be partially due to photoinduced electron transfer from the benzene moiety to the BODIPY core, as some phenyl BODIPY derivatives have previously reported.43 In addition, the fluorescence intensity was significantly enhanced in PB containing bovine serum albumin (BSA) as a model protein, indicating that the fluorogenic behavior was triggered on the hydrophobic binding cavities within BSA (Fig. 2c). The reason for the increase in fluorescence intensity of Trp–BODIPY–FF surrounded by BSA is considered to be two factors; (1) a decrease in polarity by being adjacent to the hydrophobic protein surface, and (2) the sterically inhibited C–C bond rotation between BODIPY and phenyl groups. These photophysical properties were also confirmed in the control Trp–BODIPY without FF, indicating that its fluorogenicity was not affected by the introduction of FF (Fig. S4†). The absorption spectra and fluorescence quantum yield of Trp–BODIPY and Trp–BODIPY–FF were also measured (Fig. S5, S6, Tables S2 and S3†). The fluorescence quantum yield in each solvent between Trp–BODIPY and Trp–BODIPY–FF are almost the same value, indicating that open-ring form of FF does not affect the fluorescence intensity of Trp–BODIPY. The fluorescence quantum yield of Trp–BODIPY(–FF) is lower than previously reported Trp–BODIPY derivatives. This is mainly due to the absence of the methyl group at the 7-position in the BODIPY cores (Fig. S1†).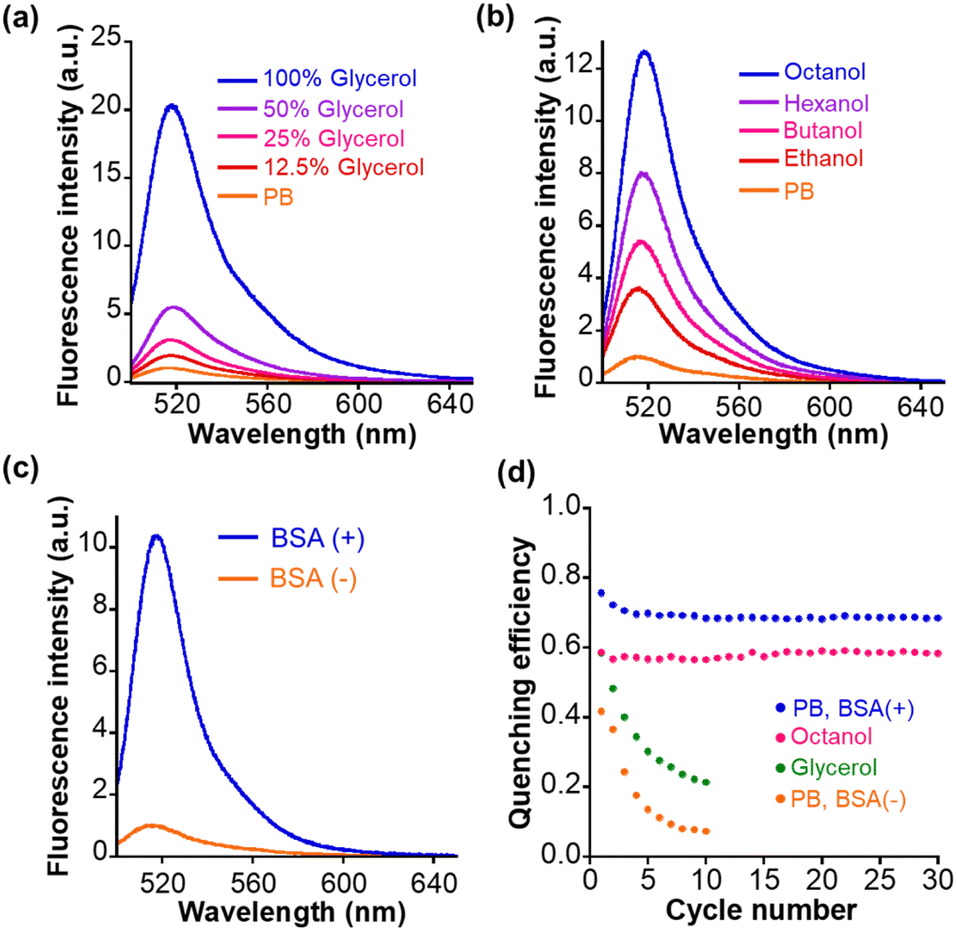 | ||
| Fig. 2 Fluorescence spectra of 1.0 μM Trp–BODIPY in (a) 100 mM phosphate buffer (PB) (pH 7.4) with increasing glycerol concentrations (from top to bottom: 100, 50, 25, 12.5, and 0%), in (b) n-octanol (blue line), n-hexanol (purple line), n-butanol (magenta line), ethanol (red line), and PB (orange line) in (c) 100 mM PB (pH 7.4) with/without 5.0 μM BSA (blue/orange line). λex: 470 nm, 37 °C. (d) Quenching efficiency of Trp–BODIPY–FF over repetitive photoirradiation cycles in PB (orange), glycerol (green), n-octanol (magenta), and PB with 5.0 μM BSA (blue). λex/em: 490/520 nm, 37 °C. | ||
Light irradiation experiments were performed for each solution in PB, glycerol, n-octanol, and PB in the presence of BSA. In all samples, the absorbance at 550 nm was increased following first-order kinetics upon 365 nm irradiation and decreased following first-order kinetics after 530 nm irradiation (Fig. S6†). It clearly indicated that Trp–BODIPY–FF undergoes cyclization and cycloreversion reactions at 365 and 530 nm irradiation. The detailed photophysical parameters including cyclization/cycloreversion reaction kinetics per light irradiation intensity (koc/co/Iirr) and quantum yields (Φoc/co) are determined following ESI experiments (Table S3†). More importantly, the fluorescence intensity of Trp–BODIPY–FF was decreased after 365 nm irradiation and completely recovered after 530 nm irradiation (Fig. S7†), indicating that FF works as a photoswitchable FRET quencher. However, in PB and glycerol, the fluorescence intensity after 365 nm irradiation was gradually suppressed with increasing cycle number (Fig. S8a and b†). Meanwhile, Trp–BODIPY–FF in both n-octanol and PB including BSA showed persistent repeatability (Fig. S8c and d†). The difference in the photostability in each solution is due to the ease of aggregate formation. Dissolution in low-polarity solvent (n-octanol) or solution containing hydrophobic proteins circumvents the formation of aggregate, while in higher-polarity solvents (PB or glycerol), Trp–BODIPY–FF readily aggregates to reduce its photostability during light irradiation. To quantify the photofatigue progress over repetitive photoirradiation cycles, we calculated quenching efficiency of Trp–BODIPY–FF in each solvent conditions. Quenching efficiency was determined by dividing the difference in fluorescence intensity before and after 365 nm irradiation by the fluorescence intensity before 365 nm irradiation. Trp–BODIPY–FF in both n-octanol and PB including BSA showed no photofatigue over 30 photoirradiation cycles (Fig. 2d). In addition, we prepared the bioconjugate of Trp–BODIPY–FF with BSA, named Trp–BODIPY–FF–BSA, by conjugating with the carboxylic acid of Trp–BODIPY–FF and lysine residues on the BSA surface. A detailed preparation procedure and characterization are described in ESI and Fig. S9a.† Trp–BODIPY–FF–BSA also shows persistent reversibility even after 10 cycles of photoswitching (Fig. S9b†). These results are consistent with our previous findings that showed improved photostability of PFSM in hydrophobic environments, including protein surfaces, thus circumventing intermolecular aggregation among PSFMs.25 As a next step, we conjugated Trp–BODIPY–FF to a Halo-tag ligand to evaluate whether Trp–BODIPY–FF would display fluorescence switching upon binding to the Halo-tag protein.
Synthesis and photophysical properties of HTL–Trp–BODIPY–FF
We synthesized the compound HTL–Trp–BODIPY–FF by conjugation of the chloroalkane Halo-tag ligand to the terminal amine of Trp–BODIPY–FF (Fig. 1a). The detailed synthetic scheme and characterization data are included in ESI (Fig. S3c).† We verified that HTL–Trp–BODIPY–FF had increased fluorescence intensity in high viscosity environments as with its precursor compound, Trp–BODIPY–FF (Fig. S10a and Table S4†). We further measured the viscosity dependence of HTL–Trp–BODIPY–FF and compared it to Trp–BODIPY and Trp–BODIPY–FF by plotting the fluorescence intensity against dynamic viscosity (Fig. S11†). HTL–Trp–BODIPY–FF clearly showed a strong fluorescence emission with increasing dynamic viscosity, consistent with the results of Trp–BODIPY and Trp–BODIPY–FF. We corroborated that the fluorescence intensity of HTL–Trp–BODIPY–FF was enhanced in non-polar environments in the same manner as Trp–BODIPY–FF (Fig. S10b and Table S4†). We also checked the affinity of Trp–BODIPY–FF and HTL–Trp–BODIPY–FF with BSA (Fig. S12†). The dissociation constant of Trp–BODIPY–FF and HTL–Trp–BODIPY–FF were determined to be 6.9 ± 2.8 μM and 11.8 ± 3.9 μM, respectively and the fluorescence increase of HTL–Trp–BODIPY–FF was suppressed compared to Trp–BODIPY–FF in higher concentrations of BSA. It may be due to the Halo-tag ligand (HTL) bypassing the interaction of Trp–BODIPY moiety with BSA.The absorption spectrum and fluorescence quantum yield of HTL–Trp–BODIPY–FF were also determined (Fig. S13 and Table S4†). The photophysical properties including excitation maximum, extinction coefficient, and fluorescence quantum yield between Trp–BODIPY–FF and HTL–Trp–BODIPY–FF are almost the same value. Importantly, in the presence of Halo-tag in PB, the fluorescence intensity increased 12-fold as shown in Fig. 3a and Table S4.† Furthermore, we performed SDS–PAGE gel analysis and observed a fluorescent band between 25 and 37 kDa, indicating that Halo-tag (34 kDa) had been coupled to HTL–Trp–BODIPY–FF (Fig. 3b and S14†). Fluorogenicity has great advantages for live cell imaging because it suppresses background fluorescence derived from free probes and allows for no-wash labeling. We further described the optimized conformation of HTL–Trp–BODIPY–FF labeled with Halo-tag using computational calculation software (Macromodel: Schrödinger Maestro v13.1) as presented in the ESI.† As shown in Fig. S15,† no specific binding interaction was observed except for the HTL region and Trp–BODIPY was partially exposed in hydrophobic environments surrounded by several hydrophobic residues. It is reported that the crystal structure of HTL-bearing tetramethylrhodamine demonstrates multiple interaction modes at the protein surface.44 Although we do not exclude the possibility that HTL–Trp–BODIPY–FF may have similar interactions, the result of the simulation indicates that Trp–BODIPY is confined in hydrophobic environments, supporting the mechanism that fluorogenic enhancement is induced by hydrophobic interaction and steric suppression of phenyl ring rotation.
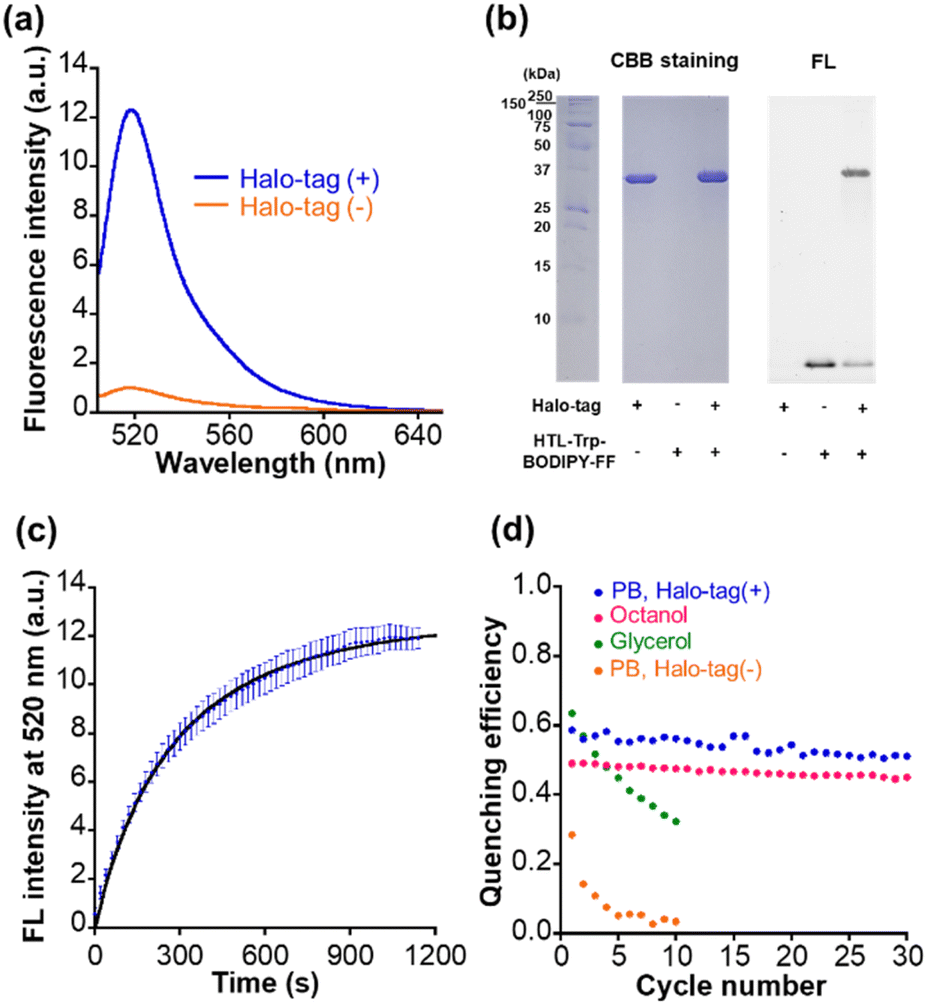 | ||
| Fig. 3 (a) Fluorescence spectra of 1.0 μM HTL–Trp–BODIPY–FF in PB (pH 7.4), after (blue)/before (orange) incubating with 2.0 μM Halo-tag for 20 min. λex: 490 nm, 37 °C. (b) The results of SDS–PAGE of HTL–Trp–BODIPY–FF with Halo-tag. The left and right image shows CBB-stained and fluorescence images, respectively. 2.0 μM HTL–Trp–BODIPY–FF was incubated with 4.0 μM Halo-tag at 37 °C for 1 h. The first column at the left represents a ladder to comfirm protein size (Halo-tag: 34 kDa). Fluorescence images were obtained with excitation at 488 nm. (c) Time course of fluorescence intensities of 1.0 μM HTL–Trp–BODIPY–FF after incubating with 2.0 μM Halo-tag in PB (pH 7.4). N = 3. (d) Quenching efficiency of HTL–Trp–BODIPY–FF over repetitive photoirradiation cycles in PB (orange), glycerol (green), n-octanol (magenta), and PB after incubating with 2 μM Halo-tag for 30 min (blue). λex/em: 490/520 nm, 37 °C. | ||
We determined the labeling rate constant k2 to be 1.5 × 103 M−1 s−1 by the second-order approximation, and the increase in fluorescence intensity was mostly completed within 20 min of the addition of 1 μM HTL–Trp–BODIPY–FF to 2 μM Halo-tag solution (Fig. 3c). The labeling kinetics of common Halo-tag ligands conjugated with fluorophores show different values (around 103 to 108 M−1 s−1 order) because the affinity is strongly influenced by the substrate structure.45 The labeling kinetics of HTL–Trp–BODIPY–FF with Halo-tag is relatively slow compared to other fluorophores bearing HTL. Thus, it may be partly due to the negative charge on the carboxylic acid of HTL–Trp–BODIPY–FF, which has less affinity to the Halo-tag protein surface, resulting in low labeling kinetics.45 In the molecular simulation, no interactions were observed, and the carboxylic acid was exposed to the solvent (water) away from the Halo-tag residues as shown in Fig. S16b.† According to the latest reviews on bioorthogonal click chemistry, one of the fastest click reactions is the inverse electron demand Diels–Alder (IEDDA) reaction.46 The kinetics of IEDDA reactions used for protein labeling reach about 104 M−1 s−1.46–48 The rate constant is higher than that of HTL–Trp–BODIPY–FF with Halo-tag; however, IEDDA reactions have been reported that fluorophore–tetrazine conjugates shows slow labeling and takes more than 1 hour until the completion in living cells.49,50 On the other hand, HTL–Trp–BODIPY–FF completed Halo-tag labeling within 20 min in live cells, which is comparable to the result of the in vitro experiment, as discussed in the next section.
Light irradiation experiments were performed for each solution in glycerol, n-octanol, and before and after Halo-tag binding in PB. In all samples, the absorbance at 550 nm was increased following first-order kinetics upon 365 nm irradiation and decreased following first-order kinetics after 530 nm irradiation (Fig. S13†), and the fluorescence intensity was decreased after 365 nm irradiation followed by recovery after 530 nm irradiation (Fig. S16†). The detailed photoswitching parameters were determined following a ESI experiment (Table S4†). The photophysical properties of HTL–Trp–BODIPY–FF labeled with Halo-tag were compared with other representative green photoswitchable fluorescent proteins applicable for super-resolution imaging (rsEGFP2,51 Dronpa,52 and Padron2 (ref. 53)) in Table S5.† Although, the fluorescence quantum yield and brightness of HTL–Trp–BODIPY–FF are lower than those of photoswitchable fluorescent proteins, both OFF-to-ON and ON-to-OFF photoswitching quantum yields (ΦOFF–ON and ΦOFF–ON) are higher than those of proteins (ΦOFF–ON of Dronpa shows higher value; however, ΦON–OFF is over 200 times lower than that of HTL–Trp–BODIPY–FF. ΦON–OFF of Padron2 shows a slightly higher value; however, ΦOFF–ON is 10 times lower than that of HTL–Trp–BODIPY–FF). Overall, the photoswitching efficiency of HTL–Trp–BODIPY–FF is superior to photoswitchable fluorescent proteins. The photofatigue progress was observed in glycerol over 10 cycles of photoirradiation, while not in n-octanol even after 30 cycles of photoirradiation (Fig. 3d and S17†). Most importantly, the pre-labeled HTL–Trp–BODIPY–FF showed a rapid reduction of quenching efficiency over repeated light irradiation cycles, whereas the labeled HTL–Trp–BODIPY–FF obtains persistent fluorescence switching performance maintaining over 50% quenching efficiency after repeated 30 cycles of light irradiation (Fig. 3d). It was demonstrated that the protein surface-assisted strategy is effective for site-specific labeling with Halo-tag and contributes to the improved photostability of a PSFM.
Live-cell imaging studies using HTL–Trp–BODIPY–FF
As a next step, we performed live cell imaging with HTL–Trp–BODIPY–FF. The fluorescence signals of HTL–Trp–BODIPY–FF were visualized in HeLa cells transfected with maltose binding protein (MBP) fused to Halo-tag in order to stain intracellular cytosol. The constructs were also fused with the red fluorescent protein, mCherry, to both confirm the colocalization of Halo-tag and HTL–Trp–BODIPY–FF and to quantitatively evaluate the fluorescence intensity changes of HTL–Trp–BODIPY–FF during light irradiation experiments. The plasmids used for live-cell imaging (pcDNA3.1(+)–MBP–Halo–mCherry) were prepared according to the procedures described in the ESI.† After incubation of the cells with HTL–Trp–BODIPY–FF for 20 min, fluorescence images were acquired under a confocal laser scanning microscope. The green fluorescence signals from HTL–Trp–BODIPY–FF co-localized with the red fluorescence signal from mCherry in the cytosol of HeLa cells expressing Halo–MBP–mCherry, (Fig. S18a†). In contrast, cells transfected with an empty vector (pcDNA3.1(+)) were devoid of fluorescent signals (Fig. S18b†). To demonstrate the stability of HTL–Trp–BODIPY–FF in living cells, we have performed extended live cell imaging and monitored fluorescence signals in transfected cells and background signals in non-transfected cells, which show no signal from mCherry in the same field of view with multiple cells. After 20 min of incubation from probe addition, the fluorescence signal from HTL–Trp–BODIPY–FF was fully enhanced in transfected cells (Fig. S19a†). Importantly, as seen in Fig. S19b,† the fluorescence signals in transfected cells were maintained without any attenuation even after prolonged incubation time (up to 600 min after probe addition), while the background signal from non-transfected cells (surrounded by yellow circles) was negligible in this visualization.We performed light irradiation experiments in Halo–MBP–mCherry expressing HeLa cells after treatments with HTL–Trp–BODIPY–FF. Cells were repeatedly irradiated at 365 nm for 10 s and 488 and 561 nm for 20 s, respectively, using LED light and excitation light from confocal microscopy. Although the 488 and 561 nm lasers were set up for excitation in the green channel (HTL–Trp–BODIPY–FF) and the red channel (mCherry), these light sources are sufficient to induce the cycloreversion reaction because the closed-ring form of FF has a large absorption band in the visible region (Fig. S2†) that covers both 488 and 561 nm. Detailed experimental conditions are described in the ESI (Fig. S20).† Under these conditions, the fluorescence intensity of HTL–Trp–BODIPY–FF decreased upon 365 nm irradiation and rapidly recovered upon 488 and 561 nm light stimulation, whereas the fluorescence intensity of mCherry showed little changes during light irradiation (Fig. 4a and S21†). It indicates that the effect of FRET from HTL–Trp–BODIPY–FF to mCherry was negligible. This resulted in the fluorescence intensity ratio of HTL–Trp–BODIPY–FF to mCherry to decrease after 365 nm irradiation followed by recovery after 488 and 561 nm excitation light (Fig. 4a). As shown in Fig. 4b, it was confirmed that changes in fluorescence intensity induced by light stimulation were repeated over more than 10 cycles in a random selection of 10 cells. The fluorescence intensity ratios did not change in the absence of 365 nm light irradiation (Fig. S22b†). Moreover, we calculated the quenching efficiency of HTL–Trp–BODIPY–FF in living cells transfected with Halo–MBP–mCherry by dividing the difference in fluorescence intensity ratio before and after 365 nm irradiation by the fluorescence intensity ratio before 365 nm irradiation. It showed persistent reversibility even after 10 cycles of photoswitching, maintaining over 15% averaged quenching efficiency (Fig. 4c).
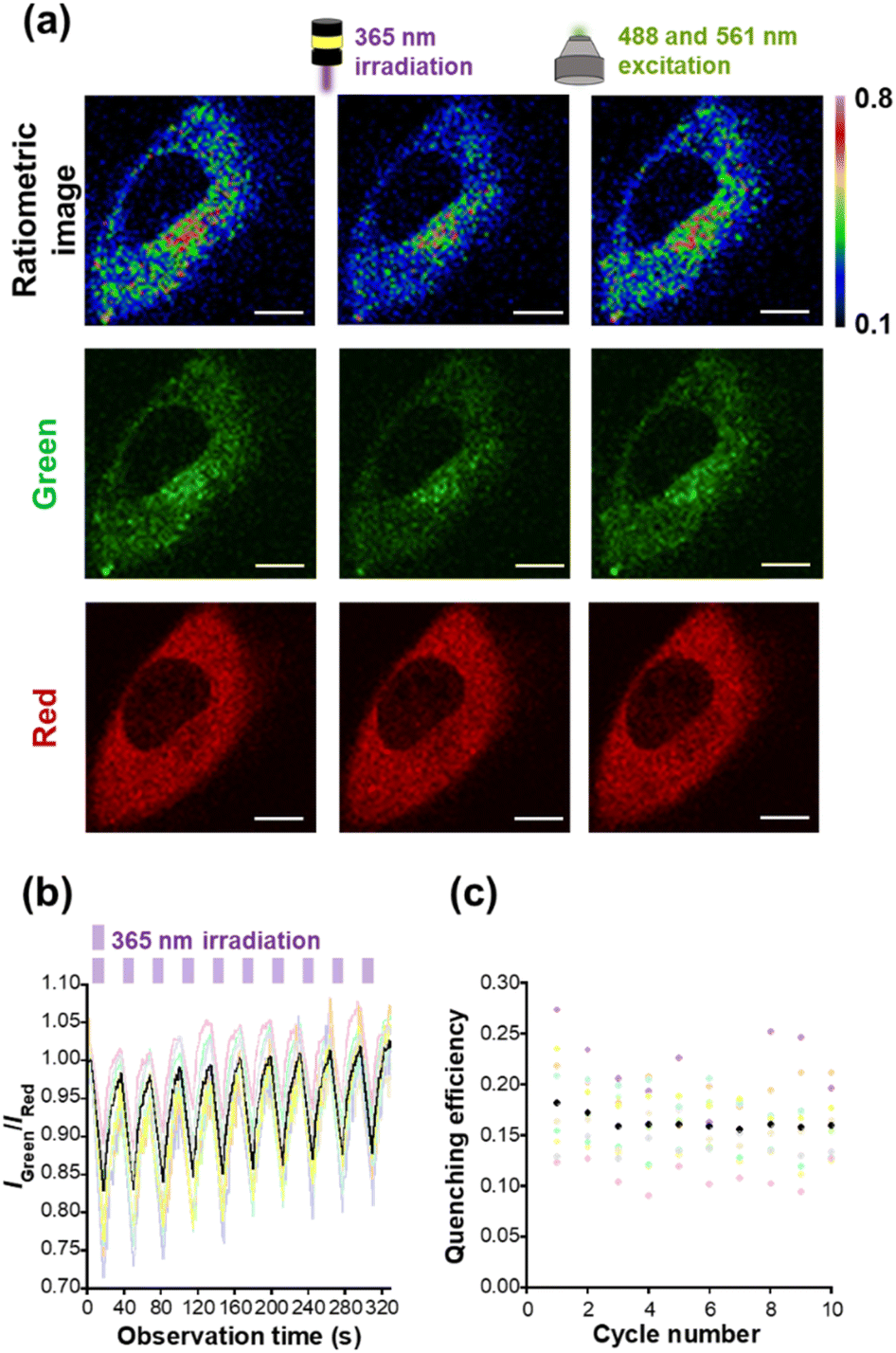 | ||
| Fig. 4 (a) Ratiometric imaging of living HeLa cells transfected with pcDNA3.1(+)–MBP–Halo–mCherry, using confocal microscope (Ti2-E). After adding 1.0 μM, HTL–Trp–BODIPY–FF (0.1% DMSO), cells were incubated for 20 min. Samples were alternately irradiated at 365 nm (10 mW cm−2 for 10 s) and 488 and 561 nm excitation light exposure (20 s). Scale bar: 10 μm. Green channel: λex = 488 nm, λem = 499–551 nm. Red channel: λex = 561 nm, λem = 571–625 nm. Ratiometric images were generated by dividing the intensity of green channel by that of red channel in each pixel. Maximum ratio: 0.8; minimum ratio: 0.1. (b) Time course of the ratio for fluorescence intensities of green channel to red channel with 365 nm irradiation (10 cycles). The fluorescence intensity ratio (IGreen/IRed) was obtained from a random selection of 10 cells in three different cell plates (the number of replicates: N = 3). (c) Quenching efficiency calculated from the data of (b). The black (b) line and (c) dots represent the average ratio intensity and quenching efficiency from ten different cells. Other pale-colored (b) lines and (c) dots describe each ratio intensity/quenching efficiency from each cell. | ||
We have also investigated the photoswitching behavior of unlabeled HTL–Trp–BODIPY–FF in non-transfected cells (Fig. S23†). The signal intensity of non-transfected cells is about 3 times lower than that of non-transfected cells (Fig. S23c†). Fig. S23d† shows representative photoswitching behavior of transfected and non-transfected cells in the same field of view (Fig. S23a and b†). As seen in Fig. S23e,† the fluorescence signals from non-transfected cells were repeatedly changed upon light irradiation.
To confirm the photoswitching performance for other targets in living cells, we have visualized both plasma membrane and mitochondria using the plasmids of pcDNA3.1(+)–Halo–EGFR–mCherry and pcDNA3.1(+)–Tom20–Halo–mCherry, respectively. We confirmed that the HTL–Trp–BODIPY–FF localized to the plasma membrane and mitochondria, respectively (Fig. S24 and S25†). Moreover, we have performed the photoirradiation experiments as with cytosol imaging. Both plasma membrane and mitochondria imaging shows reputative fluorescence switching upon light irradiations (Fig. S26 and S27†). These experiments demonstrated that our protein-surface-assisted strategy can be applied to live cell imaging and enables reversible fluorescence photoswitching in living cells.
Conclusions
In this study, we have developed a PSFM, named Trp–BODIPY–FF, which has both fluorogenic and photoswitching properties. Trp–BODIPY–FF exhibited fluorogenicity in hydrophobic environments including on protein-surfaces. Trp–BODIPY–FF enables reversible fluorescence switching upon repetitive light irradiation cycles when interacting with protein surfaces. To adapt Trp–BODIPY–FF to a protein labeling system for live-cell imaging, we have further developed a PSFM, named HTL–Trp–BODIPY–FF, which was created by the conjugation of a Halo-tag ligand to Trp–BODIPY–FF. HTL–Trp–BODIPY–FF successfully labeled Halo-tag with a significant increase in fluorescence intensity. Importantly, HTL–Trp–BODIPY–FF exhibits reversible fluorescence photoswitching in Halo-tag expressing live cells upon light irradiation and for more than 10 cycles. This work is the first example of a photoswitchable fluorogenic probe being used as a Halo tag system for reversible fluorescence control in living cells. Although there are concerns about the phototoxic effects of UV light irradiation on live cells with visible light responsive photochromic molecules such as indolyl fulgides,38 hemi-indigos,54 spironaphthoxazines,55 and azobenzene derivatives,56,57 we believe that these molecules have the potential to reduce the phototoxicity of PSFMs. The switching contrast of Trp–BODIPY–FF derivatives is still challenging for our final purpose of super-resolution imaging due to the low fluorescence switching contrast between ON and OFF state. Even though this drawback, we have demonstrated that synthetic PSFMs exhibit excellent fluorogenicity and photoswitching efficiency that is superior to representative photoswitchable fluorescent proteins applicable for super-resolution imaging.We demonstrated the methodology for reversible fluorescence control using no-wash fluorogenic labeling system. It is in principle applicable to other protein tags for versatile live-cell imaging technology. For example, SNAP58 and CLIP tag59 which may provide hydrophobic environments are also applicable for using our fluorogenic labeling system if the ligand is modified for each protein-tag. Moreover, we can also explore the fluorogenic fluorophores that can bind in the hydrophobic cavity of specific proteins as seen in PYP-tag60 and would provide more compact probe designs. In conclusion, this study demonstrates a new scaffold for fluorescence photoswitching and opens avenues in the design of future live-cell super-resolution imaging.
Data availability
The data supporting the findings of this study can be found in both the main text and the ESI.†Author contributions
All authors contributed to the preparation of the manuscript. All authors approved the final version of the manuscript.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
This work was supported by the JSPS KAKENHI (Grant Numbers: JP17H06409 “Frontier Research on Chemical Communications”, JP18H03935, JP19K22255, and JP21H04706 to K. K. and JP20H02879, JP21K19048, and JP21H05075 to Y. H.), JSPS A3 Foresight Program, JSPS Asian CORE Program, “Asian Chemical Biology Initiative”, Japan (JSPS)-UK (RSC) Research Cooperative Program (JPJSBP120195705 to K. K.), AMED-CREST, Toray Science Foundation (19-6008), Grant-in-Aid for JSPS Fellows (JP21J20593 to K. T.), the Uehara Memorial Foundation to Y. H., and Grant-in-Aid for Transformative Research Area (A)“Latent Chemical Space” (23H04880 and 23H004881 to K. K.) from the Ministry of Education, Culture, Sports, Science and Technology, Japan. M. V. acknowledges funds from EPSRC (EP/W015706/1) and an ERC Consolidator Grant (DYNAFLUORS, 771443). We are grateful to the Nikon Imaging Center at Osaka University for being very helpful with confocal microscopy (Ti2-E), image acquisition, and analysis (NIS-Elements software).Notes and references
- D. M. Chudakov, V. V. Verkhusha, D. B. Staroverov, E. A. Souslova, S. Lukyanov and K. A. Lukyanov, Nat. Biotechnol., 2004, 22, 1435–1439 CrossRef CAS PubMed.
- J. Kwon, J. Hwang, J. Park, G. R. Han, K. Y. Han and S. K. Kim, Sci. Rep., 2015, 5, 17804 CrossRef CAS PubMed.
- B. Roubinet, M. L. Bossi, P. Alt, M. Leutenegger, H. Shojaei, S. Schnorrenberg, S. Nizamov, M. Irie, V. N. Belov and S. W. Hell, Angew. Chem., Int. Ed., 2016, 55, 15429–15433 CrossRef CAS PubMed.
- X. Chai, H.-H. Han, A. C. Sedgwick, N. Li, Y. Zang, T. D. James, J. Zhang, X.-L. Hu, Y. Yu, Y. Li, Y. Wang, J. Li, X.-P. He and H. Tian, J. Am. Chem. Soc., 2020, 142, 18005–18013 CrossRef CAS PubMed.
- A. T. Frawley, K. G. Leslie, V. Wycisk, S. Galiani, D. Shrestha, C. Eggeling and H. L. Anderson, ChemPhysChem, 2023, e202300125 CrossRef CAS PubMed.
- A. T. Frawley, V. Wycisk, Y. Xiong, S. Galiani, E. Sezgin, I. Urbančič, A. V. Jentzsch, K. G. Leslie, C. Eggeling and H. L. Anderson, Chem. Sci., 2020, 11, 8955–8960 RSC.
- J. B. Grimm, B. P. English, H. Choi, A. K. Muthusamy, B. P. Mehl, P. Dong, T. A. Brown, J. Lippincott-Schwartz, Z. Liu, T. Lionnet and L. D. Lavis, Nat. Methods, 2016, 13, 985–988 CrossRef CAS PubMed.
- M. Weber, T. A. Khan, L. J. Patalag, M. Bossi, M. Leutenegger, V. N. Belov and S. W. Hell, Chem.–Eur. J., 2021, 27, 451–458 CrossRef CAS PubMed.
- X. Zhang, D. Guan, Y. Liu, J. Liu, K. Sun, S. Chen, Y. Zhang, B. Zhao, T. Zhai, Y. Zhang, F. Li and Q. Liu, Angew. Chem., Int. Ed., 2022, 61, e202211767 CrossRef CAS PubMed.
- Y. Zhang, Y. Zheng, A. Tomassini, A. K. Singh and F. M. Raymo, ACS Appl. Opt. Mater., 2023, 1, 640–651 CrossRef CAS PubMed.
- A. Aktalay, T. A. Khan, M. L. Bossi, V. N. Belov and S. W. Hell, Angew. Chem., Int. Ed., 2023, 62, e202302781 CrossRef CAS PubMed.
- R. Lincoln, M. L. Bossi, M. Remmel, E. D'Este, A. N. Butkevich and S. W. Hell, Nat. Chem., 2022, 14, 1013–1020 CrossRef CAS PubMed.
- M. Hofmann, C. Eggeling, S. Jakobs and S. W. Hell, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17565–17569 CrossRef CAS PubMed.
- S. Habuchi, R. Ando, P. Dedecker, W. Verheijen, H. Mizuno, A. Miyawaki and J. Hofkens, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 9511–9516 CrossRef CAS PubMed.
- T. Grotjohann, I. Testa, M. Leutenegger, H. Bock, N. T. Urban, F. Lavoie-Cardinal, K. I. Willig, C. Eggeling, S. Jakobs and S. W. Hell, Nature, 2011, 478, 204–208 CrossRef CAS PubMed.
- W. Sheng, S. T. Nick, E. M. Santos, X. Ding, J. Zhang, C. Vasileiou, J. H. Geiger and B. Borhan, Angew. Chem., Int. Ed., 2018, 57, 16083–16087 CrossRef CAS PubMed.
- L. Wang, M. S. Frei, A. Salim and K. Johnsson, J. Am. Chem. Soc., 2019, 141, 2770–2781 CrossRef CAS PubMed.
- Z. Ye, H. Yu, W. Yang, Y. Zheng, N. Li, H. Bian, Z. Wang, Q. Liu, Y. Song, M. Zhang and Y. Xiao, J. Am. Chem. Soc., 2019, 141, 6527–6536 CrossRef CAS PubMed.
- Y. Zheng, Z. Ye, X. Zhang and Y. Xiao, J. Am. Chem. Soc., 2023, 145, 5125–5133 CrossRef CAS PubMed.
- K. Uno, M. L. Bossi, M. Irie, V. N. Belov and S. W. Hell, J. Am. Chem. Soc., 2019, 141, 16471–16478 CrossRef CAS PubMed.
- D. Kim, A. Aktalay, N. Jensen, K. Uno, M. L. Bossi, V. N. Belov and S. W. Hell, J. Am. Chem. Soc., 2022, 144, 14235–14247 CrossRef CAS PubMed.
- K. Uno, A. Aktalay, M. L. Bossi, M. Irie, V. N. Belov and S. W. Hell, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2100165118 CrossRef CAS PubMed.
- K. Uno, M. L. Bossi, V. N. Belov, M. Irie and S. W. Hell, Chem. Commun., 2020, 56, 2198–2201 RSC.
- K. Torii, Y. Hori, K. Watabe and K. Kikuchi, Bull. Chem. Soc. Jpn., 2020, 93, 821–824 CrossRef CAS.
- K. Torii, Y. Hori and K. Kikuchi, Anal. Chem., 2023, 95, 8834–8841 CrossRef CAS PubMed.
- K. Ally, L. Öztürk and M. Köse, Turk. J. Chem., 2020, 44, 1031–1042 CrossRef CAS PubMed.
- G. Lukinavičius, L. Reymond, K. Umezawa, O. Sallin, E. D'Este, F. Göttfert, H. Ta, S. W. Hell, Y. Urano and K. Johnsson, J. Am. Chem. Soc., 2016, 138, 9365–9368 CrossRef PubMed.
- L. Wang, M. Tran, E. D'Este, J. Roberti, B. Koch, L. Xue and K. Johnsson, Nat. Chem., 2020, 12, 165–172 CrossRef CAS PubMed.
- Y. Hori, T. Norinobu, M. Sato, K. Arita, M. Shirakawa and K. Kikuchi, J. Am. Chem. Soc., 2013, 135, 12360–12365 CrossRef CAS PubMed.
- G. V. Los, L. P. Encell, M. G. McDougall, D. D. Hartzell, N. Karassina, C. Zimprich, M. G. Wood, R. Learish, R. F. Ohana, M. Urh, D. Simpson, J. Mendez, K. Zimmerman, P. Otto, G. Vidugiris, J. Zhu, A. Darzins, D. H. Klaubert, R. F. Bulleit and K. V. Wood, ACS Chem. Biol., 2008, 3, 373–382 CrossRef CAS PubMed.
- L. Mendive-Tapia, C. Zhao, A. R. Akram, S. Preciado, F. Albericio, M. Lee, A. Serrels, N. Kielland, N. D. Read, R. Lavilla and M. Vendrell, Nat. Commun., 2016, 7, 10940 CrossRef CAS PubMed.
- L. Mendive-Tapia, R. Subiros-Funosas, C. Zhao, F. Albericio, N. D. Read, R. Lavilla and M. Vendrell, Nat. Protoc., 2017, 12, 1588–1619 CrossRef CAS PubMed.
- R. Subiros-Funosas, V. C. L. Ho, N. D. Barth, L. Mendive-Tapia, M. Pappalardo, X. Barril, R. Ma, C.-B. Zhang, B.-Z. Qian, M. Sintes, O. Ghashghaei, R. Lavilla and M. Vendrell, Chem. Sci., 2020, 11, 1368–1374 RSC.
- L. Mendive-Tapia, L. Miret-Casals, N. D. Barth, J. Wang, A. de Bray, M. Beltramo, V. Robert, C. Ampe, D. J. Hodson, A. Madder and M. Vendrell, Angew. Chem., Int. Ed., 2023, 62, e202302688 CrossRef CAS PubMed.
- L. Mendive-Tapia, D. Mendive-Tapia, C. Zhao, D. Gordon, S. Benson, M. J. Bromley, W. Wang, J. Wu, A. Kopp, L. Ackermann and M. Vendrell, Angew. Chem., Int. Ed., 2022, 61, e202117218 CrossRef CAS PubMed.
- N. D. Barth, R. Subiros-Funosas, L. Mendive-Tapia, R. Duffin, M. A. Shields, J. A. Cartwright, S. T. Henriques, J. Sot, F. M. Goñi, R. Lavilla, J. A. Marwick, S. Vermeren, A. G. Rossi, M. Egeblad, I. Dransfield and M. Vendrell, Nat. Commun., 2020, 11, 4027 CrossRef CAS PubMed.
- R. Subiros-Funosas, L. Mendive-Tapia, J. Sot, J. D. Pound, N. Barth, Y. Varela, F. M. Goñi, M. Paterson, C. D. Gregory, F. Albericio, I. Dransfield, R. Lavilla and M. Vendrell, Chem. Commun., 2017, 53, 945–948 RSC.
- D. Lachmann, R. Lahmy and B. König, Eur. J. Org Chem., 2019, 2019, 5018–5024 CrossRef CAS.
- Y. Yokoyama, Chem. Rev., 2000, 100, 1717–1740 CrossRef CAS PubMed.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- H. Takakusa, K. Kikuchi, Y. Urano, H. Kojima and T. Nagano, Chem.–Eur. J., 2003, 9, 1479–1485 CrossRef CAS PubMed.
- X. Liu, W. Chi, Q. Qiao, S. V. Kokate, E. P. Cabrera, Z. Xu, X. Liu and Y.-T. Chang, ACS Sens., 2020, 5, 731–739 CrossRef CAS PubMed.
- H. Sunahara, Y. Urano, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2007, 129, 5597–5604 CrossRef CAS PubMed.
- C. Deo, A. S. Abdelfattah, H. K. Bhargava, A. J. Berro, N. Falco, H. Farrants, B. Moeyaert, M. Chupanova, L. D. Lavis and E. R. Schreiter, Nat. Chem. Biol., 2021, 17, 718–723 CrossRef CAS PubMed.
- J. Wilhelm, S. Kühn, M. Tarnawski, G. Gotthard, J. Tünnermann, T. Tänzer, J. Karpenko, N. Mertes, L. Xue, U. Uhrig, J. Reinstein, J. Hiblot and K. Johnsson, Biochemistry, 2021, 60, 2560–2575 CrossRef CAS PubMed.
- M. M. A. Mitry, F. Greco and H. M. I. Osborn, Chem.–Eur. J., 2023, 29, e202203942 CrossRef CAS PubMed.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- R. J. Blizzard, D. R. Backus, W. Brown, C. G. Bazewicz, Y. Li and R. A. Mehl, J. Am. Chem. Soc., 2015, 137, 10044–10047 CrossRef CAS PubMed.
- A. Borrmann, S. Milles, T. Plass, J. Dommerholt, J. M. M. Verkade, M. Wießler, C. Schultz, J. C. M. van Hest, F. L. van Delft and E. A. Lemke, ChemBioChem, 2012, 13, 2094–2099 CrossRef CAS PubMed.
- K. Lang, L. Davis, S. Wallace, M. Mahesh, D. J. Cox, M. L. Blackman, J. M. Fox and J. W. Chin, J. Am. Chem. Soc., 2012, 134, 10317–10320 CrossRef CAS PubMed.
- S. Duwé, E. De Zitter, V. Gielen, B. Moeyaert, W. Vandenberg, T. Grotjohann, K. Clays, S. Jakobs, L. Van Meervelt and P. Dedecker, ACS Nano, 2015, 9, 9528–9541 CrossRef PubMed.
- R. Ando, H. Mizuno and A. Miyawaki, Science, 2004, 306, 1370–1373 CrossRef CAS PubMed.
- T. Konen, D. Stumpf, T. Grotjohann, I. Jansen, M. Bossi, M. Weber, N. Jensen, S. W. Hell and S. Jakobs, ACS Nano, 2021, 15, 9509–9521 CrossRef CAS PubMed.
- D. V. Berdnikova, Chem. Commun., 2019, 55, 8402–8405 RSC.
- Y. Xiong, A. V. Jentzsch, J. W. M. Osterrieth, E. Sezgin, I. V. Sazanovich, K. Reglinski, S. Galiani, A. W. Parker, C. Eggeling and H. L. Anderson, Chem. Sci., 2018, 9, 3029–3040 RSC.
- J. Volarić, J. Buter, A. M. Schulte, K.-O. van den Berg, E. Santamaría-Aranda, W. Szymanski and B. L. Feringa, J. Org. Chem., 2022, 87, 14319–14333 CrossRef PubMed.
- M. Gao, D. Kwaria, Y. Norikane and Y. Yue, Nat. Sci., 2023, 3, e220020 CrossRef CAS.
- A. Keppler, S. Gendreizig, T. Gronemeyer, H. Pick, H. Vogel and K. Johnsson, Nat. Biotechnol., 2003, 21, 86–89 CrossRef CAS PubMed.
- A. Gautier, A. Juillerat, C. Heinis, I. R. Corrêa, M. Kindermann, F. Beaufils and K. Johnsson, Chem. Biol., 2008, 15, 128–136 CrossRef CAS PubMed.
- N. Kumar, Y. Hori, M. Nishiura and K. Kikuchi, Chem. Sci., 2020, 11, 3694–3701 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04953a |
| This journal is © The Royal Society of Chemistry 2024 |