
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced solid-state phosphorescence of organoplatinum π-systems by ion-pairing assembly†
Yohei
Haketa
 a,
Kaifu
Komatsu
a,
Hiroi
Sei
b,
Hiroki
Imoba
b,
Wataru
Ota
c,
Tohru
Sato
de,
Yu
Murakami
a,
Hiroki
Tanaka
a,
Nobuhiro
Yasuda
f,
Norimitsu
Tohnai
b and
Hiromitsu
Maeda
*a
a,
Kaifu
Komatsu
a,
Hiroi
Sei
b,
Hiroki
Imoba
b,
Wataru
Ota
c,
Tohru
Sato
de,
Yu
Murakami
a,
Hiroki
Tanaka
a,
Nobuhiro
Yasuda
f,
Norimitsu
Tohnai
b and
Hiromitsu
Maeda
*a
aDepartment of Applied Chemistry, College of Life Sciences, Ritsumeikan University, Kusatsu 525-8577, Japan. E-mail: maedahir@ph.ritsumei.ac.jp
bDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, Suita 565-0871, Japan
cMOLFEX, Inc., Kyoto 606-8103, Japan
dFukui Institute for Fundamental Chemistry, Kyoto University, Kyoto 606-8103, Japan
eDepartment of Molecular Engineering, Graduate School of Engineering, Kyoto University, Kyoto 615-8510, Japan
fBeamline Division, Japan Synchrotron Radiation Research Institute, Sayo 679-5198, Japan
First published on 5th December 2023
Abstract
Anion binding and ion pairing of dipyrrolyldiketone PtII complexes as anion-responsive π-electronic molecules resulted in photophysical modulations, as observed in solid-state phosphorescence properties. Modifications to arylpyridine ligands in the PtII complexes significantly impacted the assembling behaviour and photophysical properties of anion-free and anion-binding (ion-pairing) forms. The PtII complexes, in the presence of guest anions and their countercations, formed various anion-binding modes and ion-pairing assembled structures depending on constituents and forms (solutions and crystals). The PtII complexes emitted strong phosphorescence in deoxygenated solutions but showed extremely weak phosphorescence in the solid state owing to self-association. In contrast, the solid-state ion-pairing assemblies with tetraalkylammonium cations exhibited enhanced phosphorescence owing to the formation of hydrogen-bonding 1D-chain PtII complexes dispersed by stacking with aliphatic cations. Theoretical studies revealed that the enhanced phosphorescence in the solid-state ion-pairing assembly was attributed to preventing the delocalisation of the electron wavefunction over PtII complexes.
Introduction
π-Electronic molecules in ordered arrangements demonstrate fascinating electronic and electrooptical properties that are not observed in single molecules.1 Introducing multiple building units in assemblies induces properties that can be modulated by constituent species. Among organic materials, solid-state luminescent materials have received significant attention owing to their applications in light-emitting diodes, sensors and photonics.2 In particular, crystals of square-planar organoplatinum(II) complexes exhibit fascinating photoluminescence properties,3,4 such as triplet energy transfer and phosphorescence anisotropy amplification.4h,j The solid-state luminescence of π-electronic species is frequently quenched or weakened by self-association (Fig. 1a left), primarily because of exciton coupling between neighbouring molecules.5,6 To prevent this, introducing bulky groups to interfere with the molecular contact is an effective strategy for enhancing solid-state luminescence by electronic decoupling of organoplatinum π-systems.4c,e,j,7 Therefore, appropriate isolation arrangements for emissive species have been highly demanded. Furthermore, it is important to maintain rigid structures to decrease the rates of non-radiative decay processes that interfere with phosphorescent emission.3c,8 Thus, both isolation and robustness in packing structures are required for effective luminescence.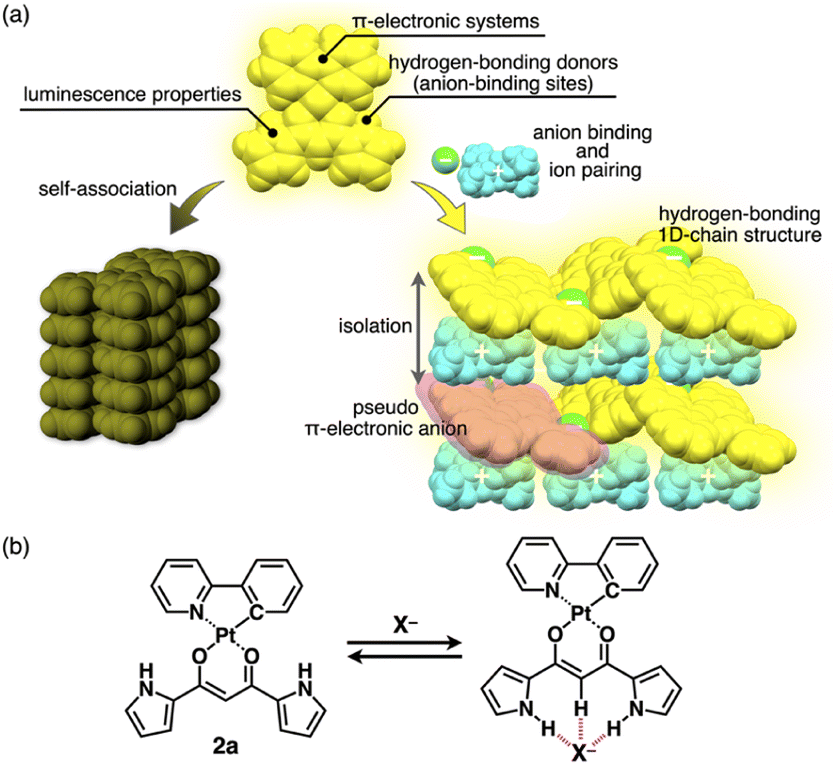 | ||
| Fig. 1 (a) Conceptual diagram of anion binding and ion pairing for emission enhancement: stacking structure of anion-responsive luminescent molecules (left) and charge-by-charge assembly with hydrogen-bonding 1D-chain structures of receptor–anion complexes as pseudo π-electronic anions (right) and (b) anion complexation of dipyrrolyldiketone PtII complex 2a. | ||
Ion pairs, which comprise complementarily associated positively and negatively charged species, can be employed for an isolation strategy.9,10 Isolation and electronic decoupling of luminescent cations have been achieved by introducing bulky counteranions.11 Furthermore, ion complexation by ion-responsive π-electronic molecules (receptors) followed by pairing with counterions is appropriate for preparing π-electronic ion pairs. This method can be used to create pseudo-π-electronic anions in the form of receptor–anion complexes. Spatially and electronically isolated charged fluorophores (cationic dyes) exhibit fluorescence emission in the solid state when combined with well-designed counter species (anion complexes).12 In contrast, including photo-functional anion-responsive π-electronic systems would also provide luminescent crystalline states through anion complexation with hydrogen bonding and alternate stacking with countercations (Fig. 1a right). The anion complexation would give rise to rigid structures by hydrogen bonds tightly connecting building units (pseudo-π-electronic anions comprising luminescent receptors) with the support of the charge-by-charge arrangement by electrostatic and dispersion forces. Designing luminescent anion receptors and combining them with countercations for isolation enable the control of the photophysical properties.
As luminescent π-electronic systems, dipyrrolyldiketone PtII complexes (e.g., 2a, Fig. 1b), with phenylpyridine (ppy) as a C^N ligand, have been synthesized as phosphorescent anion sensors, exhibiting absorption and emission maxima (λmax and λem, respectively) at 410 and 510 nm, respectively, with an emission quantum yield (Φem) of 42% for 2a in CH2Cl2.13 Planar geometries around PtII with a ppy ligand are suitable for stacking structures by themselves and in the form of ion-pairing assemblies.14 The PtII complexes would exhibit luminescent properties according to the introduced C^N ligands in solution and ion-pairing assemblies. Furthermore, the solid-state luminescent properties, whose control by countercations is also challenging, have not been elucidated. In this study, enhanced solid-state phosphorescence was achieved by ion pairing and the isolation of emissive anion complex moieties by countercations, for diverse PtII complexes.
Results and discussion
Synthesis and characterization of PtII complexes
Four different C^N ligands were used to produce PtII complexes 2b–e in 7.0–27% yields by treating dipyrrolyldiketone 1 with the mixture of the ligands and [(PtMe2)2(SMe2)2]15 at r.t. in the presence of trifluoromethanesulfonic acid (TfOH) (1 equiv.) and K2CO3 (1.5 equiv.) (Fig. 2). The obtained PtII complexes were characterized by 1H and 13C NMR and ESI-TOF-MS. Conformations without pyrrole inversions (py0i conformations) were suggested by the theoretically optimized structures: doubly pyrrole-inverted (py2i) conformations of 2b–e, suitable for anion binding, were less stable by 6.84, 6.79, 7.10 and 6.15 kcal mol−1, respectively, due to the enhanced molecular electric dipoles.16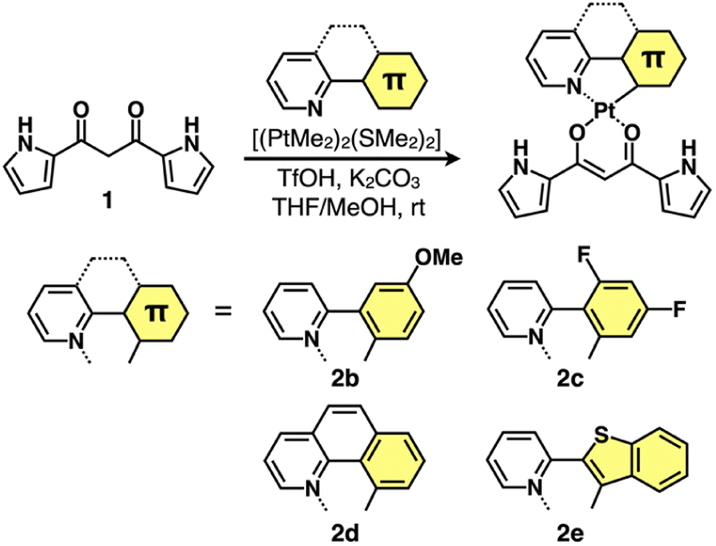 | ||
| Fig. 2 Synthesis of dipyrrolyldiketone PtII complexes 2b–e. | ||
Solution-state properties
The UV/vis absorption spectra of 2b–e in CH2Cl2 exhibited the λmax at ∼310–450 nm, and the spectral features, λmax values and absorbance intensities, were complicated depending on the introduced ligand moieties (Fig. 3 and Table 1). For example, 2b displayed the λmax at 368, 390 and 412 nm, and similar spectral features were observed in 2c,d, whereas 2e exhibited a distinctive spectrum with the λmax at 388 and 409 nm along with those at 318 and 455 nm. Similarly to 2a,13 the main absorption bands of 2b–e were assigned as the lowest-lying singlet states, originating primarily from the HOMO-to-LUMO transition with a significant contribution from ligand-to-ligand charge transfer (LLCT) from the dipyrrolyldiketone unit to the arylpyridine ligands and metal-to-ligand charge transfer (MLCT) from PtII to arylpyridine ligands; the theoretical study was conducted using time-dependent (TD)-DFT calculations at the CAM-B3LYP level using the 6-31+G(d,p) basis set with the LanL2DZ basis set and associated effective core potentials for Pt, which were used for the calculations in the following parts.16 Furthermore, characteristic absorption bands of 2e at 318 and 455 nm can be attributed to the π–π* transition of the benzothienyl-pyridine ligand and intraligand charge transfer from the π orbitals of the benzo unit to the π* orbitals of the pyridyl group, respectively.17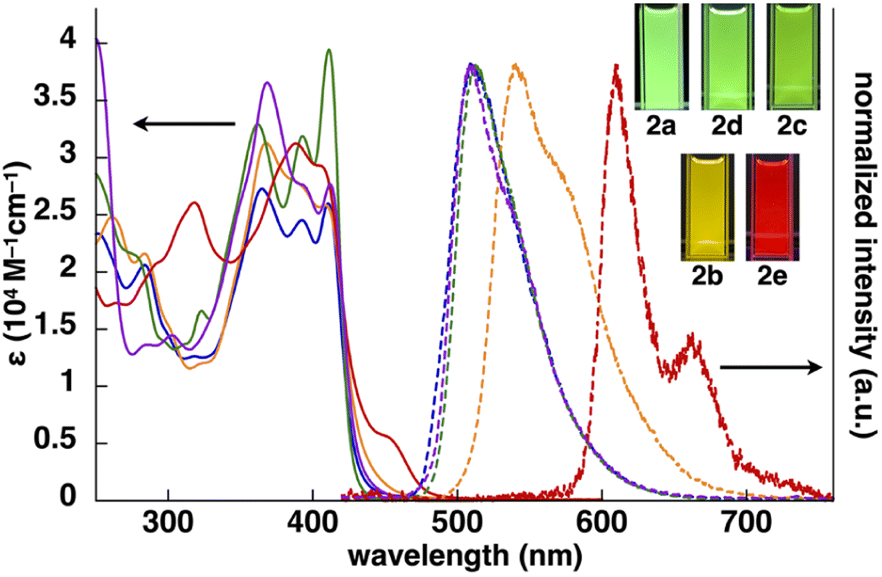 | ||
| Fig. 3 UV/vis absorption spectra (solid lines, CH2Cl2) and normalized emission spectra (dashed lines, deoxygenated CH2Cl2) with the excitation at 410, 368, 411, 368 and 388 nm of 2a (blue), 2b (orange), 2c (green), 2d (purple) and 2e (red) (inset: photographs of PtII complexes under UV365 (0.03 mM)). | ||
The phosphorescence spectra of 2b–e in deoxygenated CH2Cl2 exhibited broad emission bands with a λem of 542, 512, 508 and 610/662(sh) nm, respectively (Fig. 3 and Table 1), suggesting the red-shifted emissions for 2b,e compared to 2a. The Φem of 2b–d were 68%, 50% and 50%, respectively, which were greater than that of 2e (16%). The phosphorescence emissions of 2b–e originating from the triplet states were suggested by the emission lifetimes (τ) of 4.6, 9.6, 4.2 and 6.8 μs, respectively, similar to that of 2a (5.1 μs).13 TD-DFT calculations for the optimized T1 structures at the PCM-M06-2X level in CH2Cl2 showed theoretically estimated emission maxima at 499, 524, 495 and 714 nm for 2b–e, respectively, which are close to the observed values (Fig. S56†). The phosphorescence emissions of 2c,e were mainly derived from the LUMO-to-HOMO (84% and 96%, respectively) transitions, whereas those of 2b,d were ascribed to the LUMO-to-HOMO−1 (68%) and LUMO+1-to-HOMO (60%) transitions, respectively.
Anion-binding behaviours were revealed by 1H NMR spectral changes in CD2Cl2 (1.0 mM) (Fig. S74–S77†). Upon the addition of 3.3 equiv. of tetrabutylammonium chloride (TBACl) to, as an example, 2c, the signals of the pyrrole NH and bridging CH at 9.35/9.31 and 6.41 ppm, respectively, at −50 °C disappeared, whereas the corresponding new signals appeared at 12.55/12.49 and 7.42 ppm, respectively. The downfield shifts were caused by hydrogen bonding with Cl−, implying the formation of [1 + 1]-type Cl− complexes, as demonstrated by theoretical studies (Fig. S39–S42†).16
The anion-binding constants (Ka) of 2b–e in a [1 + 1]-binding mode were evaluated by the changes in UV/vis absorption spectra caused by the addition of anions (Cl−, Br− and CH3CO2−) as TBA salts in CH2Cl2 (Table 2 and Fig. S70–S73†). In all the derivatives discussed in this study, the Ka values were in the order of CH3CO2− > Cl− > Br− correlating with the basicity. Cl− complexation produced phosphorescence emissions (λem at 542, 505, 511 and 610/660 nm for 2b–e, respectively) with Φem (54%, 32%, 41% and 14%, respectively) and τ (4.3–14.2 μs), which were comparable to those of 2a·Cl− (λem: 490/520 nm, Φem: 48%, τ: 2.4 μs) (Table 3). It should be noted that anion complexation of the PtII complexes in solution did not significantly affect the emission properties. PtII complexes with such emission properties can be used as building blocks for solid-state materials whose packing structures and resulting luminescent properties are modulated by anion complexation and ion pairing with countercations.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000
000Solid-state luminescence and packing structures
In contrast to the phosphorescence emission in the dispersed solution state, the solid-state PtII complexes obtained from the single crystals (vide infra) exhibited extremely weak luminescence: 2a–e exhibited λem at 523/682, 591, 673, 652 and 627 nm with Φem of 0.7%, ∼0.1%, 1.8%, ∼0.1% and ∼0.2%, respectively (Fig. S86–S90†).18 Such red-shifted emission with emission quenching can be caused by the excimer formation of excited PtII complexes,19 as theoretically discussed in the following section. Notably, the Φem of solid-state 2a–e are lower than those of 1,3-diphenyl-1,3-propanedione PtII complexes.4h The details of the assembled structures were revealed by X-ray analysis for the single crystals of 2b–e obtained by vapour diffusion of n-hexane into CH2Cl2 solutions (Fig. 4 and S19–S23†).202b–e exhibited planar pyrrole-non-inverted (py0i) conformations in stacking structures, similar to 2a,13 with mean-plane deviations (defined by the core atoms without hydrogen atoms) of 0.103–0.174 Å. The PtII complexes 2b–e formed columnar π–π stacking structures with stacking distances of 3.21–3.38 Å. Among them, 2b,e showed stacked-dimer alignment with Pt⋯Pt distances of 3.46 and 3.29 Å, respectively. The π–π stacking structures were identified by Hirshfeld surface analysis (Fig. S33–S37†).20 There are no notable intermolecular interactions on the lateral side of the columnar π–π stacking structures except for 2c, which showed N–H⋯π interactions between pyrrole rings.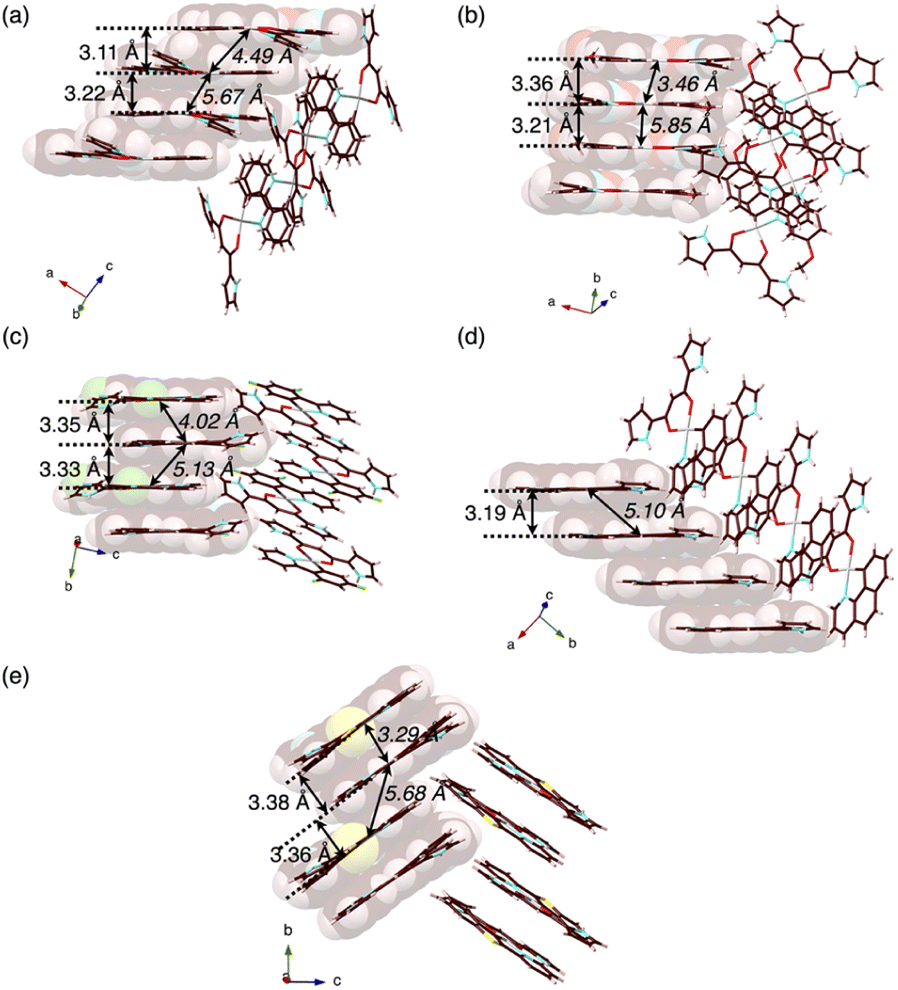 | ||
| Fig. 4 Single-crystal X-ray structures, with selected stacking and Pt⋯Pt (italic) distances, of (a) 2a as a reference,13 and (b) 2b, (c) 2c, (d) 2d and (e) 2e as packing diagrams. Atom colour codes in Fig. 4 and the following figures: brown, pink, blue, red, yellow green, yellow and grey refer to carbon, hydrogen, nitrogen, oxygen, fluorine, sulfur and platinum, respectively. | ||
The extremely weak crystal-state emissive properties were modulated by isolating the PtII complexes without stacking by themselves. Single crystals of the ion pairs 2a·Cl−-TBA+ and 2a·Cl−-TPeA+ (TPeA+: tetrapentylammonium) were prepared by vapour diffusion of n-hexane into the THF solutions of 2a and corresponding tetraalkylammonium salts (Fig. 5). The ion pair 2a·Cl−-TPA+ (TPA+: tetrapropylammonium) was also prepared as precipitates by adding n-hexane to a mixture of 2a and TPACl in CH2Cl2.21 Compositions of the Cl−-binding PtII complex and countercations in all the solid-state samples were fully characterized by using 1H NMR. In contrast to the anion-free states, solid-state 2a·Cl−-TPA+, 2a·Cl−-TBA+ and 2a·Cl−-TPeA+ exhibited phosphorescence with the λem at 654, 522 and 509 nm with Φem (relative intensities to the solid-state 2a) of 3.6% (5.1), 6.2% (8.9) and 2.6% (3.7), respectively, indicating enhanced phosphorescence properties (Table 4, Fig. 6a, b and S86†). The λem of 2a·Cl−-TBA+ and 2a·Cl−-TPeA+ were similar to the solution-state λem of 2a·Cl−, suggesting the phosphorescence derived from the monomeric Cl− complex as suggested by single-crystal packing structures (vide infra). Notably, 2a·Cl−-TPA+ exhibited a distinctive red-shifted emission. The emission lifetimes, such as 440 μs for 2a·Cl−-TPeA+, were nearly 200 times longer than those of the monomers in solution (Fig. S82†).
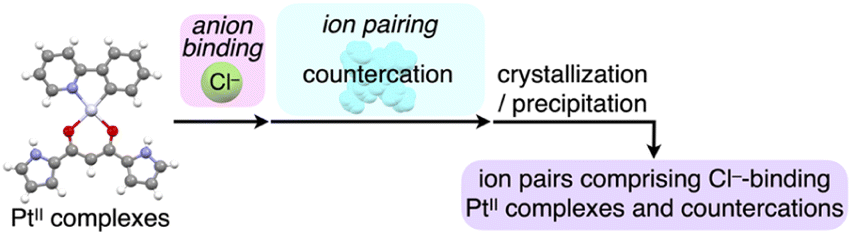 | ||
| Fig. 5 Preparation procedures of ion-pairing assemblies comprising Cl−-binding PtII complexes and countercations. | ||
| λ em [nm] | τ 1 [μs]/f1 [%] | τ 2 [μs]/f2 [%] | τ 3 [μs]/f3 [%] | Φ em [%] | |
|---|---|---|---|---|---|
| 2a | 523 | 140/31 | 550/69 | — | 0.7 |
| 2a·Cl−-TPA+ | 654 | 110/24 | 440/76 | — | 3.6 |
| 2a·Cl−-TBA+ | 522 | 2.9/43 | 23/25 | 210/32 | 6.2 |
| 2a·Cl−-TPeA+ | 509 | 6.3/8 | 86/24 | 440/68 | 2.6 |
 | ||
| Fig. 6 (a) (i) Photographs and (ii) quantum yields of solid-state 2a,132a·Cl−-TPA+, 2a·Cl−-TBA+ and 2a·Cl−-TPeA+, (b) solid-state emission spectra of 2a (grey), 2a·Cl−-TPA+ (red), 2a·Cl−-TBA+ (green) and 2a·Cl−-TPeA+ (yellow) and (c) single-crystal X-ray structures of (i) 2a·Cl−-TBA+ and (ii) 2a·Cl−-TPeA+ as top 1D chain and side packing structures. The atom colour code in (c): green (spherical) refers to chlorine. Magenta and cyan denote the receptor–Cl− complex 1D-chain and cation parts, respectively. For the crystals of 2a in (a), reflections of excitation light at UV365 nm were observed in partially pink colour. | ||
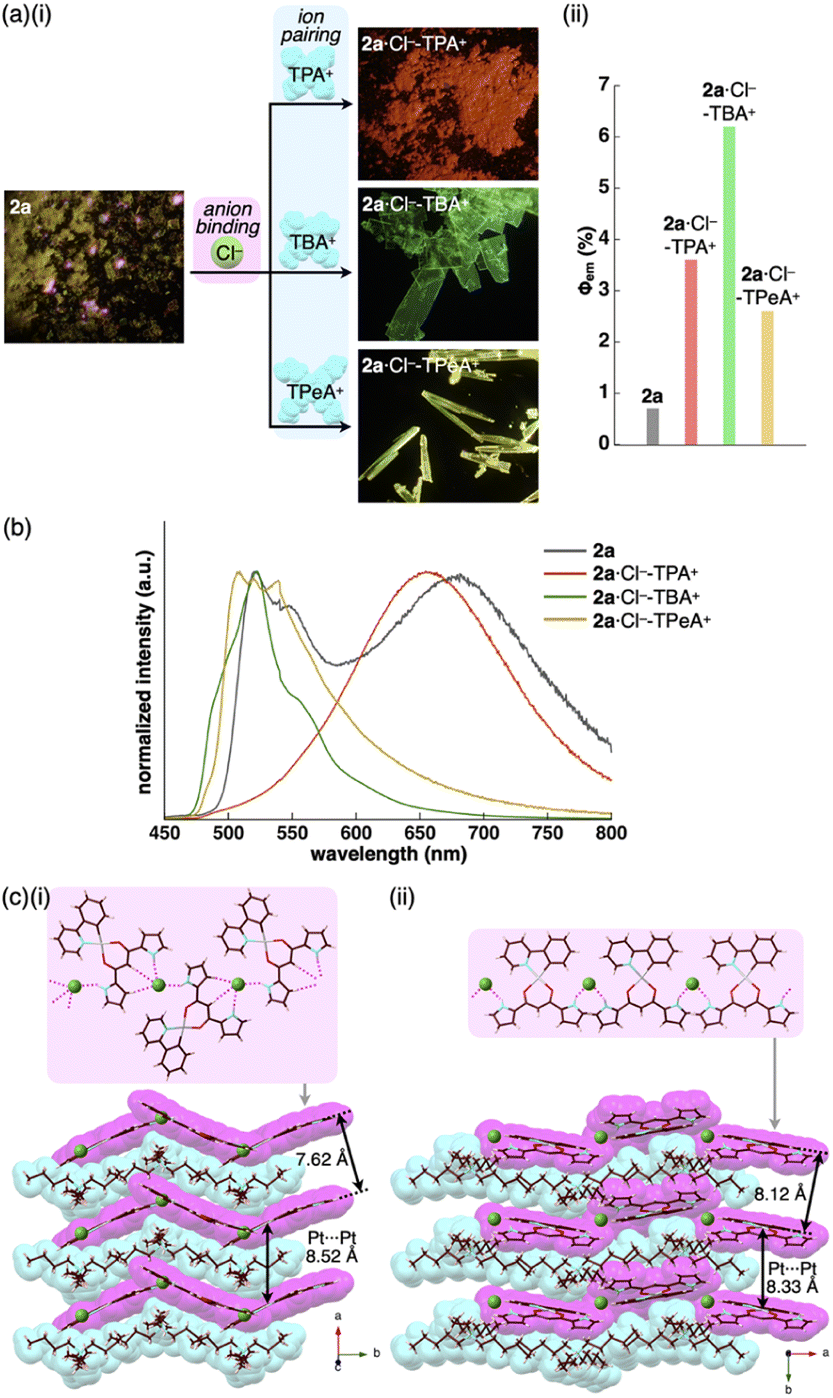
The enhanced phosphorescence intensities in the ion-pairing assemblies were investigated using solid-state packing structures revealed by single-crystal X-ray analysis. The ion pair 2a·Cl−-TBA+ exhibited an anion-binding mode with hydrogen bonding of the singly inverted pyrrole NH, bridging CH and pyrrole CH (Fig. 6c(i), S24† and Table 5). The Cl− also interacted with the pyrrole NH of neighbouring 2a, resulting in a Cl−-bridged 1D-chain structure based on the singly pyrrole-inverted (py1i) conformation. Importantly, 2a·Cl− and TBA+ are alternately arranged on the a-axis to form a charge-by-charge assembly with a Pt⋯Pt distance of 8.52 Å. In contrast, crystal-state 2a·Cl−-TPeA+ exhibited a packing structure with a py0i conformation (Fig. 6c(ii) and S25†). The pyrrole NHs of 2a formed hydrogen bonds independently, resulting in a Cl−-bridged chain structure. 2a·Cl− and TPeA+ were alternately arranged to form a charge-by-charge assembly with a Pt⋯Pt distance of 8.34 Å. The assembling modes, with different numbers of inverted pyrrole rings, are determined by the alkyl chain lengths of the countercations (TBA+ and TPeA+), resulting in different nearest Cl−···Cl− distances of 8.90 and 10.72 Å, respectively. Hirshfeld surface analysis of the crystal structures indicated no characteristic close contacts between the PtII complex and cation, suggesting that both cations exhibited similar interactions. This is also supported by the energy decomposition analysis (EDA) in the framework of the fragment molecular orbital method at the FMO2-MP2 using mixed basis sets including NOSeC-V-DZP with MCP with TZP for Pt, demonstrating that the dispersion forces were effective between the PtII complex and cations (Fig. S57 and S58†).22–25 Although the exact assembling structure for the precipitates of 2a·Cl−-TPA+ could not be determined by single-crystal X-ray analysis, synchrotron XRD analysis revealed no characteristic diffraction pattern, suggesting the less ordered arrangement of 2a·Cl− and TPA+ (Fig. S83†). The speculated slipped stacking of 2a·Cl− in the solid-state 2a·Cl−-TPA+ could be correlated with the red-shifted phosphorescence.
In any ion pairs of 2a·Cl−, the isolation of the PtII complexes by aliphatic cations, required for enhancing phosphorescence intensities, was clearly indicated by the X-ray structures of the ion-pairing assemblies. Furthermore, the rigidification of packing structures by Cl−-bridged 1D-chain structures is vital for enhancing the phosphorescence intensities. The larger Φem of 2a·Cl−-TBA+ than that of 2a·Cl−-TPeA+ can be ascribed to the stabilization of the 1D-chain structure by hydrogen bonding. The electrostatic energy, revealed by EDA, originating mainly from hydrogen-bonding interactions for the 2a⋯Cl−···2a structure in the 1D-chain of 2a·Cl−-TBA+ possessed a larger absolute value by 10.7 kcal mol−1 than that of 2a·Cl−-TPeA+ (Fig. S57 and S58†).
Charge-by-charge assemblies were observed in the single-crystal packing structures of 2c,d with tetraalkylammonium cations (Fig. 7a, S28–S31† and Table 6). A crystal of 2c·Cl−-TBA+, prepared from CH2ClCH2Cl/n-hexane, for example, demonstrated a py1i conformation with Cl− binding by pyrrole NH, β-CH and bridging CH. A Cl−-bridged chain structure similar to 2a·Cl−-TBA+ was formed by the interaction between uninverted pyrrole NH and neighbouring Cl−. In contrast, a single crystal of 2c·Cl−-TPeA+, prepared from CH2ClCH2Cl/n-hexane, exhibited a py0i conformation and a chain structure (Fig. 7b and S29†) as observed in 2a·Cl−-TPeA+. Furthermore, crystallization of 2d·Cl−-TBA+ from CH2Cl2/n-hexane (2d·Cl−-TBAM+) and CH2ClCH2Cl/n-hexane (2d·Cl−-TBAE+) produced two pseudo polymorphs, with 2d exhibiting py1i conformations that form Cl−-bridged chain structures in both (Fig. 7c and S31†). In all the ion-pairing assemblies revealed by X-ray analysis, PtII complexes in Cl−-bridged polymers were isolated by aliphatic cations in charge-by-charge arrangements.
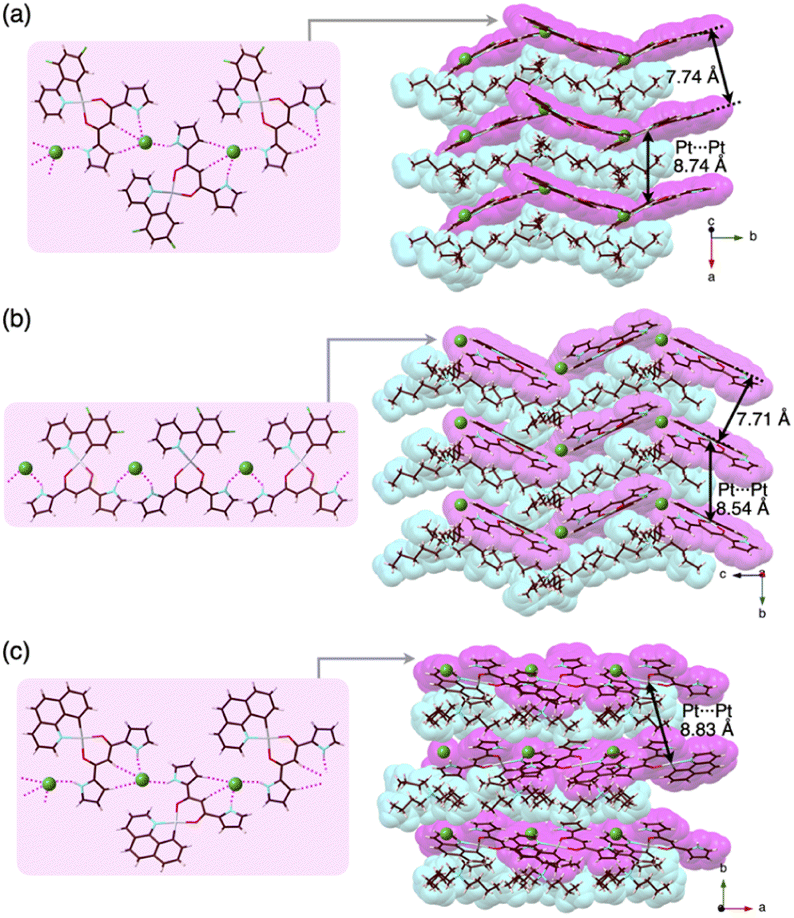 | ||
| Fig. 7 Single-crystal X-ray structures of (a) 2c·Cl−-TBA+, (b) 2c·Cl−-TPeA+ and (c) 2d·Cl−-TBA+E as top 1D-chain and side packing structures. | ||
| 2c·Cl−-TBA+ | 2c·Cl−-TPeA+ | 2d·Cl−-TBAE+ | |
|---|---|---|---|
| a Distances in charge-by-charge assemblies. b Distances in Cl−-bridged 1D-chain structures. | |||
| N(–H)⋯Cl− [Å] | 3.14, 3.18, 3.19, 3.20 | 3.14, 3.18 | 3.10, 3.16 |
| Cbridging(–H)⋯Cl− [Å] | 3.84, 3.85 | — | 3.82 |
| Cβ(–H)⋯Cl− [Å] | 3.65, 3.72 | — | 3.73 |
| Pt⋯Pta [Å] | 8.74 | 8.33 | 8.83 |
| Cl−···Cl−b [Å] | 9.01 | 10.94 | 8.87 |
As expected from the crystal packing structures, emission enhancement of the solid-state ion-pairing assemblies of 2c,d was observed, as was a similar tendency for 2a (Table 7, Fig. 8a, b, S88 and S89†). For example, solid-state ion pairs 2c·Cl−-TPA+,262c·Cl−-TBA+ and 2c·Cl−-TPeA+ showed the λem at 507, 504 and 515 nm with comparable and enhanced Φem of 1.7%, 3.2% and 2.4%, respectively, due to the formation of charge-by-charge assemblies.27 Similar to the XRD pattern of 2a·Cl−-TPA+, the synchrotron XRD pattern of 2d·Cl−-TPA+/TPeA+ precipitates exhibited less clear diffraction patterns with broad peaks, suggesting less ordered arrangements of 2d·Cl− and countercations. The Φem of 2d·Cl−-TPA+ was estimated to be 1.7%, which is greater than that of the other 2d·Cl− ion pairs. Substituents at π-electronic ligands controlled emissive properties, as observed in the red-shifted phosphorescence at 606–617 nm with enhanced quantum yields of 1.1–2.7% for the ion pairs of 2e·Cl− with tetraalkylammonium cations as precipitates (Table 7, Fig. 8c and S90†). Furthermore, the 2b·Cl−-TPA+ precipitate obtained from CH2Cl2/n-hexane exhibited enhanced phosphorescence with a Φem of 7.5%, which is 75 times greater than that of 2b (Fig. S87†).28 The solid-state phosphorescence properties, λem and Φem, in ion-pairing assemblies of the anion-responsive PtII complexes were modulated by the introduced π-electronic C^N ligands and coexisting cations.
| λ em [nm] | τ 1 [μs]/f1 [%] | τ 2 [μs]/f2 [%] | τ 3 [μs]/f3 [%] | Φ em [%] | |
|---|---|---|---|---|---|
| a Accurate values could not be determined due to low Φem. | |||||
| 2c | 673 | 61/90 | 450/10 | — | 1.8 |
| 2c·Cl−-TPA+ | 507 | 49/15 | 390/85 | — | 1.7 |
| 2c·Cl−-TBA+ | 504 | 6.3/41 | 80/14 | 410/44 | 3.2 |
| 2c·Cl−-TPeA+ | 515 | 7.7/10 | 57/25 | 390/65 | 2.4 |
| 2d | 652 | 120/28 | 500/72 | — | ∼0.1a |
| 2d·Cl−-TPA+ | 627 | 29/36 | 190/64 | — | 1.7 |
| 2d·Cl−-TBA+ | 563 | 120/26 | 490/74 | — | 0.7 |
| 2d·Cl−-TPeA+ | 594 | 30/17 | 330/83 | — | 0.8 |
| 2e | 627 | 100/21 | 440/79 | — | ∼0.2a |
| 2e·Cl−-TPA+ | 617 | 130/28 | 510/72 | — | 1.9 |
| 2e·Cl−-TBA+ | 606 | 130/27 | 510/73 | — | 1.1 |
| 2e·Cl−-TPeA+ | 606 | 96/18 | 420/82 | — | 2.7 |
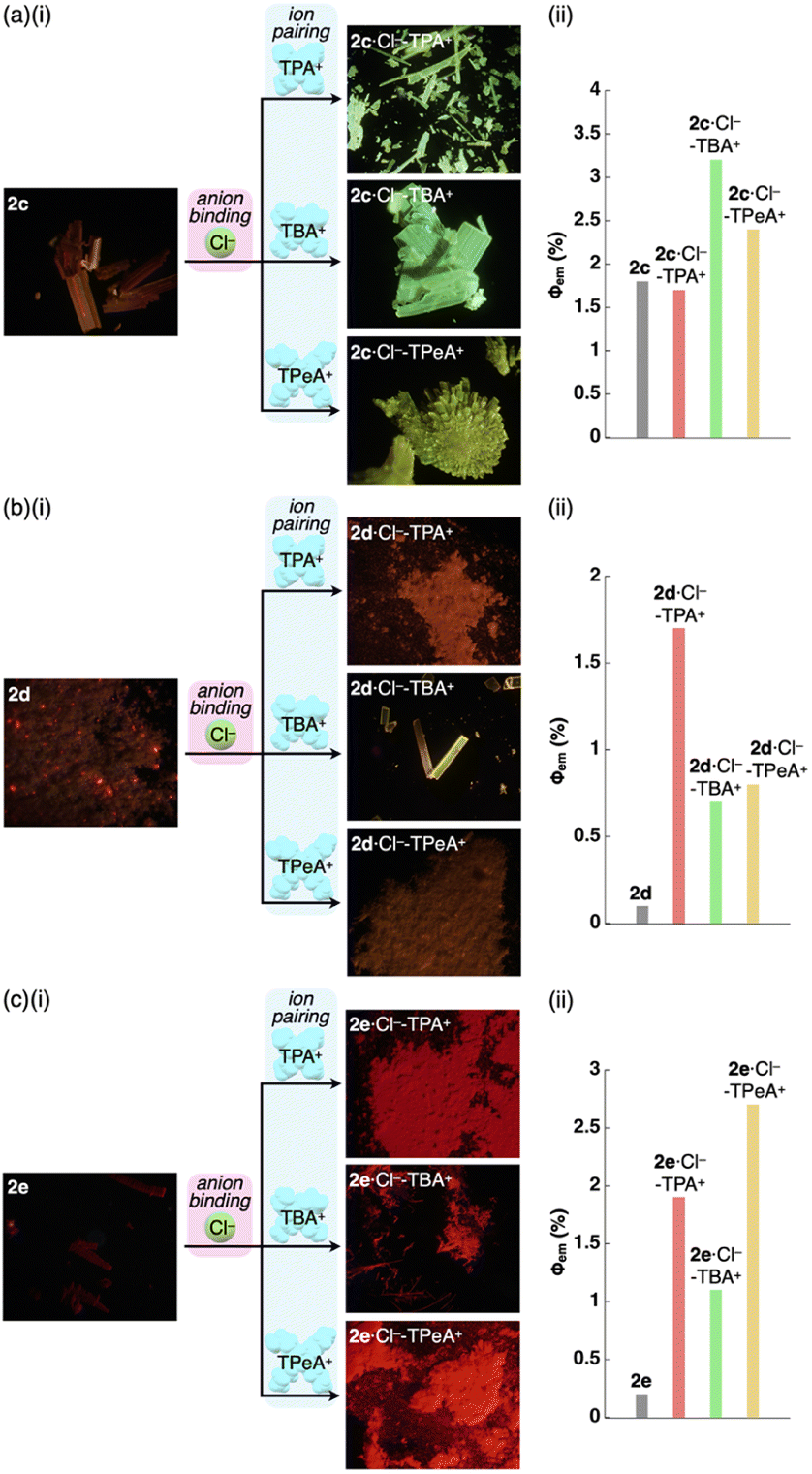 | ||
| Fig. 8 (i) Photographs and (ii) quantum yields of the PtII complexes (a) 2c, (b) 2d and (c) 2e and their Cl− complexes as ion pairs with TPA+, TBA+ (2d·Cl−-TBAE+ for 2d) and TPeA+ as solid-state samples. | ||
Mechanism for phosphorescence enhancement
Enhancing the phosphorescence in the ion-pairing assembly of PtII complexes was elucidated by DFT calculations based on an ONIOM approach29,30 for the packing structures of 2a and 2a·Cl−-TBA+. Computational models for solid-state 2a and 2a·Cl−-TBA+ were constructed by cutting out the 1 × 1 × 2 and 3 × 2 × 2 unit cells (Fig. S61 and S67†), where the centre parts and surroundings were treated as quantum mechanics (QM) regions at the DFT level and molecular mechanics (MM) regions at the UFF level, respectively. The electronic structures of the QM region were calculated at the CAM-B3LYP/6-31+G(d,p) and 3-21G with LanL2DZ for Pt in 2a and 2a·Cl−-TBA+, respectively. In both cases, the QM regions exhibited Ci site symmetry.The optimized S0 geometry of solid-state 2a showed pseudo-generate T1 (Ag) and T2 (Au), where Ag and Au denote the irreducible representations of the excited electronic states, as well as T3 (Au) and T4 (Ag) states, whose wavefunctions were symmetrically delocalized over the 2a dimer (Fig. S62†). The geometries of T1 (Ag) and T4 (Ag) were further optimized owing to the distribution of their wavefunctions at the dipyrrolyldiketone and ppy ligand, respectively. The optimized T1 and T4 states with lower symmetries of C1 owing to a symmetry breaking of pseudo-Jahn–Teller distortion31 exhibited the lowest excited states with excitation energies of 2.25 eV/551 nm and 1.95 eV/636 nm, respectively (Fig. S63†). The S0–T1 electron density difference at the T1 optimized structure (Fig. 9a(i)) demonstrated that T1 was an excited state localized on the single molecule. In contrast, the S0–T4 electron density difference at the T4 optimized structure (Fig. 9a(ii)) indicated that T4 was an excited state with the wavefunction asymmetrically delocalized over the dimer. The calculated phosphorescence spectrum from T1 reproduced the sharp shape of the experimental spectrum in the short-wavelength region, whereas that from T4 reproduced the broad shape in the long-wavelength region (Fig. S64†). The T4 state mainly comprised the HOMO–LUMO transition (the CI coefficient: 0.599) and HOMO-8–LUMO transition (0.234). The HOMO and HOMO-8 originated from the intermolecular interaction between the Pt dz2 orbital in one molecule and the π orbital of the ppy ligand in the other (Fig. 9b).32 Thus, this intermolecular interaction was responsible for the energetically stable T4 formation.
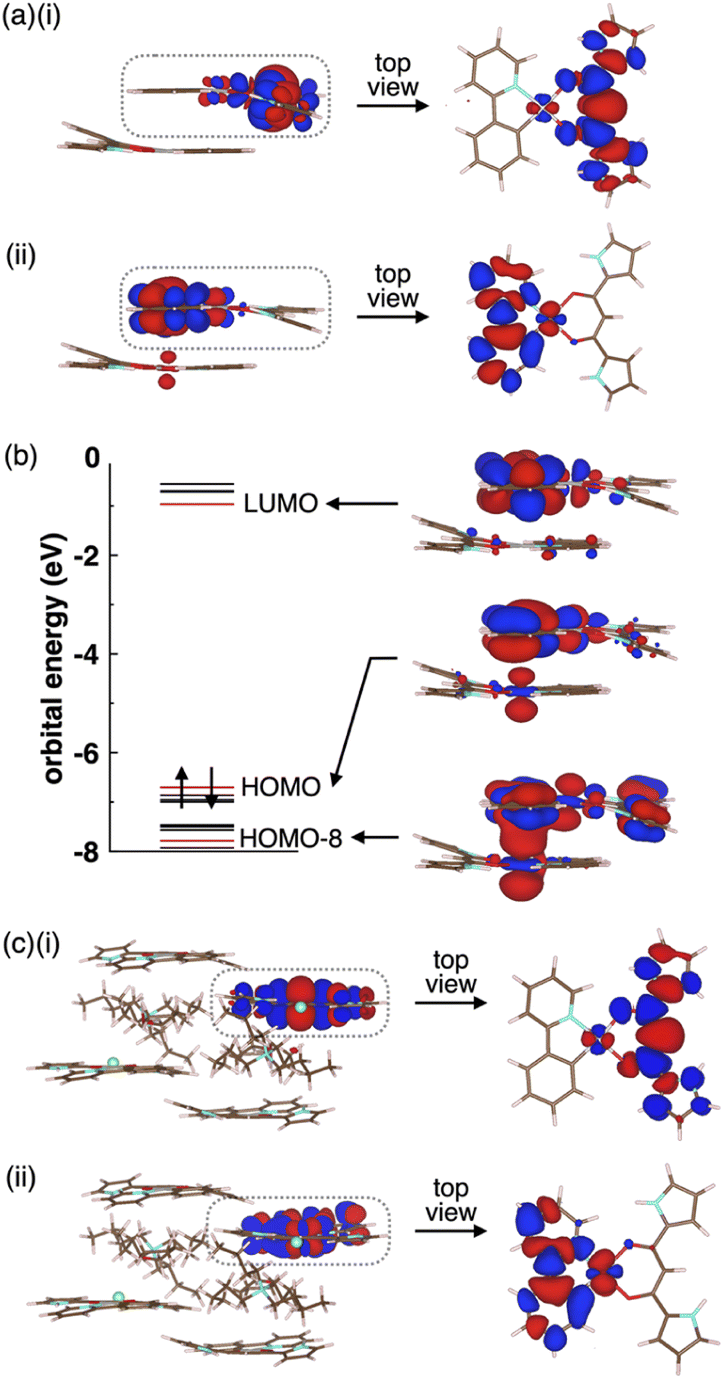 | ||
| Fig. 9 (a) Electron density differences for solid-state 2a (i) between S0 and T1 at the T1 optimized structure and (ii) between S0 and T4 at the T4 optimized structure (isosurface value: 5 × 10−4 a.u), (b) orbital levels and molecular orbitals at the T4 optimized structure (isosurface value: 2 × 10−2 a.u.) and (c) electron density differences for solid-state 2a·Cl−-TBA+ (i) between S0 and T1 at the T1 optimized structure and (ii) between S0 and T6 at the T6 optimized structure (isosurface value: 5 × 10−4 a.u.). The red and blue regions are positive and negative in electron density differences, respectively. Only the QM region is shown for simplicity. | ||
The Φem values depend on nonradiative rate constants from the excited to the ground states. The rate constant significantly increases with diagonal vibronic coupling constants (VCCs) of the final electronic state because of easier acceptance of electronic excitation energy.33 For the solid-state 2a, the diagonal VCCs of S0 at the T4 optimized structure were larger than those at the T1-optimized structure (Fig. S65†), suggesting that the nonradiative transition from the T4 state was fast. Vibronic coupling density (VCD)33,34 elucidated that the large VCCs arose from the strong coupling between the electronic state and vibrational modes distributed over the ppy ligand (Fig. S66†). Thus, the low Φem of the solid-state 2a was attributed to T4 with large diagonal VCCs.
The charge-by-charge packing structure significantly affected the electronic states of 2a. In 2a·Cl−-TBA+, the pseudo-degenerate T1 (Ag) and T2 (Au) as well as T3 (Au) and T4 (Ag) wavefunctions at the S0 optimized structure were distributed over the dipyrrolyldiketone unit, whereas the T5 (Au) and T6 (Ag) wavefunctions were distributed over the ppy ligand (Fig. S68†). The optimized geometries of T1 (Ag) and T6 (Ag) exhibited the wavefunctions localized on the single molecule by the pseudo-Jahn–Teller distortion (Fig. 9c and S69†). In contrast to the solid-state 2a, the electronic wavefunction was not delocalized over dimer for these states because of the presence of TBA+ between 2a·Cl−. The TBA+ in the charge-by-charge assembly clearly decoupled the electronic interaction, resulting in enhanced phosphorescence derived from the monomeric PtII complex. Although the Φem values of the ion-pairing assemblies are lower than those in the solution state because of the reduced interactions in the solvated monomeric forms, introduction of aliphatic cations is efficient for inhibiting the self-association and enhancing the phosphorescence properties.
Conclusions
An ion-pairing strategy has been applied for fabricating solid-state phosphorescent materials by isolating dipyrrolyldiketone PtII complexes, as emissive anion-responsive molecules, in the form of anion complexes with aliphatic cations. Charge compensation between the anion complexes, in anion-bridged rigid chain structures, and countercations interfered with self-association, resulting in enhanced phosphorescence emission. Solid-state arrangement of the PtII complexes and their photophysical properties were influenced by the introduced arylpyridine ligands and coexisting cations. Furthermore, the facile recrystallization procedures in the ion-pairing strategy for preparing luminescent materials can be applied for large-scale production. The ion-pairing strategy used in this study does not require the solution-state anion-binding mode. To the best of our knowledge, such a room-temperature phosphorescence enhancement by anion binding and ion-pairing assembly has not been demonstrated thus far. The mechanism of the phosphorescence intensity modulated by anion binding and ion pairing was clearly revealed by theoretical studies for stacking structures. Assembly systems with multiple components, based on the combination of host systems and cationic species, would provide diverse materials with controllable emission wavelengths, quantum yields and lifetimes. Introducing more bulky and robust countercations would further improve the solid-state emission properties. Moreover, introduction of π-electronic cations35 would also result in the formation of materials with intriguing photophysical properties. Further studies on ion-pairing luminescent materials are currently being conducted by designing and synthesizing charged building units and precursors (ion-responsive molecules).Data availability
Data supporting the work in this publication are available via the ESI and associated crystallographic data.†Author contributions
H. M. designed and conducted the project. Y. H., K. K., Y. M. and H. T. carried out the synthesis, characterization and property examinations. H. S., H. I. and N. T. evaluated the solid-state absorption and emission spectra. W. O. and T. S. conducted the theoretical calculations. Y. H. and N. Y. analyzed the single-crystal X-ray structures.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers JP18H01968 and JP22H02067 for Scientific Research (B), JP19K05444 and JP22K05253 for Scientific Research (C), JP20J22745 for JSPS Fellows and JP20H05863 for Transformative Research Areas (A) “Condensed Conjugation” and the Ritsumeikan Global Innovation Research Organization (R-GIRO) project (2017–22 and 2022–27). Theoretical calculations were partially performed using the Research Center for Computational Science, Okazaki, Japan (Projects: 19-IMS-C085, 20-IMS-C079, 21-IMS-C077 and 22-IMS-C077) and Information Initiative Center, Hokkaido University. The synchrotron radiation experiments were performed at BL02B1 (2019B1638), BL40XU (2021B1703) and BL40B2 (2021B1828) with the approval of the Japan Synchrotron Radiation Research Institute (JASRI). We thank Prof. Atsuhiro Osuka, Dr Takayuki Tanaka, Dr Akito Nakai and Dr Koki Kise, Kyoto University, for single-crystal X-ray analysis, Dr Kunihisa Sugimoto, Kindai University, for synchrotron radiation single-crystal X-ray analysis, Dr Noboru Ohta, JASRI/SPring-8, for synchrotron radiation XRD analysis, Prof. Shigeki Kuwata, Ritsumeikan University, for elemental analysis and Prof. Hitoshi Tamiaki, Ritsumeikan University, for various measurements.Notes and references
- Selected books and reviews on supramolecular assemblies for electronic materials: (a) Functional Supramolecular Architectures: For Organic Electronics and Nanotechnology, ed. P. Samorì and F. Cacialli, Wiley, 2011 Search PubMed; (b) Supramolecular Materials for Opto-Electronics, ed. N. Koch, RSC, 2015 Search PubMed; (c) T. Wöhrle, I. Wurzbach, J. Kirres, A. Kostidou, N. Kapernaum, J. Litterscheidt, J. C. Haenle, P. Staffeld, A. Baro, F. Giesselmann and S. Laschat, Chem. Rev., 2016, 116, 1139–1241 CrossRef PubMed; (d) P. Yu, Y. Zhen, H. Dong and W. Hu, Chem, 2019, 5, 2814–2853 CrossRef CAS; (e) C. J. Kousseff, R. Halaksa, Z. S. Parr and C. B. Nielsen, Chem. Rev., 2022, 122, 4397–4419 CrossRef CAS PubMed; (f) J. Chen, W. Zhang, L. Wang and G. Yu, Adv. Mater., 2023, 35, 2210772 CrossRef CAS PubMed.
- J. Gierschner, J. Shi, B. Milián-Medina, D. Roca-Sanjuán, S. Varghese and S. Y. Park, Adv. Opt. Mater., 2021, 9, 2002251 CrossRef CAS.
- (a) V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed; (b) T. Strassner, Acc. Chem. Res., 2016, 49, 2680–2689 CrossRef CAS PubMed; (c) L. Ravotto and P. Ceroni, Coord. Chem. Rev., 2017, 346, 62–76 CrossRef CAS; (d) E. V. Puttock, M. T. Walden and J. A. G. Williams, Coord. Chem. Rev., 2018, 367, 127–162 CrossRef CAS; (e) M. Yoshida and M. Kato, Coord. Chem. Rev., 2020, 408, 213194 CrossRef CAS; (f) Y. Han, Z. Gao, C. Wang, R. Zhong and F. Wang, Coord. Chem. Rev., 2020, 414, 213300 CrossRef CAS.
- (a) V. W.-W. Yam, K. M.-C. Wong and N. Zhu, J. Am. Chem. Soc., 2002, 124, 6506–6507 CrossRef CAS PubMed; (b) Y. Sun, K. Ye, H. Zhang, J. Zhang, L. Zhao, B. Li, G. Yang, B. Yang, Y. Wang, S.-W. Lai and C.-M. Che, Angew. Chem., Int. Ed., 2006, 45, 5610–5613 CrossRef CAS PubMed; (c) N. Komiya, M. Okada, K. Fukumoto, D. Jomori and T. Naota, J. Am. Chem. Soc., 2011, 133, 6493–6496 CrossRef CAS PubMed; (d) C.-H. Chen, F.-I. Wu, Y.-Y. Tsai and C.-H. Cheng, Adv. Funct. Mater., 2011, 21, 3150–3158 CrossRef CAS; (e) N. Komiya, N. Itami and T. Naota, Chem. – Eur. J., 2013, 19, 9497–9505 CrossRef CAS PubMed; (f) M. Mauro, A. Aliprandi, C. Cebrián, D. Wang, C. Kübel and L. De Cola, Chem. Commun., 2014, 50, 7269–7272 RSC; (g) K. Ohno, S. Yamaguchi, A. Nagasawa and T. Fujihara, Dalton Trans., 2016, 45, 5492–5503 RSC; (h) M.-J. Sun, Y. Liu, W. Zeng, Y. S. Zhao, Y.-W. Zhong and J. Yao, J. Am. Chem. Soc., 2019, 141, 6157–6161 CrossRef CAS PubMed; (i) H. Yang, H. Li, L. Yue, X. Chen, D. Song, X. Yang, Y. Sun, G. Zhou and Z. Wu, J. Mater. Chem. C, 2021, 9, 2334–2349 RSC; (j) F.-F. Xu, W. Zeng, M.-J. Sun, Z.-L. Gong, Z.-Q. Li, Y. S. Zhao, J. Yao and Y.-W. Zhong, Angew. Chem., Int. Ed., 2022, 61, e202116603 CrossRef CAS PubMed.
- M. Kasha, H. R. Rawls and M. Ashraf El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CAS.
- D. L. Dexter and J. H. Schulman, J. Chem. Phys., 1954, 22, 1063–1070 CrossRef CAS.
- Pt⋯Pt interaction is another factor to enhance emissions for the PtII complexes that have appropriate electronic structures, although resulting metal-metal-to-ligand charge transfer (MMLCT) gives rise to the photophysical properties differing from those of monomeric states. See also ref. 4h.
- (a) Z. Cheng, H. Shi, H. Ma, L. Bian, Q. Wu, L. Gu, S. Cai, X. Wang, W.-w. Xiong, Z. An and W. Huang, Angew. Chem., Int. Ed., 2018, 57, 678–682 CrossRef CAS PubMed; (b) S. Cai, H. Ma, H. Shi, H. Wang, X. Wang, L. Xiao, W. Ye, K. Huang, X. Cao, N. Gan, C. Ma, M. Gu, L. Song, H. Xu, Y. Tao, C. Zhang, W. Yao, Z. An and W. Huang, Nat. Commun., 2019, 10, 4247 CrossRef PubMed.
- (a) C. F. J. Faul, Acc. Chem. Res., 2014, 47, 3428–3438 CrossRef CAS PubMed; (b) K. Goossens, K. Lava, C. W. Bielawski and K. Binnemans, Chem. Rev., 2016, 116, 4643–4807 CrossRef CAS PubMed.
- Reviews and the first report by our group: (a) Y. Haketa, K. Urakawa and H. Maeda, Mol. Syst. Des. Eng., 2020, 5, 757–771 RSC; (b) Y. Haketa, K. Yamasumi and H. Maeda, Chem. Soc. Rev., 2023, 52, 7170–7198 RSC; (c) Y. Haketa, S. Sasaki, N. Ohta, H. Masunaga, H. Ogawa, N. Mizuno, F. Araoka, H. Takezoe and H. Maeda, Angew. Chem., Int. Ed., 2010, 49, 10079–10083 CrossRef CAS PubMed.
- (a) Z.-m. Ou, H. Yao and K. Kimura, J. Photochem. Photobiol., A, 2007, 189, 7–14 CrossRef CAS; (b) D. K. Bwambok, B. El-Zahab, S. K. Challa, M. Li, L. Chandler, G. A. Baker and I. M. Warner, ACS Nano, 2009, 4, 3854–3860 CrossRef PubMed; (c) Y. Yang, C. Sun, S. Wang, K. Yan, M. Zhao, B. Wu and F. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202117436 CrossRef CAS PubMed.
- (a) C. R. Benson, L. Kacenauskaite, K. L. VanDenburgh, W. Zhao, B. Qiao, T. Sadhukhan, M. Pink, J. Chen, S. Borgi, C.-H. Chen, B. J. Davis, Y. C. Simon, K. Raghavachari, B. W. Laursen and A. H. Flood, Chem, 2020, 6, 1978–1997 CrossRef CAS; (b) L. Kacenauskaite, S. G. Stenspil, A. H. Olsson, A. H. Flood and B. W. Laursen, J. Am. Chem. Soc., 2022, 144, 19981–19989 CrossRef CAS PubMed.
- A. Kuno, G. Hirata, H. Tanaka, Y. Kobayashi, N. Yasuda and H. Maeda, Chem. – Eur. J., 2021, 27, 10068–10076 CrossRef CAS PubMed.
- Negatively charged luminescent species can be obtained by anion binding by electronically neutrally charged π-electronic systems. Actually, since the first report on π-electronic ion-pairing assemblies based on dipyrrolyldiketone boron complexes as anion-responsive π-electronic systems (ref. 10c), new properties and functions derived from the ordered arrangement of charged π-systems have been revealed (ref. 10a and b). Replacement of boron with other metal ions would induce new π-electronic systems and ion-pairing assemblies. However, dipyrrolyldiketones form complexes, comprising a single diketone unit, with only PtII in a square planar fashion (ref. 13).
- Z. M. Hudson, B. A. Blight and S. Wang, Org. Lett., 2012, 14, 1700–1703 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- (a) J. Brooks, Y. Babayan, S. Lamansky, P. I. Djurovich, I. Tsyba, R. Bau and M. E. Thompson, Inorg. Chem., 2002, 41, 3055–3066 CrossRef CAS PubMed; (b) D. Wang, X. Chen, H. Yang, D. Zhong, B. Liu, X. Yang, L. Yue, G. Zhou, M. Ma and Z. Wu, Dalton Trans., 2020, 49, 15633–15645 RSC; (c) A. Haque, H. E. Moll, K. M. Alenezi, M. S. Khan and W.-Y. Wong, Materials, 2021, 14, 4236 CrossRef CAS PubMed.
- The photophysical properties were examined for the single crystals, lightly crushed with a spatula, on a quartz plate. The crystals showed no significant changes in the XRD patterns.
- (a) B. Ma, P. I. Djurovich and M. E. Thompson, Coord. Chem. Rev., 2005, 249, 1501–1510 CrossRef CAS; (b) T. Shigehiro, S. Yagi, T. Maeda, H. Nakazumi, H. Fujiwara and Y. Sakurai, J. Phys. Chem. C, 2013, 117, 532–542 CrossRef CAS; (c) Y.-J. Cho, S.-Y. Kim, H.-J. Son, D. W. Cho and S. O. Kang, Phys. Chem. Chem. Phys., 2017, 19, 5486–5494 RSC.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- Crystal polymorphs were observed for 2a in the photograph, and the details will be discussed elsewhere.
- Articles for GAMESS: (a) M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery Jr, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS; (b) M. S. Gordon, and M. W. Schmidt, in Theory and Applications of Computational Chemistry: The First Forty Years, ed. C. E. Dykstra, G. Frenking, K. S. Kim and G. E. Scuseria, Elsevier, 2005 Search PubMed.
- M. J. S. Phipps, T. Fox, C. S. Tautermann and C.-K. Skylaris, Chem. Soc. Rev., 2015, 44, 3177–3211 RSC.
- Report for FMO: K. Kitaura, E. Ikeo, T. Asada, T. Nakano and M. Uebayasi, Chem. Phys. Lett., 1999, 313, 701–706 CrossRef CAS.
- Report for pair interaction energy decomposition analysis (PIEDA): D. G. Fedorov and K. Kitaura, J. Comput. Chem., 2007, 28, 222–237 CrossRef CAS PubMed.
- The preliminarily examined single-crystal X-ray analysis of 2c·Cl−-TPA+ for the crystal of low quality revealed the formation of a charge-by-charge assembly.
- The electrostatic energy, revealed by EDA, originating mainly from hydrogen-bonding interactions for the 2c⋯Cl−⋯2c structure in the 1D chain of 2c·Cl−-TBA+ was larger in the absolute value by 13.5 kcal/mol than that of 2c·Cl−-TPeA+, resulting in a larger Φem for 2c·Cl−-TBA+.
- 2b and TPACl were included in 2b·Cl−-TPA+ in a 1:1 ratio as determined by 1H NMR. Notably, 2b, in the py1i conformation, formed a [4 + 1]-type anion complex 2b4·Cl− as ion pairs with TBA+ and TPeA+ (Fig. S26 and 27†). Their solid-state emission properties would be discussed elsewhere owing to the difficulty of preparing crystals.
- M. J. Frisch, et al., Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- L. W. Chung, W. M. C. Sameera, R. Ramozzi, A. J. Page, M. Hatanaka, G. P. Petrova, T. V. Harris, X. Li, Z. Ke, F. Liu, H.-B. Li, L. Ding and K. Morokuma, Chem. Rev., 2015, 115, 5678–5796 CrossRef CAS PubMed.
- I. B. Bersuker and V. Z. Polinger, Vibronic Interactions in Molecules and Crystals, Springer, 1989 Search PubMed.
- (a) D. Kim and J.-L. Brédas, J. Am. Chem. Soc., 2009, 131, 11371–11380 CrossRef CAS PubMed; (b) W.-C. Chen, C. Sukpattanacharoen, W.-H. Chan, C.-C. Huang, H.-F. Hsu, D. Shen, W.-Y. Hung, N. Kungwan, D. Escudero, C.-S. Lee and Y. Chi, Adv. Funct. Mater., 2020, 30, 2002494 CrossRef CAS; (c) P. Pander, A. Sil, R. J. Salthouse, C. W. Harris, M. T. Walden, D. S. Yufit, J. A. G. Williams and F. B. Dias, J. Mater. Chem. C, 2022, 10, 15084–15095 RSC.
- (a) M. Uejima, T. Sato, D. Yokoyama, K. Tanaka and J.-W. Park, Phys. Chem. Chem. Phys., 2014, 16, 14244–14256 RSC; (b) W. Ota, M. Uejima and T. Sato, Bull. Chem. Soc. Jpn., 2023, 96, 582–590 CrossRef CAS.
- (a) T. Sato, K. Tokunaga and K. Tanaka, J. Phys. Chem. A, 2008, 112, 758–767 CrossRef CAS PubMed; (b) T. Kato, N. Haruta and T. Sato, Vibronic Coupling Density: Understanding Molecular Deformation, Springer, 2021 CrossRef.
- (a) [2 + 1]-type complex 2d2·Cl− formed an ion pair with tetraphenylporphyrin AuIII complex as a π-electronic cation (Fig. S32†); (b) Y. Haketa, Y. Bando, Y. Sasano, H. Tanaka, N. Yasuda, I. Hisaki and H. Maeda, iScience, 2019, 14, 241–256 CrossRef CAS PubMed; (c) H. Tanaka, Y. Kobayashi, K. Furukawa, Y. Okayasu, S. Akine, N. Yasuda and H. Maeda, J. Am. Chem. Soc., 2022, 144, 21710–21718 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, analytical data, computational details and CIF files for the single-crystal X-ray structural analysis. CCDC 2158662–2158674. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04564a |
| This journal is © The Royal Society of Chemistry 2024 |