
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCooperatively designed aptamer-PROTACs for spatioselective degradation of nucleocytoplasmic shuttling protein for enhanced combinational therapy†
Ran
Liu
 a,
Zheng
Liu
a,
Mohan
Chen
a,
Hang
Xing
b,
Penghui
Zhang
*c and
Jingjing
Zhang
*a
a,
Zheng
Liu
a,
Mohan
Chen
a,
Hang
Xing
b,
Penghui
Zhang
*c and
Jingjing
Zhang
*a
aState Key Laboratory of Analytical Chemistry for Life Science, School of Chemistry and Chemical Engineering, Chemistry and Biomedicine Innovation Center (ChemBIC), Nanjing University, Nanjing 210023, China. E-mail: jing15209791@nju.edu.cn
bInstitute of Chemical Biology and Nanomedicine, State Key Laboratory of Chemo/Biosensing and Chemometrics, Hunan Provincial Key Laboratory of Biomacromolecular Chemical Biology, College of Chemistry and Chemical Engineering, Hunan University, Changsha 410082, China
cZhejiang Cancer Hospital, The Key Laboratory of Zhejiang Province for Aptamers and Theranostics, Hangzhou Institute of Medicine (HIM), Chinese Academy of Sciences, Hangzhou 310022, China. E-mail: phzhang@ucas.ac.cn
First published on 29th November 2023
Abstract
Nucleocytoplasmic shuttling proteins (NSPs) have emerged as a promising class of therapeutic targets for many diseases. However, most NSPs-based therapies largely rely on small-molecule inhibitors with limited efficacy and off-target effects. Inspired by proteolysis targeting chimera (PROTAC) technology, we report a new archetype of PROTAC (PS-ApTCs) by introducing a phosphorothioate-modified aptamer to a CRBN ligand, realizing tumor-targeting and spatioselective degradation of NSPs with improved efficacy. Using nucleolin as a model, we demonstrate that PS-ApTCs is capable of effectively degrading nucleolin in the target cell membrane and cytoplasm but not in the nucleus, through the disruption of nucleocytoplasmic shuttling. Moreover, PS-ApTCs exhibits superior antiproliferation, pro-apoptotic, and cell cycle arrest potencies. Importantly, we demonstrate that a combination of PS-ApTCs-mediated nucleolin degradation with aptamer–drug conjugate-based chemotherapy enables a synergistic effect on tumor inhibition. Collectively, PS-ApTCs could expand the PROTAC toolbox to more targets in subcellular localization and accelerate the discovery of new combinational therapeutic approaches.
Introduction
Nucleocytoplasmic shuttling, one of the major mechanisms to transport a variety of molecules in living cells, plays critical roles in diverse cellular functions.1 In particular, the dysregulation of the nucleocytoplasmic shuttling has been found to invoke various diseases including cancer.2 For example, the nucleocytoplasmic shuttling proteins (NSPs) that constantly shuttle between the nucleus and cytoplasm play an important role in promoting abnormal cell survival, tumor progression, and drug resistance.3 Given the importance of NSPs in tumor research, many efforts have been made to develop small molecule-based inhibitors capable of targeting NSPs for potential intervention of nucleocytoplasmic shuttling.4 Despite significant progress, intrinsic or acquired resistance limits the therapeutic efficacy of small molecule-based inhibitors in patients.3 In addition, the “druggable proteome” of NSPs is hampered by the competitive- or occupancy-driven mechanism of traditional inhibitors. Therefore, there is still an urgent need to discover new agents to the aberrant nucleocytoplasmic shuttling.Proteolysis targeting chimeras (PROTACs) are a heterobifunctional class of small molecules that simultaneously recruit a target protein and an E3 ubiquitin ligase complex to trigger target polyubiquitination and subsequent proteasomal degradation.5–7 Despite substantial progress,8–15 membrane proteins are considered less ideal targets for PROTACs due to the distribution of the ubiquitin system in the cytoplasm. Additionally, the complexity, diversity, and heterogeneity of tumors have propelled the shift of treatment from monotherapy to polytherapy for an enhanced therapeutic outcome. This concept also has great significance for the future development and clinical translation of PROTACs but has rarely been explored.
With these considerations in mind, herein we present a conceptual approach for the design of a new archetype of PROTAC (denoted as PS-ApTCs) by introducing phosphorothioate-modified aptamers to an E3 ligand (Fig. 1a). Different from previous aptamer-PROTAC conjugates, PS-aptamer serves as a versatile scaffold for both cell-specific PROTAC delivery and efficient recruitment of target protein. Using nucleolin (NCL) as a model NSP, we demonstrated that PS-ApTCs can recruit E3 ligase CRBN to NCL in human cervical cancer cells and potently induce the degradation of NCL in vitro and in vivo (Fig. 1b). Our results indicate that PS-ApTCs can effectively block the aberrant shuttling of NCL protein between the cell nucleus, cytoplasm, and cell membrane through the ubiquitin–proteasome pathway, effectively reducing the expression levels of NCL in the cell membrane and cytoplasm. Moreover, the anti-tumor superiority of PS-ApTCs was further highlighted by integrating PS-ApTCs with aptamer–drug conjugate-based chemotherapy (Fig. 1c). Since in vitro selection can obtain aptamers selective for many NSP targets, such a conceptual PROTAC design might also possess the ability to expand the PROTAC toolbox to an even wider range of targets in subcellular localization and accelerate the discovery of new combinational therapeutic approaches.
 | ||
| Fig. 1 PS-ApTCs-based PROTAC strategy for enhanced combinational therapy. (a) Rational design of PS-ApTCs, which contains an E3 ligand, a linker and a phosphorothioate modified aptamer for targeting proteins of interest (POI). (b) PS-ApTCs simultaneously recruit the E3 ubiquitin ligase to ubiquitinate the POI, which is subsequently degraded by the 26S proteasome. (c) Combination of PS-ApTCs-mediated NCL degradation and aptamer–drug conjugate-based chemotherapy for enhanced tumor therapy. | ||
Results and discussion
Rational design and characterization of phosphorothioate-modified aptamer-PROTACs
A typical PS-ApTCs is composed of three components: a phosphorothioate-modified aptamer warhead to recruit the protein of interest (POI), a ligand to recruit the CRBN E3 ubiquitin ligase, and a linker between these two fragments (Fig. 1a). The critical difference between traditional small-molecule PROTACs and PS-ApTCs is the aptamer targeting warhead enables PS-ApTCs to specifically recognize target cancer cells, while PS modification enhances the stability of PS-ApTCs from nuclease-mediated degradation.16 These features are generally considered to be a requirement for improving the therapeutic index.17,18 To evaluate this experimentally, we first chose the NCL as the POI, and AS1411 as the NCL-binding domain (Fig. S1a†). The former is a shuttling protein overexpressed in the cytoplasm and on the cell surface of various cancer cells, while the latter is a well-defined guanine-rich aptamer and has been extensively validated as a highly specific NCL binder in vitro and in vivo.19,20 Meanwhile, to ensure that the PS-ApTCs design is generally applicable, pomalidomide, one of the most extensively used CRBN recruiting ligands,21 was installed in PS-ApTCs (Fig. S1a†). Thus, PS-ApTCs are capable of specifically recruiting both the NCL and the E3 ubiquitin ligase to form a ternary complex. The propinquity allows E3 ubiquitin ligase to induce the polyubiquitination of the NCL and lead to the protein degradation by proteasome (Fig. 1b).The PS-ApTCs was synthesized through a copper-free click reaction to conjugate azide-modified pomalidomide (pom-PEG4-azide) to DBCO-modified PS-AS1411 (Fig. S2a†). The molecular weight from MALDI-TOF MS is similar to the theoretical value (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 326.9) (Fig. S2b†), indicating the successful synthesis of PS-ApTCs. Furthermore, ApTCs, a negative control, was designed and synthesized by replacing the DBCO-modified PS-AS1411 with a DBCO-modified AS1411 (Fig. S1b†). Firstly, the phosphorothioate modification provided PS-ApTCs with enhanced resistance to degradation. As shown in polyacrylamide gel electrophoresis (PAGE) results (Fig. 2a), the corresponding bands of PS-ApTCs are still observed after incubation with 10% fetal bovine serum (FBS) for 24 h, while the corresponding bands of ApTCs completely disappear after 12 h. The calculated half-lives of PS-ApTCs and ApTCs in a 10% serum solution are 32 h and 5 h, respectively. Then, we verified whether the modifications of pomalidomide and phosphorothioate groups would affect the binding ability of the AS1411 aptamer towards recombinant NCL. Surprisingly, biolayer interferometry (BLI) confirmed that PS-ApTCs exhibited higher binding affinity for NCL compared to ApTCs (Fig. S3a†).22 In fact, kinetic measurements further demonstrated that the binding affinity of PS-ApTCs (KD = 8.7 nM) was approximately 15-fold higher than that of wild-type AS1411 (Fig. S3b†).23 This is mainly attributed to the fact that phosphorothioate-modified aptamers can interact with target proteins through electrostatic and hydrophobic interaction networks,24 leading to a significant enhancement of the affinity of PS-AS1411 for NCL protein. Next, to validate the effective binding of the pomalidomide on PS-ApTCs to CRBN, we immobilized PS-ApTCs on a BLI tip, followed by subsequent addition of CRBN protein. The BLI binding signals increased in a dose-dependent manner with a KD of 25 nM (Fig. S3c†), indicating that PS-ApTCs retained high affinity binding to CRBN.25 Furthermore, to investigate the feasibility of PS-ApTCs-mediated ternary complex formation, we conducted a similar BLI test by successive incubation in CRBN and NCL solutions. As expected, two successive increases in the BLI signal were observed, confirming that PS-ApTCs can recruit both CRBN and NCL concurrently (Fig. 2b). Given that the linker length is essential for the formation of sufficiently stable ternary complexes, we further optimized the linker length using two-step BLI characterization (Fig. S4 and S5†).
326.9) (Fig. S2b†), indicating the successful synthesis of PS-ApTCs. Furthermore, ApTCs, a negative control, was designed and synthesized by replacing the DBCO-modified PS-AS1411 with a DBCO-modified AS1411 (Fig. S1b†). Firstly, the phosphorothioate modification provided PS-ApTCs with enhanced resistance to degradation. As shown in polyacrylamide gel electrophoresis (PAGE) results (Fig. 2a), the corresponding bands of PS-ApTCs are still observed after incubation with 10% fetal bovine serum (FBS) for 24 h, while the corresponding bands of ApTCs completely disappear after 12 h. The calculated half-lives of PS-ApTCs and ApTCs in a 10% serum solution are 32 h and 5 h, respectively. Then, we verified whether the modifications of pomalidomide and phosphorothioate groups would affect the binding ability of the AS1411 aptamer towards recombinant NCL. Surprisingly, biolayer interferometry (BLI) confirmed that PS-ApTCs exhibited higher binding affinity for NCL compared to ApTCs (Fig. S3a†).22 In fact, kinetic measurements further demonstrated that the binding affinity of PS-ApTCs (KD = 8.7 nM) was approximately 15-fold higher than that of wild-type AS1411 (Fig. S3b†).23 This is mainly attributed to the fact that phosphorothioate-modified aptamers can interact with target proteins through electrostatic and hydrophobic interaction networks,24 leading to a significant enhancement of the affinity of PS-AS1411 for NCL protein. Next, to validate the effective binding of the pomalidomide on PS-ApTCs to CRBN, we immobilized PS-ApTCs on a BLI tip, followed by subsequent addition of CRBN protein. The BLI binding signals increased in a dose-dependent manner with a KD of 25 nM (Fig. S3c†), indicating that PS-ApTCs retained high affinity binding to CRBN.25 Furthermore, to investigate the feasibility of PS-ApTCs-mediated ternary complex formation, we conducted a similar BLI test by successive incubation in CRBN and NCL solutions. As expected, two successive increases in the BLI signal were observed, confirming that PS-ApTCs can recruit both CRBN and NCL concurrently (Fig. 2b). Given that the linker length is essential for the formation of sufficiently stable ternary complexes, we further optimized the linker length using two-step BLI characterization (Fig. S4 and S5†).
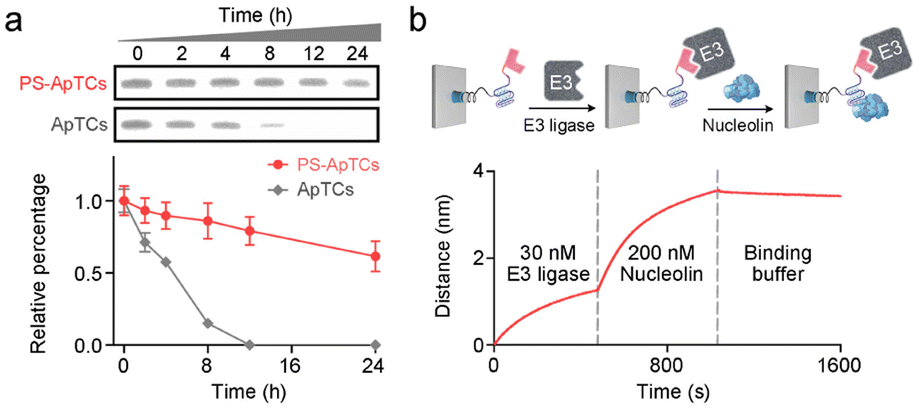 | ||
| Fig. 2 Characterization of PS-ApTCs. (a) Native polyacrylamide gel analysis of the stability of PS-ApTCs and ApTCs in 10% FBS for different times. Quantitative degradation curves showing PS-ApTCs has a significantly prolonged serum half-life. Error bars represent the standard deviations of three independent measurements. (b) Two-step BLI experiment showing that PS-ApTCs can simultaneously bind to both CRBN and NCL. Biotin-modified PS-ApTCs and ApTCs were captured by streptavidin BLI probes. | ||
After confirming the functional activity of the two ligands on PS-ApTCs, we attempted to test whether PS-ApTCs could target and internalize into the membrane NCL-positive cells. We incubated the FAM-labeled PS-ApTCs with membrane NCL-positive HeLa cells and NCL-negative L-02 cells at 4 °C for 60 min. Confocal laser scanning microscopy (CLSM) images showed bright green fluorescence evenly distributed on the membrane of HeLa cells, while negligible green fluorescence was observed on the membrane of L-02 cells following an identical protocol (Fig. S6a†). Flow cytometry analysis further confirmed the efficient targeting of NCL protein on the cell membrane by PS-ApTCs (Fig. S6b†), consistent with the results obtained from FAM-AS1411-treated cells (Fig. S6c and d†). Subsequently, the time-dependent internalization and intracellular localization of PS-ApTCs was evaluated by CLSM. Fig. S6e† demonstrates that FAM-labeled PS-ApTCs (green fluorescence) exhibited rapid accumulation on the cell membrane within 10 min of co-incubation with HeLa cells at 37 °C. Furthermore, over an extended period (10–240 min), PS-ApTCs predominantly accumulated in the cytoplasm. Together, these results demonstrated the vital role of PS-modification in improving the serum stability and binding affinity of PS-ApTCs, while retaining the specific targeting and internalization capabilities of AS1411 towards NCL-positive cells.26
PS-ApTCs preferentially degrades membrane and cytoplasmic NCL in targeted cancer cells
We next asked whether PS-ApTCs can be used to degrade NCL in cells. Given that NCL was extensively located in the cell membrane, cytoplasm, and nucleus in most cancer cells,2 we first incubated HeLa cells with 200 nM PS-ApTCs for different durations, followed by staining the cell membrane NCL with FAM-AS1411 at 4 °C to assess the degradation of NCL on the membrane. The CLSM imaging results demonstrate that the levels of membrane-bound NCL exhibited a time-dependent decrease during the incubation with PS-ApTCs (Fig. 3a and S7†). In addition, quantitative analysis using flow cytometry revealed that after co-incubation with PS-ApTCs for 12 h, the expression level of membrane NCL in HeLa cells was reduced to less than 10% and remained at this level without recovery until 24 h (Fig. 3b).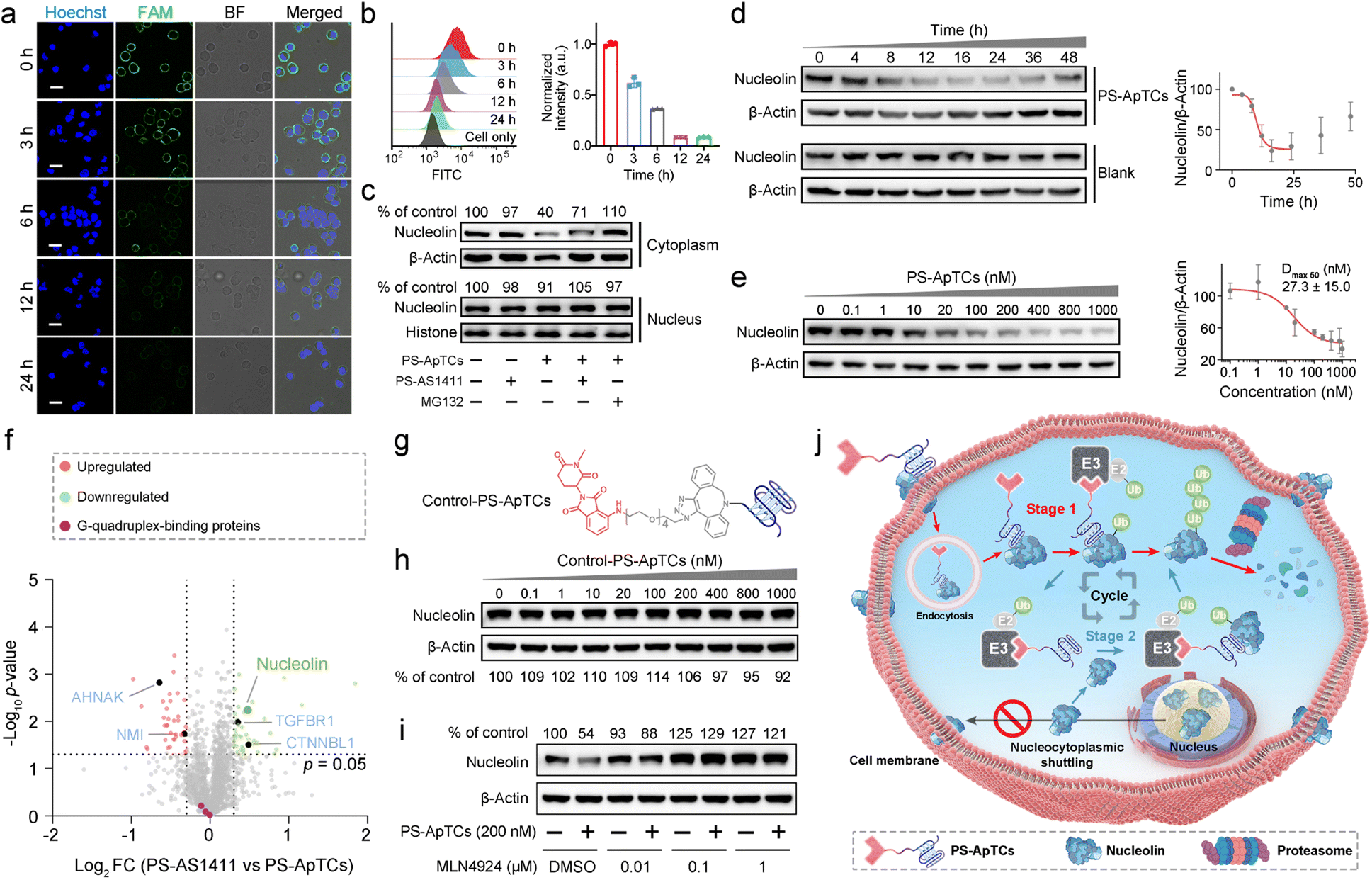 | ||
| Fig. 3 PS-ApTCs efficiently degraded membrane and cytoplasmic NCL in targeted cancer cells. Confocal microscopy images (a) and flow cytometry analysis (b) showing a time-dependent decrease of membrane NCL levels in HeLa cells. The scale bar is 20 μm. (c) Western blot analysis showing PS-ApTCs preferentially degraded NCL in the cytoplasm but not in the nucleus. (d) Quantitative western blot analysis showing a time-dependent degradation of cytoplasmic NCL in HeLa cells post-incubation with 200 nM PS-ApTCs. (e) Dose-dependent experiment showing PS-ApTCs efficiently degraded cytoplasmic NCL after 24 h treatment. The quantification of NCL levels relative to the control was performed, and the values for the half-maximal degradation concentration (Dmax 50) were determined. (f) Quantitative MS-based proteomics comparing protein abundance profiles of HeLa cells treated with PS-AS1411 or PS-ApTCs (200 nM) for 24 h. (g) Chemical structures of Control-PS-ApTCs. (h) Western blot analysis of cytoplasmic NCL levels in HeLa cells treated with increasing concentrations of Control-PS-ApTCs for 24 h. (i) Effect of MLN4924 on neddylation and cytoplasmic NCL degradation in HeLa cells. (j) Schematic representation of PS-ApTCs for targeted degradation of NCL in the cell membrane and cytoplasm. | ||
The aberrant nucleocytoplasmic shuttling of NCL protein in cancer cells occurs between the cell nucleus, cytoplasm, and cell membrane, with the cytoplasm playing a crucial role among the three compartments. Importantly, the ubiquitination and degradation processes of proteins primarily occur in the cytoplasm. Therefore, we next evaluated the ability of PS-ApTCs to degrade NCL in the cytoplasm. The western blot analysis results showed a significant decrease in cytoplasmic NCL levels in cells treated with PS-ApTCs compared to the untreated control group (Fig. 3c), while PS-AS1411 had no effect on the expression levels of NCL in the cytoplasm. When co-treated with PS-ApTCs and PS-AS1411, the degradation of cytoplasmic NCL was partially inhibited. This observation can be attributed to the binding of PS-AS1411 to NCL in the cytoplasm, which prevents the ubiquitination degradation of NCL by PS-ApTCs. Notably, the degradation ability of PS-ApTCs was not significantly affected with a shorter PEG chain (Fig. S8†). Moreover, the degradation of cytoplasmic NCL by PS-ApTCs could be completely blocked by the proteasome inhibitor MG132 (Fig. 3c). However, we found that the expression of NCL in the cell nucleus was not affected by PS-ApTCs (Fig. 3c). This result may be attributed to the less effective proteasome degradation of NCL in the nucleus where additional steps or factors are required.27 To evaluate the degradation efficiency of PS-ApTCs, we further treated HeLa cells with 200 nM PS-ApTCs for different times. Fig. 3d shows the time-dependent degradation profiles of cytoplasmic NCL, and we found that the degradation could begin as early as 4 h and plateau at 12 h, giving a lowest cytoplasmic NCL level of 23.8 ± 13.5% of the initial value. Notably, a trend toward recovery of cytoplasmic NCL was also observed after 24 h, indicating that PS-ApTCs-induced sustained degradation is also dependent on continued exposure of PS-ApTCs. Furthermore, we also found that PS-ApTCs degraded NCL in HeLa cells in a concentration-dependent manner (Fig. 3e), with a half-maximal degradation concentration (Dmax 50) of 27.3 ± 15.0 nM and maximum degradation (Dmax) of approximately 70%.
To assess whether the expression levels of other cytoplasmic proteins are affected by PS-ApTCs, we next performed quantitative dimethyl-based proteomic studies.28 Briefly, the proteomic samples were obtained by treating HeLa cells with PS-ApTCs, or PS-AS1411 at 200 nM for 24 h, followed by extraction of cytoplasmic proteins. A total of 2064 proteins were quantified in the two groups of samples. Compared to the control group (PS-AS1411), 43 proteins (2.08%) were significantly downregulated and 36 proteins (1.74%) showed increased expression in the PS-ApTCs-treated group (Fig. 3f). Moreover, NCL was one of the most significantly downregulated protein, consistent with the immunoblotting results. Notably, the 10 proteins with a significantly reduced expression level in PS-ApTCs-treated samples were closely involved in the NCL signaling network/pathway (Fig. S9†), including the cell proliferation modulator of TGFBR1 and CTNNBL1.29,30 Meanwhile, the 25 up-regulated proteins associated with cell cycle arrest, antiproliferation or apoptosis, including AHNAK and NMI were found to a have functional relationship with NCL,31,32 indicating a potential antitumor mechanism. Moreover, PS-ApTCs treatment did not lead to a significant change in the protein levels of other G-quadruplex-binding proteins such as CIRBP, RHAU, and SLIRP (Fig. 3f).33–35 These results suggest that PS-ApTCs is relatively selective for NCL degradation. In conclusion, PS-ApTCs effectively decreased NCL protein expression in the membrane and cytoplasm of HeLa cells.
Mechanisms of targeted degradation of NCL by PS-ApTCs
While we observed potent NCL degradation in the membrane and cytoplasm by PS-ApTCs, it was still not clear how PS-ApTCs contributes to the degradation process, and importantly, how they affect the expression level of NCL on the cell membrane. To study the degradation pathway, two negative controls, including a PROTAC synthesized using a randomized DNA sequence (R-PROTAC) and PS-AS1411 (Fig. S1b†), were also designed and tested with an identical protocol. Successful synthesis was confirmed through MALDI-TOF MS (Fig. S10†). Both fluorescence microscopy and flow cytometry showed minimal change in membrane NCL levels when HeLa cells were subjected to either R-PROTACs (Fig. S11†) or PS-AS1411 (Fig. S12†). We also prepared Control-PS-ApTCs containing a methylated glutarimide ring (Fig. S13–20†), which greatly reduces the affinity for CRBN and retains the identical NCL binding moiety and linker as PS-ApTCs (Fig. 3g and S5†).36,37 Western blot analysis confirmed that Control-PS-ApTCs did not reduce the cytoplasmic NCL level in HeLa cells at concentrations up to 1000 nM for 24 h (Fig. 3h). These results indicated that both components of PS-AS1411 and pomalidomide are necessary for the overall function of the PS-ApTCs degrader. In addition, MLN4924, a neddylation inhibitor that disrupts CRBN E3 ligase function,37 could effectively restore the cytoplasmic NCL level in PS-ApTCs-treated HeLa cells (Fig. 3i). Of note, when HeLa cells were co-incubated with PS-ApTCs and a lysosome inhibitor (chloroquine, CQ), the degradation of NCL was not significantly inhibited (Fig. S21†), indicating that lysosomes are not involved in the degradation of NCL. However, no significant degradation of cytoplasmic NCL was observed in L-02 cells treated with PS-ApTCs for up to 48h (Fig. S22†), indicating the targeted degradation of NCL by PS-ApTCs in membrane NCL-positive cells. These findings collectively confirm that PS-ApTCs can effectively and selectively degrade membrane and cytoplasmic NCL in membrane NCL-positive cells in a CRBN- and proteasome-dependent manner.Overall, these findings illustrate the potential mechanism of spatially selective degradation, particularly in membrane protein degradation, of the nucleocytoplasmic shuttling protein NCL (Fig. 3j): PS-ApTCs selectively bind to the NCL membrane on tumor cells and are internalized into the cytoplasm. Then, they recruit CRBN E3 ligase to induce ubiquitination of NCL, followed by degradation of ubiquitinated NCL by the proteasome (Stage 1). Subsequently, PS-ApTCs continued to degrade the cytoplasmic NCL protein, blocking the aberrant shuttling of NCL protein between the cytoplasm and cell membrane, effectively reducing the expression level of NCL on the cell membrane (Stage 2). These two stages cooperatively facilitate the spatioselective proteolysis of NCL through the disruption of nucleocytoplasmic NCL shuttling. Importantly, PS-ApTCs selectively target and activate the ubiquitin-proteasome system in tumor cells, rather than normal cells, which is crucial for the clinical translation of PROTACs.
PS-ApTCs shows enhanced antiproliferation, pro-apoptotic and cycle arrest potencies
The aberrant shuttling process of NCL protein between the cytoplasm and cell membrane plays a significant role in tumor growth and progression. Therefore, effectively blocking the aberrant shuttling of NCL protein in cancer cells may be a promising therapeutic approach for tumors. We further examined the anti-proliferative activity of PS-ApTCs using the CCK-8 assay. As shown in Fig. 4a, 200 nM PS-ApTCs displayed a time-dependent response in inhibiting HeLa cell viability during 48 h post-incubation. Our data demonstrate that the inhibitory effect of PS-ApTCs on the proliferation of HeLa cells is in a dose-dependent manner (Fig. 4b), with an IC50 value of 33.7 nM and a maximum inhibition (Imax) of approximately 42.0%. In addition, the effect of PS-ApTCs on HeLa cell apoptosis was analyzed by flow cytometry analysis. As shown in Fig. 4c, PS-ApTCs promoted the largest population of HeLa cells to undergo early- and late-stage apoptosis, and the result was in accordance with the antiproliferation test. Next, we sought to ask whether NCL degradation affects cell cycle. Flow cytometry analysis revealed that PS-ApTCs (200 nM) resulted in cell cycle arrest by increasing the accumulation of HeLa cells in the G2/M phase while reducing the accumulation of cells in the G0/G1 and S phases (Fig. 4d).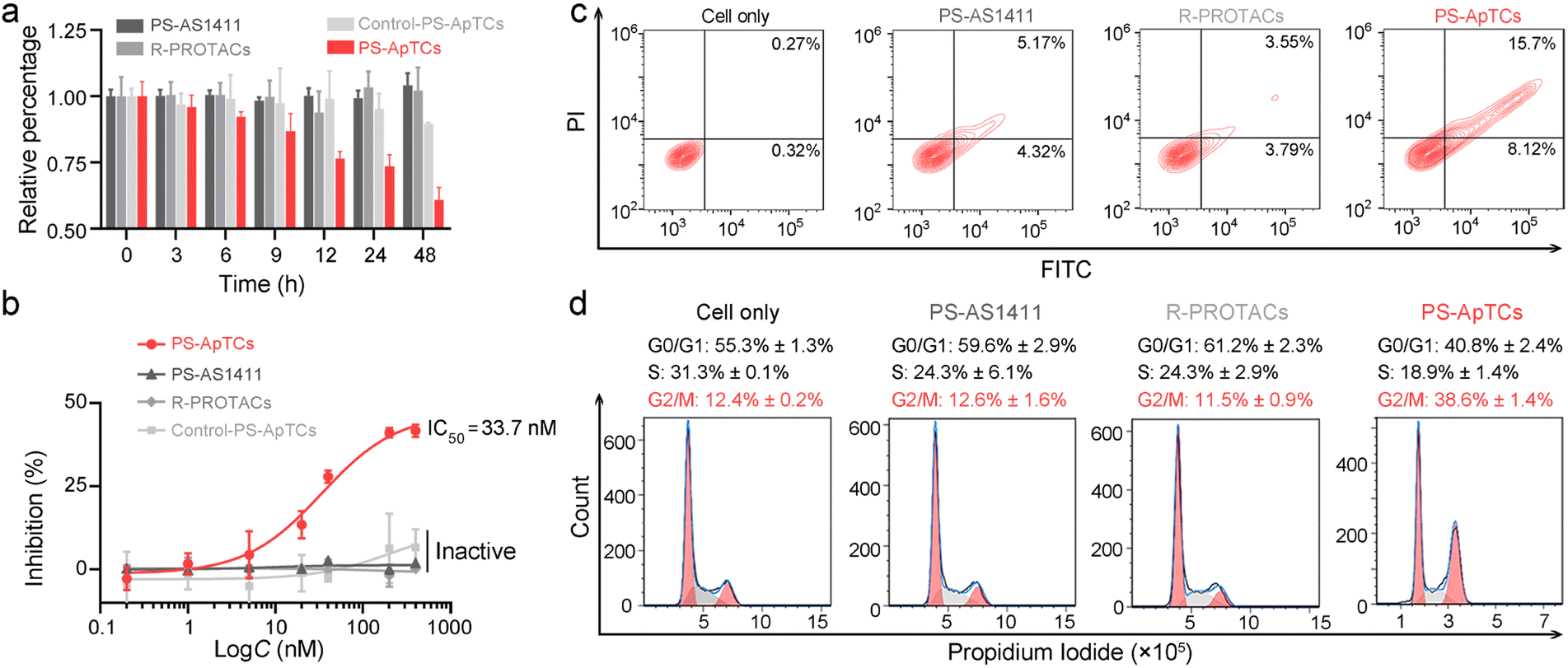 | ||
| Fig. 4 PS-ApTCs showed enhanced antiproliferation, pro-apoptotic and cycle arrest potencies. (a) Cell viability of HeLa cells treated with 200 nM of different PROTACs as indicated in cell culture medium at 37 °C. (b) Quantitative dose-dependent experiment showing PS-ApTCs has excellent antiproliferation activity with an IC50 of 33.7 nM and Imax of 42.0%. HeLa cells were treated with different concentrations of PROTACs as indicated for 48 h. (c) Flow cytometry analysis of HeLa cells post-incubation with 200 nM of different PROTACs for 48 h, followed by staining with Annexin V-FITC and propidium iodide. (d) Cell cycle analysis of HeLa cells treated with 200 nM of different PROTACs, as determined by flow cytometry using propidium iodide staining. | ||
Meanwhile, we also examined the cytotoxicity of PS-AS1411, R-PROTACs and Control-PS-ApTCs. As shown in Fig. 4a and b, none of these compounds demonstrated significant cytotoxicity in HeLa cells, suggesting that the antiproliferation of PS-ApTCs relies on the combined action of the CRBN recruiting ligand (pomalidomide) and NCL recruiting ligand (AS1411). Correspondingly, R-PROTACs, PS-AS1411, and Control-PS-ApTCs at the same concentrations did not exhibit any pro-apoptotic (Fig. 4c) or cell cycle arrest activity (Fig. 4d). To further confirm the inhibition of interfering with aberrant nucleocytoplasmic shuttling of NCL on cancer cell proliferation, we co-cultured PS-ApTCs with membrane NCL-negative L-02 cells (Fig. S23†). As shown in Fig. S23A,† the PS-ApTCs treatment failed to decrease cell viability, and all three PROTACs as indicated displayed no obvious inhibition effect even at a high concentration of 400 nM. In addition, there was no significant effect on apoptosis activation (Fig. S24†) or cell cycle distribution (Fig. S25†) in L-02 cells for all groups tested. Therefore, PS-ApTCs should only induce cytotoxicity in membrane NCL-positive tumor cells. Taken together, these results demonstrated that the anti-proliferative and pro-apoptotic activities of PS-ApTCs in HeLa cells were attributed to the effective inhibition of aberrant nucleocytoplasmic shuttling of NCL protein through the ubiquitin–proteasome pathway.
Through systematic evolution of ligands by exponential enrichment (SELEX), it is possible to obtain aptamers that exhibit selectivity towards numerous targets. Therefore, this concept of aptamer-PROTAC design may have the potential to expand the PROTAC toolbox to a broader range of targets. To explore the generalizability of the aptamer-PROTAC concept, estrogen receptor alpha (ERα), a member of the nuclear hormone receptor superfamily,38,39 was further selected as a model target. First, we conjugated the DBCO-modified ERα aptamer to pom-PEG4-azide to generate PS-ApTCs/ERα (Fig. S26a†). The successful synthesis of this degrader was confirmed by MALDI-TOF MS (Fig. S26b†). Then, we validated the degradation ability of PS-ApTCs/ERα on ERα using MCF-7 breast cancer cells, which exhibit high membrane ERα expression. The CLSM imaging results showed a significant decrease in membrane ERα levels in cells treated with different concentration of PS-ApTCs/ERα (Group II and III) compared to the untreated control group (Fig. S27a†). Meanwhile, pre-treatment with MG132 (Group 4) or MLN4924 (Group 5) inhibited the decrease of ERα levels caused by PS-ApTCs/ERα, indicating that the degradation occurred in a proteasome and CRBN-dependent manner. Further flow cytometry and western blot analysis displayed a trend similar to that of this CLSM analysis (Fig. S27b and c†). These results demonstrated that our aptamer-PROTAC concept could be applied for degrading other membrane protein. Finally, we also conjugated DBCO-modified PS-AS1411 with another E3 ubiquitin ligase (VHL) recruiting ligand, (S,R,S)-AHPC, and generated the PS-ApTCs/AHPC (Fig. S28†). Further CLSM, flow cytometry and western blot analysis confirmed that PS-ApTCs/AHPC could effectively reduce the expression levels of NCL in HeLa cells (Fig. S29†). In summary, these findings suggest that our aptamer-PROTACs represent a universal approach for the targeted degradation of proteins, demonstrating their wide applicability.
Design and characterization of GSH-responsive AS1411 aptamer–drug conjugates (ApDCs)
Unlike traditional small molecule drugs, PROTAC drugs do not require tight and prolonged binding with disease-causing targets to induce their degradation, making them likely to effectively address drug resistance issues.7 However, the limited universality and repeated administration of monotherapy restrict their clinical efficacy. This prompted us to further investigate whether the anticancer activity triggered by PS-ApTCs could be combined with other treatment modalities to achieve systemic cancer therapy. As a proof-of-principle experiment, ApDCs, an innovative targeted drug delivery strategy for enhancing the efficacy of chemotherapy was chosen for an initial investigation. As illustrated in Fig. 5a, the typical ApDCs is composed of three modules: (1) a DBCO-modified AS1411 aptamer, which contains a disulfide bond (−S–S−) in the middle of the aptamer sequence that can be cleaved in a GSH-responsive manner within cells, (2) an azide-modified paclitaxel (PTX) that acts as an antitumor drug, and (3) a linker containing a hydrophilic PEG segment with a molecular weight of approximately 1000 Da. Considering that GSH levels are relatively low in the extracellular matrix but high in the cytoplasm,40 we reasoned that PS-ApTCs could remain in the –S–S− state before it enters the cells, enabling efficient NCL receptor-mediated uptake into the cytoplasm. Moreover, it can be subsequently cleaved by intracellular GSH to release the active PTX for chemotherapy, and importantly, disrupt the G-quadruplex structure of AS1411 to minimize the competition with PS-ApTCs for intracellular NCL occupancy when polytherapy was applied. The proposed ApDCs was synthesized via the copper-free click reaction (Fig. S30a†), and confirmed by mass spectrometry (Fig. S30b†). Prior to undertaking intracellular evaluation of ApDCs, it was important to first characterize its GSH-responsive properties. To this end, we conducted a PAGE-based cleavage assay in the presence of GSH (2 mM) to mimic the reducing environment. As shown in Fig. S31a,† GSH-treated DBCO-AS1411-intSH results in two DNA fragments in perfect agreement with the expected cleavage products upon disulfide bond cleavage in the GSH-responsive aptamer, confirming the GSH-responsive releasing feature. In addition, the release of PTX from GSH-treated ApDCs was verified by high-performance liquid chromatography (HPLC) (Fig. S31b†). These results indicate that the ApDCs retains GSH-responsive properties of the DBCO-AS1411, liberating the PTX for the subsequent chemotherapy. In addition, we confirmed that ApDCs had a negligible impact on the degradation activity of PS-ApTCs (Fig. S32†), enabling potential applications of these two distinct agents for synergistically enhanced therapy.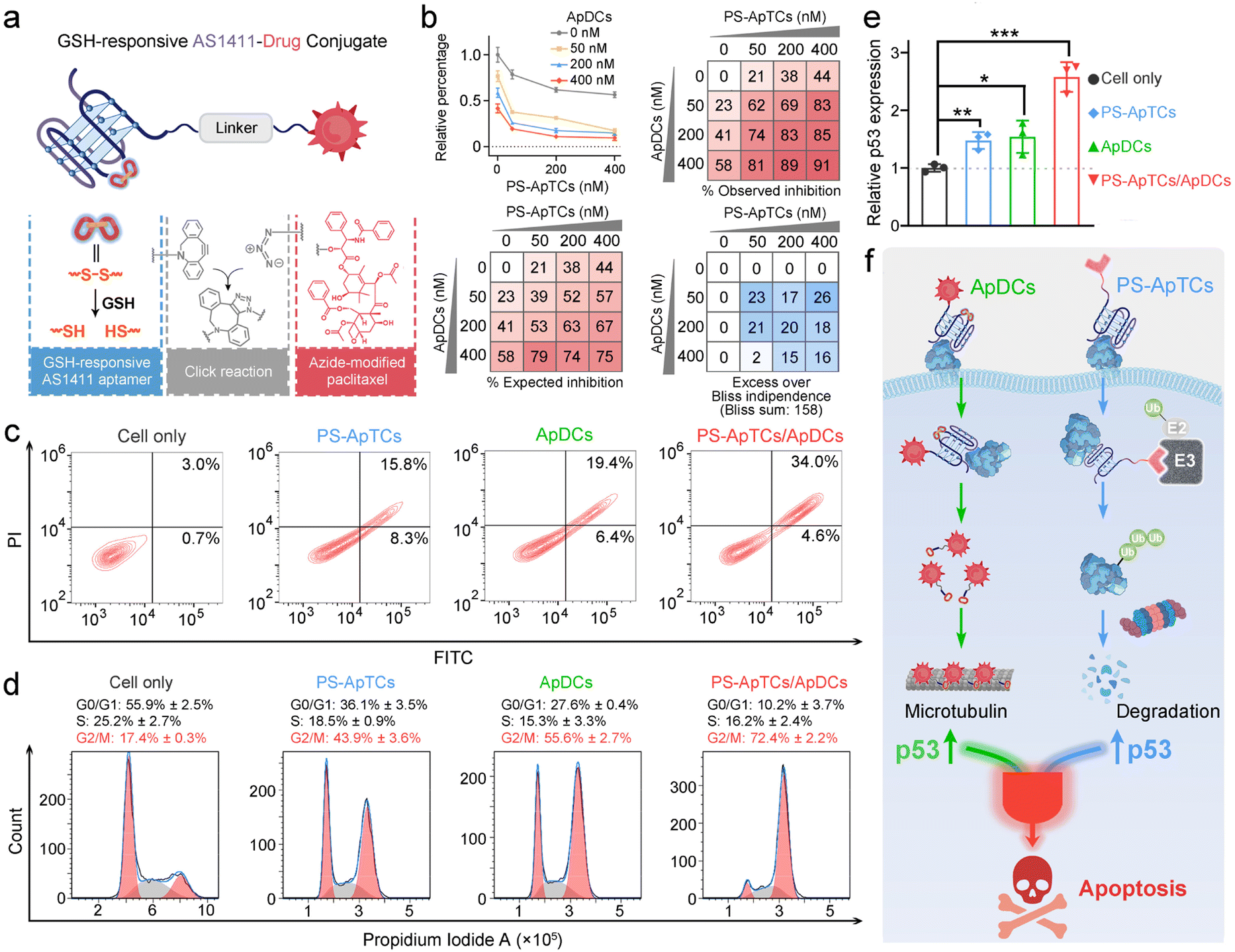 | ||
| Fig. 5 PS-ApTCs and ApDCs resulted in a synergistic therapeutic effect by upregulating p53. (a) Design of GSH-responsive aptamer–drug conjugates (ApDCs), which incorporates an AS1411 aptamer containing a cleavable disulfide bridge into an azide-modified paclitaxel via a copper-free click reaction. (b) Drug synergy test of PS-ApTCs and ApDCs using the Bliss independence model. HeLa cells were treated with PS-ApTCs and ApDCs alone and in combination at the indicated concentrations for 24 h. (c) Flow cytometry analysis of HeLa cells post-incubation with 200 nM of different compounds as indicated for 48 h, followed by staining with Annexin V-FITC and propidium iodide. (d) Cell cycle analysis of HeLa cells treated with 200 nM of different compounds, as determined by flow cytometry using propidium iodide staining. (e) Quantitative reverse transcription-PCR study showing PS-ApTCs and ApDCs displayed apparent positive synergy for upregulating the p53 mRNA expression level in HeLa cells. (f) Schematic illustration of synergistically designed PS-ApTCs and ApDCs for enhanced therapeutic purposes. | ||
PS-ApTCs and ApDCs possess a synergistic therapeutic effect by upregulating p53
We further investigated whether PS-ApTCs could be used in combination with ApDCs. PTX on ApDCs is a widely used chemotherapy drug for various malignant tumors. It has been reported that PTX interferes with microtubule protein stability and dynamics within cells, activating the p53 signaling pathway, inducing cell cycle arrest at the G2/M phase, and ultimately leading to apoptosis of tumor cells.41 Therefore, before assessing the therapeutic efficacy of PS-ApTCs and ApDCs in vivo, it is necessary to examine whether there is any synergistic or antagonistic interaction between PS-ApTCs and ApDCs. For this purpose, we evaluated the drug synergy between PS-ApTCs and ApDCs using the Bliss independence model.42 The analysis showed an average synergistic effect greater than 15% within the dosage range of 50–400 nM for both PS-ApTCs and ApDCs (Fig. 5b). And the sum of individual Bliss scores in the 3 × 3 drug dosage matrix (Bliss sum) was 158, indicating a strong synergy between PS-ApTCs and ApDCs for enhanced combinational therapy. Notably, at a concentration of 200 nM, the combined treatment resulted in a proliferation inhibition of over 80% in HeLa cells. And the Bliss synergy score was determined to be 19.1 (Fig. S33†), further confirming the synergistic antiproliferative effect of PS-ApTCs and ApDCs in HeLa cells. In contrast, no synergistic effect was observed between Control-PS-ApTCs and ApDCs (Fig. S34†).To verify the proliferation inhibition of HeLa cells is really induced by apoptosis, flow cytometry-based apoptosis assay was performed after 48 h incubation of the cells with PS-ApTCs, ApDCs, or PS-ApTCs/ApDCs (Fig. 5c). Compared with untreated cells, apoptosis rates induced by the above three therapeutic agents increased to 24.1%, 25.8%, and 38.6%, respectively. Meanwhile, early and late apoptotic induction between different treatments was compared, and, notably, cotreatment of PS-ApTCs and ApDCs induced a much greater degree of late apoptosis than all individual drug-treated groups. To investigate the biological effect associated with the aforementioned enhanced antiproliferation and pro-apoptotic effects, cell cycle analysis was performed on the same four groups by flow cytometry. As shown in Fig. 5d, untreated HeLa cells showed a typical cell cycle distribution pattern with the majority of the cells in the G0/G1 phase and a small proportion of approximately 17.4% in the G2/M phase. Following treatment with either PS-ApTCs or ApDCs, a dramatic decrease in the G0/G1 phase was observed, while the proportion of cells in the G2/M phase increases to 43.9 ± 3.6% and 55.6 ± 2.7%, respectively. Importantly, PS-ApTCs/ApDCs codosed resulted in a more significant increase in G2/M phase and decrease in G0/G1 phase than individual treatment, indicating a synergistic strengthening of cell cycle arrest at the G2/M phase by PS-ApTCs and ApDCs.
We subsequently investigated the potential mechanisms of their synergistic cytotoxicity. It has been reported that NCL binds to the 5′ untranslated region of p53 mRNA, stabilizing its stem-loop structure and inhibiting p53 translation, thus maintaining low levels of p53 protein in undisturbed cells, while downregulation of nucleolar proteins promotes p53 expression.43 We therefore hypothesized that p53 may participate in a critical functional axis of synergistic cytotoxicity. To test this hypothesis, we further performed a quantitative reverse transcription-PCR (qRT-PCR) study to evaluate the p53 mRNA levels in HeLa cells treated with 200 nM of ApDCs, PS-ApTCs, or PS-ApTCs/ApDCs for 48 h (Fig. S35†). Compared with untreated cells, the mRNA levels of p53 in cells treated with either PS-ApTCs or ApDCs increased similarly by about 1.5-fold (Fig. 5e). Notably, cotreatment of PS-ApTCs and ApDCs resulted in a further increase of the mRNA level of p53 to about 2.5-fold. These results indicated that the synergistic cytotoxicity of PS-ApTCs and ApDCs might arise from the up-regulation of p53 through two distinct intracellular pathways (Fig. 5f). That is, as confirmed in Fig. 3, PS-ApTCs efficiently inhibited the aberrant nucleocytoplasmic shuttling process of NCL protein in cancer cells, rendering the NCL ineffective in its function towards p53 mRNA, resulting in the upregulation of p53 in the cytoplasm. On the other hand, the intracellular GSH-activated release of PTX from ApDCs could impair the function of microtubules, which in turn would upregulate the expression of p53. As a result, these two intracellular pathways represent synergistic functions targeting the up-regulation of p53 (Fig. 5f). Taken together, these qRT-PCR data, together with the above in vitro cellular results, successfully demonstrate the ability to synergistically design PS-ApTCs and ApDCs for enhanced therapeutic purposes.
Combinational delivery of PS-ApTCs and ApDCs results in enhanced synergistic therapy in a mouse xenograft model
The tumor-targeting properties were first examined using Cy5-modified PS-ApTCs (PS-ApTCs-Cy5) as a model by intravenously administering to HeLa xenografted tumor-bearing nude mice, followed by fluorescence imaging with the IVIS animal imaging system. Free Cy5 and Cy5-modified R-PROTACs (R-PROTACs-Cy5) were applied as the controls. The fluorescence signal at the tumor site of the PS-ApTCs-Cy5 treated group increased in a time-dependent manner, and importantly, with much longer retention time than the control groups (Fig. 6a). Then, we examined the colocalization of PS-ApTCs-Cy5 or R-PROTACs-Cy5 with NCL-expressing cells in the tumor cryosections by immunofluorescence staining and more co-localization of the Cy5 fluorescent signal with NCL-expressing cells was found in PS-ApTCs-Cy5 treated mice as compared to R-PROTACs-Cy5 treated mice (Fig. S36†). Ex vivo imaging was further performed in harvested tumors and major organs at 8 h post injection (Fig. 6b). The fluorescence signal in the tumor tissue collected from PS-ApTCs-Cy5 treated mice was significantly higher than that from either Cy5 or R-PROTACs-Cy5 treated mice, demonstrating a superior high intratumoral accumulation of PS-ApTCs-Cy5. Moreover, strong fluorescence signals in the liver and kidneys and negligible fluorescence signals in the heart and lungs were observed in all treated groups, which were consistent with previous findings.19 Taken together, these results demonstrated that the proposed PS-ApTCs and ApDCs possess excellent tumor-targeting capability, which thus promotes the accumulation of PS-ApTCs and ApDCs at tumor sites for subsequent internalization and therapy.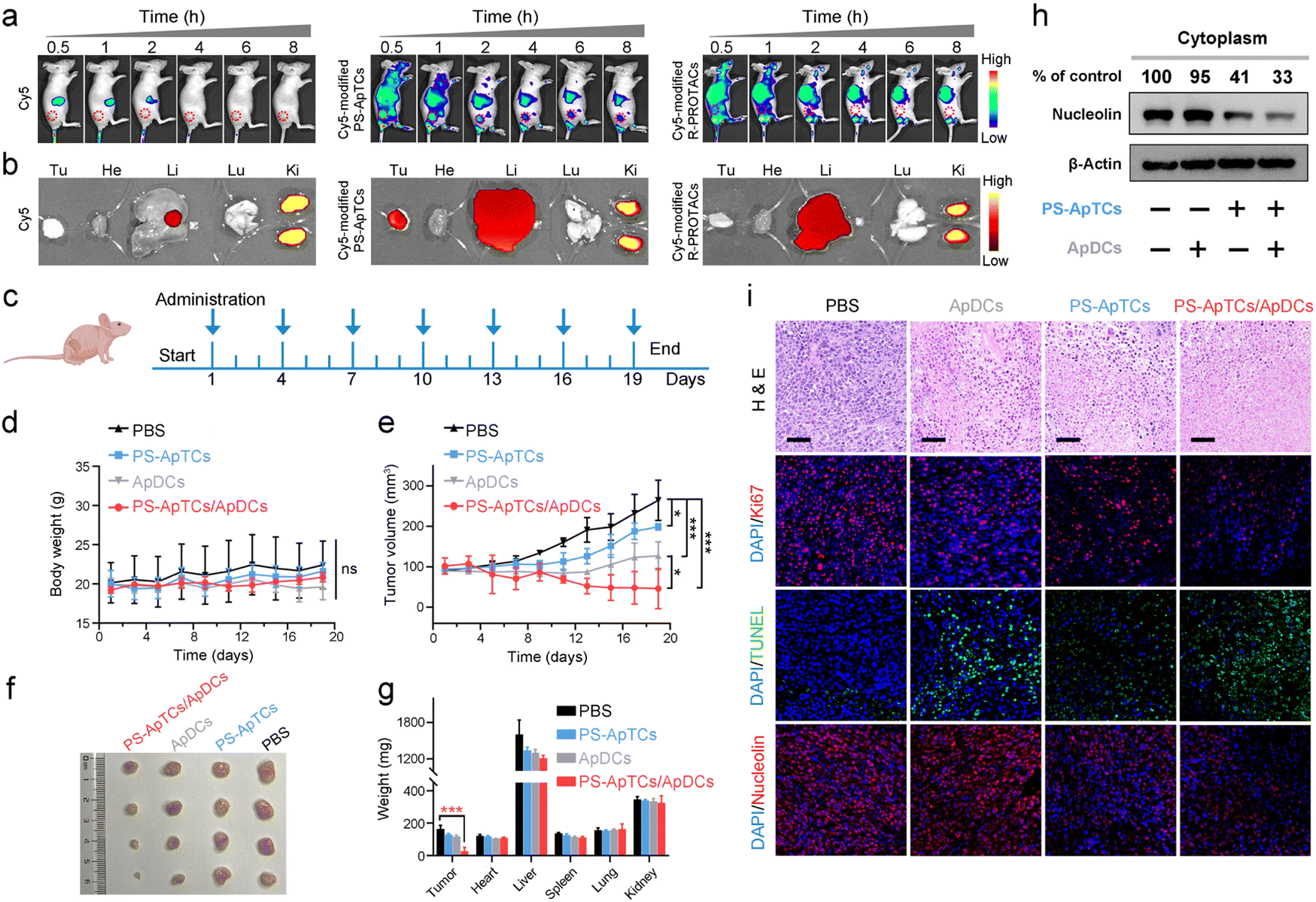 | ||
| Fig. 6 Combinational delivery of PS-ApTCs and ApDCs results in enhanced synergistic therapy in vivo. (a) Time-dependent in vivo fluorescence imaging showing PS-ApTCs-Cy5 exhibited active tumor-targeting properties. (b) Distribution of Cy5, Cy5-modified PS-ApTCs, and R-PROTACs in the tumor (Tu) and major viscera (He, heart; Li, liver; Lu, lung; and Ki, kidney) at the organ level 8 h post intravenous injection. (c) Schematic displaying the treatment implemented in the mouse model. The dosing points are indicated with blue arrows. (d) Body weight and (e) tumor volume of mice with different treatment during therapy. (f) Macroscopic views of the xenografted tumor after 19 days of different treatments from the groups indicated. (g) Analysis of tumor and organ weight after 19 days of different treatments at a dosing frequency of every three day via intravenous injection. The data are presented as the mean ± s.d., n = 4; ns: no significant difference; *p < 0.05; ***0.0001 < p < 0.001. (h) Expression levels of cytoplasmic NCL in tumor tissue, as determined via western blotting. (i) Microphotographs of H&E, Ki67, TUNEL, and NCL stained tumor tissues from different treatment groups. Scale bars, 50 μm. | ||
Next, we evaluated the in vivo pharmacokinetic (PK) properties of PS-ApTCs using a HeLa xenograft mouse model. After drug administration, the blood concentration of PS-ApTCs was quantified using a ligation-qPCR assay (Fig. S37†). As shown in Fig. S38,† PS-ApTCs exhibited a maximum blood concentration (Cmax) of 42.82 ± 2.46 nM at 10 min, and was rapidly cleared from the blood and became undetectable within 24 h with a circulation half-life (t1/2) of 4.44 ± 0.35 h. Other PK parameters (e.g. AUC, MRT) are also summarized in Table S2.† It should be noted that the rapid clearance rate of PS-ApTCs may be attributed to the AS1411-mediated intratumoral accumulation of PS-ApTCs, which has been verified using in vivo fluorescence imaging (Fig. 6a and b). To further evaluate the tumor levels of PS-ApTCs in real time, Cy5-modified PS-ApTCs was intravenously injected to HeLa xenograft mice, and the in vivo fluorescence images were collected at the indicated time points (Fig. S38a†). The fluorescence signal at the tumor sites increased gradually in a time-dependent manner (Fig. S38b†), and the signal intensity peaked at 2 h after drug administration (Fig. S38c†). Although the tumor levels of PS-ApTCs decreased gradually after 2 h, the stable fraction remained above 80% in the tumor site after 24 h, which is sufficient to ensure continuous availability of PS-ApTCs over the treatment period. The high tumor accumulation and long tumor retention of PS-ApTCs could be attributed to the tumor-specific targeting ability of AS1411 (Fig. S6†) and its good serum stability by PS-modification (Fig. 2a). This result is also consistent with the rapid blood clearance rate of PS-ApTCs as mentioned above.
Encouraged by the enhanced tumor accumulation and retention capacity of PS-ApTCs, we further evaluated the in vivo therapeutic efficacy of PS-ApTCs and ApDCs using a HeLa xenograft mouse model. The treatment process was performed via seven tail vein injections over 19 days (Fig. 6c), while the body weight and tumor size were monitored every two days. No significant change in the body weight of mice was observed for all the groups (Fig. 6d), indicating the low systemic toxicity of these treatments. In contrast, compared with PBS treated groups (Fig. 6e), the tumor growth in PS-ApTCs treated groups was slightly inhibited (p < 0.05), while ApDCs displayed a significant inhibition of tumor growth (p < 0.001). Consistent with this, the tumor size of mice treated with ApDCs for 19 days was reduced to 126.9 ± 34.8 mm3, compared to the 264.2 ± 50.0 mm3 for the PBS group and 198.9 ± 8.0 mm3 for the PS-ApTCs group (Fig. 6f). These results indicated that both PS-ApTCs and ApDCs had a limited tumor growth inhibition effect with a tumor growth inhibition (TGI) of 32% and 79%, respectively, consistent with the in vitro data presented in Fig. 5. Notably, the tumor growth in the PS-ApTCs/ApDCs group was efficiently inhibited with a TGI of 132% (p < 0.001), leading to a significant decrease of the tumor weight (Fig. 6g, p < 0.001). This indicated that the combination of the two types of therapeutics was more effective at suppressing the tumor growth than either individuals. In addition, no obvious pathological changes were observed in the major organs of the mice after treatment with different groups (Fig. S39†), which further verify the low toxic side effects of PS-ApTCs and ApDCs. Moreover, our western blotting results showed that PS-ApTCs or PS-ApTCs/ApDCs codosed could effectively degrade the cytoplasmic NCL in tumor tissues (>50%), compared with the two negative control groups (Fig. 6h). Next, we analyzed the tumor pathological changes related to NCL degradation using H&E sections, Ki67 immunofluorescence, and TUNEL staining (Fig. 6i). Compared with PBS-treated groups, the H&E staining sections of PS-ApTCs/ApDCs-treated groups showed obvious nuclear shrinkage and fragmentation, and extensive necrotic areas. However, PS-ApTCs- or ApDCs-treated groups displayed reduced areas of necrosis. In addition, Ki67 immunofluorescence and TUNEL staining results showed that combination therapy of PS-ApTCs/ApDCs displayed the weakest cell proliferation signal but the strongest apoptotic cell signal. Meanwhile, immuno-histochemical analysis revealed that PS-ApTCs- and PS-ApTCs/ApDCs-treated groups significantly downregulated the NCL expression in tumor tissues, while ApDCs-treated groups showed a negligible effect. Thus, PS-ApTCs-mediated NCL degradation coupled with ApDCs-based targeted chemotherapy elicited potent antitumor response, indicating enhanced synergistic therapy.
Conclusions
In summary, we developed an effective and cell-type-specific delivery strategy for PROTACs to degrade nucleocytoplasmic shuttling proteins. Using overexpressed NCL in cancer cells as a model, PS-ApTCs efficiently blocked the aberrant nucleocytoplasmic shuttling process of NCL, resulting in spatioselective degradation of NCL in the cell membrane and cytoplasm, thereby enhancing the anti-proliferative and pro-apoptotic capabilities against target cells. Notably, unlike the lysosomal degradation mechanism by AbTACs, GluTAC, and bispecific aptamer chimeras,22,44,45 PS-ApTCs effectively degrades membrane POI through the disruption of the nucleocytoplasmic shuttling process (Fig. 3j and S40†). In addition, phosphorothioate-modified aptamers offer excellent versatility for aptamer-based PROTACs.46,47 Moreover, we demonstrated for the first time that combination of PS-ApTCs-mediated NCL degradation with ApDCs-based targeted chemotherapy could produce a synergistic effect on tumor inhibition. This is particularly attractive when monotherapy has limited anti-tumor efficacy and tends to induce drug resistance. Overall, in addition to highlighting new strategies for PROTAC design, our results provided a starting point for the evaluation of PROTAC-based combinational anti-cancer therapy with the ultimate goal of clinical translation.Ethical statement
Animal husbandry and the experimental protocols used in this study were approved by the Institutional Animal Care and Use Committee (IACUC) of Nanjing (animal protocol number: IACUC-2204009). The experiments also comply with the ‘Animal Research: Reporting In Vivo Experiments’ (ARRIVE) 2.0 guidelines (https://arriveguidelines.org/arrive-guidelines).Data availability
The datasets supporting this article have been uploaded as part of the ESI, including the synthetic strategy, fluorescence imaging, flow cytometry analysis, western blot analysis, RT-qPCR, pharmacokinetics study, histology and uncropped full western blot images.Author contributions
R. L., P. Z. and J. Z. designed the experiments. R. L. performed the synthesis, characterization, and assays. Z. L. and M. C. assisted with the animal experiments. Z. L. helped to perform part of the additional experiment. All authors contributed to the analysis and discussion of the results. R. L., H. X., P. Z. and J. Z. wrote the manuscript. J. Z. and H. X. conceived and supervised this research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (no. 22274072) and Natural Science Foundation of Jiangsu Province (no. 20200303). Dr Xing acknowledges the financial support from the National Natural Science Foundation of China (no. 21877032).Notes and references
- L. Xu and A. Massague, Nucleocytoplasmic Shuttling of Signal Transducers, Nat. Rev. Mol. Cell Biol., 2004, 5, 209–219 CrossRef CAS PubMed.
- X. K. Fu, C. Liang, F. F. Li, L. Y. Wang, X. Q. Wu, A. P. Lu, G. Z. Xiao and G. Zhang, The Rules and Functions of Nucleocytoplasmic Shuttling Proteins, Int. J. Mol. Sci., 2018, 19, 1445 CrossRef.
- F. Conforti, Y. Wang, J. A. Rodriguez, A. T. Alberobello, Y.-W. Zhang and G. Giaccone, Molecular Pathways: Anticancer Activity by Inhibition of Nucleocytoplasmic Shuttling, Clin. Cancer Res., 2015, 21, 4508–4513 CrossRef CAS.
- N. Kudo, N. Matsumori, H. Taoka, D. Fujiwara, E. P. Schreiner, B. Wolff, M. Yoshida and S. Horinouchi, Leptomycin B Inactivates CRM1/exportin 1 by Covalent Modification at a Cysteine Residue in the Central Conserved Region, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9112–9117 CrossRef CAS.
- K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews and R. J. Deshaies, Protacs: Chimeric Molecules that Target Proteins to the Skp1-Cullin-F Box Complex for Ubiquitination and Degradation, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8554–8559 CrossRef CAS.
- Y. Naro, K. Darrah and A. Deiters, Optical Control of Small Molecule-Induced Protein Degradation, J. Am. Chem. Soc., 2020, 142, 2193–2197 CrossRef CAS.
- M. Bekes, D. R. Langley and C. M. Crews, PROTAC Targeted Protein Degraders: the Past is Prologue, Nat. Rev. Drug Discovery, 2022, 21, 181–200 CrossRef CAS.
- G. E. Winter, D. L. Buckley, J. Paulk, J. M. Roberts, A. Souza, S. Dhe-Paganon and J. E. Bradner, Phthalimide Conjugation as a Strategy for in vivo Target protein degradation, Science, 2015, 348, 1376–1381 CrossRef CAS PubMed.
- G. M. Burslem, J. Y. Song, X. Chen, J. Hines and C. M. Crews, Enhancing Antiproliferative Activity and Selectivity of a FLT-3 Inhibitor by Proteolysis Targeting Chimera Conversion, J. Am. Chem. Soc., 2018, 140, 16428–16432 CrossRef CAS PubMed.
- G. Xue, K. Wang, D. L. Zhou, H. B. Zhong and Z. Y. Pan, Light-Induced Protein Degradation with Photocaged PROTACs, J. Am. Chem. Soc., 2019, 141, 18370–18374 CrossRef CAS PubMed.
- A. Ryan, J. H. Liu and A. Deiters, Targeted Protein Degradation through Fast Optogenetic Activation and Its Application to the Control of Cell Signaling, J. Am. Chem. Soc., 2021, 143, 9222–9229 CrossRef CAS.
- J. W. Shao, Y. Q. Yan, D. L. Ding, D. J. Wang, Y. D. He, Y. Q. Pan, W. Yan, A. Kharbanda, H. Y. Li and H. J. Huang, Destruction of DNA-Binding Proteins by Programmable Oligonucleotide PROTAC (O'PROTAC): Effective Targeting of LEF1 and ERG, Adv. Sci., 2021, 8, 2102555 CrossRef CAS PubMed.
- H. T. Zhang, R. Peng, S. Chen, A. Shen, L. X. Zhao, W. Tang, X. H. Wang, Z. Y. Li, Z. G. Zha, M. M. Yi and L. M. Zhang, Versatile Nano-PROTAC-Induced Epigenetic Reader Degradation for Efficient Lung Cancer Therapy, Adv. Sci., 2022, 9, 2202039 CrossRef CAS.
- B. H. Ma, Y. Z. Fan, D. Z. Zhang, Y. Wei, Y. L. Jian, D. H. Liu, Z. X. Wang, Y. Gao, J. Ma, Y. L. Chen, S. Xu and L. Li, De Novo Design of an Androgen Receptor DNA Binding Domain-Targeted peptide PROTAC for Prostate Cancer Therapy, Adv. Sci., 2022, 9, 2201859 CrossRef CAS.
- F. Liu, X. Wang, J. L. Duan, Z. J. Hou, Z. M. Wu, L. L. Liu, H. Q. Lei, D. Huang, Y. F. Ren, Y. Wang, X. Y. Li, J. X. Zhuo, Z. J. Zhang, B. He, M. Yan, H. M. Yuan, L. H. Zhang, J. S. Yan, S. J. Wen, Z. F. Wang and Q. T. Liu, A Temporal PROTAC Cocktail-Mediated Sequential Degradation of AURKA Abrogates Acute Myeloid Leukemia Stem Cells, Adv. Sci., 2022, 9, 2104823 CrossRef CAS PubMed.
- Y. Y. Wang, D. L. Wang, J. C. Lin, Z. D. Lyu, P. R. Chen, T. Y. Sun, C. Y. Xue, M. Mojtabavi, A. Vedadghavami, Z. Y. Zhang, R. M. Wang, L. Zhang, C. Park, G. S. Heo, Y. J. Liu, S. J. Dong and K. Zhang, A Long-Circulating Vector for Aptamers Based upon Polyphosphodiester-Backboned Molecular Brushes, Angew. Chem., Int. Ed., 2022, 61, e202204576 CrossRef CAS PubMed.
- D. L. Wang, J. Q. Lin, F. Jia, X. Y. Tan, Y. Y. Wang, X. Y. Sun, X. Y. Cao, F. Y. Che, H. Lu, X. M. Gao, J. C. Shimkonis, Z. Nyoni, X. G. Lu and K. Zhang, Bottlebrush-Architectured Poly(Ethylene Glycol) as an Efficient Vector for RNA Interference in vivo, Sci. Adv., 2019, 5, eaav9322 CrossRef CAS PubMed.
- W. Shen, C. L. De Hoyos, M. T. Migawa, T. A. Vickers, H. Sun, A. Low, T. A. Bell, M. Rahdar, S. Mukhopadhyay, C. E. Hart, M. Bell, S. Riney, S. F. Murray, S. Greenlee, R. M. Crooke, X. H. Liang, P. P. Seth and S. T. Crooke, Chemical Modification of PS-ASO Therapeutics Reduces Cellular Protein-binding and Improves the Therapeutic Index, Nat. Biotechnol., 2019, 37, 640–650 CrossRef CAS PubMed.
- J. X. He, T. H. Peng, Y. B. Peng, L. L. Ai, Z. Y. Deng, X. Q. Wang and W. H. Tan, Molecularly Engineering Triptolide with Aptamers for High Specificity and Cytotoxicity for Triple-Negative Breast Cancer, J. Am. Chem. Soc., 2020, 142, 2699–2703 CrossRef CAS.
- L. P. Qiu, C. C. Wu, M. X. You, D. Han, T. Chen, G. Z. Zhu, J. H. Jiang, R. Q. Yu and W. H. Tan, A Targeted, Self-Delivered, and Photocontrolled Molecular Beacon for mRNA Detection in Living Cells, J. Am. Chem. Soc., 2013, 135, 12952–12955 CrossRef CAS PubMed.
- H. Y. Gao, X. Y. Sun and Y. Rao, PROTAC Technology: Opportunities and Challenges, ACS Med. Chem. Lett., 2020, 11, 237–240 CrossRef CAS PubMed.
- A. D. Cotton, D. P. Nguyen, J. A. Gramespacher, I. B. Seiple and J. A. Wells, Development of Antibody-Based PROTACs for the Degradation of the Cell-Surface Immune Checkpoint Protein PD-L1, J. Am. Chem. Soc., 2021, 143, 593–598 CrossRef CAS.
- A. Miranda, T. Santos, J. Carvalho, D. Alexandre, A. Jardim, C. R. F. Caneira, V. Vaz, B. Pereira, R. Godinho, D. Brito, V. Chu, J. P. Conde and C. Cruz, Aptamer-based Approaches to Detect Nucleolin in Prostate Cancer, Talanta, 2021, 226, 122037 CrossRef CAS.
- M. Hyjek-Skladanowska, T. A. Vickers, A. Napiorkowska, B. A. Anderson, M. Tanowitz, S. T. Crooke, X. H. Liang, P. P. Seth and M. Nowotny, Origins of the Increased Affinity of Phosphorothioate-Modified Therapeutic Nucleic Acids for Proteins, J. Am. Chem. Soc., 2020, 142, 7456–7468 CrossRef CAS PubMed.
- G. Petzold, E. S. Fischer and N. H. Thoma, Structural Basis of Lenalidomide-Induced CK1α Degradation by the CRL4(CRBN) Ubiquitin Ligase, Nature, 2016, 532, 127–130 CrossRef CAS.
- V. Stepanova, T. Lebedeva, A. Kuo, S. Yarovoi, S. Tkachuk, S. Zaitsev, K. Bdeir, I. Durnler, M. S. Marks, Y. Parfyonova, V. A. Tkachuk, A. A.-R. Higazi and D. B. Cines, Nuclear Translocation of Urokinase-type Plasminogen Activator, Blood, 2008, 112, 100–110 CrossRef CAS PubMed.
- A. Ghidini, A. Clery, F. Halloy, F. H. T. Allain and J. Hall, RNA-PROTACs: Degraders of RNA-Binding Proteins, Angew. Chem., Int. Ed., 2021, 60, 3163–3169 CrossRef CAS PubMed.
- P. J. Boersema, R. Raijmakers, S. Lemeer, S. Mohammed and A. J. R. Heck, Multiplex Peptide Stable Isotope Dimethyl Labeling for Quantitative Proteomics, Nat. Protoc., 2009, 4, 484–494 CrossRef CAS.
- Q. Hu, T. Hisamatsu, M. Haemmerle, M. S. Cho, S. Pradeep, R. Rupaimoole, C. Rodriguez-Aguayo, G. Lopez-Berestein, S. T. Wong and A. K. Sood, Role of Platelet-Derived Tgfb1 in the Progression of Ovarian Cancer, Clin. Cancer Res., 2017, 23, 5611–5621 CrossRef CAS PubMed.
- Y. W. Li, H. Y. Guo, C. J. Jin, C. P. Qiu, M. Gao, L. Zhang, Z. J. Liu and B. H. Kong, Spliceosome-Associated Factor CTNNBL1 Promotes Proliferation and Invasion in Ovarian Cancer, Exp. Cell Res., 2017, 357, 124–134 CrossRef CAS PubMed.
- I. H. Lee, M. Sohn, H. J. Lim, S. Yoon, H. Oh, S. Shin, J. H. Shin, S. H. Oh, J. Kim, D. K. Lee, D. Y. Noh, D. S. Bae, J. K. Seong and Y. S. Bae, Ahnak Functions as a Tumor Suppressor via Modulation of TGF beta/Smad Signaling Pathway, Oncogene, 2014, 33, 4675–4684 CrossRef CAS.
- B. Chen, J. Wang, D. N. Dai, Q. Y. Zhou, X. F. Guo, Z. Tian, X. J. Huang, L. Yang, H. L. Tang and X. M. Xie, AHNAK Suppresses Tumour Proliferation and Invasion by Targeting Multiple Pathways in Triple-negative Breast Cancer, J. Exp. Clin. Cancer Res., 2017, 36, 65 CrossRef PubMed.
- Z. L. Huang, J. Dai, W. H. Luo, X. G. Wang, J. H. Tan, S. B. Chen and Z. S. Huang, Identification of G-Quadruplex-Binding Protein from the Exploration of RGG Motif/G-Quadruplex Interactions, J. Am. Chem. Soc., 2018, 140, 17945–17955 CrossRef CAS PubMed.
- K. M. Patil, D. Chin, H. L. Seah, Q. Shi, K. W. Lim and A. T. Phan, G4-PROTAC: Targeted Degradation of a G-quadruplex Binding Protein, Chem. Commun., 2021, 57, 12816–12819 RSC.
- X. Y. Zhang, J. Spiegel, S. M. Cuesta, S. Adhikari and S. Balasubramanian, Chemical Profiling of DNA G-quadruplex-Interacting Proteins in Live Cells, Nat. Chem., 2021, 13, 626–633 CrossRef CAS PubMed.
- C. Steinebach, S. Lindner, N. D. Udeshi, D. C. Mani, H. Kehm, S. Kopff, S. A. Carr, M. Gutschow and J. Kronke, Homo-PROTACs for the Chemical Knockdown of Cereblon, ACS Chem. Biol., 2018, 13, 2771–2782 CrossRef CAS PubMed.
- J. Jang, C. To, D. J. H. De Clercq, E. Park, C. M. Ponthier, B. H. Shin, M. Mushajiang, R. P. Nowak, E. S. Fischer, M. J. Eck, P. A. Janne and N. S. Gray, Mutant-Selective Allosteric EGFR Degraders are Effective Against a Broad Range of Drug-Resistant Mutations, Angew. Chem., Int. Ed., 2020, 59, 14481–14489 CrossRef CAS.
- A. Sett, B. B. Borthakur, J. D. Sharma, A. C. Kataki and U. Bora, DNA Aptamer Probes for Detection of Estrogen Receptor α Positive Carcinomas, Transl. Res., 2017, 183, 104–120 CrossRef CAS PubMed.
- H. Tsujimura, M. Naganuma, N. Ohoka, T. Inoue, M. Naito, G. Tsuji and Y. Demizu, Development of DNA Aptamer-Based PROTACs That Degrade the Estrogen Receptor, ACS Med. Chem. Lett., 2023, 14, 827–832 CrossRef CAS.
- Y. E. Kurtoglu, R. S. Navath, B. Wang, S. Kannan, R. Romero and R. M. Kannan, Poly(amidoamine) Dendrimer-drug Conjugates with Disulfide Linkages for Intracellular Drug Delivery, Biomaterials, 2009, 30, 2112–2121 CrossRef CAS PubMed.
- F. F. Li, J. Lu, J. Liu, C. Liang, M. L. Wang, L. Y. Wang, D. F. Li, H. Z. Yao, Q. L. Zhang, J. Wen, Z. K. Zhang, J. Li, Q. X. Lv, X. J. He, B. S. Guo, D. G. Guan, Y. Y. Yu, L. Dang, X. H. Wu, Y. S. Li, G. F. Chen, F. Jiang, S. G. Sun, B. T. Zhang, A. P. Lu and G. Zhang, A Water-soluble Nucleolin Aptamer-Paclitaxel Conjugate for Tumor-specific Targeting in Ovarian Cancer, Nat. Commun., 2017, 8, 1390 CrossRef.
- K. Han, E. E. Jeng, G. T. Hess, D. W. Morgens, A. Li and M. C. Bassik, Synergistic Drug Combinations for Cancer Identified in a CRISPR Screen for Pairwise Genetic Interactions, Nat. Biotechnol., 2017, 35, 463–474 CrossRef CAS.
- M. Takagi, M. J. Absalon, K. G. McLure and M. B. Kastan, Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin, Cell, 2005, 123, 49–63 CrossRef CAS.
- H. Zhang, Y. Han, Y. F. Yang, F. Lin, K. X. Li, L. H. Kong, H. X. Liu, Y. J. Dang, J. Lin and P. R. Chen, Covalently Engineered Nanobody Chimeras for Targeted Membrane Protein Degradation, J. Am. Chem. Soc., 2021, 143, 16377–16382 CrossRef CAS.
- Y. Y. Miao, Q. Q. Gao, M. H. Mao, C. Zhang, L. Q. Yang, Y. Yang and D. Han, Bispecific Aptamer Chimeras Enable Targeted Protein Degradation on Cell Membranes, Angew. Chem., Int. Ed., 2021, 60, 11267–11271 CrossRef CAS PubMed.
- S. P. He, F. Gao, J. H. Ma, H. Q. Ma, G. Q. Dong and C. Q. Sheng, Aptamer-PROTAC Conjugates (APCs) for Tumor-Specific Targeting in Breast Cancer, Angew. Chem., Int. Ed., 2021, 60, 23299–23305 CrossRef CAS.
- L. Zhang, L. Li, X. Wang, H. Liu, Y. Zhang, T. Xie, H. Zhang, X. Li, T. Peng, X. Sun, J. Dai, J. Liu, W. Wu, M. Ye and W. Tan, Development of a Novel PROTAC Using the Nucleic Acid Aptamer as a Targeting Ligand for Tumor Selective Degradation of Nucleolin, Mol. Ther.–Nucleic Acids, 2022, 30, 66–79 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04249a |
| This journal is © The Royal Society of Chemistry 2024 |