
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand-controlled regiodivergent Ni-catalyzed trans-hydroboration/carboboration of internal alkynes with B2pin2†
Zunsheng
Chen‡
a,
Biao
Nie‡
b,
Xiaoning
Li
a,
Teng
Liu
a,
Chunsheng
Li
c and
Jiuzhong
Huang
 *a
*a
aKey Laboratory of Prevention and Treatment of Cardiovascular and Cerebrovascular Diseases (Gannan Medical University), Ministry of Education, School of Pharmacy, Gannan Medical University, Ganzhou 341000, P. R. China. E-mail: huangjz@gmu.edu.cn
bState Key Laboratory of Anti-Infective Drug Development, Sunshine Lake Pharma Company, Ltd, Dongguan 523871, P. R. China
cSchool of Chemistry and Chemical Engineering, Zhaoqing University, Zhaoqing 526060, P. R. China
First published on 4th January 2024
Abstract
Unprecedented regioselective trans-hydroboration and carboboration of unbiased electronically internal alkynes were realized via a nickel catalysis system with the aid of the directing group strategy. Furthermore, the excellent α- and β-regioselectivity could be accurately switched by the nitrogen ligand (terpy) and phosphine ligand (Xantphos). Mechanistic studies provided an insight into the rational reaction process, that underwent the cis-to-trans isomerization of alkenyl nickel species. This transformation not only expands the scope of transition-metal-catalyzed boration of internal alkynes but also, more particularly, portrays the vast prospects of the directing group strategy in the selective functionalization of unactivated alkynes.
Introduction
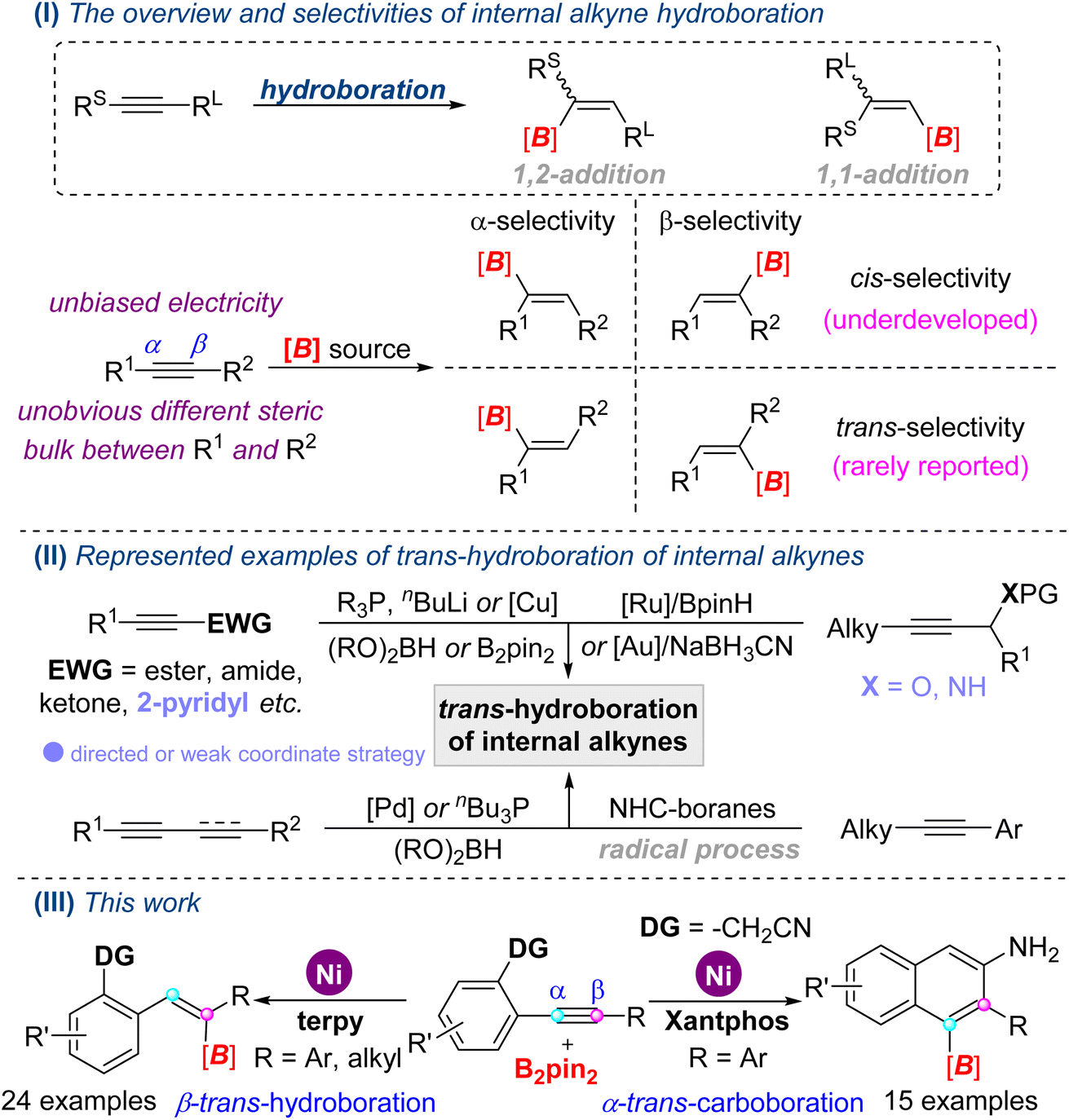
Organoboron compounds are versatile building blocks in the pharmaceutical industry and materials science, and could be rapidly and conveniently converted into more complicated molecules via cross-coupling and other well-established reactions, in which the stereochemistry of organoboron compounds are always retained.1 Undeniably, the hydroboration reaction of easily available unsaturated hydrocarbons is still a straightforward and indispensable method to synthesize boron-containing compounds.2Generally, the hydroboration of alkynes with trivalent boranes provides synthetically useful alkenyl boron compounds, including 1,2-addition3a–c and 1,1-addition products;3d however, the cis-addition mode is strictly complied in the transition metal-catalyzed 1,2-hydroboration reactions (Scheme 1, I). Even so, trans-selective hydroboration of terminal alkynes was firstly realized through the metal vinylidene intermediate two decades ago.4 With respect to the hydroboration of internal alkynes, regio- and stereoselectivity would give mixtures of α- and β-addition, cis- and trans-addition products, due to isomerization and other related processes.5
 | ||
| Scheme 1 Background and project synopsis. | ||
Since Früstner's group reported the first real trans-hydroboration of internal alkynes with [Cp*Ru(MeCN)3]PF6 and HBpin,6 a few examples of trans-hydroboration reactions have been disclosed in the past decade (Scheme 1, II). However, these work particularly well with symmetrical internal alkynes,6 activated internal alkynes,7 1,3-enynes8 or 1,3-diynes.9 For the unsymmetrical internal alkynes with unbiased electricity, few examples were reported, which were limited to a weak coordinate group (propargyl amines and ether)10 and NHC-boryl radical process.11 To the best of our knowledge, ubiquitous electronically unbiased internal alkynes with unobvious different steric bulks between R1 and R2 rarely underwent trans-hydroboration reactions because of unmanageable regio- and stereoselectivity. Therefore, the development of feasible, efficient methods for the hydroboration of widespread unsymmetrical internal alkynes is highly desirable. As part of our continuing studies on transition-metal catalyzed boronation reactions of multiple bonds with the hydrostable and easily handled diboron(4) compounds (B2(OR)4) as a boron source,12 herein, we developed nickel-catalyzed trans-hydroboration of electronically unbiased internal alkynes. Furthermore, opposite regioselectivity in a cyclization carboboration reaction was also established depending on the regulation of the ligand (Scheme 1, III).
Results and discussion
In order to acquire high regioselectivity, we attempted to utilize the directing group strategy in the hydroboration of internal alkynes.13 And optimization of the directing group and reaction conditions was carried out in parallel (Table 1). Firstly, due to the good coordinating function of cyano-containing groups, the cyano group (–CN) and cyanomethyl (–CH2CN) were installed next to the triple bond and showcased disparate outcomes (entries 1 and 2). Furthermore, the electron-deficient sulfamide with lower pKa values of N–H bonds was unreactive (entry 3). An oxygen atom as a coordinating site was considered and diaryl alkynes with methyl ketone (–COCH3) among oxygen-containing directing groups (entries 4–6) afford the trans-hydroboration product with 47% yield. Obviously, diphenyl acetylene only generated unassigned geometry alkenylborate B in very low yield without the aid of an auxiliary group (entry 7). Taken together, after initial screening revealed that a cyanomethyl (–CH2CN) directing group (as in 1) provided the highest yield and selectivity, reaction conditions were optimized with this substrate as a model substrate (1). From the reaction conditions (entry 1) as a reference, this transformation could also work by other nickel salts catalysis, albeit with lower yields of 50–73% (entries 8–12). However, changing the ligands has an effect on the reaction so that quite a few other products are formed. The carboboration product 1-boryl naphthylamine 3 was generated while xantphos acted as a ligand.14 Next, various kinds of base were screened, and phosphate salts were still the optimized choice (entries 19–21). Lastly, a single solvent system was beneficial for the production of 2 and 3, respectively (entries 22–23). Noticeably, trans-hydroboration product 2 could not be obtained without the limited detection amount under anhydrous conditions (entry 24). Lastly, quantitative water was added in the anhydrous solvent for better reproducibility (entry 25).|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | DG | [Ni] | L | Base | Solvent | B (%) | 3 (%) |
a Conditions: substrate A (0.2 mmol), nickel catalyst (5 mol%), ligand (10 mol%), B2pin2 (1.5 equiv.), base (1.5 equiv.), cyclohexane (Cyh)/toluene (Tol) (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 2.0 mL) under an N2 atmosphere at 130 °C for 24 h in a sealed tube; yields were determined by 1H NMR with CH2Br2 as an internal standard, isolated yield is in parentheses; ND = not detected.
b Absolute dry cyclohexane.
c Absolute dry cyclohexane, H2O (5 equiv.).
d Absolute dry toluene, H2O (5 equiv.). :1, 2.0 mL) under an N2 atmosphere at 130 °C for 24 h in a sealed tube; yields were determined by 1H NMR with CH2Br2 as an internal standard, isolated yield is in parentheses; ND = not detected.
b Absolute dry cyclohexane.
c Absolute dry cyclohexane, H2O (5 equiv.).
d Absolute dry toluene, H2O (5 equiv.).
|
|||||||
| 1 | –CH2CN (1) | Ni(PPh3)2Cl2 | Terpy | K3PO4 | Cyh/Tol | 78 | ND |
| 2 | –CN (4) | Ni(PPh3)2Cl2 | Terpy | K3PO4 | Cyh/Tol | <10 | — |
| 3 | –CH2NHTs (5) | Ni(PPh3)2Cl2 | Terpy | K3PO4 | Cyh/Tol | ND | — |
| 4 | –COCH3 (6) | Ni(PPh3)2Cl2 | Terpy | K3PO4 | Cyh/Tol | 47 | — |
| 5 | –CHO (7) | Ni(PPh3)2Cl2 | Terpy | K3PO4 | Cyh/Tol | <10 | — |
| 6 | –CH2OH (8) | Ni(PPh3)2Cl2 | Terpy | K3PO4 | Cyh/Tol | ND | — |
| 7 | None (9) | Ni(PPh3)2Cl2 | Terpy | K3PO4 | Cyh/Tol | <10 | — |
| 8 | 1 | Ni(OAc)2 | Terpy | K3PO4 | Cyh/Tol | 58 | ND |
| 9 | 1 | Ni(dppe)Cl2 | Terpy | K3PO4 | Cyh/Tol | 50 | ND |
| 10 | 1 | Ni(dppp)Cl2 | Terpy | K3PO4 | Cyh/Tol | 53 | ND |
| 11 | 1 | Ni(acac)2 | Terpy | K3PO4 | Cyh/Tol | 55 | ND |
| 12 | 1 | Ni(cod)2 | Terpy | K3PO4 | Cyh/Tol | 73 | ND |
| 13 | 1 | Ni(PPh3)2Cl2 | Bpy | K3PO4 | Cyh/Tol | <10 | ND |
| 14 | 1 | Ni(PPh3)2Cl2 | t Bu-terpy | K3PO4 | Cyh/Tol | 23 | ND |
| 15 | 1 | Ni(PPh3)2Cl2 | DAF | K3PO4 | Cyh/Tol | 65 | ND |
| 16 | 1 | Ni(PPh3)2Cl2 | Xantphos | K3PO4 | Cyh/Tol | ND | 76 |
| 17 | 1 | Ni(PPh3)2Cl2 | DPEPhos | K3PO4 | Cyh/Tol | <10 | 57 |
| 18 | 1 | Ni(PPh3)2Cl2 | None | K3PO4 | Cyh/Tol | <5 | 21 |
| 19 | 1 | Ni(PPh3)2Cl2 | Terpy | Na3PO4 | Cyh/Tol | <10 | ND |
| 20 | 1 | Ni(PPh3)2Cl2 | Terpy | K2CO3 | Cyh/Tol | 34 | ND |
| 21 | 1 | Ni(PPh3)2Cl2 | Terpy | MeOK | Cyh/Tol | 15 | ND |
| 22 | 1 | Ni(PPh3)2Cl2 | Terpy | K3PO4 | Cyh | 88 (85) | ND |
| 23 | 1 | Ni(PPh3)2Cl2 | Xantphos | K3PO4 | Tol | ND | 79 (74) |
| 24b | 1 | Ni(PPh3)2Cl2 | Terpy | K3PO4 | Cyh | ND | ND |
| 25c | 1 | Ni(PPh3)2Cl2 | Terpy | K3PO4 | Cyh | 89 | ND |
| 26d | 1 | Ni(PPh3)2Cl2 | Xantphos | K3PO4 | Tol | ND | 78 |
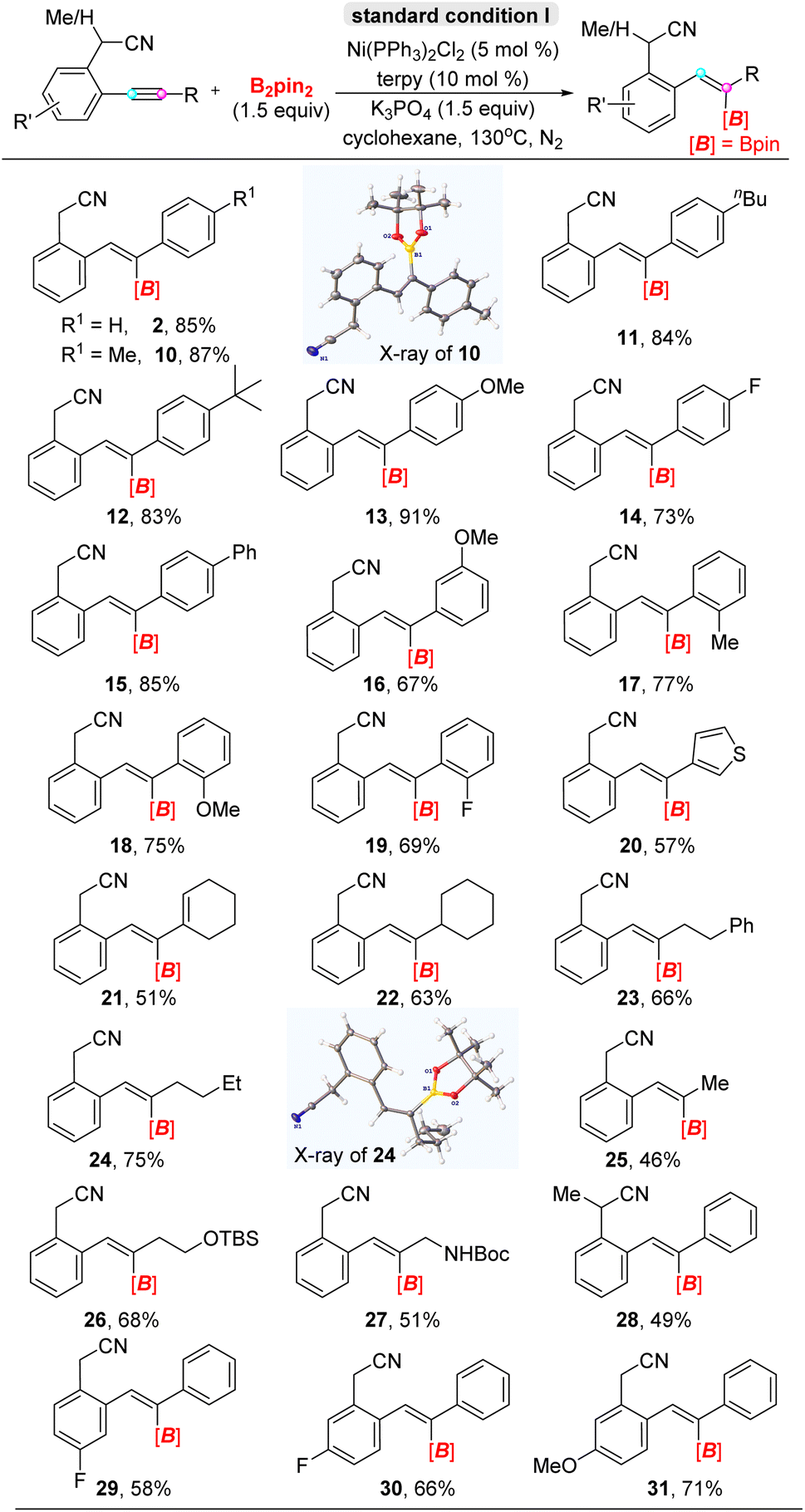
Having ascertained the optimized conditions with 2-(2-(phenylethynyl)phenyl)acetonitrile 1 as the standard substrate, we conducted trans-hydroboration reaction of a series of ortho-acetonitrile internal aryl alkynes with B2pin2 as the integrated partner (Table 2). In terms of the electronic properties, biaryl internal alkynes ranging with substitutions in para- and meta-positions including electron-donating and electron-withdrawing groups were well-tolerated and afforded the corresponding alkenyl boronates in moderate to excellent yields (10–16). Significantly, highly regioselective trans-alkenylborates transforming from biaryl alkynes with ortho-position substituents in moderate yields suggested that steric-hindrance couldn't influence regioselectivity under the present system (17–19). Furthermore, heteroaromatic aryl alkyne and conjugated enyne could be tolerated and converted into desired products (20). Of particular interest is that the reaction with high trans-addition selectivity was compatible with aryl–alkyl alkynes, that could give the moderate yields, that was compatible with 1-cyclohexenyl (21), cyclohexyl (22), phenethyl (23), n-butyl (24), methyl (25), siloxane (26) and carbamate (27). However, a secondary nitrile as a directing group was less beneficial for the reaction than a primary nitrile due to weaker coordination ability, which further supported the directing role of the –CH2CN group (28). On the other hand, the variation of substitutions on the acetonitrilyl phenyl ring was acceptable with slightly deceasing yields (29–31). Besides, the structures of alkenyl boronates 10 (CCDC: 2265346) and 24 (CCDC: 2265348)† in the solid state identified the constitution and assignment of the double bond geometry, and elaborative NMR analysis confirmed that either regio-isomer incorporated an E-olefin moiety. According to our knowledge, high stereoselectivity values (>20:1) have not previously been developed for any trans-addition hydroboration reactions of diaryl internal alkynes. Moreover, the trisubstituted alkenylborates obtained through the hydroboration of diaryl alkynes are difficult to obtain by known methods.
|
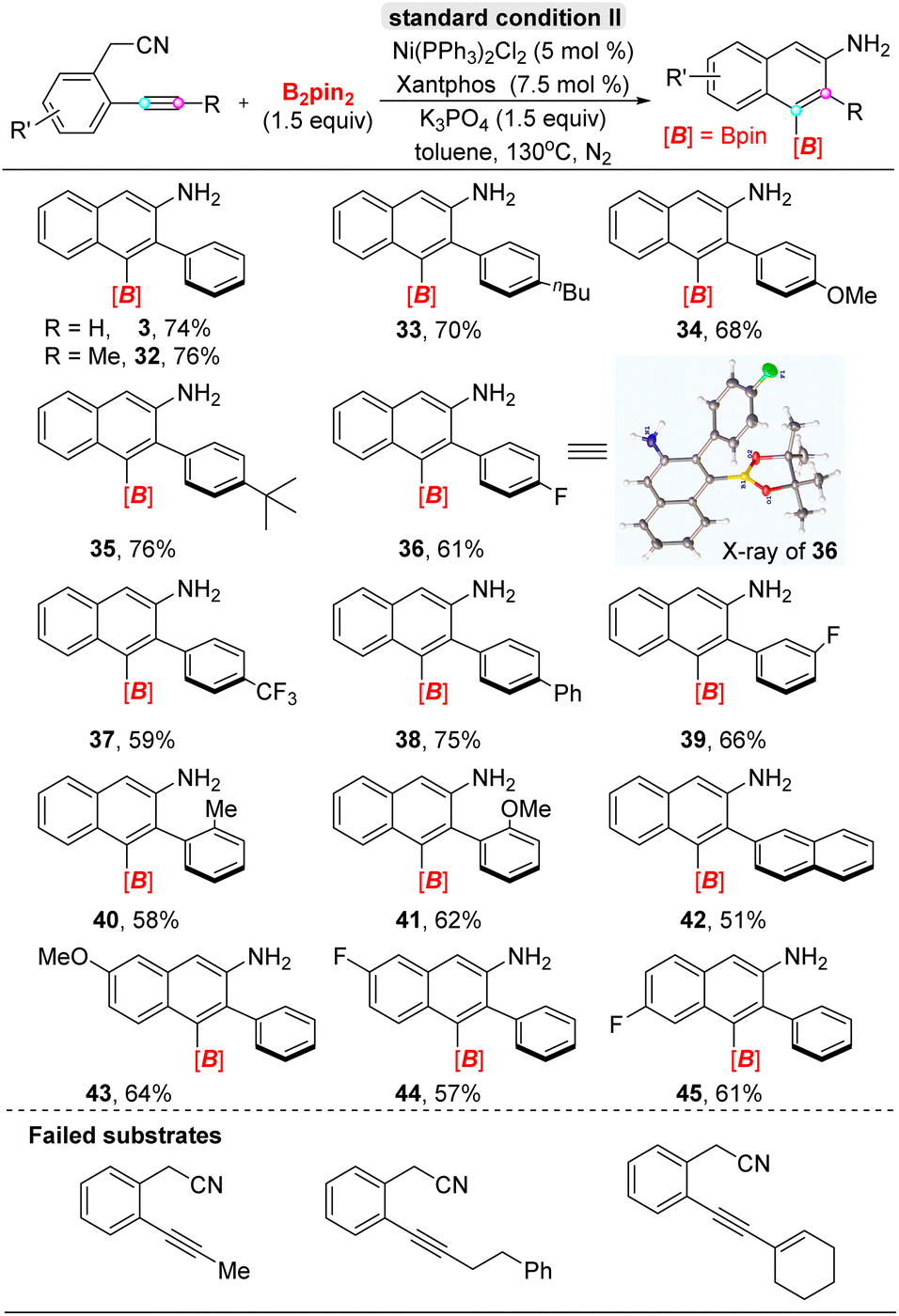
Next, we turned to evaluate the scope of ortho-acetonitrile internal aryl alkynes for the α-selective cyclization carboboration in the presence of xantphos ligand (Table 3), in which the –CH2CN group participated in the formation of naphthylamine derivatives and acted as a directing group and reaction partner. Generally, a wide range of diaryl alkynes, bearing electron-donating or withdrawing substituents on the para-, meta-, or ortho-positions of aromatic rings (32–41), all ortho-acetonitrile internal aryl alkynes, underwent efficient cyclization to furnish 1-boryl-2-aryl-3-naphthylamines with excellent regioselectivity. Even the hindrance effect of the ortho-position substituent group didn't influence reaction results (40–41), including 2-naphthyl-phenyl alkyne (42). Additionally, the electron-donating or withdrawing substitutions on the acetonitrilyl phenyl ring was compatible under standard conditions (43–45). Furthermore, the structure of 1-boryl-2-aryl-3-naphthylamine 36 was confirmed by X-ray analysis (CCDC: 2265345†); interestingly, single-crystal analysis of 36 showed phenyl and dioxaborolane rings are all vertical with naphthyl planes, and a formal dihedral angle existed between the adjacent two groups. Regretfully, aryl–alkyl alkynes failed in the cyclizative carboboration reaction for a special electronic effect.
|
To exhibit the practicality of this strategy, a gram-scale synthesis was performed under β-selective trans-hydroboration standard conditions with lower amounts of catalyst (3 mol%) and ligand (6 mol%) (Scheme 2). In view of the importance of alkenyl boronate in organic synthesis, we conducted some further transformations on the target product. As the examples show, 2 could be smoothly converted to a ketone (46) and polysubstituted alkene (47) via oxidation and cross-coupling processes. Intriguingly, 3-phenyl isoquinoline (48) instead of alkenyl azide was generated under the reaction of alkenyl boronate 2 with NaN3.15 Moreover, 2-amino-3-phenylnaphthalene (49) was acquired through an approach of rhodium catalysis.
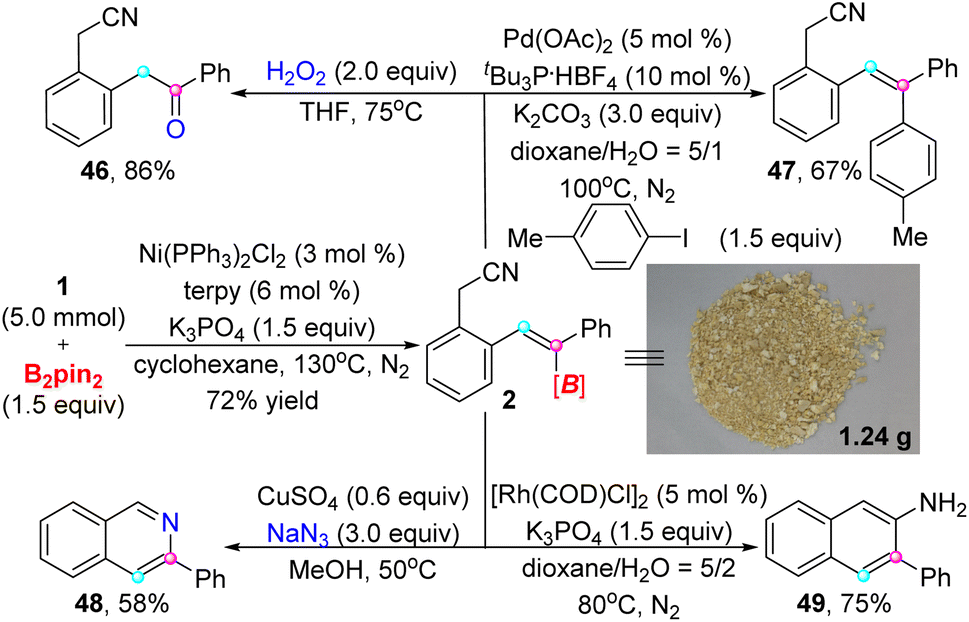 | ||
| Scheme 2 Gram-scale reaction and the transformation of alkenyl boronate product 2. | ||
Next, in order to gain insight into the detailed reaction mechanism, a series of control experiments were conducted (Scheme 3). Firstly, there was 51% proportion of deuterium detected at the double bond of d-24 and 67% proportion of deuterium detected in the activated methylene of d-24, indicating that water was the hydrogen source in the deuterium labeling experiments (eqn (I)). Meanwhile, deuterated starting materials d-50 locating in the position of activated methylene delivered the desired product d-24 without deuterium labeling in the double-bond position (eqn (II)). Furthermore, the Z-isomer of diaryl alkenyl boronate Z-2 and Z-24 couldn't transform into the corresponding E-isomer under the standard conditions I (eqn (III)).16 Additionally, BpinH was not suitable for the hydroboration and carboboration reactions instead of B2pin2 (eqn (IV)). Based on the above results, nickel hydride species could be ruled out in the hydroboration/carboboration process.17 In order to assess the importance of the directing group, diphenyl acetylene afforded the target product 51 with very low efficiency. And 2-acetyl diphenyl acetylene could convert into alkenyl boronate product 52 with 53% yield, since ketone may act as the directing group (eqn (V)). Last but not least, meta-CH2CN diphenyl acetylene 53 could convert into α/β-trans-isomers with the ratio of 84:16 in major/minor (see details in the ESI†),18 and meta-propionitrile diphenyl acetylene 54 was unreactive under the standard conditions II, and exhibited poor regioselectivity and reactivity, respectively (eqn (VI)).
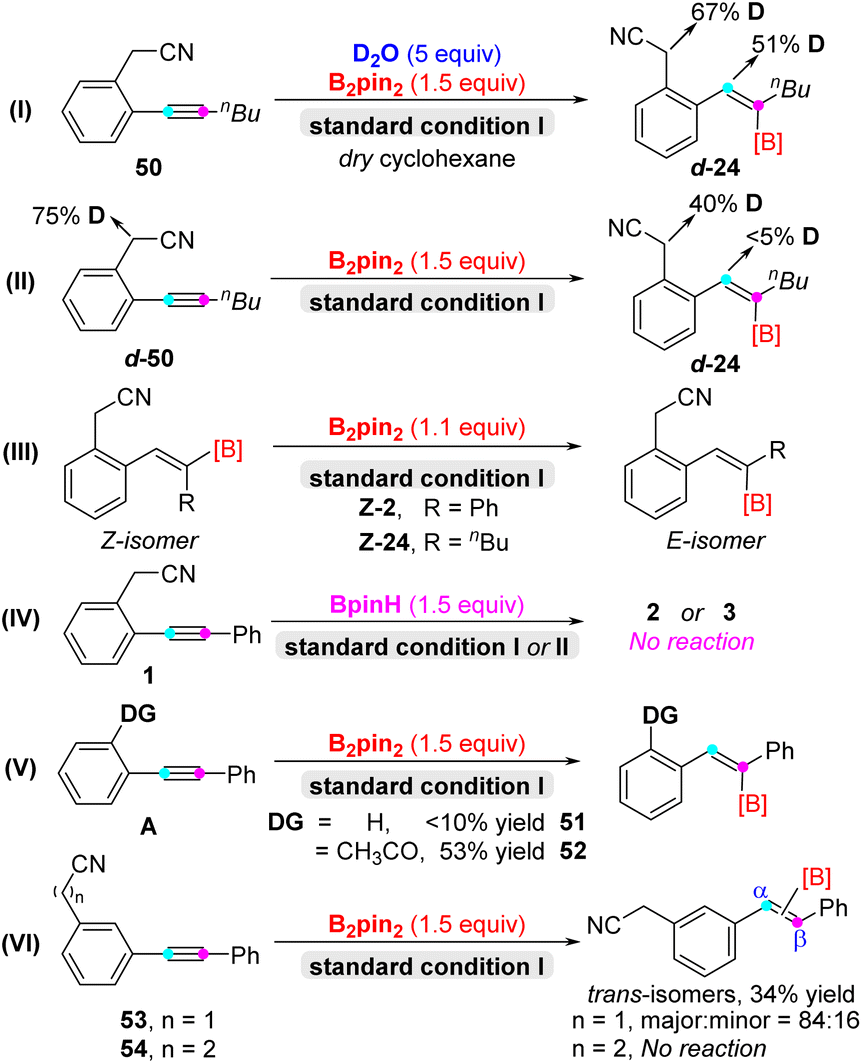 | ||
| Scheme 3 Control experiments. | ||
On the basis of the experimental results as well as previous reports,19 a plausible mechanistic pathway for the trans-hydroboration is tentatively proposed in Scheme 4 (left). Initially, Ni(PPh3)2Cl2 exchanges the phosphorus ligand with terpy to afford new nickel species (Bpin)Ni(terpy)Cl, that undergoes transmetalation with B2pin2 to generate (Bpin)Ni(terpy)Cl. Then, Int-1 is obtained through the coordination of (Bpin)Ni(terpy)Cl with the alkyne and cyano group of substrate 1 and experiences a cis-insertion process to deliver alkenylnickel species Int-2. Noticeably, reversible cis-to-trans isomerization between Int-2 and Int-3 is essential for the formation of trans-hydroboration. Finally, water-participation protolysis reaction of Int-3 gives the target 2 and releases the active nickel species (terpy)NiCl2. Furthermore, the mechanism of cis-to-trans isomerization may involve the intermediacy of zwitterionic carbene-type species TS-A according to primary DFT calculation (Fig. 1). Meanwhile, trans-Int-3 is more stable than cis-Int-2 due to removal of the huge steric-hindrance between the Bpin and tpy ligand, that may be the inherent driving force for selective formation of the trans-isomer (see details in the ESI†).20
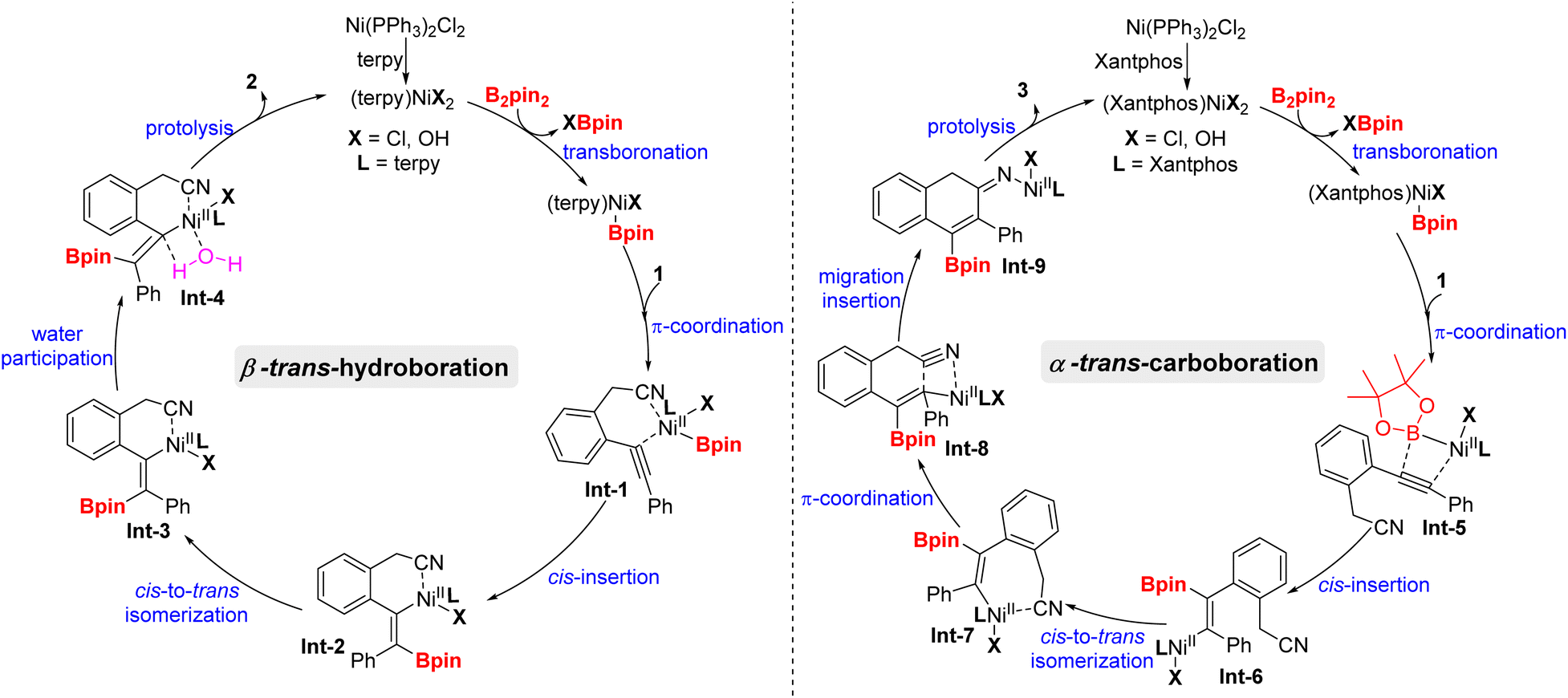 | ||
| Scheme 4 Plausible mechanistic pathways. | ||
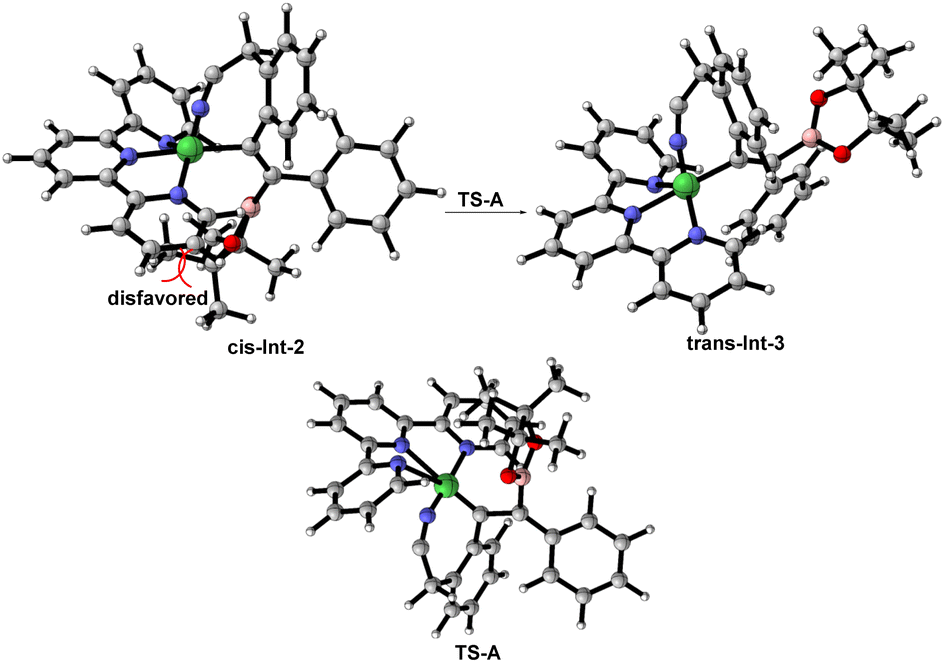 | ||
| Fig. 1 DFT calculation to investigate cis-to-trans isomerization. | ||
On the other hand (Scheme 4, right), when the nitrogen ligand terpy was replaced with phosphine ligand xantphos, the similar nickel complex (Bpin)Ni(Xantphos)Cl coordinates with substrate 1 without the aid of the cyano group due to the stronger coordination and steric hindrance effect of xantphos. Next, cis-nickelboration of Int-5 produces α-alkenyl borate Int-6, that undergoes cis-to-trans isomerization with the induction of the cyano group. Spontaneously, migration insertion of alkenyl nickel to the carbon–nitrogen triple bond in Int-7 of the cyano group generates cyclizative intermediate Int-9. Lastly, the successive protolysis and imine isomerization process of Int-9 produce the 1-boryl-2-aryl-3-naphthylamine product 3.
Conclusions
In conclusion, we have successfully developed a facile and efficient approach for the synthesis of high-value alkenyl and 1-naphthyl boronates with high stereo- and regioselectivity via ligand-controlled Ni-catalyzed trans-hydroboration/carboboration of unactivated internal alkynes. In the case of carboboration of alkynes, the cyano group played dual roles of directing group and reaction partner, delivering synthetically useful 1-boryl-2-aryl-3-naphthylamines. The detailed mechanism suggested that Ni-Bpin species should be the key intermediate and cis-to-trans isomerization of the alkenyl nickel intermediate was a crucial procedure for the unusual stereoselectivity. This system provides a potentially powerful strategy to access the trans-boration reaction of alkynes that may find applications in other trans-functionalization processes of internal alkynes.Data availability
General information, detailed experimental procedures, characterization data for all new compounds, NMR spectra, and X-ray crystallographic data are provided in the ESI.†Author contributions
J. Huang and C. Li conceived the idea. J. Huang and X. Li directed the project. Z. Chen, T. Liu and B. Nie performed the experiments. J. Huang and B. Nie wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Prof. Liang Xu (Shihezi University) for helpful suggestions on the DFT calculation. This work was supported by the National Natural Science Foundation of China (22361003), the Jiangxi Provincial Natural Science Foundation (20224BAB216005), Guangdong Basic and Applied Basic Research Foundation (2020A1515111156) and the Fundamental Research Funds for Gannan Medical University (QD202001).Notes and references
- (a) A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 RSC; (b) J. Jiao and Y. Nishihara, J. Organomet. Chem., 2012, 721, 3–16 CrossRef; (c) D. Shi, L. Wang, C. Xia and C. Liu, Chin. J. Org. Chem., 2020, 40, 3605–3619 CrossRef CAS.
- (a) M. Magre, M. Szewczyk and M. Rueping, Chem. Rev., 2022, 122, 8261–8312 CrossRef CAS PubMed; (b) A. Whyte, A. Torelli, B. Mirabi, A. Zhang and M. Lautens, ACS Catal., 2020, 10, 11578–11622 CrossRef CAS; (c) J. Hu, M. Ferger, Z. Shi and T. B. Marder, Chem. Soc. Rev., 2021, 50, 13129–13188 RSC.
- (a) S. Rej, A. Das and T. K. Panda, Adv. Synth. Catal., 2021, 363, 4818–4840 CrossRef CAS; (b) X. Wang, Y. Wang, W. Huang, C. Xia and L. Wu, ACS Catal., 2021, 11, 1–18 CrossRef CAS; (c) J. Altarejos, A. Valero, R. Manzano and J. Carreras, Eur. J. Org Chem., 2022, 2022, e202200521 CrossRef CAS; (d) Q. Feng, H. Wu, X. Li, L. Song, L. W. Chung, Y.-D. Wu and J. Sun, J. Am. Chem. Soc., 2020, 142, 13867–13877 CrossRef CAS PubMed; (e) Y.-X. Tan, S. Li, L. Song, X. Zhang, Y.-D. Wu and J. Sun, Angew. Chem., Int. Ed., 2022, 61, e202204319 CrossRef CAS PubMed.
- (a) T. Ohmura, Y. Yamamoto and N. Miyaura, J. Am. Chem. Soc., 2000, 122, 4990–4991 CrossRef CAS; (b) C. Gunanathan, M. Hölscher, F. Pan and W. Leitner, J. Am. Chem. Soc., 2012, 134, 14349–14352 CrossRef CAS PubMed.
- A. Fürstner, Isr. J. Chem., 2023, 63, e202300004 CrossRef.
- B. Sundararaju and A. Fürstner, Angew. Chem., Int. Ed., 2013, 52, 14050–14054 CrossRef CAS PubMed.
- (a) H. R. Kim and J. Yun, Chem. Commun., 2011, 47, 2943–2945 RSC; (b) K. Yuan, N. Suzuki, S. K. Mellerup, X. Wang, S. Yamaguchi and S. Wang, Org. Lett., 2016, 18, 720–723 CrossRef CAS PubMed; (c) R. Fritzemeier, A. Gates, X. Guo, Z. Lin and W. L. Santos, J. Org. Chem., 2018, 83, 10436–10444 CrossRef CAS PubMed; (d) Y. Zi, F. Schömberg, F. Seifert, H. Görls and I. Vilotijevic, Org. Biomol. Chem., 2018, 16, 6341–6349 RSC; (e) K. Nagao, A. Yamazaki, H. Ohmiya and M. Sawamura, Org. Lett., 2018, 20, 1861–1865 CrossRef CAS PubMed; (f) R. J. Grams, R. G. Fritzemeier, C. Slebodnick and W. L. Santos, Org. Lett., 2019, 21, 6795–6799 CrossRef CAS PubMed; (g) J. Corpas, M. Gomez-Mendoza, J. Ramírez-Cárdenas, V. A. de la Peña O'Shea, P. Mauleón, R. G. Arrayás and J. C. Carretero, J. Am. Chem. Soc., 2022, 144, 13006–13017 CrossRef CAS PubMed.
- S. Xu, Y. Zhang, B. Li and S.-Y. Liu, J. Am. Chem. Soc., 2016, 138, 14566–14569 CrossRef CAS PubMed.
- S. Jos, C. Szwetkowski, C. Slebodnick, R. Ricker, K. L. Chan, W. C. Chan, U. Radius, Z. Lin, T. B. Marder and W. L. Santos, Chem.–Eur. J., 2022, 28, e202202349 CrossRef CAS PubMed.
- (a) Q. Wang, S. E. Motika, N. G. Akhmedov, J. L. Petersen and X. Shi, Angew. Chem., Int. Ed., 2014, 53, 5418–5422 CrossRef CAS PubMed; (b) L. E. Longobardi and A. Fürstner, Chem.–Eur. J., 2019, 25, 10063–10068 CrossRef CAS PubMed.
- M. Shimoi, T. Watanabe, K. Maeda, D. P. Curran and T. Taniguchi, Angew. Chem., Int. Ed., 2018, 57, 9485–9490 CrossRef CAS PubMed.
- (a) X. Li, Z. Chen, W. Chen, X. Xie, H. Zhou, Y. Liao, F. Yu and J. Huang, Org. Lett., 2022, 24, 7372–7377 CrossRef CAS PubMed; (b) X. Li, Z. Chen, Y. Liu, N. Luo, W. Chen, C. Liu, F. Yu and J. Huang, J. Org. Chem., 2022, 87, 10349–10358 CrossRef PubMed; (c) J. Huang, L. Ouyang, J. Li, J. Zheng, W. Yan, W. Wu and H. Jiang, Org. Lett., 2018, 20, 5090–5093 CrossRef CAS PubMed; (d) J. Huang, W. Yan, C. Tan, W. Wu and H. Jiang, Chem. Commun., 2018, 54, 1770–1773 RSC.
- (a) J.-C. Wan, J.-M. Huang, Y.-H. Jhan and J.-C. Hsieh, Org. Lett., 2013, 15, 2742–2745 CrossRef CAS PubMed; (b) C. Lin, Z. Chen, Z. Liu and Y. Zhang, Org. Lett., 2017, 19, 850–853 CrossRef CAS PubMed; (c) T. Kang, N. Kim, P. T. Cheng, H. Zhang, K. Foo and K. M. Engle, J. Am. Chem. Soc., 2021, 143, 13962–13970 CrossRef CAS PubMed.
- (a) G. Xia, X. Han and X. Lu, Org. Lett., 2014, 16, 6184–6187 CrossRef CAS PubMed; (b) F. Yoshimura, T. Abe and K. Tanino, Org. Lett., 2016, 18, 1630–1633 CrossRef CAS PubMed; (c) M. Jash, B. Das and C. Chowdhury, J. Org. Chem., 2016, 81, 10987–10999 CrossRef CAS PubMed; (d) M. Jash, S. De, S. Pramanik and C. Chowdhury, J. Org. Chem., 2019, 84, 8959–8975 CrossRef CAS PubMed; (e) S. Pramanik, M. Jash, D. Mondal and C. Chowdhury, Adv. Synth. Catal., 2019, 361, 5223–5238 CrossRef CAS.
- X. Yang, C. Yuan and S. Ge, Chem, 2023, 9, 198–215 CAS.
- (a) S. Rao and K. R. Prabhu, Chem.–Eur. J., 2018, 24, 13954–13962 CrossRef CAS PubMed; (b) C.-Q. Zhao, Y.-G. Chen, H. Qiu, L. Wei, P. Fang and T.-S. Mei, Org. Lett., 2019, 21, 1412–1416 CrossRef CAS PubMed.
- (a) C. Wu and S. Ge, Chem. Sci., 2020, 11, 2783–2789 RSC; (b) A. Brzozowska, V. Zubar, R.-C. Ganardi and M. Rueping, Org. Lett., 2020, 22, 3765–3769 CrossRef CAS PubMed; (c) E. E. Touney, R. V. Hoveln, C. T. Buttke, M. D. Freidberg, I. A. Guzei and J. M. Schomaker, Organometallics, 2016, 35, 3436–3439 CrossRef CAS.
- The hydroboration products converted from meta-CH2CN diphenyl acetylene 53 were the α-trans-isomer and β-trans-isomer in comparison with the α-cis-isomer (CCDC: 2312562) and β-cis-isomer (CCDC: 2312563)†.
- (a) X. Liu, J. A. Henderson, T. Sasaki and Y. Kishi, J. Am. Chem. Soc., 2009, 131, 16678–16680 CrossRef CAS PubMed; (b) C. Yap, G. M. J. Lenagh-Snow, S. N. Karad, W. Lewis, L. J. Diorazio and H. W. Lam, Angew. Chem., Int. Ed., 2017, 56, 8216–8220 CrossRef CAS PubMed; (c) J. Jiang, H. Liu, L. Cao, C. Zhao, Y. Liu, L. Ackermann and Z. Ke, ACS Catal., 2019, 9, 9387–9392 CrossRef CAS; (d) Y. Yang, J. Jiang, H. Yu and J. Shi, Chem.–Eur. J., 2018, 24, 178–186 CrossRef CAS PubMed.
- Primary DFT calculations on the cis to trans isomerization are shown in the ESI† (a) T. Sperger, C. M. Le, M. Lautens and F. Schoenebeck, Chem. Sci., 2017, 8, 2914–2922 RSC; (b) C. Shan, M. He, X. Luo, R. Lia and T. Zhang, Org. Chem. Front., 2023, 10, 4243–4249 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2265345, 2265346, 2265348, 2312562 and 2312563. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04184k |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |