
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of altered backbone composition on the folding kinetics and mechanism of an ultrafast-folding protein†
Jacqueline R.
Santhouse‡
,
Jeremy M. G.
Leung‡
 ,
Lillian T.
Chong
* and
W. Seth
Horne
*
,
Lillian T.
Chong
* and
W. Seth
Horne
*
Department of Chemistry, University of Pittsburgh, Pittsburgh, PA 15260, USA. E-mail: ltchong@pitt.edu; horne@pitt.edu
First published on 4th December 2023
Abstract
Sequence-encoded protein folding is a ubiquitous biological process that has been successfully engineered in a range of oligomeric molecules with artificial backbone chemical connectivity. A remarkable aspect of protein folding is the contrast between the rapid rates at which most sequences in nature fold and the vast number of conformational states possible in an unfolded chain with hundreds of rotatable bonds. Research efforts spanning several decades have sought to elucidate the fundamental chemical principles that dictate the speed and mechanism of natural protein folding. In contrast, little is known about how protein mimetic entities transition between an unfolded and folded state. Here, we report effects of altered backbone connectivity on the folding kinetics and mechanism of the B domain of Staphylococcal protein A (BdpA), an ultrafast-folding sequence. A combination of experimental biophysical analysis and atomistic molecular dynamics simulations performed on the prototype protein and several heterogeneous-backbone variants reveal the interplay among backbone flexibility, folding rates, and structural details of the transition state ensemble. Collectively, these findings suggest a significant degree of plasticity in the mechanisms that can give rise to ultrafast folding in the BdpA sequence and provide atomic level insights into how protein mimetic chains adopt an ordered folded state.
Introduction
The idea expressed by Feynman that “what I cannot create, I do not understand”1 has inspired numerous efforts in nanoscience, including the creation of artificial systems that reproduce biological processes vital to life. One such process is protein folding. Sequence-encoded folding is not the exclusive purview of natural polypeptide chain connectivities,2 and diverse synthetic oligomeric molecules of artificial backbone composition are able to adopt discrete folded conformations.3–6 One approach to defined folding behavior is to engineer backbone composition in a natural peptide or protein sequence through replacement of a subset of α-amino acid residues with artificial monomers to produce a heterogeneous-backbone analogue.7,8 This approach can yield secondary structure mimetics and their assemblies as well as artificial analogues of complex tertiary folding patterns.9As the sophistication of folded structure in protein mimetics has increased, questions arise as to how the folding process of these entities compares to that of natural biomacromolecules. The mechanism by which an unfolded protein finds its way to a precise three-dimensional folded structure has long been a subject of intense study.10 Among many remarkable aspects of protein folding is the apparent disconnect between the vast number of conformational states theoretically possible in a given chain and the fact that most proteins in nature fold in seconds or much faster – the so-called Levinthal paradox.11 Despite revolutionary breakthroughs in the de novo prediction of protein folded structure from sequence,12–14 important gaps in knowledge remain surrounding the fundamental chemical principles that dictate the speed and mechanism of natural protein folding.15,16
While the structural and functional versatility of heterogeneous-backbone protein mimetics is well established, the fundamental folding behavior of these molecules is less explored. Most work to this end has focused on determining how changes to backbone composition influence folding thermodynamics.17–25 In contrast, efforts to probe folding kinetics in these systems are rare. In one pioneering example, a series of artificial β-turn mimetics were introduced in a hairpin loop found in the WW domain of the human protein Pin1.26,27 Altering local conformational behavior and thus β-turn nucleation had significant effects on folding rates; however, rigidification alone was not sufficient to achieve faster folding than the native.27 Another study measuring kinetic effects of backbone modification in β-turns focused on the consequences of enhanced conformational freedom through introduction of a β-dipeptide moiety as a reverse turn surrogate in ribonuclease A, where increased chain flexibility led to a faster unfolding rate.28 The kinetic effects of backbone alteration have also been examined in β-sheet secondary structure in the form of amide-to-ester substitution, which perturbs a backbone hydrogen bond.19 An important finding in that work was a pronounced context dependence—a single substitution could dramatically slow folding or have no measurable effect, depending on the exact position in sequence.
As part of an ongoing program seeking insights into fundamental folding behavior of protein tertiary structure mimetics,21,22,24 we recently explored the effects of altered backbone composition on the folded structure and thermodynamic stability of the B domain of protein A from Staphylococcal bacteria (BdpA), which adopts a compact tertiary fold consisting of three antiparallel α-helices.29 A combination of experimental biophysical analysis and atomistic simulation yielded insights into the folded states, unfolded states, and equilibrium folding thermodynamics of BpdA and heterogeneous-backbone variants in which artificial monomers of different types were introduced in different regions of the domain.30 Folded structures of the variants were virtually identical to the prototype natural protein; however, pronounced context-dependent effects of altered backbone composition on folding energetics and the conformational properties of the unfolded state ensemble were observed.30 Based on the above findings, we hypothesized altered backbone composition would impact the folding kinetics and mechanism of BdpA. Motivated by the demonstrated power of applying experiment and simulation in concert for the study of natural protein folding,31–33 we employed both methods in concert to gain an atomically detailed picture of how the folding process of BdpA is influenced by backbone alteration.
Results
Experimental analysis of folding kinetics
BdpA is an extremely fast folding protein sequence,34 and a G29A mutant has an even faster reported folding time (τf = 3 μs at 37 °C)35 that is near the theoretical “speed limit” for a protein of its size.36,37 BdpAG29A (WT, Fig. 1A) served as the prototype sequence in our prior work to understand structural and thermodynamic impacts of backbone alteration on the stable states (folded and unfolded) that define the folding equilibrium.30 Among variants we prepared and characterized in that study were two sequences in which identical side-chain retaining α→β3 residue substitutions were made in two different contexts of the native sequence. Replacing four α-residues at solvent exposed sites in helix 2 with the corresponding β3-residue analogue led to variant β3-H2, while four α→β3 substitutions at solvent-exposed sites in helix 3 led to variant β3-H3 (Fig. 1A). The rationale for selecting solvent-exposed sites for incorporation of artificial residues was to minimize the impact of altered backbone composition on the long-range contacts that stabilize the tertiary fold. Experimental analysis of folded structures by NMR showed that β3-H2 and β3-H3 adopt tertiary folds virtually identical to the prototype natural backbone (Fig. 1B).30 Compared to a canonical all-α-peptide backbone, each β3-residue substitution adds a freely rotatable Csp3–Csp3 bond to the chain. Curious to see the effects of such altered conformational flexibility on the folding kinetics of BdpA, we undertook experiments to that end.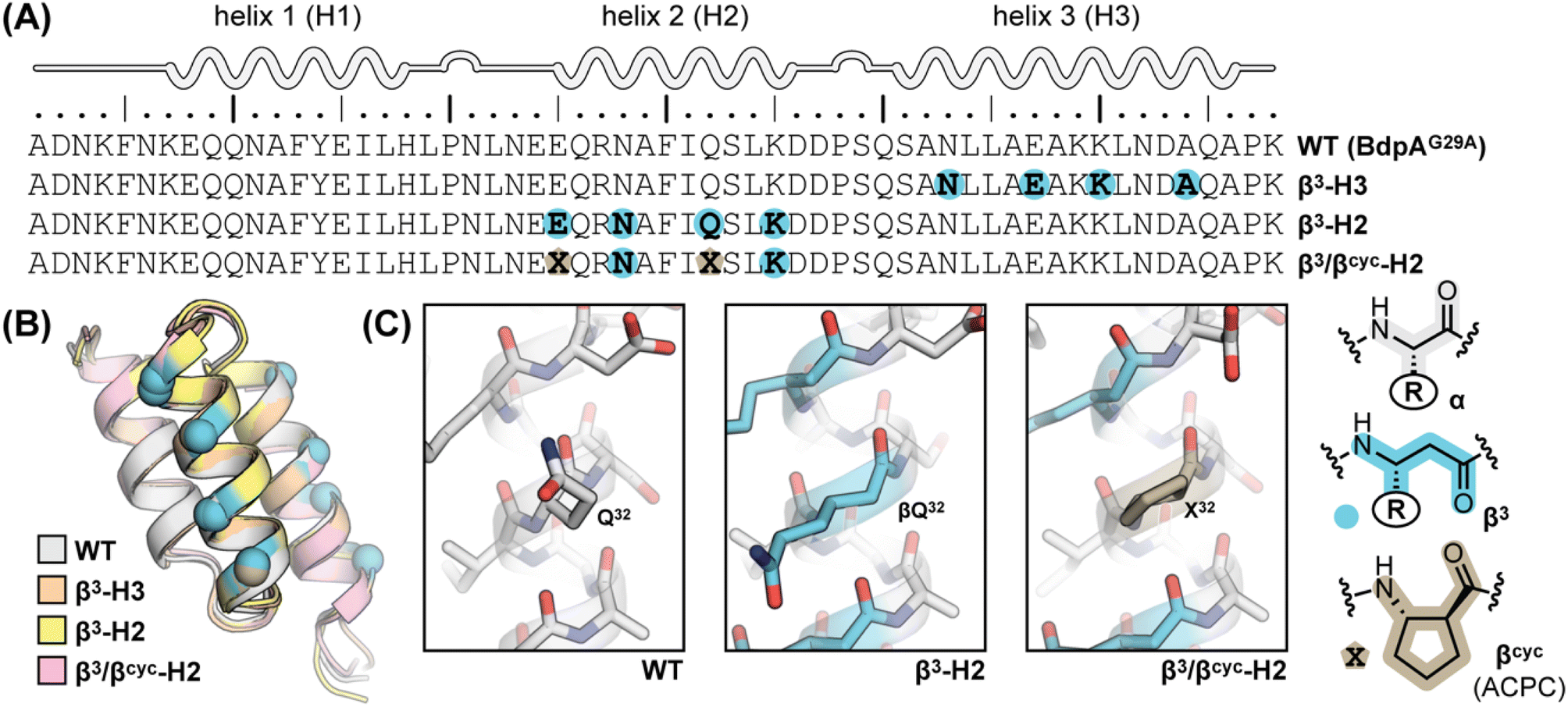 | ||
| Fig. 1 (A) Sequences of BdpA and variants. “R” groups in β3-residues match that of the α-residue specified by the corresponding single letter code. (B) Overlay of a representative model from the NMR structure ensembles of each indicated protein. Structures for WT (PDB 7TIO), β3-H2 (PDB 7TIP), and β3-H3 (PDB 7TIR) were previously reported,31 while that of β3/βcyc-H2 was determined in the present study. (C) Zoomed view around sequence position 32 in WT, β3-H2, and β3/βcyc-H2 showing the structural similarity among the native α-residue, β3 analogue, and cyclic β-residue ACPC in the corresponding proteins. | ||
The folding kinetics of BdpA and mutants have been assessed in prior work by a variety of experimental methods, including dynamic NMR,34,35 temperature jump fluorescence,38–40 and single molecule FRET.41 Among these, we were drawn to NMR due to the lack of need for an extrinsic label as well as local availability of suitable instrumentation. Pioneering efforts by Oas and co-workers have shown that an Hε resonance in the side chain of residue His18 of BdpA is a highly sensitive probe for the folding transition.34,35 When measurements are performed in D2O, this signal is a readily resolved singlet in a region of the 1HNMR spectrum otherwise devoid of signals. Further, it undergoes a large shift in frequency between the folded and unfolded states. Analysis of 1HNMR line shape for this signal over a range of concentrations of chemical denaturant yields folding (kf) and unfolding (ku) rate constants over the transition region that can be used to extrapolate corresponding folding and unfolding rate constants under benign conditions.34,35 Here, we applied the same NMR-based approach to BdpA β3-substitution variants β3-H2 and β3-H3. Experiments were carried out in 20 mM sodium acetate-d3, 100 mM NaCl in D2O at pH 5 (uncorrected for presence of deuterium). A mixture of urea and thiourea was employed as the denaturant, matching conditions used previously for WT, to avoid complications arising from the use of the ionic denaturant guanidinium chloride in the experiment.35 As the heterogeneous-backbone variants have lower thermal stability than WT,30 we performed NMR experiments at 10 °C rather than the 37 °C used previously for the native protein.
Based on NMR analysis of the His18 Hε signal for β3-H2 and β3-H3 (Fig. S1†), the apparent kf and ku values obtained showed the expected linear correlations in semi-log plots versus concentration of denaturant across the transition region (Fig. 2). Fits of these data sets provided kinetic parameters for the folding processes (Table 1 and Fig. 3). One finding apparent in comparing results for the two variants is that, despite their close structural similarity, the proteins have dramatically different folding rates. Variant β3-H3 (τf = 23 μs) folds on a timescale similar to WT reported at the same temperature (τf = 12 μs, as determined by temperature-jump fluorescence for a F13W mutant), while the folding process of β3-H2 is slowed dramatically (τf = 2.5 ms). Recall, β3-H2 and β3-H3 are regioisomeric molecules differing only in the placement of four methylene (–CH2–) units along the backbone of a 58-residue chain and are virtually indistinguishable in their folded states. Considering this close structural similarity, the >100-fold difference in folding rate is remarkable and suggests context dependent effects of enhanced chain flexibility on the folding process.
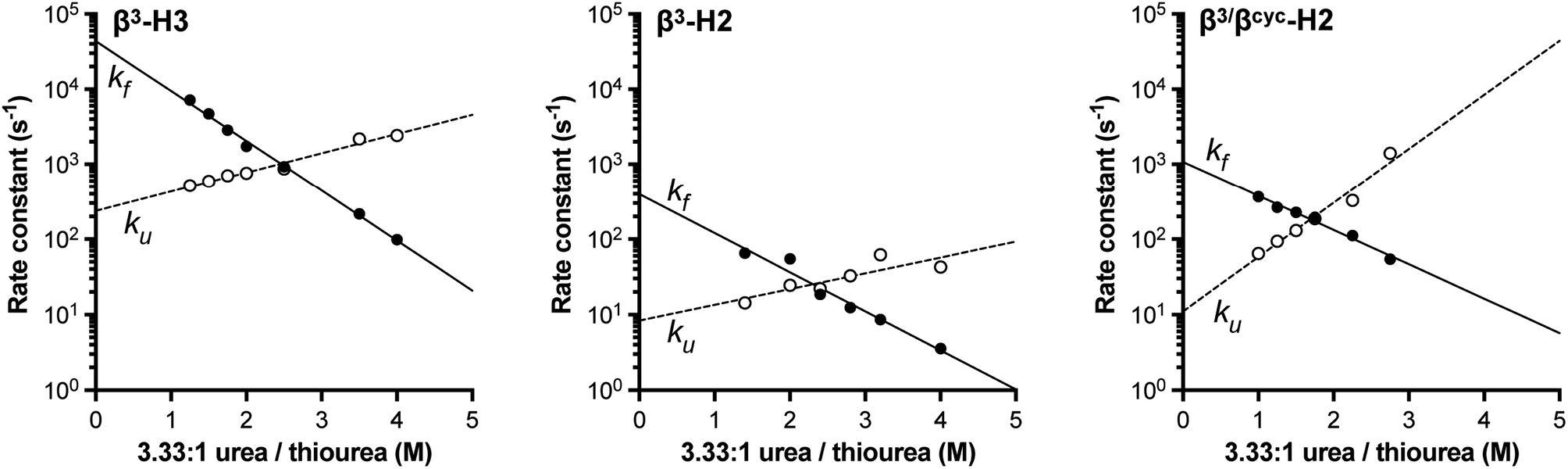 | ||
| Fig. 2 Folding (filled circles) and unfolding (open circles) rate constants determined for BdpA variants β3-H2, β3-H3, and β3/βcyc-H2 as a function of denaturant concentration by 1HNMR line shape analysis of the signal for the Hε resonance in residue His18 (Fig. S1, S2 and S4†). Lines depict linear regression fits of the natural logarithm of the rate constant versus denaturant concentration used to determine kinetic parameters reported in Table 1. Experiments were carried out in 20 mM sodium acetate-d3, 100 mM NaCl, pH 5 (uncorrected for presence of deuterium) at 10 °C. | ||
| WT | β3-H3 | β3-H2 | β3/βcyc-H2 | |
|---|---|---|---|---|
| a Conditions: 20 mM sodium acetate, 100 mM NaCl, pH 5 at 10 °C. b Values were reported previously38 and determined by temperature-jump fluorescence on a sequence with a F13W substitution relative to WT shown in Fig. 1. c Values determined in the present study by NMR with d3-acetate and in D2O. | ||||
| k f × 10−3 (s−1) | 83 ± 12 | 43.7 ± 4.3 | 0.39 ± 0.13 | 1.1 ± 0.1 |
| k u (s−1) | 2.5 ± 0.4 | 240 ± 30 | 8 ± 3 | 11 ± 3 |
| τ f (μs) | 12 ± 1 | 23 ± 2 | 2500 ± 800 | 900 ± 100 |
| m f (kcal mol−1 M−1) | 0.93 ± 0.19 | 0.85 ± 0.02 | 0.67 ± 0.06 | 0.59 ± 0.04 |
| m u (kcal mol−1 M−1) | 0.55 ± 0.19 | 0.33 ± 0.02 | 0.27 ± 0.08 | 0.93 ± 0.11 |
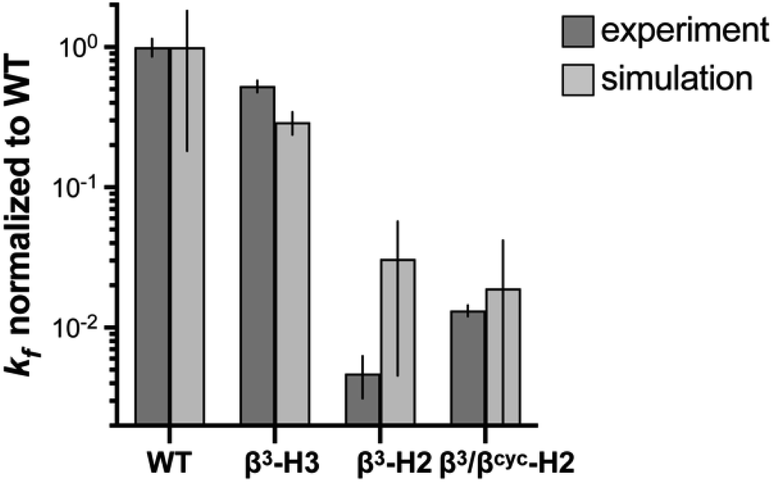 | ||
| Fig. 3 Comparison of relative folding rate constants for BdpA and variants determined by experiment and simulation. All values normalized to WT. Experimental values and uncertainties depict best fit parameters and parameter errors obtained from NMR analysis (Table 1). Simulation values depict averages from three independent haMSM estimates and uncertainties represent 95% confidence intervals (see Methods). | ||
Based on the reduced folding rate resulting from flexibility enhancing backbone alterations in β3-H2, we designed and synthesized an additional BdpA variant intended to rigidify the helix that had been altered. Thus, we replaced two of the β3-residues in β3-H2 with the conformationally constrained cyclic β-residue ACPC42 to generate BdpA variant β3/βcyc-H2. The trans-substituted five-membered ring in the ACPC monomer sets the central torsional angle at a value that promotes helical secondary structure in heterogeneous α/β-peptide backbones.22,23,43β3/βcyc-H2 was synthesized and purified following methods applied previously to WT and the other variants (Fig. S2†).30 While two side-chains are lost as a result of β3→ACPC substitution in β3/βcyc-H2 relative to β3-H2, these residues are solvent exposed and not engaged in long-range polar contacts in the structure of WT. To confirm the altered backbone composition in β3/βcyc-H2 did not have a significant effect on the folded state of the protein, we acquired 1H/1H COSY, TOCSY, and NOESY spectra, completed a full 1H resonance assignment, and determined the folded structure by simulated annealing with NMR derived restraints (Table S1 and Fig. S3†). These results support the innocuous nature of the two β3→ACPC substitutions with respect to the tertiary fold of the domain (Fig. 1B and C).
Subjecting β3/βcyc-H2 to the same dynamic NMR experiments described above (Fig. 2 and S4†) and comparison of the resulting kinetic parameters to those for β3-H2 (Table 1) revealed the enhanced backbone rigidity resulting from two β3→ACPC substitutions led to a ∼2.5-fold faster folding rate (decrease in τf from 2.5 ms to 900 μs). We attempted to rigidify the backbone further through replacement of all four β3-residues in β3-H2 with ACPC; however, the resulting protein showed evidence for aggregation in solution and was not amenable to biophysical analysis (data not shown).
The above comparison of folding behavior among WT and heterogeneous-backbone variants β3-H2, β3-H3, and β3/βcyc-H2 focused on folding rates; however, unfolding rate constants also differ considerably across the series. β3-H3, which folds on a timescale within 2-fold of WT, has an unfolding rate that is ∼100-fold faster. The origins of this effect are not clear. In contrast, data for β3-H2 and β3/βcyc-H2 show that impacts of altered backbone composition in helix 2 on the unfolding rate are modest.
Characterization of folding mechanism by simulation
To determine the impact of altered backbone composition and corresponding changes to chain flexibility on the BdpA folding mechanism at the molecular level, we applied all-atom molecular dynamics (MD) simulation using the weighted ensemble (WE) path sampling strategy,44,45 as implemented in the WESTPA 2.0 software package,46 to simulate complete folding pathways and corresponding rate constants for wild-type BdpA and the three heterogeneous-backbone variants. The WE strategy enhances the sampling of rare events by running multiple weighted trajectories in parallel and iteratively applying a resampling procedure in which promising trajectories are split and less-promising trajectories are merged while maintaining rigorous kinetics.44,45 This strategy has been demonstrated to be orders of magnitude more efficient than conventional MD simulations in generating pathways and rates for rare events such as protein folding47 and protein binding.48,49 The WE simulations employed the AMBER ff15ipq-m force field we recently developed and validated for simulation of protein-mimetic chains of the type under study here.50 The overall trend in the relative kf values for BdpA and variants observed in NMR experiments is reproduced by the simulations (Fig. 3 and Table S2†). WT folds the fastest, variant β3-H3 folds on a similar time scale to WT, and variants β3-H2 and β3/βcyc-H2 fold dramatically slower.One difference between the two methods was that relative kf values obtained for the slowest folding variant pair from simulation were within error of each other, while NMR analysis suggests β3/βcyc-H2 folds slightly faster. However, the uncertainty in the kf values from simulations for this variant pair is much larger compared to WT and β3-H3 as a result of the greater challenge in reaching converged non-equilibrium properties (i.e., rate constants) for the slower folding processes. The absolute kf values determined for all four proteins from simulations were similar to experimental observations despite differences in conditions for the two approaches. In particular, the simulations were performed at 25 °C, while experimental measurements made at 10 °C. In addition, simulations were initiated from an unfolded state that was sampled under non-denaturing conditions,30 while experimental kinetic parameters were obtained from extrapolation to non-denaturing conditions from measurements in the presence of urea.
A significant motivation in applying simulation in the present study was to gain atomically detailed information on how the folding process of BdpA was impacted by introduction of artificial backbone composition. To this end, we generated pairwise residue contact maps for the transition state ensemble of each protein and compared these maps to those of the corresponding unfolded and folded state ensembles (Fig. 4). For WT and all three variants, the helices are largely preformed in the transition-state ensemble, consistent with a diffusion–collision folding mechanism (Fig. 4A).34,51 For WT, helix 1 is the most conformationally flexible helix in the unfolded state, forming the fewest number of intra-helical contacts. This behavior holds for the unfolded states of β3-H2 and β3/βcyc-H2. For β3-H3, a greater degree of preorganization of helix 1 is seen in both the unfolded state and transition state, as evident by a large number of native contacts within that helix (Fig. 4B). This may contribute to the faster folding compared to the regioisomeric β3-H2 variant.
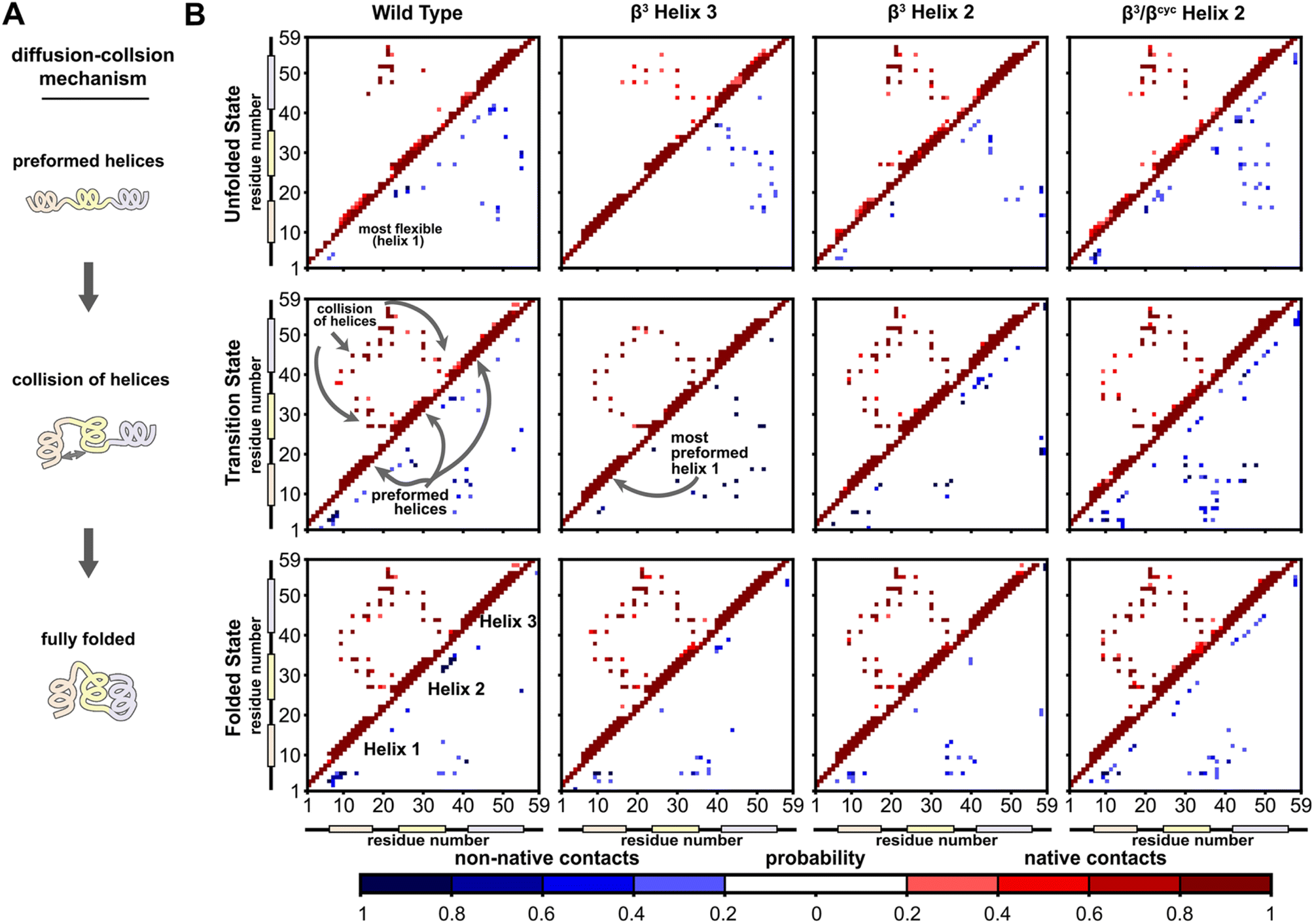 | ||
| Fig. 4 Folding simulations of BdpA and all three variants support a diffusion–collision mechanism. (A) Illustration of the diffusion–collision mechanism. (B) Probability maps of residue-level contacts for the unfolded state, transition state, and folded state ensembles for each BdpA variant. The region above and left of the diagonal shows probabilities in shades of red for contacts present in the reference folded structure (i.e., “native”) while the region below and right of the diagonal shows probabilities in shades of blue for contacts absent in the reference structure (i.e., “non-native”). Residues are considered in contact when the residue pair contains heavy atoms within 5 Å. See also Fig. S6–S9.† | ||
Overall, the transition-state contact maps of the helix 2 modified variants β3-H2 and β3/βcyc-H2 are similar to that of the WT, differing mainly by exact probabilities of specific pairwise interactions as well as the number of non-native contacts. The transition state ensemble for β3/βcyc-H2 shows multiple non-native contacts involving helix 2, which may be due to the loss of hydrophilic side chains upon substitution of Glu25 and Gln32 with the more hydrophobic ACPC. The transition state ensemble of β3-H3 bears the least resemblance to that of WT among the variant series, with the absence of multiple native interhelical interactions involving the loop regions at both ends of helix 3 as well as the formation of non-native intrahelical contacts within both helix 2 and helix 3 (Fig. S5†). In contrast, non-native contacts for the WT transition state ensemble are less probable and localized to flexible loops and terminal regions of the protein.
To identify kinetically important residues in the folding process for each BdpA variant, we searched for high probability contacts between helices that were formed in the transition state but not in the unfolded state of a given protein (Fig. 5). Results for WT reveal 19 residues are involved in such contacts. Of these 19 residues, 13 consisted of the BdpA hydrophobic core as defined from previous mutagenesis studies (Ala12, Phe13, Ile16, Leu17, Leu19, Leu22, Phe30, Ile31, Leu34, Leu44, Leu45, Ala48, and Leu51), while 6 others were interspersed elsewhere throughout the protein (Glu15, Pro20, Arg27, Ser41, Glu47, Gln55). The set of kinetically important residues observed for each heterogeneous-backbone BdpA variant is largely similar to the natural protein, though with some notable differences (Fig. S10†). β3-H2 is most like WT, sharing 17/19 kinetically important residues; the only native hydrophobic-core contact that is missing in a substantial portion (∼60%) of the transition-state ensemble of this variant is between Gln55 and Leu22. Furthermore, four additional kinetically important residues are seen for this variant in the vicinity of helix 2, the region where the backbone was altered. Variant β3-H3 shares 14/19 kinetically important residues with WT; however, numerous high probability non-native helix 1 to helix 3 and helix 2 to helix 3 contacts from are observed, many which are not present in the WT transition-state ensemble. For β3/βcyc-H2, 12/19 kinetically important residues are shared with WT alongside four new positions predominantly in helix 2.
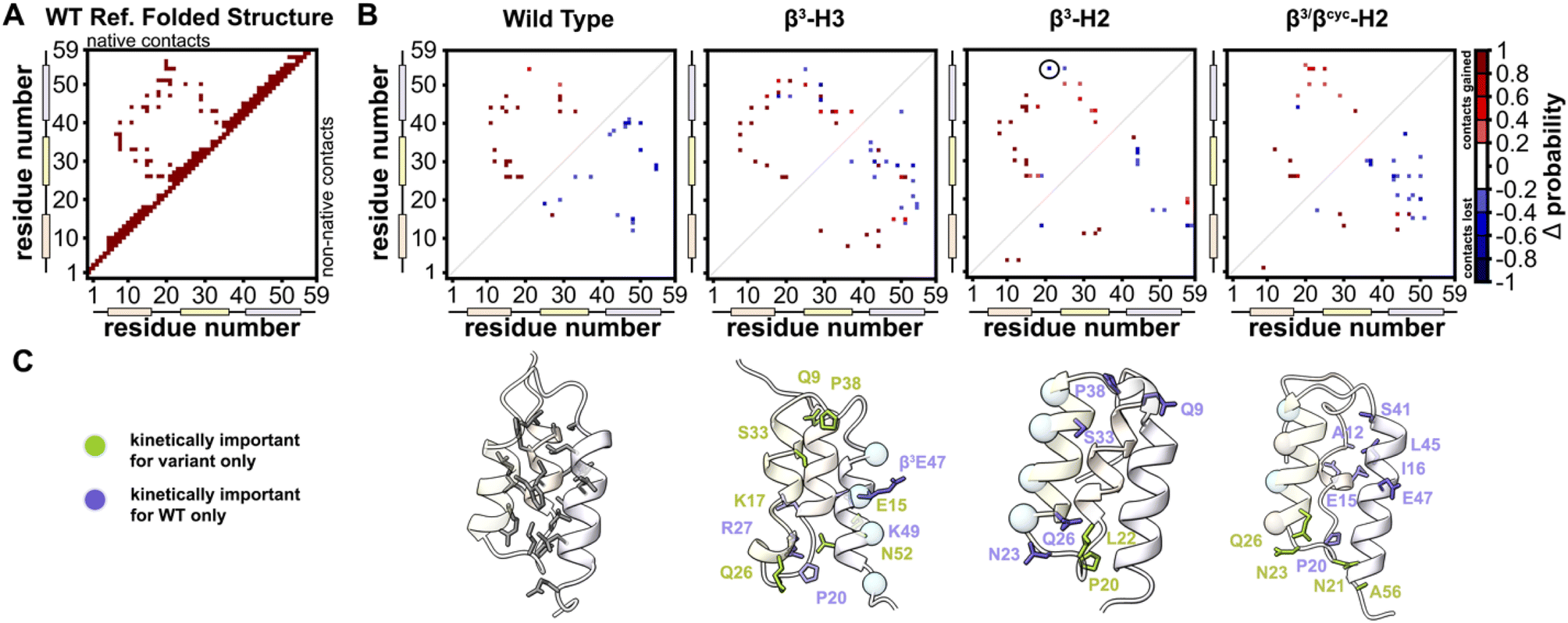 | ||
| Fig. 5 Identification of kinetically important residues in the folding mechanism of BdpA and variants. (A) Map of native contacts, as defined using an equilibrated folded structure of WT as reference. (B) Difference probability maps (Δ probability) of native (above diagonal line) and non-native (below diagonal line) interactions formed in the transition state ensemble (committor values between 0.45–0.55) relative to those formed in the unfolded state ensemble. Only gained contacts with ≥0.8 probability or lost contacts with ≤0.2 probability in the transition state ensemble are shown. Contacts involving atoms within ≤5 residues are omitted. Of note is a native contact between Gln55 and Leu22 in β3-H2 (circled) that has a ∼60% probability of being lost in the transition state; this is the only native hydrophobic-core contact missing in the transition state for this variant. (C) Ribbon diagrams of a representative transition state structure for each protein with kinetically important residues shown as sticks. For the heterogeneous-backbone variants, highlighting indicates kinetically important residues that are shared with WT (purple) or unique to the variant (green). | ||
Discussion
We have reported here an analysis of the effects of altered backbone composition on the folding kinetics and mechanism of the ultrafast-folding protein domain BdpA through a combination of biophysical experiments and atomistic MD simulations enabled by the WE path sampling strategy. Overall, good quantitative agreement was seen in relative folding rates obtained from the simulations and experiments. Backbone modification in BdpA through replacement of α-residues with β3-residue analogues led to two variants with the same side-chain sequence displayed on two isomeric backbone compositions. The added carbon resulting from each α→β3 substitution results in a new degree of conformational freedom and, thus, greater flexibility in the chain. How this chemical change influences the folding rate of the protein depends on context for the modifications.Incorporation of four β3-residues at solvent-exposed sites in the third helix of BdpA led to ultrafast folding behavior similar to the wild-type domain. In contrast, the same changes to backbone covalent structure made at solvent-exposed sites in the second helix led to an isomeric analogue with folding times two orders of magnitude slower. The observation that β3-residue incorporation slows folding is reasonable from first principles, given the resulting increase in conformational freedom in the backbone; however, the context-dependence of these effects is stark. The unfavorable kinetic consequences of enhancing flexibility in the second but not the third helix of BdpA is in accord with prior experimental as well as computational studies, which suggest that formation of the second helix may be a rate limiting step in folding of the domain.39,40,52 Rigidification of the slow-folding variant by substitution of two β3-residues in the second helix with cyclically constrained counterparts led to an increase in folding rate; however, the magnitude of the change was modest. This may result from an altered transition state ensemble, due to the incorporation of new hydrophobic moieties at solvent-exposed sites resulting from replacement of β3-residues bearing hydrophilic proteinogenic side chains with carbocyclic analogues.
Computational analysis of the folding process of BdpA and variants provided additional atomistic information on the effects of altered backbone composition on the folding mechanism of the protein. Unique insights gleaned from the simulations include the finding that the location of altered backbone composition in BdpA influences the extent to which specific residues are kinetically important to the folding process. In general, substitutions in the third helix of the domain result in a greater number of contacts in the transition state ensemble that are not present in the folded structure, while substitutions in the second helix reduce the number of native hydrophobic-core contacts in the transition state. Some prior studies have concluded that native contacts determine folding mechanisms in natural proteins.53,54 Interestingly, no correlation is apparent in the present data set between the extent to which the transition state of a given variant resembles that of the wild-type domain and the folding rate of the variant. Indeed, the fastest folding heterogeneous-backbone BdpA analogue folds nearly as fast as the prototype natural protein; however, this variant shows numerous non-native contacts in the transition state. These results suggest that a fast-folding sequence can retain that characteristic even when relying on a folding mechanism that involves critical long-range contacts that are not present in the folded state. In contrast, the slowest folding variant exhibits a transition state most like that of the prototype protein among the analogous examined yet folds on the order of ms rather than μs. This further supports the conclusion that the folding pathway of the domain is dramatically impeded by enhanced flexibility in the critical second helix.
Conclusions
Collectively, our findings suggest a degree of plasticity in the folding mechanisms that can give rise to ultrafast folding in the well-studied BdpA sequence. Enhanced chain flexibility introduced through backbone alteration in a helix can be accommodated without compromising folding rate but only in certain regions of the protein. Further, retention of fast folding after backbone modification in the case where this was observed was accompanied by significant changes to folding mechanism in the variant relative to the prototype domain. Our observations suggest a complex interplay exists among backbone composition, folding rates, and folding mechanism in artificial backbone protein mimetics. More broadly, the results of the present effort suggest protein backbone engineering in α-helices as a potentially general tool to control chain flexibility and establish the effects of such changes on the folding process of other systems – both fast folding sequences as well as chains that fold at a much slower rate. We anticipate ongoing application of backbone modification to this end will provide new fundamental insights into the importance of conformational freedom as a general chemical feature that determines the timescale and mechanism by which an unfolded protein chain adopts an ordered folded state.Data availability
Additional data supporting the findings of this study can be found in the ESI† for the article. Coordinates and associated experimental data for the NMR structure of β3/βcyc-H2 are deposited in the PDB (7URJ) and BMRB (31012). Other data are available from the corresponding authors upon request.Author contributions
J. R. S. and J. M. G. L. performed research; J. R. S., J. M. G. L., L. T. C. and W. S. H. analyzed data and wrote the paper.Conflicts of interest
L. T. C. is a member of the Scientific Advisory Board of OpenEye Scientific, Cadence Molecular Sciences; and an Open Science Fellow with Psivant Therapeutics.Acknowledgements
Funding for this work was provided by NSF (CHE-1807301 to W. S. H. and L. T. C.) and NIH (R01GM115805 to L. T. C., R35GM149220 to W. S. H.). Computational resources were provided through NSF XSEDE Award MCB-100109, MCB-180038 and ACCESS BIO-220144 for the use of PSC's Bridges-2 supercomputer and by the University of Pittsburgh Center for Research Computing (RRID: SCR_022735) for use of the H2P cluster, funded by NSF award number OAC-2117681. We are grateful for helpful discussions with Daniel Zuckerman and John Russo (OHSU) on the use of their haMSM restart plugin for the WESTPA 2.0 software package.References
- Richard Feynman’s blackboard at the time of his death, 1988, 1.10-67, Caltech Photographs, California Institute of Technology Archives and Special Collections, https://collections.archives.caltech.edu/repositories/2/archival_objects/106392, accessed June 9, 2023.
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173–180 CrossRef CAS.
- D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893–4011 CrossRef CAS PubMed.
- A. D. Bautista, C. J. Craig, E. A. Harker and A. Schepartz, Curr. Opin. Chem. Biol., 2007, 11, 685–692 CrossRef CAS PubMed.
- C. M. Goodman, S. Choi, S. Shandler and W. F. DeGrado, Nat. Chem. Biol., 2007, 3, 252–262 CrossRef CAS PubMed.
- G. Guichard and I. Huc, Chem. Commun., 2011, 47, 5933–5941 RSC.
- W. S. Horne and S. H. Gellman, Acc. Chem. Res., 2008, 41, 1399–1408 CrossRef CAS.
- K. L. George and W. S. Horne, Acc. Chem. Res., 2018, 51, 1220–1228 CrossRef CAS.
- W. S. Horne and T. N. Grossmann, Nat. Chem., 2020, 12, 331–337 CrossRef CAS.
- K. A. Dill and J. L. MacCallum, Science, 2012, 338, 1042–1046 CrossRef CAS.
- C. Levinthal, in Mossbauer Spectroscopy in Biological Systems: Proceedings of a meeting held at Allerton House, March 17 and 18, 1969, Monticello, Ill, ed. P. P. Debrunner, J. C. M. Tsibris and E. Münck, University of Illinois, Urbana, 1969, pp. 22–24 Search PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS.
- K. Tunyasuvunakool, J. Adler, Z. Wu, T. Green, M. Zielinski, A. Žídek, A. Bridgland, A. Cowie, C. Meyer, A. Laydon, S. Velankar, G. J. Kleywegt, A. Bateman, R. Evans, A. Pritzel, M. Figurnov, O. Ronneberger, R. Bates, S. A. A. Kohl, A. Potapenko, A. J. Ballard, B. Romera-Paredes, S. Nikolov, R. Jain, E. Clancy, D. Reiman, S. Petersen, A. W. Senior, K. Kavukcuoglu, E. Birney, P. Kohli, J. Jumper and D. Hassabis, Nature, 2021, 596, 590–596 CrossRef CAS PubMed.
- M. Baek, F. DiMaio, I. Anishchenko, J. Dauparas, S. Ovchinnikov, G. R. Lee, J. Wang, Q. Cong, L. N. Kinch, R. D. Schaeffer, C. Millán, H. Park, C. Adams, C. R. Glassman, A. DeGiovanni, J. H. Pereira, A. V. Rodrigues, A. A. van Dijk, A. C. Ebrecht, D. J. Opperman, T. Sagmeister, C. Buhlheller, T. Pavkov-Keller, M. K. Rathinaswamy, U. Dalwadi, C. K. Yip, J. E. Burke, K. C. Garcia, N. V. Grishin, P. D. Adams, R. J. Read and D. Baker, Science, 2021, 373, 871–876 CrossRef CAS.
- T. R. Sosnick and D. Barrick, Curr. Opin. Struct. Biol., 2011, 21, 12–24 CrossRef CAS PubMed.
- S.-J. Chen, M. Hassan, R. L. Jernigan, K. Jia, D. Kihara, A. Kloczkowski, S. Kotelnikov, D. Kozakov, J. Liang, A. Liwo, S. Matysiak, J. Meller, C. Micheletti, J. C. Mitchell, S. Mondal, R. Nussinov, K.-i. Okazaki, D. Padhorny, J. Skolnick, T. R. Sosnick, G. Stan, I. Vakser, X. Zou and G. D. Rose, Proc. Natl. Acad. Sci. U.S.A., 2023, 120, e2214423119 CrossRef CAS.
- W. Lu, M. A. Qasim, M. Laskowski and S. B. H. Kent, Biochemistry, 1997, 36, 673–679 CrossRef CAS.
- E. Chapman, J. S. Thorson and P. G. Schultz, J. Am. Chem. Soc., 1997, 119, 7151–7152 CrossRef CAS.
- S. Deechongkit, H. Nguyen, E. T. Powers, P. E. Dawson, M. Gruebele and J. W. Kelly, Nature, 2004, 430, 101–105 CrossRef CAS PubMed.
- D. Bang, A. V. Gribenko, V. Tereshko, A. A. Kossiakoff, S. B. Kent and G. I. Makhatadze, Nat. Chem. Biol., 2006, 2, 139–143 CrossRef CAS PubMed.
- Z. E. Reinert and W. S. Horne, Chem. Sci., 2014, 5, 3325–3330 RSC.
- N. A. Tavenor, Z. E. Reinert, G. A. Lengyel, B. D. Griffith and W. S. Horne, Chem. Commun., 2016, 52, 3789–3792 RSC.
- B. F. Fisher, S. H. Hong and S. H. Gellman, J. Am. Chem. Soc., 2018, 140, 9396–9399 CrossRef CAS PubMed.
- S. R. Rao and W. S. Horne, Pept. Sci., 2020, 112, e24177 CrossRef CAS.
- V. Bauer, B. Schmidtgall, G. Gógl, J. Dolenc, J. Osz, Y. Nominé, C. Kostmann, A. Cousido-Siah, A. Mitschler, N. Rochel, G. Travé, B. Kieffer and V. Torbeev, Chem. Sci., 2021, 12, 1080–1089 RSC.
- F. Liu, D. Du, A. A. Fuller, J. E. Davoren, P. Wipf, J. W. Kelly and M. Gruebele, Proc. Natl. Acad. Sci. U.S.A., 2008, 105, 2369–2374 CrossRef CAS.
- A. A. Fuller, D. Du, F. Liu, J. E. Davoren, G. Bhabha, G. Kroon, D. A. Case, H. J. Dyson, E. T. Powers, P. Wipf, M. Gruebele and J. W. Kelly, Proc. Natl. Acad. Sci. U.S.A., 2009, 106, 11067–11072 CrossRef CAS.
- U. Arnold, B. R. Huck, S. H. Gellman and R. T. Raines, Protein Sci., 2013, 22, 274–279 CrossRef CAS PubMed.
- H. Gouda, H. Torigoe, A. Saito, M. Sato, Y. Arata and I. Shimada, Biochemistry, 1992, 31, 9665–9672 CrossRef CAS.
- J. R. Santhouse, J. M. G. Leung, L. T. Chong and W. S. Horne, Chem. Sci., 2022, 13, 11798–11806 RSC.
- C. D. Snow, H. Nguyen, V. S. Pande and M. Gruebele, Nature, 2002, 420, 102–106 CrossRef CAS.
- U. Mayor, N. R. Guydosh, C. M. Johnson, J. G. Grossmann, S. Sato, G. S. Jas, S. M. Freund, D. O. Alonso, V. Daggett and A. R. Fersht, Nature, 2003, 421, 863–867 CrossRef CAS PubMed.
- A. J. Wirth, Y. Liu, M. B. Prigozhin, K. Schulten and M. Gruebele, J. Am. Chem. Soc., 2015, 137, 7152–7159 CrossRef CAS.
- J. K. Myers and T. G. Oas, Nat. Struct. Biol., 2001, 8, 552–558 CrossRef CAS.
- P. Arora, T. G. Oas and J. K. Myers, Protein Sci., 2004, 13, 847–853 CrossRef CAS.
- S. J. Hagen, J. Hofrichter, A. Szabo and W. A. Eaton, Proc. Natl. Acad. Sci. U.S.A., 1996, 93, 11615–11617 CrossRef CAS.
- J. Kubelka, J. Hofrichter and W. A. Eaton, Curr. Opin. Struct. Biol., 2004, 14, 76–88 CrossRef CAS.
- G. Dimitriadis, A. Drysdale, J. K. Myers, P. Arora, S. E. Radford, T. G. Oas and D. A. Smith, Proc. Natl. Acad. Sci. U.S.A., 2004, 101, 3809–3814 CrossRef CAS.
- S. Sato, T. L. Religa, V. Daggett and A. R. Fersht, Proc. Natl. Acad. Sci. U.S.A., 2004, 101, 6952–6956 CrossRef CAS.
- D. M. Vu, E. S. Peterson and R. B. Dyer, J. Am. Chem. Soc., 2004, 126, 6546–6547 CrossRef CAS PubMed.
- H. Oikawa, K. Kamagata, M. Arai and S. Takahashi, J. Phys. Chem. B, 2015, 119, 6081–6091 CrossRef CAS PubMed.
- D. H. Appella, L. A. Christianson, D. A. Klein, D. R. Powell, X. Huang, J. J. Barchi and S. H. Gellman, Nature, 1997, 387, 381–384 CrossRef CAS.
- W. S. Horne, J. L. Price and S. H. Gellman, Proc. Natl. Acad. Sci. U.S.A., 2008, 105, 9151–9156 CrossRef CAS.
- G. A. Huber and S. Kim, Biophys. J., 1996, 70, 97–110 CrossRef CAS PubMed.
- D. M. Zuckerman and L. T. Chong, Annu. Rev. Biophys., 2017, 46, 43–57 CrossRef CAS PubMed.
- J. D. Russo, S. Zhang, J. M. G. Leung, A. T. Bogetti, J. P. Thompson, A. J. DeGrave, P. A. Torrillo, A. J. Pratt, K. F. Wong, J. Xia, J. Copperman, J. L. Adelman, M. C. Zwier, D. N. LeBard, D. M. Zuckerman and L. T. Chong, J. Chem. Theory Comput., 2022, 18, 638–649 CrossRef CAS PubMed.
- U. Adhikari, B. Mostofian, J. Copperman, S. R. Subramanian, A. A. Petersen and D. M. Zuckerman, J. Am. Chem. Soc., 2019, 141, 6519–6526 CrossRef CAS PubMed.
- M. C. Zwier, A. J. Pratt, J. L. Adelman, J. W. Kaus, D. M. Zuckerman and L. T. Chong, J. Phys. Chem. Lett., 2016, 7, 3440–3445 CrossRef CAS.
- A. S. Saglam and L. T. Chong, Chem. Sci., 2019, 10, 2360–2372 RSC.
- A. T. Bogetti, H. E. Piston, J. M. G. Leung, C. C. Cabalteja, D. T. Yang, A. J. DeGrave, K. T. Debiec, D. S. Cerutti, D. A. Case, W. S. Horne and L. T. Chong, J. Chem. Phys., 2020, 153, 064101 CrossRef CAS.
- J. K. Myers and T. G. Oas, Annu. Rev. Biochem., 2002, 71, 783–815 CrossRef CAS PubMed.
- S. Chowdhury, H. Lei and Y. Duan, J. Phys. Chem. B, 2005, 109, 9073–9081 CrossRef CAS PubMed.
- R. B. Best, G. Hummer and W. A. Eaton, Proc. Natl. Acad. Sci. U.S.A., 2013, 110, 17874–17879 CrossRef CAS.
- K. Lindorff-Larsen, S. Piana, R. O. Dror and D. E. Shaw, Science, 2011, 334, 517–520 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S19, Tables S1, S2, Materials and methods. See DOI: https://doi.org/10.1039/d3sc03976e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |