
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCluster dynamics of heterometallic trinuclear clusters during ligand substitution, redox chemistry, and group transfer processes†
Cristin E.
Juda
 ,
Rex C.
Handford
,
Amymarie K.
Bartholomew
,
Tamara M.
Powers
,
Nina X.
Gu
,
Elisabeth
Meyer
,
Nikolaj
Roth
,
Yu-sheng
Chen
,
Shao-Liang
Zheng
and
Theodore A.
Betley
*
,
Rex C.
Handford
,
Amymarie K.
Bartholomew
,
Tamara M.
Powers
,
Nina X.
Gu
,
Elisabeth
Meyer
,
Nikolaj
Roth
,
Yu-sheng
Chen
,
Shao-Liang
Zheng
and
Theodore A.
Betley
*
Department of Chemistry and Chemical Biology, Harvard University, Cambridge, MA 02139, USA. E-mail: betley@chemistry.harvard.edu
First published on 15th February 2024
Abstract
Stepwise metalation of the hexadentate ligand tbsLH6 (tbsLH6 = 1,3,5-C6H9(NHC6H4-o-NHSiMe2tBu)3) affords bimetallic trinuclear clusters (tbsL)Fe2Zn(thf) and (tbsL)Fe2Zn(py). Reactivity studies were pursued to understand metal atom lability as the clusters undergo ligand substitution, redox chemistry, and group transfer processes. Chloride addition to (tbsL)Fe2Zn(thf) resulted in a mixture of species including both all-zinc and all-iron products. Addition of ArN3 (Ar = Ph, 3,5-(CF3)2C6H3) to (tbsL)Fe2Zn(py) yielded a mixture of two trinuclear products: (tbsL)Fe3(μ3-NAr) and (tbsL)Fe2Zn(μ3-NAr)(py). The two imido species were separated via crystallization, and outer sphere reduction of (tbsL)Fe2Zn(μ3-NAr)(py) resulted in the formation of a single product, [2,2,2-crypt(K)][(tbsL)Fe2Zn(μ3-NAr)]. These results provide insight into the relationship between heterometallic cluster structure and substitutional lability and could help inform both future catalyst design and our understanding of metal atom lability in bioinorganic systems.
1 Introduction
Metalloenzyme cofactors (e.g., FeMoco in nitrogenase,1–4 oxygen evolving complex (OEC) in photosystem II5–9) and heterogeneous catalysts (e.g., Haber–Bosch,10,11 Fischer–Tropsch12) are known to utilize polynuclear reaction sites to afford multi-electron redox chemistry. However, full mechanistic understanding of these catalytic reactions remains elusive primarily due to the challenges associated with reaction monitoring.2,3,12,13 Consequently, synthetic polynuclear clusters, readily studied using a variety of spectroscopic techniques, have been developed as models for polynuclear active sites of biological14–22 and abiological23 catalysts. The breadth of spectroscopic methods for monitoring the reactivity of synthetic clusters allows one to probe cluster dynamics in a variety of chemical reactions. Specifically, we are interested in probing metal atom lability and changes in coordination geometry during reaction processes. The extent to which these processes occur can provide insight into how cluster integrity changes during chemical transformations.We are particularly interested in investigating metal atom lability in synthetic heterometallic clusters given the prevalence of metalloenzyme heterometallic cofactors (e.g., nitrogenase cofactors,1–4,24–26 carbon monoxide dehydrogenase,27–30 OEC,5–9,31,32 and the Mn/Fe cofactor in ribonucleotide reductase,33–35 see Fig. 1) as well as their conformational lability in during reactivity (e.g., nitrogenase, OEC).24,26,29–32 Given these motivations we sought to use a mixed-metal system to uncover fundamental principles of how cluster binding geometry and reactivity relate to metal atom lability within synthetic analogues.
 | ||
| Fig. 1 Example heterometallic cofactors in nature: (a) nitrogenase, (b) carbon monoxide dehydrogenase, (c) oxygen evolving complex, and (d) MnFe ribonucleotide reductase. | ||
Our lab has previously employed a hexadentate ligand, 1,3,5-C6H9-(NHC6H4-o-NHSitBuMe2)3 (tbsLH6), to yield both homo-21,36–38 and hetero-trinuclear39 clusters, including reactive, high spin (tbsL)Fe3(thf) (1) and (tbsL)Fe3(py) (2). Treatment of 1 with small molecule substrates results in multi-electron bond activation processes.36 Previous work by our lab has also demonstrated that reactive, high-spin clusters can undergo metal atom metathesis.40 Therefore, to test the metal atom lability of our clusters during reactions, we sought to explore reaction chemistry on a heterometallic trinuclear cluster. In this work, we describe the synthesis of [Fe2Zn] clusters and assess their substitutional integrity as they undergo substrate binding, oxidative group transfer, and outer sphere redox chemistry. In both substrate binding and group transfer chemistry, we observe redistribution of metals, whereas in the presence of a μ3-imido cap on the clusters, the cluster core maintains its integrity during redox processes. The results presented herein illustrate that for open-shell clusters, metal atom exchange can be facile even when starting with substitutionally homogeneous materials. This point is especially important given the formation of heterometallic enzymatic cofactors that do not necessarily feature electronically differentiated binding sites. Thus, formation of heterometallic species might form under mild conditions.
2 Results and discussion
2.1. Synthesis of diiron zinc trinuclear clusters
A transmetalation strategy was employed to synthesize the targeted [Fe2Zn] clusters. The Fe/Zn combination allows for the facile determination of metal atom composition via X-ray fluorescence (XRF) spectroscopy, as the Kα lines differ significantly in energy. Additionally, the large difference in atomic scattering factors for Fe and Zn potentially enables their discrimination through single-crystal X-ray diffraction (SC-XRD) analysis. In the absence of coordinating solvent, metalation of tbsLH6 with Fe2(N(SiMe3)2)4 forms (tbsLH2)Fe2 (3; Scheme 1). Deprotonation of 3 with Na(N(SiMe3)2) (2 equiv.) in thawing THF yielded [Na(thf)][(tbsL)Fe2Na(thf)] (4). Addition of 4 to a frozen suspension of ZnCl2 in THF afforded (tbsL)Fe2Zn(thf) (5), which was crystallized from a mixture of hexane and benzene at −33 °C. Similarly, addition of ZnCl2py2 (ref. 41) to a frozen suspension of 4 in benzene afforded (tbsL)Fe2Zn(py) (6). Crystals suitable for SC-XRD analysis were obtained from a concentrated solution of 6 in diethyl ether at −33 °C (Scheme 1). | ||
| Scheme 1 Synthesis of (tbsL)Fe2Zn(thf) (5) and (tbsL)Fe2Zn(py) (6). | ||
SC-XRD studies of 5 (Fig. 2a) and 6 (Fig. S37†) reveals C1-symmetric complexes containing a [Fe2Zn] core, isostructural to 1 and 2. The average M–M distances of 5 (dFe–Fe: 2.7354(9) Å, dFe–Zn: 3.051(1) Å) and 6 (dFe–Fe: 2.8141(7) Å, dFe–Zn: 2.9366(8) Å) are longer than the average M–M distances in the tri-iron cores of 1 (2.577(6) Å)11 and 2 (2.608(1) Å). Stabilization of the triiron core via direct exchange pathways results from short Fe–Fe contacts, thus incorporation of zinc, which does not participate in this exchange, increases the average M–M separation.37 We assigned the two metal sites with the shortest M–M contact as iron, leaving zinc at the three-coordinate site (Fig. 2a). X-ray fluorescence (XRF) spectroscopy was used to determine the bulk metal composition of the crystalline samples. Using a calibration curve to correct for the differential absorption power of the two metals, the Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zn molar ratios were measured as 2.2:1 for 5 and 2.1:1 for 6. These values are consistent with the expected 2:1 ratio.
:Zn molar ratios were measured as 2.2:1 for 5 and 2.1:1 for 6. These values are consistent with the expected 2:1 ratio.
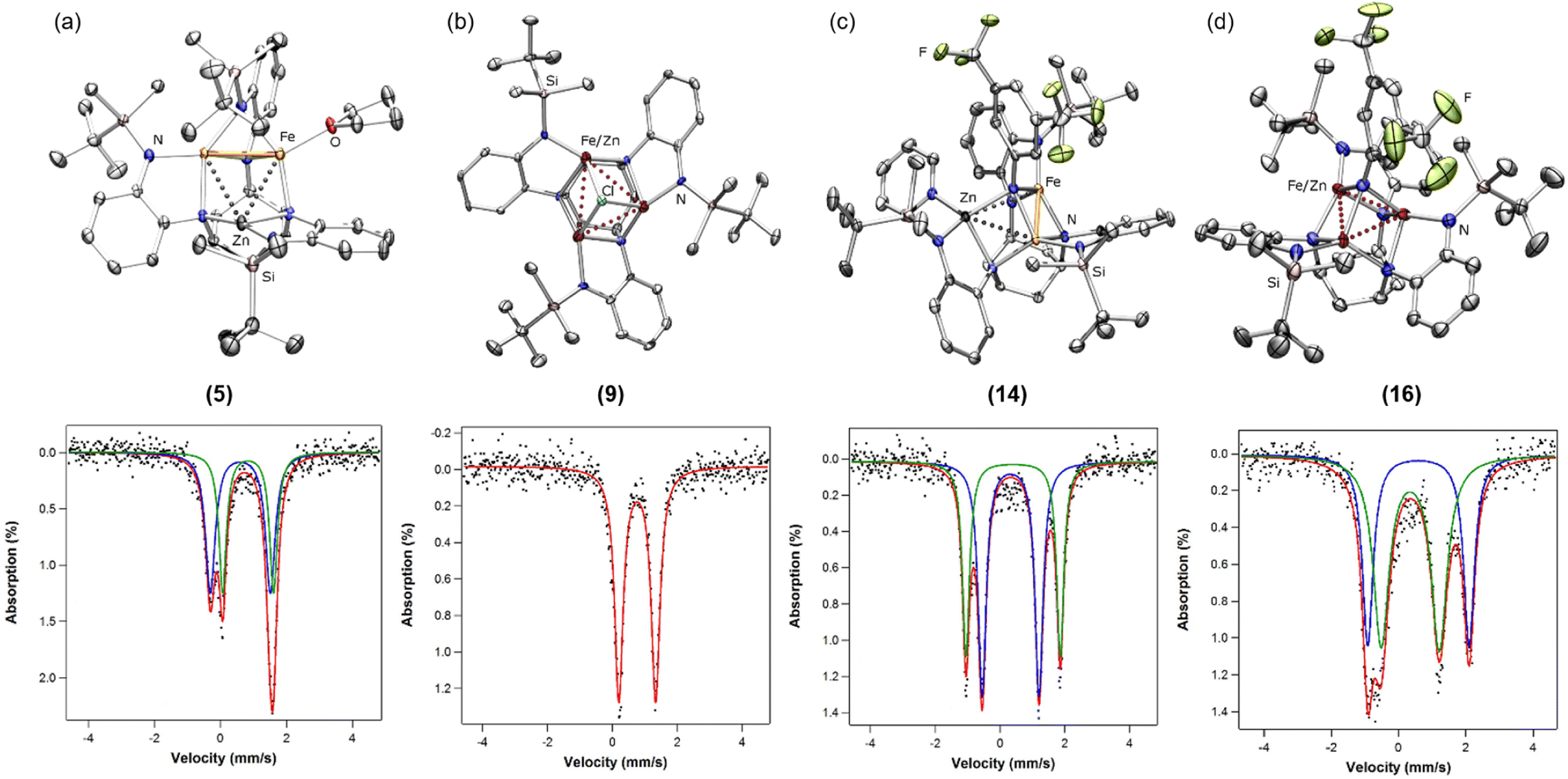 | ||
| Fig. 2 Solid-state molecular structures and zero-field 57Fe MB spectra of (a) (tbsL)Fe2Zn(thf) (5), (b) [NBu4][(tbsL)FenZn3−n(μ3-Cl)] (9), (c) (tbsL)Fe2Zn(μ3-NArF)(py) (14), (d) [2,2,2-crypt(K)][(tbsL)Fe2Zn(μ3-NArF)] (16) with 50% probability ellipsoids. Hydrogen atoms and counter ions omitted for clarity. Zero-field 57Fe MB spectra collected at 90 K. Fitting parameters: δ, |ΔEQ| (mm s−1) for (a): 0.60, 1.83 (blue, 50%); 0.84, 1.55 (green, 50%) (b): 0.77, 1.13 (red) (c): 0.33, 1.76 (green, 46%); 0.40, 2.92 (blue, 54%) (d): 0.35, 1.73 (green, 50%); 0.59, 3.01 (blue, 50%). | ||
Zero-field 57Fe Mössbauer spectroscopy (57Fe MB) analysis of 1 and 2 reveals three iron environments in each (δ, |ΔEQ| (mm s−1) (%) for 1:36–38 0.88, 1.29 (28%); 0.37, 1.99 (36%); 0.46, 1.52 (36%); for 2: 0.33, 1.84 (38%); 0.55, 1.76 (28%); 0.74, 1.39 (33%); Fig. S1†). However, only two quadrupole doublets are observed for 5 and 6, indicating site-isolated insertion of zinc into the trinuclear core (δ, |ΔEQ| (mm s−1) for 5: 0.60, 1.83 (50%); 0.84, 1.55 (50%); for 6: 0.61, 1.68 (50%); 0.79, 1.63 (50%); Fig. 2a and S4†). Compared to 1 and 2, 5 and 6 lack a quadrupole doublet with an isomer shift near 0.3 mm s−1, corresponding to the three-coordinate site in the core.
The electronic structure of 5 was further interrogated by study of its magnetic susceptibility via SQUID magnetometry. A plot of XMT vs T indicates that 5 possesses a low-spin ground state (XMT (2.0 K) = 0.73 cm3 K mol−1). Beyond 100 K, the susceptibility increases monotonically with temperature, reaching a value of 3.99 cm3 K mol−1 at 300 K (Fig. 3a). Low-temperature magnetization studies of 5 reveal a saturation magnetization value of 0.58 NAμB, suggestive of a diamagnetic ground state for 5 with a small population of paramagnetic states remaining. The electronic structure of 5 stands in contrast to the structurally analogous, maximally high-spin S = 6 triferrous cluster (tbsL)Fe3(thf) (1).21 The susceptibility of 1 increases rapidly from 1.8 to 20 K from an initial value of 13.75 cm3 K mol−1, reaching a maximum of 19.33 cm3 K mol−1 at 150 K, and diminishing moderately to 17.80 cm3 K mol−1 at 300 K (Fig. 3a). A low-temperature magnetization study of triiron 1 reveals a saturation magnetization value of 8.32 NAμB, which is below the value anticipated for a maximally high-spin S = 6 system, in line with the presence of zero-field splitting (Fig. 3b). The susceptibility and magnetization data of 1 were best fit to a spin-Hamiltonian describing three coupled spins in an equilateral arrangement, providing J = 40(1) cm−1, D = 20.0(9) cm−1, |E/D| = 0.082(7) cm−1, and g = 1.92(1) (see ESI†). These data reveal that in 5, magnetic coupling of the two iron centers is dominated by antiferromagnetic superexchange through the dianilido bridge, and further suggests that the high-spin ground-state of isostructural (tbsL)Fe3(thf) (1) is strongly influenced by the incorporation of a third ferrous ion.
 | ||
| Fig. 3 (a) Plot of χMT vs T for 1 (blue circles) and 5 (red circles). (b) Plot of reduced magnetization for 1 from 1.8–10 K; data are represented as circles, and the fit is represented as black lines. | ||
2.2. Redox neutral substrate binding
To test the cluster integrity upon redox neutral substrate binding, tetrabutylammonium chloride was added to a solution of 5 in benzene (Scheme 2). Up to four isostructural products may be formed in this reaction: [NBu4][(tbsL)Fe3(μ3-Cl)] (7), [NBu4][(tbsL)Fe2Zn(μ3-Cl)], [NBu4][(tbsL)FeZn2(μ3-Cl)], and [NBu4][(tbsL)Zn3(μ3-Cl)] (8); of these, 7 and 8 were independently synthesized. Crystallization was achieved by diffusion of hexanes into a concentrated solution of the reaction product of 5 and tetrabutylammonium chloride in THF at −33 °C. The XRF spectrum of the crystalline product gave an Fe:Zn ratio of 2.1:1 (Fig. S16†). SC-XRD analysis revealed a pseudo C3-symmetric chloride-bridged cluster (Fig. 2b). Due to the symmetric ligand environment, the metal identity at each site could not be assigned, but the average M–M distance of 2.945(2) Å is longer than in 7 (2.772(1) Å) and shorter than in 8 (2.985(1) Å), consistent with formation of a species containing a mixture of Fe and Zn within the trinuclear core, which we nominally formulate as [NBu4][(tbsL)FenZn3−n(μ3-Cl)] (9; n = 1,2). The 57Fe MB spectrum of 9 (δ, |ΔEQ| (mm s−1): 0.77, 1.13) is similar to that of 7 (δ, |ΔEQ| (mm s−1): 0.72, 1.31), with spectra of both materials showing just one quadrupole doublet (Table S1† and Fig. 2b). Despite the solid-state structure of 7 revealing three distinct Fe–Fe distances, only one quadrupole doublet in the 57Fe MB spectrum is observed. The similarity of the spectral parameters for 7 and 9 suggest the iron sites are in similar electronic environments, and any Zn in 9 is evenly distributed amongst the three sites, and the diminished symmetry is not adequately resolved by 57Fe MB spectroscopy.
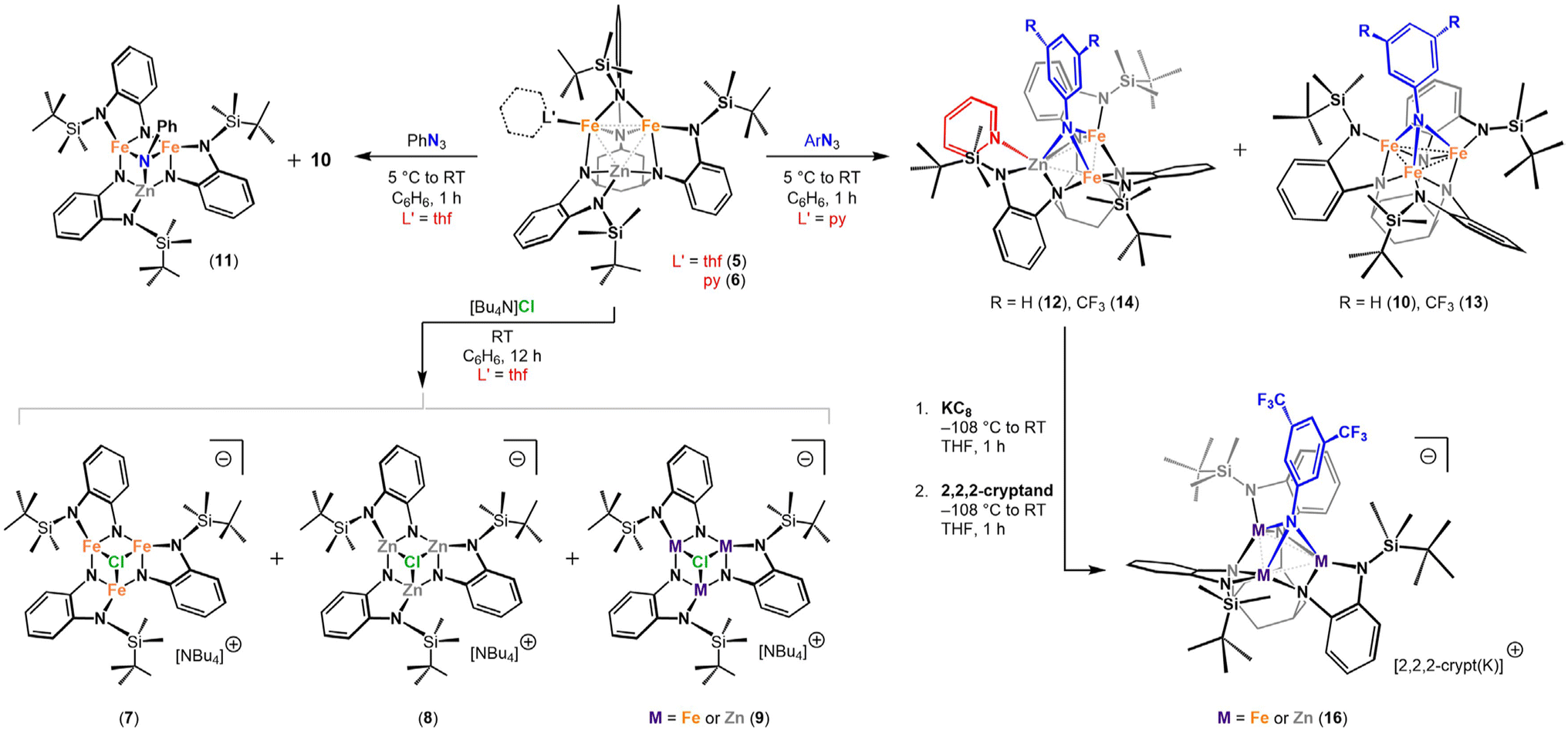 | ||
| Scheme 2 Reactivity of (tbsL)Fe2Zn(thf) (5) and (tbsL)Fe2Zn(py) (6). | ||
While the bulk metal ratio of (tbsL)Fe2Zn(thf) (5) was conserved upon chloride ligation, the 1H NMR spectrum of redissolved crystalline material (Fig. S20†) shows the all-iron and all-zinc clusters [NBu4][(tbsL)Fe3(μ3-Cl)] (7) and [NBu4][(tbsL)Zn3(μ3-Cl)] (8), alongside mixed-metal cluster 9. These data indicate that chloride anion substitution of THF in 5 results in metal atom redistribution. Indeed, the substitution of a labile ligand for chloride seems to be a determining factor in facilitating metal atom redistribution. Notably, such redistribution reactivity is hindered in an equimolar mixture of the μ3-halide capped complexes 7 and 8 (Scheme 3). In THF solution, 7 and 8 showed no evidence of metal atom redistribution after 24 h at room temperature, with only trace amounts of mixed-metal products forming after heating to 80 °C, according to 1H NMR spectroscopy (Fig. S21†). In contrast, 1H NMR analysis of a solution containing 5 and tri-ferrous 7 revealed complete consumption of 5 to generate 9, with some 7 persisting in solution (Scheme 3 and Fig. S22†). In contrast, reaction of 5 with all-zinc 8 produced only trace quantities of 9 at room temperature, with only modestly greater conversion upon heating to 60 °C (Fig. S23†). The qualitatively slower redistribution reactivity of 5 with 8 (compared to 7) is likely a manifestation of the greater Lewis acidity of the [Zn3] core relative to the [Fe3] core, which results in stronger binding of the μ3-chloride cap.
 | ||
| Scheme 3 Metal-atom scrambling screens. | ||
2.3. Reactivity with organic azides
Complexes 5 and 6 were treated with organic azides to probe the lability of the metal sites during oxidative group transfer (Scheme 2). The 1H NMR spectrum of the reaction of 5 with phenyl azide (PhN3) revealed the presence of (tbsL)Fe3(μ3-NPh) (10) as well as a new paramagnetic species, tentatively assigned as (tbsL)Fe2Zn(μ3-NPh) (11). The 57Fe MB spectrum of the crude reaction mixture containing 11 is best fit to a single quadrupole doublet (δ, |ΔEQ| (mm s−1): 0.28, 2.31; Fig. S7†), indicating a major product distinct from 10 (ref. 36) (δ, |ΔEQ| (mm s−1): 0.42, 1.97 (67%); 0.42, 1.09 (33%)).The structure of 11 was confirmed via a SC-XRD study. As in 10, the imido moiety is bound μ3 to the face of the trinuclear core, and tbsL is arranged in a C3-symmetric orientation. An elongation of the average M–M contact in 11 (2.701(1) Å vs. 2.530(1) Å in 10), supports the presence of zinc. Treatment of the py-bound species 6 with PhN3 also resulted in a mixture of a new product and 10 by 1H NMR. Unlike the reaction of 5 with PhN3, crystallization from hexane at −33 °C cleanly separates the new paramagnetic product from 10.
SC-XRD analysis of this new product reveals a C1-symmetric cluster, (tbsL)Fe2Zn(μ3-NPh)(py) (12) (Fig. S42†). In contrast to the reaction of 2 with PhN3, which produces unsolvated 10, pyridine remains bound to one metal center in 12. We assigned the py-bound site as the zinc site as it also features the longest M–M separations (2.7599(6), 3.2059(7) Å). The remaining two atoms are assigned as the iron positions, supported by the short M–M contact between these sites (2.4880(6) Å). The Fe–Fe contact in this species is shorter than all three Fe–Fe contacts in 10. This shortened Fe–Fe distance is suggestive of an increased M–M interaction, consistent with localization of two-electron oxidation across two iron sites. The 57Fe MB spectrum of 12 exhibits two quadrupole doublets due to the differences in coordination environment at the two iron centers (δ, |ΔEQ| (mm s−1): 0.35, 2.76 (49%); 0.33, 1.80 (51%); Fig. S8†). However, the similar isomer shift of the two signals confirms that both iron centers are in the 3+ oxidation state. The XRF spectrum of crystalline 12 gave an Fe:Zn ratio of 2.1:1, consistent with a compositionally homogenous [Fe2Zn] cluster (Fig. S17†).
To quantify metal atom redistribution, 6 was treated with 3,5-bistrifluoromethyl-azidobenzene (3,5-(CF3)2C6H3N3), which allowed for integration of the product ratio via19F NMR spectroscopy. Two resonances were observed in the 19F NMR spectrum of the reaction mixture in a 7.8:1 ratio, where the less intense peak at −69.4 ppm was identified by independent synthesis as the triiron imido (tbsL)Fe3(μ3-N(3,5-(CF3)2C6H3)) (13), confirming formation of (tbsL)Fe2Zn(μ3-N(3,5-(CF3)2C6H3))(py) (14) as the major product. Akin to 12, cluster 14 can be selectively crystallized and SC-XRD analysis of 14 revealed a structure directly analogous to 12 (Fig. 2c). The XRF spectrum of crystalline 14 yielded an Fe:Zn ratio of 2.1:1.
2.4. Outer-sphere electron transfer
The aforementioned experiments demonstrated that metal atom redistribution in open shell clusters can be triggered by ligand substitution and group transfer reactions. We next sought to establish whether outer-sphere electron transfer reactions would elicit the same metal-atom redistribution. To this end, 14 was treated with KC8 in thawing THF (Scheme 2) and [2.2.2]-cryptand (2,2,2-crypt) was added to sequester the counterion. The 1H and 19F NMR spectra of the reaction mixture contain no signals corresponding to the analogous, independently prepared triiron product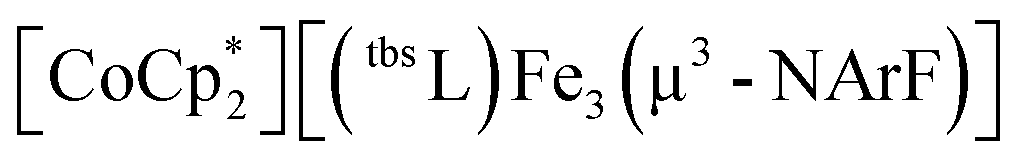 (15) (Fig. S27†). The observation of a single peak in the 19F NMR spectrum suggests the formation of an exclusive imido-bearing cluster; consistently, XRF analysis of this reduction product yielded a Fe/Zn ratio of 1.9:1 (Fig. S19†).
(15) (Fig. S27†). The observation of a single peak in the 19F NMR spectrum suggests the formation of an exclusive imido-bearing cluster; consistently, XRF analysis of this reduction product yielded a Fe/Zn ratio of 1.9:1 (Fig. S19†).
Crystals of the product were formed by diffusion of hexanes into a concentrated THF solution at −33 °C. SC-XRD analysis confirmed that the imido ligand remains μ3-bound, while pyridine has been expelled to form the mixed-metal imido cluster [2,2,2-crypt(K)][(tbsL)Fe2Zn(μ3-NArF)] (16) (Fig. 2d). The three metal sites span a modest range with respect to M–Nimido distances from the two molecules in the asymmetric unit (2.010(2)–2.101(3) Å) and M–M separation (2.6660(7)–2.7843(6) Å). Despite its geometric symmetry, 16 exhibits two quadrupole doublets in its 57Fe MB spectrum. While one doublet has a similar isomer shift to that of the two quadrupole doublets in the starting material (0.35 mm s−1), the other increases to 0.59 mm s−1 (Fig. 2d), consistent with site-isolated reduction.42 As it is impossible to discern the metal identity of the three sites by traditional X-ray diffraction, we are currently investigating 16 using resonant X-ray and neutron diffraction to evaluate the metal occupancy at each site.
2.5. Potential mechanisms of metal-atom exchange
We propose the mechanism of metal exchange from the cluster is facilitated by halide addition. While the chloride-capped all-ferrous [NBu4][(tbsL)Fe3(μ3-Cl)] (7) and all-zinc [NBu4][(tbsL)Zn3(μ3-Cl)] (8) do not undergo metal-atom redistribution upon mixing in solution, we propose that terminal halide coordination to one metal site facilitates exchange. This hypothesis is bolstered by the observed reactivity of (tbsL)Fe2Zn(thf) (5) with either 7 or 8, which results in variable extents of metal-atom redistribution. Thus, exchange of L′ = thf or py for chloride can lead to ligand reorganization about the mixed metal core to facilitate metal atom excision. Similar reactivity was observed with the related (PhL)Fe3 cluster43 that was found to undergo ligand reorganization to form extended structures, as well as undergo both degenerative iron exchange (as evidenced by isotope exchange) or cobalt for iron exchange upon addition of MCl2 (M = 57Fe, Co; Scheme 4). The open-shell electronic structure of these complexes makes ligand exchange facile, ultimately favoring formation of products that maximize the stability of the resulting cluster. | ||
| Scheme 4 Metal-atom metathesis from mixed valent cluster (PhL)Fe3Cl(thf) see ref. 43. | ||
Azide binding likely facilitates metal atom extrusion akin to the chloride addition, particularly in instances where the azide ligand is bound in a terminal, μ1-fashion.44 While the mixed-metal imido clusters 12 and 14 could be trapped and selectively crystallized, monitoring of the reaction between 6 and ArN3 indicated metal atom redistribution, as evidenced by the formation of the triiron imido, (tbsL)Fe3(μ3-NAr). Again, exchange of L′ for NAr, followed by (tbsL) reorganization about the trinuclear core, can expose the azide-bound metal to facilitate exchange. For example, organic azide binding in the structurally analogous [Cr3] cluster (tbsL)Cr3(thf) produces different end products (i.e., bridging nitride vs. terminal imido, Scheme 5) depending on how the reacting azide binds to the all-chromous cluster.44
 | ||
| Scheme 5 Solvent directed change in azide breakdown. See ref. 44. | ||
We propose that open-faced clusters (e.g., non-symmetrical binding of the templating ligand about the trinuclear core) are necessary to facilitate metal-atom exchange. This is bolstered by the observation that there appears to be little accumulation of free ligand following the metal-atom redistribution. However, we cannot rule out that small amounts of in situ generated MX2 species are aiding in the metal atom redistribution reactions; notably, added MX2 salts have been shown to catalyze metal-atom exchange in related clusters.43 Indeed, treatment of 5 with FeCl2 (one equiv.) in THF solution resulted in rapid metal redistribution to form 9 at room temperature (Fig. S24†), although additional unidentified paramagnetic species were also generated, according to 1H NMR spectroscopy. Surprisingly, no redistribution was observed from a mixture of 5 with ZnCl2 (one equiv.) in THF solution, although a diminution of overall resonance intensity in the 1H NMR spectrum (Fig. S25†), as well as a color change from brown to purple, suggested that a transformation had occurred.
3 Conclusions
The foregoing results present the synthesis of [Fe2Zn] clusters via a transmetalation strategy that is more general than our previous transamination strategy for synthesizing bimetallic clusters.39 We then canvassed the behavior of the [Fe2Zn] clusters with respect to key reaction types: ligand binding, oxidative group transfer, and outer-sphere electron transfer. Chloride addition to 5 resulted in a mixture of species, where the Fe:Zn of the reaction mixture was maintained, but all-iron 7 and all-zinc 8 were observed in the reaction mixture. Addition of ArN3 (Ar = Ph; 3,5-(CF3)2C6H3) to 5 or 6 yielded a mixture of two products: the triiron imido product and a mixed-metal imido cluster. In the case of py-bound 6, the mixed-metal imido included a bound pyridine, allowing for the isolation of [Fe2Zn] imidos 12 and 14 by selective crystallization. The substitutional homogeneity of 12 and 14 were determined by 1H/19F NMR and 57Fe MB spectroscopies, XRF spectroscopy, and SC-XRD analysis. Finally, outer sphere reduction of py-bound, imido-capped cluster 14 formed reduced mixed-metal imido 16, with no evidence of metal atom lability from the cluster core.
While metal-atom shuffling was observed upon both neutral substrate-binding and oxidative group transfer chemistry, the integrity of our mixed-metal clusters was maintained upon outer sphere electron transfer when stabilized by an imido capping group. This stabilization is consistent with previous work in cluster chemistry, which has employed bridging ligands to prevent cluster degradation during reactivity.45–47 The results of this inquiry allow us to demonstrate the dynamic nature of cluster reaction sites. Moreover, knowledge of the factors which confer stability to these clusters can provide valuable information to be used in future catalyst design. Finally, these results illustrate the dynamic nature of bimetallic reaction site substitutions, which may be relevant not only for synthetic cofactor analogues, but also for biological cofactors themselves.
Data availability
Crystallographic data have been deposited at the CCDC under deposition numbers 1993457–1993469. Experimental information and images of experimental data can be found in the ESI.†Author contributions
C. E. J. and T. A. B. conceived of the hypothesis and experimental designs. C. E. J. performed the experiments, characterization, data collection, and analysis. R. C. H. assisted in synthesis, characterization, and susceptibility measurements. A. K. B., T. M. P., N. X. G., and E. M. provided feedback in data interpretation and carried out several syntheses. N. R., Y. S. C., and S. L. Z. assisted and performed single-crystal X-ray diffraction analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the NIH (GM-098395), DOE (DE-SC0019144), and Harvard University. C. E. J. is grateful for an NSF Predoctoral Fellowship. We thank the support to the X-ray core facility from the Major Research Instrumentation (MRI) Program of the National Science Foundation (NSF) under Award Number 2216066. Solid-state structures of 2, 5, 6, 12, and 16 were obtained at ChemMatCARS Sector 15. NSF's ChemMatCARS Sector 15 is supported by the Divisions of Chemistry (CHE) and Materials Research (DMR), National Science Foundation, under grant number NSF/CHE-1834750. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357.Notes and references
- B. K. Burgess and D. J. Lowe, Chem. Rev., 1996, 96, 2983–3012 CrossRef CAS PubMed.
- P. C. Dos Santos, R. Y. Igarashi, H. I. Lee, B. M. Hoffman, L. C. Seefeldt and D. R. Dean, Acc. Chem. Res., 2005, 38, 208–214 CrossRef CAS PubMed.
- B. M. Hoffman, D. R. Dean and L. C. Seefeldt, Acc. Chem. Res., 2009, 42, 609–619 CrossRef CAS PubMed.
- J. B. Howard and D. C. Rees, Chem. Rev., 1996, 96, 2965–2982 CrossRef CAS PubMed.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831–1838 CrossRef CAS PubMed.
- T. J. Wydrzynski, K. Satoh and J. A. Freeman, Photosystem II The Light Driven Water: Plastoquinone Oxidoreductase, Springer, Dordrecht, The Netherlands, 2005 Search PubMed.
- J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455–4483 CrossRef CAS PubMed.
- Y. Umena, K. Kawakami, J. R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- N. Cox, D. A. Pantazis, F. Neese and W. Lubitz, Acc. Chem. Res., 2013, 46, 1588–1596 CrossRef CAS PubMed.
- W. Guo and D. G. Vlachos, Nat. Commun., 2015, 6, 8619 CrossRef CAS PubMed.
- J. Humphreys, R. Lan and S. Tao, Adv. Energy Sustainability Res., 2021, 2, 2000043 CrossRef CAS.
- F. Morales and B. M. Weckhuysen, Catalysis, 2006, 19, 1–40 CAS.
- R. H. Holm, P. Kennepohl and E. I. Solomon, Chem. Rev., 1996, 96, 2239–2314 CrossRef CAS PubMed.
- A. M. Barrios and S. J. Lippard, J. Am. Chem. Soc., 2000, 122, 9172–9177 CrossRef CAS.
- W. Z. Lee and W. B. Tolman, Inorg. Chem., 2002, 41, 5656–5658 CrossRef CAS PubMed.
- J. S. Kanady, E. Y. Tsui, M. W. Day and T. Agapie, Science, 2011, 333, 733–736 CrossRef CAS PubMed.
- E. Y. Tsui, J. S. Kanady and T. Agapie, Inorg. Chem., 2013, 52, 13833–13848 CrossRef CAS PubMed.
- S. Zhang, M. M. Melzer, S. N. Sen, N. Celebi-Olcum and T. H. Warren, Nat. Chem., 2016, 8, 663–669 CrossRef CAS PubMed.
- E. Kim, E. E. Chufan, K. Kamaraj and K. D. Karlin, Chem. Rev., 2004, 104, 1077–1133 CrossRef CAS PubMed.
- A. J. Jasniewski and L. Que Jr, Chem. Rev., 2018, 118, 2554–2592 CrossRef CAS PubMed.
- T. M. Powers, A. R. Fout, S. L. Zheng and T. A. Betley, J. Am. Chem. Soc., 2011, 133, 3336–3338 CrossRef CAS PubMed.
- R. B. Ferreira and L. J. Murray, Acc. Chem. Res., 2019, 52, 447–455 CrossRef CAS PubMed.
- E. L. Muetterties, Science, 1977, 196, 839–848 CrossRef CAS PubMed.
- O. Einsle and D. C. Rees, Chem. Rev., 2020, 120, 4969–5004 CrossRef CAS PubMed.
- Y. Hu and M. W. Ribbe, Coord. Chem. Rev., 2011, 255, 1218–1224 CrossRef CAS PubMed.
- W. Kang, C. C. Lee, A. J. Jasniewski, M. W. Ribbe and Y. Hu, Science, 2020, 368, 1381–1385 CrossRef CAS PubMed.
- M. Can, F. A. Armstrong and S. W. Ragsdale, Chem. Rev., 2014, 114, 4149–4174 CrossRef CAS PubMed.
- E. C. Wittenborn, S. E. Cohen, M. Merrouch, C. Léger, V. Fourmond, S. Dementin and C. L. Drennan, J. Biol. Chem., 2019, 294, 13017–13026 CrossRef CAS PubMed.
- E. C. Wittenborn, C. Guendon, M. Merrouch, M. Benvenuti, V. Fourmond, C. Leger, C. L. Drennan and S. Dementin, ACS Catal., 2020, 10, 7328–7335 CrossRef CAS PubMed.
- E. C. Wittenborn, M. Merrouch, C. Ueda, L. Fradale, C. Leger, V. Fourmond, M. E. Pandelia, S. Dementin and C. L. Drennan, Elife, 2018, 7, e39451 CrossRef PubMed.
- M. Chrysina, E. Heyno, Y. Kutin, M. Reus, H. Nilsson, M. M. Nowaczyk, S. DeBeer, F. Neese, J. Messinger, W. Lubitz and N. Cox, Proc. Natl. Acad. Sci., 2019, 116, 16841–16846 CrossRef CAS PubMed.
- J. Kern, R. Chatterjee, I. D. Young, F. D. Fuller, L. Lassalle, M. Ibrahim, S. Gul, T. Fransson, A. S. Brewster, R. Alonso-Mori, R. Hussein, M. Zhang, L. Douthit, C. de Lichtenberg, M. H. Cheah, D. Shevela, J. Wersig, I. Seuffert, D. Sokaras, E. Pastor, C. Weninger, T. Kroll, R. G. Sierra, P. Aller, A. Butryn, A. M. Orville, M. Liang, A. Batyuk, J. E. Koglin, S. Carbajo, S. Boutet, N. W. Moriarty, J. M. Holton, H. Dobbek, P. D. Adams, U. Bergmann, N. K. Sauter, A. Zouni, J. Messinger, J. Yano and V. K. Yachandra, Nature, 2018, 563, 421–425 CrossRef CAS PubMed.
- M. Carboni and J.-M. Latour, Coord. Chem. Rev., 2011, 255, 186–202 CrossRef CAS.
- E. C. Kisgeropoulos, J. J. Griese, Z. R. Smith, R. M. Branca, C. R. Schneider, M. Högbom and H. S. Shafaat, J. Am. Chem. Soc., 2020, 142, 5338–5354 CrossRef CAS PubMed.
- E. K. Miller, N. E. Trivelas, P. T. Maugeri, E. J. Blaesi and H. S. Shafaat, Biochemistry, 2017, 56, 3369–3379 CrossRef CAS PubMed.
- T. M. Powers and T. A. Betley, J. Am. Chem. Soc., 2013, 135, 12289–12296 CrossRef CAS PubMed.
- R. Hernández Sánchez, A. K. Bartholomew, T. M. Powers, G. Ménard and T. A. Betley, J. Am. Chem. Soc., 2016, 138, 2235–2243 CrossRef PubMed.
- T. M. Powers, A. R. Fout, S. L. Zheng and T. A. Betley, J. Am. Chem. Soc., 2011, 133, 3336–3338 CrossRef CAS PubMed.
- T. M. Powers, N. X. Gu, A. R. Fout, A. M. Baldwin, R. Hernandez Sanchez, D. M. Alfonso, Y. S. Chen, S. L. Zheng and T. A. Betley, J. Am. Chem. Soc., 2013, 135, 14448–14458 CrossRef CAS PubMed.
- E. V. Eames, R. Hernández Sánchez and T. A. Betley, Inorg. Chem., 2013, 52, 5006–5012 CrossRef CAS PubMed.
- J. R. Allan, D. H. Brown, R. H. Nuttall and D. W. A. Sharp, J. Chem. Soc. A, 1966, 1031–1034 RSC.
- F. Menil, J. Phys. Chem. Solids, 1985, 46, 763–789 CrossRef CAS.
- E. V. Eames, R. l. Hernández Sánchez and T. A. Betley, Inorg. Chem., 2013, 52, 5006–5012 CrossRef CAS PubMed.
- A. K. Bartholomew, C. E. Juda, J. N. Nessralla, B. Lin, S. G. Wang, Y. S. Chen and T. A. Betley, Angew. Chem., Int. Ed., 2019, 58, 5687–5691 CrossRef CAS PubMed.
- R. C. Ryan and C. U. Pittman, J. Am. Chem. Soc., 1977, 99, 1986–1988 CrossRef CAS.
- R. J. Haines, N. D. C. T. Steen and R. B. English, J. Chem. Soc., Dalton Trans., 1984, 515–525 RSC.
- J. A. Cabeza, J. M. Fernandez-Colinas, A. Llamazares, V. Riera, S. Garcia-Granda and J. F. Van der Maelen, Organometallics, 1994, 13, 4352–4359 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1993457–1993469. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03606e |
| This journal is © The Royal Society of Chemistry 2024 |