
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Kinetics of thermal dry reforming of methane for syngas production and solid carbon capture†
Manas
Mokashi
a,
Akash Bhimrao
Shirsath
a,
Sinan
Demir
a,
Ahmet
Çelik
 a,
Patrick
Lott
*a,
Steffen
Tischer
b and
Olaf
Deutschmann
ab
a,
Patrick
Lott
*a,
Steffen
Tischer
b and
Olaf
Deutschmann
ab
aInstitute for Chemical Technology and Polymer Chemistry, Karlsruhe Institute of Technology (KIT), Engesserstr. 20, 76131 Karlsruhe, Germany. E-mail: patrick.lott@kit.edu
bInstitute of Catalysis Research, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 6th August 2024
Abstract
Dry reforming of CH4, either by co-feeding CH4 and CO2 from waste streams or directly using biogas, has potential as a CO2-sink. This study investigates entirely thermal, catalyst-free dry reforming in a tubular flow reactor, aiming for syngas production with concurrent carbon capture. Kinetic modelling couples an elementary step-based gas-phase mechanism with a carbon deposition model. One-dimensional numerical simulations of the flow reactor are compared with experimental measurements. For this, operating conditions are widely varied, in particular temperature (1273 K to 1873 K), residence time (1 to 7 seconds), and CH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO2 molar feed ratio (1 to 4). Two temperature regimes are identified, with varying dominance of the reverse water-gas shift and CH4 pyrolysis reactions. Above 1673 K, CO2 is fully consumed, independent of residence time and feed composition. Optimized operating parameters result in a H2/CO ratio of 2 in the effluent gas stream, e.g. as commonly desired for methanol and oxo-alcohol synthesis. Notably, under such optimized conditions, only a minor share of carbonaceous species remains in the gas-phase as hydrocarbons, while 33% of the CH4-borne carbon is transformed into CO and 48% of CH4-borne carbon is captured as solid carbon.
:CO2 molar feed ratio (1 to 4). Two temperature regimes are identified, with varying dominance of the reverse water-gas shift and CH4 pyrolysis reactions. Above 1673 K, CO2 is fully consumed, independent of residence time and feed composition. Optimized operating parameters result in a H2/CO ratio of 2 in the effluent gas stream, e.g. as commonly desired for methanol and oxo-alcohol synthesis. Notably, under such optimized conditions, only a minor share of carbonaceous species remains in the gas-phase as hydrocarbons, while 33% of the CH4-borne carbon is transformed into CO and 48% of CH4-borne carbon is captured as solid carbon.
Introduction
In the context of reducing greenhouse gas emissions, hydrogen (H2) is projected to play a pivotal role as a carbon-free energy carrier.1,2 It is also essential for carbon capture and utilization (CCU) technologies such as direct hydrogenation of carbon dioxide (CO2) into methanol3–5 and olefins.6,7 However, with regard to large-scale production of H2 that is necessary to cover the increasing demand, the entirely carbon-free production process through water electrolysis faces challenges such as high initial investment costs and substantial energy requirements.8,9 Therefore, methane (CH4) pyrolysis is gaining increasing attention as a bridging technology for H2 production on the pathway towards a sustainable future.10–14 This endothermic process that does not exhibit any direct CO2 emissions involves the decomposition of CH4, resulting in the production of H2 and the capture of solid carbon2,10 according to eqn (1).| CH4 → 2H2 + C ΔRH° = 75 kJ mol−1 | (1) |
| CH4 + CO2 ⇌ 2CO + 2H2 ΔRH° = 247 kJ mol−1 | (2) |
An alternative strategy to directly use CO2 in chemical processes and circumvent the challenges associated with catalyst deactivation is through a non-catalytic high-temperature approach.48–53 In this context, Angeli et al.51 reported the utilization of steelwork off-gases through an entirely thermal DRM process and Blanck et al.53 studied the influence of operating parameters for steelwork off-gases in a pilot plant. They found that 97% CH4 conversion and 94% CO2 conversion can be obtained at 1721 K. In addition to CH4 and CO2, steelwork off-gases contain high levels of CO, nitrogen (N2), and water (H2O). Furthermore, Chen and Gan49 investigated non-catalytic thermal DRM in the presence of oxygen (O2) and found that a feed ratio of CH4, O2, and CO2 of 5:4:1 results in a conversion of 99.88% and 46.85% for CH4 and CO2, respectively, with the resulting syngas exhibiting a H2/CO ratio of 1.19. Savchenko et al.50 carried out a kinetic analysis of non-catalytic DRM involving C1–C4 hydrocarbons in a temperature range spanning from 1400 K to 1800 K. Their findings highlighted the central role played by acetylene (C2H2) in the non-catalytic DRM process.
In the context of entirely sustainable feedstocks, Shapovalova et al.52 employed volumetric matrix reformers to transform biogas with a CO2 content exceeding 60 mol% into syngas. Furthermore, Çelik et al.48 conducted comprehensive experimental investigations on biogas pyrolysis in a flow reactor setup with a feed mixture of H2-diluted CH4/CO2 mixtures for temperatures ranging from 1273 K to 1873 K, with residence times (τ) spanning from 1 to 7 seconds, and CH4:CO2 molar feed ratios varying from 1 to 4. In addition to carbon deposition that enables the process to act as a carbon sink, syngas formation was observed, resulting in an essentially full exploitation of the feed gas stream.
Despite promising prior research efforts, profound mechanistic studies of syngas production and simultaneous capture of solid carbon in thermal, non-catalytic DRM lack so far. At the elevated temperatures necessary for thermal DRM, and with the presence of additional species such as CO2 and H2 in the feed, in addition to CH4 pyrolysis the reverse water-gas shift reaction (RWGS) (eqn (3)) becomes relevant. It is important to note that the RWGS reaction is reversible; therefore, it is highly sensitive to changes of feed composition, temperature, and pressure.54
| H2 + CO2 ⇌ CO + H2O ΔRH° = 41 kJ mol−1 | (3) |
Experimental
The experiments were performed in a high-temperature and high-pressure flow reactor setup that was already described in previous work.10,51 The reactor consists of ceramic α-Al2O3 (DEGUSSIT AL23 by Friatec/Aliaxis) and has a length of 1 m and an inner diameter of 0.02 m. The reaction mixture was pre-heated to 443 K and fed to the reactor via mass flow controllers. The effluent products were analyzed with an online HPR-20 mass spectrometer (Hiden Analytical). The experimental setup is schematically illustrated in Fig. 1.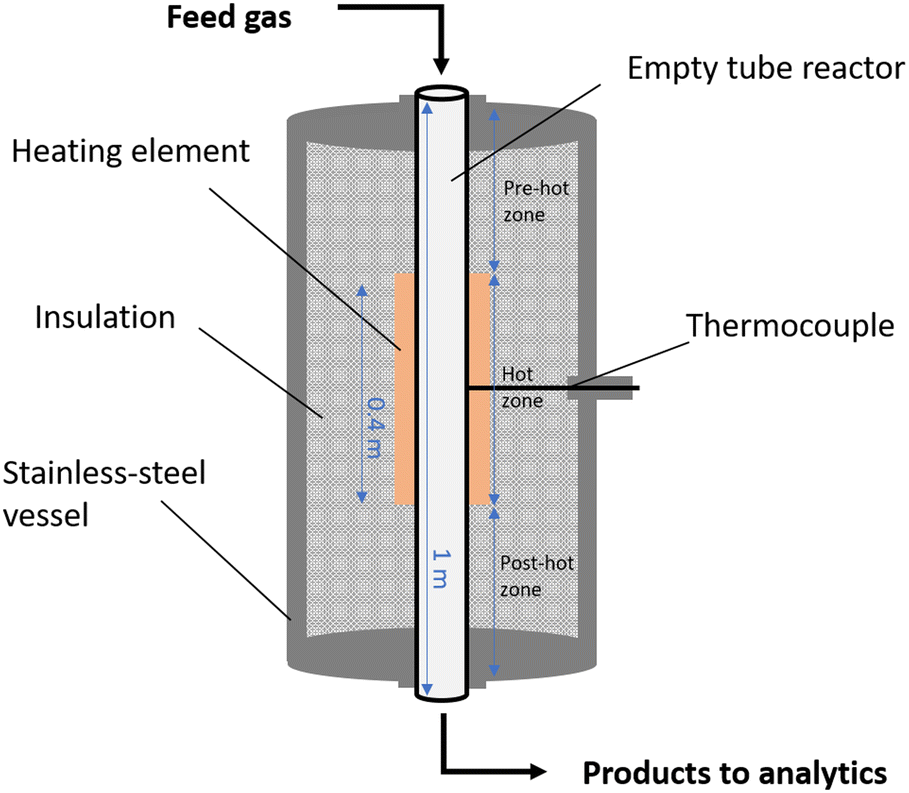 | ||
| Fig. 1 Experimental flow reactor setup. | ||
The procedure for the experiments that serve for model validation and evaluation herein is based on the approach chosen during a recently published study by our group.48 In particular, the reactor was operated under reaction conditions for 20 minutes while monitoring the composition of the effluent gas stream. Subsequently, any carbon deposited in the reactor was oxidized at temperatures above 1273 K in order to recover the reactor for the next experiment. The recovery was considered to be complete once the end-of-pipe CO and CO2 levels reached zero. The operating conditions and inlet feed compositions used for model development and validation are summarized in Tables 1 and 2, respectively. Specific flow rates that were chosen during the experiments in order to achieve the desired residence times are provided in the supplementary material (Tables S1 and S2†).
| Process parameter | Variation range |
|---|---|
| Temperature/K | 1273–1373–1473–1573–1673–1773–1873 |
| Residence time/s | 1–3–5–7 |
| Pressure/bar | 1 |
| Molar feed ratio | CH4 | CO2 | H2 |
|---|---|---|---|
| CH4:CO2 ratio 1 |
0.167 | 0.167 | 0.667 |
| CH4:CO2 ratio 2 |
0.222 | 0.111 | 0.667 |
| CH4:CO2 ratio 4 |
0.2664 | 0.0666 | 0.667 |
Modelling approach
Flow reactor model
Thermal DRM experiments were modeled and simulated with the DETCHEM software package.11,55,56 To simulate the experiments, a one-dimensional (in flow direction) plug flow reactor model based on a continuum model approach was used. Assuming steady-state and non-dispersive flow conditions, the model solves mass, species, and pressure drop equations in one dimension. Eqn (4) represents the continuity equation. | (4) |
 | (5) |
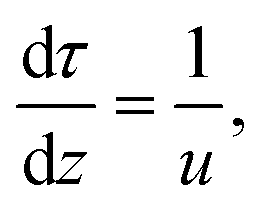 | (6) |
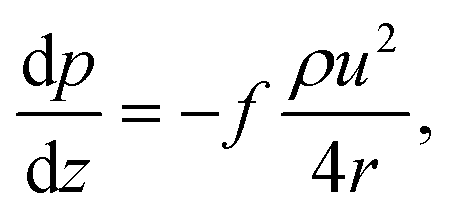 | (7) |
In this study, the deposition reaction is examined as a function of time. For this, transient simulations were conducted using the computer code DETCHEMPFR_transient, which acts as a transient wrapper for the one-dimensional plug flow reactor model (DETCHEMPFR). The DETCHEMPFR_transient simulation assumes that the time scale for deposition reactions substantially exceeds the residence time that is sufficient for gas-phase reactions. In this model, the transient wrapper operates the DETCHEMPFR simulation in an iterative manner for each time step. In each step, it performs a near-steady-state analysis to determine the changing concentrations of gas-phase species, while keeping the concentrations of deposition species constant. The coverage of these species can differ based on their position within the reactor. After calculating these local concentrations in the gas-phase, this information is fed back into the transient wrapper, which then updates the concentrations according to eqn (8) through a time-integration step. To finalize the simulation, the process involves a repeated steady-state plug flow simulation, but with revised inlet conditions and an updated surface state.
 | (8) |
Reaction mechanisms
Detailed gas-phase kinetic mechanisms by Appel et al.57 (hereinafter referred to as ABF2000), Blanquart et al.58 (Caltechmech), and Porras et al.59 (Polymech) were reviewed in the current work. Moreover, the present study focuses on a temperature range between 1273 to 1673 K, where solid carbon formation can take place as well.10,48 Therefore, a carbon deposition mechanism60 was coupled to the above-mentioned gas-phase mechanisms.ABF2000 was developed for describing the combustion and pyrolysis of light hydrocarbons. The mechanism consists of oxidation and pyrolysis reactions of C1 and C2 species and comprises also carbon coupling reactions as well as the formation of polycyclic aromatic hydrocarbons (PAHs) up to pyrene (C16H10). Additionally, the oxidation of aromatic species is incorporated. The mechanism consists of 99 species and 538 reactions.
Caltechmech was developed for describing the combustion of small as well as large hydrocarbons along with the prediction of soot precursors. It starts from reactions of smaller hydrocarbons such as CH4, C2H2, ethylene (C2H4), and ethane (C2H6), as well as intermediate hydrocarbons such as propadiene (C3H4), propylene (C3H6), and propane (C3H8). It also considers larger hydrocarbons such as n-heptane, iso-octane, benzene, and toluene. Furthermore, the mechanism considers the formation of PAHs until cyclopenta(cd)pyrene (C18H10). The comprehensive mechanism consists of 149 species and 1651 reactions.
Polymech was developed in order to describe dimethyl-ether and CH4 combustion. The oxidation and pyrolysis of C1–C4 species exists as a sub-mechanism along with a dimethyl-ether sub-mechanism. The mechanism consists of 83 reactions and 558 reactions and comprises an extensive set of reactions for smaller hydrocarbons up to C6.
Although all three gas-phase mechanisms discussed above are in principle suitable for describing processes taking place under the operating conditions considered in our present work, a brief comparison of the three mechanisms (cf. Fig. S1†) uncovers some differences. ABF2000 consists of reactions from light hydrocarbons to PAHs and is computationally the fastest among the three mechanisms considered herein. However, it lacks certain C3 species intermediates such as C3H6, which can play a role according to Polymech. In turn, Polymech considers these intermediates and their corresponding reaction steps, however, species larger than benzene (C6H6) are missing. Finally, Caltechmech is the most comprehensive mechanism among the three and is therefore considered for coupling with deposition reactions.
Apart from the gas-phase mechanisms, also a deposition mechanism was considered in this work. The by-products formed in the gas-phase mechanisms were coupled to deposition reactions in order to describe the formation of solid carbon. It consists of 6 species, namely CH4, C2H6, C2H4, C2H2, C4H6, and C6H6. After coupling, gas-phase and deposition reactions take place in parallel as illustrated in Fig. 2. Solid carbon deposits on the reactor wall while H2 is simultaneously released from the gas-phase precursors.61 Furthermore, H2 acts as an inhibitor to deposition reactions due to the formation of stable hydrogen–carbon surface complexes.62 Therefore, inhibition functions were also considered for each deposition reaction. More details about the deposition reactions and their coupling with gas-phase reactions is available in our previous publication.60 All the mechanisms and kinetic parameters used herein are provided in the supplementary information (Table S3†).
 | ||
| Fig. 2 Schematic of gas-phase reactions and carbon deposition reactions. | ||
Results and discussion
Thermodynamic analysis
Various hydrocarbon gas-phase species along with H2, CO, and solid carbon can be formed during thermal dry reforming. Using DETCHEMEQUIL from the DETCHEM software package,55,56 the equilibrium product distribution was calculated for feed mixtures with a CH4:CO2 ratio of either 1 or 4 under isothermal and isobaric conditions. Fig. 3 displays the respective equilibrium product distribution of major species, considering gas-phase species from Caltechmech and additional graphitic carbon species.
 | ||
| Fig. 3 Effect of different CH4:CO2 ratios and pressure on the equilibrium product distribution. | ||
At 1 bar, the CH4 from the feed remains mostly unconverted only at lower temperatures, whereas it is entirely consumed at temperatures above 873 K. The formation of the gaseous products H2 and CO is energetically favored at higher temperatures. Notably, the thermodynamic stability of graphitic carbon depends significantly on the CH4:CO2 ratio. For a CH4:CO2 ratio of 4, the formation of solid graphitic carbon is thermodynamically possible throughout the entire temperature range investigated in this study. In contrast, for a CH4:CO2 ratio of 1, the formation of graphitic carbon is favored only at temperatures below 1673 K. A pressure increase from 1 bar to 5 bar impacts the product composition particularly at temperatures between 673 K and 1373 K: the pressure increase suppresses CH4 conversion and thus the formation of graphitic carbon as well as H2 formation.
In contrast, the influence of pressure on the CO mole fractions is negligible. In fact, due to its exothermic nature, the Boudouard reaction, as depicted in eqn (9), is known to result in graphitic carbon deposition primarily at temperatures below 1273 K.51,63
| 2CO ⇌ C + CO2 ΔRH° = −172 kJ mol−1 | (9) |
In summary, the thermodynamic analysis reveals that elevated temperatures benefit thermal DRM. Furthermore, a CH4:CO2 ratio above 1 can result in the formation of solid carbon across the entire range of temperatures subject to this study and the CH4:CO2 ratio also significantly influences the H2/CO ratio in the resulting syngas. The influence of pressure is of particular relevance below 1373 K, as thermodynamics indicate that intermediate to low temperatures and high pressure are disadvantageous for the DRM reaction.
Comparison of experiments with simulations
:CO2 ratio of 2, τ of 5 s, and 1 bar pressure are used for validation.
Fig. 4 illustrates the end-of-pipe mole fractions for both experiments and simulations. Note that the experimental data are presented with error bars set to 5% in order to account for uncertainties in gas species quantification by mass spectrometry.48 The fraction of the feed gas component CH4 steadily decreases with temperature and reaches negligible levels above 1673 K at the reactor outlet. In the temperature range of 1273 K to 1373 K, CO forms rapidly as CO2 is consumed. However, CO formation increases steadily above 1473 K even when CO2 is fully depleted. H2 shows a unique trend in both experiments and simulations: first it is consumed up to 1373 K, but then hydrogen forms in increasing quantity above 1423 K. Other hydrocarbon by-products such as C2H2, C2H4, C2H6, and C6H6 are formed in very small quantities and remain below 1% for all the temperatures considered herein (cf. Fig. S3†). Given the very small quantities, which are near the detection limit of the analytics, the model matches the experimental end-of-pipe measurement data fairly well.
 | ||
| Fig. 4 End-of-pipe experiments vs. simulations at different hot zone temperatures at constant CH4:CO2 ratio = 2, τ = 5 s, and p = 1 bar. | ||
Since the lab reactor used for the present study consists of a pre-heating zone, an isothermal hot zone, and a post-cooling zone that results in a non-isothermal temperature profile that can have a significant impact on the process (cf.Fig. 1), an accurate analysis of the system requires species concentration data with axial resolution.
Fig. 5 illustrates such axial temperature profiles along with the main species mole fractions at hot zone temperatures of 1373 K (Fig. 5(a)) and 1673 K (Fig. 5(b)) as predicted by means of simulations. At 1373 K (Fig. 5(a)), CH4 and CO2 consumption start at the beginning of the hot zone. As the reaction progresses along the reactor length, CO and H2O are formed at a nearly equal rate, yielding almost identical molar fractions. H2 is consumed as well, however, its consumption rate is lower compared to CH4 and CO2. When the temperature is increased to 1673 K while keeping all other parameters unchanged (Fig. 5(b)), the trends change considerably. Once the temperature in the heating zone exceeds approx. 1300 K (corresponding to a reactor length of ∼0.26 m), CH4 and CO2 consumption sets in, which is well before the reaction gas mixture reaches the actual isothermal hot zone that starts at 0.35 m. Notably, CO2 is fully depleted already before the hot zone starts. Furthermore, H2 exhibits a minimum in front of the hot zone, but rapidly increases afterwards, resulting in net H2 production at the reactor outlet. H2O formation is observed upon the onset of feed gas species conversion. In contrast to the overall increasing water levels along the reactor length observed at 1373 K, the H2O concentration peaks after approx. 0.33 m if the hot zone temperature is set to 1673 K; subsequently, the molar fraction of H2O depletes again and reaches almost zero at the reactor outlet. CO formation is most pronounced right in front of the isothermal hot zone and its level continues to increase only marginally towards the end of the hot zone.
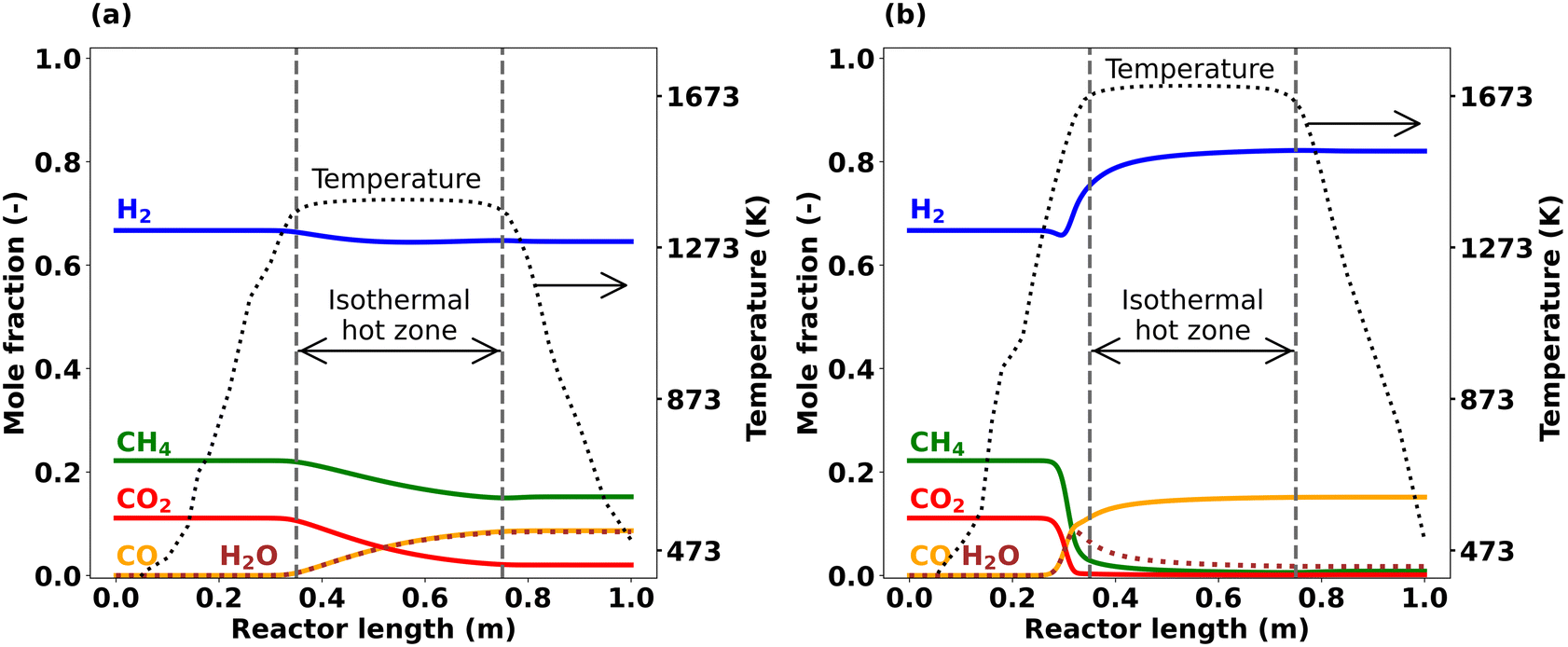 | ||
| Fig. 5 Simulated axial profiles of main species at CH4:CO2 ratio = 2, τ = 5 s, and p = 1 bar; (a) T = 1373 K, (b) T = 1673 K. | ||
In addition to these main products, hydrocarbon by-products as well as carbon deposition play a major role at 1673 K, as underscored by the axial profiles depicted in Fig. 6. According to the simulation data plotted in Fig. 6(a), C2H2 is the most important hydrocarbon formed at 1673 K, whose mole fraction reaches a sharp maximum of about 0.036 just before the start of the hot zone (at about 0.33 m). The C2H4 and C6H6 mole fractions exhibit maxima around the same axial position, however at significantly lower levels (less than 0.005). None of these hydrocarbons survives until the reactor outlet, since they are consumed during carbon formation and deposition reactions that take place particularly on the reactor wall. Fig. 6(b) shows the deposited carbon amount along the reactor length after 1800 s of reactor operation. The data suggest that carbon deposition is most pronounced right in front of the hot zone, which is the same position where the hydrocarbon concentrations peak according to the data plotted in Fig. 6(a). To understand the different trends at different temperatures, a detailed analysis of the reaction system is mandatory. Therefore, an integral reaction flow analysis (RFA)64,66 was performed for the Caltechmech gas-phase mechanism, which reveals the consumption and formation of each species.
 | ||
| Fig. 6 Simulated axial profiles at 1673 K, CH4:CO2 ratio = 2, τ = 5 s and p = 1 bar; (a) hydrocarbon by-products, (b) deposited moles of carbon along the reactor axis after 1800 s of reactor operation. | ||
Fig. 7 shows the results for an RFA conducted for a temperature of 1373 K, a residence time τ of 5 s, and a CH4:CO2 ratio of 2. According to the RFA, all the feed species, namely CO2, CH4, and H2, are consumed under these operating conditions. CO2 is reduced by an H radical originating either from H2 or CH4 decomposition, which results in the formation of CO and the release of an OH radical; this OH radical attacks the H2 molecule, which yields H2O as main product. Moreover, the OH radical plays an important role in CH4 dissociation into a CH3 (and an H) radical, which is crucial for the following sequential C–C coupling reactions. The first step of C–C coupling reactions is the combination of two CH3 radicals to form C2 species, namely C2H6, which dehydrogenates to C2H4. In the next step, the CH3 radical attacks C2H4 and results in C3 species formation. However, since the reaction barely progresses further than C3H6, C3 species formation is a rather dead end of the reaction sequence – only 5% of the C3H6 molecules react to allyl radicals (C3H5) – and H2O and CO represent the main products at 1373 K. Propylene (C3H6) formation has been reported previously to take place during the CH4 pyrolysis reaction sequence if instead of H2 argon was used to dilute the feed.65 In the current work, RWGS, which consumes H2, creates similar process conditions, which enable the formation of C3H6 intermediates. The progression of the reaction beyond C3H6 to C2H2 and aromatic rings is kinetically limited at 1373 K. C2H2 and C5 rings that are formed in negligible quantities beyond C3H5 do not remain stable and are completely opened back to C2H4.
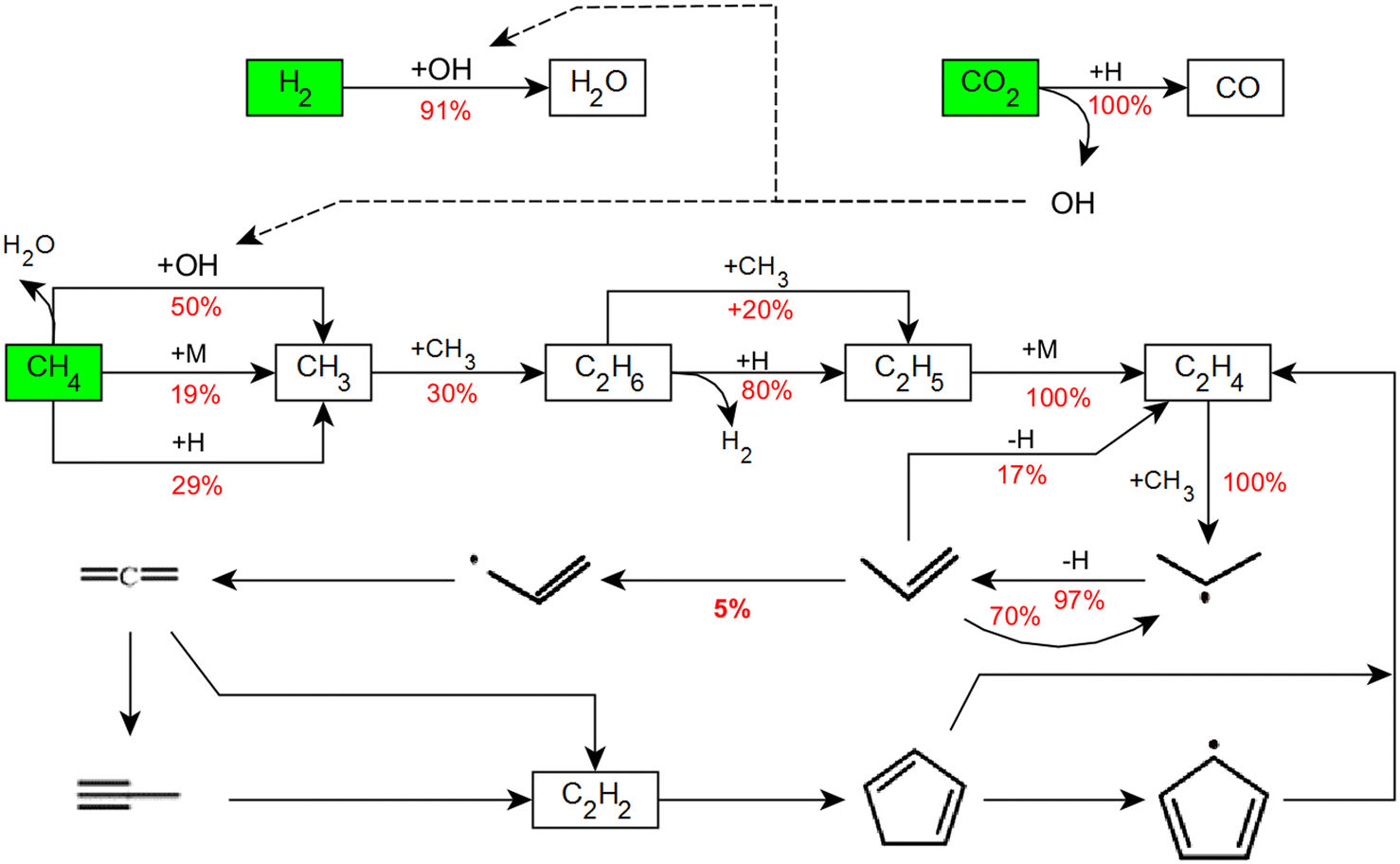 | ||
| Fig. 7 Integral reaction flow analysis at 1373 K for τ = 5 s, CH4:CO2 ratio = 2, and p = 1 bar. | ||
In contrast, the RFA that was performed at 1673 K while keeping both the residence time τ (5 s) and the CH4:CO2 ratio (2) constant suggests that the reaction pathways become substantially more complex if the temperature is increased by 300 K (Fig. 8). At this temperature, CH4 and CO2 are consumed while there is a net rate of production for H2. CO2 is fully converted to CO under release of an OH radical. Note that compared to 1373 K, the role of the OH radical in the activation of CH4 is much less pronounced at 1673 K. Furthermore, CH4 dissociation into CH3 and H radicals primarily takes place via an H radical attack at 1673 K. The reaction steps that lead to C2H2 formation via a C–C coupling reaction are similar to those at lower temperature. However, the reaction progression from C3H6 onwards is more than doubled (5% at 1373 K versus 11% at 1673 K). Although the formation of the first aromatic ring (mostly benzene) takes place via C3 intermediates, C2H2 is the central species according to the RFA: C3 intermediates are partially cracked to C2H2 and most of the aromatic rings also produce C2H2 when opened. Furthermore, the RFA suggests that OH radicals react with C2H2 molecules to significant extent, which enables the formation of CO from hydrocarbons. From C6H6 onwards, the addition of a CH3 radical leads to toluene (C7H8) formation. In the following steps, PAHs are formed via the addition of C3H3 or benzyl radicals. Although OH radicals also attack aromatic molecules such as C6H6 and naphthalene to form phenol and 2-naphthol, respectively, these molecules are unstable and quickly form hydrocarbon products, hereby releasing CO.
 | ||
| Fig. 8 Integral reaction flow analysis at 1673 K for τ = 5 s, CH4:CO2 ratio = 2, and p = 1 bar. | ||
Overall, the temperature is a parameter that greatly influences both the DRM reaction progression as well as the product distribution. At lower temperatures up to 1373 K, H2 is consumed as underscored by the data plotted in Fig. 4 and 5(a). As revealed by the RFA results (Fig. 7), H2 is consumed during H2O formation via the RWGS reaction (eqn (3)) while at the same time CO2 is converted into CO. On the other hand, the progress of C–C coupling reactions that release H2 are limited. C2H2, which is the key species with regard to carbon deposition reactions,10,58,60,64 is also formed only in negligible quantity. Therefore, there is a net consumption of H2 at 1373 K. In contrast, at 1673 K there is a net production of H2 as depicted in Fig. 4. However, the axial profile simulated for a reaction temperature of 1673 K (Fig. 5(b)) suggests different reaction regimes along the reactor length. It uncovers that H2 is consumed in the pre-heating zone at temperatures between 1273 K and 1423 K, and then rises rapidly. Between 1273 K and 1423 K, the RWGS reaction (eqn (3)) is dominant and results in H2O and CO production. As underscored by the RFA results plotted in Fig. 8, a temperature increase to 1673 K facilitates C–C coupling reactions. The coupling reactions and subsequent gas-phase dehydrogenation reactions generate all the species involved in the carbon deposition mechanism. The surface deposition reactions further release the H2 in the gas-phase while forming the solid carbon layer on the reactor wall. Therefore, net H2 formation is observed at 1673 K. After all, RWGS is the dominating reaction in the temperature regime between 1273 K and 1423 K, and CH4 pyrolysis dominates above 1473 K. Consequently, the temperature regime governs the predominant reaction that activates the CH4 molecule in the gas-phase and thus directly affects C–C coupling reactions and ultimately carbon deposition as well. This becomes particularly clear when the two small peaks in the deposition profiles in Fig. 6(b) are considered. At higher temperature H2O is split into H and OH radicals. These OH radicals can then attack aromatic hydrocarbons, which are cracked to release CO. Sustained CO formation even after complete depletion of CO2 is a result of this reaction sequence and explains the continuous rise of the CO mole fraction in Fig. 5(b). At 1673 K, τ = 5 s, and a CH4:CO2 ratio of 2, 38% of CH4 from the feed react to CO, whereas 52% of CH4 are captured in the form of solid carbon that deposits on the reactor walls and at the bottom of the reactor. These findings underscore the major importance of both, gas-phase and deposition reactions. Considering the experimental error bars of 5%, the simulations are in good agreement with the experiments. Notably, beyond a mere description of the end-of-pipe data, the axially resolved species profiles as well as the RFA allow to explain the complex reaction chemistry during thermal DRM.
:CO2 ratios is studied in this section. Fig. 9 illustrates the end-of-pipe mole fractions of both experiments and numerical simulations for different CH4:CO2 ratios in the feed gas, while keeping temperature (T = 1673 K), residence time (τ = 5 s), and pressure (p = 1 bar) constant. Analogous to the data discussed in the previous section, the experimental data exhibit error bars of 5%. Overall, decreasing the CH4:CO2 ratio from 4 to 1 leads to increased CO formation and a suppression of H2 formation. CO2 is completely consumed in all cases, whereas traces of CH4 can be seen at the reactor outlet for the CH4:CO2 ratio of 3 and higher. Hydrocarbon by-products remain below 0.2% under all conditions (cf. Fig. S4†).
 | ||
| Fig. 9 End-of-pipe experiments vs. simulations at different CH4:CO2 ratios keeping the T = 1673 K, τ = 5 s, and p = 1 bar constant. | ||
Since analyzing axial profiles is essential for a comprehensive understanding of the impact of the CH4:CO2 ratio, Fig. 10 shows axially resolved mole fraction profiles for the main gas-phase species H2, CH4, CO2, and CO as well as for the deposition of carbon that were simulated for CH4:CO2 ratios of 1 and 4. For a CH4:CO2 ratio of 1, H2 shows a sharp drop prior to the hot zone, which points to a consumption of H2. Subsequently, the H2 mole fraction increases along the reactor length and results in a net H2 production by the reactor outlet (Fig. 10(a)). For a higher CH4:CO2 ratio of 4, the minimum of H2 disappears and H2 steadily increases along the reactor length, resulting in a pronounced net H2 production at the reactor outlet. Since the feed gas contains more CH4 if a CH4:CO2 ratio of 4 is chosen, C–C coupling reactions and thus C2H2 formation are promoted (Fig. S5†). Higher C2H2 levels ultimately lead to higher carbon deposition, which is supported by the data depicted in Fig. 10(b).
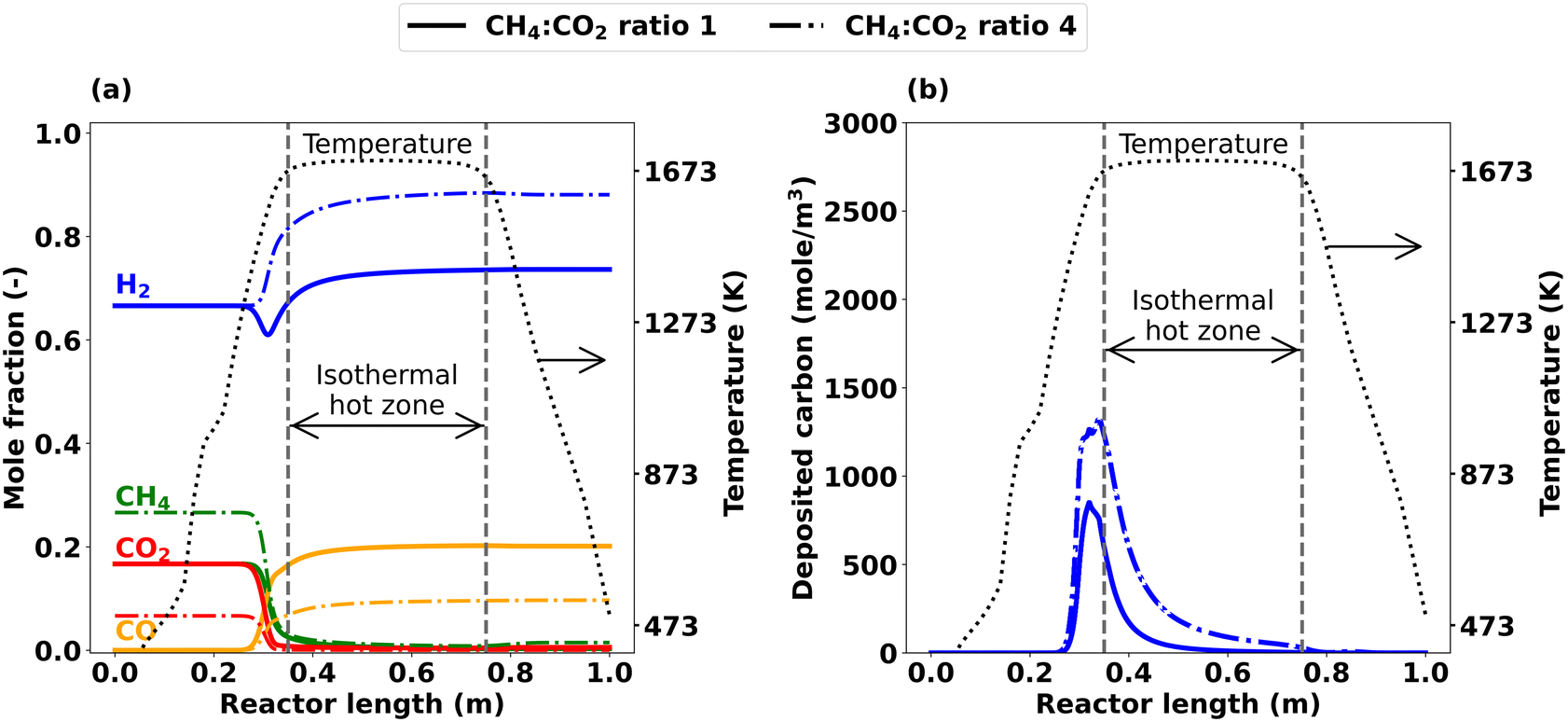 | ||
| Fig. 10 Simulated axial profiles for CH4:CO2 ratio 1 and 4 at T = 1673 K, τ = 5 s and p = 1 bar; (a) main species, (b) carbon deposition after 1800 s. | ||
Overall, the CH4:CO2 ratio is the key process parameter to control the H2/CO ratio of the effluent syngas. For a CH4:CO2 ratio of 1 in the feed, the end-of-pipe H2/CO syngas ratio is 3.6, but it increases to 9 when a CH4:CO2 ratio of 4 is chosen instead. However, note that the H2-rich syngas at the reactor outlet is also a result of the H2 content in the feed gas stream, as the reaction gases are diluted by 66.7% H2 prior to entering the reactor. Typical processes in chemical industry that rely on syngas, e.g. the synthesis of methanol, oxo-alcohols, and the Fischer–Tropsch process, require a H2/CO ratio of approx. 2.21 Therefore, integrating syngas production into chemical production would call for an adjustment of the H2/CO ratio. Notably, most of the worldwide syngas demand is currently covered by production via SMR,15 which commonly results in syngas with an H2/CO ratio between 2.8 and 4.8.21,67 Therefore, syngas produced by thermal DRM from feeds with comparably low CH4:CO2 ratios might be used to replace syngas produced in SMR plants, which enables the direct use of existing downstream infrastructure.
As an alternative, a mixture of only CH4 and CO2 without H2 dilution could be used as feed. Under the assumption of a hypothetical CH4:CO2 ratio of 1.86, a syngas H2/CO ratio of 2 can be attained end-of-pipe according to the axially resolved simulation data shown in Fig. 11. However, the higher CH4 levels in the feed gas go along with a higher carbon formation and deposition on the reactor walls as well as with a relatively high H2O formation. In general, thermal DRM represents a remarkably versatile reaction system. By considering downstream gas processing units and the constraints of reactor design, it is possible to obtain a wide range of syngas qualities based on a rational choice of the feed gas composition.
 | ||
| Fig. 11 Simulated axial profiles with CH4:CO2 molar ratio of 1.86, without H2 in feed and T = 1673 K, τ = 5 s and p = 1 bar; (a) gas-phase species, (b) carbon deposition after 1800 s. From the CH4-borne carbon, only a minor share remains in the gas-phase as hydrocarbons, while 33% of the CH4-borne carbon is transformed into CO and 48% of CH4-borne carbon is captured as solid carbon. | ||
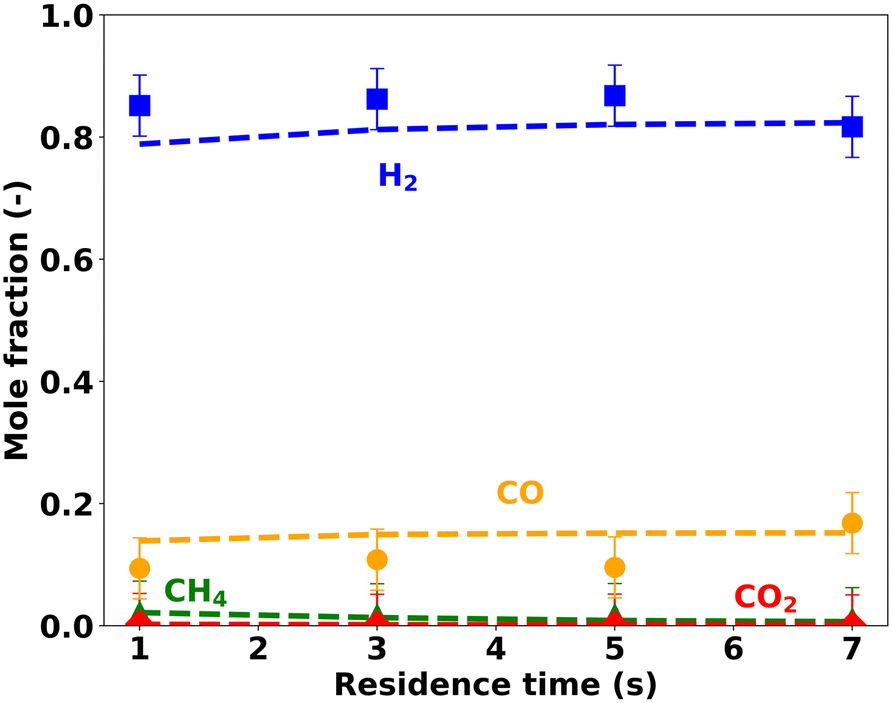 | ||
| Fig. 12 End-of-pipe experiments vs. simulations at different residence times keeping T = 1673 K, CH4:CO2 ratio = 2, and p = 1 bar. | ||
The axially resolved mole fraction profiles illustrated in Fig. 13 enable a more detailed understanding and ultimately offer an explanation for the trends observed. The profiles obtained at a residence time τ of 1 s and 7 s differ most at a reactor length of approx. 0.32 m, as more CH4 and CO2 are consumed during the onset of the reaction if a residence time of 7 s is chosen instead of 1 s (Fig. 13(a)). Similarly, H2 and CO are formed earlier in the reactor for τ = 7 s. These residence time-induced differences become smaller towards the reactor outlet. Although the end-of-pipe gas-phase concentrations deviate only to a minor extent, residence time variations exhibit a significant impact on carbon deposition: as shown by the simulation data in Fig. 13(b), the carbon formation and deposition along the reactor axis is much higher at τ of 1 s compared to 7 s.
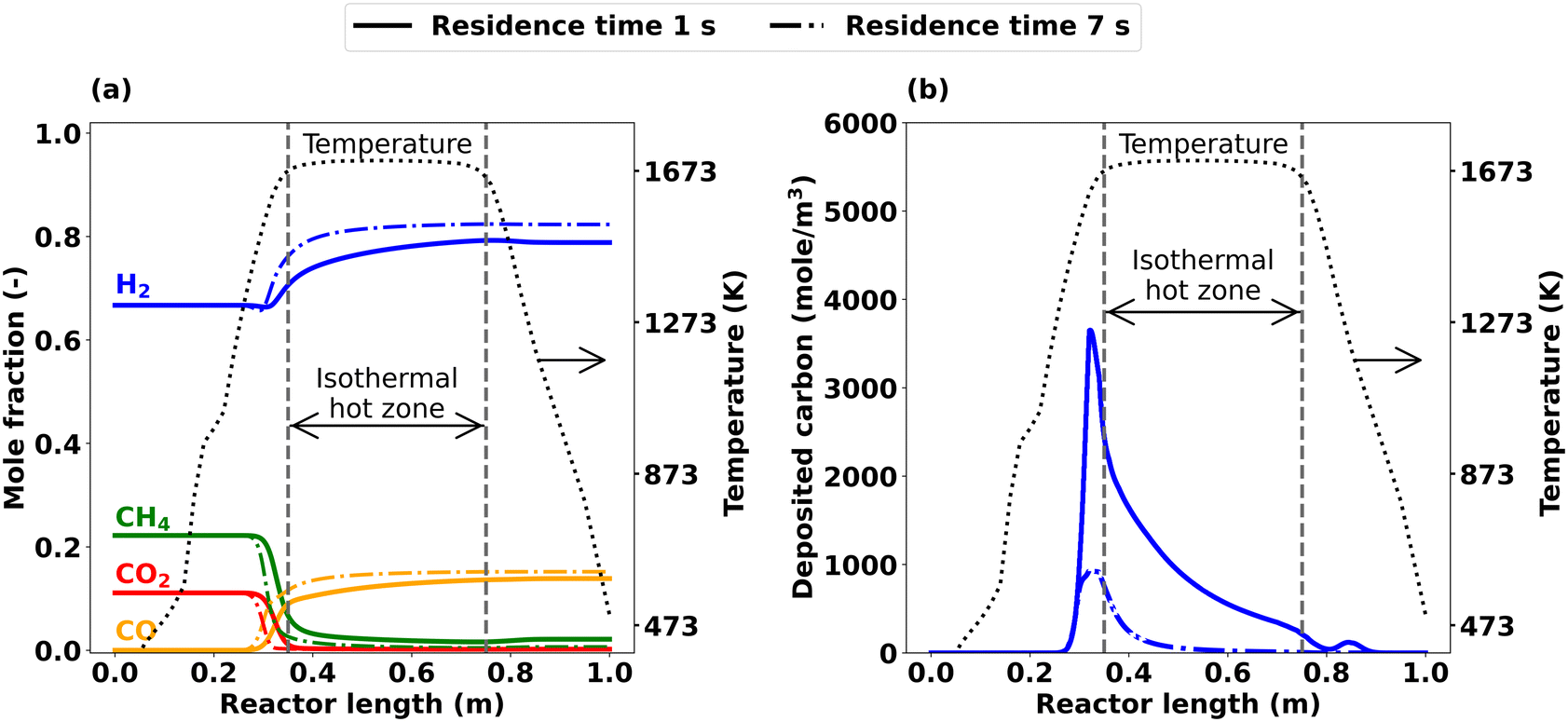 | ||
| Fig. 13 Simulated axial profiles for τ of 1 s and 7 s at CH4:CO2 ratio = 2, T = 1673 K and p = 1 bar; (a) main species, (b) carbon deposition after 1800 s. | ||
Notably, a lower residence time is achieved by increasing the mass flow rate of the feed gas (cf. Table S1†). Consequently, more CH4 molecules enter the reactor at τ of 1 s compared to 7 s, which results in a substantial increase in deposited carbon from 0.35 g at 7 s to 1.95 g at 1 s. With regard to technical applications, these findings are of high relevance as carbon deposition can cause reactor clogging. However, innovative reactor designs such as the use of a carbon moving bed, e.g. as proposed by BASF SE in the context of methane pyrolysis, might be a useful concept for thermal DRM processes as well.68
Since carbon formation is a consequence of C–C-coupling and dehydrogenation reactions, also the by-products, C2 species in particular, are strongly influenced by variations of the residence time τ, which is illustrated in Fig. 14. At τ = 7 s, the share of C2 species by the end of the reactor is 0.04%, which rises to 1.4% when τ is reduced to 1 s. Previous studies have focused on the pyrolysis of natural gas at short residence times, typically below 1 s, and especially investigated C2 species formation.18,69,70 Our present results indicate that τ is a crucial process parameter regarding carbon deposition also in the 1 s to 7 s range. However, the primary products at these conditions are H2 and CO, whereas at smaller residence times the C2 fraction in the products increase. Moreover, the fraction of CH4 converted into CO and solid carbon also changes considerably with τ. At 1 s, 27% CH4 are converted into CO, 42% into captured deposited carbon, and 32% remain in the gas-phase in the form of other hydrocarbons. Increasing τ to 7 s enables to capture 53% of the CH4 in the form of elemental carbon that deposits in the reactor, while 38% are converted into CO, and only 8% remain in the gas-phase in the form of other hydrocarbons.
 | ||
| Fig. 14 Simulated axial profiles of C2H2 and C2H4 for τ of 1 s and 7 s at CH4:CO2 ratio = 2, T = 1673 K and p = 1 bar. | ||
Conclusions
In the present study, thermal dry reforming of methane was studied for a wide range of operating conditions by combining numerical simulations and experiments. A one-dimensional plug flow reactor model was used to study coupled gas-phase and deposition reaction systems. Simulated gas-phase species were compared to the end-of-pipe measurements for model validation.The deposition model applied herein offers insights into the distribution of carbon deposits along the length of the reactor. Two different temperature-dependent reaction regimes were identified. For lower temperatures between 1273 K and 1373 K, the reverse water-gas shift reaction plays a key role, resulting in the consumption of H2 and the formation of CO and H2O. In contrast, methane pyrolysis is the dominating reaction pathway at 1473 K and above and thus facilitates carbon deposition and H2 production. For a CH4:CO2 ratio of 2, a residence time of 5 s, and a pressure of 1 bar, CO2 was completely consumed when the temperature exceeded 1473 K, mostly forming CO via the reverse water-gas shift reaction. In contrast, CH4 conversion requires higher temperatures. Above 1673 K, CH4 is fully consumed due to CO formation from dry reforming and carbon deposition from pyrolytic reaction pathways. While under such harsh conditions only small amounts of hydrocarbon species remain in the gas-phase, 38% of the CH4 from the feed stream are transformed into CO and 52% are captured as solid carbon. The composition of the feed gas, specifically in terms of the CH4:CO2 ratio and additional H2 dilution, is a critical parameter for tuning the H2/CO ratio in the effluent syngas product gas stream. Thus, optimizing the feed gas composition offers great potential to meet specific requirements, such as achieving a particular H2/CO syngas ratio while simultaneously minimizing or maximizing solid carbon deposition. Although residence time variations between 1 s and 7 s have an only marginal impact on the H2/CO syngas ratio, reducing the residence time resulted in higher carbon deposition due to higher mass flow rates. In conclusion, all the process parameters varied herein have a relevant influence on the distribution of carbon derived from CH4, determining the proportion converted into CO and the amount sequestrated as solid deposits on the reactor walls.
Thermal dry reforming of CO2-containing biogas possesses a great potential to act as a CO2-negative process while simultaneously producing sustainable syngas as feedstock for valuable chemicals. The combined gas-phase and deposition model used in this study accurately describes the thermal DRM process. Beyond simply serving as a means to validate against experimental data, our model offers an essential advantage by providing detailed axial profiles and quantitative information on deposited carbon, which are challenging to obtain through experimental approaches. Despite the comprehensive chemistry considered in the mechanism used herein, the simulations can be run on a desktop computer within a reasonable time frame. This framework and the insights gained from this work lay a strong foundation for reactor design and scale-up of the thermal dry reforming process, which may be an industrially viable production route for sustainable syngas.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Manas Mokashi: writing – original draft, visualization, validation, methodology, investigation, formal analysis, data curation, conceptualization. Akash Bhimrao Shirsath: writing – review & editing, software, formal analysis. Sinan Demir: investigation, formal analysis. Ahmet Çelik: investigation, formal analysis. Patrick Lott: writing – review & editing, supervision, project administration, methodology, data curation, conceptualization. Steffen Tischer: writing – review & editing, supervision, software, formal analysis. Olaf Deutschmann: writing – review & editing, supervision, resources, project administration, funding acquisition, data curation, conceptualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the omegadot software & consulting GmbH for a cost-free academic license of DETCHEM and H. Müller (ITCP, KIT) for fruitful discussion.References
- K. Oshiro and S. Fujimori, Appl. Energy, 2022, 313, 118803 CrossRef CAS.
- P. Lott and O. Deutschmann, Proc. Combust. Inst., 2023, 39, 3183–3215 CrossRef CAS.
- A. González-Garay, M. S. Frei, A. Al-Qahtani, C. Mondelli, G. Guillén-Gosálbez and J. Pérez-Ramírez, Energy Environ. Sci., 2019, 12, 3425–3436 RSC.
- A. A. Kiss, J. J. Pragt, H. J. Vos, G. Bargeman and M. T. de Groot, Chem. Eng. J., 2016, 284, 260–269 CrossRef CAS.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112–3135 RSC.
- C. Kim, C. J. Yoo, H. S. Oh, B. K. Min and U. Lee, J. CO2 Util., 2022, 65, 102239 CrossRef CAS.
- G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711–1731 RSC.
- O. Schmidt, A. Gambhir, I. Staffell, A. Hawkes, J. Nelson and S. Few, Int. J. Hydrogen Energy, 2017, 42, 30470–30492 CrossRef CAS.
- O. Machhammer, A. Bode and W. Hormuth, Chem. Ing. Tech., 2016, 39, 1185–1193 CAS.
- P. Lott, M. B. Mokashi, H. Müller, D. J. Heitlinger, S. Lichtenberg, A. B. Shirsath, C. Janzer, S. Tischer, L. Maier and O. Deutschmann, ChemSusChem, 2023, 16, e202300301 CrossRef PubMed.
- A. B. Shirsath, M. Mokashi, P. Lott, H. Müller, R. Pashminehazar, T. Sheppard, S. Tischer, L. Maier, J.-D. Grunwaldt and O. Deutschmann, J. Phys. Chem. A, 2023, 127, 2136–2147 CrossRef CAS PubMed.
- T. Becker, F. Keuchel and D. W. Agar, Chem. Ing. Tech., 2021, 93, 762–770 CrossRef CAS.
- N. Sánchez-Bastardo, R. Schlögl and H. Ruland, Ind. Eng. Chem. Res., 2021, 60, 11855–11881 CrossRef.
- T. Geißler, A. Abánades, A. Heinzel, K. Mehravaran, G. Müller, R. K. Rathnam, C. Rubbia, D. Salmieri, L. Stoppel, S. Stückrad, A. Weisenburger, H. Wenninger and T. Wetzel, Chem. Eng. J., 2016, 299, 192–200 CrossRef.
- G. A. Olah, A. Goeppert and G. K. Surya Prakash, Methanol Economy, John Wiley & Sons Ltd, 2009, pp. 143–178 Search PubMed.
- E. Bartholomé, Chem. Eng. Sci., 1954, 3, 94–104 CrossRef.
- D. Chen, X. Chen, C. Luo, Z. Liu and L. H. Gan, Chem. Eng. J., 2021, 426, 130871 CrossRef CAS.
- B. Baek, B. Nair, I. Lengyel, L. Chen, S. Pannala, R. Vm and D. West, Ind. Eng. Chem. Res., 2021, 60, 6993–7002 CrossRef CAS.
- S. Gudiyella, Z. J. Buras, T. C. Chu, I. Lengyel, S. Pannala and W. H. Green, Ind. Eng. Chem. Res., 2018, 57, 7404–7420 CrossRef CAS.
- R. Reimert, F. Marschner, H.-J. Renner, W. Boll, E. Supp, M. Brejc, W. Liebner and G. Schaub, Gas Production, 2. Processes, in Ullmann's Encyclopedia of Industrial Chemistry, John Wiley & Sons Ltd, 2011, pp. 423–482 Search PubMed.
- E. Schwab, A. Milanov, S. A. Schunk, A. Behrens and N. Schödel, Chem. Ing. Tech., 2015, 87, 347–353 CrossRef CAS.
- K. Wittich, M. Krämer, N. Bottke and S. A. Schunk, ChemCatChem, 2020, 12, 2130–2147 CrossRef CAS.
- J. R. Rostrup-Nielsen, J. Sehested and J. K. Nørskov, Adv. Catal., 2002, 47, 65–139 CAS.
- M. H. Halabi, M. H. J. M. de Croon, J. van der Schaaf, P. D. Cobden and J. C. Schouten, Chem. Eng. J., 2008, 137, 568–578 CrossRef CAS.
- R. Schwiedernoch, S. Tischer, C. Correa and O. Deutschmann, Chem. Eng. Sci., 2003, 58, 633–642 CrossRef CAS.
- O. Deutschmann and L. D. Schmidt, AIChE J., 1998, 44, 2465–2477 CrossRef CAS.
- S. Hanf, S. Angeli, D. Dussol, C. Fritsch, L. Maier, M. Müller, O. Deutschmann and S. A. Schunk, Chapter 9: Methane Dry Reforming, in: Chemical Valorisation of Carbon Dioxide, ed. G. Stefanidis and A. Stankiewicz, The Royal Society of Chemistry, 2022, pp. 187–207 Search PubMed.
- A. Giehr, L. Maier, S. Angeli, S. A. Schunk and O. Deutschmann, Ind. Eng. Chem. Res., 2020, 59, 18790–18797 CrossRef CAS.
- L. A. Schulz, L. C. S. Kahle, K. H. Delgado, S. A. Schunk, A. Jentys, O. Deutschmann and J. A. Lercher, Appl. Catal., A, 2015, 504, 599–607 CrossRef CAS.
- A. Calbry-Muzyka, H. Madi, F. Rüsch-Pfund, M. Gandiglio and S. Biollaz, Renewable Energy, 2022, 181, 1000–1007 CrossRef CAS.
- D. Zambrano, J. Soler, J. Herguido and M. Menéndez, Top. Catal., 2019, 62, 456–466 CrossRef CAS.
- N. Gokon, Y. Yamawaki, D. Nakazawa and T. Kodama, Int. J. Hydrogen Energy, 2011, 36, 203–215 CrossRef CAS.
- N. E. McGuire, N. P. Sullivan, O. Deutschmann, H. Zhu and R. J. Lee, Appl. Catal., A, 2011, 394, 257–265 CrossRef CAS.
- M. Maestri, D. G. Vlachos, A. Beretta, G. Groppi and E. Tronconi, J. Catal., 2008, 259, 211–222 CrossRef CAS.
- B. Steinhauser, M. R. Reddy, J. Radnik and A. Martin, Appl. Catal., A, 2009, 366, 333–341 CrossRef.
- Y. Kathiraser, U. Oemar, E. T. Saw, Z. Li and S. Kawi, Chem. Eng. J., 2015, 278, 62–78 CrossRef CAS.
- T. Roussière, L. Schulz, K. M. Schelkle, G. Wasserschaff, A. Milanov, E. Schwab, O. Deutschmann, A. Jentys, J. Lercher and S. A. Schunk, ChemCatChem, 2014, 6, 1447–1452 CrossRef.
- C. Fritsch, J. Titus, T. Roussière, C. Lizandara-Pueyo, R. Müller, R. Gläser and S. A. Schunk, Chem. Ing. Tech., 2022, 94, 1727–1738 CrossRef CAS.
- Y. Lin, C. Yang, W. Zhang, H. Machida and K. Norinaga, Chem. Eng. Sci., 2023, 268, 118380 CrossRef CAS.
- S. Chen, J. Zaffran and B. Yang, Appl. Catal., B, 2020, 270, 118859 CrossRef CAS.
- M. Usman, W. M. A. W. Daud and H. F. Abbas, Renewable Sustainable Energy Rev., 2015, 45, 710–744 CrossRef CAS.
- C. Pizzolitto, E. Pupulin, F. Menegazzo, E. Ghendini, A. D. Michele, M. Mattarelli, G. Cruciani and M. Signoretto, Int. J. Hydrogen Energy, 2019, 44, 28065–28076 CrossRef CAS.
- J. O. Ighalo and P. B. Amama, J. CO2 Util., 2024, 81, 102734 CrossRef CAS.
- B. Stolze, J. Titus, S. A. Schunk, A. Milanov, E. Schwab and R. Gläser, Front. Chem. Sci. Eng., 2016, 10, 281–293 CrossRef CAS.
- J. H. Bitter, K. Seshan and J. A. Lercher, J. Catal., 1999, 183, 336–343 CrossRef CAS.
- C. Fukuhara, Y. Matsui, M. Tanebayashi and R. Watanabe, Chem. Eng. J. Adv., 2021, 5, 100057 CrossRef CAS.
- L. C. S. Kahle, T. Roussière, L. Maier, K. Herrera Delgado, G. Wasserschaff, S. A. Schunk and O. Deutschmann, Ind. Eng. Chem. Res., 2013, 52, 11920–11930 CrossRef CAS.
- A. Çelik, I. Ben Othman, H. Müller, P. Lott and O. Deutschmann, React. Chem. Eng., 2023, 9, 108–118 RSC.
- D. Chen and L. H. Gan, Chem. Eng. J., 2022, 439, 135732 CrossRef CAS.
- V. I. Savchenko, Y. S. Zimin, A. V. Nikitin, I. V. Sedov and V. S. Arutyunov, J. CO2 Util., 2021, 47, 101490 CrossRef CAS.
- S. D. Angeli, S. Gossler, S. Lichtenberg, G. Kass, A. K. Agrawal, M. Valerius, K. P. Kinzel and O. Deutschmann, Angew. Chem., Int. Ed., 2021, 60, 11852–11857 CrossRef CAS PubMed.
- O. V. Shapovalova, Y. N. Chun, M. S. Lim, V. M. Shmelev and V. S. Arutyunov, Int. J. Hydrogen Energy, 2012, 37, 14040–14046 CrossRef CAS.
- P. Blanck, G. Kass, K. P. Kinzel and O. Deutschmann, Energy Adv., 2024, 3, 123–130 RSC.
- F. Bustamante, R. M. Enick, A. V. Cugini, R. P. Killmeyer, B. H. Howard, K. S. Rothenberger, M. V. Ciocco, B. D. Morreale, S. Chattopadhyay and S. Shi, AIChE J., 2004, 50, 1028–1041 CrossRef CAS.
- A. B. Shirsath, M. L. Schulte, B. Kreitz, S. Tischer, J.-D. Grunwaldt and O. Deutschmann, Chem. Eng. J., 2023, 469, 143847 CrossRef CAS.
- O. Deutschmann, S. Tischer, S. Kleditzsch, V. Janardhanan, C. Correa, D. Chatterjee, N. Mladenov, H. D. Minh, H. Karadeniz, M. Hettel, V. Menon, A. Banerjee, H. Gossler, A. Shirsath and E. Daymo, DETCHEM, 2022, https://www.detchem.com Search PubMed.
- J. Appel, H. Bockhorn and M. Frenklach, Combust. Flame, 2000, 121, 122–136 CrossRef CAS.
- G. Blanquart, P. Pepiot-Desjardins and H. Pitsch, Combust. Flame, 2009, 156, 588–607 CrossRef CAS.
- S. Porras, D. Kaczmarek, J. Herzler, S. Drost, M. Werler, T. Kasper, M. Fikri, R. Schießl, B. Atakan, C. Schulz and U. Maas, Combust. Flame, 2020, 212, 107–122 CrossRef CAS.
- M. Mokashi, A. B. Shirsath, A. Çelik, P. Lott, H. Müller, S. Tischer, L. Maier, J. Bode, D. Schlereth, S. Frederik, D. Flick, M. Bender, K. Ehrhardt and O. Deutschmann, Chem. Eng. J., 2024, 485, 149684 CrossRef CAS.
- K. J. Hüttinger, Chem. Vap. Deposition, 1998, 4, 151–158 CrossRef.
- A. Becker, Z. Hu and K. J. Hüttinger, Fuel, 2000, 79, 1573–1580 CrossRef CAS.
- A. Shamsi and C. D. Johnson, Catal. Today, 2003, 84, 17–25 CrossRef CAS.
- M. Mokashi, A. B. Shirsath, P. Lott, H. Müller, S. Tischer, L. Maier and O. Deutschmann, Chem. Eng. J., 2024, 479, 147556 CrossRef CAS.
- A. Çelik, A. B. Shirsath, F. Syla, H. Müller, P. Lott and O. Deutschmann, J. Anal. Appl. Pyrolysis, 2024, 181, 106628 CrossRef.
- H. Gossler and O. Deutschmann, Int. J. Hydrogen Energy, 2015, 40, 11046–11058 CrossRef CAS.
- S. Luo, L. Zeng, D. Xu, M. Kathe, E. Chung, N. Deshpande, L. Qin, A. Majumder, T. L. Hsieh, A. Tong, Z. Sun and L. S. Fan, Energy Environ. Sci., 2014, 7, 4104–4117 RSC.
- BASF SE, New technologies: Methane pyrolysis, 2022, https://www.basf.com/global/en/who-we-are/sustainability/we-produce-safely-and-efficiently/energy-and-climate-protection/carbon-management/innovations-for-a-climate-friendly-chemical-production.html#text-1002215085. Access: August 4, 2024 Search PubMed.
- A. Holmen, O. Olsvik and O. A. Rokstad, Fuel Process. Technol., 1995, 42, 249–267 CrossRef CAS.
- F. G. Billaud, F. Baronnet and C. P. Gueret, Ind. Eng. Chem. Res., 1993, 32, 1549–1554 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details on mechanism and additional data on by-products. See DOI: https://doi.org/10.1039/d4re00312h |
| This journal is © The Royal Society of Chemistry 2024 |