
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of a solid-compatible continuous flow reactor for the paraformaldehyde slurry mediated α-hydroxymethylation of methyl vinyl ketone†
Bavo
Vandekerckhove‡
*a,
Lise
Van Coillie‡
*a,
Bert
Metten
 b,
Thomas S. A.
Heugebaert
a and
Christian V.
Stevens
a
b,
Thomas S. A.
Heugebaert
a and
Christian V.
Stevens
a
aSynBioC Research Group, Department of Sustainable Organic Chemistry and Technology, Department of Green Chemistry and Technology, Faculty of Bioscience Engineering, Ghent University, Coupure Links 653, B-9000 Ghent, Belgium. E-mail: Chris.Stevens@UGent.be
bAjinomoto Bio-Pharma Services, Cooppallaan 91, 9230 Wetteren, Belgium
First published on 29th May 2024
Abstract
The α-hydroxymethylation reactions hold a significant position within the pharmaceutical industry due to their intriguing nature. Despite numerous reported methods, they often entail prolonged reaction times and moderate yields. Moreover, the prevalent use of aqueous formaldehyde restricts the applicability of this chemistry to water-compatible substrates. Gaseous formaldehyde remains largely avoided due to its toxicity, hazards, and requirement for substantial excess. Within this context, paraformaldehyde emerges as a promising alternative for the C1 building block, offering safety and ease of handling. Continuous flow methodology is employed to facilitate the in situ depolymerization of paraformaldehyde under optimized conditions, enabling direct utilization of the released formaldehyde gas. This research explores the use of a paraformaldehyde slurry in continuous flow for α-hydroxymethylation reactions, with methyl vinyl ketone serving as a proof-of-concept substrate. A solid-compatible continuous flow reactor was self-constructed and the hydroxymethylation of methyl vinyl ketone could successfully be optimised, resulting in a STY of 2040 kg h−1 m−3.
Introduction
Hydroxymethyl motifs are often found in the core structure of natural products and in various important active pharmaceutical ingredients (APIs). For example, α-hydroxymethyl ketones are found in anticholinergic medication (atropine), antiproliferative agents, antitumor agents (olivomycin A and chromomycin A3), possible anti-cancer substances etc.1,2 Moreover, the –CH2OH unit is easily converted into many other functional groups, which contributes to the further expansion of its synthetic potential. In addition, the introduction of small substituents, such as a hydroxymethyl group, has the capacity to modulate molecular properties such as solubility, bioavailability, receptor binding affinity and metabolic stability. These modifications have a pivotal role in optimizing the pharmacological and pharmacokinetic properties.3–7The synthesis of α-hydroxymethyl ketones is most often performed with formaldehyde as a C1 source. Under normal circumstances, formaldehyde exists as a colorless gas with flammable properties, marked by a high reactivity and characterized by a pungent, irritating odor. The levels at which gaseous formaldehyde is carcinogenic and causes toxic irritations are lower than its odor threshold, generating a substantial safety hazard. Theoretically, formaldehyde gas could be used in batch reactors by bubbling it through the reaction mixture. In practice, however, formaldehyde gas is avoided in industry because of these safety concerns. Therefore, it is mostly used as a solution in water, known as formalin. However, this limits the substrate scope tremendously since only water compatible substrates and reactive intermediates can be used.8,9 In addition, both aqueous and gaseous formaldehyde most often require very long reaction times or high stoichiometric excesses, leading to low efficiencies, low atom economy and/or low yields.10–13
To address the limitations of aqueous and/or gaseous formaldehyde, the solid paraformaldehyde (PFA) polymer could be an interesting alternative. Due to its polymeric nature, PFA is easily transported and stored, and can be handled safely. Under appropriate reaction conditions, it enables a gradual release of monomeric formaldehyde gas. The latter can be used directly to perform the hydroxymethylation reaction of interest. Several indicators show that continuous flow chemistry could be advantageous for this type of chemistry.14 At elevated temperatures and pressures, the depolymerization efficiency can be increased while maintaining the containment of the in situ generated formaldehyde, thus strongly reducing the dangers associated with the use of this carcinogenic gas. In addition, the reactivity of the monomeric formaldehyde molecules can be increased and thus reaction times can be shortened. Nevertheless, paraformaldehyde is a solid and handling suspensions/slurries is known to be detrimental for continuous flow applications.15–17 Literature showed alternative reaction setups regarding the ex situ generation of formaldehyde gas from paraformaldehyde and its use within a Teflon AF 2400 tube-in-tube design.18 However, this setup is often limited as well by difficult heating, relatively low gas loading, insufficient radial mixing and rather challenging scale-up.19
Until the present, the use of solids or slurries in continuous flow has posed significant challenges, primarily due to issues such as clogging, accumulation and sedimentation, subsequently leading to blockage of the setup. A number of studies attempt to address this, but place a predominant focus on immobilized catalyst chemistry, often in packed bed-type reactors.20–23 Studies addressing the use of solid reagents requiring accurate dosing into the flow reactor or handling formed precipitations are scarce.24,25 This study presents the efforts to design a novel continuous flow setup to deal with the conditions required for paraformaldehyde slurry mediated chemistry.
Scope
α-Hydroxymethylation of Michael acceptor systems using formaldehyde embodies a simple, yet important reaction with respect to the products' vital role in total synthesis and polymer science. The Morita–Baylis–Hillman (MBH) reaction has been generally described for vinyl ketones using formaldehyde for the synthesis of hydroxymethyl α-unsaturated ketones. Mantel et al. reported an efficient one-pot MBH-type α-hydroxymethylation of vinyl ketones followed by the convenient, temperature-controlled etherification using alcohols.26 Even though this type of chemistry is generally known in literature, long reaction times are often required, while using toxic solvents/reagents and a disfavorable atom economy.27–29 The two-step α-hydroxymethylation consist of preliminary paraformaldehyde depolymerization, followed by addition of the vinyl ketone (Fig. 1). | ||
| Fig. 1 Two-step α-hydroxymethylation of methyl vinyl ketone in batch according to Mantel et al.26 | ||
The α-hydroxymethylation of methyl vinyl ketone (MVK) was chosen as a proof-of-concept reaction to evaluate paraformaldehyde slurry chemistry in continuous flow (Fig. 1). Although the reported batch reaction time is reasonable (±3.5 h), there is potential to significantly reduce it to the minute range. Hence, this reaction is a good model to monitor the efficiency of a paraformaldehyde slurry mediated continuous flow process. The focus is set on the reduction of reaction times, yield optimisation and safety improvement of the procedure. If PFA depolymerization can be performed efficiently in the flow reactor, the applicability of the MBH reaction can be widened and scale-up of this type of chemistry can be performed safely.
Results and discussion
Batch control experiments
Before attempting the paraformaldehyde (PFA) mediated α-hydroxymethylation reaction in a continuous flow setup, a short batch optimisation was performed to validate literature reports and tune the reaction conditions towards a flow compatible process. Therefore, sealed vials were used in a batch setup to withstand some possible pressure build-up. According to the Arrhenius law, increasing reaction temperatures can decrease the reaction time. Nevertheless, Mantel et al. showed that at elevated temperatures, the hydroxyl group undergoes an etherification reaction with the alcohol that is present (as solvent). Since higher temperatures will be employed in further optimisation, it may be of interest to avoid alcoholic solvents to minimise the etherification. A variety of solvents were tested using the reaction conditions outlined by Mantel et al., involving the two-step process (Table 1). It is apparent that the alcoholic solvents such as EtOH and IPA exhibited the most favourable reaction performance. Only 1,4-dioxane provided a moderate yield when employed as a non-alcoholic solvent. Consequently, these will be revisited during the subsequent reaction time screening phase. Moreover, Robiette et al. already demonstrated the beneficial impact of protic solvents on the kinetics of the MBH reaction.30| Entry | Temperature (°C) | Reaction time (min) | Solvent | PFA (eq.) | NMR yieldc (%) |
|---|---|---|---|---|---|
| a Literature conditions by Mantel et al. b Isolated yield after column chromatography. c NMR yield calculated relative to internal standard (trimethoxybenzene). d Depolymerisation time and temperature were increased to ensure complete depolymerisation of PFA. | |||||
| 1a | 85/rt | 20/180 | EtOH | 1.4 | 71 (68)b |
| 2 | 85–115/rt | 20–60/180 | THFd | 1.4 | 2 |
| 3 | 85–115/rt | 20–60/180 | THF + 5% EtOHd | 1.4 | 37 |
| 4 | 85–115/rt | 20–60/180 | THF + 10% EtOHd | 1.4 | 49 |
| 5 | 85/rt | 20/180 | IPA | 1.4 | 71 |
| 6 | 85–115/rt | 20–60/180 | EtOAcd | 1.4 | 0 |
| 7 | 85/rt | 20/180 | H2O | 1.4 | 14 |
| 8 | 85–115/rt | 20–60/180 | Toluened | 1.4 | 6 |
| 9 | 85–115/rt | 20–60/180 | ACNd | 1.4 | 7 |
| 10 | 85/rt | 20/180 | 1,4-Dioxane | 1.4 | 57 |
| 11 | 85/rt | 20/180 | DME | 1.4 | 50 |
Following the solvent screening, it becomes crucial to decrease the reaction time to maintain an acceptable material throughput using continuous flow. Initially, ethanol was used to start the reaction time optimisation. As a first improvement, depolymerisation of paraformaldehyde and the subsequent reaction could be combined in a single step (Table 2). A short optimisation resulted in a decrease of the reaction time from 3.5 h to 8 minutes at 100 °C, giving similar NMR spectra and isolated yields to those obtained using the literature conditions. It was expected that less etherification side product would be formed when using isopropanol, however a lower yield was obtained under these conditions. Furthermore, alternative degradation mechanisms took precedence when 1,4-dioxane was utilized as a solvent, leading to a substantially lower yield.
| Entry | Temperature (°C) | Reaction time (min) | Solvent | PFA (Eq.) | NMR yieldc (%) |
|---|---|---|---|---|---|
| a Literature conditions by Mantel et al. b Isolated yield after column chromatography. c NMR yield calculated relative to internal standard (trimethoxybenzene). | |||||
| 1a | 85/25 | 20/180 | EtOH | 1.4 | 71 (68)b |
| 2 | 60 | 180 | EtOH | 1.4 | 71 |
| 3 | 60 | 60 | EtOH | 1.4 | 56 |
| 4 | 60 | 50 | EtOH | 1.4 | 51 |
| 5 | 60 | 40 | EtOH | 1.4 | 65 |
| 6 | 60 | 30 | EtOH | 1.4 | 66 |
| 7 | 60 | 20 | EtOH | 1.4 | 60 |
| 8 | 80 | 10 | EtOH | 1.4 | 68 |
| 9 | 80 | 20 | EtOH | 1.4 | 39 |
| 10 | 100 | 10 | EtOH | 1.4 | 53 |
| 11 | 100 | 8 | EtOH | 1.4 | 74 (70)b |
| 12 | 100 | 8 | IPA | 1.4 | 65 |
| 13 | 100 | 8 | 1,4-Dioxane | 1.4 | 27 |
| 14 | 100 | 5 | EtOH | 1.4 | 65 |
| 15 | 120 | 5 | EtOH | 1.4 | 59 |
| 16 | 120 | 3 | EtOH | 1.4 | 66 |
| 17 | 120 | 1 | EtOH | 1.4 | 57 |
| 18 | 100 | 8 | EtOH | 1.0 | 66 |
Experimental setup
As mentioned earlier, solids or slurries are acknowledged to pose challenges within continuous flow systems. This research utilizes a paraformaldehyde slurry, thus requiring a flow reactor design compatible with such a slurry. This has the potential to expand the scope of continuous flow chemistry, encompassing solids as reagents, catalysts, or products.Initially, tests were performed with a high pressure Teledyne ISCO 500d module with a Büchi mixer, a semi-continuous syringe pump with internal mixing to prevent solid sedimentation. However, it quickly became evident that this pump was underperforming with a significant portion of the solids remaining in the syringe, leading to a notably low paraformaldehyde output. Hence, a peristaltic pump (Vapourtec, SF-10) was employed as an alternative to accurately dose suspensions into the flow reactor. Conversely, this restricts the process window to pressures up to 10 bar. To avoid sedimentation of solids and eventually clogging of the flow setup, several alternative techniques have been reported: pulsatile flow, sonication, mechanical agitation, segmented flow etc.31–34 Within our reactor design, an oscillatory flow was used to keep solid particles suspended. Therefore, the Lewa Ecosmart diaphragm metering pump was selected and subsequently, its valves were removed to enable its operation as a pulsator (Fig. 2).35 This device is capable of handling system pressures up to 80 bar and the stroke amplitude of the oscillating flow can be manually adjusted. A frequency controller was installed and connected to the Lewa Ecosmart pump to allow adjustments of the stroke frequency of the oscillation as well, enabling complete control of the parameters governing the oscillatory flow.
 | ||
| Fig. 2 Lewa Ecosmart diaphragm metering pump. | ||
The stainless steel Uniqsis Hotcoil, a standalone heated reactor module with an internal volume of 40 mL was used as reactor, allowing reaction temperatures up to 300 °C and maximum pressures up to 100 bar.36 To interconnect all components, 1/8′′ stainless steel tubing and Swagelok connections were used to comply with the demands of the process window. Back pressure regulators (BPR), such as dome pressurized (Equilibar, Zaiput) and spring loaded cartridges, prove problematic when dealing with solids. Therefore, a blockage-resistant BPR was constructed, based on the research of Deadman et al.37 The design consists of a pressurised collection vessel with a tube-in-tube design (Fig. 3). To permit freely flowing chemical slurries, the flow paths had to be sufficiently wide-bore without restrictions to prevent blockages from occurring. At the bottom of the pressure chamber, a large-bore quarter-turn valve allows the contents of the chamber to be expelled whilst maintaining back pressure in the flow system. Inert nitrogen gas is employed for vessel pressurization, while Swagelok connections guarantee a system pressure capped at 100 bar.35
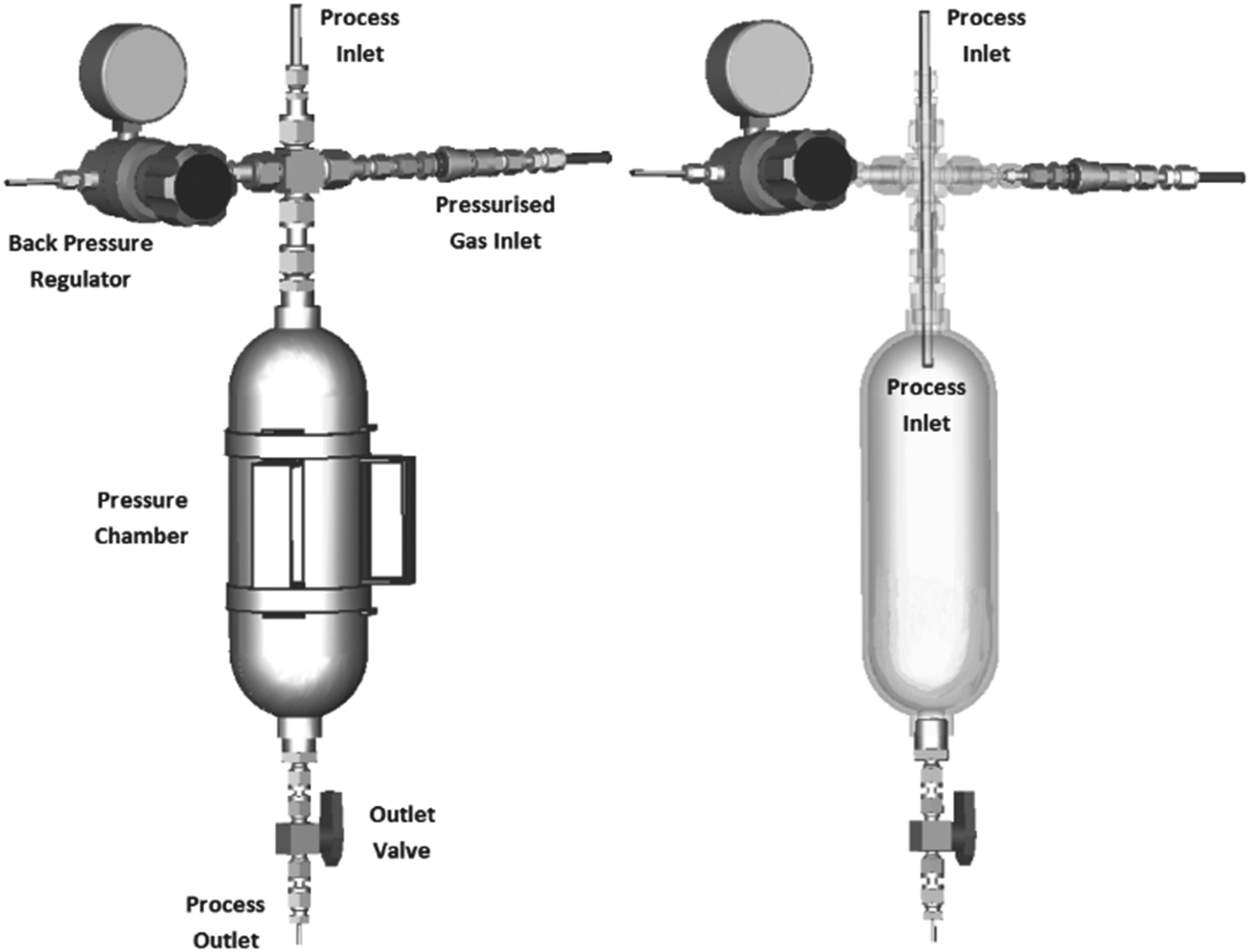 | ||
| Fig. 3 Design of the blockage-resistant back pressure regulator based on the research of Deadman et al.37 | ||
The integration of the aforementioned systems results in a slurry-compatible continuous flow reactor. Despite the initial focus of the reactor design being on high temperature/pressure chemistry (300 °C, 100 bar), the current process window is restricted to 10 bar due to the absence of a slurry-compatible high-pressure pump (TBoil,EtOH = 144 °C@10 bar). The applicability and efficiency of this self-constructed CØPE reactor§ (C = Celsius (temperature), Ø = particle size (slurry), P = pressure, E = extreme) will be monitored with the paraformaldehyde slurry mediated α-hydroxymethylation of methyl vinyl ketone.
In a continuous flow system, residence time distribution (RTD) is introduced as an important parameter to describe the plug flow character. Narrow RTDs decrease the probability of side reactions or incomplete conversion to occur. With the addition of a pulsator to prevent solid particle sedimentation, the amplitude and frequency of pulsations emerge as new process parameters significantly impacting the RTD. Symmetrical oscillations will be applied generating vortices (eddies), leading to improved radial mixing, whilst aiming to maintain a plug flow character (minimal axial mixing). Therefore, mixing is decoupled from the net flow rate and mainly depends on the oscillation conditions. Axial dispersion previously proved to be very sensitive to oscillatory conditions at low net flow rates.38 As such, the RTD of our assembly was measured for different pulsator amplitudes and frequencies to gain insights in the mixing behaviour (Table 3, Fig. 4).
| Entry | Oscillation frequency (Hz) | Oscillation amplitudea (%) | ReOsc | Bo |
|---|---|---|---|---|
| a Pulsator amplitude is expressed in percentage of the maximum stroke volume of 0.76 mL. | ||||
| 1 | 3 | 15 | 542 | 51 |
| 2 | 3 | 30 | 1134 | 63 |
| 3 | 3 | 50 | 1874 | 59 |
| 4 | 3 | 70 | 2614 | 69 |
| 5 | 3 | 95 | 3551 | 68 |
| 6 | 1 | 15 | 181 | 54 |
| 7 | 1 | 30 | 378 | 57 |
| 8 | 1 | 50 | 625 | 51 |
| 9 | 1 | 70 | 871 | 43 |
| 10 | 1 | 95 | 1184 | 49 |
 | ||
| Fig. 4 Residence time distribution for different pulsator settings within the CØPE reactor with a pulsator frequency of 3 Hz (a) and 1 Hz (b). | ||
For all pulsator amplitudes, the Bodenstein number (Bo) exhibits consistency for every frequency, with a slightly elevated Bo number observed at a 3 Hz frequency. Although all RTD curves are nearly symmetrical, the Bo number indicates that no fully ideal plug flow behaviour is observed (Bo < 100) within this tubular system (Fig. 4).
Continuous flow optimisation
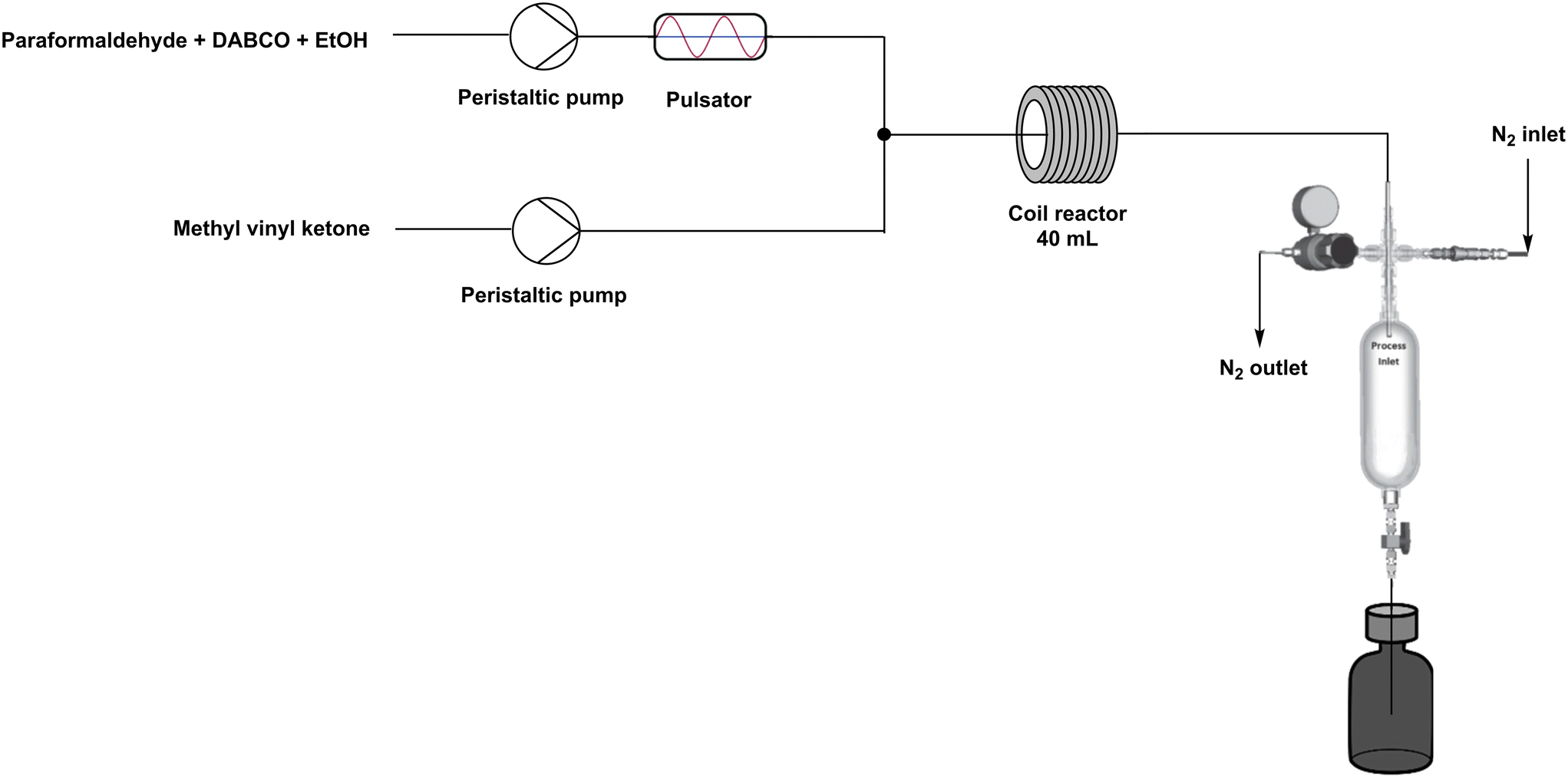
Equipped with these optimised batch conditions and the self-constructed slurry-compatible flow reactor, the transition towards a continuous flow process in the CØPE reactor was initiated. Originally, all reagents were mixed in a continuously stirred feed vessel and used as such within setup A (Fig. 5). | ||
| Fig. 5 Schematic overview of continuous flow setup A. | ||
Although the residence time distribution was measured for different pulsator settings, it seems valuable to study their effect on the conversion of methyl vinyl ketone. However, Table 4 shows similar NMR derived yields for all pulsator settings. All experiments were conducted using a large premixed feed vessel (2 L), leading to possible preliminary reaction/degradation before entering the flow reactor. Particularly, considering the literature's specified conditions of a 3.5 hour reaction time at room temperature, pre-mixing all reagents in a feed vessel could cloud the interpretation of these obtained yields. NMR analysis indeed confirmed a partial preliminary conversion (55%) after 2 hours within the feed vessel, indicating that only a fraction of the MVK remained available for the reaction within the flow reactor. To increase controllability of the process and reliability of the results, the setup was adapted to a two-stream flow process (Fig. 6). This setup modification significantly enhanced the obtained yields, achieving results similar to or slightly better than those obtained in the high temperature batch experiments. With conversions ranging from 89% to 92%, it is evident that the reactions are much cleaner, with a significant reduction in side products. It is important to highlight that even when low yields are obtained, all paraformaldehyde undergoes depolymerization and dissolution at these elevated temperatures. Based on Table 4, an oscillation frequency of 3 Hz and an amplitude of 50% were determined to be the most suitable for further experiments.
| Entry | Oscillation frequency (Hz) | Oscillation amplitudea (%) | NMR yieldb (%) setup A | NMR yieldb (%) setup B |
|---|---|---|---|---|
| a Pulsator amplitude is expressed in percentage of the maximum stroke volume of 0.76 mL. b NMR yield calculated relative to internal standard (trimethoxybenzene). c NMR yield of multiple samples vary, creating a non-representative average. | ||||
| 1 | 3 | 15 | 36 | 80 |
| 2 | 3 | 30 | 34 | 70 |
| 3 | 3 | 50 | 52 | 84 |
| 4 | 3 | 70 | 62 | 76 |
| 5 | 3 | 95 | 47 | 86c |
| 6 | 1 | 15 | 43 | 56 |
| 7 | 1 | 30 | 44 | 80 |
| 8 | 1 | 50 | 48 | 83 |
| 9 | 1 | 70 | 47 | 82c |
| 10 | 1 | 95 | 44 | 82c |
 | ||
| Fig. 6 Schematic overview of continuous flow setup B. | ||
Since full conversion was not obtained in the two-feed approach yet, the residence time and temperature were varied to push the reaction towards completion and maximize the yield (Table 5). However, prolonging the residence time at 100 °C led to a higher conversion rate but a significant drop in NMR yield, accompanied by increased formation of side products (see further).39 Similar patterns were noted when the residence time was reduced at higher temperatures (110 °C). These trends were consistently observed during the batch screening phase as well (Table 2). Reducing the paraformaldehyde equivalents to stoichiometric amounts resulted in a decrease of the NMR yield. Thus, a residence time of 8 minutes at 100 °C resulted in the most optimal yield for the α-hydroxymethylation of methyl vinyl ketone in continuous flow. While parallel trends are visible in both batch and flow screening, reactions conducted in flow tend to exhibit less side product formation and a higher yield compared to batch processes.
| Entry | Temperature (°C) | Residence time (min) | PFA (eq.) | Conversion (%) | NMR yielda (%) |
|---|---|---|---|---|---|
| a NMR yield calculated relative to internal standard (trimethoxybenzene). b Isolated yield after column chromatography. | |||||
| 1 | 100 | 8 | 1.4 | 89 | 84 (80)b |
| 2 | 100 | 10 | 1.4 | 96 | 62 |
| 3 | 100 | 12 | 1.4 | 97 | 62 |
| 4 | 110 | 5 | 1.4 | 90 | 61 |
| 5 | 110 | 4 | 1.4 | 88 | 49 |
| 6 | 100 | 8 | 1.0 | 93 | 60 |
As outlined above, both elevated temperatures and extended residence times led to a reduction in yield, albeit with an increase in conversion. Analysis via1H-NMR and GC-MS revealed the formation of three significant side products under these conditions. As anticipated, further etherification with ethanol (B1) was evident. As mentioned earlier, a solvent switch to isopropanol to suppress this side reaction did not lead to an improved yield. Additionally, B3 resulted from the introduction of a second formaldehyde unit, followed by etherification. Also the free hemiacetal was detected by NMR (B2). Lastly, the Diels–Alder type dimer of methyl vinyl ketone was observed (B4, Fig. 7). Post-treatment with acidic water failed to convert B1, B2 and B3 into the desired product.
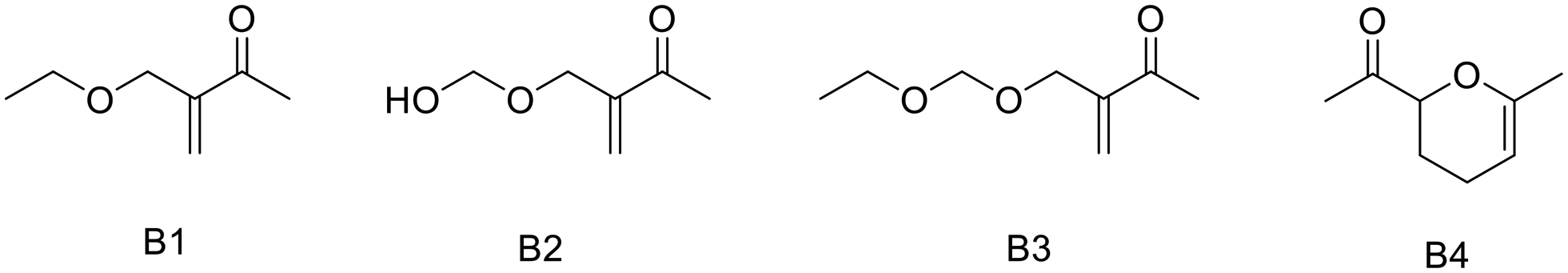 | ||
| Fig. 7 Side products formed during the α-hydroxymethylation of methyl vinyl ketone at suboptimal reaction conditions. | ||
Productivity comparison
Mantel et al. performed the hydroxymethylation of MVK only on a milligram scale (166 mg) within a batch setup. With a reaction time of 200 minutes and an isolated yield of 85%, the reaction's productivity was calculated to be 0.06 g h−1. Similarly, this calculation was applied to the developed flow process under optimal conditions (8 min, 100 °C, flow rate = 5 mL min−1, isolated yield = 80%), yielding a productivity of 81.6 g h−1 (eqn (1)). This represents a significant increase in productivity compared to previously reported literature, with >3 orders of magnitude improvement.
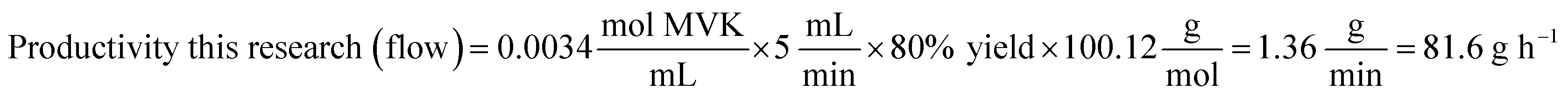 | (1) |
The space–time yield (STY) was calculated as well to provide valuable insights into reactor performance of the CØPE reactor (eqn (2)). The achieved STY significantly surpasses that of the batch procedure (STY batch = 3  ).
).
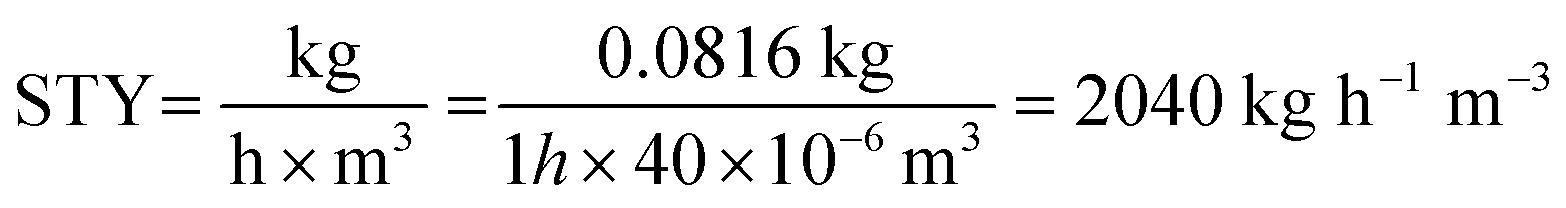 | (2) |
Conclusions
This research introduces a novel approach to reactor design, employing a solid-compatible continuous flow reactor utilizing pulsatile flow to maintain solid suspension and featuring a self-constructed blockage-resistant back pressure regulator. The efficacy of this innovative setup was evaluated through the hydroxymethylation reaction of methyl vinyl ketone using a paraformaldehyde slurry. Initially, significant improvements of the reaction conditions were achieved in a batch setting. The reaction was streamlined into a single step, eliminating the need for a separate depolymerization stage, while reducing the reaction time from hours to minutes. Subsequently, optimisation of the reaction was successfully performed in continuous flow, resulting in even cleaner reactions and a higher yield compared to the optimised batch conditions. Furthermore, hydroxymethylation reactions were performed through in situ depolymerization of a paraformaldehyde slurry, allowing direct use of the formed formaldehyde. This approach allows a continuous and safe operation of formaldehyde chemistry with a remarkable increase in productivity of 3 orders of magnitude.Author contributions
Bavo Vandekerckhove – University of Ghent (Belgium), Faculty of Bioscience Engineering, Department of Sustainable Organic Chemistry and Technology, Coupure links 653, B-9000 – designed the project, reactor assembly, conducted wet lab experiments, analysis and calculations, writing of the manuscript. Lise Van Coillie – University of Ghent (Belgium), Faculty of Bioscience Engineering, Department of Green Chemistry and Technology, Coupure links 653, B-9000 Ghent – conducted wet lab experiments, analysis and calculations. Bert Metten – Ajinomoto Bio-Pharma Services Belgium, Cooppallaan 97, 9230 Wetteren, Belgium – designed and supervised the project, technical input. Thomas S. A. Heugebaert – University of Ghent (Belgium), Faculty of Bioscience Engineering, Department of Green Chemistry and Technology, Coupure links 653, B-9000 Ghent – designed and supervised the project, contributed to the implementation of the research, to the analysis of the results, to the writing of the manuscript. Christian V. Stevens – University of Ghent (Belgium), Faculty of Bioscience Engineering, Department of Green Chemistry and Technology, Coupure links 653, B-9000 Ghent – designed and supervised the project, contributed to the implementation of the research, to the analysis of the results, to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are indebted to VLAIO – Belgium (Baekeland HBC.2021.0198) for financial support.References
- L. K. Kohn, C. H. Pavam, D. Veronese, F. Coelho, J. E. De Carvalho and W. P. Almeida, Antiproliferative effect of Baylis-Hillman adducts and a new phthalide derivative on human tumor cell lines, Eur. J. Med. Chem., 2006, 41(6), 738–744, DOI:10.1016/j.ejmech.2006.03.006.
- L. N. Solano, G. L. Nelson, C. T. Ronayne, E. A. Lueth, M. A. Foxley, S. K. Jonnalagadda, S. Gurrapu and V. R. Mereddy, Synthesis, in vitro, and in vivo evaluation of novel functionalized quaternary ammonium curcuminoids as potential anti-cancer agents, Bioorg. Med. Chem. Lett., 2015, 25(24), 5777–5780, DOI:10.1016/j.bmcl.2015.10.061.
- E. Papadaki, L. Delaude and V. Magrioti, Microwave-Assisted Synthesis of Hydroxymethyl Ketones Using Azolium-2- Carboxylate Zwitterions as Catalyst Precursors, Tetrahedron, 2017, 73(52), 7295–7300, DOI:10.1016/j.tet.2017.11.024.
- P. Hoyos, J. V. Sinisterra, F. Molinari, A. R. Alcántara and P. D. De María, Biocatalytic Strategies for the Asymmetric Synthesis of α-Hydroxy Ketones, Acc. Chem. Res., 2010, 43(2), 288–299, DOI:10.1021/ar900196n.
- S. S. Santos, R. V. Gonzaga, C. B. Scarim, J. Giarolla, M. C. Primi, C. M. Chin and E. I. Ferreira, Drug/Lead Compound Hydroxymethylation as a Simple Approach to Enhance Pharmacodynamic and Pharmacokinetic Properties, Front. Chem., 2022, 9 DOI:10.3389/fchem.2021.734983.
- L. Guillemard, N. Kaplaneris, L. Ackermann and M. J. Johansson, Late-Stage C–H Functionalization Offers New Opportunities in Drug Discovery, Nat. Rev. Chem., 2021, 522–545, DOI:10.1038/s41570-021-00300-6.
- L. Caiger, C. Sinton, T. Constantin, J. J. Douglas, N. S. Sheikh, F. Juliá and D. Leonori, Radical Hydroxymethylation of Alkyl Iodides Using Formaldehyde as a C1 Synthon, Chem. Sci., 2021, 12(31), 10448–10454, 10.1039/d1sc03083c.
- A. Duong, C. Steinmaus, C. M. McHale, C. P. Vaughan and L. Zhang, Reproductive and Developmental Toxicity of Formaldehyde: A Systematic Review, Mutat. Res., Rev. Mutat. Res., 2011, 118–138, DOI:10.1016/j.mrrev.2011.07.003.
- Formaldehyde General Description, WHO Regional Office for Europe, 2001, ch. 5.8 Search PubMed.
- R. K. Boeckmann and J. R. Miller, Direct Enantioselective Organocatalytic Hydroxymethylation of Aldehydes Catalyzed by a,a Diphenylprolinol Trimethylsilyl Ether, Org. Lett., 2009, 4544–4547, DOI:10.1055/s-0029-1218409.
- S. Nishimura and A. Shibata, Hydroxymethylation of Furfural to HMF with Aqueous Formaldehyde over Zeolite Beta Catalyst, Catalysts, 2019, 9, 314, DOI:10.3390/catal9040314.
- S. Nishimura, A. Shibata and K. Ebitani, Direct Hydroxymethylation of Furaldehydes with Aqueous Formaldehyde over a Reusable Sulfuric Functionalized Resin Catalyst, ACS Omega, 2018, 3(6), 5988–5993, DOI:10.1021/acsomega.8b00120.
- J. Cong-Bin, L. Yun-Lin, C. Zhong-Yan, Z. Yi-Yu and Z. Jian, Hydroxymethylation of α-substituted nitroacetates, Tetrahedron Lett., 2011, 6118–6121, DOI:10.1016/j.tetlet.2011.09.020.
- M. Movsisyan, E. I. P. Delbeke, J. K. E. Berton, C. Battilocchio, S. Ley and C. V. Stevens, Taming Hazardous Chemistry by Continuous Flow Technology, Chem. Soc. Rev., 2016, 4892–4928, 10.1039/C5CS00902B.
- W. Li and X. F. Wu, The Applications of (Para)Formaldehyde in Metal-Catalyzed Organic Synthesis, Adv. Synth. Catal., 2015, 3393–3418, DOI:10.1002/adsc.201500753.
- World Health Organization, IARC Working Group on the Evaluation of Carcinogenic Risks to Humans and International Agency for Research on Cancer, Formaldehyde, 2-Butoxyethanol and 1-Tert-Butoxypropan-2-Ol, International Agency for Research on Cancer, 2006 Search PubMed.
- X. L. Liu, Y. H. Liao, Z. J. Wu, L. F. Cun, X. M. Zhang and W. C. Yuan, Organocatalytic Enantioselective Hydroxymethylation of Oxindoles with Paraformaldehyde as C1 Unit, J. Org. Chem., 2010, 4872–4875, DOI:10.1021/jo100769n.
- A. E. Buba, S. Koch, H. Kunz and H. Löwe, Fluorenylmethoxycarbonyl-N-methylamino Acids Synthesized in a Flow Tube-in-Tube Reactor with a Liquid–Liquid Semipermeable Membrane, Eur. J. Org. Chem., 2013, 4509–4513, DOI:10.1002/ejoc.201300705.
- L. Yang and K. F. Jensen, Mass Transport and Reactions in the Tube-in-Tube Reactor, Org. Process Res. Dev., 2013, 17(6), 927–933, DOI:10.1021/op400085a.
- A. Simoens, T. Scattolin, T. Cauwenbergh, G. Pisanò, C. S. J. Cazin, C. V. Stevens and S. P. Nolan, Continuous Flow Synthesis of Metal–NHC Complexes, Chem. – Eur. J., 2021, 27, 5653, DOI:10.1002/chem.202100190.
- L. Huck, A. de la Hoz, A. Díaz-Ortiz and J. Alcázar, Grignard Reagents on a Tab: Direct Magnesium Insertion under Flow Conditions, Org. Lett., 2017, 19(14), 3747–3750, DOI:10.1021/acs.orglett.7b01590.
- M. P. Thompson, I. Peñafiel, S. C. Cosgrove and N. J. Turner, Biocatalysis Using Immobilized Enzymes in Continuous Flow for the Synthesis of Fine Chemicals, Org. Process Res. Dev., 2019, 23(1), 9–18, DOI:10.1021/acs.oprd.8b00305.
- C. Len, S. Bruniaux, F. Delbecq and V. S. Parmar, Palladium-Catalyzed Suzuki–Miyaura Cross-Coupling in Continuous Flow, Catalysts, 2017, 7, 146, DOI:10.3390/catal7050146.
- K. Matcha, A. Huygaerts, L. Moens, M. Peeters, D. Gala and D. Depré, Exploratory Process Development of a High-Temperature Thermal (3 + 2) Cycloaddition in Continuous Flow for the Synthesis of Seltorexant, Org. Process Res. Dev., 2023, 28(5), 1494–1503, DOI:10.1021/acs.oprd.3c00113.
- B. Vandekerckhove, B. Ruttens, B. Metten, C. V. Stevens and T. S. A. Heugebaert, Merging Inline Crystallization and Pulsed Flow Operation to enable Enantiospecific Solid State Photodecarbonylation, React. Chem. Eng., 2024 10.1039/D4RE00058G.
- M. Mantel, M. Guder and J. Pietruszka, Simple organocatalysts in multi-step reactions: An efficient one-pot Morita-Baylis-Hillman-type α-hydroxymethylation of vinyl ketones followed by the convenient, temperature-controlled one-pot etherification using alcohols, Tetrahedron, 2018, 5442–5450, DOI:10.1016/j.tet.2018.05.026.
- B. Schwartz, A. Porzelle, K. Jack, J. Faber, I. Gentle and C. Williams, Cyclic Enones as Substrates in the Morita–Baylis–Hillman Reaction: Surfactant Interactions, Scope and Scalability with an Emphasis on Formaldehyde, Adv. Synth. Catal., 2009, 1148–1154, DOI:10.1002/adsc.200800739.
- J. Gu, B.-X. Xiao, Q. Ouyang, W. Du and Y.-C. Chen, Phosphine-Catalyzed Interrupted Morita–Baylis–Hillman Reaction and Switchable Domino Reactions of α-Substituted Activated Olefins with Formaldehyde and Mechanism Elucidation, Chin. J. Chem., 2019, 155–160, DOI:10.1002/cjoc.201800466.
- Y. Furukawa and M. Ogura, A Unique Heterogeneous Nucleophilic Catalyst Comprising Methylated Nitrogen-Substituted Porous Silica Provides High Product Selectivity for the Morita–Baylis–Hillman Reaction, J. Am. Chem. Soc., 2014, 136(1), 119–121, DOI:10.1021/ja410781z.
- R. Robiette, V. K. Aggarwal and J. N. Harvey, Mechanism of the Morita−Baylis−Hillman Reaction: A Computational Investigation, J. Am. Chem. Soc., 2007, 129(50), 15513–15525, DOI:10.1021/ja0717865.
- J. Claes, A. Vancleef, M. Segers, B. Brabants, M. E. Leblebici, S. Kuhn, L. Moens and L. C. J. Thomassen, Synthesis of amines: From a microwave batch reactor to a continuous milliflow reactor with heterogeneous feed and product, Chem. Eng. Process., 2023, 183 DOI:10.1016/j.cep.2022.109252.
- Z. Dong, S. D. A. Zondag, M. Schmid, Z. Wen and T. Noël, A meso-scale ultrasonic milli-reactor enables gas–liquid-solid photocatalytic reactions in flow, Chem. Eng. J., 2022, 1385–8947, DOI:10.1016/j.cej.2021.130968.
- D. L. Browne, B. J. Deadman, R. Ashe, I. R. Baxendale and S. V. Ley, Continuous Flow Processing of Slurries: Evaluation of an Agitated Cell Reactor, Org. Process Res. Dev., 2011, 693–697, DOI:10.1021/op2000223.
- Z. Peng, G. Wang, B. Moghtaderi and E. Doroodchi, A review of microreactors based on slurry Taylor (segmented) flow, Chem. Eng. Sci., 2022, 0009–2509, DOI:10.1016/j.ces.2021.117040.
- Lewa group, accessed 23 February 2024, https://www.lewa.com/en/pumps/metering-pumps/lewa-ecoflow-diaphragm-metering-pump.
- Uniqsis accessible flow chemistry, accessed 23 February 2024, https://www.uniqsis.com/paProductsDetail.aspx?ID=HotCoil.
- B. J. Deadman, D. L. Browne, I. R. Baxendale and S. V. Ley, Back Pressure Regulation of Slurry-Forming Reactions in Continuous Flow, Chem. Eng. Technol., 2015, 259–264, DOI:10.1002/ceat.201400445.
- P. Bianchi, J. Williams and C. O. Kappe, Oscillatory flow reactors for synthetic chemistry applications, J. Flow Chem., 2020, 475–490, DOI:10.1007/s41981-020-00105-6.
- I. M. Mándity, S. B. Ötvös and F. Fülöp, Strategic Application of Residence-Time Control in Continuous-Flow Reactors, ChemistryOpen, 2015, 4, 212–223, DOI:10.1002/open.201500018.
Footnotes |
| † Electronic supplementary information (ESI) available: A detailed description of the experimental work is provided in the ESI. See DOI: https://doi.org/10.1039/d4re00220b |
| ‡ These authors contributed equally. |
| § This system was constructed as a down-scaled model of the CØPE reactor of Ajinomoto Bio-Pharma Services, Belgium. |
| This journal is © The Royal Society of Chemistry 2024 |