
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrofluidic synthesis of PLGA nanoparticles enabled by an ultrasonic microreactor†
Aniket Pradip
Udepurkar
 a,
Laura
Mampaey
a,
Christian
Clasen
b,
Victor
Sebastián Cabeza
*c and
Simon
Kuhn
*a
a,
Laura
Mampaey
a,
Christian
Clasen
b,
Victor
Sebastián Cabeza
*c and
Simon
Kuhn
*a
aKU Leuven, Department of Chemical Engineering, Process Engineering for Sustainable Systems (ProcESS), Celestijnenlaan 200F, 3001 Leuven, Belgium. E-mail: simon.kuhn@kuleuven.be
bKU Leuven, Department of Chemical Engineering, Soft Matter, Rheology and Technology (SMaRT), Celestijnenlaan 200J, 3001 Leuven, Belgium
cDepartment of Chemical Engineering and Environmental Technologies, Instituto de Nanociencia y Materiales de Aragon (INMA), University of Zaragoza, Zaragoza, 50018, Spain. E-mail: victorse@unizar.es
First published on 21st May 2024
Abstract
We present an ultrasonic microreactor for synthesising poly(lactic-co-glycolic) acid (PLGA) nanoparticles through the emulsion-solvent evaporation technique. Monodispersed PLGA nanoparticles (polydispersity index (PDI) < 0.3) in the size range of 20–300 nm are desired for biomedical applications. An ultrasonic microreactor with rough microchannels is utilised for the synthesis of PLGA nanoparticles. Through a comprehensive parametric investigation, we identify the optimal ultrasonic power, PLGA concentration, and aqueous-to-organic phase flow rate ratio, to minimise the size of the PLGA nanoparticles. By varying the operational parameters and the concentration of PLGA, the mean hydrodynamic diameter of the monodispersed PLGA nanoparticles (PDI of 0.1–0.2) can be varied within the range of 115–150 nm. Furthermore, the successful encapsulation of a hydrophobic dye, Nile Red, is demonstrated, where a dye loading (DL) of up to 0.34% is achieved, which is in agreement with the previously reported loading of Nile Red. The in vitro release study performed for the Nile Red-loaded PLGA nanoparticles (NR-PLGA) reveals a triphasic release profile of Nile Red. In summary, this work highlights the potential of the ultrasonic microreactor as a versatile platform for the synthesis of PLGA nanoparticles suitable for biomedical applications.
1. Introduction
With the growing number of hydrophobic drugs with low bioavailability entering the market, efforts are focused on increasing their bioavailability and delivering drugs to the targeted site.1–3 Poly(lactic-co-glycolic) acid (PLGA), a biodegradable synthetic polymer, is used widely for drug encapsulation and delivery applications.4,5 PLGA is US FDA and European Medicine Agency (EMA) approved for various drug delivery systems.6,7 PLGA-based drug delivery systems are favoured to protect the drug from biochemical degradation, control drug release, and possibly target cells or tissues.3,8,9 In addition, they are utilised for the encapsulation and delivery of proteins, RNA, nucleic acids, vaccines, and plasmids.3,10–12 PLGA offers the possibility to obtain a desired release profile by tuning the molecular weight of the polymer, particle size, surface functionalisation, etc.13–15Nanoparticles in the size range of 20–200 nm are considered optimal for drug delivery applications as the particles in this size range are retained longest in the bloodstream and cross biological and physiological barriers for drug delivery.16,17 Etheridge et al. recommended a cut-off value of 300 nm for the nanoparticles aimed at drug encapsulation and delivery.18 In addition, a narrow particle size distribution, defined by the polydispersity index (PDI), is desired for drug encapsulation and delivery.19,20 For biomedical applications, a PDI below 0.3 is preferred.20,21
The two common techniques employed for the generation of PLGA nanoparticles are nanoprecipitation and emulsion-solvent evaporation.22–24 Nanoprecipitation involves the rapid mixing of a water-miscible organic phase stream (e.g. acetonitrile, DMSO) containing PLGA and an aqueous stream to nucleate, grow, and synthesise PLGA nanoparticles.8,25–30 Nanoprecipitation in batch systems can result in large particle size, wide particle size distribution, and batch-to-batch variability.8,25,31–33 Microreactors can address these drawbacks with their narrow channel size (typically <500 μm), small diffusion lengths, and rapid mixing.14,27–30,34 For instance, Karnik et al. synthesised monodispersed PLGA-PEG nanoparticles (mean diameter 24–40 nm, PDI < 0.3) utilising a PDMS microchannel with a hydrodynamic flow-focusing geometry.25
Passive (e.g. mixing element) or active (e.g. ultrasound) mixing techniques employed in the microreactors can be exploited to reduce the mixing time, control the nanoparticle size, and increase the throughput.3,35,36 Ultrasonic microreactors are a common microfluidic platform employed to synthesise PLGA nanoparticles by the nanoprecipitation technique.37,38Table 1 lists the previous studies that employed ultrasonic microreactors for PLGA nanoparticle synthesis. The fast mixing achieved due to cavitation microstreaming or acoustic streaming is beneficial for synthesising monodisperse PLGA nanoparticles of a desired size.12,37,38
| Synthesis technique | Frequency [kHz] | Voltage/powera | MDb [nm] | PDIc [—] | Ref. |
|---|---|---|---|---|---|
| a Applied voltage in Vpp (peak to peak voltage) or applied ultrasound power in W. b Mean diameter. c Polydispersity index. | |||||
| Nanoprecipitation | 205 | 20 Vpp | 65.0 | 0.05 | 38 |
| Nanoprecipitation | 608 | 3.33 W | 52.2 | 0.44 | 39 |
| Nanoprecipitation | 4 | 30 Vpp | 64.7 | 0.13 | 40 |
| Nanoprecipitation | 4.9 | 56 Vpp | 64.5 | 0.06 | 41 |
| Nanoprecipitation | 74.2 | 40 Vpp | 51.9 | 0.07 | 42 |
| Nanoprecipitation | 4 | 38 Vpp | 101.0 | 0.17 | 37 |
| Nanoprecipitation | 80 | 100 W | 157.2 | 0.19 | 43 |
| Nanoprecipitation | 20 | 30 W | 45.0 | 0.06 | 12 |
| Emulsion-solvent evaporation | 24 | 32 W | 490 | — | 44 |
A major drawback of nanoprecipitation lies in the utilisation of class 2 solvents (e.g. acetonitrile, DMSO, and THF) which are undesirable.45 The EMA recommends limiting class 2 solvents in the synthesis of drug products whenever possible.45 Another drawback is the separation of a water-miscible solvent from the nanoparticle suspension, which could require complex post-processing steps (e.g. dialysis) to meet the permissible limit (e.g. 410 ppm for acetonitrile).45,46 Emulsion-solvent evaporation, a technique involving a less toxic solvent (class 3, e.g. ethyl acetate), is a viable alternative for the microfluidic synthesis of PLGA nanoparticles.
Emulsion-solvent evaporation is a two-step process for the synthesis of PLGA nanoparticles.47–49 In the first step, an oil-in-water (O/W) emulsion is generated with the organic phase droplets containing PLGA dispersed in a continuous aqueous phase. In the second step, the organic phase (solvent) is evaporated to obtain an aqueous suspension of PLGA nanoparticles. Batch ultrasonic emulsification is a common technique utilised for the generation of O/W emulsion for PLGA nanoparticle synthesis.28,32,50 The batch PLGA nanoparticle synthesis faces issues with batch-to-batch variability, wide particle size distribution, and the possibility of contamination due to the erosion of the ultrasound horn.44,51
Microreactors have enabled the generation of O/W emulsions with precise control over the droplet size, low polydispersity, and excellent reproducibility.52–54 For instance, De Solorzano et al. utilised an interdigital micromixer for the synthesis of PLGA nanoparticles by emulsion-solvent evaporation technique.51 However, the smallest nanoparticle diameter they could achieve with the setup was 220 ± 54 nm. Freitas et al. utilised a contamination-free glass ultrasonic microreactor coupled with a micromixer for the synthesis of PLGA nanoparticles.44 They synthesised PLGA nanoparticles with a mean diameter of 490 nm, significantly larger than the size desired for intravenous drug delivery.
Staff et al. highlighted that the O/W emulsion droplet size is crucial in the preparation of nanoparticles in the emulsion-solvent evaporation technique.55 They noted that the droplet re-coalescence during the solvent evaporation stage did not significantly contribute to the final nanoparticle size.55 The main challenge for the microfluidic synthesis of PLGA nanoparticles is the generation of monodispersed O/W emulsions of desired size. We aim to address this challenge by employing an ultrasonic microreactor to synthesise monodispersed PLGA nanoparticles.
Our previous work highlighted the effective generation of monomodal O/W emulsion utilising the water-jet cut (WJR) ultrasonic microreactor.56 Taking advantage of the monomodal O/W emulsion generation, this study demonstrates the synthesis of monodispersed (PDI < 0.3) PLGA nanoparticles of the desired size (diameter < 300 nm) utilising the WJR ultrasonic microreactor.
Ethyl acetate, a class 3 solvent, was employed as the organic phase to synthesise PLGA nanoparticles by the emulsion-solvent evaporation technique. The objective of the study is three-fold. Firstly, a parametric investigation of the operating parameters (ultrasound power, frequency, organic-to-aqueous phase flow rate ratio, outlet temperature) and PLGA concentration is performed to identify the optimum operating parameters for the synthesis of monodispersed PLGA nanoparticles. Secondly, as a proof of concept, the encapsulation of Nile Red, a hydrophobic dye, is demonstrated at the optimum operating parameters. Thirdly, the Nile Red release from the Nile Red-loaded PLGA nanoparticles (NR-PLGA) for 10 days is quantified. The versatility of the ultrasonic microreactor in synthesising monodispersed PLGA nanoparticles in various size ranges is demonstrated.
2. Materials and methods
2.1. Materials
PLGA (Resomer® RG 504 H, acid terminated, L![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :G 50:50, MW 38000–54000), surfactant poloxamer 407 (purified, non-ionic), ethyl acetate (≥99.5%), and Nile Red (technical grade) were purchased from Sigma-Aldrich. Phosphate buffer (0.01 M, pH 7.4) was prepared by adding phosphate buffered saline (powder, pH 7.4, Sigma-Aldrich) to 1 L Milli-Q water. Acetone (Uvasol® for spectroscopy) was purchased from VWR, Belgium. Milli-Q water was utilised for all the experiments.
:G 50:50, MW 38000–54000), surfactant poloxamer 407 (purified, non-ionic), ethyl acetate (≥99.5%), and Nile Red (technical grade) were purchased from Sigma-Aldrich. Phosphate buffer (0.01 M, pH 7.4) was prepared by adding phosphate buffered saline (powder, pH 7.4, Sigma-Aldrich) to 1 L Milli-Q water. Acetone (Uvasol® for spectroscopy) was purchased from VWR, Belgium. Milli-Q water was utilised for all the experiments.
2.2. Experimental setup
In this study, a water-jet cut (WJR) ultrasonic microreactor (Little Things Factory GmbH, Germany) coupled to a piezoelectric plate transducer was employed for the synthesis of the PLGA nanoparticles by emulsion-solvent evaporation technique (see Fig. 1(b)). Further details on the ultrasonic microreactor can be found in our previous work.56 A piezoelectric plate transducer (Pz26, Ferroperm, 80 × 40 × 1.67 mm3) was bonded to the microreactor using epoxy glue (EPO-TEK® 301, Epotek) to drive the reactor at resonance frequencies of 48 kHz and 142 kHz. The reactor was bonded with a piezoelectric plate transducer (Pz26, Ferroperm, 80 × 40 × 4 mm3) to drive the reactor at frequencies of 310 kHz and 540 kHz.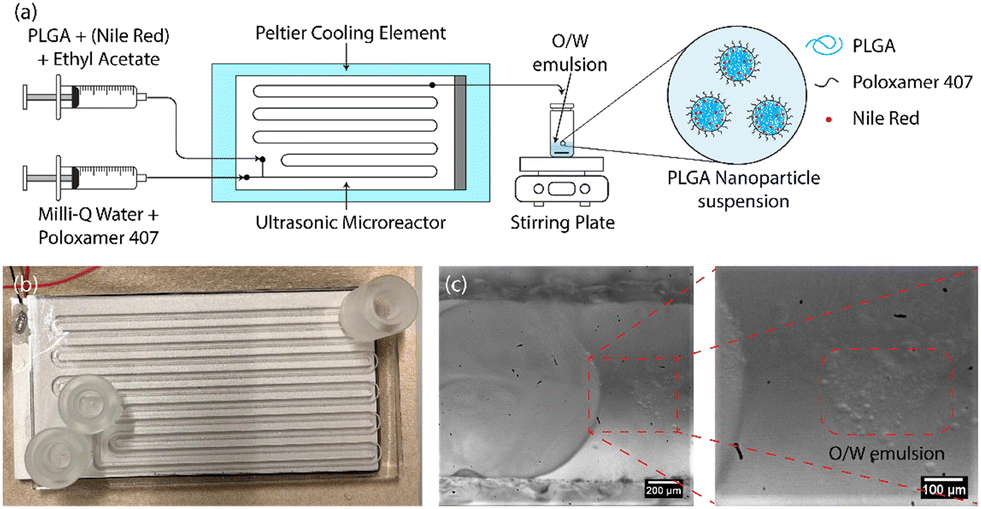 | ||
| Fig. 1 (a) Schematic representation of the procedure for the synthesis of the (Nile Red-loaded) PLGA nanoparticles, (b) the ultrasonic microreactor, (c) the O/W emulsion generated in the microchannel on the actuation of ultrasound. | ||
The ultrasonic microreactor was placed on a Peltier cooling element (RS components) connected to a DC power supply (Velleman) to regulate the temperature. The temperature was measured at the outlet of the reactor and was regulated to 30 °C during the emulsification unless otherwise stated. Syringes filled with the aqueous and organic phases were mounted onto syringe pumps (Fusion 200, KR Analytical) to deliver the phases to the microreactor. A glass syringe (FORTUNA optima, 5 mL, luer lock tip) was employed for the delivery of the organic phase, while a plastic syringe (Terumo, 20 mL, 3-part syringe) was employed for the aqueous phase. The syringes were connected to the inlet of the microreactor with PFA tubing (inner diameter 0.5 mm, outer diameter 1/16′′, IDEX).
The resonance frequency of the ultrasonic microreactor was determined by measuring the admittance using an impedance analyzer (16777 k, SinePhase). A signal generator (33500B, Keysight) coupled with an amplifier (RF 1040 L, 400 W, E&I) was connected to the piezoelectric plate transducer to actuate the ultrasound at the desired frequency and power. The reactor was operated at the resonance frequency of 48 kHz unless otherwise stated.
2.3. PLGA nanoparticle synthesis
The schematic of the PLGA nanoparticle synthesis procedure is shown in Fig. 1(a). For the synthesis of blank PLGA nanoparticles, the organic phase of PLGA in ethyl acetate (6–24 mg mL−1) and the aqueous phase of the surfactant poloxamer 407 in Milli-Q water (5 mg mL−1) were introduced to the ultrasonic microreactor. The total flow rate and the residence time were set to 250 μL min−1 and 4 min respectively for all the experiments. The organic and aqueous phase flow rates were 50 μL min−1 and 200 μL min−1 respectively unless otherwise stated. A segmented flow was established in the microchannel and the ultrasound was actuated at a desired power. On the actuation of ultrasound, the organic phase underwent emulsification leading to the generation of an oil-in-water (O/W) emulsion (see Fig. 1(c)). The emulsification mechanism for the generation of O/W emulsion is described in detail in a previous work.56 The sample was collected after 3 reactor residence times in a vial for 10 min (2.5 mL) and stirred gently (400 rpm) overnight on a stirring plate (MIXdrive 15, 2MAG) to evaporate the organic solvent and obtain a PLGA nanoparticle suspension. For the synthesis of NR-PLGA, Nile Red was also dissolved in the organic phase (1 mg mL−1) and the aforementioned procedure was followed.2.4. PLGA nanoparticle characterisation
The particle size of the PLGA nanoparticles was measured by 3D cross-correlation dynamic light scattering (DLS) (LS instruments). The DLS setup is comprised of a 660 nm cobalt laser, two Glan-Thompson polarisers, a beam splitter, a decalin-filled sample chamber, two photon-counting modules mounted on a rotatable goniometer arm, and a correlator box. For the particle size measurement, 30–60 μL nanoparticle suspension was pipetted in a cylindrical cuvette filled with 3 mL Milli-Q water. For the NR-PLGA nanoparticles, the sample was filtered with a syringe filter (Whatman, 0.45 μm) to remove the excess precipitated Nile Red crystals. The particle size was measured at a scattering angle of 120° for 60 seconds and the measurement repeated 5 times for each sample. The mean hydrodynamic diameter (MHD) and the polydispersity index (PDI) of the particle size were determined by the CUMULANT method. The average particle size of the three independent experiments for each condition is reported.The particle morphology was characterised by scanning electron microscopy (SEM) (InspectF50A, FEI, Eindhoven, the Netherlands) and transmission electron microscopy (TEM) (FEI T20 Company, Hillsboro, OR, USA). The PLGA nanoparticles were washed and centrifuged before the SEM and TEM analysis to remove excess surfactant. PLGA nanoparticle suspension (250 μL) was pipetted in a 1.5 mL vial (Eppendorf) filled with Milli-Q water (750 μL). The PLGA nanoparticles were washed and centrifuged (Eppendorf 5804) at 10000g for 10 min to settle the nanoparticles. The supernatant (750 μL) was replaced with fresh Milli-Q water (750 μL) and the washing and centrifugation step was repeated 5 times to remove the excess surfactant and concentrate the particles. At the end of the washing and centrifugation cycle, the pellet was sonicated in an ultrasonic bath to resuspend the PLGA nanoparticles and break agglomerates. Similar to the DLS measurements, the NR-PLGA nanoparticles were filtered before the first washing and centrifugation cycle to remove the Nile Red crystals.
For the SEM analysis, a drop of the washed PLGA nanoparticle suspension was placed on a silicon substrate and air-dried. The dried sample was coated with Au–Pd coating before the SEM analysis. An accelerating voltage of 10–15 kV was employed for the SEM analysis. For the TEM analysis, approximately 2.5 μL of PLGA nanoparticle suspension was pipetted onto a transmission electron microscopy copper grid with a continuous carbon film. The sample was air-dried before the TEM analysis.
2.5. Encapsulation efficiency and dye loading
The encapsulation efficiency (EE) and the dye loading (DL) are defined by eqn (1) and (2). The EE gives the fraction of the dye in the starting solution encapsulated in the nanoparticles. The DL is defined as the fraction of dye per unit mass of dye-loaded nanoparticles. | (1) |
 | (2) |
The Nile Red loading in the PLGA nanoparticles was determined by measuring the Nile Red encapsulated in the PLGA nanoparticles. The NR-PLGA nanoparticles were first filtered with a syringe filter of 0.45 μm to remove the excess precipitated Nile Red crystals. Subsequently, the nanoparticles were washed and centrifuged employing the aforementioned protocol to remove the excess surfactant. A known amount of dried pellet was added to 3 mL acetone to dissolve nanoparticles and release the encapsulated Nile Red. The absorbance of the solution was measured using UV-vis spectroscopy (Perkin-Elmer Lambda 365) and the concentration of Nile Red was determined from the calibration of the Nile Red concentration in acetone (see Fig. S8†). The experiments were performed in triplicate.
2.6. In vitro release study
The in vitro release of encapsulated Nile Red from the NR-PLGA nanoparticles was studied by the dialysis-bag diffusion method. The NR-PLGA nanoparticles were washed and freeze-dried. Phosphate buffer (PBS, 0.01 M, pH 7.4) was prepared as a release medium. A known quantity of the freeze-dried NR-PLGA nanoparticles was dispersed in PBS (5 mL) in a dialysis bag (molecular weight cut-off ∼12400 Da) and the bag was placed in a closed glass bottle filled with PBS (45 mL). The glass bottle was placed on a magnetic stirrer and the temperature was maintained at 37 ± 1 °C. The contents were gently stirred at 100 rpm during the experiment. A sample (1 mL) was drawn at the determined time steps and replaced with fresh PBS (1 mL). The sample (1 mL) was diluted with acetone (2 mL) and the Nile Red concentration in the sample was measured using fluorescence spectrometry (Edinburgh FLS980). A calibration curve for Nile Red in PBS–acetone (1:2 volume ratio) was determined (see Fig. S10†). The release experiments were performed in triplicate and the average is reported.
3. Results and discussion
On the actuation of ultrasound, the cavitation bubbles generated in the crevices of the rough microchannel contributed to the emulsification of the organic phase in the continuous aqueous phase.56 The residence time of 4 min was sufficient to emulsify the organic phase completely.3.1. Blank PLGA nanoparticles
Table 2 shows the mean hydrodynamic diameter (MHD) and polydispersity index (PDI) of the blank PLGA nanoparticles synthesised utilising the ultrasonic microreactor.| Frequency [kHz] | Power [W] | Wa [μL min−1] | Ob [μL min−1] | P [mg mL−1] | T [°C] | MHDe [nm] | PDIf [—] |
|---|---|---|---|---|---|---|---|
| a Aqueous phase flow rate. b Organic phase flow rate. c PLGA concentration in ethyl acetate. d Outlet temperature. e Mean hydrodynamic diameter. f Polydispersity index. | |||||||
| 48 | 5 | 200 | 50 | 12 | 30 | 150.63 ± 0.28 | 0.11 ± 0.05 |
| 48 | 10 | 200 | 50 | 12 | 30 | 117.63 ± 2.21 | 0.11 ± 0.07 |
| 48 | 15 | 200 | 50 | 12 | 30 | 134.81 ± 0.91 | 0.19 ± 0.04 |
| 48 | 20 | 200 | 50 | 12 | 30 | 126.17 ± 1.99 | 0.18 ± 0.08 |
| 142 | 10 | 200 | 50 | 12 | 30 | 120.93 ± 1.16 | 0.12 ± 0.06 |
| 310 | 10 | 200 | 50 | 12 | 30 | 133.46 ± 1.28 | 0.15 ± 0.02 |
| 540 | 10 | 200 | 50 | 12 | 30 | 116.29 ± 0.91 | 0.11 ± 0.07 |
| 48 | 10 | 225 | 25 | 12 | 30 | 113.26 ± 1.86 | 0.16 ± 0.07 |
| 48 | 10 | 237.5 | 12.5 | 12 | 30 | 231.49 ± 1.03 | 0.23 ± 0.03 |
| 48 | 10 | 200 | 50 | 6 | 30 | 131.43 ± 3.27 | 0.28 ± 0.05 |
| 48 | 10 | 200 | 50 | 9 | 30 | 123.59 ± 1.81 | 0.18 ± 0.06 |
| 48 | 10 | 200 | 50 | 24 | 30 | 153.86 ± 3.51 | 0.17 ± 0.04 |
| 48 | 10 | 200 | 50 | 12 | 20 | 127.38 ± 2.93 | 0.18 ± 0.02 |
| 48 | 10 | 200 | 50 | 12 | 25 | 129.82 ± 2.08 | 0.12 ± 0.06 |
First, the influence of ultrasonic power on the synthesis of PLGA nanoparticles was investigated for an ultrasound frequency of 48 kHz. The ultrasound power was varied between 5–20 W for the generation of the O/W emulsion. The power of 5 W resulted in PLGA nanoparticles with MHD and PDI of 150.63 nm and 0.11 respectively. Increasing the power to 10 W led to a decrease in the MHD to 117.63 nm with a PDI of 0.11. Further increasing the power to 15 W and 20 W resulted, however, in PLGA nanoparticles with larger MHD and PDI (see Table 2). This is contrary to the previous reports of a decrease in the O/W emulsion droplet size with an increase in ultrasonic power for an ultrasonic microreactor.57–59 It is important to note that the PLGA nanoparticles synthesised at a power of 5–20 W have a PDI below 0.3, i.e. they are monodispersed, which is desired for biomedical applications. The SEM analysis of the blank PLGA nanoparticles revealed a spherical morphology of the particles with a smooth surface (see Fig. 2). In addition, as seen in Fig. 2, it is evident that sonication at a higher power of 15 W and 20 W resulted in wider particle size distribution compared to PLGA nanoparticles synthesised at 5 W and 10 W.
 | ||
| Fig. 2 Representative SEM images of the blank PLGA nanoparticles synthesised for ultrasonic powers of (a) 5 W, (b) 10 W, (c) 15 W, and (d) 20 W. The ultrasonic microreactor was operated at a frequency of 48 kHz. The PLGA concentration was 12 mg mL−1. | ||
Previous reports have suggested that above a certain threshold of ultrasonic power, a further increase in ultrasonic power could increase the probability of droplet collision and eventual coalescence.59–62 In addition, the increase in the number and size of the cavitation bubbles at higher power could dampen the ultrasound, thereby resulting in larger emulsion droplets.63 The combination of these factors, namely, droplet re-coalescence and ultrasound damping at higher power, could lead to a broader droplet size distribution resulting in a broader PLGA nanoparticle size distribution for higher ultrasound power. The results point to optimal ultrasound power as a key parameter in PLGA nanoparticle synthesis.
In our previous study, we demonstrated a decrease in the O/W emulsion droplet size with an increase in the frequency.56 PLGA nanoparticle synthesis at higher frequencies could be beneficial for synthesising smaller nanoparticles. The ultrasonic microreactor coupled with the piezoelectric plate of thickness 1.67 mm was operated at the second resonance frequency of 142 kHz and the microreactor coupled with a piezoelectric plate of thickness 4 mm was operated at resonance frequencies of 310 kHz and 540 kHz for PLGA nanoparticle synthesis.
The frequency of 540 kHz resulted in the synthesis of the smallest nanoparticles (see Table 2). However, the SEM analysis of the nanoparticles revealed the presence of a small fraction of large particles (diameter > 300 nm) (see Fig. S2†). The presence of the large particles could be explained by inefficient emulsification of the organic phase at higher frequencies, which was observed for the emulsification of a high dispersed phase volume fraction at a higher frequency (525 kHz) in our previous study.56 The large nanoparticles (diameter > 300 nm) are not desirable as they are not suitable for intravenous drug delivery.51 Further investigation to avoid the large nanoparticles at higher frequencies could be beneficial for decreasing the nanoparticle size.
Previous studies have reported a decrease in the emulsion droplet size on decreasing the organic phase volume fraction.64,65 Hence, the influence of organic phase volume fraction of 5–20% on PLGA nanoparticle size was investigated. As expected, decreasing the organic phase volume fraction from 20% to 10% resulted in a decrease in MHD. However, a further decrease in the organic phase volume fraction to 5% resulted in significantly larger PLGA nanoparticles (see Table 2 and Fig. S3†). The solubility of ethyl acetate in water at 30 °C is 7.7 g per 100 g water, which amounts to a volume fraction of 7.8%.66 Amanatchi et al. performed ethyl acetate extraction on a microfluidic chip and did not report nanoprecipitation or nanoparticle formation.67 Thus, for a higher volume fraction of ethyl acetate, only the organic fraction migrates to the aqueous phase. However, at a 5% volume fraction, ethyl acetate is completely miscible with the aqueous phase. Cavitation microstreaming likely resulted in the mixing of the two phases and nanoprecipitation of the PLGA as opposed to the generation of an O/W emulsion. Nanoprecipitation and inefficient mixing in the microchannel could have resulted in the synthesis of large and polydisperse nanoparticles at a 5% volume fraction.
Next, the PLGA concentration in ethyl acetate varied between 6–24 mg mL−1. A higher PLGA concentration resulted in a larger viscosity of the organic phase (see Table S1†). The smallest nanoparticle size was obtained for the PLGA concentration of 12 mg mL−1 (see Table 2). Surprisingly, the lower viscosity at the concentration of 6 mg mL−1 and 9 mg mL−1 resulted in larger PLGA nanoparticles (see Table 2), which is contrary to the previous studies suggesting a decrease in droplet size at higher viscosity.56,59,68 Kamp et al. outlined that lowering the droplet viscosity increased the probability of droplet coalescence.69 The increase in the emulsion droplets' re-coalescence could have resulted in the larger droplet size at lower PLGA concentrations.69 The interplay between droplet generation due to emulsification and their re-coalescence could have played a crucial role in determining the final droplet size, and thus the final particle size, at a lower viscosity of the organic phase. For the range of viscosity investigated in this work, an optimum exists for the PLGA concentration of 12 mg mL−1, resulting in the smallest PLGA nanoparticles. Additionally, lowering the reactor temperature led to larger PLGA nanoparticles.
Moreover, it is crucial to analyse the degradation of PLGA due to the cavitation activity in the ultrasonic microreactor. The collapse of transient cavitation bubbles in the close vicinity of a polymer chain can cleave the polymer from the middle.70–72 The cleavage of the polymer would result in a smaller polymer chain length (and a lower molecular weight). It is a well-reported fact that the PLGA molecular weight is critical for the drug release profile, with a lower PLGA molecular weight resulting in a faster release of the drug molecules.13,73 The PLGA degradation during the synthesis of the PLGA nanoparticles could potentially influence the desired release profile due to a shorter polymer chain length (and a lower molecular weight).
The PLGA degradation was evaluated with gel permeation chromatography (GPC). A solution of PLGA in ethyl acetate (12 mg mL−1) was sonicated in the ultrasonic microreactor at 48 kHz and 10 W for 20 min. The GPC analysis was conducted for the sonicated sample and compared to the non-sonicated sample. From Fig. S6,† which shows the molecular weight distribution of PLGA, it is evident that the PLGA molecular weight distribution did not undergo a significant change (change in MW ∼ 1.66%).
The parametric investigation of the PLGA nanoparticle synthesis revealed that the ultrasonic power and the organic phase viscosity are the main parameters influencing the PLGA nanoparticle size. The re-coalescence of the emulsion droplets during ultrasonic emulsification, which is influenced either by lowering the viscosity of the organic phase or increasing the power input played a crucial role in determining the PLGA nanoparticle size. The ultrasonic microreactor employed in this work successfully synthesised spherical PLGA nanoparticles with an MHD of 115–150 nm and PDI of 0.1–0.2. The results point to the versatility of the ultrasonic microreactor in the size-tuneable synthesis of PLGA nanoparticles with good reproducibility targeting various biomedical applications (diameter < 300 nm, PDI < 0.3). In addition, it was seen that the sonication does not result in any significant degradation of the polymer.
3.2. Nile Red-loaded PLGA nanoparticles
Nile Red is a hydrophobic fluorescent dye with poor solubility in water (solubility < 1 μg mL−1).74 It is utilised to track the PLGA nanoparticles in vivo and their cellular uptake.74,75 The hydrophobic nature of Nile Red coupled with its utility in in vivo studies make it a molecule of interest for encapsulation. The NR-PLGA nanoparticles were synthesised for ultrasound powers of 5 W, 10 W, and 15 W. After solvent evaporation, needle-shaped Nile Red crystals were seen in the aqueous phase. Nile Red molecules, not encapsulated in the PLGA nanoparticles during solvent evaporation, precipitated to form long needle-shaped crystals (see Fig. S9†). The crystals were removed from the NR-PLGA suspension by filtering the suspension with a 0.45 μm syringe filter.NR-PLGA nanoparticles synthesised at 5 W and 10 W were larger than the blank PLGA nanoparticles (see Table 3). The slight increase in the particle size can be attributed to Nile Red encapsulated in the nanoparticles. However, the NR-PLGA nanoparticles were smaller than the blank PLGA nanoparticles for the power of 15 W. The possible explanation could lie with the reduction or absence of droplet re-coalescence due to an increase in the organic phase viscosity on the addition of Nile Red. Overall, no significant change in the NR-PLGA nanoparticle size was observed compared to the blank PLGA nanoparticles while achieving a desirable PDI (PDI < 0.3). The NR-PLGA nanoparticles were spherical with a smooth surface, similar to the blank PLGA nanoparticles (see Fig. 3).
| PLGA nanoparticles | Power [W] | MHDa [nm] | PDIb [−] | EEc [%] | DLd [%] |
|---|---|---|---|---|---|
| a Mean hydrodynamic diameter. b Polydispersity index. c Encapsulation efficiency. d Dye loading. | |||||
| Blank PLGA | 5 | 150.63 ± 0.28 | 0.11 ± 0.05 | — | — |
| Blank PLGA | 10 | 117.63 ± 2.21 | 0.11 ± 0.07 | — | — |
| Blank PLGA | 15 | 134.81 ± 0.91 | 0.19 ± 0.04 | — | — |
| NR-PLGA | 5 | 168.91 ± 2.22 | 0.12 ± 0.01 | 2.27 ± 0.51 | 0.19 ± 0.04 |
| NR-PLGA | 10 | 119.71 ± 8.81 | 0.13 ± 0.03 | 4.08 ± 1.87 | 0.34 ± 0.16 |
| NR-PLGA | 15 | 116.88 ± 3.63 | 0.12 ± 0.04 | 4.13 ± 1.67 | 0.34 ± 0.13 |
 | ||
| Fig. 3 Representative SEM image of (a) blank PLGA nanoparticles and (b) Nile Red-loaded PLGA nanoparticles synthesised at an ultrasonic frequency and power of 48 kHz and 10 W respectively. | ||
Next, the encapsulation efficiency (EE) and dye loading (DL) of the NR-PLGA nanoparticles were analysed. The DL and EE increased with an increase in the ultrasonic power applied for the synthesis of the NR-PLGA nanoparticles. The highest DL and EE of 0.34% and 4.13% were achieved at a power of 15 W. The increase in the EE and DL can be attributed to the NR-PLGA nanoparticle size.
From the TEM images of the NR-PLGA nanoparticles (see Fig. 4), it is evident that the Nile Red molecules, indicated by the dark ring (high atomic density) around the nanoparticles, were primarily encapsulated closer to the nanoparticle surface.76 In addition, a major fraction of the nanoparticles has successfully encapsulated Nile Red (see Fig. 4(c)). The poor Nile Red–PLGA interaction could have resulted in the dye molecules diffusing slowly through the polymer matrix towards the outer edges of the droplets to migrate away from the polymeric chains during solvent evaporation. This would explain a major fraction of Nile Red present close to the particle surface and the low encapsulation efficiency and dye loading for NR-PLGA nanoparticles. Previous studies also report low dye loading (DL < 1%) for Nile Red in nanoparticles.10,77 Li et al. achieved the highest DL of 0.42% in PLGA nanoparticles with a mean diameter of 205 nm.77 The DL achieved in this is comparable to the previous reports for comparatively smaller NR-PLGA nanoparticles (MHD ∼ 120 nm).
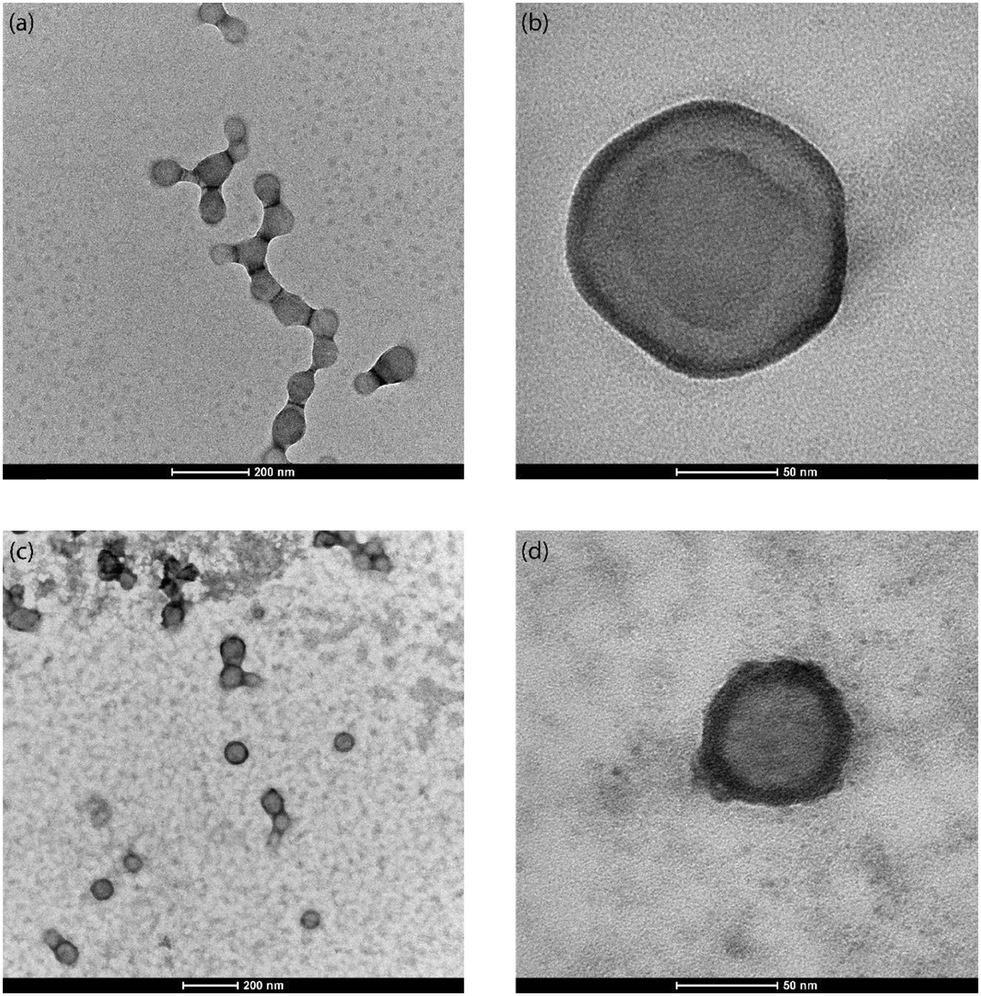 | ||
| Fig. 4 Representative TEM image of (a and b) blank PLGA nanoparticles and (c and d) Nile Red-loaded PLGA nanoparticles synthesised at the ultrasonic frequency and power of 48 kHz and 10 W respectively. | ||
3.3. In vitro release
The in vitro release profile of the NR-PLGA nanoparticles in phosphate buffer (PBS, pH 7.4) is shown in Fig. 5. A fraction of the dye was released by the initial burst during the initial hours of the release experiment (48 h) (see Fig. 5(a)). The initial burst, characterised by the rapid release of Nile Red, occurred due to the release of dye molecules encapsulated close to the nanoparticle surface. The highest release rate in the initial hours was observed for NR-PLGA nanoparticles synthesised at 15 W, where the release of 17.5% was achieved at 6 h. The release slowed down significantly after 6 h. The release rate was lowest for NR-PLGA nanoparticles synthesised at 5 W (21% in 48 h). The particle size (and in turn the surface area) is detrimental to the release rate of the drug or dye. Smaller particles exhibit a higher release rate during the initial burst phase of the drug/dye release.13,73 This is also evident for the NR-PLGA particles.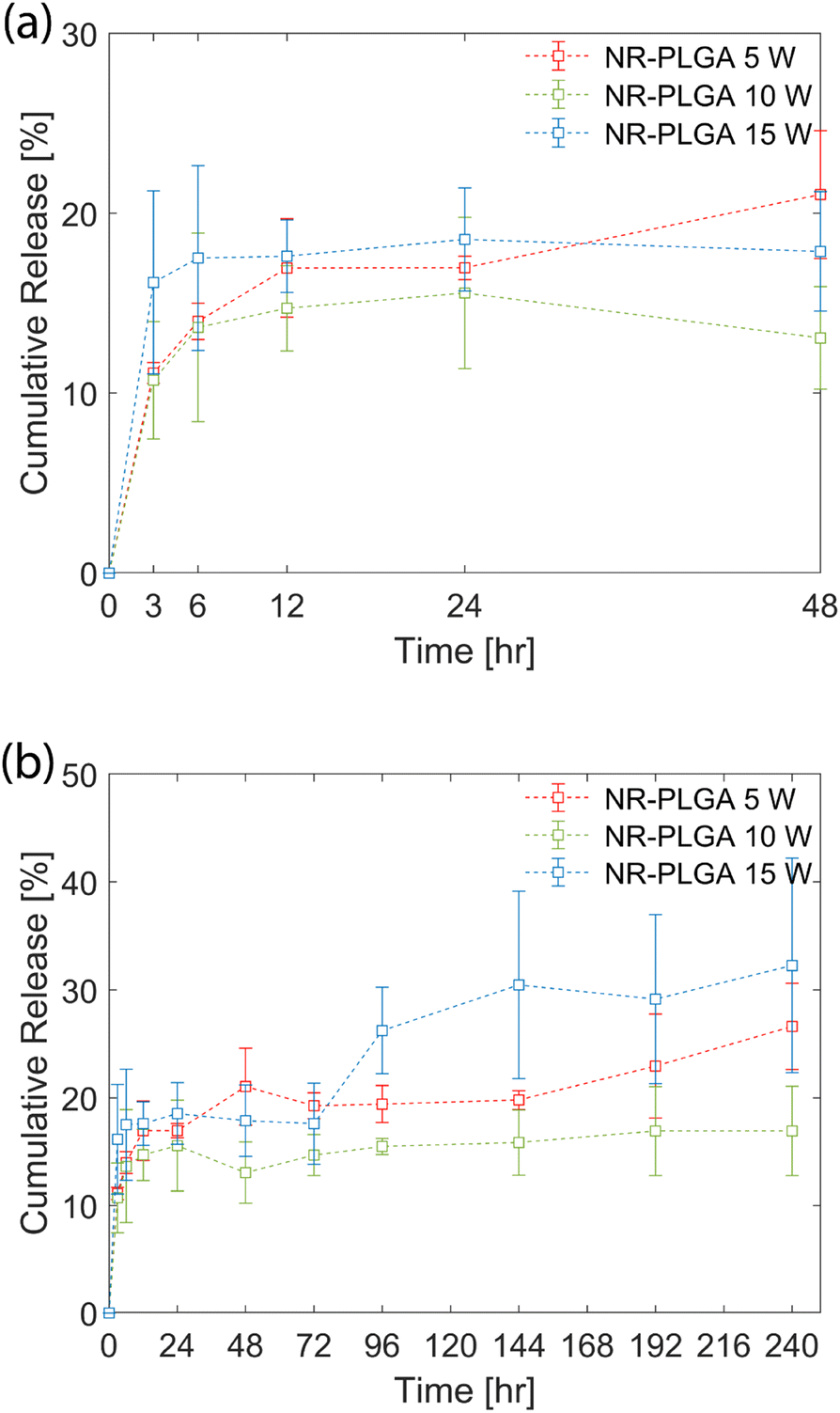 | ||
| Fig. 5 In vitro release profile of the NR-PLGA nanoparticles synthesised at ultrasonic powers of 5 W, 10 W, and 15 W. (a) The release profile for the first 48 hours and (b) the release profile for 240 hours. | ||
After the first 48 h, the release rate dropped significantly for NR-PLGA nanoparticles synthesised at 5 W and 10 W until 144 h. The release rate increased again after 144 h for both cases. For the NR-PLGA nanoparticles synthesised at 15 W, the release rate increased after 72 h. This release profile is known as type III or triphasic release profile.13,73 The type III or triphasic release profile is characterised by an initial burst in the first stage, a slow release in the second stage, and another release in the next stage. The second release phase occurs mainly due to the slow diffusion of the drug/dye through the polymer matrix and pores formed due to the hydrolysis of PLGA. A similar triphasic release profile of Nile Red from nanoparticles was reported by Delmas et al. and Vij et al.10,78
The lower MHD and PDI could be a factor for the higher initial release rate and the early onset of secondary release in NR-PLGA nanoparticles synthesised at 15 W compared to 5 W and 10 W. However, factors such as the Nile Red distribution in the nanoparticles, particle degradation or erosion rate, pore formation, etc. could also play a role in determining the release rate. The influence of these factors could offer a possible explanation for the observed release kinetics but is not explored in this work.
4. Conclusions
The WJR ultrasonic microreactor successfully synthesised PLGA nanoparticles within the size range desirable for biomedical applications (diameter < 300 nm, PDI < 0.3). The synthesis protocol did not require extra steps for the generation of a crude emulsion. The MHD of the monodispersed PLGA nanoparticles (PDI < 0.3) could be tuned between 115–150 nm by varying the operating parameters. The parametric investigation revealed an optimum power, aqueous-to-organic phase flow rate ratio, and PLGA concentration to minimise the PLGA nanoparticle size. In addition, no significant degradation of PLGA due to sonication in the ultrasonic microreactor.The successful encapsulation of Nile Red was demonstrated, achieving an encapsulation efficiency and a dye loading of 4% and 0.34% respectively. The dye loading achieved in this study is in line with the previously reported loading of 0.01–0.4% for smaller PLGA nanoparticles.10,77 The TEM images reveal that a major fraction of Nile Red was encapsulated close to the nanoparticle surface. Nile Red release from the NR-PLGA nanoparticles was triphasic, which involved an initial burst in the first 48 h and a second burst after 72–144 h. The initial burst could have resulted from the dye encapsulated close to the surface. Further erosion/degradation of the nanoparticles could have contributed to the second release.
In conclusion, this work demonstrates that the emulsion-solvent evaporation technique utilising the ultrasonic microreactor is a viable alternative to microfluidic nanoprecipitation for PLGA nanoparticle synthesis and eliminates the need for class 2 undesirable solvents and extra steps for their removal.
Author contributions
Aniket Pradip Udepurkar: conceptualization, methodology, investigation, writing – original draft. Laura Mampaey: methodology, investigation, writing – review & editing. Christian Clasen: supervision, writing – review & editing. Victor Sebastián Cabeza: conceptualization, methodology, investigation, writing – review & editing. Simon Kuhn: conceptualization, funding acquisition, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from the Research Foundation Flanders (G0B5921N, G088922N). The authors would like to thank Suqi Zhang for his assistance with the GPC measurements and Prof. Erin Koos and Yanshen Zhu for the access and assistance with the DLS measurements. VSC acknowledges the financial support by the Spanish Ministry of Science and Innovation (grant number PID2021-127847OB-I00), NextGenerationEU/PRTR project: PDC2022-133866-I00, CIBER-BBN and ELECMI and NANBIOSIS ICTSs.References
- J. Ghitman, E. I. Biru, R. Stan and H. Iovu, Mater. Des., 2020, 193, 108805 CrossRef CAS
.
- J. M. Lü, X. Wang, C. Marin-Muller, H. Wang, P. H. Lin, Q. Yao and C. Chen, Expert Rev. Mol. Diagn., 2009, 9, 325–341 CrossRef PubMed
- A. Fabozzi, F. Della Sala, M. di Gennaro, M. Barretta, G. Longobardo, N. Solimando, M. Pagliuca and A. Borzacchiello, Lab Chip, 2023, 23, 1389–1409 RSC
- R. A. Jain, Biomaterials, 2000, 21, 2475–2490 CrossRef CAS PubMed
- M. Mir, N. Ahmed and A. ur Rehman, Colloids Surf., B, 2017, 159, 217–231 CrossRef CAS PubMed
- I. Bala, S. Hariharan and M. N. V. R. Kumar, Crit. Rev. Ther. Drug Carrier Syst., 2004, 21, 387–422 CrossRef CAS PubMed
- S. Sharma, A. Parmar, S. Kori and R. Sandhir, TrAC, Trends Anal. Chem., 2016, 80, 30–40 CrossRef CAS
- Y. Liu, G. Yang, Y. Hui, S. Ranaweera and C.-X. Zhao, Small, 2022, 18, 2106580 CrossRef CAS PubMed
- F. Danhier, E. Ansorena, J. M. Silva, R. Coco, A. Le Breton and V. Préat, J. Controlled Release, 2012, 161, 505–522 CrossRef CAS PubMed
- T. Delmas, A. Fraichard, P.-A. Bayle, I. Texier, M. Bardet, J. Baudry, J. Bibette and A.-C. Couffin, J. Colloid Sci. Biotechnol., 2012, 1, 16–25 CrossRef CAS
- K. Derakhshandeh, M. Erfan and S. Dadashzadeh, Eur. J. Pharm. Biopharm., 2007, 66, 34–41 CrossRef CAS PubMed
- Z. Liu, M. Yang, Z. Dong, C. Yao and G. Chen, AIChE J., 2023, 69, 1–15 Search PubMed
- J. Yoo and Y. Y. Won, ACS Biomater. Sci. Eng., 2020, 6, 6053–6062 CrossRef CAS PubMed
- J. Xu, S. Zhang, A. MacHado, S. Lecommandoux, O. Sandre, F. Gu and A. Colin, Sci. Rep., 2017, 7, 1–12 CrossRef PubMed
- J. H. Lee and Y. Yeo, Chem. Eng. Sci., 2015, 125, 75–84 CrossRef CAS PubMed
- B. L. Banik, P. Fattahi and J. L. Brown, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016, 8, 271–299 Search PubMed
- T. L. Doane and C. Burda, Chem. Soc. Rev., 2012, 41, 2885–2911 RSC
- M. L. Etheridge, S. A. Campbell, A. G. Erdman, C. L. Haynes, S. M. Wolf and J. McCullough, Nanomedicine, 2013, 9, 1–14 CrossRef CAS PubMed
-
N. Raval, R. Maheshwari, D. Kalyane, S. R. Youngren-Ortiz, M. B. Chougule and R. K. Tekade, Importance of physicochemical characterization of nanoparticles in pharmaceutical product development, Elsevier Inc., 2018 Search PubMed
- M. Danaei, M. Dehghankhold, S. Ataei, F. Hasanzadeh Davarani, R. Javanmard, A. Dokhani, S. Khorasani and M. R. Mozafari, Pharmaceutics, 2018, 10 Search PubMed
- S. Mahmood, U. K. Mandal, B. Chatterjee and M. Taher, Nanotechnol. Rev., 2017, 6, 355–372 CAS
- T. Pulingam, P. Foroozandeh, J.-A. Chuah and K. Sudesh, Nanomaterials, 2022, 12 Search PubMed
- C. J. Martínez Rivas, M. Tarhini, W. Badri, K. Miladi, H. Greige-Gerges, Q. A. Nazari, S. A. Galindo Rodríguez, R. Á. Román, H. Fessi and A. Elaissari, Int. J. Pharm., 2017, 532, 66–81 CrossRef
-
F. B. Schappo, L. S. Assunção, C. D. F. Ribeiro and I. L. Nunes, in Micro and Nano Technologies, ed. D. D. Kesharwani, Elsevier, 2023, pp. 477–506 Search PubMed
- R. Karnik, F. Gu, P. Basto, C. Cannizzaro, L. Dean, W. Kyei-Manu, R. Langer and O. C. Farokhzad, Nano Lett., 2008, 8, 2906–2912 CrossRef CAS
- Y. Bao, M. Maeki, A. Ishida, H. Tani and M. Tokeshi, ACS Omega, 2022, 7, 33079–33086 CrossRef CAS PubMed
- P. H. Huang, S. Zhao, H. Bachman, N. Nama, Z. Li, C. Chen, S. Yang, M. Wu, S. P. Zhang and T. J. Huang, Adv. Sci., 2019, 6, 1900913 CrossRef CAS PubMed
- M. C. Operti, Y. Dölen, J. Keulen, E. A. W. van Dinther, C. G. Figdor and O. Tagit, Pharmaceutics, 2019, 11, 1–17 CrossRef PubMed
- M. C. Operti, A. Bernhardt, S. Grimm, A. Engel, C. G. Figdor and O. Tagit, Int. J. Pharm., 2021, 605, 120807 CrossRef CAS PubMed
- W. Huang and C. Zhang, Biotechnol. J., 2018, 13, 1–8 Search PubMed
- S. Streck, H. Neumann, H. M. Nielsen, T. Rades and A. McDowell, Int. J. Pharm.: X, 2019, 1, 100030 CAS
- K. Y. Hernández-Giottonini, R. J. Rodríguez-Córdova, C. A. Gutiérrez-Valenzuela, O. Peñuñuri-Miranda, P. Zavala-Rivera, P. Guerrero-Germán and A. Lucero-Acuña, RSC Adv., 2020, 10, 4218–4231 RSC
- Z. Liu, F. Fontana, A. Python, J. T. Hirvonen and H. A. Santos, Small, 2020, 16, 1–24 Search PubMed
- A. Agha, W. Waheed, I. Stiharu, V. Nerguizian, G. Destgeer, E. Abu-Nada and A. Alazzam, Discover Nano, 2023, 18, 18 CrossRef CAS PubMed
- V. Kamat, P. Dey, D. Bodas, A. Kaushik, A. Boymelgreen and S. Bhansali, J. Mater. Chem. B, 2023, 11, 5650–5667 RSC
- V. Sebastian, Nanoscale, 2022, 14, 4411–4447 RSC
- S. Zhao, P. H. Huang, H. Zhang, J. Rich, H. Bachman, J. Ye, W. Zhang, C. Chen, Z. Xie, Z. Tian, P. Kang, H. Fu and T. J. Huang, Lab Chip, 2021, 21, 2453–2463 RSC
- A. Ozcelik and Z. Aslan, Biomicrofluidics, 2022, 16, 14103 CrossRef CAS PubMed
- A. Pourabed, J. Brenker, T. Younas, L. He and T. Alan, Ultrason. Sonochem., 2022, 83, 105936 CrossRef CAS PubMed
- P.-H. Huang, S. Zhao, H. Bachman, N. Nama, Z. Li, C. Chen, S. Yang, M. Wu, S. P. Zhang and T. J. Huang, Adv. Sci., 2019, 6, 1900913 CrossRef CAS PubMed
- H. Bachman, C. Chen, J. Rufo, S. Zhao, S. Yang, Z. Tian, N. Nama, P. H. Huang and T. J. Huang, Lab Chip, 2020, 20, 1238–1248 RSC
- M. R. Rasouli and M. Tabrizian, Lab Chip, 2019, 19, 3316–3325 RSC
- C. Liu, W. Zhang, Y. Li, J. Chang, F. Tian, F. Zhao, Y. Ma and J. Sun, Nano Lett., 2019, 19, 7836–7844 CrossRef CAS PubMed
- S. Freitas, G. Hielscher, H. P. Merkle and B. Gander, Ultrason. Sonochem., 2006, 13, 76–85 CrossRef CAS PubMed
- ICH guideline Q3C (R8) on impurities: guideline for residual solvents, European Medicines Agency, 2022, pp. 1–51 Search PubMed.
- Y. Liu, G. Yang, D. Zou, Y. Hui, K. Nigam, A. P. J. Middelberg and C. X. Zhao, Ind. Eng. Chem. Res., 2020, 59, 4134–4149 CrossRef CAS
- S. Mohammadi-Samani and B. Taghipour, Pharm. Dev. Technol., 2015, 20, 385–393 CrossRef CAS PubMed
- C. E. Astete and C. M. Sabliov, J. Biomater. Sci., Polym. Ed., 2006, 17, 247–289 CrossRef CAS PubMed
- M. C. Operti, D. Fecher, E. A. W. van Dinther, S. Grimm, R. Jaber, C. G. Figdor and O. Tagit, Int. J. Pharm., 2018, 550, 140–148 CrossRef CAS PubMed
- S. Schiller, A. Hanefeld, M. Schneider and C. M. Lehr, Pharm. Res., 2015, 32, 2995–3006 CrossRef CAS PubMed
- I. O. De Solorzano, L. Uson, A. Larrea, M. Miana, V. Sebastian and M. Arruebo, Int. J. Nanomed., 2016, 11, 3397–3416 CrossRef CAS PubMed
- P. Zhu and L. Wang, Lab Chip, 2017, 17, 34–75 RSC
- K. Doufène, C. Tourné-Péteilh, P. Etienne and A. Aubert-Pouëssel, Langmuir, 2019, 35, 12597–12612 CrossRef
-
X. Zhao, J. Li and Y. Zhao, in Nanotechnology and Microfluidics, 2020, pp. 1–46 Search PubMed
- R. H. Staff, D. Schaeffel, A. Turshatov, D. Donadio, H. J. Butt, K. Landfester, K. Koynov and D. Crespy, Small, 2013, 9, 3514–3522 CrossRef CAS PubMed
- A. P. Udepurkar, C. Clasen and S. Kuhn, Ultrason. Sonochem., 2023, 94, 106323 CrossRef CAS PubMed
- E. Nieves, G. Vite, A. Kozina and L. F. Olguin, Ultrason. Sonochem., 2021, 74, 105556 CrossRef CAS PubMed
- S. Zhao, C. Yao, L. Liu and G. Chen, Chem. Eng. J., 2022, 450, 138185 CrossRef CAS
- C. Yao, S. Zhao, L. Liu, Z. Liu and G. Chen, Front. Chem. Sci. Eng., 2022, 16, 1560–1583 CrossRef
- S. G. Gaikwad and A. B. Pandit, Ultrason. Sonochem., 2008, 15, 554–563 CrossRef CAS PubMed
- J. P. Canselier, H. Delmas, A. M. Wilhelm and B. Abismaïl, J. Dispersion Sci. Technol., 2002, 23, 333–349 CrossRef CAS
- S. M. Jafari, E. Assadpoor, Y. He and B. Bhandari, Food Hydrocolloids, 2008, 22, 1191–1202 CrossRef CAS
- A. Brotchie, F. Grieser and M. Ashokkumar, Phys. Rev. Lett., 2009, 102, 1–4 CrossRef PubMed
- S. M. M. Modarres-Gheisari, R. Gavagsaz-Ghoachani, M. Malaki, P. Safarpour and M. Zandi, Ultrason. Sonochem., 2019, 52, 88–105 CrossRef CAS PubMed
-
S. M. T. Gharibzahedi and S. M. Jafari, in Nanoemulsions, ed. S. M. Jafari and D. J. B. T.-N. McClements, Academic Press, 2018, pp. 233–285 Search PubMed
- A. P. Altshuller and H. E. Everson, J. Am. Chem. Soc., 1953, 75, 1727 CrossRef CAS
- N. Amanatchi, I. Bihi, M. Briet, J. Stichelmans, W. De Malsche and K. H. Hellemans, SSRN, 2023, preprint, DOI:10.2139/ssrn.4625184.
- J. J. John, S. Kuhn, L. Braeken and T. Van Gerven, Chem. Eng. Process., 2016, 102, 37–46 CrossRef CAS
- J. Kamp, J. Villwock and M. Kraume, Rev. Chem. Eng., 2017, 33, 1–47 CrossRef
- C. Clasen and W. M. Kulicke, Prog. Polym. Sci., 2001, 26, 1839–1919 CrossRef CAS
- T. G. McKenzie, F. Karimi, M. Ashokkumar and G. G. Qiao, Chem. – Eur. J., 2019, 25, 5372–5388 CrossRef CAS PubMed
- A. V. Mohod and P. R. Gogate, Ultrason. Sonochem., 2011, 18, 727–734 CrossRef CAS
- Y. Xu, C. S. Kim, D. M. Saylor and D. Koo, J. Biomed. Mater. Res., Part B, 2017, 105, 1692–1716 CrossRef CAS
- P. Greenspan and S. D. Fowler, J. Lipid Res., 1985, 26, 781–789 CrossRef CAS
- P. Xu, E. Gullotti, L. Tong, C. B. Highley, D. R. Errabelli, T. Hasan, J.-X. Cheng, D. S. Kohane and Y. Yeo, Mol. Pharmaceutics, 2009, 6, 190–201 CrossRef CAS PubMed
- T. Alejo, V. Sebastian, G. Mendoza and M. Arruebo, J. Colloid Interface Sci., 2022, 607, 1466–1477 CrossRef CAS PubMed
- Q. Li, C. Li and W. Tong, J. Nanosci. Nanotechnol., 2016, 16, 5569–5576 CrossRef CAS PubMed
- N. Vij, T. Min, R. Marasigan, C. N. Belcher, S. Mazur, H. Ding, K. T. Yong and I. Roy, J. Nanobiotechnol., 2010, 8, 1–18 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4re00107a |
| This journal is © The Royal Society of Chemistry 2024 |