Automated kinetic model identification via cloud services using model-based design of experiments†
Received
24th January 2024
, Accepted 25th March 2024
First published on 28th March 2024
Abstract
Industry 4.0 has birthed a new era for the chemical manufacturing sector, transforming reactor design and integrating digital twin into process control. To bridge the gap between autonomous chemistry development, on-demand manufacturing and real-time optimization, we developed a cloud-based platform driven by model-based design of experiment (MBDoE) algorithms integrated in a simulation software for model identification (SimBot) to remotely coordinate a smart flow reactor, also known as the LabBot, sited in a different location. With real-time data and setpoints synchronization, MBDoE was able to identify kinetic models using a limited number of experimental runs. Within this platform, two pharmaceutically relevant syntheses were investigated as case studies: amide formation and nucleophilic aromatic substitution. A new kinetic model providing statistically adequate data description within the whole investigated experimental design space was identified for the amide formation reaction. The model for the nucleophilic aromatic substitution with a well-known but complex mechanism was accurately identified ensuring a statistically precise estimation of kinetic parameters.
1 Introduction
Recent advances in automated chemistry development,1 chemical manufacturing on-demand2 and real time optimization3,4 indicate that data-driven modelling approaches can be used to autonomously explore chemistry development in a closed loop. With the goal of automation to chemical synthesis, closed-loop reaction systems have become more commonplace, providing advantages like control and self-optimisation of reactions in automated platforms. The development of automated platforms can help the automation of different tasks through a computer network that schedules them. Self-optimising reactors have proven to be effective5,6 and have shown the possibility to conduct experimental campaigns in a reactor with minimum human interaction. However, these automated platforms do not necessarily aim to develop physics-based models. The use of physics-based models would increase process understanding, enhance platform robustness, and explore different conditions, in comparison to self-optimization platforms where optimised reaction conditions are identified through a black-box approach. The development of such models aligns with the increasing interest in the greener synthesis of active pharmaceutical ingredients (APIs) using continuous flow chemistry to reduce the drug development timeline.2 Continuous-flow chemical reactors have attracted a significant attention in the pharmaceutical industry for reaction development and scale-up.7 Flow reactors are efficient in acquiring the reaction information from minimal amount of resources alongside with the ability for feedback control into the continuous system.1 Online analysis of flow reactor systems has shown a great potential for full automation. In this work, we take a further step on this development where the closed-loop integration and online analysis is automated through a cloud-based platform, a cloud service enabling communication to remotely control the reactor system which allows flexibility on the location of platforms and the hosts of the software, enabling the use of proprietary algorithms and computationally data-efficient approaches.
To achieve physics-based models for use in the cloud-based platform, analyses of relevant experimental designs, that typically require extensive amounts of time and resources, need to be performed. Examples of experimental designs include factorial experiments,8 randomised experiments,9 and model-based design of experiments (MBDoE).10 Factorial experiments consider all possible combinations of various additive levels in different factors (e.g. reactor temperature and reactant concentration) affecting the chemical system. Although such designs are easy to generate and execute, the experimental cost is prohibitive especially in the case of multiple experimental factors. Even where fractional factorial experiments11 are selected to reduce the cost, the design may result in scarcely informative experiments. Randomized experiments involve fewer combinations of levels of factors generated by statistical methods. As the number of experiments is predefined, random experiments can achieve lower experimental costs than factorial experiments, while rapidly exploring the experimental design space for informative experiments. The application of MBDoE requires one or more candidate physics-based models and to design optimal experimental conditions resulting in the most informative data to perform 1) parameter identification or/and 2) model discrimination. With physics-based models, the MBDoE techniques can design optimum experiments that match available resources and provide feedback to improve the model performance.
Algorithms for parameter estimation and optimal MBDoE have been employed online to drive experimental campaigns with the aim of selecting the best model among a set of given model structures (i.e. model discrimination)12,13 and/or improving the statistical quality of the parameter estimates for a given model structure.14,15 In conventional MBDoE frameworks, the model identification process is performed in a purely sequential manner involving: 1) design of experiments at the available parameter estimates, 2) execution of experiments and 3) parameter estimation and evaluation of a posteriori statistics using the available measurements. This process is repeated until parameters are estimated with minimum uncertainty or until the maximum experimental budget (number of experiments) is achieved. It is evident that such a procedure is sub-optimal as the initial parameter estimates are affected by parametric uncertainty. This drawback has been resolved by online redesign of experiments,16 where a new design of experiment can be computed every time as soon as a new measurement becomes available. The online MBDoE techniques will therefore be used in this work to drive the cloud-based platform when scheduling a sequence of experiments to be executed remotely in steady-state in a smart flow reactor (LabBot).
While MBDoE techniques have been applied in a number of fields, a recent review17 alluded to paucity of applications in the pharmaceutical industry. In anticipation, McMullen and Jensen13 demonstrated with an organic chemistry synthesis the use of MBDoE for model discrimination and parameter estimation, delivering a scalable kinetic expression using Bayesian statistical selection studying a homogeneous single-step Diels–Alder transformation discriminating 3 alternative kinetic models.
In this paper we propose the use of a cloud-based platform based on a MBDoE-driven software for model identification (“SimBot”), to optimally design experiments executed remotely in an automated flow reaction system (“LabBot”). In this work, while the LabBot site for generating understanding of the case studied is located at the University of Leeds, the MBDoE site for set-point generation is located at the University College London. We validate the effectiveness of our MBDoE-driven platform using two case studies: the pharmaceutically relevant synthesis of N-alkyl amides18 as well as a multistep reaction, a nucleophilic aromatic substitution.5 A key feature of this research is the integrated multisite approach, where all partners contribute to the evolution of the series of experiments. The research centres interacted via a cloud-based experimental design and analysis system (EDAS) that facilitated the exchange of processing conditions and experimental results. Advanced automated flow chemistry was integrated with MBDoE to update information in the Bayesian sense. Although in this work we have used a single LabBot, our vision is for the use of multiple computational services that exchange information with multiple automated laboratory robots, integrating experimental set-point generation, experimental execution, analysis of experiments and optimisation, so that eventually the FAIR (findability, accessibility, interoperability and reusability) guiding principles for scientific data across distributed laboratories are satisfied.19
The structure of this paper is as follows. Section 2 presents in details materials and method employed in this work from the two main aspects of the work: experimental and computational, as well as their cloud-based communication protocol. Section 3 describes the two case studies and discusses the results on application of the cloud-based platform and Section 4 concludes the paper.
2 Materials and methods
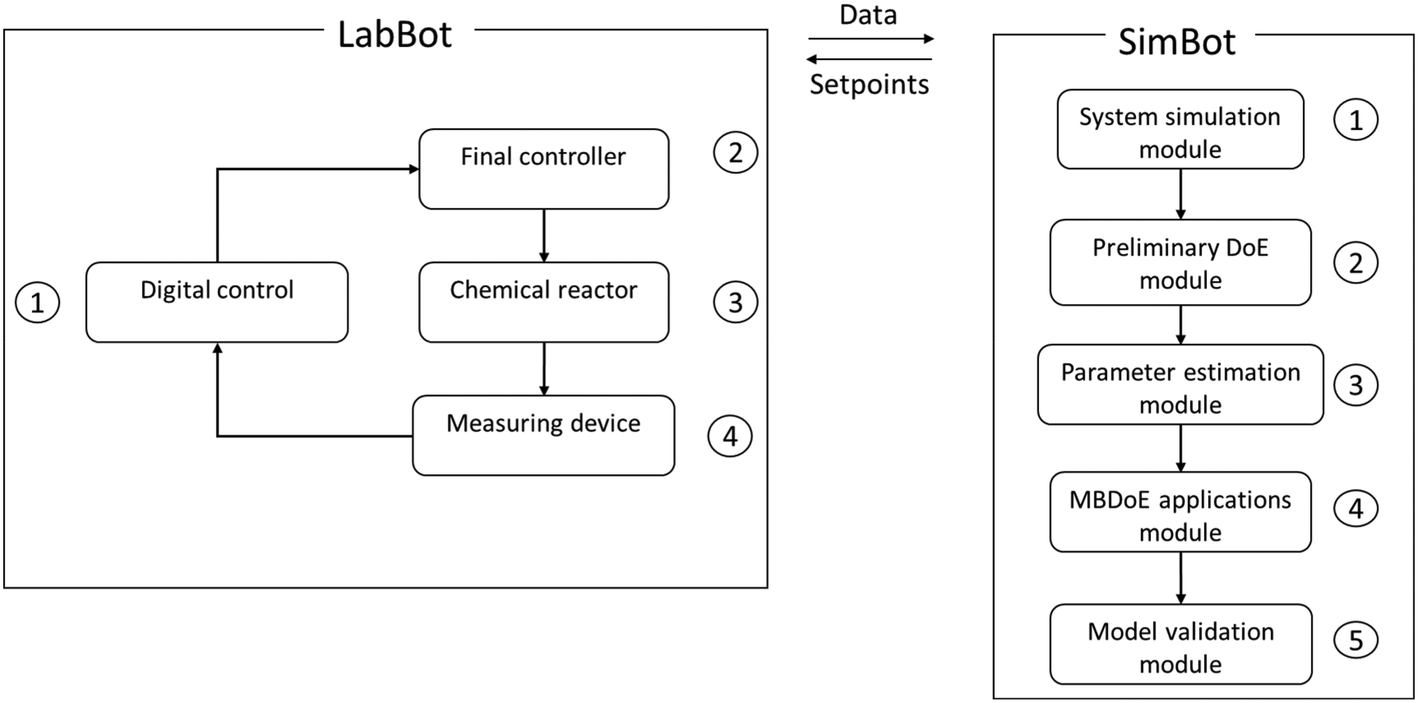
In this section, we discuss in detail the LabBot – the experimental setup, the SimBot – the modelling and simulation software, and the communication protocol between the two systems via the cloud. The LabBot comprises compartments for reaction, control, analysis, and flow while the SimBot comprises modules for simulation, sampling, parameter estimation, MBDoE applications and validation. These two systems exchange information as shown in Fig. 1: the LabBot receives experimental setpoints to produce experimental data while the SimBot computes new experimental setpoints from past experimental data.
 |
| | Fig. 1 An illustration of exchange of experimental data and setpoints required between LabBot and SimBot with the enabling compartments and modules of the two systems also shown numbered according to the sequence of activities in each system. | |
2.1 LabBot reactor system
Fig. 2 shows a sketch of the LabBot, a smart flow reactor system comprising a tubular reactor, a series of control instruments and an automation network. The reactor is a 4′′ circular aluminium block with the tubing wrapped around it, and the heating provided by a Eurotherm 3200 temperature controller.20 Three pumps (HPLC dual piston reciprocating pumps – JASCO PU2800) supply feed A, feed B and neat solvent into the reactor, and the supply flows are connected through 2 tee-pieces (Upchurch Scientific P-207) as shown. The reactor pressure is controlled with a back-pressure regulator (BPR, 100 psi, Upchurch Scientific P-505). The reactor system is connected to an online LC, using an in-line 4 port sampling valve (VICI Valco EUDA-CI4W.06) equipped with 0.06 μL sample loop and connected to the HPLC (Agilent 1260 equipped with Sigma Ascentis Express C18 reverse phase column, 5 cm, 4.6 mm ID and 2.7 μm particle size).
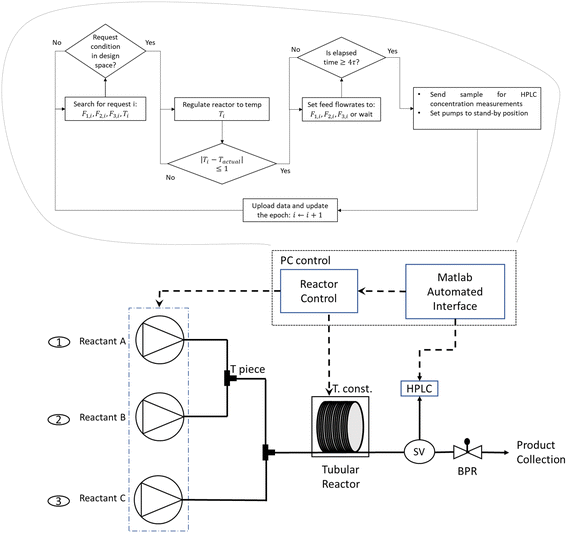 |
| | Fig. 2 Automated continuous reactor system equipped with software for controlled flow ramps. The sequence of actions with timer objects in the PC control is also displayed. | |
The LabBot is an automated system controlled by Matlab algorithms. The system, requiring expertise in reactor initialisation, mechanism analysis and experimentation campaign, operates based on four timer objects to schedule the execution of commands. The four timer objects are: 1) the request, 2) the temperature, 3) the steady state and 4) the sample analysis. The communication between the external client and the LabBot is achieved via a cloud service. When the LabBot needs to be used by an external user, a new campaign is generated with a directory in the cloud. In this directory the external user will submit requests and the LabBot its output files related to the process monitoring and the sample analysis results. The request and the output files are in the comma separated value (csv) format. The request and output files need to have a unique identification number to be distinguished one from each other. This is achieved by adding a sequential number to the name of request and output files which we call “epoch”. Further information about the operation of the LabBot using the four timer objects can be found in Section S1 in the ESI.†
Data from distributed laboratories is required to satisfy four objectives, which are contained in the FAIR guiding principles agreed globally for scientific data management.19 These four objectives are findability, accessibility, interoperability and reusability, as depicted in the acronym ‘FAIR’. The data management procedure used this work satisfies three of these objectives: 1) findability, because each data is correctly labelled, 2) accessibility, because the csv file format used can be accessed using a computer input program, and 3) re-usability, because the data is secured in the cloud managed by Dropbox. The data, however, does not satisfy interoperability, which is necessary if the data originates from multiple laboratories. A single LabBot location has been used in this work.
2.2 SimBot software system
The SimBot software system generates experimental designs that can be employed to control the operation of the LabBot reactor. To generate experimental designs, the software system uses mathematical modelling, computer programming and statistical analysis.21 Mathematical modelling describes the reaction system by developing models, which could be physics-based, machine learning or a combination of both (hybrid models).22 The SimBot encodes the mathematical models in Python, a popular programming language with a library of packages for fast and reliable solution of complex differential and algebraic equations for reactor simulation and/or robust optimization algorithms as well as packages for statistical analysis.23,24 Key Python packages imported include CasAdi for model implementation;25 Scipy, Ipopt and Pylab for model simulation and optimisation;26–28 pyDOE and Stats for preliminary design of experiment and statistical analysis.29,30 Within this programming environment, the SimBot performs computations that are arranged into modules for reactor system simulation, preliminary design of experiment, parameter estimation, MBDoE, and model validation. A systematic framework leveraged by MBDoE techniques is applied to maximize information gain from experiments while minimising time and resource costs.
2.2.1 Reaction system model.
The flow reactor used in this work is modelled as:| |  | (1) |
where ci is the ith species concentration, rj is the reaction rate (mol s−1 L−1) of the jth reaction with the vij the stoichiometric coefficient, t is the dynamic time and τ is the residence time expressed as the ratio of the reactor volume V and the volumetric flowrate vo. In this work, only steady-state measurements are acquired and the term  .
.
Rate of reactions expressions are derived from the mechanism proposed in a preliminary study or literature. A preliminary study, which is usually conducted on a newly introduced chemical synthesis, helps with setting up the LabBot system and acquiring information about experimental design space and preliminary rate expressions for the chemical synthesis. Stable noisy optimization by Branch and Fit (Snobfit), an optimizer developed for optimization problems with noisy and expensive to compute objective functions,31,32 is used in this preliminary study. For a discussion on the application of Snobfit for a LabBot preliminary study, we refer the reader to Section S2 of the ESI.†
Reaction rates in reparametrized form are expressed as:33
| | 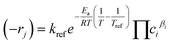 | (2) |
kref is the rate constant at a reference temperature
Tref,
T the reactor temperature,
Ea the activation energy,
R universal gas constant equal to 8.31 kJ mol
−1 K
−1, and
βi the reaction order for component
i.
Eqn (1) together with the expressions for the reaction rates in eqn (2) can be written as a system of differential and algebraic equations (DAEs):
| | 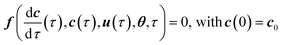 | (3) |
with
c ∈
Nnc being the vector of the state variables (concentrations),
u ∈
Nnu being the vector of manipulated variables (reactants' inlet concentrations, and reactor temperature),
θ ∈
Nnθ being the vector of the kinetic parameters to be identified. Where
c0 and
θ are specified, the DAEs system is integrated numerically in Python using the Adams method (for non-stiff problems) and the method based on backward differentiation formulas (BDF) (for stiff problems) reported
34 and available in the Scipy Library. However, the set of kinetic parameter
θ is not known and will be determined using a dedicated parameter estimation module (Section 2.2.3).
2.2.2 Preliminary DoE module.
Once the experimental design space is defined by specifying upper and lower bounds on experimental decision variables, the SimBot software commences operations using a preliminary Design of Experiment module. The module is used to generate a first set of experimental data for prior parameter estimation in each proposed kinetic model. Parameter estimation of the Arrhenius pre-exponential factor and activation energy in a kinetic model describing a single-step synthesis requires a minimum of two independent measurements. For modelling multiple-step synthesis, minimum data requirements for parameter estimation increase linearly with the number of parameters.35 The preliminary DoE aims at sampling the design space for a minimum number of points that are distant enough to capture the maximum variations in the system behaviour. The two case studies in this contribution employed Latin hypercube sampling, a near-random technique for generating a specified number of points while ensuring maximum distance among the points in the design space.36
2.2.3 Parameter estimation module.
Using the measured data, the model parameters can be estimated via nonlinear optimisation. To account for the uncertainty in experimental data, the objective function for parameter estimation is defined using the negative log-likelihood function and optimized by minimizing the variable term as:35| |  | (4) |
subject to the model equations:| | f(ẋ(τ),![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) x(τ),u(τ),θ,τ) = 0 x(τ),u(τ),θ,τ) = 0 | (5) |
| | | φ = [uT,τ,x0T]T | (8) |
where x ∈ ![[Doublestruck R]](https://www.rsc.org/images/entities/char_e175.gif) Nx is the vector of state variables, ẋ is the first derivative of the state variables, u ∈ Nu is the vector of inputs or control variables that define the condition of an experiment, θ ∈ Nθ is the vector of model parameters, y ∈ Nŷ is the vector of model predictions for measurements y, Vy is the measurement error covariance matrix, and Nexp is the total number of samples. Eqn (5) is the vector of differential equations resulting from the material balance on the components while eqn (6) are the algebraic equations relating measurements to the state variables. Eqn (7)–(9) define the initial conditions, experimental design, and state space, respectively.
Nx is the vector of state variables, ẋ is the first derivative of the state variables, u ∈ Nu is the vector of inputs or control variables that define the condition of an experiment, θ ∈ Nθ is the vector of model parameters, y ∈ Nŷ is the vector of model predictions for measurements y, Vy is the measurement error covariance matrix, and Nexp is the total number of samples. Eqn (5) is the vector of differential equations resulting from the material balance on the components while eqn (6) are the algebraic equations relating measurements to the state variables. Eqn (7)–(9) define the initial conditions, experimental design, and state space, respectively.
With the estimated values ![[small straight theta, Greek, circumflex]](https://www.rsc.org/images/entities/b_i_char_e12d.gif) , statistical analysis can be performed on the model to assess model-data adequacy, parameter precision and hence potential in MBDoE applications.
, statistical analysis can be performed on the model to assess model-data adequacy, parameter precision and hence potential in MBDoE applications.
The model adequacy is assessed for a candidate kinetic model by employing a χ2 (chi-square) lack-of-fit test.37 The chi-square value is defined as:
| |  | (10) |
where
ysk,
ŷsk is the
k-th entry of
y at the
s-th experiment of the measured and predicted response, respectively. Additionally,
σ2kk is the variance of the
k-th measured response and the
k-th diagonal entry of the measurement's variance–covariance. The
![[small chi, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e10c.gif) 2
2 is compared with a reference value
χ2ref, which is the inverse of the cumulative distribution function of
χ2 distribution at 1 −
α confidence level (usually
α = 0.05% or 0.01%) with
NexpNy −
Nθ degrees of freedom. The value of the
2 needs to be as small as possible and ideally lower than
χ2ref.
The precision in the estimation of kinetic parameters after the execution of the designed experiments is evaluated employing a t-test.10 The ith kinetic parameter is considered to be statistically precise if its ti > tref, where
| | 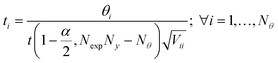 | (11) |
ti is the inverse of cumulative distribution function of Student's-
t distribution at

confidence level with
NexpNy −
Nθ degrees of freedom, and its corresponding
tref is
t(1 −
α,
NexpNy −
Nθ).
Vii is the
ith diagonal entry of the parameter variance-covariance matrix
V computed using the estimated parameter and expressed as:
| | | V(,φ) = [H(,φ) + V−10]−1 | (12) |
with
| | 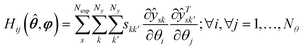 | (13) |
V0 is the preliminary approximation of the variance–covariance matrix of the parameters, which contains the initial information on parametric uncertainty,
ŝkk′ is the
kk′ element of the
Ny ×
Ny inverse of the variance–covariance matrix of the measurement errors,
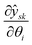
is the parameter sensitivity of
ŷsk the
kth entry of
ŷ at the
sth experiment with respect to
θi. It should be noted the inverse of the variance–covariance matrix corresponds to the Fisher information matrix
H.
With only diagonal entries, the t-test considers each parameter in isolation and would be insufficient to confirm parameter precision with highly correlated parameters indicated by the off-diagonal elements in the parameter covariance matrix. Highly correlated sets of model parameters can make the objective function in the MBDoE application ill-conditioned.38 A reliable model precision test would therefore combine the t-test with a parameter correlation test to confirm model precision. To test for parameter correlation, we analyse the correlation matrix R(, φ), which is the normalised parameter covariance matrix where the ij-element is computed as:
| |  | (14) |
with
Rij values in the interval [0, ±1].
Rij values approaching zero indicates parameters
θi and
θj as completely uncorrelated while values approaching +1 indicates the parameters as perfectly correlated (or perfectly anti-correlated for values approaching −1).
2.2.4 Model-based design of experiments module.
Activities required for robust MBDoE application can be classified into three stages: model identification, model calibration and model validation.10 Model identification involves identifying the right model structure with a fully determined set of parameters corresponding to that structure.35 Parameter estimation, and model testing, discussed in the previous section, are therefore fundamental in successfully identifying the right model.39 Model calibration, also involving parameter (re)estimation and statistical analysis, seeks to determine the parameter space that minimises prediction uncertainty of the right model even as further uncertainty may emerge with new experimental conditions.40 Model validation using statistical analysis evaluates the appropriateness of the model in describing the chemical system. While model calibration employs optimal experimental points generated using MBDoE, model validation requires testing the model at conditions not exploited in model calibration by employing experimental points generated from other techniques (in our case Snobfit and factorial designs of experiments).
In the MBDoE optimisation structure, the constraints are the DAEs in eqn (5)–(9), but the objective function depends on the task, which may be MBDoE for model discrimination, MBDoE for model precision, or a combination thereof. The two case studies to be considered in Section 3 require MBDoE for model precision in computing the design vector φ = [uT,τ,cT0]T ∈ Φ ⊆ Nnφ to improve the precision of the kinetic parameters.41 The improvement of parametric precision is equivalent to the shrinkage of the elements of the variance covariance matrix of the model parameters. The expected marginal posterior covariance VNexp+1(,φ) at a new experimental design can be obtained using:
| | | VNexp+1(,φ) = [HNexp+1(,φ) + V−10]−1 | (15) |
Eqn (15) represents an upper bound on information as dictated by the Cramer Rao Theorem
35 as it strongly depends on the unknown parameters
θ. The design of experiments is performed using the current estimate for the parameters
and the variance covariance is computed at this estimate. To obtain the optimum experimental design the design vector
φ is computed that minimizes or maximizes a relevant metric
J(·) of the
V or
H respectively. Different metrics have been proposed in the literature
10 including the minimization of the determinant of
V (D-optimal), the minimization of the trace of
V (A-optimal) or the minimization of largest eigenvalue of
V (E-optimal). These metrics on the variance–covariance matrix of model parameters can equally be used to rank and compare the information content of the proposed experimental design to the
Nexp past experiments. For example, a trace-based relative Fisher information index (RFI) can be computed as:
42| | 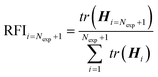 | (16) |
In this work, the design of experiments for model precision will be performed in a closed-loop fashion using online MBDoE. In standard MBDoE, the design is conducted either offline for all the experiments or sequentially after each experiment. Both strategies may result in a waste of the bulk materials as the former designs all experiments with poor prior knowledge and the latter has a waiting period where the continuous flow reactor is still running. Here, an online redesign is proposed,
16 where the bulk waste is kept to the minimum. Initially, the first
n preliminary experiments are designed simultaneously using the method of Latin Hypercube sampling
36 and the
ith experiment has duration Δ
i, that includes transient (unmeasured) behaviour and measurement delay. The online strategy for designing the experiments in the flow reactor is illustrated in
Fig. 3. After the availability of observations from
n − 1 experiments and the corresponding parameter estimates, the
n + 1 experiment is designed using MBDoE. During this period the
nth designed experiment is running in the reactor. Then data from the
nth experiment are collected and the (
n + 1)
th experiment is ready to be executed. It should be noted that the parameter estimation and the MBDoE should efficiently be computed in a time frame faster than the duration Δ
i using fast optimization techniques.
43 The online redesign step is as fast as the offline design, where the information gathered is exploited only at the end of preliminary experiments, but faster than the sequential design, where preliminary experimentation is paused for a time duration to redesign the subsequent experiment. Specifically, where 4 experiments are initially designed, after the availability of the first 2, experiments are redesigned using MBDoE, while experiment #3 is running.
 |
| | Fig. 3 Strategies for redesign of experiments. | |
The progressive availability of measurements supports the statistical evaluation of the model, ensuring at the same time a precise estimation of kinetic parameters. The procedure stops when the statistics on lack of fit and parameter precision are satisfied or when the maximum number of experiments has been reached.
2.2.5 Numerical solution.
MBDoE objectives present complex mathematical formulations that can only be solved using computational programming. Parameter estimation, for example, encountered within the MBDoE framework, aims at computing parametric values that would optimize the likelihood function while obeying the differential and algebraic equations. Computational programming for maximum likelihood parameter estimation optimisation problems begins with a set of values (initial guesses) and proceeds to generate a sequence of parametric values that would converge to a parameter set where the likelihood function is maximised. An optimization problem embedded with differential and algebraic equations, can be solved simultaneously using orthogonal collocation44 and solved computationally using nonlinear programming. Orthogonal collocation divides the domain of the independent variable (in our case, residence time τ) into finite elements and connects each element using collocation points to describe different states of the system as a polynomial function. The polynomial transcription describing initial, intermediate, and final states, included in the objective function and the constraints, can then be solved as a stationary optimization problem, consistent with the real reactor where measurements are taken at steady state. CasADi, a module in Python for open-source numerical optimization, provides an environment to implement the parameter estimation problem while IPOPT, a software library for nonlinear optimization, solves for the parametric values.25,27
2.3 Communication protocol – cloud-based communication
The LabBot and SimBot systems operating from University of Leeds (site 1) and University College London (site 2), respectively, communicate via Dropbox, a popular cloud-based technology.45 The technology provides a platform for multiple concurrent users, online storage of files, and real-time synchronization and sharing of the files. The cloud-based communication protocol (CP) between the two sites is shown in Fig. 4 and summarized as follows:
 |
| | Fig. 4 Operation of the cloud-based platform connecting the LabBot and SimBot sites displaying the communication protocol (CP) among the sites, and the expertise and framework or scheme that each site requires. | |
CP 1: site 1 selects chemical synthesis of interest and specifies the experimental design space of the control variables during experimentation. The chemical synthesis dictates the model structure along with the kinetic parameters, developed using the plug flow reactor model and the reaction rate expression of the synthesis mechanism proposed from a previous study or literature.
CP 2: site 2 generates a number preliminary design of experiments (DoEs) from the experimental design space that would create sufficient initial data to estimate the model parameters.
CP 3: site 1 operates the LabBot as shown in Fig. 4 to execute the experiments and generate experimental data in terms of concentration of relevant chemical species that can be employed in parameter estimation.
CP 4: site 2 employs the experimental data to assesses the mechanistic models and integrates the set of candidate models in the MBDoE framework if models are identifiable.
CP 5: site 1 executes the experiments at the conditions dictated by MBDoE using the LabBot operation scheme and generates new experimental data.
CP 6: site 2 recalibrates the synthesis model to improve parameter precision for model validation with previously unseen experimental data.
The procedure is iterated until the parameters of the best model are estimated with minimum uncertainty or until the maximum experimental budget (number of experiments) is achieved.
3 Results and discussion
In this section, we demonstrate the developed MBDoE-driven cloud-based platform on two pharmaceutical case studies: nucleophilic aromatic substitution (case study 1) and homogeneous amide formation (case study 2). The first case study, characterised by a unique model structure, involves the precise identification of kinetic parameters for a complex reaction mechanism43 while the second case study requires the development and identification of a new kinetic model.
3.1 Case study 1
To evaluate the cloud-based platform, a first benchmark case study was used related to the nucleophilic aromatic substitution (SNAr) of 2,4-difluoronitrobenzene{1} with morpholine{2} in ethanol (EtOH) to give a mixture of the desired product ortho-substituted {3}, para-substituted {4} and bis-adduct {5} as side products. This reaction was chosen because it produces 3 different major products in parallel and consecutive steps, and there is available data published by both our and other groups,46–49 as well as being relevant for pharmaceuticals and fine chemicals.50 In the LabBot setup illustrated by Fig. 2, feed A contains a solution of 2,4-difluoronitrobenzene while feed B contains a solution of morpholine. There are four experimental design variables in this system: residence time, temperature of the reactor, and inlet concentrations of 2,4-difluoronitrobenzene and morpholine solutions. It was noticed that the bis-substituted product has a low solubility in ethanol, and if in higher concentrations could lead to blockages. Under the described conditions boundaries, the system was operational for 18 hours without issues. The experimental design space employed for this case study is reported in Table 1. Further details about the feed preparation and chemical analytics can be found in the ESI†, Section S2.
Table 1 Experimental design space of control variables employed in the nucleophilic aromatic substitution case study
| Limits |
c
1(0) (M) |
c
equiv2
|
τ (min) |
Temp (°C) |
| Lower |
0.0967 |
0.2054 |
0.3 |
60 |
| Upper |
1.6917 |
2.54 |
3.0 |
130 |
The scheme in Fig. 5 describes the aromatic substitution reaction mechanism showing parallel and consecutive chemical steps of reactants and intermediates.
 |
| | Fig. 5 Scheme for nucleophilic aromatic substitution reaction mechanism.47 | |
The molar balance equations for the reactor in the form given by eqn (1) (using the chemistry from the scheme in Fig. 5, where each step is assumed as an elementary step) can be written as:
| |  | (17) |
The values of the design variables that were implemented for LabBot experimentation in this case study are shown in
Fig. 6. The experimental conditions used in experiments 1–4 were the preliminary ones generated by a LH factorial design, while the ones used in experiments 5–9 were generated by the MBDoE; the last 10 and 11 runs were validation experiments. The measurements obtained by the latter conditions were generated from Snobfit optimisation and were not included in model calibration but specifically used to validate the performance of the kinetic model in an unseen environment. For this case study, we approximated the MBDoE formulation using a surrogate model that is based on the Gaussian process (GP) and optimized using Bayesian optimisation. The GP employs a prior mean function on the D-optimal MBDoE criterion and a squared exponential kernel function with the variation frequency and amplitude being the hyperparameters optimised during GP training. The approach is necessary in the MBDoE optimisation when handling Fisher information profiles with discontinuities. More detail about this MBDoE computational approach has been reported.
43
 |
| | Fig. 6 Design variables for the performed experiments: 1–4 are the experiments design using LH sampling. The experiments 5–9 are design using the online MBDoE method. The experiments 10 & 11 are unseen from our algorithm and used to validate the kinetic model. | |
To compare the results of MBDoE, the t-values for the kinetic parameters calculated before and after parameter estimation are provided in Table 2. It should be noticed that originally kinetic parameters for the reaction from para-product to bis-product (i.e., k4,ref and Ea4) were not statistically significant. However, after MBDoE the t-values for these critical parameters were significantly higher. Notice that the parameters k3,ref and Ea3 are determined to be 0 by the parameter estimation (i.e. the corresponding step is not active).
Table 2 Parameter values and their respective t-values calculated before and after MBDoE. Tref = 90 °C
|
k
i,ref (M−1 min−1)/Eai (kJ mol−1) |
k
1,ref
|
E
a1
|
k
2,ref
|
E
a2
|
k
3,ref
|
E
a3
|
k
4,ref
|
E
a4
|
| Note that * means that the true values converged to 0. |
| Parameter values |
1.21 |
34.53 |
0.21 |
27.84 |
0.* |
0.* |
0.057 |
42.49 |
|
t-value before MBDoE (tref (99%) = 2.68) |
9.61 |
39.04 |
16.91 |
5.89 |
0.* |
0.* |
0.45 |
0.23 |
|
t-value after MBDoE (tref (99%) = 2.40) |
55.04 |
260.76 |
136.65 |
73.08 |
0.* |
.0* |
27.60 |
15.60 |
To test the model adequacy, the χ2 was computed according to eqn (10). It is noticeable that even though χ2 is not small enough to pass the statistical test (see Table 3), its value has been significantly reduced using MBDoE. This is evident by comparing the prediction for the concentrations with the experimental measurements before and after MBDoE. The parity plot is depicted in Fig. 7, where the red markers correspond to the predictions using the kinetic parameters before the MBDoE was applied. The figure shows that there are a few points where the prediction with the initial parameters is not adequate.
Table 3 The χ2 values for the whole data-set before and after MBDoE
|
χ
2 – Before MBDoE (χ2ref = 78) |
1687 |
|
χ
2 – After MBDoE (χ2ref = 78) |
272 |
 |
| | Fig. 7 Parity plot for the predictions of models using the kinetic parameters before and after the MBDoE. ▼ ● ■ + correspond to starting material, ortho, para and bis product, respectively. | |
This result is also illustrated in Fig. 8, where the model after the MBDoE has a significantly reduced uncertainty on the predicted concentrations (see reduced error bars) as a result of a more precise parameter estimation achieved.
 |
| | Fig. 8 Comparison of model predictions with experimental data before and after MBDoE. Predictions after MBDoE are more accurate and precise: A. concentration of starting material (SM); B. concentration of ortho-substituted product; C. concentration of para-substituted product; D. concentration of bis-substituted product. | |
3.2 Case study 2
In the second case study, we demonstrate the cloud-based platform on an amide synthesis. Amides are a promising group of organic compounds, which can act as important lead substances for producing biocidal products, functional food, cosmeceuticals, and drugs.18
A simple synthesis method involves reacting an amine with an ester. With the LabBot setup illustrated by Fig. 2, feed A contains the amine solution and feed B contains the ester solution. While keeping the inlet concentration of amine c1(0) constant at 0.100 M, we designed a set of experiments considering 3 experimental design variables, namely inlet concentration of ester c2(0), reactor temperature, and residence time, from which the LabBot calculated the flowrates of the reactants. The experimental design space employed for this case study is reported in Table 4. The online HPLC analysis provided the concentration of starting material left-over after the reaction. Details about sources of reagents with their chemical analysis, and preparation of stock solutions with the expression for flowrates are included in the ESI.†
Table 4 Experimental design space for the control variables employed in the amide formation case study
| Limits |
c
1(0) (M) |
c
equiv2(0) |
τ (min) |
Temp (°C) |
| Lower |
0.100 |
1.000 |
0.5 |
40 |
| Upper |
0.100 |
5.000 |
7.0 |
150 |
Two mechanisms can be inferred from the literature. Nakajima and Ikada51 reported that this synthesis is a single-step, forward reaction to form amide and alcohol. Clark et al.,52 on the other hand, reported the products can also react and revert to form amine and ester. Therefore, two mechanisms have been considered: 1) single-step, forward reaction and 2) a single-step, reversible reaction. Modelling for these mechanisms, tested using the cloud-based platform, are reported in Table 5.
Table 5 Models 1 and 2 describing the kinetics of single-step forward and reversible mechanisms, respectively, in the amide formation case study
|
|
Chemical equations |
Rate equations |
Component balances |
Components |
| Model 1 |
RCOOR′ + R′′NH2 → RCONH2 + R′′OR′ |
r
f = kfc1c2 |
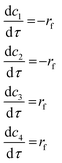 (18) (18) |
c
1 = RCOOR′ |
|
c
2 = R′′NH2 |
|
c
3 = RCONH2 |
|
c
4 = R′′OR′ |
| Model 2 |
RCOOR′ + R′′NH2 ⇌ RCONH2 + R′′OR′ |
r
f = kfc1c2 |
 (19) (19) |
|
r
b = kbc3c4 |
Model 2, which has the highest number of parameters to be estimated (4), determines the minimum number of experiments that should be conducted initially for parameter estimation. Thus, 4 preliminary design of experiments (PDoEs) were generated by a Latin Hypercube sampling and these experiments were conducted in the LabBot at Leeds. Fig. 9 shows the experimental design space and the 4 PDoEs, evenly dispersed in the design space. Using the data from the four experiments (whose amide yields have been shown by the bubble size), the model parameters were estimated, and the relative model performance has been investigated. Fig. 10, in the A and B panels, shows the parity plots for models 1 and 2, respectively. The former fails the χ2-distribution adequacy test: χ2 = 494.1, significantly higher than the reference χ2ref = 23.7. Thus, model 1 does not adequately describe the amide synthesis and is consequently rejected. Model 2, on the other hand, satisfies the χ2-distribution adequacy test: χ2 is close to zero illustrating that this model (and associated mechanism) accurately describes the synthesis as a reversible single-step reaction.
 |
| | Fig. 9 Locations of the preliminary design of experiments (PDoEs) in the design space and the percent amide yield obtained in the LabBot indicated by the bubble size. | |
 |
| | Fig. 10 Parity plots for c3 = RCONH2 by model 1 (in A) and model 2 (in B) with the χ2 values being 494 and ∼0, respectively, model 2 accurately describing the amide synthesis (experimental error {= ±2σ; σ = 0.0003 mol dm−3} is also indicated). | |
Because the reaction is reversible, we can estimate its equilibrium constants, given as:
| |  | (20) |
Fig. 11 shows the profile of the equilibrium constant with temperature, showing that the synthesis obeys the van't Hoff equation.
53
 |
| | Fig. 11 Equilibrium constant dependence on temperature with R2 = 1.00 illustrating that the synthesis follows the van't Hoff equation. | |
The kinetic parameters were used in predicting the equilibrium conditions. The amide synthesis had not reached equilibrium for the conditions reported in the study. Thus, the reported kinetic information could be extracted. We can illustrate this assertion using the reaction characteristic time, which we estimated as ∼30 minutes. This value is longer than the upper bound of 7 minutes residence time used in the experimental design space (Table 4).
Nevertheless, the synthesis requires MBDoE to improve the model performance as some of the kinetic parameters are still poorly estimated. Table 6 shows the Student's t-test value for the model 2 parameters before and after MBDoE. The value of the activation energies before applying MBDoE are not precise. On applying MBDoE for parameter precision to generate a new experiment, conducting the experiment in the LabBot, and recalibrating the model, the parameter statistics improved illustrating that the new parameter values have been precisely estimated. D-optimal criterion was used by the SimBot to design the experiment. In Fig. 12, panel A shows the location of the D-new experiment on the Fisher information map as the global optimum in the experimental design space. This MBDoE is equally the most informative when compared with the PDoEs using the relative Fisher information index. We had tested in silico the three metrics of the Fisher information matrix, that is A-, D-, and E-optimal criteria, and found that only the D-criterion could significantly improve the parameter precision because of parameter correlation. Fig. 13 shows the correlation matrices before and after MBDoE (with A-, D-, and E-design criteria) from the in silico testing, illustrating that parameter correlation could reduce significantly by using the D-optimality criterion. The two other experimental design criteria could not significantly improve the parameter precision in this specific case study.
Table 6 Values of the estimated pre-exponential factors and activation energies for the forward and backward steps in model 2 {θ = [lnkf,ref, 0.1Ea,f (kJ mol−1 K−1), lnkb,ref, 0.1Ea,b (kJ mol−1K−1)]T} and their corresponding statistics improving on application of MBDoE for parameter precision. Tref = 75 °C
| Design |
Before MBDoE |
After MBDoE |
|
Superscript asterisk (*) indicates t-value failing the t-test.
|
| Param. estimate |
θ
= [3.090, 0.997, 3.939, 1.096]T |
θ
= [3.031, 1.234, 3.871, 1.348]T |
| Conf. interval (95%) |
[±0.207, ±0.804, ±0.276, ± 0.9036]T |
[±0.078, ±0.235, ±0.167, ± 0.395]T |
|
t-valuea (tref = 1.78) |
[14.96, 1.24*, 14.25, 1.21*]T |
[38.99, 5.23, 23.14, 3.42]T |
|
χ
2
|
7.29 × 10−9(χ2ref = 21.03) |
3.03 (χ2ref = 26.30) |
 |
| | Fig. 12 Distribution of experimental information among the five experiments employed using RFI, the MBDoE experiment being the most informative experiment, hence improving the parameter statistics (panel A). D-optimal Fisher information map in the design space showing the location of the new experiment (panel B). | |
 |
| | Fig. 13 Normalized parameter covariance matrices in the amide synthesis calculated before (panel A) and after the MBDoE of different criteria: A- (panel B), D- (panel C), and E-optimality (panel D), D-optimality being the criterion that ensured parameter precision after one new experiment. | |
To validate the newly developed kinetic model, we employed new test data from experiments designed by a full factorial design of experiments generated at the control bounds (CBDoEs) of the experimental design space. Fig. 14 illustrates the corner locations of the validation experiments in the design space (panels A and B showing the yield and % RFI, respectively by the bubble size) while panel C shows the model performance at these corner conditions. All the model predictions are within the 95% confidence interval, that is, ±2σ (σ = 0.0003 mol dm−3).
 |
| | Fig. 14 Corner locations of the validation experiments (CBDoEs) as well previous experiments in the design space, yield indicated by the bubble size in panel A, % RFI indicated by the bubble size in panel B, and parity plot in panel C for the validation experiments and previous experimental designs showing all model predictions for c3 = RCONH2 within the experimental confidence interval (experimental error {= ±2σ; σ = 0.0003 mol dm−3} is also indicated). | |
4 Conclusions
In this work, we developed a novel cloud-based platform to remotely drive experimentation in a smart flow reactor situated in the University of Leeds using optimal experiment design algorithms operated from University College London. Communicating via the Dropbox cloud technology, the smart reactor, called the LabBot, receives experimental designs from the SimBot, a Python-based experimental design and data analysis software. Through automation, the LabBot sets the process conditions, conducts experimentation, and sends data files to the cloud.
SimBot, on the other hand, interrogates Dropbox for process data and employs kinetic modelling to simulate the experimental setup. Constituted of modules for preliminary DoE, parameter estimation, and model-based design of experiment (MBDoE), the SimBot software, initially starting with minimal experimental designs for preliminary parameter estimation, computes new optimal design of solutions as soon as a new measurement becomes available using online MBDoE techniques to improve model performance in real-time. Efficient numerical solvers for dynamic optimization and statistical analysis for model testing available in Python packages (CasADi, IPOPT, Scipy) have been used.
The SimBot, in tandem with the LabBot, autonomously identified synthesis chemistry in just a few experiments, as demonstrated on the two pharmaceutical case studies employed in this work. While in the first case study (nucleophilic aromatic substitution) the cloud-based platform identified well-known complex kinetics guaranteeing the minimum uncertainty on kinetic parameters, the platform supported the development of new kinetic models in the second case study (homogeneous amide formation).
Future work will explore other pharmaceutical systems to: 1) maximise information gained from experimental campaigns, 2) increase process understanding by minimising the number of required experiments, 3) advance the applicability of the developed cloud-based platform to reduce time and cost in process development as well as deliver on-demand in drug manufacturing by incorporating a Pilot Bot to scale up the optimised chemistry from the LabBot and 4) extend the communication protocol to multiple automated platforms that satisfy interoperability within the FAIR guiding principles for scientific data management.
Symbols
Latin symbols
|
c
i
|
ith species concentration |
|
E
a
| The activation energy |
|
g
| Rate master curve function |
|
k
0
| Arrhenius pre-exponential factor |
|
k
ref
| Reparametrized pre-exponential factor applicable at Tref |
|
k
obs
| Assumed first-order rate constant |
|
k
eq
| Equilibrium constant |
|
r
j
| Is the reaction rate (mol s−1 L−1) of the jth reaction |
|
R
| Universal gas constant equal to 8.31 kJ mol−1 K−1 |
|
t
| Time |
|
t
i
| Student-t distribution value |
|
T
| Reactor temperature |
|
v
ij
| The stoichiometric coefficient |
|
v
z
| Speed of fluid flow in z-direction |
|
V
| Volume of the reactor |
|
v
o
| Volumetric flowrate |
|
z
| Reactor axial coordinate |
Greek symbols
|
β
i
| Reaction order for component i |
|
χ
2
| Chi-square distribution value |
|
σ
2
kk
| Variance of the kth measured response and the kth diagonal entry of the measurement's variance–covariance |
|
τ
| Residence time |
|
λ
| Eigen value of covariance matrix V |
Matrices and vectors
|
c
∈ Nnc | Vector of the state variables (concentrations) |
|
u
∈ Nnu | Vector of manipulated variables |
|
V
y
| Measurement covariance matrix |
|
V
(, φ) | Expected marginal posterior covariance |
|
x
∈ Nx | Is the vector of state variables |
|
ŷ(τ) | Matrix of the model predictions of measurements 2-D in number of measurement response variables Ny and τ |
|
H
| Corresponds to the Fisher information matrix |
|
R
(, φ) | The correlation matrix |
|
θ
∈ Nnθ | Vector of the kinetic parameters to be identified, |
|
| Vector of estimated parameter values |
|
φ
| Design vector [uT,τ,cT0]T |
Acronym
| CBDoE: | Control-bounds DoE |
| DoE: | Design of experiment |
| MBDoE: | Model-based DoE |
| PDoE: | Preliminary DoE |
| RFI: | Relative Fisher information index |
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The project has received funding from EPSRC (EP/R032807/1). The support is gratefully acknowledged.
References
- Y. Li, L. Xia, Y. Fan, Q. Wang and M. Hu, Recent advances in autonomous synthesis of materials, ChemPhysMater, 2022, 1(2), 77–85, DOI:10.1016/j.chphma.2021.10.002 , ISSN 2772-5715.
- S. Kuehn, Pharmaceutical manufacturing: Current trends and what's next, Chem. Eng. Prog., 2018, 114, 23 Search PubMed , Retrieved from https://www.proquest.com/magazines/pharmaceutical-manufacturing-current-trends-whats/docview/2159927208/se-2.
- W. S. Yip and T. E. Marlin, The effect of model fidelity on real-time optimization performance, Comput. Chem. Eng., 2004, 28(1–2), 267–280, DOI:10.1016/S0098-1354(03)00164-9 , ISSN 0098-1354.
-
L. T. Biegler, Technology Advances for Dynamic Real-Time Optimization, in Computer Aided Chemical Engineering, ed. R. M. de Brito Alves, C. A. O. do Nascimento and E. C. Biscaia, Elsevier, 2009, vol. 27, pp. 1–6, ISSN 1570-7946, ISBN 9780444534729, DOI:10.1016/S1570-7946(09)70220-2.
- C. A. Hone, N. Holmes, G. R. Akien, R. A. Bourne and F. L. Muller, Rapid multistep kinetic model generation from transient flow data, React. Chem. Eng., 2017, 2, 103–108, 10.1039/D0RE00066C.
- A. Echtermeyer, Y. Amar, J. Zakrzewski and A. Lapkin, Self-optimisation and model-based design of experiments for developing a C–H activation flow process, Beilstein J. Org. Chem., 2017, 13, 150–163, DOI:10.3762/bjoc.13.18.
- M. Baumann, T. S. Moody, M. Smyth and S. Wharry, A Perspective on Continuous Flow Chemistry in the Pharmaceutical Industry, Org. Process Res. Dev., 2020, 24(10), 1802–1813, DOI:10.1021/acs.oprd.9b00524.
-
C. Yuangyai and H. B. Nembhard, Design of Experiments: A Key to Innovation in Nanotechnology, in Micro and Nano Technologies, Emerging Nanotechnologies for Manufacturing, ed. W. Ahmed and M. J. Jackson, William Andrew Publishing, 2010, pp. 207–234, DOI:10.1016/B978-0-8155-1583-8.00008-9.
-
T. Caliński and S. Kageyama, Block designs: A Randomization approach, Volume I: Analysis, in Lecture Notes in Statistics, Springer-Verlag, New York, 2000, vol. 150, ISBN 0-387-98578-6 Search PubMed.
- G. Franceschini and S. Macchietto, Model-based design of experiments for parameter precision: State of the art, Chem. Eng. Sci., 2008, 63(19), 4846–4872, DOI:10.1016/j.ces.2007.11.034 , ISSN 0009-2509.
-
D. C. Montgomery, Technology & Engineering, Design and Analysis of Experiments, John Wiley & Sons, 10th edn, 2020, ISBN: 978-1-118-14692-7 Search PubMed.
- C. Waldron, A. Pankajakshan, M. Quaglio, E. Cao, F. Galvanin and A. Gavriilidis, Closed-Loop Model-Based Design of Experiments for Kinetic Model Discrimination and Parameter Estimation: Benzoic Acid Esterification on a Heterogeneous Catalyst, Ind. Eng. Chem. Res., 2019, 58(49), 22165–22177, DOI:10.1021/acs.iecr.9b04089.
- J. P. McMullen and K. F. Jensen, Rapid determination of reaction kinetics with an automated microfluidic system, Org. Process Res. Dev., 2011, 15, 398–407, DOI:10.1021/op100300.
- M. Quaglio, C. Waldron, A. Pankajakshan, E. Cao, A. Gavriilidis, E. S. Fraga and F. Galvanin, An online reparameterization approach for robust parameter estimation in automated model identification platforms, Comput. Chem. Eng., 2019, 124, 270–284 CrossRef CAS.
- A. Pankajakshan, S. G. Bawa, A. Gavriilidis and F. Galvanin, Autonomous kinetic model identification using optimal experimental design and retrospective data analysis: methane complete oxidation as a case study, React. Chem. Eng., 2023, 8, 3000–3017, 10.1039/D3RE00156C.
- F. Galvanin, M. Barolo and F. Bezzo, Online model-based redesign of experiments for parameter estimation in dynamic systems, Ind. Eng. Chem. Res., 2009, 48, 4415–4427 CrossRef CAS.
- F. Destro and M. Barolo, A review on the modernization of pharmaceutical development and manufacturing – Trends, perspectives, and the role of mathematical modelling, Int. J. Pharm., 2022, 620, 121715, DOI:10.1016/j.ijpharm.2022.121715.
- J. Boonen, A. Bronselaer, J. Nielandt, L. Veryser, G. De Tré and B. De Spiegeleer, Alkamid database: Chemistry, occurrence and functionality of plant N-alkylamides, J. Ethnopharmacol., 2012, 142(3), 563–590, DOI:10.1016/j.jep.2012.05.038 , Epub 2012 May 30. PMID: 22659196.
- M. D. Wilkinson,
et al., The FAIR Guiding Principles for scientific data management and stewardship, Sci. Data, 2016, 3, 160018, DOI:10.1038/sdata.2016.18.
- O. J. Kershaw, A. D. Clayton, J. A. Manson, A. Barthelme, J. Pavey, P. Peach, J. Mustakis, R. M. Howard, T. W. Chamberlain, N. W. Warren and R. A. Bourne, Machine learning directed multi-objective optimization of mixed variable chemical systems, Chem. Eng. J., 2023, 451(Part 1), 138443, DOI:10.1016/j.cej.2022.138443.
- J. Wang and A. W. Dowling, Pyomo.DOE: An open-source package for model-based design of experiments in Python, AIChE J., 2022, 68(12) DOI:10.1002/aic.17813 , ISSN 1385-8947.
- J. Sansana, M. N. Joswiak, I. Castillo, Z. Wang, R. Rendall, L. H. Chiang and M. S. Reis, Recent trends on hybrid modeling for Industry 4.0, Comput. Chem. Eng., 2021, 151, 107365, DOI:10.1016/j.compchemeng.2021.107365 , ISSN 0098-1354.
- T. E. Oliphant, Python for Scientific Computing, Comput. Sci. Eng., 2007, 9(3), 10–20, DOI:10.1109/MCSE.2007.58.
-
E. Matthes, Python Crash Course: A Hands-On, Project-Based Introduction to Programming Paperback, No Starch Press, San Francisco, 25 Nov. 2015 Search PubMed.
- J. A. E. Andersson, J. Gillis, G. Horn, J. B. Rawlings and M. Diehl, CasADi: a software framework for nonlinear optimization and optimal control, Math. Program. Comput., 2019, 11, 1–36, DOI:10.1007/s12532-018-0139-4.
- P. Virtanen, R. Gommers, T. E. Oliphant, M. Haberland, T. Reddy, D. Cournapeau, E. Burovski, P. Peterson, W. Weckesser, J. Bright, S. J. van der Walt, M. Brett, J. Wilson, K. J. Millman, N. Mayorov, A. R. J. Nelson, E. Jones, R. Kern, E. Larson, C. J. Carey, I. Polat, Y. Feng, E. W. Moore, J. VanderPlas, D. Laxalde, J. Perktold, R. Cimrman, I. Henriksen, E. A. Quintero, C. R. Harris, A. M. Archibald, A. H. Ribeiro, F. Pedregosa, P. van Mulbregt and SciPy 1.0 Contributors, SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python, Nat. Methods, 2020, 17, 261–272, DOI:10.1038/s41592-019-0686-2.
- A. Wächter and L. Biegler, On the implementation of a primal-dual interior point filter line search algorithm for large-scale nonlinear programming, Math. Program., 2006, 106(1), 25–57 CrossRef.
-
N. P. Rougier, Scientific Visualization: Python + Matplotlib, 2021, 978-2-9579901-0-8. hal-03427242 Search PubMed.
-
A. Lee, pyDoE: The Experimental Design Package for Python. Pythpn Package Version 0.3, 2015, p. 8, https://pythonhosted.org/pyDOE/ Search PubMed.
-
T. Haslwanter, An Introduction to Statistics with Python With Applications in the Life Sciences, Springer Cham, 2022, DOI:10.1007/978-3-030-97371-1.
- W. Huyer and A. Neumaier, Snobfit – Stable Noisy Optimization by Branch and Fit, ACM Trans. Math. Softw., 2008, 35, 9, DOI:10.1145/1377612.1377613.
- D. R. Morrison, S. H. Jacobson, J. J. Sauppe and E. C. Sewell, Branch-and-bound algorithms: A survey of recent advances in searching, branching, and pruning, Discrete Optim., 2016, 19, 79–102, DOI:10.1016/j.disopt.2016.01.005 , ISSN 1572-5286.
- M. Schwaab, L. P. Lemos and J. C. Pinto, Optimum reference temperature for reparameterization of the Arrhenius equation. Part 2: Problems involving multiple reparameterizations, Chem. Eng. Sci., 2008, 63(11), 2895–2906, DOI:10.1016/j.ces.2008.03.010 , ISSN 0009-2509.
-
E. Hairer, S. P. Norsett and G. Wanner, Solving Ordinary Differential Equations i. Nonstiff Problems, Springer Series in Computational Mathematics, Springer-Verlag, 2nd edn, 1993 Search PubMed.
-
Y. Bard, Nonlinear parameter estimation, Academic Press, 1974 Search PubMed.
- M. D. McKay, R. J. Beckman and W. J. Conover, A Comparison of Three Methods for Selecting Values of Input Variables in the Analysis of Output from a Computer Code, Technometrics, 1979, 21(2), 239–245, DOI:10.2307/1268522.
- W. E. Stewart, Y. Shon and G. E. P. Box, Discrimination and goodness of fit of multiresponse mechanistic models, AIChE J., 1998, 44, 1404–1412 CrossRef CAS.
- G. Franceschini and S. Macchietto, Novel anticorrelation criteria for model-based experiment design: Theory and formulations, AIChE J., 2008, 54(8), 1009–1024, DOI:10.1002/aic.11429.
- S. P. Asprey and S. Macchietto, Statistical Tools for Optimal Dynamic Model Building, Comput. Chem. Eng., 2000, 24, 1261–1267 CrossRef CAS.
-
T. V. Daele, S. V. Hoey and I. Nopens, pyIDEAS: an Open Source Python Package for Model Analysis, in Computer Aided Chemical Engineering, ed. K. V. Gernaey, J. K. Huusom and R. Gani, Elsevier, 2015, vol. 37, pp. 569–574, ISSN 1570-7946, ISBN 9780444634290, DOI:10.1016/B978-0-444-63578-5.50090-6.
-
F. Galvanin, E. Cao, N. Al-Rifai, A. Gavriilidis and V. Dua, Model-based design of experiments for the identification of kinetic models in microreactor platforms, in Computer Aided Chemical Engineering, ed. K. V. Gernaey, J. K. Huusom and R. Gani, Elsevier, 2015, vol. 37, pp. 323–328, ISSN 1570-7946, ISBN 9780444634290, DOI:10.1016/B978-0-444-63578-5.50049-9.
- F. Galvanin, E. Cao, N. Noor Al-Rifai, A. Gavriilidis and V. Dua, A joint model-based experimental design approach for the identification of kinetic models in continuous flow laboratory reactors, Comput. Chem. Eng., 2016, 95, 202–215, DOI:10.1016/j.compchemeng.2016.05.009 , ISSN 0098-1354.
- P. Petsagkourakis and F. Galvanin, Safe model-based design of experiments using Gaussian processes, Comput. Chem. Eng., 2021, 151, 107339, DOI:10.1016/j.compchemeng.2021.107339.
-
L. T. Biegler, Nonlinear Programming – Concepts, Algorithms, and Applications to Chemical Processes, Computer Science, MOS-SIAM Series on Optimization, 2010 Search PubMed.
- R. Metz, How Dropbox Could Rule a Multi-Platform World, MIT Technology Review, 2013 Search PubMed , https://www.technologyreview.com/2013/07/09/177416/how-dropbox-could-rule-a-multi-platform-world/ [Accessed 6 July 2023].
- A. M. Schweidtmann, A. D. Clayton, N. Holmes, E. Bradford, R. A. Bourne and A. A. Lapkin, Machine learning meets continuous flow chemistry: Automated optimization towards the Pareto front of multiple objectives, Chem. Eng. J., 2018, 352, 277–282, DOI:10.1016/j.cej.2018.07.031 , ISSN 1385-8947.
- C. A. Hone, A. Boyd, A. O'Kearney-McMullan, R. A. Bourne and F. L. Muller, Definitive screening designs for multistep kinetic models in flow, React. Chem. Eng., 2019, 4, 1565–1570, 10.1039/C9RE00180H.
- A. G. O'Brien, Z. Horváth, F. Lévesque, J. W. Lee, A. Seidel-Morgenstern and P. H. Seeberger, Continuous Synthesis and Purification by Direct Coupling of a Flow Reactor with Simulated Moving-Bed Chromatography, Angew. Chem., Int. Ed., 2012, 51, 7028–7030, DOI:10.1002/anie.20120279.
- M. D. Wendt and A. R. Kunzer, Ortho-selectivity in SNAr substitutions of 2,4-dihaloaromatic compounds, Reactions with anionic nucleophiles, Tetrahedron Lett., 2010, 51(23), 3041–3044, DOI:10.1016/j.tetlet.2010.03.124 , ISSN 0040-4039.
- D. G. Brown and J. Boström, Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reactions Gone?, J. Med. Chem., 2016, 59(10), 4443–4458, DOI:10.1021/acs.jmedchem.5b01409.
- N. Nakajima and Y. Ikada, Mechanism of Amide Formation by Carbodiimide for Bioconjugation in Aqueous Media, Bioconjugate Chem., 1995, 6, 123–130 CrossRef CAS PubMed.
-
J. Clark, S. Farmer, D. Kennepohl and L. Morsch, Chemistry of Amides, shared under a CC BY-SA 4.0 license, 2022 Search PubMed.
-
P. Atkins and J. De Paula, Physical Chemistry, W. H. Freeman and Company, 8th edn, 2006, p. 212, ISBN 978-0-7167-8759-4 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Panagiotis
Petsagkourakis
a,
Muhammad
Yusuf
b,
Ricardo
Labes
b,
Thomas
Chamberlain
a,
Panagiotis
Petsagkourakis
a,
Muhammad
Yusuf
b,
Ricardo
Labes
b,
Thomas
Chamberlain


 .
.





 confidence level with NexpNy − Nθ degrees of freedom, and its corresponding tref is t(1 − α, NexpNy − Nθ). Vii is the ith diagonal entry of the parameter variance-covariance matrix V computed using the estimated parameter and expressed as:
confidence level with NexpNy − Nθ degrees of freedom, and its corresponding tref is t(1 − α, NexpNy − Nθ). Vii is the ith diagonal entry of the parameter variance-covariance matrix V computed using the estimated parameter and expressed as: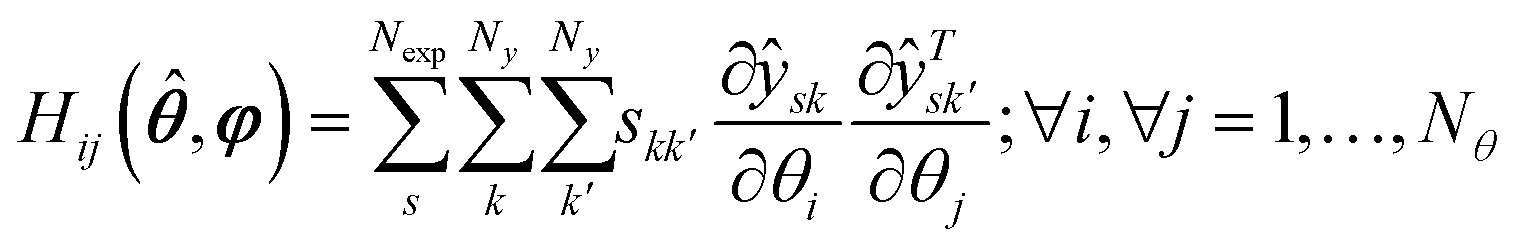
 is the parameter sensitivity of ŷsk the kth entry of ŷ at the sth experiment with respect to θi. It should be noted the inverse of the variance–covariance matrix corresponds to the Fisher information matrix H.
is the parameter sensitivity of ŷsk the kth entry of ŷ at the sth experiment with respect to θi. It should be noted the inverse of the variance–covariance matrix corresponds to the Fisher information matrix H.









 (18)
(18)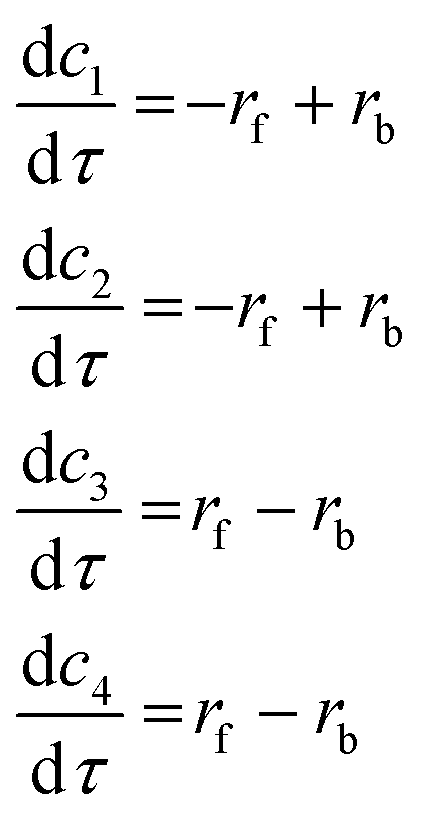 (19)
(19)





