
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidative cleavage of lignin model substrates with Co(salen) catalyst: an experimental study on the effect of different reaction parameters in batch and continuous flow†
Jonas
Mortier
 a,
Christian V.
Stevens
a,
Joseph J.
Bozell
b and
Thomas S. A.
Heugebaert
*a
a,
Christian V.
Stevens
a,
Joseph J.
Bozell
b and
Thomas S. A.
Heugebaert
*a
aDepartment of Green Chemistry and Technology, Faculty of Bioscience Engineering, Ghent University, Coupure Links 653, 9000 Ghent, Belgium. E-mail: Thomas.Heugebaert@UGent.be
bCenter for Renewable Carbon, University of Tennessee, 2506 Jacob Drive, Knoxville, TN 37996, USA
First published on 7th March 2024
Abstract
This study investigates the Co(salen)-catalyzed oxidative cleavage of monomeric lignin model substrates to benzoquinones in a continuous flow system and maps the impact of various reaction parameters on the selectivity and yield of the cleavage. Our findings highlight the crucial role of precise oxygen dosing and its interplay with product solubility to achieve a successful reaction. Exposing the substrates to excess oxygen in a continuous flow system resulted in lower yields, while product precipitation was shown to be crucial in batch systems. Additionally, we explored the effects of added bases, oxygen pressure, solvents, and reaction time in a batch set-up. Overall, this work presents a valuable overview of what conditions are favourable when conversion towards benzoquinones is desired and what conditions should be avoided.
Introduction
Lignin is a renewable feedstock that is abundantly present in the cell wall of different plant cells. Depending on the biomass source, it is formed via polycondensation of coniferyl alcohol (G), p-coumaryl alcohol (H) or sinapyl alcohol (S) in different ratios. Despite the potentially valuable phenolic moieties present in the structure, valorization techniques are still limited and most lignin is burned as fuel.1 In native lignin, most aromatic units are linked through β-O-4 ether bonds. (Fig. 1) Most literature research dedicated to lignin depolymerization, aims to effectively cleave these ether bonds yielding various aromatic compounds.2–6 These compounds can then undergo hydrodeoxygenation to obtain a high-quality bio-oil.7 | ||
| Fig. 1 Reaction sites of Co(salen)-mediated cleavage and traditional β-O-4 cleavage. | ||
Although β-O-4 ether bonds account for the majority of the interunit linkages in native lignin, biorefining of biomass results in many of these β-O-4 ether bonds being broken.8
Further, native lignin consists of approximately 10% free phenolic groups.9 However, after lignin is removed from its matrix via various biorefining processes, the amount of free phenolic units can increase to up to 70% of the total aromatic units.10 Focusing on transforming these substituted phenol groups would more accurately address the challenge of industrial lignin valorization.
A range of Co-Schiff base complexes (e.g. Co(salen), 1) in the presence of O2 have been shown to catalyze the oxidative cleavage of C-C bonds between the α-carbon and the aromatic ring in phenolic lignin model substrates (2: H model, 3: G model and 4: S model), yielding benzoquinones 5, 7 and 9.11–14 Latter benzoquinones can be used for the synthesis of anthraquinones through diels-alder reaction (H2O2 production),15 precursor for hydroquinones (polyether ether ketone synthesis),16 polyaminoquinones (coatings, adhesion agent)17 or as high value intermediate for other applications.18 The proposed mechanism of the oxidation starts with the activation of the Co(salen) complex with triplet oxygen, forming a Co(III) superoxo complex, which will initiate the reaction (Scheme 1, a). Initial coordination of O2 typically requires the presence of a coordinating base as an axial ligand, for example, pyridine, to form the superoxo complex.19 The superoxo complex abstracts a phenolic hydrogen from the lignin model, forming a phenoxy radical (Scheme 1, b). This radical then reacts towards the desired benzoquinones, breaking the C–C bond and forming an intermediate cobalt hydroxo species (Scheme 1, cI). A competing pathway is the formation of the corresponding aldehydes, where an intermediate cobalt hydroperoxo species is formed. (Scheme 1, cII).
 | ||
| Scheme 1 Proposed reaction mechanism of Co(salen)-catalyzed oxidation of lignin model substrates towards benzoquinones and benzaldehydes, via (a) oxygen uptake by Co(salen); (b) phenolic hydrogen abstraction; and (cI) C–C cleavage or (cII) benzaldehyde formation. | ||
Previous experiments have shown that the oxidation can be highly effective for S lignin models, leading to nearly quantitative yields of the corresponding dimethoxybenzoquinone from model substrate 4 within 1 hour of reaction time. Oxidative conversion of the less reactive G-type substrates 3 to the corresponding monomethoxybenzoquinone however, only occurs in 68% yield with a reaction time of 22 hours when enhanced by the addition of a non-coordinating base, such as N,N-diisopropylethylamine (DIPEA).19 It was proposed that the non-coordinating base abstracts the phenolic proton, affording a more easily oxidized phenoxide anion.20 H type model substrates such as 2 were not converted to the corresponding benzoquinone in any of the experiments.
Batch processes can control time, temperature, concentration, catalyst level, etc. However, dosing oxygen while maintaining the pressure during a batch reaction would be difficult. Continuous flow chemistry with its enhanced gas–liquid mass transfer,21 could improve these results. With these intensified oxidation conditions and the ability to monitor the oxygen supply towards the reaction more precisely, we aimed to improve overall productivity of lignin model oxidation, eventually leading to improved oxidation of lignin itself. Accordingly, this research compares Co-catalyzed aryl-Cα cleavage of monomeric lignin models vanillyl alcohol 3 and syringyl alcohol 4 under both continuous flow and conventional batch conditions to better understand key reaction parameters in both systems.
Results and discussion
G type model substrate in continuous flow
Continuous flow operation was first evaluated for vanillyl alcohol 3 (VA), using the set-up described in the ESI.† As this set-up requires that all compounds remain in solution during the reaction to avoid clogging of the reactor tubing, proper solvent selection was needed. It was observed that Co(salen) dissolved well in dichloromethane, but VA 3 did not. A 4/1 DCM/alcohol (MeOH or EtOH) mixture ensured that the catalyst, VA 3 and methoxybenzoquinone product 5 (MBQ) remained dissolved. In addition, we observed that replacing EtOH with MeOH in the solvent mixture increases the product yield. Further, higher yields were obtained adding DIPEA to the reaction mixture. These first experiments displayed promising results, approaching the 68% yield obtained in the current best batch process while lowering the reaction time from 22 hours to 45 minutes.19 (Table 1, entries 1–4).|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Base (0.1 eq.) | O2 (eq.) | Conversiond (%) | MBQ 5 (%) |
| a Conversion = 100 – isolated starting material (%). b Isolated yield. c Yield obtained via quantitative HPLC analysis of crude reaction mixture. d Yield obtained via1H-NMR integration, unless specified otherwise. | |||||
| 1 | DCM/EtOH (4/1) | Pyridine | 4.46 | 81a | 42b |
| 2 | DCM/EtOH (4/1) | DIPEA | 4.46 | 100 | 57b |
| 3 | DCM/MeOH (4/1) | DIPEA | 4.46 | 100 | 64b |
| 4 | DCM/MeOH (4/1) | DIPEA + pyridine | 4.46 | 100 | 64b/73c |
| 5 | DCM/MeOH (4/1) | DIPEA + pyridine | 2.82 | 100 | 87c |
| 6 | DCM/MeOH (4/1) | DIPEA + pyridine | 2.62 | 100 | 94 |
| 7 | DCM/MeOH (4/1) | DIPEA + pyridine | 2.45 | 92 | 89c |
| 8 | DCM/MeOH (4/1) | DIPEA + pyridine | 2.23 | 78 | 76c |
| 9 | DCM/MeOH (4/1) | DIPEA | 2.73 | 100 | 74c |
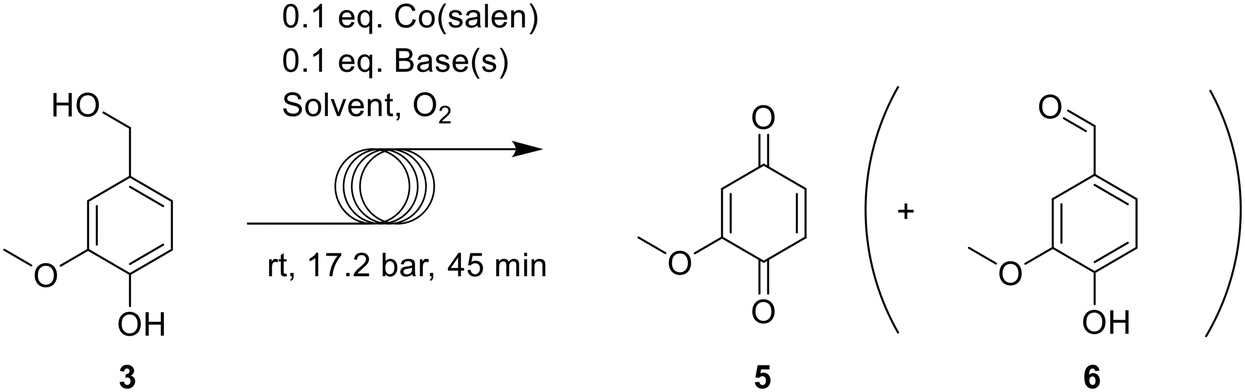
Importantly, lowering the amount of oxygen gave significantly higher yields for substrate 3 than those previously reported19 (Table 1, entries 4–8). Addition of 2.62 equivalents of oxygen was found to be optimal, resulting in a yield of 94%. A further reduction to 2.45 maintained the observed selectivity, but incomplete conversion was once again observed. Repeating the optimal conditions using only DIPEA as base gave lower yields, proving that the combination of both bases performs better (entry 9). As such, controlled oxygen dosing proved to be of utmost importance to ensure reaction selectivity. For G model substrates, the corresponding aldehyde 6 was never formed in significant amounts, but traces of the product could be seen via1H-NMR analysis.
S type model substrate in continuous flow
In contrast to the more recalcitrant G model, literature indicates that syringyl alcohol 4 can be easily converted to 2,6-dimethoxybenzoquinone 7 (DMBQ) in batch. Within 1 hour of reaction time when using pyridine as additive in methanol, quantitative yields were reached.19 Performing this reaction in a continuous flow system, however, required the use of a DCM/MeOH (4/1) mixture to maintain solubility of the reagents and DMBQ product. The experiments showed that, in contrast to the batch reaction, a significant amount of syringaldehyde 8 (SAld) is obtained. An interesting observation was the drop of 46% to 39% DMBQ yield when increasing the reaction time from 10 to 45 minutes, while the yield of Sald increased from 7 to 30%. As the reaction proceeds, the selectivity shifts towards SAld away from DMBQ. However, a conversion of DMBQ towards SAld was excluded. In fact, under the applied conditions syringaldehyde 8 was converted in low yields towards DMBQ 7 (entry 6). A later batch experiment using formaldehyde (which is released in the reaction medium during the oxidation) as a one-carbon synthon additive showed that the opposite reaction did not occur (entry 7). These observations indicate a slow degradation of the initially formed benzoquinone and in addition, since full conversion was not achieved in 10 minutes, a selectivity shift towards syringaldehyde for the remaining substrate. A possible explanation for the latter is the occurrence of product inhibition, as it was shown that dissolved quinone products could inhibit the desired oxidation process.22 (Table 2, entries 3–4).|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Starting product | Base | O2 (eq.) | Pressure (bar) | Reaction time (min) | DMBQ 7a (%) | SAld 8a (%) |
| a Yield obtained via quantitative HPLC analysis of crude reaction mixture. b No full conversion. c One equivalent of formaldehyde was added to this reaction as possible carbon source to form syringaldehyde since former product is obtained after cleavage of syringyl alcohol in the proposed mechanism and can thus play a role in the interconversion (Scheme 1, cI). d Reaction performed in batch with a high-pressure Parr reactor. e Reaction performed with only 0.7 eq. of substrate instead of 1 eq. to avoid precipitation problems. | |||||||
| 1 | 4 | DIPEA | 2.73 | 17.2 | 45 | 42 | 27 |
| 2 | 4 | DIPEA | 2.73 | 17.2 | 10 | 45 | 28 |
| 3 | 4 | DIPEA + pyridine | 2.73 | 17.2 | 45 | 39 | 30 |
| 4b | 4 | DIPEA + pyridine | 2.73 | 17.2 | 10 | 46 | 7 |
| 5 | 4 | DIPEA + pyridine | 2.73 | 5.1 | 45 | 48 | 24 |
| 6 | 8 | DIPEA + pyridine | 2.73 | 17.2 | 45 | 8 | 91 |
| 7c,d | 7 | DIPEA + pyridine | — | 17.2 | 60 | 100 | 0 |
| 8e | 7 | DIPEA + pyridine | 2.73 | 17.2 | 45 | 100 | 0 |

To investigate the extent of product degradation in the flow set-up, a degradation experiment was performed starting from pure DMBQ. However, no degradation of the benzoquinone occurred. (entry 8) As such, the exact nature of the drop in q-HPLC yields of DMBQ upon increasing reaction times (entries 3–4) remains somewhat ambiguous. Although degradation when utilizing pure DMBQ under these conditions could not be proven, it should be noted that intermediate complexes formed when starting from syringyl alcohol19 were not considered in these experiments.
When compared to the fast and selective batch process, the most likely reason for the lower yields and altered selectivity in the flow reactor is the solvent choice. The use of methanol in a batch process, which precipitates the poorly soluble 2,6-dimethoxybenzoquinone 7 (DMBQ) as soon as it is formed, could avoid selectivity alteration. In continuous flow, however, homogeneous process conditions are imperative.
H type model substrate in continuous flow
A third phenol derivative relevant to lignin models is 4-hydroxybenzyl alcohol 2. In literature, conversion of 2 to 1,4-benzoquinone (BQ) generally delivers no quinone product even after 22 hours of reaction time.19 The reason for the low conversion is due to the higher activation energy needed for the oxidation compared to the other model substrates,19 as well as the pronounced capacity to deactivate the catalyst of this substrate.22Although the H subunit rarely makes up more than 5% of the total subunits present in different types of plants,23 we briefly examined the reactivity of 2 under continuous flow conditions. When exposing 4-hydroxybenzyl alcohol to the same conditions as the previously discussed for vanillyl alcohol, a modest but unprecedented 10% yield was obtained (Scheme 2).
 | ||
| Scheme 2 Reaction of 4-hydroxybenzyl alcohol towards 1,4-benzoquinone in continuous flow. aYield obtained via quantitative HPLC analysis of crude reaction mixture. | ||
Comparative oxidation of lignin model substrates under batch conditions
Given the ultimate interest in lignin oxidation, conditions that result in high yielding conversion of both S and G model substrates are desirable. Given our newly established observations that careful oxygen dosing is crucial for G model oxidation, and precipitation of DMBQ helps drive effective S model substrate oxidation, we decided to examine batch reaction conditions that may combine the strengths of both processes, thus increasing overall product yield. Although dosing the amount of oxygen is more challenging in batch, we established that careful control of oxygen pressure and reaction time is essential. These parameters, however, have received surprisingly little attention in current literature. In parallel, we also examined a range of different solvent mixtures and added bases to control or enhance DMBQ precipitation.S type model substrate in batch
In batch, previous work showed that nearly quantitative yields of DMBQ are obtained from 4 as an insoluble solid within 60 minutes in methanol.19 To perform the batch reactions, a high pressure Parr reactor was used. Switching the set-up made it possible to change solvent mixtures, to an extent that product precipitation occurs. This type of reactor also makes it possible to investigate the impact of different oxygen pressures.In contrast to G model substrates, S model substrates can be transformed to a significant amount of corresponding aldehyde 8 with respect to DMBQ 7. Starting from the homogeneous DCM/MeOH (4/1) solvent mixture used in our flow experiments, we examined how varying the oxygen pressure over a range of 3.5–60 bar affects the oxidation process (Table 3, entries 1–5). Increasing the O2 pressure, consistently reduced the amount of aldehyde in the final product mixture. However, this reduction is not accompanied by an increase in benzoquinone yield, which remained relatively constant over these experiments. Whether the improved product ratio is due to faster breakdown of the aldehyde, or a higher intrinsic selectivity for the formation of DMBQ remains unclear, but syringaldehyde yield clearly dropped as the reaction time was increased, and is accompanied by a small increase of the quinone yield (entry 3b).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Base | Pressure (bar) | DMBQ 7a (%) | SAld 8a (%) |
| a Yield obtained via quantitative HPLC analysis of crude reaction mixture. | |||||
| 1 | DCM/MeOH (4/1) | DIPEA + pyridine | 3.5 | 76 | 22 |
| 2 | DCM/MeOH (4/1) | DIPEA + pyridine | 10 | 73 | 21 |
| 3a | DCM/MeOH (4/1) | DIPEA + pyridine | 17 | 78 | 18 |
| 3b | 81 (17 h) | 13 (17 h) | |||
| 4 | DCM/MeOH (4/1) | DIPEA + pyridine | 35 | 77 | 13 |
| 5 | DCM/MeOH (4/1) | DIPEA + pyridine | 60 | 79 | 9 |
| 6 | DCM/MeOH (3/2) | DIPEA + pyridine | 17 | 84 | 6 |
| 7 | DCM/MeOH (2/3) | DIPEA + pyridine | 17 | 91 | Traces |
| 8 | DCM/MeOH (1/4) | DIPEA + pyridine | 17 | 94 | Traces |
| 9 | MeOH | DIPEA + pyridine | 17 | 97 | 0 |
| 10 | MeOH | Pyridine | 17 | 99 | 0 |
| 11 | MeOH | None | 17 | 38 | 21 |

It can be noted that this batch reaction already outperformed the continuous flow system under similar conditions. However, even though acceptable yields for 2,6-DMBQ were obtained (73–81%), the previously reported quantitative yields (MeOH, 4 bar, 1 h) were far from reached. The observation that an increase in DCM results in lower DMBQ yield is in accordance with prior research where other cobalt catalysts were used for the conversion of syringyl alcohol.12 Since precipitation no longer restricts the solvent choice in a batch system, its effect on these reactions at elevated pressures was further investigated. When changing the solvent mixture to DCM/MeOH (3/2), precipitation of benzoquinone 7 was observed at the bottom of the reactor. The experiments showed that increasing the amount of MeOH drastically increases the selectivity towards the desired DMBQ while also increasing the yield. When pure MeOH is used as solvent, no more aldehyde is observed and DMBQ is formed with a yield of 97% (entries 3, 6–9). For this model substrate, the solvent plays a major role in both the selectivity and the yield of the reaction, once again confirming product induced inhibition of the catalytic system. Importantly, the pressure increase to 17 bar as compared to literature is not detrimental to the isolated yield. When pyridine is used without DIPEA (entry 10), near quantitative results are obtained which is in accordance with previous results.19 Using no base (entry 11) results in low yields for DMBQ while significant amounts of syringaldehyde are formed, an effect which was previously observed by Bozell et al.19
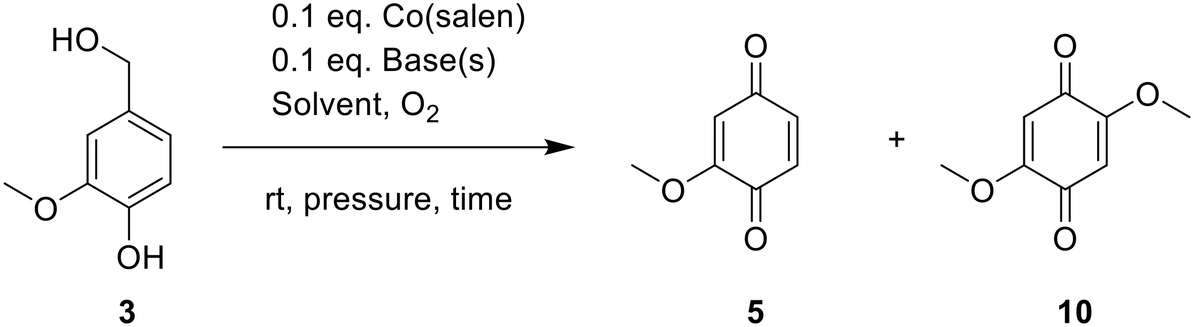
G type model substrate in batch
To allow for an efficient simultaneous oxidation of G and S model substrates, we next examined the effect of different reaction parameters on oxidation of substrate 3 in batch. The optimized reaction conditions of the continuous flow reaction (Table 1, entry 7) were mimicked in the batch set-up, and intermediate samples were taken periodically to evaluate the reaction rate. To our surprise, an MBQ yield of 80% was obtained after only 5 minutes, and full conversion of starting product was observed after 30 minutes, resulting in a qHPLC yield of 97%. These observations were very promising as literature reports required 22 hours of stirring to afford only 68% yield.19 The overtime decrease in MBQ yield at longer reaction times (higher oxygen levels) was less pronounced as compared to the flow experiments, and only 3% yield was lost after 19 hours. (Table 4. entry 1) Furthermore, the influence of the added bases was examined when DCM/MeOH (4/1) was used as solvent. The main observation of these screening experiments is that the combination of DIPEA and pyridine works better than either base individually. (Table 4, entries 1, 3, 5, 7) It looks like the combination of both additives simultaneously enhances the oxygen uptake of Co(salen) via axial coordination of the pyridine to the Co center in parallel to deprotonation of the starting product to the more reactive phenoxide anion.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Base | Reaction time | Conversionb (%) | MBQ 5a (%) | 2,5-DMBQ 10b (%) |
| a Yield obtained via quantitative HPLC analysis of crude reaction mixture. b Conversion obtained via1H-NMR integration. | ||||||
| 1 | DCM/MeOH (4/1) | DIPEA + pyridine | 5 min | 80 | 80 | 0 |
| 10 min | 92 | 85 | 0 | |||
| 20 min | 97 | 91 | 0 | |||
| 30 min | 100 | 97 | 0 | |||
| 3 h | 100 | 97 | 0 | |||
| 19 h | 100 | 94 | 0 | |||
| 2 | MeOH | DIPEA + pyridine | 30 min | 86 | 21 | 12 |
| 1 h | 97 | 14 | 10 | |||
| 2 h | Traces left | 12 | 10 | |||
| 3 | DCM/MeOH (4/1) | DIPEA | 30 min | 100 | 75 | 0 |
| 1 h | 100 | 81 | 0 | |||
| 4 h | 100 | 83 | 0 | |||
| 4 | MeOH | DIPEA | 30 min | 100 | 26 | 9 |
| 1 h | 100 | 24 | 11 | |||
| 17 h | 100 | 7 | 6 | |||
| 5 | DCM/MeOH (4/1) | Pyridine | 30 min | 93 | 72 | 0 |
| 1 h | 95 | 80 | 0 | |||
| 4 h | 100 | 77 | 0 | |||
| 6 | MeOH | Pyridine | 30 min | 83 | 67 | 3 |
| 1 h | 86 | 68 | 3 | |||
| 2 h | 87 | 71 | 3 | |||
| 17 h | Traces left | 68 | 5 | |||
| 7 | DCM/MeOH (4/1) | None | 30 min | 87 | 53 | 0 |
| 1 h | 91 | 64 | 0 | |||
| 8 | MeOH | None | 30 min | 92 | 85 | Traces |
| 1 h | 95 | 86 | Traces | |||

Conversely, using MeOH as the solvent, several different trends were observed. Under additive-free conditions, better yields of MBQ were observed (entry 8 vs. 7). It is known that methanol itself can act as an axial ligand on the Co(salen) complex to enhance the oxygen uptake properties which could explain why high yields are obtained without additives.12 MeOH in combination DIPEA however, promotes product degradation over time. (entries 2, 4, 6, 8) 1H-NMR analysis also indicated the presence of 2,5-DMBQ 10 as a side product. This compound was only formed when MeOH was used as solvent and its formation was increased when basic additives were present.
We also examined the influence of solvent ratios when using pyridine and DIPEA as additives. A few important trends could be observed. Firstly, using more MeOH lowers the overall yield while also slowing the conversion. Secondly, the more MeOH present, the faster the decline in yield of MBQ. These experiments again indicate that MeOH promotes product degradation in the presence of additives, while DCM does not. Lastly, a higher MeOH content in the solvent mixture results in more 2,5-DMBQ formation (Table 5, entries 1–5).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Solvent | Base | Pressure (bar) | Reaction time | Conversionb (%) | MBQ 5a (%) | 2,5-DMBQ 10b (%) |
| a Yield obtained via quantitative HPLC analysis of crude reaction mixture. b Yield obtained via1H-NMR integration. | |||||||
| 1 | DCM/MeOH (4/1) | DIPEA + pyridine | 17 | 5 min | 80 | 80 | 0 |
| 10 min | 92 | 85 | 0 | ||||
| 20 min | 97 | 91 | 0 | ||||
| 30 min | 100 | 97 | 0 | ||||
| 3 h | 100 | 97 | 0 | ||||
| 19 h | 100 | 94 | 0 | ||||
| 2 | DCM/MeOH (3/2) | DIPEA + pyridine | 17 | 30 min | 100 | 74 | 3 |
| 1 h | 100 | 69 | 3 | ||||
| 3 | DCM/MeOH (2/3) | DIPEA + pyridine | 17 | 30 min | Traces left | 48 | 4 |
| 1 h | 100 | 41 | 6 | ||||
| 2 h | 100 | 35 | 6 | ||||
| 4 | DCM/MeOH (1/4) | DIPEA + pyridine | 17 | 30 min | 93 | 27 | 7 |
| 1 h | 98 | 22 | 7 | ||||
| 5 | MeOH | DIPEA + pyridine | 17 | 30 min | 86 | 21 | 12 |
| 1 h | 97 | 14 | 10 | ||||
| 2 h | Traces left | 12 | 10 | ||||
| 6 | MeOH | Pyridine | 17 | 30 min | 83 | 67 | 3 |
| 1 h | 86 | 68 | 3 | ||||
| 2 h | 87 | 71 | 3 | ||||
| 17 h | Traces left | 68 | 5 | ||||
| 7 | MeOH | Pyridine | 10 | 30 min | 72 | 44 | 6 |
| 1 h | 80 | 50 | 7 | ||||
| 8 | MeOH (our catalyst) | Pyridine | 3.5 | 30 min | 54 | 31 | 6 |
| 1 h | 62 | 38 | 7 | ||||
| 18 h | 87 | 39 | 9 | ||||
| 9 | MeOH (commercial catalyst) | Pyridine | 3.5 | 30 min | 80 | 25 | 9 |
| 1 h | 89 | 25 | 11 | ||||
| 18 h | 98 | 20 | 6 | ||||
Another interesting observation was the ability of pyridine to convert the G lignin model to MBQ, since literature suggested that a non-coordinating base was needed to achieve conversion.19 When the reaction was performed with pyridine as additive in methanol in literature, 0% yield was achieved after 22 hours of reaction time while our observation proved that lower yields were obtained after 17 hours of reaction time (entry 6).19 The only difference in our reaction set-up is the use of a higher pressure (17 vs. 3.5 bar). By testing the reaction at various pressures, it was, as expected, found that the reaction rate declined as the pressure decreased, and lower overall yields were obtained. When performing the reaction at 3.5 bar however, we still observed moderate yields after 18 hours of reaction time, which does not match the literature observation (entries 6–8). As a potential explanation, the performance of the home-made Co(salen) catalyst (which was used for all previous reactions) was compared to commercially available Co(salen). Commercially available catalyst demonstrated a marked difference in outcome with a faster conversion of the starting material but a decreased MBQ yield (20% vs. 39%) after 1 hour, thus displaying a much poorer selectivity for MBQ (entries 8–9). The exact difference between these two catalysts could not be pinpointed.
Given the large solvent effects observed, and in an attempt to move away from the halogenated solvent DCM, a range of greener solvents were evaluated as well. Although some attempts gave promising results (up to 51% yield) none could match the yields obtained when DCM was used. The most promising case could be made for CH3CN mixtures containing polar co-solvents (EtOH, cyrene). (Table S1, ESI†).
H type model substrate in batch
Since S and G type lignin models are most abundantly present in plants, less attention was given to H type lignin model substrate 4-hydroxybenzyl alcohol. Nonetheless, continuous flow experiments showed that some conversion to the corresponding benzoquinone 9 occurred, in contrast with current literature conditions, where no conversion was observed. The same reaction conditions as in the continuous flow experiment were utilized and a surprisingly high yield of 41% was obtained after 17 hours of reaction time. To obtain higher yields, the pressure was increased to 50 bar. Unfortunately, the same substrate conversion was obtained while the yield of 1,4-benzoquinone (BQ) dropped, indicating product degradation. The reaction was repeated in methanol, but in this case only traces of product were formed. (Table 6).|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Base | Pressure (bar) | Reaction time | BQ 9a (%) |
| a Yield obtained via quantitative HPLC analysis of crude reaction mixture. b Conversion obtained via1H-NMR integration. c Formation of 2,5-DMBQ was observed via1H-NMR. | |||||
| 1 | DCM/MeOH (4/1) | DIPEA + pyridine | 17 | 30 min | 15 |
| 1 h | 24 | ||||
| 17 h | 41 (52b conversion) | ||||
| 2 | DCM/MeOH (4/1) | DIPEA + pyridine | 50 | 18 h | 31 (52b conversion) |
| 3 | MeOH | DIPEA + pyridine | 17 | 18 h | Tracesc |
| 4 | MeOH | Pyridine | 17 | 18 h | Tracesc |

Organosolv lignin in batch
After examining the cleavage in model substrates, it was examined how lignin would react to these conditions. As substrate, commercially available organosolv lignin was used and a number of reaction conditions were tested. The reaction mixture was analyzed by quantifying the low weight aromatic fraction after column chromatography. Only the presence of benzoquinones and aldehydes derived from model substrates was considered. (Table 7).|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Base | Reaction time | Products |
| a Yield obtained via quantitative HPLC analysis of crude reaction mixture. b Yield obtained via1H-NMR integration. c Not detected via HPLC analysis, traces seen on 1H-NMR. d Detected via1H-NMR analysis. | ||||
| 1 | DCM/MeOH (4/1) | DIPEA (32 mg)+ pyridine (20 mg) | 22 h | 7 (3.4 w%)a |
| 8 (1.1 w%)b | ||||
| 6 (0.8 w%)b | ||||
| 2 | MeOH | DIPEA (32 mg) + pyridine (20 mg) | 18 h | 7 (1.1 w%)a |
| 6 (0.3 w%)b | ||||
| 8 (0.2 w%)b | ||||
| 5 (0.2 w%)b | ||||
| 3 | MeOH | Pyridine (20 mg) | 18 h | 7 (0.9 w%)a |
| 8 (0.5 w%)b | ||||
| 6 (0.4 w%)b | ||||
| 4 | Aqueous NaOH (0.04 M) | — | 17 h | 7 (traces)c |
| 8 (traces)d | ||||
| 6 (traces)d | ||||

The main product obtained after the reaction was 2,6-DMBQ 7 with a maximum yield of 3.4% (w/w). Minor products also present were syringaldehyde 8, MBQ 5 and vanillin 6, albeit in lower concentrations, resulting in a total depolymerization yield of 5.3% (m/m). It should be noted that while the total yield remains low, the simplest of Co–salen catalysts under our improved operational conditions performs beyond the most advanced reported catalyst derivatives under standard conditions (3.5% mass yield from a cyclohexyl diamine linked Co–salen complex, aptly decorated with benzylated piperazines, 72 h, rt, 3.4 bar O2, MeOH/DMSO).11 Given the fast conversions of the model substrates, temperature effects were not included in the original screening of the reaction conditions. When dealing with polymeric lignin fragments, however increased conversion may be achieved at higher temperatures. Entries 1–3 were repeated at 70 °C (Table S3, ESI†), but unfortunately lower overall yields were obtained while no significant change in selectivity occurred. Reduced oxygen solubility may be at the root of this observation.
Conclusions
A continuous flow approach of the Co(salen)-catalyzed oxidation of lignin model substrates was examined because it was believed that intensified oxidation conditions and especially the ability to control the oxygen dosing were vital parameters to limit suspected degradation and obtain higher yields. When the oxidation of the G lignin model substrate was examined, the latter presumption was confirmed and the amount and pressure of oxygen dosed to the substrate had a major impact on the obtained yield. Too little oxygen resulted in incomplete conversion while too much oxygen gave a decrease in yield. The continuous flow approach was able to reduce the residence time from 22 hours to 45 minutes and increase the yield from 68% to 94% compared to the current best Co(salen)-catalyzed batch process. We observed that the continuous flow approach was able to convert some of the H model substrate, where previously reported batch processes failed completely. Despite this success, when performing the reaction with the S model substrate however, physical barriers which are intrinsic to flow chemistry arose and made it impossible to obtain quantitative yields as reported for the batch process. The inability to leverage product precipitation in continuous flow resulted in mediocre benzoquinone yields and selectivity, and while heterogeneous operation in continuous flow is feasible,24 this was outside the scope of our current study.When re-evaluating batch operation in light of the previous findings, the improved yields for G model substrates at increased pressure operation could be easily translated: up to 97% of MBQ was formed in the homogeneous DCM/MeOH solvent system. In addition, quantitative conversions could still be obtained for S model substrates at these higher oxygen pressures. Unfortunately however, the optimal solvent system for each of these conversions are mutually exclusive. As such, a compromise must be made: homogeneous operation in DCM/MeOH delivers near quantitative MBQ yields but reduces DMBQ yields to the 70–80% range. Heterogeneous operation in MeOH on the other hand only delivers around 70% of MBQ while achieving quantitative yields of DMBQ.
Overall, we have shown that for each of the model substrates conversion potentials are much higher than literature reports. When the reaction parameters are properly controlled, quantitative yields are achievable for both S and G models. However, it remains challenging to unify the optimized conditions (solvent, additive, time) for each of the lignin subunits, thus resulting in low depolymerization yields of organosolv lignin. Clearly, future novel catalyst systems should be screened at their optimal oxygen dosing conditions. In addition however, a major focus on solvent compatibility and the interplay with product inhibition seems essential. Ideally, catalysts which are free of product inhibition by DMBQ could allow to unify the oxidation conditions for both lignin subunits.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We would like to thank the research group of prof. Frederik Ronsse for providing organosolv lignin. Joseph J. Bozell gratefully acknowledges financial support for this publication by the Fullbright U.S. Scholar Program, which is sponsored by the U. S. Department of State and the Belgian Fulbright Commission. Its contents are solely the responsibility of the author and do not necessarily represent the official views of the Fulbright Program, the Government of the United States, or the Belgian Fulbright Commission. Jonas Mortier gratefully acknowledges financial support for this publication by Ghent University Special Research Fund; grant nr. BOF/STA/202102/003.Notes and references
- R. Behling, S. Valange and G. Chatel, Green Chem., 2016, 18, 1839–1854 RSC.
- S. Liu, L. Bai, A. P. Van Muyden, Z. Huang, X. Cui, Z. Fei, X. Li, X. Hu and P. J. Dyson, Green Chem., 2019, 21, 1974–1981 RSC.
- Z. Tang, Y. Wang, M. Chen, H. Li, L. Li, Z. Zhou, C. Li, Z. Yang and J. Wang, ACS Sustainable Chem. Eng., 2023, 11, 16722–16738 CrossRef CAS.
- D. Ando, T. Takano and F. Nakatsubo, Holzforschung, 2011, 66, 331–339 Search PubMed.
- S. Li and K. Lundquist, Holzforschung, 2005, 55, 296–301 Search PubMed.
- J. Chen, D. Wang, X. Lu, H. Guo, P. Xiu, Y. Qin, C. Xu and X. Gu, Ind. Eng. Chem. Res., 2022, 61, 1675–1683 CrossRef CAS.
- C. Chen, M. Zhou, P. Liu, B. K. Sharma and J. Jiang, New J. Chem., 2020, 44, 18906–18916 RSC.
- J. J. Bozell, C. J. O'Lenick and S. Warwick, J. Agric. Food Chem., 2011, 59, 9232–9242 CrossRef CAS PubMed.
- Y.-Z. Lai and X.-P. Guo, Wood Sci. Technol., 1991, 25, 467–472 CAS.
- D. R. Robert, M. Bardet, G. Gellerstedt and E. L. Lindfors, J. Wood Chem. Technol., 1984, 4, 239–263 CrossRef CAS.
- B. Biannic and J. J. Bozell, Org. Lett., 2013, 15, 2730–2733 CrossRef CAS PubMed.
- R. E. Key, T. Elder and J. J. Bozell, Tetrahedron, 2019, 75, 3118–3127 CrossRef CAS.
- E. C. Zuleta, G. A. Goenaga, T. A. Zawodzinski, T. Elder and J. J. Bozell, Catal. Sci. Technol., 2020, 10, 403–413 RSC.
- B. Biannic, J. J. Bozell and T. Elder, Green Chem., 2014, 16, 3635–3642 RSC.
- A. A. Ingle, S. Z. Ansari, D. Z. Shende, K. L. Wasewar and A. B. Pandit, Environ. Sci. Pollut. Res., 2022, 29, 86468–86484 CrossRef CAS PubMed.
- A. A. Ingle, S. Z. Ansari, D. Z. Shende, K. L. Wasewar and A. B. Pandit, Environ. Sci. Pollut. Res., 2022, 29, 86468–86484 CrossRef CAS PubMed.
- E. Vaccaro, C. D. Simone and D. A. Scola, J. Adhes., 2000, 72, 157–176 CrossRef CAS.
- Y. A. Rodikova, E. G. Zhizhina and Z. P. Pai, ChemistrySelect, 2016, 1, 2113–2128 CrossRef CAS.
- J. J. Bozell, J. M. Parks, C. J. Cooper, S. Alam, V. de P. N. Nziko, R. C. Johnston, A. S. Ivanov, Z. Mou, D. B. Turpin, A. W. Rudie and T. J. Elder, ACS Sustainable Chem. Eng., 2020, 8, 7225–7234 CrossRef.
- D. Cedeno and J. J. Bozell, Tetrahedron Lett., 2012, 53, 2380–2383 CrossRef CAS.
- N. J. W. Straathof, Y. Su, V. Hessel and T. Noël, Nat. Protoc., 2016, 11, 10–21 CrossRef CAS PubMed.
- E. C. Zuleta, G. A. Goenaga, T. A. Zawodzinski, T. Elder and J. J. Bozell, Catal. Sci. Technol., 2020, 10, 403–413 RSC.
- J. Ralph, C. Lapierre and W. Boerjan, Curr. Opin. Biotechnol., 2019, 56, 240–249 CrossRef CAS PubMed.
- R. Ciriminna, M. Pagliaro and R. Luque, Green Energy Environ., 2021, 6, 161–166 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3re00638g |
| This journal is © The Royal Society of Chemistry 2024 |