
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe novel quinoline derivative SKA-346 as a KCa3.1 channel selective activator†
Brandon Han Siang Wong ab,
Heesung Shimc,
Stephanie Shee Min Goayad,
Seow Theng Ongad,
Nur Ayuni Binte Muhammad Taibe,
Kelila Xin Ye Chaie,
Kerry Lime,
Dachuan Huangef,
Choon Kiat Ongef,
Thamil Selvan Vaiyapurig,
Yeong Cheng Cheahh,
Yulan Wangh,
Heike Wulffj,
Richard D. Websterk,
Vishalkumar G. Shelatai and
Navin Kumar Verma*a
ab,
Heesung Shimc,
Stephanie Shee Min Goayad,
Seow Theng Ongad,
Nur Ayuni Binte Muhammad Taibe,
Kelila Xin Ye Chaie,
Kerry Lime,
Dachuan Huangef,
Choon Kiat Ongef,
Thamil Selvan Vaiyapurig,
Yeong Cheng Cheahh,
Yulan Wangh,
Heike Wulffj,
Richard D. Websterk,
Vishalkumar G. Shelatai and
Navin Kumar Verma*a
aLee Kong Chian School of Medicine, Nanyang Technological University Singapore, Singapore. E-mail: nkverma@ntu.edu.sg
bInterdisciplinary Graduate Programme, NTU Institute for Health Technologies (HealthTech NTU), Nanyang Technological University Singapore, Singapore
cPhysical and Life Sciences, Lawrence Livermore National Laboratory, Livermore, CA, USA
dLKCMedicine-ICE Collaborative Platform, Lee Kong Chian School of Medicine, Nanyang Technological University Singapore, Singapore
eLymphoma Translational Research Laboratory, Division of Cellular and Molecular Research, National Cancer Centre, Singapore
fDuke-NUS Medical School, Singapore
gMMD Lab, Institute of Molecular and Cell Biology, A*STAR, Singapore
hSingapore Phenome Center, Lee Kong Chian School of Medicine, Nanyang Technological University Singapore, Singapore
iDepartment of General Surgery, Tan Tock Seng Hospital, Singapore
jDepartment of Pharmacology, University of California Davis, CA, USA
kSchool of Chemistry, Chemical Engineering and Biotechnology, Nanyang Technological University Singapore, Singapore
First published on 4th December 2024
Abstract
The calcium-activated KCa3.1 channel plays a crucial role in T-cell immune response. Genetic manipulation of T-cells to upregulate the expression of K+ channels has been shown to boost T-cell cytotoxicity in cancer. Here, we aimed to identify and characterize an activator that would augment KCa3.1 currents without affecting other channels. We synthesized five quinoline derivatives and used electrophysiology to screen them on KCa3.1 and a panel of 14 other ion channels. One quinoline derivative, SKA-346, activated KCa3.1 with an EC50 of 1.9 μM and showed selectivity against the other channels. In silico analysis using RosettaLigand and GLIDE demonstrated a well-converged pose of SKA-346 in a binding pocket at the interface between the calmodulin N-lobe and the S45A helix in the S4–S5 linker of the KCa3.1 channel. SKA-346 (30 mg kg−1), tolerated by mice after intra-peritoneal administration, exhibited a peak plasma concentration of 6.29 μg mL−1 (29.2 μM) at 15 min and a circulating half-life (t1/2) of 2.8 h. SKA-346 could serve as a template for the development of more potent KCa3.1 activators to enhance T-cell cytotoxicity in cancer.
1 Introduction
Potassium (K+) channels play a key role in the immune function. Human T-cells use the voltage-dependent KV1.3 channel and the intermediate-conductance Ca2+-activated KCa3.1 channel to regulate membrane potential and K+ efflux.1 Following activation, both naïve and central memory T-cells transcriptionally up-regulate KCa3.1 channels (<20 channels per cell to >500 channels per cell) without a change in KV1.3 expression, while effector memory T-cells upregulate KV1.3 channels (200 channels per cell to 1500 channels per cell) without altering KCa3.1 expression. Consequently, KCa3.1 plays an important functional role in activated naïve and central memory T-cells, while KV1.3 modulates the function of effector memory T-cells.2 KCa3.1 channels are also expressed in other cell types, including macrophages,3 mast cells,4 microglia,5 fibroblasts,6 dedifferentiated proliferating smooth muscles,7,8 airway epithelium9 and vascular endothelium.10Unlike large-conductance Ca2+-activated K+ channels which directly bind Ca2+, intracellular Ca2+ interacts indirectly with KCa3.1,11 through calmodulin, constitutively bound to the C-terminal region of KCa3.1. Binding of Ca2+ to channel-bound calmodulin induces a conformational change that opens the channel pore and increases K+ efflux.12 K+ efflux results in hyperpolarization of the membrane potential, which provides the electrochemical gradient needed to drive Ca2+ influx into the cell. Ca2+-dependent cellular processes,13 important for T-cell activation and effector functions involving proliferation, differentiation, and migration, are thereby regulated by the dynamic and precise control of the membrane potential.14–16
The death of cancer cells in tumours is associated with poor patient prognosis.17 Dying tumour cells release K+, the main intracellular cation, which accumulates in the tumour microenvironment (TME), reaching 5–10 times the levels encountered by T-cells in the bloodstream. T-cells in the TME exposed to the K+-rich fluid accumulate K+ intracellularly due to greater entry through ‘pump’ or ‘leak’ channels than exit through KCa3.1 and KV1.3 K+ channels.18,19 The increased intracellular K+ suppresses genes involved in a myriad of pathways in T-cells, nutrient consumption and metabolic changes are altered, the transition of T-cells from the resting to the tumour-killing effector stage is inhibited, and the transition to regulatory T-cells that dampen immune responses is promoted. These alterations occur without affecting the viability of T-cells.18,19 Overexpression of KV1.3 or KCa3.1 K+ channels in T-cells rescues T-cells from the immunosuppressive effects of the K+-rich TME.18 Moreover, defects in KCa3.1 activity hinder T-cell recruitment in the adenosine-rich TME, thereby promoting tumour immune escape.14,20 Pharmacological activation of the KCa3.1 channels in T-cells thus represents a promising approach to boost anti-tumour T-cell immunity within the TME.21
While KCa3.1 inhibitors are in pre-clinical development to dampen T-cell hyperactivation in autoimmune diseases, there is limited information on KCa3.1 channel activators. A previously developed KCa3.1 activator, SKA-31, potently augmented KCa3.1 activity and lowered blood pressure in experimental mice,22 but it lacked selectivity over closely related small-conductance Ca2+-activated KCa2 channels.23 Here, we synthesized a panel of five compounds structurally related to SKA-31 and demonstrate that one analogue, SKA-346, effectively activates KCa3.1 and exhibits selectivity for KCa3.1 over related channels. We further show in vivo tolerability, pharmacokinetic profiles, and tissue distribution of SKA-346 in mice. These findings will be helpful for evaluating SKA-346 as a novel therapy for conditions requiring specific activation of the KCa3.1 channel such as alleviating the immunosuppressive effects on tumour-infiltrating lymphocytes in the necrotic microenvironment of solid tumours.
2 Results and discussion
2.1. Chemistry of quinoline derivatives
The synthesis steps of the five quinoline derivative compounds (i) 5-methyl-[1,3]thiazolo[4,5-f]quinolin-2-amine (SKA-346), (ii) 8-(trifluoromethyl)thiazolo[4,5-f]quinolin-2-amine (NKV-101), (iii) 5-methyl-8-(trifluoromethyl)thiazolo[4,5-f]quinolin-2-amine (NKV-102), (iv) 5-fluoro-[1,3]thiazolo[4,5-f]quinolin-2-amine (NKV-103), and (v) 5-(trifluoromethyl)thiazolo[4,5-f]quinolin-2-amine (NKV-104) are provided in Scheme 1. Final products of all the five quinoline derivatives were confirmed by proton nuclear magnetic resonance spectroscopy (1H NMR) and liquid chromatography-mass spectrometry (LC-MS) (ESI Fig. 1–5†).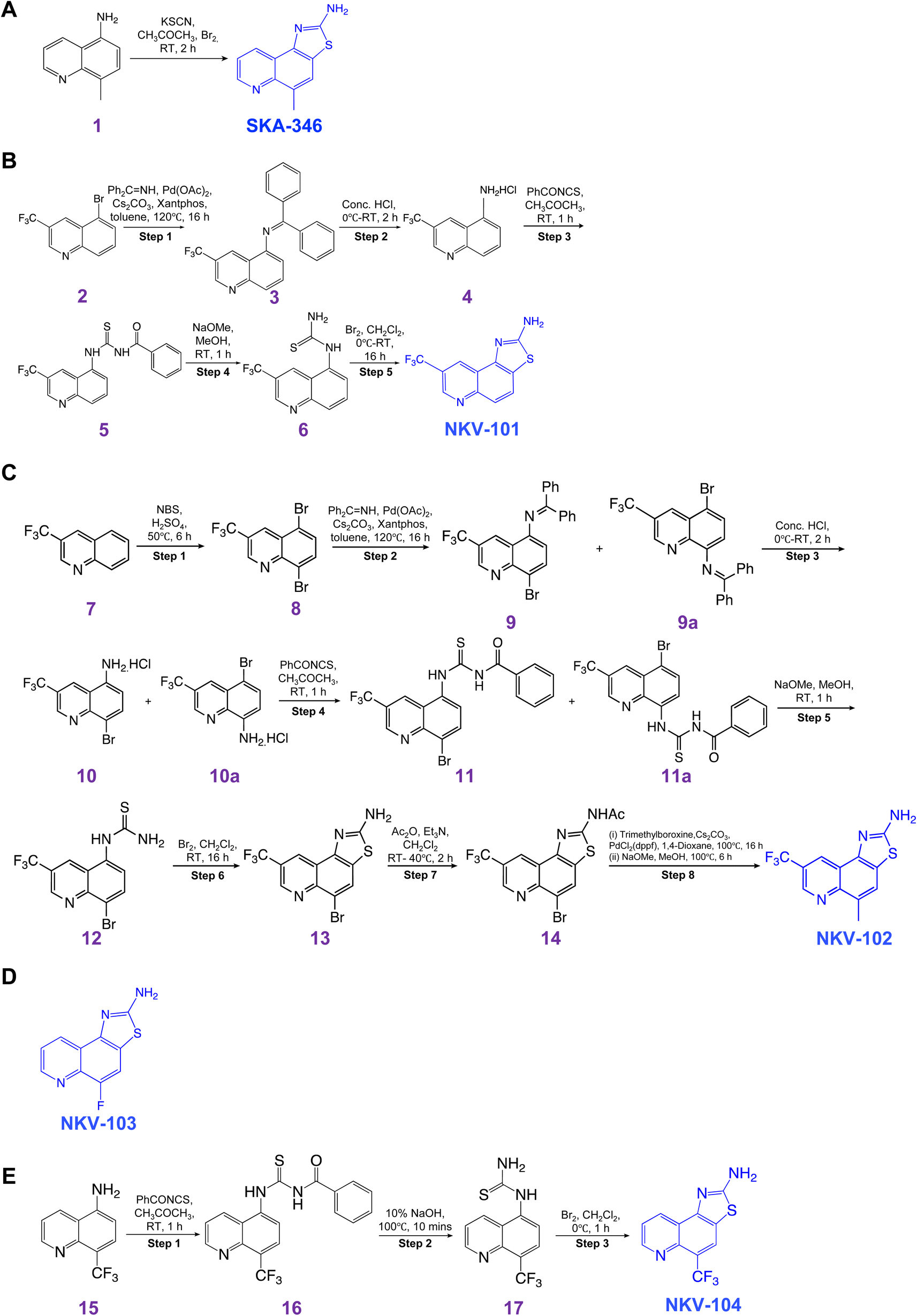 | ||
| Scheme 1 | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH) (1 equivalent, 0.19 g, 1.086 mmol) and cesium carbonate (Cs2CO3) (2 equivalent, 0.70 g, 2.173 mmol) were added. The reaction mixture was degassed with argon for 10 min prior to the addition of palladium acetate (Pd(OAc)2) (0.1 equivalent, 0.04 g, 0.217 mmol) and Xantphos (0.2 equivalent, 0.0031 g, 0.005 mmol) and subsequently stirred at 120 °C for 16 h. After completion (monitored by thin layer chromatography, TLC), the mixture was quenched with ice-cold H2O (10 mL), extracted with ethyl acetate (EtOAc) (2 × 50 mL) and washed with brine (10 mL). The organic layer was then dried over anhydrous sodium sulfate (Na2SO4) and concentrated under reduced pressure. The crude was then purified by combi-flash column chromatography (70% EtOAc/heptane) to afford 1,1-diphenyl-N-(3-(trifluoromethyl)quinolin-5-yl)methanimine (3; 0.20 g, 0.531 mmol, 48.89%). Compound 3 (0.03 g, 0.079 mmol), in a stirred solution of HCl (1 mL) at 0 °C, was stirred at room temperature for 2 h. Upon completion (monitored by TLC), the reaction mixture was quenched with ice-cold H2O (5 mL) and extracted with EtOAc (2 × 10 mL). The aqueous layer was basified with saturated sodium bicarbonate (NaHCO3; pH 8.0), washed with brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure to afford 3-(trifluoromethyl)quinolin-5-amine hydrochloride (4; 0.015 g, 0.070 mmol, 88.70%). Benzoyl isothiocyanate (PhCONCS) (2 equivalent, 0.19 g, 1.206 mmol) was then added to 4 (0.15 g, 0.603 mmol) in a stirred solution of acetone (CH3COCH3; 12 mL). Sequentially, the reaction mixture was stirred at room temperature for 1 h. After completion (monitored by TLC), the mixture was diluted with EtOAc and H2O. The organic layer was separated, washed with brine, and concentrated under reduced pressure. The crude was purified by combi-flash column chromatography (40–60% EtOAc/hexane) to afford N-((3-(trifluoromethyl)quinolin-5-yl)carbamothioyl)benzamide (5; 0.18 g, 0.479 mmol, 79.50%). Sodium methoxide (NaOMe) (2 equivalent, 0.08 g, 1.598 mmol) was then added to the stirred solution N-((3-(trifluoromethyl)quinolin-5-yl)carbamothioyl) benzamide (0.30 g, 0.799 mmol) in methanol (MeOH, 10 mL) and stirred at room temperature for 1 h. Upon completion (monitored by TLC), the mixture was concentrated under reduced pressure and diluted with EtOAc and H2O. The organic layer was separated, washed with brine, and concentrated under reduced pressure. The crude was then purified by combi-flash column chromatography (30–50% EtOAc/hexane) to afford 1-(3-(trifluoromethyl)quinolin-5-yl)thiourea (6; 0.20 g, 0.737 mmol, 92.29%). Br2 (0.10 mL) was then added to a stirred solution of 6 (0.15 g, 0.552 mmol) in dichloromethane (CH2Cl2, 10 mL) and stirred at room temperature for 16 h. After completion (monitored by TLC), the mixture was quenched in ice-cold H2O. The organic layer was separated, washed with brine, and concentrated under reduced pressure. The crude was purified by combi-flash column chromatography (50–80% EtOAc/hexane) to afford 8-(trifluoromethyl)thiazolo[4,5-f]quinolin-2-amine (NKV-101; 0.06 g, 0.222 mmol, 40.32%) as a brown solid. LC-MS calculated for C11H6F3N3S: 269.02; found: 270 (M+1); 1H NMR (400 MHz, DMSO-d6) δ ppm 9.13 (d, J = 2.25 Hz, 1H), 8.93–8.95 (m, 1H), 8.27 (d, J = 8.88 Hz, 1H), 7.96 (s, 2H), 7.78 (d, J = 8.88 Hz, 1H) (ESI Fig. 2†).NH (0.8 equivalent, 0.28 g, 1.577 mmol) and Cs2CO3 (2 equivalent, 1.28 g, 3.944 mmol) was then added to a stirred solution of 8 (0.70 g, 1.972 mmol) in toluene (8 mL). The reaction mixture was degassed with argon for 10 min prior to the addition of Pd(OAc)2 (0.2 equivalent, 0.08 g, 0.394 mmol) and Xantphos (0.4 equivalent, 0.45 g, 0.788 mmol). The reaction mixture was then stirred at 120 °C for 16 h. After completion (monitored by TLC), the mixture was quenched with ice-cold H2O (10 mL) and extracted with EtOAc (2 × 50 mL) and washed with brine (10 mL). The organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was purified by combi-flash column chromatography (70% EtOAc/Heptane) to afford mixture of N-(8-bromo-3-(trifluoromethyl)quinolin-5-yl)-1,1-diphenylmethanimine (9) and N-(5-bromo-3-(trifluoromethyl)quinolin-8-yl)-1,1-diphenylmethanimine (9a; 0.70 g, 1.537 mmol, 77.98%). In a mixture of 9 and 9a (0.70 g, 1.537 mmol) stirred in concentrated HCl (6 mL) at 0 °C, the reaction mixture was left to be stirred at room temperature for 2 h. Upon completion (monitored by TLC), the mixture was quenched with ice-cold H2O (5 mL) and extracted with EtOAc (2 × 10 mL). The aqueous layer was basified with saturated NaHCO3 (pH 8), washed with brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure to afford mixture of 8-bromo-3-(trifluoromethyl)quinolin-5-amine hydrochloride (10) and 5-bromo-3-(trifluoromethyl)quinolin-8-amine hydrochloride (10a; 0.30 g, 0.915 mmol, 59.58%, HCl salt). PhCONCS (1 equivalent, 0.14 g, 0.915 mmol) was then added to the stirred solution of 10 and 10a (0.30 g, 0.915 mmol) in CH3COCH3 (10 mL). Following which, the reaction mixture was stirred at room temperature for 1 h. After completion (monitored by TLC), the mixture was diluted with EtOAc and H2O. The organic layer was separated, washed with brine and concentrated under reduced pressure. The crude was purified by combi-flash column chromatography (30–50% EtOAc/hexane) to afford mixture of N-((8-bromo-3-(trifluoromethyl)quinolin-5-yl)carbamothioyl)benzamide (11) and N-((5-bromo-3-(trifluoromethyl)quinolin-8-yl)carbamothioyl)benzamide (11a; 0.40 g, 0.880 mmol, 96.15%). In a stirred solution of mixture of 11 and 11a; (0.40 g, 0.880 mmol) in MeOH (10 mL), NaOMe (2 equivalent, 0.09 g, 1.761 mmol) was added to it and stirred at room temperature for 1 h. Upon completion (monitored by TLC), the mixture was concentrated under reduced pressure and diluted with EtOAc and H2O. The organic layer was separated, washed with brine and concentrated under reduced pressure to afford the crude. The crude was then purified by combi-flash column chromatography (30–50% EtOAc/hexane) to yield 1-(8-bromo-3-(trifluoromethyl)quinolin-5-yl)thiourea (12; 0.30 g, 0.856 mmol, 97.30%) which was directly used in the next step without purification. Br2 (0.20 mL) was added to the stirred solution of 12 (0.30 g, 0.856 mmol) in CH2Cl2 (10 mL) and stirred at room temperature for 16 h. After completion (monitored by TLC), the mixture was quenched with ice-cold H2O and diluted with CH2Cl2. The organic layer was separated, washed with brine and concentrated under reduced pressure. The crude was purified by combi-flash column chromatography (50–80% EtOAc/hexane) to afford 5-bromo-8-(trifluoromethyl)thiazolo[4,5-f]quinolin-2-amine (13; 0.12 g, 0.344 mmol, 40.24%). Triethylamine (Et3N) (5 equivalent, 0.30 mL, 2.154 mmol) and acetic anhydride (Ac2O) (3 equivalent, 0.12 mL, 1.292 mmol) were then added to the stirred solution of 13 (0.15 g, 0.430 mmol) in CH2Cl2 (10 mL) at room temperature and stirred at 40 °C for 2 h. After completion (monitored by TLC), the mixture was concentrated under reduced pressure and diluted with CH2Cl2 and H2O. The organic layer was separated and concentrated under reduced pressure. The crude was purified by combi-flash column chromatography (70–80% EtOAc/hexane) to afford N-(5-bromo-8-(trifluoromethyl)thiazolo[4,5-f]quinolin-2-yl)acetamide (14; 0.11 g, 0.281 mmol, 65.43%). Trimethylboroxine (4 equivalent, 0.14 g, 1.127 mmol) and Cs2CO3 (2 equivalent, 2.93 g, 9.021 mmol) were then added to the stirred solution of 14 (0.11 g, 0.281 mmol) in 1,4-dioxane (5 mL) and degassed with argon for 5 min prior to the addition of PdCl2 (dppf) (0.1 equivalent, 0.02 g, 0.028 mmol). The reaction mixture was then stirred at 100 °C for 16 h before it was diluted with EtOAc and H2O. The organic layer was separated and concentrated under reduced pressure. The crude was dissolved in MeOH (2 mL) followed by the addition of NaOMe (1.5 equivalent, 0.02 g, 0.422 mmol) and stirred at 100 °C for 16 h. After completion (monitored by TLC), the mixture was concentrated under reduced pressure, quenched with ice-cold H2O, and extracted with EtOAc (2 × 10 mL). The organic layer was separated and concentrated under reduced pressure. The crude was purified by combi-flash column chromatography (70% EtOAc/hexane) to afford 5-methyl-8-(trifluoromethyl)thiazolo[4,5-f]quinolin-2-amine (NKV-102; 0.013 g, 0.045 mmol, 16.28%) as a solid. LC-MS calculated for C12H8F3N3S: 283.04; found: 284.10 (M1+); 1H NMR (400 MHz, DMSO-d6) δ ppm 9.18 (d, J = 2.32 Hz, 1H), 8.93 (d, J = 1.34 Hz, 1H), 8.15 (s, 1H), 7.84 (s, 2H), 2.73 (s, 3H) (ESI Fig. 3†).
NH) (1 equivalent, 0.19 g, 1.086 mmol) and cesium carbonate (Cs2CO3) (2 equivalent, 0.70 g, 2.173 mmol) were added. The reaction mixture was degassed with argon for 10 min prior to the addition of palladium acetate (Pd(OAc)2) (0.1 equivalent, 0.04 g, 0.217 mmol) and Xantphos (0.2 equivalent, 0.0031 g, 0.005 mmol) and subsequently stirred at 120 °C for 16 h. After completion (monitored by thin layer chromatography, TLC), the mixture was quenched with ice-cold H2O (10 mL), extracted with ethyl acetate (EtOAc) (2 × 50 mL) and washed with brine (10 mL). The organic layer was then dried over anhydrous sodium sulfate (Na2SO4) and concentrated under reduced pressure. The crude was then purified by combi-flash column chromatography (70% EtOAc/heptane) to afford 1,1-diphenyl-N-(3-(trifluoromethyl)quinolin-5-yl)methanimine (3; 0.20 g, 0.531 mmol, 48.89%). Compound 3 (0.03 g, 0.079 mmol), in a stirred solution of HCl (1 mL) at 0 °C, was stirred at room temperature for 2 h. Upon completion (monitored by TLC), the reaction mixture was quenched with ice-cold H2O (5 mL) and extracted with EtOAc (2 × 10 mL). The aqueous layer was basified with saturated sodium bicarbonate (NaHCO3; pH 8.0), washed with brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure to afford 3-(trifluoromethyl)quinolin-5-amine hydrochloride (4; 0.015 g, 0.070 mmol, 88.70%). Benzoyl isothiocyanate (PhCONCS) (2 equivalent, 0.19 g, 1.206 mmol) was then added to 4 (0.15 g, 0.603 mmol) in a stirred solution of acetone (CH3COCH3; 12 mL). Sequentially, the reaction mixture was stirred at room temperature for 1 h. After completion (monitored by TLC), the mixture was diluted with EtOAc and H2O. The organic layer was separated, washed with brine, and concentrated under reduced pressure. The crude was purified by combi-flash column chromatography (40–60% EtOAc/hexane) to afford N-((3-(trifluoromethyl)quinolin-5-yl)carbamothioyl)benzamide (5; 0.18 g, 0.479 mmol, 79.50%). Sodium methoxide (NaOMe) (2 equivalent, 0.08 g, 1.598 mmol) was then added to the stirred solution N-((3-(trifluoromethyl)quinolin-5-yl)carbamothioyl) benzamide (0.30 g, 0.799 mmol) in methanol (MeOH, 10 mL) and stirred at room temperature for 1 h. Upon completion (monitored by TLC), the mixture was concentrated under reduced pressure and diluted with EtOAc and H2O. The organic layer was separated, washed with brine, and concentrated under reduced pressure. The crude was then purified by combi-flash column chromatography (30–50% EtOAc/hexane) to afford 1-(3-(trifluoromethyl)quinolin-5-yl)thiourea (6; 0.20 g, 0.737 mmol, 92.29%). Br2 (0.10 mL) was then added to a stirred solution of 6 (0.15 g, 0.552 mmol) in dichloromethane (CH2Cl2, 10 mL) and stirred at room temperature for 16 h. After completion (monitored by TLC), the mixture was quenched in ice-cold H2O. The organic layer was separated, washed with brine, and concentrated under reduced pressure. The crude was purified by combi-flash column chromatography (50–80% EtOAc/hexane) to afford 8-(trifluoromethyl)thiazolo[4,5-f]quinolin-2-amine (NKV-101; 0.06 g, 0.222 mmol, 40.32%) as a brown solid. LC-MS calculated for C11H6F3N3S: 269.02; found: 270 (M+1); 1H NMR (400 MHz, DMSO-d6) δ ppm 9.13 (d, J = 2.25 Hz, 1H), 8.93–8.95 (m, 1H), 8.27 (d, J = 8.88 Hz, 1H), 7.96 (s, 2H), 7.78 (d, J = 8.88 Hz, 1H) (ESI Fig. 2†).NH (0.8 equivalent, 0.28 g, 1.577 mmol) and Cs2CO3 (2 equivalent, 1.28 g, 3.944 mmol) was then added to a stirred solution of 8 (0.70 g, 1.972 mmol) in toluene (8 mL). The reaction mixture was degassed with argon for 10 min prior to the addition of Pd(OAc)2 (0.2 equivalent, 0.08 g, 0.394 mmol) and Xantphos (0.4 equivalent, 0.45 g, 0.788 mmol). The reaction mixture was then stirred at 120 °C for 16 h. After completion (monitored by TLC), the mixture was quenched with ice-cold H2O (10 mL) and extracted with EtOAc (2 × 50 mL) and washed with brine (10 mL). The organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was purified by combi-flash column chromatography (70% EtOAc/Heptane) to afford mixture of N-(8-bromo-3-(trifluoromethyl)quinolin-5-yl)-1,1-diphenylmethanimine (9) and N-(5-bromo-3-(trifluoromethyl)quinolin-8-yl)-1,1-diphenylmethanimine (9a; 0.70 g, 1.537 mmol, 77.98%). In a mixture of 9 and 9a (0.70 g, 1.537 mmol) stirred in concentrated HCl (6 mL) at 0 °C, the reaction mixture was left to be stirred at room temperature for 2 h. Upon completion (monitored by TLC), the mixture was quenched with ice-cold H2O (5 mL) and extracted with EtOAc (2 × 10 mL). The aqueous layer was basified with saturated NaHCO3 (pH 8), washed with brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure to afford mixture of 8-bromo-3-(trifluoromethyl)quinolin-5-amine hydrochloride (10) and 5-bromo-3-(trifluoromethyl)quinolin-8-amine hydrochloride (10a; 0.30 g, 0.915 mmol, 59.58%, HCl salt). PhCONCS (1 equivalent, 0.14 g, 0.915 mmol) was then added to the stirred solution of 10 and 10a (0.30 g, 0.915 mmol) in CH3COCH3 (10 mL). Following which, the reaction mixture was stirred at room temperature for 1 h. After completion (monitored by TLC), the mixture was diluted with EtOAc and H2O. The organic layer was separated, washed with brine and concentrated under reduced pressure. The crude was purified by combi-flash column chromatography (30–50% EtOAc/hexane) to afford mixture of N-((8-bromo-3-(trifluoromethyl)quinolin-5-yl)carbamothioyl)benzamide (11) and N-((5-bromo-3-(trifluoromethyl)quinolin-8-yl)carbamothioyl)benzamide (11a; 0.40 g, 0.880 mmol, 96.15%). In a stirred solution of mixture of 11 and 11a; (0.40 g, 0.880 mmol) in MeOH (10 mL), NaOMe (2 equivalent, 0.09 g, 1.761 mmol) was added to it and stirred at room temperature for 1 h. Upon completion (monitored by TLC), the mixture was concentrated under reduced pressure and diluted with EtOAc and H2O. The organic layer was separated, washed with brine and concentrated under reduced pressure to afford the crude. The crude was then purified by combi-flash column chromatography (30–50% EtOAc/hexane) to yield 1-(8-bromo-3-(trifluoromethyl)quinolin-5-yl)thiourea (12; 0.30 g, 0.856 mmol, 97.30%) which was directly used in the next step without purification. Br2 (0.20 mL) was added to the stirred solution of 12 (0.30 g, 0.856 mmol) in CH2Cl2 (10 mL) and stirred at room temperature for 16 h. After completion (monitored by TLC), the mixture was quenched with ice-cold H2O and diluted with CH2Cl2. The organic layer was separated, washed with brine and concentrated under reduced pressure. The crude was purified by combi-flash column chromatography (50–80% EtOAc/hexane) to afford 5-bromo-8-(trifluoromethyl)thiazolo[4,5-f]quinolin-2-amine (13; 0.12 g, 0.344 mmol, 40.24%). Triethylamine (Et3N) (5 equivalent, 0.30 mL, 2.154 mmol) and acetic anhydride (Ac2O) (3 equivalent, 0.12 mL, 1.292 mmol) were then added to the stirred solution of 13 (0.15 g, 0.430 mmol) in CH2Cl2 (10 mL) at room temperature and stirred at 40 °C for 2 h. After completion (monitored by TLC), the mixture was concentrated under reduced pressure and diluted with CH2Cl2 and H2O. The organic layer was separated and concentrated under reduced pressure. The crude was purified by combi-flash column chromatography (70–80% EtOAc/hexane) to afford N-(5-bromo-8-(trifluoromethyl)thiazolo[4,5-f]quinolin-2-yl)acetamide (14; 0.11 g, 0.281 mmol, 65.43%). Trimethylboroxine (4 equivalent, 0.14 g, 1.127 mmol) and Cs2CO3 (2 equivalent, 2.93 g, 9.021 mmol) were then added to the stirred solution of 14 (0.11 g, 0.281 mmol) in 1,4-dioxane (5 mL) and degassed with argon for 5 min prior to the addition of PdCl2 (dppf) (0.1 equivalent, 0.02 g, 0.028 mmol). The reaction mixture was then stirred at 100 °C for 16 h before it was diluted with EtOAc and H2O. The organic layer was separated and concentrated under reduced pressure. The crude was dissolved in MeOH (2 mL) followed by the addition of NaOMe (1.5 equivalent, 0.02 g, 0.422 mmol) and stirred at 100 °C for 16 h. After completion (monitored by TLC), the mixture was concentrated under reduced pressure, quenched with ice-cold H2O, and extracted with EtOAc (2 × 10 mL). The organic layer was separated and concentrated under reduced pressure. The crude was purified by combi-flash column chromatography (70% EtOAc/hexane) to afford 5-methyl-8-(trifluoromethyl)thiazolo[4,5-f]quinolin-2-amine (NKV-102; 0.013 g, 0.045 mmol, 16.28%) as a solid. LC-MS calculated for C12H8F3N3S: 283.04; found: 284.10 (M1+); 1H NMR (400 MHz, DMSO-d6) δ ppm 9.18 (d, J = 2.32 Hz, 1H), 8.93 (d, J = 1.34 Hz, 1H), 8.15 (s, 1H), 7.84 (s, 2H), 2.73 (s, 3H) (ESI Fig. 3†).2.2. Electrophysiology analysis identifies SKA-346 and NKV-104 as potential KCa3.1 activators
Using an automated QPatch-HTX electrophysiology platform (Sophion Biosciences), we performed initial screening of all five quinoline derivatives for their potential activity on the KCa3.1 channel (Fig. 1). Two of the five quinoline derivatives, SKA-346 and NKV-104, augmented KCa3.1 currents with EC50 values 1.9 μM and 1.7 μM, respectively (Fig. 1A and E). We therefore prioritized these two compounds for more extensive characterization.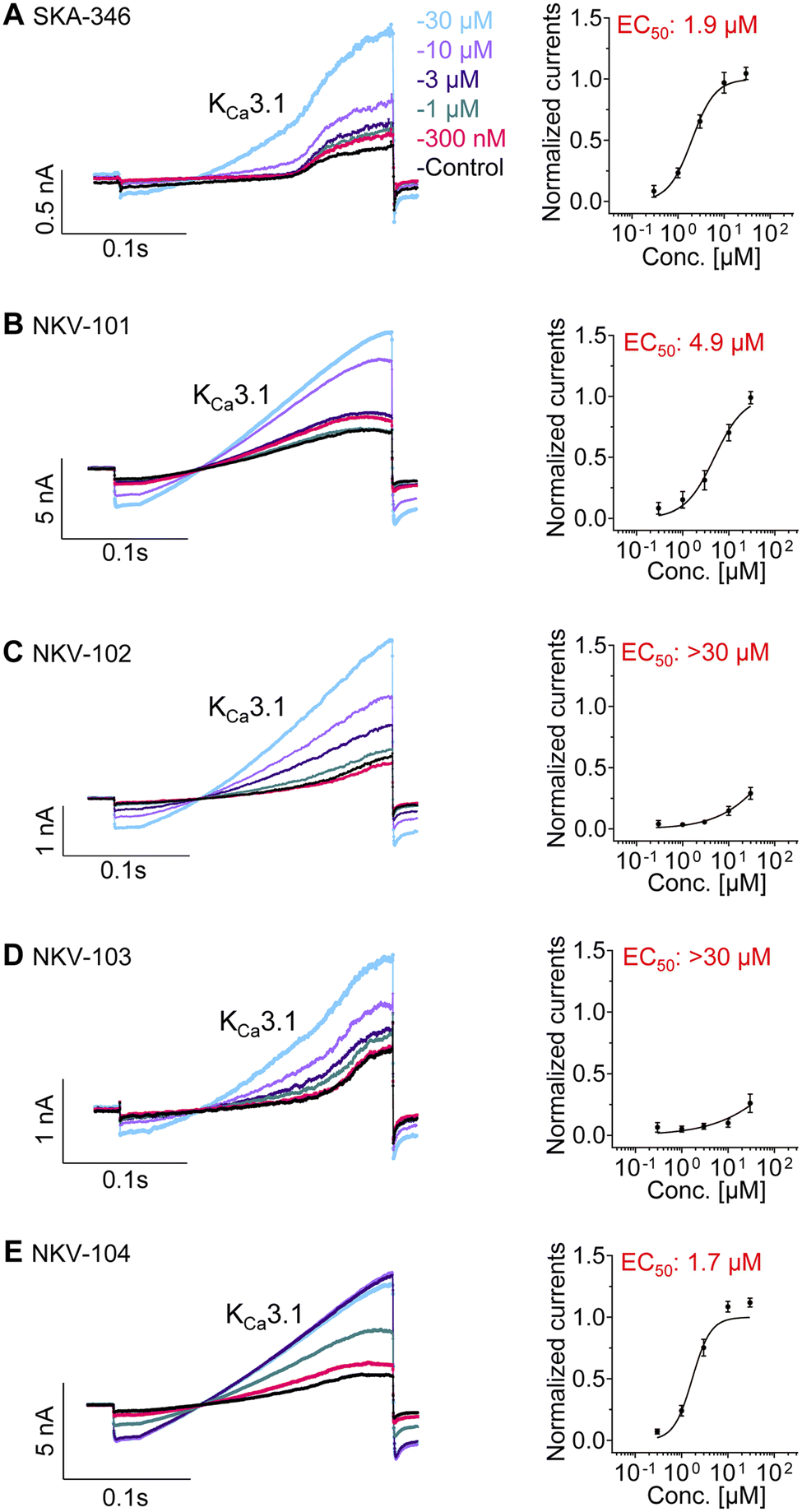 | ||
| Fig. 1 Patch-clamp recordings of the five quinoline derivatives in activating KCa3.1 channel. Representative current recordings and concentration response curves of the five quinoline derivatives towards the KCa3.1 channel are shown in the left and right columns, respectively; (A) SKA-346, (B) NKV-101, (C) NKV-102, (D) NKV-103, (E) NKV-104. Concentrations of each quinoline derivatives (between 300 nM to 30 μM) were log transformed and plotted against the normalized current values derived from at least three individual cells to generate the concentration–response curves. The EC50 values in activating KCa3.1 were computationally determined via GraphPad Prism v10.1.2. EC50, effective concentration 50%; EC50: >30 μM, EC50 could not be achieved at the highest tested concentration of 30 μM. Data are mean ± SEM of at least three independent experiments. | ||
2.3. SKA-346 shows selectivity for the KCa3.1 channel
There are multiple K+ and other ion channels that are differentially expressed on various cell types and tissues.24 Non-specific activation of ion channels may lead to potential detrimental side effects, underscoring the need for drug selectivity testing. We therefore examined SKA-346 and NKV-104 on three closely related calcium-activating K+ channels [KCa1.1, expressed mainly in brain, smooth muscle, and skeletal muscle cells; KCa2.2, expressed mainly in neuronal and skeletal muscle cells; and KCa2.3, expressed mainly in neurons and vascular endothelial cells] and two voltage-gated K+ channels [KV1.3, expressed in T-cells and various immune cell types including macrophages; and hERG/KV11.1, a cardiac channel required for the assessment of cardiac safety]25 (Fig. 2A–E). NKV-104, at a concentration of 30 μM, inhibited the hERG/KV11.1 channel (Fig. 2B, right panel) and activated the KCa2.2 and KCa2.3 channels with EC50 values of 11.9 μM and 29.3 μM, respectively (Fig. 2D and E, right panels), showing its lack of selectivity towards KCa3.1. SKA-346 did not activate any of the above channels (Fig. 2, left panels), highlighting its selectivity for KCa3.1. We therefore focused SKA-346 for further analysis. | ||
| Fig. 2 Patch-clamp recordings of SKA-346 and NKV-104 selectivity in activating various K+ channels. Representative current recordings and concentration response curves of SKA-346 and NKV-104 for various K+ channels are shown in the left and right columns, respectively; (A) KV1.3, (B) hERG/KV11.1, (C) KCa1.1, (D) KCa2.2, and (E) KCa2.3. For testing of activation effect, each cell served as its own maximal control and the increases in cell conductance were normalized to the effect obtained with a saturating activator concentration (30 μM SKA-346/NKV-104) applied during the last liquid periods before the final washout. For channels that were not activated by quinoline derivatives, cell currents were normalized to the last vehicle period applied to the cell. Increasing concentrations of SKA-346 and NKV-104 were log transformed and plotted against the normalized current values derived from at least three individual cells to generate the concentration–response curves. Note: NKV-104, at a concentration of 30 μM, caused inhibition to KV11.1 current. EC50, effective concentration 50%. EC50: >30 μM, EC50 could not be achieved at the highest tested concentration of 30 μM. Data are mean ± SEM of at least three independent experiments. | ||
Finally, we tested SKA-346 on 9 additional ion channels – (1) KV1.1, expressed in brain, heart, smooth muscle and distal convoluted tubule; (2) KV1.2, expressed in neurons; (3) KV1.4, expressed in brain, heart and pancreatic β-cells; (4) KV1.5, expressed in heart, brain, smooth muscle and pancreatic β-cells; (5) KV1.7, expressed in neuronal adrenal gland, brain and smooth muscle cells; (6) KV3.1, expressed in neuronal cells; (7) KV4.2, expressed in the heart and in neurons; (8) NaV1.5 expressed in heart, neurons, and skeletal muscle cells; and (9) Kir2.1 expressed in smooth muscle, neurons and endothelial cells.24 SKA-346 did not impact any of these ion channels at concentrations up to 30 μM (Fig. 3), confirming its selectivity towards KCa3.1 channel over 14 other ion channels.
 | ||
| Fig. 3 Patch-clamp recordings of SKA-346 selectivity in activating various ion channels. Representative current recordings and concentration response curves of SKA-346 towards 9 different ion channels are shown in the left and right columns, respectively. (A) KV1.1, (B) KV1.2, (C) KV1.4, (D) KV1.5, (E) KV1.7, (F) KV3.1, (G) KV4.2, (H) NaV1.5, and (I) Kir2.1. Concentrations of SKA-346 (between 300 nM to 30 μM) were log transformed and plotted against the normalized current values derived from at least three individual cells to generate the concentration–response curves. Note: SKA-346 did not significantly affect any of the tested channels. Data are mean ± SEM of at least three independent experiments. | ||
2.4. Molecular docking of SKA-346
The interface between the calmodulin N-lobe and the S45A helix in the S4–S5 linker of KCa3.1 is intimately involved in Ca2+-calmodulin-dependent activation of the KCa3.1 channel and has been suggested to be a binding pocket for activators of the channel.25,26 SKA-346 was docked into this critical pocket via RosettaLigand computer-aided docking.27,28 Ligand docking using the RosettaLigand method involves three stages. First, the ligand is randomly placed within a 10 Å sphere of the binding site. Conformers are generated and scored for shape compatibility with the protein, and the best models are filtered and selected for further stages. In the second stage, the ligand's position and orientation are fine-tuned using Monte Carlo minimization, with protein side chains repacked and optimized. The full-atom Rosetta energy function, including softened van der Waals interactions, is used for scoring. The final stage applies a stricter gradient-based minimization with a hard-repulsive van der Waals potential to refine the ligand and protein interactions, improving discrimination between native and non-native binding modes.29,30 A total of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 models were generated, taken through the above three stages of RosettaLigand docking, followed by the selection of the top 1000 models that attained the lowest total energy score. The models were further sieved to identify the top 10 models which were inspected for ligand/channel interaction and convergence. RosettaLigand predicted a single, well-converged pose for SKA-346 (Fig. 4A).
000 models were generated, taken through the above three stages of RosettaLigand docking, followed by the selection of the top 1000 models that attained the lowest total energy score. The models were further sieved to identify the top 10 models which were inspected for ligand/channel interaction and convergence. RosettaLigand predicted a single, well-converged pose for SKA-346 (Fig. 4A).
 | ||
| Fig. 4 In silico analysis of the docking and binding affinities of SKA-346 to the binding site of KCa3.1. (A) RosettaLigand docking models of the lowest energy-binding poses of SKA-346 in the binding site of KCa3.1. Van der Waals forces and hydrogen bonds of the quinoline derivative upon docking within the binding site of KCa3.1 are shown in the left and right columns, respectively. (B) SKA-346 was prepared in Schrodinger ligand prep Wizard and XP (extra precision) module prior to in silico analysis by GLIDE. The binding affinity between SKA-346 and the binding site of KCa3.1 are computationally predicted that gives an arbitrary number as a docking score, whereby a more negative value suggests a more ‘nature-like’ fitting of the quinoline derivative within the binding site. | ||
We next performed a secondary in silico analysis via the GLIDE program (open state −1, pdb: 6CNN) that empirically predicts the binding affinity between the compound and binding site upon docking. For this, SKA-346 was prepared in Schrodinger ligand prep Wizard and XP (extra precision) module (Schrödinger, Inc.). Various orientations of SKA-346 inputted into the binding pocket in the calmodulin/KCa3.1 interface were computationally determined and labelled as a docking score (Fig. 4B). We found a high negative docking score of −6.285, suggesting a ‘nature-like’ fitting of SKA-346 within the binding site.
2.5. Preclinical pharmacology of SKA-346
To access preclinical pharmacology of SKA-346, we first characterized its physicochemical properties by using pkCSM (https://biosig.lab.uq.edu.au/pkcsm)31 and Chemdraw 22.2.0. SKA-346, molecular formula C11H9N3S, has a molecular weight of 215.27 g mol−1 with calculated partition coefficient (logP) of 2.74 (computed using pkCSM) and 2.53 (computed using Chemdraw 22.2.0). The compound contains 1 hydrogen-bond donor and 4 hydrogen-bond acceptors. The molar refractivity of SKA-346 is 64.29 with topological polar surface area (TPSA) of 90.579 (computed using pkCSM) and 80.04 (computed using Chemdraw 22.2.0). All these parameters satisfy the generally accepted Lipinski's rule of five,32 suggesting SKA-346 to be a drug-like compound. SKA-346 solubility in dimethyl sulfoxide (DMSO) was calculated to be 40.6 mg mL−1 or 188.64 mM (ESI Fig. 6†) Notably, in vitro treatment of human primary T-cells (isolated from healthy volunteers' blood using standard protocol)33 with SKA-346 concentrations up to 10 μM (∼5 times of EC50) were non-cytotoxic (ESI Fig. 7†).
We (co-author Wulff's research group) have previously shown that up to 30 mg kg−1 of the benzothiazole compound SKA-31 is well-tolerated in Sprague-Dawley rats and in mice when administered intraperitoneally (i.p.).22 Using this dataset as a reference and to minimize animal usage, we assessed whether the same single dose of SKA-346 (30 mg kg−1 in 100 μL per animal) administered into female C57BL/6 mice (n = 6 each group) via i.p. route was tolerated over a short-term period of 14 consecutive days (Fig. 5A). When compared to the vehicle control group, we did not detect any obvious clinical sign of toxicity due to SKA-346 administration, as there was no significant mouse body weight loss (Fig. 5B). SKA-346 treatment did not cause any significant change in the levels of serum aspartate aminotransferase (AST, Fig. 5C), indicating that there was no major damage or injury to the liver. AST is a commonly used marker for liver function as any insult or damage to the liver or even skeletal and heart tissues will cause this enzyme to be released into the bloodstream resulting in its significantly elevated levels in the serum.34 Since the study was designed to provide a preliminary assessment of SKA-346 safety in vivo, we limited our study to a single dose of 30 mg kg−1 for 14 consecutive days treatment. It would be important to test both lower and higher doses of SKA-346 over extended periods and perform assessments using multiple biomarkers to fully characterize potential dose-dependent sub-chronic and chronic toxicity and further establish its safety profile.
 | ||
| Fig. 5 In vivo tolerability of SKA-346. SKA-346 solution was prepared in 3% DMSO + 97% peanut oil to achieve 6 mg mL−1 concentration. (A) Timeline of SKA-346 toxicity studies. SKA-346 (30 mg kg−1, total volume 100 μL) was administered daily into female C57BL/6 mice (n = 6 for each group) via i.p. for 14 days. (B) Body weights of control and SKA-346-treated mice recorded every 2–4 days. (C) The serum biochemical indicator ast (mU/mL) of control and SKA-346-treated mice. (D) Mice brain, kidney, and liver tissues were harvested upon termination and analysed for SKA-346 biodistribution by LC-MS. (E) H&E staining of mice tissue samples (kidney, liver, brain, heart, thymus, and spleen) harvested upon termination at day 14. Representative images are shown. The full images of the tissue sections are provided as ESI Fig. 9–14.† H&E, hematoxylin and eosin; i.p., intraperitoneal; LC-MS, liquid chromatography-mass spectrometry; nd, not detected; ns, non-significant. | ||
LC-MS analysis of harvested organ samples showed substantial amounts of SKA-346 in the kidney (60.7 pg mg−1) and liver (22.5 pg mg−1), but not in the brain (Fig. 5D). Moreover, the boiled-egg model analysis from SwissADME (http://www.swissadme.ch/), that utilizes the TPSA and logP values to evaluate the likelihood of the molecule in passing through the blood–brain barrier (BBB),35 indicated SKA-346 being unable to cross the BBB (ESI Fig. 8†). It is possible that SKA-346 distributes into other tissues such as heart, thymus, spleen, lungs, and bones, which could not be analyzed in the current study. When compared to the vehicle control, no detectable change in histological morphologies was observed in vital organs (kidney, liver, heart, brain, thymus, and spleen) of mice due to SKA-346 administration (Fig. 5E). These findings collectively demonstrate that SKA-346 did not cause any unwanted toxicity effect to the vital organs.
Finally, we determined the circulating half-life (t1/2) of SKA-346 (30 mg kg−1) administered by i.p. injection in female BALB/c mice (n = 4 each). Analysis of blood plasma samples, harvested at multiple time-points over the course of 24 h, by LC-MS (Fig. 6A) revealed that SKA-346 was absorbed rapidly into the blood, reaching its peak concentration in the blood plasma within 0.25 h (Fig. 6B, ESI Fig. 15†). An estimated circulated half-life (t1/2) of SKA-346 was 2.84 h with the peak plasma concentration (Cmax) of 6.29 μg mL−1 (29.2 μM) at 15 min, which is about 15 times higher than the EC50 for KCa3.1 activation. We also noted mean residence time from time of dosing to time of last measurable concentration (MRTlast) to be 4.82 h and the area under the curve from the time of dosing to the time of the last measurable concentration (AUClast) to be 45487.06 h*ng mL−1 (Fig. 6C; ESI Table 1†).
 | ||
| Fig. 6 In vivo pharmacological analysis of SKA-346. SKA-346 solution was prepared in 3% DMSO + 97% peanut oil to achieve 6 mg mL−1 concentration. (A) Workflow of SKA-346 treatment and sample collection over 24 h. Female BALB/c mice (n = 4/group, 2 groups) were administered at 30 mg kg−1; 100 μL of SKA-346 preparation via i.p. Blood plasma was taken from each group of mice alternately at 8 different time-points over the course of 24 h; 0 h, 0.25 h, 0.5 h, 1 h, 2 h, 4 h, 8 h, and 24 h. (B) SKA-346 concentrations in the mice blood plasma, as determined by LC-MS, were plotted against time over a period of 24 h. The circulating half-life (t1/2) of SKA-346 was calculated to be 2.8 h. (C) Key pharmacological parameters obtained from SKA-346-treated mice are tabulated. Detailed list of pharmacological parameters is provided as ESI Table 1.† i.p., intraperitoneal; LC-MS, liquid chromatography-mass spectrometry. | ||
3 Conclusions
In summary, our current study identified SKA-346 as a novel selective and safe activator of the KCa3.1 channel. SKA-346 constitutes a promising novel therapeutic for use in conditions that require increasing KCa3.1 activity.4 Materials and methods
4.1. Ion channel electrophysiology analysis
All the five quinoline derivatives were dissolved in DMSO to prepare 10 mM stock solutions and stored at −80 °C until use. For patch-clamp analysis, fixed-ratio dilutions of the above five compounds were made in extracellular solution buffers, specific for each channel as detailed below. Patch-clamp recordings were captured using an automated QPatch-HTX electrophysiology platform (Sophion Biosciences) equipped with Sophion QPatch software. Each current slope was the average of five measurements taken at the end of the respective liquid period. Data fitting to the Hill equation to obtain EC50 values was performed with GraphPad Prism v10.1.2. For testing of activation effect, each cell served as its own maximal control and the increases in cell conductance were normalized to the effect obtained with a saturating activator concentration (30 μM SKA-346) applied during the last liquid periods before the final washout.4.2. Solubility of SKA-346
SKA-346 solubility in DMSO was determined using UV-Vis spectrophotometer (Varioskan LUX Multimode Microplate Reader, ThermoFisher Scientific, USA), as described earlier.40 Briefly, SKA-346 was weighed in a 2 mL glass tube and dissolved in DMSO to achieve a stock concentration of 0.5 mg mL−1. The solution was serially diluted to get concentrations ranging from 0.5 to 0.007 mg mL−1. The absorption spectrum of 200–800 nm showed the absorption peak for SKA-346 at 345 nm (λmax). Accordingly, a calibration curve was generated by plotting the absorbance at 345 nm (after blank correction) against known concentrations. To determine SKA-346 solubility, excess amount of SKA-346 (50 mg) was added to 500 μL DMSO in a 2 mL glass tube, which was then placed on a shaker at 1000 rpm for 24 h to ensure saturation. The suspension was filtered using a 0.22 μm syringe filter to remove undissolved particles and the filtrate was diluted 100-fold in DMSO. The optical density at the absorption peak was used to determine the solubility of SKA-346 with reference to the calibration curve.4.3. SKA-346 tolerability assessment in mice
All protocols for animal treatment and care were in accordance with the guidelines for Responsible Care and Use of Laboratory Animals and were reviewed and approved by the Animal Ethics Committee of Singapore Health Services, SingHealth Singapore (IACUC #2018/SHS/1417). DMSO is a polar aprotic solvent that dissolves both polar and nonpolar compounds and is miscible in a wide range of organic solvents, including various oils. For administering SKA-346 in mice, we dissolved it in 3% DMSO + 97% peanut oil (Sigma-Aldrich, USA) to wet the surface and break up the SKA-346 crystals to allow for better dissolution of this highly lipophilic compound in peanut oil, as used earlier with a slight modification.41–43 Goal was to reduce the amount of DMSO to a minimum possible volume. Briefly, peanut oil was heated to a temperature of ∼60 °C in a sterile glass container with a mini-stir bar on a magnetic hotplate stirrer. SKA-346 in DMSO was added to oil with constant stirring to obtain the final preparation containing 6 mg per mL SKA-346 (DMSO: peanut oil = 3:97). The final SKA-346 solution was allowed to cool to room temperature, and it appeared light brownish in colour. The SKA-346 preparation was then aliquoted into autoclaved 2 mL glass vials and stored at −20 °C until use. Prior to injection, the SKA-346 preparation was thawed, heated to ∼60 °C with stirring and then cooled to room temperature. Female C57BL/6 mice (aged 6–7 weeks at the start of the treatment, n = 6) were injected (i.p.) with ∼100 μL of 6 mg mL−1 preparation (i.e., 30 mg kg−1) of SKA-346 once daily for 14 consecutive days. Similarly, control mice (n = 6) were injected with a solution of 3% DMSO in 97% peanut oil. Both groups (vehicle and SKA-346 treated) consisted of mice of similar age and weight ranges to ensure that any observed effects could be attributed primarily to the treatment rather than differences in these biological factors. Mice at the SingHealth facility (Singapore) were housed under standard conditions on a 12 h/12 h light/dark cycle with regulated temperature and humidity and fed with certified standard chow and tap water ad libitum. Mice were monitored daily by trained technicians throughout the study to check any signs of weight loss, intractable diarrhoea, or infection. Body weights were recorded every 2–4 days throughout the experiment. At the end of the treatment on day 14, mice were euthanized, and blood and organs (kidney, liver, brain, heart, thymus, and spleen) were harvested for biochemical and histological analysis.
AST levels in the mice serum samples were determined using commercially available colorimetric kits from Abcam (UK) in accordance with the instructions provided.
Harvested organs were fixed, embedded in paraffin, sectioned, and then mounted on glass slides. Tissue sections slides were placed in a 60 °C incubator for 1 h to remove excess paraffin. The section slides were stained with the histological stains hematoxylin and eosin using Leica ST5010 Autostainer (Leica, Germany). Cover glass slides were mounted onto the H&E-stained tissue sections and dried in the fume hood overnight. Slides were imaged using Zeiss Axioscan 7 microscope slide scanner (Zeiss, Germany) and presented.
4.4. In vivo pharmacokinetics profiling
Animal experiments for in vivo pharmacokinetics assessments of SKA-346 were conducted as per the protocol approved by the Animal Ethics Committee of Biological Resource Centre, A*STAR, Singapore (IACUC #201561). Female BALB/c mice (aged 7–9 weeks at the start of the treatment, n = 4 for each time-point) were injected intraperitoneally with SKA-346 at the dose of 30 mg kg−1/10 mL using 3% DMSO + 97% peanut oil formulation. Post-administration of SKA-346, blood samples were collected at 0, 0.25, 0.5, 1, 2, 4, 8, and 24 h. Plasma was separated from the blood samples and subsequently analyzed by LC-MS for SKA-346 quantification.4.5. LC-MS analysis of SKA-346 in mice blood plasma and organ samples
The LC-MS analysis was performed using Waters equipment coupled with Xevo TQ-S MS system (Waters Corporation, USA). Mice blood plasma samples (20 μL each) were prepared by rapid mixing with 60 μL LC-MS grade methanol: dichloromethane (1:1) on a vortex mixer. Tissue samples were prepared by homogenization of pre-weighed organs in 500 μL LC-MS grade methanol: dichloromethane (1:1) using a tissue homogenizer. These samples were incubated at 4 °C for 30 min, centrifuged at 14000 rpm at 4 °C for 10 min, and then injected into LC-MS system for analysis. The UHPLC system was equipped with a BEH C18 column (2.1 mm × 100 mm, 1.7 μm, Waters Corporation, USA). The column temperature was kept at 40 °C. The mice blood plasma and organs samples were eluted at a flow rate of 0.3 mL min−1. Water containing 0.1% formic acid (Solvent A) and acetonitrile containing 0.1% formic acid (Solvent B) were used as mobile phases. For blood plasma samples, the LC was operated with the gradient conditions as follows: 0 min, 20% B; 0.5 min, 20% B; 5 min, 95% B; 7 min, 95% B; 7.1 min, 20% B; 10 min, 20% B. For organ samples, the LC was operated with the gradient conditions as follows: 0 min, 20% B; 5 min, 95% B; 6 min, 95% B; 7 min, 20% B; 10 min, 20% B. The total chromatographic run time was 10 min. Mass spectrometer conditions were set as follows: capillary voltage at 0.5 kV, desolvation temperature at 350 °C, cone gas flow rate at 150 L h−1, desolvation gas flow rate at 800 L h−1, source temperature at 150 °C. The mass spectrometer was operated with an electrospray ionization source (ESI) in the positive mode and performed in the multiple reaction monitoring (MRM) mode. Data acquisition, integration and quantification were performed using MassLynx and TargetLynx (Waters Corporation, USA).
4.6. Statistical analysis
Experiments were replicated independently at least three times. Depending on specific experiments, comparison between two groups was made by paired Student's t-test and comparisons of multiple groups within a single experiment were made by one-way ANOVA. Prism 9.5.0 (GraphPad) was used to make the graphs and statistical analyses.Ethical statement
All animal procedures were performed in accordance with the Guidelines for Responsible Care and Use of Laboratory Animals and as per the protocol approved by the Animal Ethics Committee of Singapore Health Services, SingHealth Singapore (IACUC #2018/SHS/1417) as well as the Biological Resource Centre, A*STAR, Singapore (IACUC #201561). Experiments utilizing human peripheral blood samples were performed in accordance with institutional guidelines and the protocol approved by the Institutional Review Board of Nanyang Technological University Singapore (IRB-2018-05-034).Data availability
The data supporting this article have been included in the main article and the ESI.†Author contributions
Conceptualization, B. H. S. W. and N. K. V.; patch clamp analysis, B. H. S. W., S. S. M. G. and S. T. O.; SKA-346 synthesis, H. W. and H. S.; molecular docking, H. S.; animal experiments, data acquisition and analysis, N. A. B. M. T., K. X. Y. C., K. L., D. H., C. K. O. and T. S. V.; pharmacology, T. S. V., B. H. S. W., D. H. and Y. W.; LC-MS analysis, Y. C. C. and Y. W.; formal analysis, B. H. S. W., R. D. W., V. G. S., N. K. V. and H. W.; investigation, B. H. S. W. and N. K. V.; resources, N. K. V. and C. K. O.; data curation, B. H. S. W., S. T. O. and H. S.; writing—original draft, B. H. S. W. and N. K. V.; writing—review & editing, B. H. S. W., S. T. O., D. H., C. K. O., Y. W., H. W., R. D. W., V. G. S. and N. K. V.; supervision, N. K. V., R. D. W. and V. G. S.; project administration, N. K. V.; funding acquisition, N. K. V. All authors have seen and approved the final version of the manuscript.Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
Authors acknowledge Professor K. George Chandy for reading, editing, and critiquing this manuscript, and Sai Life Sciences for their help in chemical synthesis. Authors also acknowledge Dr Chang Jun Jie and Dr Loh Xian Jun at Institute of Materials Research and Engineering (IMRE), A*STAR for their assistance in determining the drug solubility. This research was supported, in part, by the Singapore Ministry of Education (MOE) under its MOE Academic Research Fund (AcRF) Tier 2 Grant (MOE2017-T2-2-004), MOE AcRF Tier 1 Grant (RG94/22), and the National Research Foundation Singapore under its Open Fund Large Collaborative Grant (OFLCG-23May0039) and administered by the Singapore Ministry of Health’s National Medical Research Council (NMRC). B. H. S. W. was provided with PhD fellowship by HealthTech NTU.References
- J. Lam and H. Wulff, The lymphocyte potassium channels Kv1.3 and KCa3.1 as targets for immunosuppression, Drug Dev. Res., 2011, 72, 573–584 CrossRef CAS PubMed.
- S. Ghanshani, H. Wulff, M. J. Miller, H. Rohm, A. Neben, G. A. Gutman, M. D. Cahalan and K. G. Chandy, Up-regulation of the IKCa1 potassium channel during T-cell activation. Molecular mechanism and functional consequences, J. Biol. Chem., 2000, 275, 37137–37149 CrossRef CAS PubMed.
- P. J. Hanley, B. Musset, V. Renigunta, S. H. Limberg, A. H. Dalpke, R. Sus, K. M. Heeg, R. Preisig-Muller and J. Daut, Extracellular ATP induces oscillations of intracellular Ca2+ and membrane potential and promotes transcription of IL-6 in macrophages, Proc. Natl. Acad. Sci. U.S.A., 2004, 101, 9479–9484 CrossRef CAS PubMed.
- S. M. Duffy, W. J. Lawley, E. C. Conley and P. Bradding, Resting and activation-dependent ion channels in human mast cells, J. Immunol., 2001, 167, 4261–4270 CrossRef CAS PubMed.
- R. Khanna, L. Roy, X. Zhu and L. C. Schlichter, K+ channels and the microglial respiratory burst, Am. J. Physiol.: Cell Physiol., 2001, 280, C796–C806 CrossRef CAS.
- T. L. Pena and S. G. Rane, The fibroblast intermediate-conductance KCa channel, FIK, as a prototype for the cell growth regulatory function of the IK channel family, J. Membr. Biol., 1999, 172, 249–257 CrossRef CAS.
- R. Kohler, H. Wulff, I. Eichler, M. Kneifel, D. Neumann, A. Knorr, I. Grgic, D. Kampfe, H. Si, J. Wibawa, R. Real, K. Borner, S. Brakemeier, H. D. Orzechowski, H. P. Reusch, M. Paul, K. G. Chandy and J. Hoyer, Blockade of the intermediate-conductance calcium-activated potassium channel as a new therapeutic strategy for restenosis, Circulation, 2003, 108, 1119–1125 CrossRef.
- C. B. Neylon, R. J. Lang, Y. Fu, A. Bobik and P. H. Reinhart, Molecular cloning and characterization of the intermediate-conductance Ca2+-activated K+ channel in vascular smooth muscle: relationship between KCa channel diversity and smooth muscle cell function, Circ. Res., 1999, 85, e33–e43 CrossRef CAS.
- J. D. McCann, J. Matsuda, M. Garcia, G. Kaczorowski and M. J. Welsh, Basolateral K+ channels in airway epithelia. I. Regulation by Ca2+ and block by charybdotoxin, Am. J. Physiol., 1990, 258, L334–L342 CAS.
- I. Grgic, B. P. Kaistha, J. Hoyer and R. Kohler, Endothelial Ca+-activated K+ channels in normal and impaired EDHF-dilator responses-relevance to cardiovascular pathologies and drug discovery, Br. J. Pharmacol., 2009, 157, 509–526 CrossRef CAS PubMed.
- L. Sforna, A. Megaro, M. Pessia, F. Franciolini and L. Catacuzzeno, Structure, gating and basic functions of the Ca2+-activated K channel of intermediate conductance, Curr. Neuropharmacol., 2018, 16, 608–617 CrossRef CAS PubMed.
- M. Zhang, C. Abrams, L. Wang, A. Gizzi, L. He, R. Lin, Y. Chen, P. J. Loll, J. M. Pascal and J. F. Zhang, Structural basis for calmodulin as a dynamic calcium sensor, Structure, 2012, 20, 911–923 CrossRef CAS PubMed.
- M. D. Cahalan and K. G. Chandy, The functional network of ion channels in T lymphocytes, Immunol. Rev., 2009, 231, 59–87 CrossRef CAS PubMed.
- N. K. Verma, B. H. S. Wong, Z. S. Poh, A. Udayakumar, R. Verma, R. K. J. Goh, S. P. Duggan, V. G. Shelat, K. G. Chandy and N. F. Grigoropoulos, Obstacles for T-lymphocytes in the tumour microenvironment: Therapeutic challenges, advances and opportunities beyond immune checkpoint, EBioMedicine, 2022, 83, 104216 CrossRef CAS PubMed.
- Y. Lin, Y. J. Zhao, H. L. Zhang, W. J. Hao, R. D. Zhu, Y. Wang, W. Hu and R. P. Zhou, Regulatory role of KCa3.1 in immune cell function and its emerging association with rheumatoid arthritis, Front. Immunol., 2022, 13, 997621 CrossRef CAS.
- L. Di, S. Srivastava, O. Zhdanova, Y. Ding, Z. Li, H. Wulff, M. Lafaille and E. Y. Skolnik, Inhibition of the K+ channel KCa3.1 ameliorates T cell-mediated colitis, Proc. Natl. Acad. Sci. U.S.A., 2010, 107, 1541–1546 CrossRef CAS.
- C. H. Richards, Z. Mohammed, T. Qayyum, P. G. Horgan and D. C. McMillan, The prognostic value of histological - necrosis in solid organ malignant disease: A systematic review, Future Oncol., 2011, 7, 1223–1235 CrossRef CAS.
- R. Eil, S. K. Vodnala, D. Clever, C. A. Klebanoff, M. Sukumar, J. H. Pan, D. C. Palmer, A. Gros, T. N. Yamamoto, S. J. Patel, G. C. Guittard, Z. Yu, V. Carbonaro, K. Okkenhaug, D. S. Schrump, W. M. Linehan, R. Roychoudhuri and N. P. Restifo, Ionic immune suppression within the tumour microenvironment limits T cell effector function, Nature, 2016, 537, 539–543 CrossRef CAS.
- S. T. Ong, A. S. Ng, X. R. Ng, Z. Zhuang, B. H. S. Wong, P. Prasannan, Y. J. Kok, X. Bi, H. Shim, H. Wulff, K. G. Chandy and N. K. Verma, Extracellular K+ dampens T cell functions: implications for immune suppression in the tumor microenvironment, Bioelectricity, 2019, 1, 169–179 CrossRef.
- A. A. Chimote, A. Balajthy, M. J. Arnold, H. S. Newton, P. Hajdu, J. Qualtieri, T. Wise-Draper and L. Conforti, A defect in KCa3.1 channel activity limits the ability of CD8+ T cells from cancer patients to infiltrate an adenosine-rich microenvironment, Sci. Signal., 2018, 11, eaaq1616 CrossRef PubMed.
- M. Chirra, H. S. Newton, V. S. Gawali, T. M. Wise-Draper, A. A. Chimote and L. Conforti, How the potassium channel response of T lymphocytes to the tumor microenvironment shapes antitumor immunity, Cancers, 2022, 14, 3564 CrossRef CAS PubMed.
- A. Sankaranarayanan, G. Raman, C. Busch, T. Schultz, P. I. Zimin, J. Hoyer, R. Kohler and H. Wulff, Naphtho[1,2-d]thiazol-2-ylamine (SKA-31), a new activator of KCa2 and KCa3.1 potassium channels, potentiates the endothelium-derived hyperpolarizing factor response and lowers blood pressure, Mol. Pharmacol., 2009, 75, 281–295 CrossRef CAS.
- H. Shim, B. M. Brown, L. Singh, V. Singh, J. C. Fettinger, V. Yarov-Yarovoy and H. Wulff, The trials and tribulations of structure assisted design of KCa channel activators, Front. Pharmacol, 2019, 10, 972 CrossRef CAS PubMed.
- C. Miller, An overview of the potassium channel family, Genome Biol., 2000, 1, REVIEWS0004 CrossRef CAS.
- S. D. Harding, J. F. Armstrong, E. Faccenda, C. Southan, S. P. H. Alexander, A. P. Davenport, M. Spedding and J. A. Davies, The IUPHAR/BPS guide to PHARMACOLOGY in 2024, Nucleic Acids Res., 2024, 52, D1438–D1449 CrossRef PubMed.
- C. Lee and R. Mackinnon, Activation mechanism of a human SK-calmodulin channel complex elucidated by cryo-EM structures, Science, 2018, 360, 508–513 CrossRef CAS.
- S. A. Combs, S. L. Deluca, S. H. DeLuca, G. H. Lemmon, D. P. Nannemann, E. D. Nguyen, J. R. Willis, J. H. Sheehan and J. Meiler, Small-molecule ligand docking into comparative models with Rosetta, Nat. Protoc., 2013, 8, 1277–1298 CrossRef CAS.
- J. Meiler and D. Baker, ROSETTALIGAND: protein-small molecule docking with full side-chain flexibility, Proteins, 2006, 65, 538–548 CrossRef CAS.
- I. W. Davis and D. Baker, RosettaLigand docking with full ligand and receptor flexibility, J. Mol. Biol., 2009, 385, 381–392 CrossRef CAS PubMed.
- I. W. Davis, K. Raha, M. S. Head and D. Baker, Blind docking of pharmaceutically relevant compounds using RosettaLigand, Protein Sci., 2009, 18, 1998–2002 CrossRef CAS PubMed.
- D. E. Pires, T. L. Blundell and D. B. Ascher, pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures, J. Med. Chem., 2015, 58, 4066–4072 CrossRef CAS.
- C. A. Lipinski, Drug-like properties and the causes of poor solubility and poor permeability, J. Pharmacol. Toxicol. Methods, 2000, 44, 235–249 CrossRef CAS.
- A. Kizhakeyil, S. T. Ong, M. H. U. T. Fazil, M. L. S. Chalasani, P. Prasannan and N. K. Verma, Isolation of human peripheral blood T-lymphocytes, Methods Mol. Biol., 2019, 1930, 11–17 CrossRef CAS.
- J. R. Senior, Alanine aminotransferase: a clinical and regulatory tool for detecting liver injury–past, present, and future, Clin. Pharmacol. Therapeut., 2012, 92, 332–339 CrossRef CAS.
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- N. Coleman, B. M. Brown, A. Oliván-Viguera, V. Singh, M. M. Olmstead, M. S. Valero, R. Köhler and H. Wulff, New positive Ca2+-activated K+ channel gating modulators with selectivity for KCa3.1, Mol. Pharmacol., 2014, 86, 342–357 CrossRef PubMed.
- S. Grissmer, A. N. Nguyen, J. Aiyar, D. C. Hanson, R. J. Mather, G. A. Gutman, M. J. Karmilowicz, D. D. Auperin and K. G. Chandy, Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines, Mol. Pharmacol., 1994, 45, 1227–1234 CAS.
- C. Beeton, M. W. Pennington, H. Wulff, S. Singh, D. Nugent, G. Crossley, I. Khaytin, P. A. Calabresi, C. Y. Chen, G. A. Gutman and K. G. Chandy, Targeting effector memory T cells with a selective peptide inhibitor of Kv1.3 channels for therapy of autoimmune diseases, Mol. Pharmacol., 2005, 67, 1369–1381 CrossRef CAS PubMed.
- M. R. Tanner, M. W. Pennington, S. S. Chauhan, T. Laragione, P. S. Gulko and C. Beeton, KCa1.1 and Kv1.3 channels regulate the interactions between fibroblast-like synoviocytes and T lymphocytes during rheumatoid arthritis, Arthritis Res. Ther., 2019, 21, 6 CrossRef PubMed.
- R. Kumar and P. F. Siril, Ultrafine carbamazepine nanoparticles with enhanced water solubility and rate of dissolution, RSC Adv., 2014, 4, 48101–48108 RSC.
- A. Mitrović, J. Završnik, G. Mikhaylov, D. Knez, U. Pečar Fonović, P. Matjan Štefin, M. Butinar, S. Gobec, B. Turk and J. Kos, Evaluation of novel cathepsin-X inhibitors in vitro and in vivo and their ability to improve cathepsin-B-directed antitumor therapy, Cell. Mol. Life Sci., 2022, 79, 34 CrossRef PubMed.
- T. Kyllo, V. Singh, H. Shim, S. Latika, H. M. Nguyen, Y. J. Chen, E. Terry, H. Wulff and J. D. Erickson, Riluzole and novel naphthalenyl substituted aminothiazole derivatives prevent acute neural excitotoxic injury in a rat model of temporal lobe epilepsy, Neuropharmacology, 2023, 224, 109349 CrossRef CAS PubMed.
- C. M. John, R. K. Mallat, R. C. Mishra, G. George, V. Singh, J. D. Turnbull, C. S. Umeshappa, D. J. Kendrick, T. Kim, F. M. Fauzi, F. Visser, P. W. M. Fedak, H. Wulff and A. P. Braun, SKA-31, an activator of Ca2+-activated K+ channels, improves cardiovascular function in aging, Pharmacol. Res., 2020, 151, 104539 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra07330d |
| This journal is © The Royal Society of Chemistry 2024 |