
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design and synthesis of novel 2-(2-(4-bromophenyl)quinolin-4-yl)-1,3,4-oxadiazole derivatives as anticancer and antimicrobial candidates: in vitro and in silico studies†
Noha Ryada,
Ayman Abo Elmaatyb,
Samy Selim *c,
Mohammed S. Almuhayawid,
Soad K. Al Jaounie,
Mohamed S. Abdel-Azizf,
Arwa Sultan Alqahtanig,
Islam Zaki*hj and
Lina M. A. Abdel Ghanyi
*c,
Mohammed S. Almuhayawid,
Soad K. Al Jaounie,
Mohamed S. Abdel-Azizf,
Arwa Sultan Alqahtanig,
Islam Zaki*hj and
Lina M. A. Abdel Ghanyi
aPharmaceutical Organic Chemistry Department, College of Pharmaceutical Sciences and Drug Manufacturing, Misr University for Science and Technology, 6th of October City, P.O. Box 77, Giza, Egypt
bMedicinal Chemistry Department, Faculty of Pharmacy, Port Said University, Port Said 42526, Egypt
cDepartment of Clinical Laboratory Sciences, College of Applied Medical Sciences, Jouf University, Sakaka 72388, Saudi Arabia. E-mail: sabdulsalam@ju.edu.sa
dDepartment of Clinical Microbiology and Immunology, Faculty of Medicine, King Abdulaziz University, Jeddah 21589, Saudi Arabia
eDepartment of Hematology/Oncology, Yousef Abdulatif Jameel Scientific Chair of Prophetic Medicine Application, Faculty of Medicine, King Abdulaziz University, Jeddah 21589, Saudi Arabia
fMicrobial Chemistry Department, Biotechnology Research Institute, National Research Centre, Cairo, Egypt
gDepartment of Chemistry, College of Science, Imam Mohammad Ibn Saud Islamic University (IMSIU), P.O. Box, 90950, Riyadh 11623, Saudi Arabia
hPharmaceutical Organic Chemistry Department, Faculty of Pharmacy, Port Said University, Port Said 42526, Egypt. E-mail: eslam.zaki@pharm.psu.edu.eg
iPharmaceutical Chemistry Department, College of Pharmaceutical Sciences and Drug Manufacturing, Misr University for Science and Technology, 6th of October City, P.O. Box 77, Giza, Egypt
jPharmaceutical Organic Chemistry Department, Clinical Pharmacy Program, East Port Said National University, Port Said 42526, Egypt
First published on 25th October 2024
Abstract
Cancer is the second leading cause of death globally, surpassed only by heart disease. Moreover, bacterial infections remain a significant global health burden, contributing substantially to morbidity and mortality, especially among hospitalized patients. EGFR has emerged as a prime therapeutic target due to its pivotal role in driving uncontrolled cell growth and survival across numerous cancer types. In addition, DNA gyrase represents a promising target for the development of novel antimicrobial agents. Therefore, we aimed to design and synthesize new multi-target quinoline hybrids (7–17e) capable of acting as anti-proliferative and antimicrobial agents by inhibiting EGFR and microbial DNA gyrase, respectively. The inhibitory potential of the synthesized compounds was determined using in vitro and in silico approaches. The antiproliferative activity of the synthesized quinoline-oxadiazole derivatives 7–17e was assessed against two cancer cell lines, namely, hepatocellular carcinoma (HepG2) and breast adenocarcinoma (MCF-7). The assessed compounds 7–17e showed considerable cytotoxic activity activities against HepG2 and MCF-7 with IC50 values of 0.137–0.332 and 0.164–0.583 μg mL−1, respectively, in comparison to erlotinib as the positive control, which showed an IC50 value of 0.308 and 0.512 μg mL−1, respectively. Moreover, an EGFR tyrosine kinase inhibition assay was conducted on the most prominent candidates. The results showed good IC50 values of 0.14 and 0.18 μM for compounds 8c and 12d, respectively, compared to lapatinib (IC50 value of 0.12 μM). Furthermore, the minimum antimicrobial inhibitory concentration was evaluated for the most prominent candidates with S. aureus, E. coli, and C. albicans. Compounds 17b, 17d and 17e displayed the most potent inhibitory activity, exhibiting 4-, 16- and 8-fold more activity, respectively, than the reference neomycin. Hence, we can conclude that the afforded compounds can be used as lead dual anticancer and antimicrobial candidates for future optimization.
1. Introduction
Despite significant advancements in cancer treatment, the disease remains a major global health challenge. The increasing cancer burden is disproportionately affecting lower- and middle-income countries, underscoring the complex interplay between socioeconomic status and health outcomes.1 Cancer is the second leading cause of death globally, surpassed only by heart disease. This stark reality underscores the urgent need for continued research and development of effective prevention, diagnostic, and therapeutic strategies.2 Furthermore, the limited selectivity of chemotherapeutic agents often results in adverse side effects, including immunosuppression, nausea, anemia, and hair loss. These off-target toxicities highlight the critical challenge of balancing efficacy against cancer cells with minimal harm to healthy tissues.3 In response to the urgent need for improved cancer treatments, researchers worldwide are actively pursuing innovative therapeutic strategies. Development of more effective and targeted therapies with reduced side effects remains a primary focus of cancer research.Protein kinases (PKs) are essential enzymes that regulate a wide range of critical cellular processes, including metabolism, cell growth, survival, and death. Their pivotal role in cellular signaling pathways has made them prime targets for therapeutic interventions, particularly in cancer research.4 Protein kinases catalyze the transfer of a phosphate group from ATP to specific hydroxyl residues of amino acids, such as serine, threonine, or tyrosine, on target proteins, a process known as phosphorylation. This crucial phosphorylation process regulates a wide range of cellular functions through intricate signaling networks.5 Consequently, aberrant kinase activity, resulting from either hyper activation or mutations, disrupts critical cellular signaling pathways, contributing to the pathogenesis of various diseases, including cancer.6
The epidermal growth factor receptor (EGFR) is a prominent protein kinase that plays a pivotal role in regulating cell proliferation and migration.7 Many solid tumors, such as non-small cell lung cancer,8 hepatocellular carcinoma,9 and breast cancer,4 overexpress EGFR. Recent advancements in cancer therapy have focused on targeting specific molecules that regulate cancer cell growth and survival.10,11 Consequently, EGFR has emerged as a prime therapeutic target due to its pivotal role in driving uncontrolled cell growth and survival across numerous cancer types.12–17
Erlotinib and gefitinib are examples of first-generation EGFR tyrosine kinase inhibitors.18–21 However, their efficacy can be compromised by the development of resistance mechanisms, such as the EGFR-T790M mutation, which diminishes their anticancer potency.22 To address the emergence of resistance associated with first-generation EGFR tyrosine kinase inhibitors, second-generation EGFR tyrosine kinase inhibitors (e.g., pelitinib and neratinib) were developed.23–28 These drugs have equal affinities towards the wild-type EGFR (WT) and mutant EGFR (EGFR-T790M), resulting in rash and diarrhea.29 Hence, the maximal tolerated dose (MTD) displayed by these drugs30,31 has led to the emergence of third-generation irreversible EGFR-tyrosine kinase inhibitors (e.g., osimertinib and olmutinib),32–35 as shown in Fig. 1. Recently, fourth-generation EGFR tyrosine kinase inhibitors (TKIs) have emerged as a novel therapeutic strategy to address the challenge of acquired resistance mediated by the EGFR C797S mutation, and were subjected to further clinical evaluations.36 Fourth-generation EGFR TKIs offer a novel approach to overcoming resistance to EGFR inhibitors by targeting a distinct binding site on the receptor (allosteric inhibitors). This allosteric mechanism of action differentiates them from previous generations of ATP-competitive inhibitors.36 The continuous emergence of resistance mechanisms underscores the urgent need for innovative strategies to develop novel EGFR inhibitors with enhanced efficacy and safety profiles.
 | ||
| Fig. 1 Some FDA-approved quinoline antibiotics and EGFR TK inhibitors, with their different generations as well as their drawbacks. | ||
However, bacterial infections remain a significant global health burden, contributing substantially to morbidity and mortality, especially among hospitalized patients.37 Despite the availability of numerous antimicrobial agents, their effectiveness is often compromised by the emergence of bacterial resistance, limiting their clinical utility.38 Antibiotic resistance is a pressing global health crisis, contributing to an estimated 700![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths annually.39 Without significant advancements in antimicrobial strategies, drug-resistant infections are projected to claim an estimated 10 million lives annually by 2050.39 Hence, to address the growing problem of bacterial infections, scientists are desperately searching for new antibiotics that can effectively fight both common and antibiotic-resistant bacteria. These new agents have attracted significant interest in medicinal chemistry research and offer a promising solution to the urgent need for more effective treatments.37 Bacterial DNA gyrase, an essential type II topoisomerase, plays a critical role in DNA replication and transcription by introducing negative supercoils into DNA.40,41 Given its pivotal role in bacterial survival, DNA gyrase represents a promising target for the development of novel antimicrobial agents. Quinolines (e.g., ciprofloxacin, moxifloxacin, and ofloxacin) have been identified as potent inhibitors of DNA gyrase (Fig. 1). By targeting this essential enzyme, quinolines can effectively disrupt bacterial DNA replication and recombination, leading to cell death.42
000 deaths annually.39 Without significant advancements in antimicrobial strategies, drug-resistant infections are projected to claim an estimated 10 million lives annually by 2050.39 Hence, to address the growing problem of bacterial infections, scientists are desperately searching for new antibiotics that can effectively fight both common and antibiotic-resistant bacteria. These new agents have attracted significant interest in medicinal chemistry research and offer a promising solution to the urgent need for more effective treatments.37 Bacterial DNA gyrase, an essential type II topoisomerase, plays a critical role in DNA replication and transcription by introducing negative supercoils into DNA.40,41 Given its pivotal role in bacterial survival, DNA gyrase represents a promising target for the development of novel antimicrobial agents. Quinolines (e.g., ciprofloxacin, moxifloxacin, and ofloxacin) have been identified as potent inhibitors of DNA gyrase (Fig. 1). By targeting this essential enzyme, quinolines can effectively disrupt bacterial DNA replication and recombination, leading to cell death.42
Furthermore, quinoline and oxadiazole, privileged scaffolds in medicinal chemistry,43 have been extensively investigated for its diverse biological properties in numerous research endeavors exhibiting a wide range of pharmacological activities, making them a versatile scaffold for drug discovery, including anticancer,44–49 anti-viral,50–52 anti-microbial,53–57 anti-diabetic,58–60 and anti-inflammatory activities.61–63 In particular, the literature revealed that quinoline oxadiazole hybrids were utilized as antimicrobial and/or anti-proliferative agents.42,64,65
1.1. Design rationale
The EGFR-TK pocket, where ATP binds, comprises five main key regions: (a) the adenine binding site, responsible for hydrogen bonding with the ATP adenine moiety; (b) the hydrophilic sugar binding region; (c) hydrophobic region I, critical for inhibitor selectivity; (d) hydrophobic region II, contributing to inhibitor specificity; and (e) the phosphate binding region, which influences inhibitor pharmacokinetics.66 A comprehensive understanding of the EGFR-TK binding pocket's structural features is essential for the rational design of potent and selective EGFR inhibitors.7 EGFR-TK inhibitors like erlotinib share specific structural features that allow them to bind effectively to the EGFR-TK enzyme. These features include a hydrophobic head fitting into hydrophobic region I, a –NH spacer, a flat heteroaromatic ring system fitting into the adenine binding site and composing hydrogen bonds with amino acids Thr854, Met793, and Thr790, and a hydrophobic tail fitting into the hydrophobic region II. These common pharmacophores enable these inhibitors to interact effectively with EGFR-TK to block its activity.7 Herein, via application of a molecular hybridization approach to attain all crucial pharmacophoric features, the hydrophobic head of erlotinib was replaced by diverse (un)substituted aryl, thio aryl, alicyclic derivatives for SAR studies. Moreover, the –NH spacer of erlotinib was replaced by a 1,3,4-oxadiazole ring, whereas the flat heteroaromatic quinazoline ring was replaced by the quinoline ring (Fig. 2). In addition, the hydrophobic tail of erlotinib was replaced by a 4-bromo phenyl moiety to afford the designed compounds (8a–17e). However, previous studies have demonstrated the promising antimicrobial potential of 2-phenylquinoline derivatives, highlighting their potential as scaffolds for novel antibacterial agents.42 Additionally, attaching the 2-phenyl quinoline scaffold with 1,3,4-oxadiazole motifs could afford molecular hybrids with broad-spectrum antimicrobial activity42 (Fig. 2). Hence, in this work, we hoped to design and synthesize a new set of 2-(2-phenylquinolin-4-yl)-1,3,4-oxadiazole hybrids that would act as both anticancer agents (targeting EGFR-TK), as well as antimicrobial agents. The synthesized quinoline-oxadiazole molecules were pursued using in vitro and in silico approaches for their anti-proliferative and antimicrobial properties. | ||
| Fig. 2 The design rationale of the synthesized compounds (7a–17e) as antiproliferative and antimicrobial agents. | ||
2. Results and discussion
2.1. Chemistry
The synthetic pathways 2 and 3 were used to create the target products 7–17a–e, with Scheme 1 showing the synthesis of the starting compounds. In Scheme 2, the acid hydrazide 3 is regarded as the important intermediate. Treatment of compound 3 with triethyl orthoformate afforded compound 6,67 which was cyclized upon heating in an oil bath at 10 °C above its melting point to give 2-(2-(4-bromophenyl)quinolin-4-yl)-1,3,4-oxadiazole (7) in 61% isolated yield. The IR spectrum of the obtained compound showed the disappearance of the NH and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching bands. The 1H NMR spectrum revealed the characteristic singlet signal at δ 9.57 ppm assigned to the proton of the oxadiazole ring. The 13C NMR spectrum showed the disappearance of signals corresponding to the ethoxy group, and appearance of two signals at δ 154.76 and 167.19 ppm that refer to C5 and C2 of the oxadiazole moiety, respectively. When the acid hydrazide 3 was reacted with appropriate aromatic carboxylic acid derivatives in the presence of phosphorous oxychloride, 5-substituted phenyl-1,3,4-oxadiazoles 8a–e were formed in 62–69% yield. The prepared target molecules were identified by 1H NMR spectra, which revealed the fading of two signals of NH and NH2 present in the spectra of the parent hydrazide 3, and appearance of signals corresponding to the protons of the added aromatic rings at the expected chemical shift. Heating the acid hydrazide 3 with carbon disulfide in an ethanolic solution of potassium hydroxide resulted in the formation of the 1,3,4-oxadiazole-2-thiol derivative 9 in 83% yield. The IR spectrum of the obtained compound showed the presence of the NH stretching band at 3155 cm−1 and a band at 1238 cm−1 corresponding to CS, while the 1H NMR spectrum showed singlet signals at δ 11.13 ppm that corresponded to the exchangeable SH proton. Furthermore, Scheme 3 describes how new molecules 10–17a–e were created using compound 9 as a crucial intermediate. S-(5-(2-(4-Bromophenyl)quinolin-4-yl)-1,3,4-oxadiazol-2-yl) benzothioate (10) was prepared in good yield by stirring oxadiazole-2-thiol 9 with benzoyl chloride in dioxane. The IR spectrum of the synthesized compound showed the absence of an absorption band corresponding to the NH group, and the presence of an absorption band at 1707 cm−1 that referred to the CO group. The 1H NMR spectrum exhibited the presence of an additional 5 aromatic protons and the absence of the SH proton, thereby indicating the presence of the benzoyl moiety. The 13C NMR spectrum detected the appearance of a signal referring to the CO group at δ 178.86 ppm as well, indicating the elevated number of aromatic carbons of the benzoyl moiety. Heating compound 9 with benzyl chloride in acetone containing K2CO3 afforded compound 11 in 70% yield. The 1H NMR spectrum exhibited the presence of a singlet signal at δ 4.62 ppm due to the CH2 protons of the benzyl moiety, in addition to signals of the aromatic protons. On the other hand, the 13C NMR spectrum showed a CH2 signal at δ 33.16 ppm, along with signals of the aromatic carbons. 2-(2-(4-Bromophenyl)quinolin-4-yl)-5-(substituted thio)-1,3,4-oxadiazole (12a–d) were prepared in 69–73% yield via alkylation of compound 9 with different alkyl halides in ethanol and KOH. The structures of the synthesized compounds were elucidated by 1H NMR and 13C NMR spectra. The 1H NMR spectra showed the disappearance of the signal corresponding to the SH group, and the appearance of a singlet signal at δ 2.87 ppm referring to the CH3 group in compound 12a. Meanwhile, compound 12b showed triplet and quartet signals at δ 1.49 and 3.31–3.44 ppm, which correspond to the CH3 and CH2- groups, respectively. Furthermore, the allyl group in compound 12c appeared as four signals, a doublet signal at δ 4.10 ppm that corresponds to the S-CH2 protons, two doublet signals of CH
O stretching bands. The 1H NMR spectrum revealed the characteristic singlet signal at δ 9.57 ppm assigned to the proton of the oxadiazole ring. The 13C NMR spectrum showed the disappearance of signals corresponding to the ethoxy group, and appearance of two signals at δ 154.76 and 167.19 ppm that refer to C5 and C2 of the oxadiazole moiety, respectively. When the acid hydrazide 3 was reacted with appropriate aromatic carboxylic acid derivatives in the presence of phosphorous oxychloride, 5-substituted phenyl-1,3,4-oxadiazoles 8a–e were formed in 62–69% yield. The prepared target molecules were identified by 1H NMR spectra, which revealed the fading of two signals of NH and NH2 present in the spectra of the parent hydrazide 3, and appearance of signals corresponding to the protons of the added aromatic rings at the expected chemical shift. Heating the acid hydrazide 3 with carbon disulfide in an ethanolic solution of potassium hydroxide resulted in the formation of the 1,3,4-oxadiazole-2-thiol derivative 9 in 83% yield. The IR spectrum of the obtained compound showed the presence of the NH stretching band at 3155 cm−1 and a band at 1238 cm−1 corresponding to CS, while the 1H NMR spectrum showed singlet signals at δ 11.13 ppm that corresponded to the exchangeable SH proton. Furthermore, Scheme 3 describes how new molecules 10–17a–e were created using compound 9 as a crucial intermediate. S-(5-(2-(4-Bromophenyl)quinolin-4-yl)-1,3,4-oxadiazol-2-yl) benzothioate (10) was prepared in good yield by stirring oxadiazole-2-thiol 9 with benzoyl chloride in dioxane. The IR spectrum of the synthesized compound showed the absence of an absorption band corresponding to the NH group, and the presence of an absorption band at 1707 cm−1 that referred to the CO group. The 1H NMR spectrum exhibited the presence of an additional 5 aromatic protons and the absence of the SH proton, thereby indicating the presence of the benzoyl moiety. The 13C NMR spectrum detected the appearance of a signal referring to the CO group at δ 178.86 ppm as well, indicating the elevated number of aromatic carbons of the benzoyl moiety. Heating compound 9 with benzyl chloride in acetone containing K2CO3 afforded compound 11 in 70% yield. The 1H NMR spectrum exhibited the presence of a singlet signal at δ 4.62 ppm due to the CH2 protons of the benzyl moiety, in addition to signals of the aromatic protons. On the other hand, the 13C NMR spectrum showed a CH2 signal at δ 33.16 ppm, along with signals of the aromatic carbons. 2-(2-(4-Bromophenyl)quinolin-4-yl)-5-(substituted thio)-1,3,4-oxadiazole (12a–d) were prepared in 69–73% yield via alkylation of compound 9 with different alkyl halides in ethanol and KOH. The structures of the synthesized compounds were elucidated by 1H NMR and 13C NMR spectra. The 1H NMR spectra showed the disappearance of the signal corresponding to the SH group, and the appearance of a singlet signal at δ 2.87 ppm referring to the CH3 group in compound 12a. Meanwhile, compound 12b showed triplet and quartet signals at δ 1.49 and 3.31–3.44 ppm, which correspond to the CH3 and CH2- groups, respectively. Furthermore, the allyl group in compound 12c appeared as four signals, a doublet signal at δ 4.10 ppm that corresponds to the S-CH2 protons, two doublet signals of CH![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2 protons at δ 5.24 and 5.43 ppm, in which J = 10 Hz and 17.2 Hz correspond to cis and trans protons, respectively, due to the vicinal coupling with these non-equivalent protons, and a multiplet signal referring to the HCH2 protons at δ 6.00–6.13 ppm. However, compound 12d was confirmed through the appearance of a doublet signal at δ 4.28 ppm due to S-CH2 protons, a multiplet signal at δ 6.50–6.54 ppm due to the H–CH2 proton, doublet signal at δ 6.80 ppm due to H-Ph, and the protons of the phenyl group appeared at δ 7.23–8.43 ppm. In the 13C NMR spectra, the signal of CH3 in compound 12a appeared at δ 14.92 ppm. In contrast, compound 12b displayed peaks at δ 14.87 and 27.80 ppm due to the CH3–CH2-group. Meanwhile, compound 12c displayed signals at δ 34.90 ppm due to the SCH2 carbon and two signals at 118.10 and 133.07 ppm due to CH2CH carbons, respectively. Lastly, the presence of signals at δ 35.35, 124.93 and 134.46 pointing to SCH2, CHCH-Ph, respectively, together with the carbons of the phenyl moiety, elucidated the structure of compound 12d. Likewise, heating compound 9 with 2-chloroacetic acid in methylene chloride containing a few drops of TEA led to the formation of compound 13. Moreover, the IR spectrum of the attained compound showed the appearance of the OH and CO stretching bands of the carboxylic group at 3419 and 1716 cm−1, respectively. The 1H NMR spectrum revealed characteristic singlet signals at δ 4.19 and 5.72 ppm assigned to CH2 protons and the exchangeable proton of OH, respectively. The 13C NMR spectrum showed two signals at δ 36.62 and 169.00 ppm that refer to CH2 and CO, respectively. Also, N-(4-acetylphenyl)-2-((5-(2-(4-bromophenyl)quinolin-4-yl)-1,3,4-oxadiazol-2-yl)thio)acetamide (14) was prepared in 86% yield via reaction of compound 9 with N-(4-acetylphenyl)-2-chloroacetamide (4). Two stretching bands at 3446 and 1670 cm−1 emerged in the IR spectrum of compound 14 pertaining to the NH and CO groups, respectively. The 1H NMR spectrum detected distinct singlet signals at δ 2.57 and 4.51 ppm assigned to COCH3 and CH2 protons, respectively, as well as an exchangeable NH proton at δ 10.90 ppm. Similarly, heating 1,3,4-oxadiazole 9 with the corresponding acetamide derivative 5a,b afforded 2-((5-(2-(4-bromophenyl)quinolin-4-yl)-1,3,4-oxadiazol-2-yl)thio)-N-arylacetamide (15a,b) in 50–55% yield. The structure of the synthesized compounds was verified by IR spectra, which showed the appearance of stretching bands from the NH and CO groups at 3429–3446 cm−1 and 1651–1672 cm−1, respectively. The 1H NMR spectrum of compound 15b revealed a singlet signal at δ 4.24–4.27 ppm assigned to CH2 protons, a singlet signal at δ 10.75–10.96 ppm pointing to the exchangeable NH proton, and a singlet signal at δ 3.83 ppm corresponding to the OCH3 protons. The 13C NMR spectrum of
compound 15b displayed two signals at δ 33.63–34.00 and 166.67–168.03 ppm that correspond to CH2 and CO, respectively, and a signal at δ 56.06 belonging to OCH3. The ester derivative 16 was prepared in 87% yield by heating compound 9 under reflux with ethyl chloroacetate and anhydrous potassium carbonate in dry acetone. The IR spectrum of the obtained compound showed a stretching band of CO groups at 1739 cm−1. Meanwhile, the 1H NMR spectrum showed signals at δ 1.20 (triplet) and 4.17–4.22 ppm (quartet), corresponding to the CH3- and CH2-groups, respectively, and a singlet signal at δ 4.41 ppm that was assigned to CH2 protons. The 13C NMR spectrum showed signals for CH3, CH2, and OCH2 at δ 14.14 ppm, 32.14 ppm, and 62.29 ppm, respectively. Finally, a new series of Mannich bases from the 1,3,4-oxadiazole derivative was synthesized in 74–82% yield by reacting the 1,3,4-oxadiazole derivative 9 with formaldehyde and an appropriate secondary amine (morpholine, piperidine, piperazine, methylpiperazine and diphenyl amine). The structure of the synthesized compounds was demonstrated by 1H NMR spectrum, which showed a singlet signal at δ 5.14–5.23 ppm corresponding to CH2 protons, along with the added protons of either morpholine, piperidine, piperazine, methylpiperazine, or diphenyl moieties at the expected chemical shift. The 13C NMR spectrum showed a signal for CH2 at δ 69.34–83.48 ppm, and the signal of the respective carbons of the prepared compounds was verified on the basis of their chemical shift. The target compound's molecular ion peaks, which matched their calculated molecular weights, also provided more evidence of their structure, along with the elemental analyses of their CHN components.
H2 protons at δ 5.24 and 5.43 ppm, in which J = 10 Hz and 17.2 Hz correspond to cis and trans protons, respectively, due to the vicinal coupling with these non-equivalent protons, and a multiplet signal referring to the HCH2 protons at δ 6.00–6.13 ppm. However, compound 12d was confirmed through the appearance of a doublet signal at δ 4.28 ppm due to S-CH2 protons, a multiplet signal at δ 6.50–6.54 ppm due to the H–CH2 proton, doublet signal at δ 6.80 ppm due to H-Ph, and the protons of the phenyl group appeared at δ 7.23–8.43 ppm. In the 13C NMR spectra, the signal of CH3 in compound 12a appeared at δ 14.92 ppm. In contrast, compound 12b displayed peaks at δ 14.87 and 27.80 ppm due to the CH3–CH2-group. Meanwhile, compound 12c displayed signals at δ 34.90 ppm due to the SCH2 carbon and two signals at 118.10 and 133.07 ppm due to CH2CH carbons, respectively. Lastly, the presence of signals at δ 35.35, 124.93 and 134.46 pointing to SCH2, CHCH-Ph, respectively, together with the carbons of the phenyl moiety, elucidated the structure of compound 12d. Likewise, heating compound 9 with 2-chloroacetic acid in methylene chloride containing a few drops of TEA led to the formation of compound 13. Moreover, the IR spectrum of the attained compound showed the appearance of the OH and CO stretching bands of the carboxylic group at 3419 and 1716 cm−1, respectively. The 1H NMR spectrum revealed characteristic singlet signals at δ 4.19 and 5.72 ppm assigned to CH2 protons and the exchangeable proton of OH, respectively. The 13C NMR spectrum showed two signals at δ 36.62 and 169.00 ppm that refer to CH2 and CO, respectively. Also, N-(4-acetylphenyl)-2-((5-(2-(4-bromophenyl)quinolin-4-yl)-1,3,4-oxadiazol-2-yl)thio)acetamide (14) was prepared in 86% yield via reaction of compound 9 with N-(4-acetylphenyl)-2-chloroacetamide (4). Two stretching bands at 3446 and 1670 cm−1 emerged in the IR spectrum of compound 14 pertaining to the NH and CO groups, respectively. The 1H NMR spectrum detected distinct singlet signals at δ 2.57 and 4.51 ppm assigned to COCH3 and CH2 protons, respectively, as well as an exchangeable NH proton at δ 10.90 ppm. Similarly, heating 1,3,4-oxadiazole 9 with the corresponding acetamide derivative 5a,b afforded 2-((5-(2-(4-bromophenyl)quinolin-4-yl)-1,3,4-oxadiazol-2-yl)thio)-N-arylacetamide (15a,b) in 50–55% yield. The structure of the synthesized compounds was verified by IR spectra, which showed the appearance of stretching bands from the NH and CO groups at 3429–3446 cm−1 and 1651–1672 cm−1, respectively. The 1H NMR spectrum of compound 15b revealed a singlet signal at δ 4.24–4.27 ppm assigned to CH2 protons, a singlet signal at δ 10.75–10.96 ppm pointing to the exchangeable NH proton, and a singlet signal at δ 3.83 ppm corresponding to the OCH3 protons. The 13C NMR spectrum of
compound 15b displayed two signals at δ 33.63–34.00 and 166.67–168.03 ppm that correspond to CH2 and CO, respectively, and a signal at δ 56.06 belonging to OCH3. The ester derivative 16 was prepared in 87% yield by heating compound 9 under reflux with ethyl chloroacetate and anhydrous potassium carbonate in dry acetone. The IR spectrum of the obtained compound showed a stretching band of CO groups at 1739 cm−1. Meanwhile, the 1H NMR spectrum showed signals at δ 1.20 (triplet) and 4.17–4.22 ppm (quartet), corresponding to the CH3- and CH2-groups, respectively, and a singlet signal at δ 4.41 ppm that was assigned to CH2 protons. The 13C NMR spectrum showed signals for CH3, CH2, and OCH2 at δ 14.14 ppm, 32.14 ppm, and 62.29 ppm, respectively. Finally, a new series of Mannich bases from the 1,3,4-oxadiazole derivative was synthesized in 74–82% yield by reacting the 1,3,4-oxadiazole derivative 9 with formaldehyde and an appropriate secondary amine (morpholine, piperidine, piperazine, methylpiperazine and diphenyl amine). The structure of the synthesized compounds was demonstrated by 1H NMR spectrum, which showed a singlet signal at δ 5.14–5.23 ppm corresponding to CH2 protons, along with the added protons of either morpholine, piperidine, piperazine, methylpiperazine, or diphenyl moieties at the expected chemical shift. The 13C NMR spectrum showed a signal for CH2 at δ 69.34–83.48 ppm, and the signal of the respective carbons of the prepared compounds was verified on the basis of their chemical shift. The target compound's molecular ion peaks, which matched their calculated molecular weights, also provided more evidence of their structure, along with the elemental analyses of their CHN components.
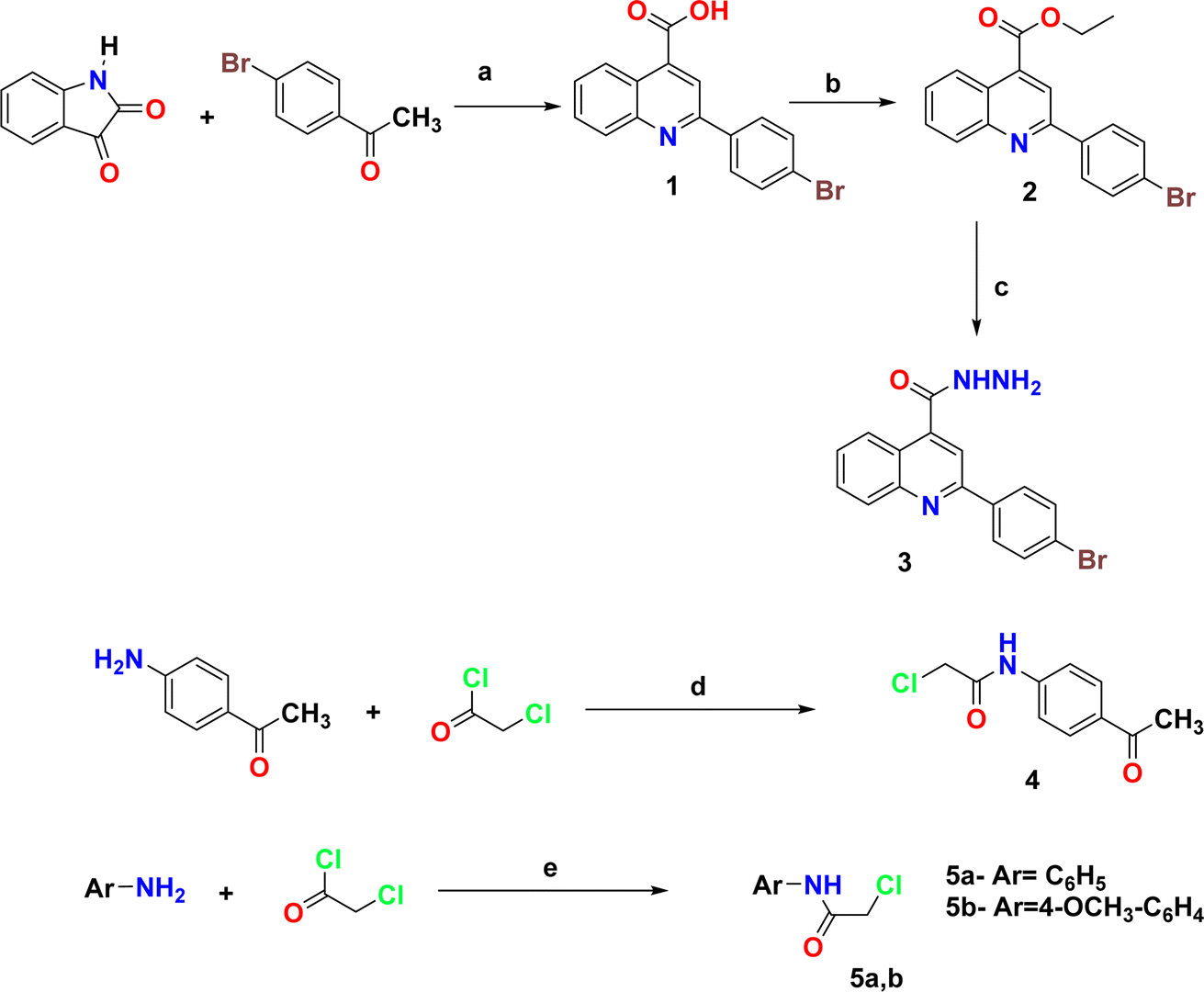 | ||
| Scheme 1 Synthesis of the starting compounds. Reagents and condition: (a) 33% KOH, 96% EtOH, reflux 12 h; (b) absolute EtOH, Conc. H2SO4, reflux 12 h; (c) NH2NH2·H2O, absolute EtOH, reflux 7 h; (d) CH2Cl2, TEA, 12, r.t.; (e) glacial AcOH, NaOAc, stirring overnight at r.t. | ||
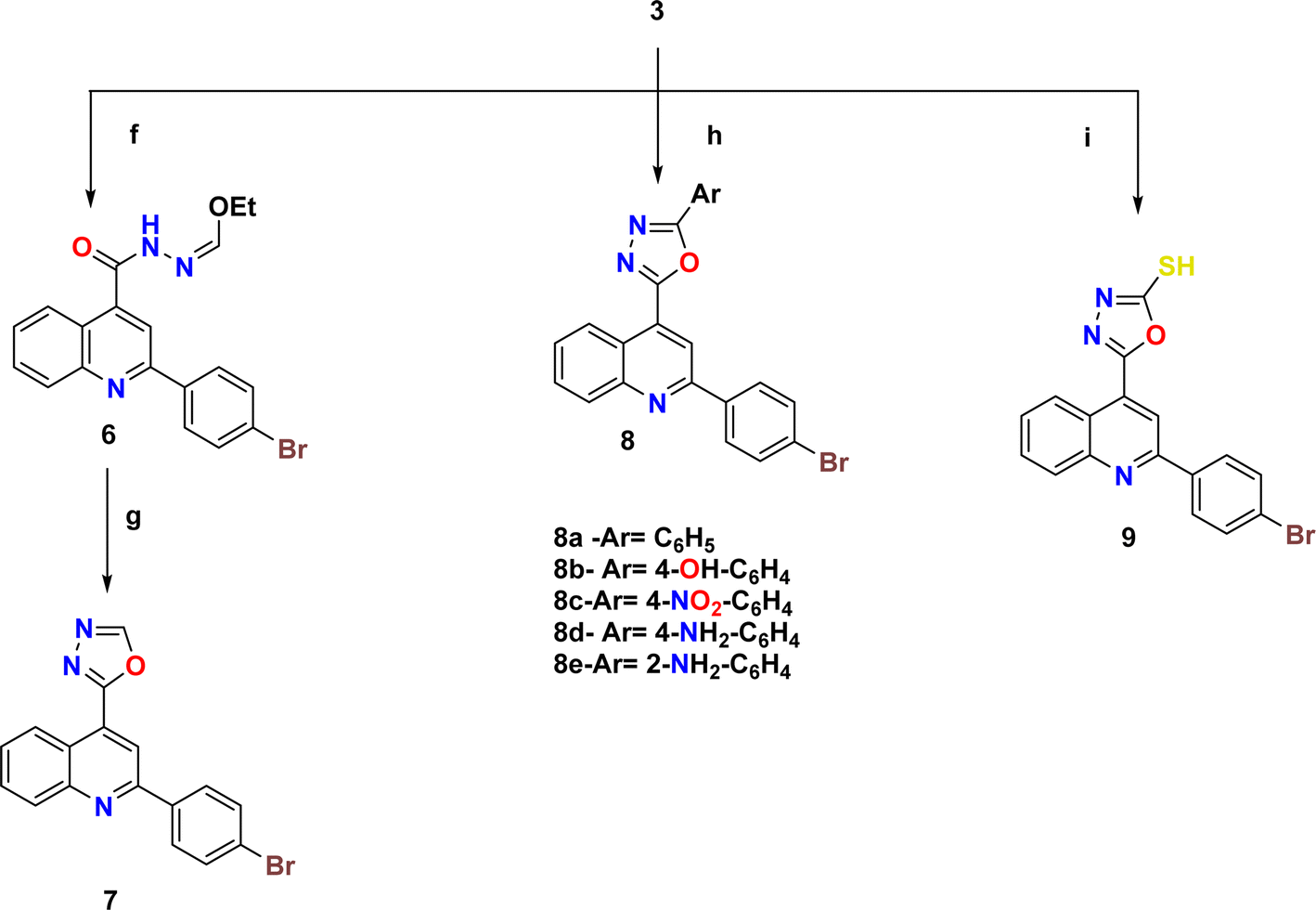 | ||
| Scheme 2 Synthetic pathway for compounds 7–9. Reagents and condition: (f) triethyl orthoformate, reflux 6 h; (g) fusion 30 min; (h) carboxylic acid derivatives, POCl3, reflux 6–8 h; (i) KOH, CS2, absolute EtOH, reflux, 12 h. | ||
 | ||
| Scheme 3 Synthetic pathway for compounds 10–17e. Reagents and conditions: (j) benzoyl chloride, dioxane, stirring overnight; (k) benzyl chloride, anhydrous K2CO3, acetone, reflux 6 h; (l) alkyl halide derivatives, KOH, aqueous EtOH, reflux 50–60 °C, 4–6 h; (m) chloroacetic acid, TEA, CH2Cl2, reflux 6 h; (n) TEA, CH2Cl2, reflux 6 h; (o) KOH, aqueous EtOH, reflux 6–8 h; (p) ethyl chloroacetate, anhydrous K2CO3, acetone, reflux 5 h; (q) 2ry amine derivatives, KOH, absolute EtOH, 36% HCHO reflux 4–6 h. | ||
2.2. Biological evaluation
| Compd no. | IC50 (μg mL−1) | |
|---|---|---|
| HepG-2 | MCF-7 | |
| 7 | 0.151 | 0.295 |
| 8a | 0.215 | 0.440 |
| 8b | 0.139 | 0.225 |
| 8c | 0.137 | 0.481 |
| 8d | 0.311 | 0.287 |
| 8e | 0.157 | 0.179 |
| 9 | 0.152 | 0.358 |
| 10 | 0.217 | 0.446 |
| 11 | 0.141 | 0.227 |
| 12a | 0.138 | 0.473 |
| 12b | 0.327 | 0.287 |
| 12c | 0.158 | 0.239 |
| 12d | 0.138 | 0.473 |
| 13 | 0.332 | 0.287 |
| 14 | 0.159 | 0.217 |
| 15a | 0.272 | 0.164 |
| 15b | 0.154 | 0.411 |
| 17a | 0.141 | 0.300 |
| 17b | 0.188 | 0.583 |
| 17c | 0.164 | 0.221 |
| 17d | 0.254 | 0.406 |
| 17e | 0.139 | 0.569 |
| Erlotinib | 0.308 | 0.512 |
 | ||
| Fig. 3 Influence of quinoline molecules 8c and 12d on the cellular cycle proportion following the HepG2 cancerous cell line treatment relative to untreated cells. | ||
 | ||
| Fig. 4 Influence of quinoline compounds 8c and 12d on the apoptosis concentration, following staining with Annexin V/PI in HepG2 cells relative to untreated cells. | ||
 | ||
| Fig. 5 The IC50 (μM) of the target quinoline molecules 8c and 12d against EGFR kinase activity compared to lapatinib. | ||
 | ||
| Fig. 6 Influence of the quinoline-oxadiazole compounds 8c and 12d on apoptosis-associated proteins after 48 h treatment in HepG2 cells. | ||
| Comp. no. | Gram+ bacteria | Gram− bacteria | Fungi | |
|---|---|---|---|---|
| S. aureus | E. coli | C. albicans | A. niger | |
| 7 | 12 | 14 | 13 | 0 |
| 8a | 0 | 0 | 0 | 0 |
| 8b | 0 | 0 | 0 | 0 |
| 8c | 0 | 0 | 0 | 0 |
| 8d | 0 | 0 | 0 | 0 |
| 8e | 0 | 0 | 0 | 0 |
| 9 | 20 | 23 | 24 | 0 |
| 10 | 25 | 30 | 28 | 0 |
| 11 | 14 | 17 | 14 | 0 |
| 12a | 0 | 0 | 0 | 0 |
| 12b | 0 | 0 | 0 | 0 |
| 12c | 0 | 0 | 0 | 0 |
| 12d | 0 | 0 | 0 | 0 |
| 13 | 0 | 0 | 0 | 0 |
| 14 | 0 | 0 | 0 | 0 |
| 15a | 0 | 0 | 0 | 0 |
| 15b | 0 | 0 | 0 | 0 |
| 16 | 0 | 0 | 0 | 0 |
| 17a | 37 | 35 | 37 | 0 |
| 17b | 33 | 34 | 36 | 0 |
| 17c | 35 | 36 | 35 | 0 |
| 17d | 34 | 37 | 36 | 0 |
| 17e | 29 | 32 | 30 | 0 |
| Neomycin | 26 | 24 | 30 | — |
| Cycloheximide | 0 | 21 | ||
| Comp. no. | MIC (μg mL−1) | ||
|---|---|---|---|
| S. aureus | E. coli | C. albicans | |
| 9 | 39.06 | 625 | 19.53 |
| 10 | 39.06 | 312.5 | 19.53 |
| 11 | 78.125 | 625 | 39.06 |
| 17a | 39.06 | 156.25 | 4.88 |
| 17b | 19.53 | 312.5 | 9.77 |
| 17c | 78.125 | 312.5 | 4.88 |
| 17d | 4.88 | 312.5 | 9.77 |
| 17e | 9.77 | 312.5 | 4.88 |
| Neomycin | 78.125 | 39.06 | 156.25 |
| Comp. no. | MBC (μg mL−1) | ||
|---|---|---|---|
| S. aureus | E. coli | C. albicans | |
| 9 | 78.125 | 1250 | 39.06 |
| 10 | 156.25 | 625 | 19.53 |
| 11 | 312.5 | 625 | 78.125 |
| 17a | 39.06 | 625 | 4.88 |
| 17b | 39.06 | 625 | 9.77 |
| 17c | 312.5 | 312.5 | 9.77 |
| 17d | 19.53 | 1250 | 39.06 |
| 17e | 19.53 | 1250 | 9.77 |
| Neomycin | 312.5 | 156.25 | 625 |
| Comp. no. | MIC of biofilm inhibition (μg mL−1) | ||
|---|---|---|---|
| S. aureus | E. coli | C. albicans | |
| 9 | 39.06 | 19.53 | 39.06 |
| 10 | 78.125 | 9.77 | 19.53 |
| 11 | 9.77 | 9.77 | 19.53 |
| 17a | 1250 | 1250 | 1250 |
| 17b | 1250 | 1250 | 1250 |
| 17c | 19.53 | 9.77 | 9.77 |
| 17d | 625 | 1250 | 1250 |
| 17e | 156.53 | 156.53 | 78.125 |
| Neomycin | 312.5 | 156.25 | 625 |
2.3. In silico studies
Accordingly, regarding their physicochemical features, except for compounds (8c, 10, 11, 12d, 14, 15a, 15b, and 17e), all synthesized compounds displayed high GIT absorption due to their feasible lipophilicity. Therefore, eligible oral bio-availabilities can be anticipated.73,74 Moreover, except for compound (7), all afforded compounds cannot pass through the blood–brain barrier. Thus, fewer CNS side effects can be assumed. Notably, compounds 7, 8c, 9, 10, 12a, 12c, 13, 14, 15a, 15b, 16, and 17e are not P-glycoprotein (Pgp-) substrates (Tables 6, 7, and 8). Hence, better bioavailability could be assured, as shown in Fig. 7. Moreover, compound 8c did not show inhibition for the common hepatic metabolizing enzymes (CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4). Except for compounds 12d, 15a, and 17e, all of the synthesized quinoline derivatives match the Lipinski's rule of five,75 assuring their oral bioavailability. To further evaluate the compounds' bioavailability, we utilized the bioavailability radar tool provided by SwissADME. This visual representation offers a comprehensive assessment of drug-like properties within a hexagonal framework. Compounds falling within the optimal physicochemical space defined by the radar are considered to have favorable oral bioavailability. These radar plots are shown in the supplementary Fig. SI5.†
| Comp. 7 | Comp. 8a | Comp. 8b | Comp. 8c | Comp. 8d | Comp. 8e | Comp. 9 | Comp. 10 | ||
|---|---|---|---|---|---|---|---|---|---|
| Molecular properties | Molar refractivity | 88.17 | 113.61 | 115.63 | 122.43 | 118.01 | 118.01 | 95.42 | 124.80 |
| TPSA (Az) | 51.81 | 51.81 | 72.04 | 97.63 | 77.83 | 77.83 | 90.61 | 94.18 | |
| logP o/w (WLOGP) | 4.71 | 6.38 | 6.09 | 6.29 | 5.97 | 5.97 | 5.00 | 6.65 | |
| Consensus logP o/w | 3.96 | 5.39 | 5.01 | 4.61 | 4.84 | 4.84 | 4.29 | 5.47 | |
| Water solubility | MS | PS | PS | PS | PS | PS | MS | PS | |
| Pharmacokinetics parameters | GI absorption | High | High | High | Low | High | High | High | Low |
| BBB permeant | Yes | No | No | No | No | No | No | No | |
| P-gp substrate | No | Yes | Yes | No | Yes | Yes | No | No | |
| CYP1A2 inhibitor | Yes | No | No | No | No | No | Yes | No | |
| CYP2C19 inhibitor | Yes | Yes | No | No | Yes | Yes | Yes | Yes | |
| CYP2C9 inhibitor | No | No | No | No | No | No | Yes | Yes | |
| CYP2D6 inhibitor | Yes | No | No | No | Yes | Yes | No | No | |
| CYP3A4 inhibitor | No | No | No | No | No | No | Yes | No | |
| Drug/lead likeness | Drug likeness (lipinski) | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Lead likeness | No | No | No | No | No | No | No | No | |
| Toxicity parameters | Ames toxicity | Yes | No | No | No | No | No | Yes | No |
| Max. tolerated dose (log mg kg−1 per day) | 0.362 | 0.649 | 0.626 | 0.596 | 0.624 | 0.633 | 0.4 | 0.671 | |
| hERG I inhibitor | No | No | No | No | No | No | No | No | |
| hERG II inhibitor | No | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Oral rat acute toxicity (LD50) (mol kg−1) | 2.229 | 2.347 | 2.77 | 2.677 | 2.993 | 3.074 | 2.512 | 2.786 | |
| Oral rat chronic toxicity (LOAEL) (log mg kg−1 bw per day) | 1.029 | 0.486 | 0.615 | 0.413 | 0.49 | 0.466 | 0.869 | 0.518 | |
| Hepatotoxicity | Yes | Yes | Yes | Yes | Yes | Yes | No | Yes | |
| Minnow toxicity (log mM) | 1.587 | −0.692 | −0.514 | −2.631 | −0.749 | −0.467 | 1.396 | −1.59 |
| Comp. 11 | Comp. 12a | Comp. 12b | Comp. 12c | Comp. 12d | Comp. 13 | Comp. 14 | Comp. 15a | ||
|---|---|---|---|---|---|---|---|---|---|
| Molecular properties | Molar refractivity | 124.38 | 99.89 | 104.70 | 109.03 | 134.31 | 106.47 | 143.92 | 133.73 |
| TPSA (Az) | 77.11 | 77.11 | 77.11 | 77.11 | 77.11 | 114.41 | 123.28 | 106.21 | |
| LogP o/w (WLOGP) | 6.85 | 5.44 | 5.83 | 5.99 | 7.41 | 4.89 | 6.46 | 6.25 | |
| Consensus logP o/w | 5.81 | 4.63 | 4.95 | 5.17 | 6.34 | 4.03 | 5.12 | 5.17 | |
| Water solubility | PS | MS | PS | PS | PS | MS | PS | PS | |
| Pharmacokinetics parameters | GI absorption | Low | High | High | High | Low | High | Low | Low |
| BBB permeant | No | No | No | No | No | No | No | No | |
| P-gp substrate | Yes | No | Yes | No | Yes | No | No | No | |
| CYP1A2 inhibitor | No | Yes | Yes | Yes | No | Yes | No | No | |
| CYP2C19 inhibitor | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| CYP2C9 inhibitor | No | Yes | Yes | Yes | No | Yes | Yes | Yes | |
| CYP2D6 inhibitor | No | No | No | No | No | No | No | No | |
| CYP3A4 inhibitor | Yes | Yes | Yes | No | Yes | No | Yes | Yes | |
| Drug/lead likeness | Drug likeness (lipinski) | Yes | Yes | Yes | Yes | No | Yes | Yes | No |
| Lead likeness | No | No | No | No | No | No | No | No | |
| Toxicity parameters | Ames toxicity | No | Yes | Yes | Yes | No | No | No | No |
| Max. tolerated dose (log mg kg−1 per day) | 0.692 | 0.346 | 0.493 | 0.574 | 0.671 | 1.035 | 0.736 | 0.764 | |
| hERG I inhibitor | No | No | No | No | No | No | No | No | |
| hERG II inhibitor | Yes | Yes | Yes | Yes | Yes | No | Yes | Yes | |
| Oral rat acute toxicity (LD50) (mol kg−1) | 2.547 | 2.317 | 2.264 | 2.213 | 2.531 | 2.604 | 3.091 | 3.009 | |
| Oral rat chronic toxicity (LOAEL) (log mg kg−1 bw per day) | 0.433 | 0.848 | 0.802 | 0.734 | 0.44 | 0.792 | 0.371 | 0.413 | |
| Hepatotoxicity | Yes | No | No | No | Yes | Yes | Yes | Yes | |
| Minnow toxicity (log mM) | −2.429 | 0.929 | 0.54 | 0.091 | −4.27 | 1.452 | −1.724 | −2.091 |
| Comp. 15b | Comp. 16 | Comp. 17a | Comp. 17b | Comp. 17c | Comp. 17d | Comp. 17e | Erlotinib | ||
|---|---|---|---|---|---|---|---|---|---|
| Molecular properties | Molar refractivity | 140.22 | 115.60 | 125.47 | 129.19 | 131.10 | 136.00 | 154.43 | 111.40 |
| TPSA (Az) | 115.44 | 103.41 | 88.41 | 79.18 | 91.21 | 82.42 | 79.18 | 74.73 | |
| LogP o/w (WLOGP) | 6.26 | 5.37 | 4.61 | 5.76 | 3.80 | 4.14 | 8.49 | 3.48 | |
| Consensus logP o/w | 5.10 | 4.77 | 4.32 | 5.17 | 4.01 | 4.26 | 6.78 | 3.28 | |
| Water solubility | PS | PS | MS | PS | MS | PS | PS | MS | |
| Pharmacokinetics parameters | GI absorption | Low | High | High | High | High | High | Low | High |
| BBB permeant | No | No | No | No | No | No | No | Yes | |
| P-gp substrate | No | No | Yes | Yes | Yes | Yes | No | No | |
| CYP1A2 inhibitor | No | Yes | Yes | Yes | Yes | Yes | No | Yes | |
| CYP2C19 inhibitor | Yes | Yes | No | Yes | Yes | No | Yes | Yes | |
| CYP2C9 inhibitor | Yes | Yes | Yes | Yes | Yes | Yes | No | Yes | |
| CYP2D6 inhibitor | Yes | No | No | No | Yes | No | No | Yes | |
| CYP3A4 inhibitor | Yes | Yes | Yes | No | Yes | Yes | No | Yes | |
| Drug/lead likeness | Drug likeness (lipinski) | Yes | Yes | Yes | Yes | Yes | Yes | No | Yes |
| Lead likeness | No | No | No | No | No | No | No | No | |
| Toxicity parameters | Ames toxicity | No | Yes | Yes | No | Yes | Yes | No | No |
| Max. tolerated dose (log mg kg−1 per day) | 0.761 | 0.537 | 0.276 | 0.31 | 0.415 | 0.434 | 0.494 | 0.355 | |
| hERG I inhibitor | No | No | No | No | No | No | No | No | |
| hERG II inhibitor | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Oral rat acute toxicity (LD50) (mol kg−1) | 3.049 | 2.406 | 2.767 | 2.725 | 2.752 | 2.755 | 3.034 | 3.058 | |
| Oral rat chronic toxicity (LOAEL) (log mg kg−1 bw per day) | 0.345 | 0.609 | 0.574 | 0.557 | 2.595 | 2.477 | −0.569 | 1.558 | |
| Hepatotoxicity | Yes | Yes | Yes | No | No | Yes | Yes | Yes | |
| Minnow toxicity (log mM) | −1.971 | 1.28 | 1.564 | 1.102 | 1.763 | 1.651 | −3.411 | −1.971 |
 | ||
| Fig. 7 The boiled-egg representation for the synthesized quinoline derivatives (7–17e), as well as erlotinib as a reference control. | ||
By employing the pkCSM descriptors algorithm protocol, it was revealed that compounds 7, 9, 12a, 12b, 12c, 16, 17a, 17c, and 17d could manifest Ames toxicity. Thus, a possible mutagenicity could be predicted.76 Additionally, all the synthesized quinoline derivatives are non-inhibitors of hERG I. Therefore, a cardiotoxic effect on the human heart's electrical activity cannot be assumed for these compounds.77 However, except for compounds 7 and 13, all synthesized compounds (including erlotinib) exhibit hERG II inhibitory activity, which raises concerns about possible cardiac arrhythmias,78 Notably, except for compounds 9, 12a, 12b, 12c, 17b, and 17c, all of the synthesized compounds are hepatotoxic.
3. Conclusions
By retaining the main pharmacophores of EGFR and DNA gyrase inhibitors, the synthesized compounds (7–17e) can emerge as promising lead anticancer and antimicrobial agents. The anti-proliferative activity was assured by the eligible cytotoxic activities of compounds (7–17e) against HepG2 and MCF-7 with IC50 values of 0.137–0.332 and 0.164–0.583 μg mL−1, respectively, in comparison to erlotinib, which showed IC50 values of 0.308 and 0.512 μg mL−1. Moreover, the investigated compounds could induce cell cycle arrest at the G1 phase. The eligible EGFR tyrosine kinase inhibitory potential of the investigated compounds (IC50 values of 0.14 and 0.18 μM for compounds 8c and 12d, respectively, compared to lapatinib (of 0.12 μM)) emphasized the paper hypothesis. Furthermore, the antimicrobial activity was assured by eligible MIC values against compounds 17b, 17d and 17e in particular, which displayed the most potent inhibitory activity with 4-, 16- and 8-fold greater potency, respectively, than the reference neomycin. The conducted molecular modeling assured the feasible binding pattern of the investigated compounds at the EGFR and DNA gyrase binding sites. Moreover, the established in silico studies assured the eligible pharmacokinetic profiles and toxicity parameters of the synthesized compounds. Accordingly, the synthesized drugs can undergo additional pre-clinical and clinical studies to determine their effectiveness as a double-edged sword for cancer and microbial infection treatment.4. Experimental
4.1. Chemistry
Compound 6 (10 mmol, 3.98 g) was heated at 10 °C above its melting point for 30 min in an oil bath. After cooling the reaction, quinoline-oxadiazole molecule 7 was attained by crystallization from ethanol (70%).
Buff crystals, yield 61%, m.p. 140–142 °C. IR (KBr, cm−1): 3138 (CH aromatic), 2993 (CH aliphatic), 1537 (CN). 1H NMR (400 MHz, DMSO-d6), δ ppm: 7.60–7.66 (m, 3H, Ar-H), 7.76 (t, 1H, Ar-H), 7.99 (d, 1H, J = 8.4 Hz, Ar-H), 8.04 (d, 2H, J = 8.4 Hz, Ar-H), 8.27 (s, 1H, Ar-H), 8.88 (d, 1H, J = 8.4 Hz, Ar-H), 9.57 (s, 1H, oxadiazole-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 121.17, 124.41, 124.78, 125.42, 127.35, 128.37, 129.33(2C), 130.00, 130.65, 132.27(2C), 137.52, 147.26, 147.93, 154.76, 167.19. MS m/z (%): 354.03 (M + 2, 17.31), 353.47 (M + 1, 12.36), 352.46 (M+, 18.72), 302.58 (100). Anal. calcd for C17H10BrN3O (352.19): C, 57.98; H, 2.86; N 11.93; found: C, 58.14; H, 3.02; N, 11.85.
A mixture of acid hydrazide 3 (10 mmol, 3.42 g), carboxylic acid derivatives (10 mmol) and phosphorous oxychloride (5 mL) was heated at 60 °C for 6–8 h, and then allowed to cool at room temperature. After the reaction mixture was added to ice-cold water, a saturated sodium bicarbonate solution was used to neutralize it. The obtained precipitate was crystallized from ethanol (70%) to give the corresponding oxadiazole product 8a–e.
Buff powder, yield 69%, m.p. 90–92 °C. IR (KBr, cm−1): 3070 (CH aromatic), 2954 (CH aliphatic), 1531 (CN). 1H NMR (400 MHz, DMSO-d6), δ ppm: 7.52 (t, 2H, Ar-H), 7.59–7.63 (m, 1H, Ar-H), 7.76–7.90 (m, 3H, Ar-H), 7.92–7.96 (m, 3H, Ar-H), 8.15–8.28 (m, 3H, Ar-H), 8.47 (s, 1H, Ar-H), 8.80 (d, 1H, J = 8 Hz, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 117.81, 123.52, 124.58, 125.56, 126.30, 127.29(2C), 128.34(2C), 129.33(2C), 130.65, 131.78, 132.04(2C), 133.39, 137.45, 140.85, 143.92, 147.99, 154.77, 165.31, 166.00. MS m/z (%): 430.40 (M + 2, 31.26), 428.20 (M+, 30.16), 292.89 (100). Anal. calcd for C23H14BrN3O (428.29): C, 64.50; H, 3.29; N 9.81; found: C, 64.32; H, 3.50; N, 10.04.
White powder, yield 63%, m.p. 190–192 °C. IR (KBr, cm−1): 3099 (CH aromatic), 2993 (CH aliphatic), 1543 (CN). 1H NMR (400 MHz, DMSO-d6), δ ppm: 5.75 (s, 1H, OH, D2O exchangeable), 7.47 (d, 2H, J = 8.4 Hz, Ar-H), 7.75–7.95 (m, 4H, Ar-H), 8.23–8.27 (m, 2H, Ar-H), 8.33 (d, 1H, J = 8.8 Hz, Ar-H), 8.39 (d, 2H, J = 7.2 Hz, Ar-H) 8.82 (s, 1H, Ar-H), 9.20 (d, 1H, J = 8.8 Hz, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 116.74(2C), 119.15(2C), 122.16, 123.27, 123.89, 124.86, 126.30, 128.33, 129.03(2C), 129.99, 131.05, 132.01(2C), 132.71, 140.52, 143.25, 147.98, 154.77, 163.65, 164.64. MS m/z (%): 446.02 (M + 2, 17.91), 443.82 (M+, 19.02), 325.62 (100). Anal. calcd for C23H14BrN3O2 (444.29): C, 62.18; H, 3.18; N 9.46; found: C, 62.40; H, 3.29; N, 9.58.
White powder, yield 66%, m.p. 288–290 °C. IR (KBr, cm−1): 3086 (CH aromatic), 2947 (CH aliphatic), 1546 (CN). 1H NMR (400 MHz, DMSO-d6), δ ppm: 7.83–7.89 (m, 3H, Ar-H), 7.97 (t, 1H, Ar-H), 8.26 (d, 1H, J = 8.8 Hz, Ar-H), 8.40 (d, 2H, J = 8.8 Hz, Ar-H), 8.50 (d, 2H, J = 8.4 Hz, Ar-H), 8.60 (d, 2H, J = 8.4 Hz, Ar-H), 8.88 (s, 1H, Ar-H), 9.20 (d, 1H, J = 8 Hz, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 116.00, 121.86, 123.90, 124.00, 125.29, 126.65(2C), 127.29(2C), 128.33(2C), 128.39, 132.03, 133.39(2C), 133.77, 139.19, 141.33, 142.18, 147.48, 155.44, 166.00, 166.44. MS m/z (%): 475.22 (M + 2, 59.19), 473.60 (M+, 60.06), 44.39 (100). Anal. calcd for C23H13BrN4O3 (473.29): C, 58.37; H, 2.77; N 11.84; found: C, 58.51; H, 2.94; N, 12.07.
Buff powder, yield 62%, m.p. 220–222 °C. IR (KBr, cm−1): 3095 (CH aromatic), 2980 (CH aliphatic), 1573 (CN). 1H NMR (400 MHz, DMSO-d6), δ ppm: 6.74 (d, 2H, J = 8.0 Hz, Ar-H), 7.82 (m, 3H, Ar-H), 7.94–8.28 (m, 5H, Ar-H + NH2 D2O exchangeable), 8.35–8.39 (m, 3H, Ar-H), 8.78 (s, 1H, Ar-H), 9.20 (d, 1H, J = 8.0 Hz, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 114.71(2C), 117.47, 122.17, 124.19, 125.93, 126.60, 127.66, 128.34, 128.62(2C), 129.03(2C), 129.68, 130.68, 132.04(2C), 142.20, 147.31, 149.06, 154.76, 164.33, 165.67. MS m/z (%): 445.95 (M + 2, 22.61), 443.72 (M+, 22.39), 400.91 (100). Anal. calcd for C23H15BrN4O (443.30): C, 62.32; H, 3.41; N 12.64; found: C, 62.56; H, 3.64; N, 12.85.
Brown powder, yield 64%, m.p. 177–179 °C, IR (KBr, cm−1): 3091 (CH aromatic), 2924 (CH aliphatic), 1537 (CN). 1H NMR (400 MHz, DMSO-d6), δ ppm: 6.83 (t, 1H, Ar-H), 6.95 (t, 1H, Ar-H), 7.07 (d, 1H, J = 8.0 Hz, Ar-H), 7.35–7.36 (m, 1H, Ar-H), 7.62–7.94 (m, 4H, Ar-H), 8.08 (d, 1H, J = 8.8 Hz, Ar-H), 8.22 (s, 2H, NH2 D2O exchangeable), 8.37 (d, 2H, J = 8.0 Hz, Ar-H), 8.78 (s, 1H, Ar-H), 9.20 (d, 1H, J = 8.0 Hz, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 117.12, 118.77, 119.84, 121.48, 122.84, 123.21, 123.88, 125.57, 126.98, 127.96, 129.69(2C), 129.99, 130.67, 132.02(2C), 133.08, 134.73, 141.21, 147.99, 154.76, 165.69, 166.66 MS m/z (%): 445.47 (M + 2, 16.37), 442.91 (M+, 16.05), 237.34 (100). Anal. calcd for C23H15BrN4O (443.30): C, 62.32; H, 3.41; N 12.64; found: C, 62.50; H, 3.57; N, 12.79.
An equimolar amount of acid hydrazide 3 (10 mmol, 3.42 g) and potassium hydroxide (0.56 g) with carbon disulfide (2 mL) in absolute ethanol (20 mL) was heated under reflux for 12 h. After reaction completion, the excess solvent was evaporated, then neutralized with dil. HCl. The crystallized solid was separated from isopropanol to attain compound 9.
Yellow powder, yield 83%, m.p. 260–262 °C. IR (KBr, cm−1): 3155 (NH), 3086 (CH aromatic), 2899 (CH aliphatic), 1543 (CN), 1238 (CS). 1H NMR (400 MHz, DMSO-d6), δ ppm: 7.75–7.86 (m, 3H, Ar-H), 7.88 (t, 1H, Ar-H), 8.15 (d, 1H, J = 8.0 Hz, Ar-H), 8.21 (d, 2H, J = 8.0 Hz, Ar-H), 8.37 (s, 1H, Ar-H), 8.76 (d, 1H, J = 8.0 Hz, Ar-H), 11.13 (s, 1H, SH, D2O exchangeable). 13C NMR (100 MHz, DMSO-d6), δ ppm: 110.33, 118.37, 122.15, 123.88, 125.33, 127.61, 128.80(2C), 129.36, 130.55, 131.10(2C), 132.49, 136.54, 141.41, 148.67, 154.66. MS m/z (%): 386.53 (M + 2, 19.17), 384.51 (M+, 20.25), 293.51 (100). Anal. calcd for C17H10BrN3OS (384.25): C, 53.14; H, 2.62; N 10.94; found: C, 53.41; H, 2.86; N, 11.17.
Benzoyl chloride (10 mmol, 1.40 mL) was added dropwise to a well-stirred solution of 1,3,4-oxadiazole-2-thiol 9 (10 mmol, 3.84 g) in dioxane (20 mL), and the reaction mixture was stirred at ambient temperature for the entire night. After adding 10% sodium carbonate solution (25 mL) to the reaction mixture, the resulting solid was crystallized from ethanol to get compound 10.
Buff powder, yield 70%, m.p. 220–222 °C. IR (KBr, cm−1): 3061 (CH aromatic), 2920 (CH aliphatic), 1707 (CO), 1589 (CN). 1H NMR (400 MHz, DMSO-d6), δ ppm: 7.50 (t, 2H, Ar-H), 7.61 (t, 1H, Ar-H), 7.76–7.81 (m, 3H, Ar-H), 7.90–7.96 (m, 3H, Ar-H), 8.19 (d, 1H, J = 8.0 Hz, Ar-H), 8.27 (d, 2H, J = 8 Hz, Ar-H), 8.45 (s, 1H, Ar-H), 8.80 (d, 1H, J = 8 Hz, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 118.48, 122.55, 123.91, 124.58, 125.26, 127.96, 128.37(2C), 128.65(2C), 129.33, 130.38, 130.68, 131.05, 131.74(2C), 132.71(2C), 136.48, 154.77, 158.84, 159.89, 167.72, 178.86. MS m/z (%): 490.09 (M + 2, 25.22), 488.91 (M+, 26.64), 363.31 (100). Anal. calcd for C24H14BrN3O2S (488.36): C, 59.03; H, 2.89; N 8.60; found: C, 59.31; H, 3.02; N, 8.87.
Benzyl chloride (10 mmol, 1.26 g) was added to a well-stirred suspension of 1,3,4-oxadiazole-2-thiol 9 (10 mmol, 3.84 g) and anhydrous potassium carbonate (10 mmol, 1.38 g) in dry acetone (20 mL). The reaction mixture was heated to reflux for 6 h, and then filtered off. Compound 11 was produced by evaporating the excess solvent and crystallizing it from ethanol.
To a well-stirred suspension of 1,3,4-oxadiazole-2-thiol 9 (10 mmol, 3.84 g) and anhydrous potassium carbonate (10 mmol, 1.38 g) in dry acetone (20 mL), benzyl chloride (10 mmol, 1.26 g) was added. The reaction mixture was heated under reflux for 6 h, and then filtered off. Excess solvent was evaporated, and the obtained solid was dried and crystallized from ethanol to give compound 11.
Yellowish green powder, yield 70%, m.p. 142–144 °C. IR (KBr, cm−1): 3099 (CH aromatic), 2929 (CH aliphatic), 1587 (CN). 1H NMR (400 MHz, DMSO-d6), δ ppm: 4.62 (s, 2H, CH2), 7.24–7.36 (m, 3H, Ar-H), 7.49 (d, 2H, J = 7.2 Hz, Ar-H), 7.67–7.69 (m, 3H, Ar-H), 7.82 (t, 1H, Ar-H), 8.06–8.29 (m, 4H, Ar-H), 8.83 (d, 1H, J = 8.4 Hz, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 33.16, 117.36, 121.99, 124.39, 124.52, 125.82, 125.91, 126.21(2C), 126.59, 129.25, 129.63(2C), 129.81(2C), 130.57, 130.83, 131.63(2C), 142.34, 143.10, 148.17, 155.38, 166.40. MS m/z (%): 476.74 (M + 2, 38.62), 474.76 (M+, 37.00), 437.47 (100). Anal. calcd for C24H16BrN3OS (474.38): C, 60.77; H, 3.40; N 8.86; found: C, 60.90; H, 3.62; N, 9.04.
The alkylating agent (20 mmol) was added to a well-stirred suspension of 1,3,4-oxadiazole-2-thiol 9 (10 mmol, 3.84 g) and potassium hydroxide (20 mmol, 1.12 g) in a mixture of ethanol (20 mL) and water (10 mL). The reaction mixture was agitated at 50–60 °C for 4–6 h. Compounds 12a–d were produced by filtering out, drying and crystallizing the produced solid from ethanol.
White powder, yield 70%, m.p. 170–172 °C. IR (KBr, cm−1): 3057 (CH aromatic), 2931 (CH aliphatic), 1571 (CN). 1H NMR (400 MHz, DMSO-d6), δ ppm: 2.87 (3, 3H, CH3), 7.75–7.79 (m, 3H, Ar-H), 7.89 (t, 1H, Ar-H), 8.16 (d, 1H, J = 8.4 Hz, Ar-H), 8.26 (d, 2H, J = 8 Hz, Ar-H), 8.51 (s, 1H, Ar-H), 9.00 (d, 1H, J = 8.4 Hz, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 14.92, 119.84, 122.17, 123.22, 126.31, 127.66, 129.31(2C), 130.02, 130.40, 132.02(2C), 138.80, 144.30, 149.04, 153.79, 163.66, 165.00. MS m/z (%): 400.18 (M + 2, 21.71), 398.73 (M+, 22.46), 258.95(100). Anal. calcd for C18H12BrN3OS (398.28): C, 54.28; H, 3.04; N 10.55; found: C, 54.56; H, 3.21; N, 10.79.
White powder, yield 73%, m.p. 121–123 °C. IR (KBr, cm−1): 3055 (CH aromatic), 2962 (CH aliphatic), 1573 (CN). 1H NMR (400 MHz, DMSO-d6), δ ppm: 1.49 (t, 3H, CH3), 3.31–3.44 (q, 2H, CH2), 7.74–7.79 (m, 3H, Ar-H), 7.89 (t, 1H, Ar-H), 8.15 (d, 1H, J = 8 Hz, Ar-H), 8.24 (d, 2H, J = 8.4 Hz, Ar-H), 8.47 (s, 1H, Ar-H), 8.98 (d, 1H, J = 8 Hz, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 14.87, 27.80, 118.11, 120.80, 122.16, 125.94, 127.29, 129.70, 129.99(2C) 130.36, 131.34, 132.03(2C), 137.15, 148.66, 155.15, 163.64, 165.31 MS m/z (%): 414.16 (M + 2, 22.22), 412.18 (M+, 22.67), 190.72(100). Anal. calcd for C19H14BrN3OS (412.31): C, 55.35; H, 3.42; N 10.19; found: C, 55.17; H, 3.64; N, 10.45.
White powder, yield 69%, m.p. 117–119 °C. IR (KBr, cm−1): 3084 (CH aromatic), 2980 (CH aliphatic), 1598 (CN). 1H NMR (400 MHz, DMSO-d6), δ ppm: 4.10 (d, 2H, J = 6.8 Hz, SCH2), 5.24 (d, 1H, Jcis = 10 Hz, CHCH![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ), 5.43 (d, 1H, Jtrans = 17.2 Hz, –CHCH), 6.00–6.13 (m, 1H, CHCH2), 7.74–7.78 (m, 3H, Ar-H), 7.88 (t, 1H, Ar-H), 8.13 (d, 1H, J = 8 Hz, Ar-H), 8.23 (d, 2H, J = 7.6 Hz, Ar-H), 8.44 (s, 1H, Ar-H), 8.97 (d, 1H, J = 8 Hz, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 34.90, 118.10, 119.84, 121.48, 121.84, 123.89, 125.57, 127.96, 129.03(2C), 129.68, 130.69, 132.41(2C), 133.07, 136.47, 148.00, 154.76, 163.65, 165.69. MS m/z (%): 426.79 (M + 2, 17.69), 424.77 (M+, 18.08), 222.87(100). Anal. calcd for C20H14BrN3OS (424.32): C, 56.61; H, 3.33; N 9.90; found: C, 56.86; H, 3.50; N, 10.14.
), 5.43 (d, 1H, Jtrans = 17.2 Hz, –CHCH), 6.00–6.13 (m, 1H, CHCH2), 7.74–7.78 (m, 3H, Ar-H), 7.88 (t, 1H, Ar-H), 8.13 (d, 1H, J = 8 Hz, Ar-H), 8.23 (d, 2H, J = 7.6 Hz, Ar-H), 8.44 (s, 1H, Ar-H), 8.97 (d, 1H, J = 8 Hz, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 34.90, 118.10, 119.84, 121.48, 121.84, 123.89, 125.57, 127.96, 129.03(2C), 129.68, 130.69, 132.41(2C), 133.07, 136.47, 148.00, 154.76, 163.65, 165.69. MS m/z (%): 426.79 (M + 2, 17.69), 424.77 (M+, 18.08), 222.87(100). Anal. calcd for C20H14BrN3OS (424.32): C, 56.61; H, 3.33; N 9.90; found: C, 56.86; H, 3.50; N, 10.14.
White powder, yield 72%, m.p. 176–178 °C. IR (KBr, cm−1): 3080 (CH aromatic), 2972 (CH aliphatic), 1587 (CN). 1H NMR (400 MHz, DMSO-d6), δ ppm: 4.28 (d, 2H, J = 6.4 Hz, SCH2), 6.50–6.54 (m, 1H, C–CH2), 6.80 (d, 1H, J = 15.6 Hz, CH-ph), 7.23–7.29 (m, 3H, Ar-H), 7.42 (d, 2H, J = 6.8 Hz, Ar-H), 7.71–7.77 (m, 3H, Ar-H), 7.85 (t, 1H, Ar-H), 8.12 (d, 1H, J = 8 Hz, Ar-H), 8.19 (d, 2H, J = 7.6 Hz, Ar-H), 8.43 (s, 1H, Ar-H), 8.96 (d, 1H, J = 8.4 Hz, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 35.35, 122.18, 122.64, 124.49, 124.93, 125.83, 126.27, 126.73, 129.23(2C), 129.71(2C), 129.93(2C), 130.35, 131.29(2C), 132.39, 134.46, 136.32, 137.17, 139.95, 148.80, 155.14, 162.89, 165.13. MS m/z (%): 502.37 (M + 2, 23.55), 500.27 (M+, 25.10), 434.76(100). Anal. calcd for C26H18BrN3OS (500.41): C, 62.41; H, 3.63; N 8.40; found: C, 62.32; H, 3.79; N, 8.67.
A mixture of 1,3,4-oxadiazole-2-thiol 9 (10 mmol, 3.84 g) and 2-chloroacetic acid (10 mmol, 0.95 g) in methylene chloride (20 mL) containing a few drops of TEA was heated under reflux for 10 h. After completion of the reaction, the excess solvent was evaporated under vacuum and the crude precipitate was crystallized from ethanol to give compound 13.
White powder, yield 69%, m.p. 258–260 °C. IR (KBr, cm−1): 3419 (OH), 3080 (CH aromatic), 2983 (CH aliphatic), 1716 (CO), 1598 (CN). 1H NMR (400 MHz, DMSO-d6), δ ppm: 4.19 (s, 2H, CH2), 5.72 (s, 1H, OH, D2O exchangeable), 7.76–7.80 (m, 3H, Ar-H), 7.90 (t, 1H, Ar-H), 8.17 (d, 1H, J = 8.4 Hz, Ar-H), 8.29 (d, 2H, J = 8.00 Hz, Ar-H), 8.54 (s, 1H, Ar-H), 9.00 (d, 1H, J = 8.0 Hz, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 36.62, 118.54, 122.91, 124.89, 125.91, 128.57, 128.93, 129.55(2C), 130.21, 131.28 132.24(2C), 136.62, 148.27, 155.29, 164.04, 166.34, 169.00. MS m/z (%): 444.62 (M + 2, 20.14), 442.56 (M+, 21.06), 326.68(100). Anal. calcd for C19H12BrN3O3S (442.29): C, 51.60; H, 2.73; N 9.50; found: C, 51.78; H, 2.90; N, 9.43.
An equimolar amount of 1,3,4-oxadiazole-2-thiol 9 (10 mmol, 3.84 g) and compound 4 (10 mmol, 2.11 g) in methylene chloride (20 mL) containing a few drops of TEA was heated under reflux for 6 h. The reaction mixture was filtered, and the resulting solid thus obtained was crystallized from ethanol to give compound 14.
White powder, yield 86%, m.p. 219–221 °C. IR (KBr, cm−1): 3446 (NH), 3061 (CH aromatic), 2927 (CH aliphatic), 1670 (CO), 1597 (CN). 1H NMR (400 MHz, DMSO-d6), δ ppm: 2.57 (s, 3H, CH3), 4.51 (s, 2H, CH2), 7.71–7.75 (m, 5H, Ar-H), 7.87–7.93 (m, 3H, Ar-H), 8.16 (d, 1H, J = 8.4 Hz, Ar-H), 8.21 (d, 2H, J = 8 Hz, Ar-H), 8.49 (s, 1H, Ar-H), 8.98 (d, 1H, J = 8.4 Hz, Ar-H), 10.90 (s, 1H, NH, D2O exchangeable). MS m/z (%): 561.96 (M + 2, 17.69), 559.09 (M+, 18.60), 404.96(100). Anal. calcd for C27H19BrN4O3S (559.44): C, 57.97; H, 3.42; N 10.01; found: C, 58.12; H, 3.61; N, 10.29.
The corresponding acetamide derivative 5a,b (10 mmol) was added to a well-stirred solution of 1,3,4-oxadiazole-2-thiol 9 (10 mmol, 3.84 g) and potassium hydroxide (20 mmol, 1.12 g) in a mixture of ethanol (20 mL) and water (10 mL). The reaction mixture was heated to reflux for 6–8 h. Compound 15a,b was produced by filtering out, drying and crystallizing the produced solid from ethanol.
White powder, yield 50% (2.4 g), m.p. 292–294 °C. IR (KBr, cm−1): 3429 (NH), 3061 (CH aromatic), 2968 (CH aliphatic), 1672 (CO), 1587 (CN). 1HNMR (400 MHz, DMSO-d6), δ ppm: 4.27 (s, 2H, CH2), 7.07 (t, 1H, Ar-H), 7.43 (d, 2H, J = 7.6 Hz, Ar-H), 7.57 (t, 1H, Ar-H), 7.70 (d, 2H, J = 7.7 Hz, Ar-H), 7.80 (t, 2H, Ar-H), 7.86 (t, 1H, Ar-H), 8.17 (d, 2H, J = 8.Hz, Ar-H), 8.26 (d, 1H, J = 8.4 Hz, Ar-H), 8.38 (s, 1H, Ar-H) 9.12 (d, 1H, J = 8.4 Hz, Ar-H), 10.96 (s, 1H, NH, D2O exchangeable). 13C NMR (100 MHz, DMSO-d6), δ ppm: 34.00, 117.20, 118.86, 119.91, 120.87(2C), 122.54, 123.89, 128.94(2C), 129.92(2C), 130.60, 131.59(2C), 133.23, 134.60, 135.56, 138.29, 141.97, 148.67, 149.94, 154.62, 163.65, 168.03. MS m/z (%): 519.48 (M + 2, 46.21), 517.38 (M+, 43.91), 195.66(100). Anal. calcd for C25H17BrN4O2S (517.40): C, 58.04; H, 3.31; N 10.83; found: C, 58.31; H, 3.47; N, 11.04.
Grayish white powder, yield 55%, m.p. 565–267 °C. IR (KBr, cm−1): 3446 (NH), 3078 (CH aromatic), 2956 (CH aliphatic), 1651 (CO), 1591 (CN). 1H NMR (400 MHz, DMSO-d6), δ ppm: 3.83 (s, 3H, OCH3), 4.24 (s, 2H, CH2), 7.10 (d, 2H, J = 6.4 Hz, Ar-H), 7.33 (d, 2H, J = 6 Hz, Ar-H), 7.61–7.78 (m, 4H, Ar-H), 8.18–8.36 (m, 4H, Ar-H), 9.13 (d, 1H, J = 7.2 Hz. Ar-H), 10.75 (s, 1H, NH, D2O exchangeable). 13C NMR (100 MHz, DMSO-d6), δ ppm: 33.63, 56.06, 114.54(2C), 116.52, 117.58, 119.90, 124.21(2C), 125.28, 129.24, 129.55, 129.91(2C), 130.60, 132.27(2C), 132.93, 137.30, 138.97, 148.64, 149.28, 153.35, 155.31, 160.05, 166.67. MS m/z (%): 549.92 (M + 2, 24.86), 547.22 (M+, 28.38), 398.33 (100). Anal. calcd for C26H19BrN4O3S (547.43): C, 57.05; H, 3.50; N 10.23; found: C, 57.29; H, 3.62; N, 10.51.
Ethyl chloroacetate (10 mmol, 1.22 g) was added to a well-stirred suspension of 1,3,4-oxadiazole-2-thiol 9 (10 mmol, 3.84 g) and anhydrous potassium carbonate (10 mmol, 1.38 g) in dry acetone (20 mL). The reaction mixture was heated to reflux for 5 h. Compound 6 was produced by filtering out, drying and crystallizing the produced precipitate from ethanol.
White powder, yield 87%, m.p. 310–312 °C. IR (KBr, cm−1): 3077 (CH aromatic), 2983 (CH aliphatic), 1739 (CO), 1588 (CN). 1H NMR (400 MHz, DMSO-d6), δ ppm: 1.20 (t, 3H, CH3), 4.17–4.22 (q, 2H, CH2), 4.41 (s, 2H, CH2), 7.79–7.83 (m, 3H, Ar-H), 7.93 (t, 1H, Ar-H), 8.21 (d, 1H, J = 8.4 Hz, Ar-H), 8.31 (d, 2H, J = 8.8 Hz, Ar-H), 8.59 (s, 1H, Ar-H), 9.00 (d, 1H, J = 8.4 Hz, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 14.14, 32.14, 62.29, 118.74, 122.04, 124.58, 125.55, 126.22, 129.01, 129.66(2C), 130.38, 130.68, 132.04(2C), 135.47, 141.22, 148.62, 155.22, 166.38, 167.66. MS m/z (%): 472.95 (M + 2, 23.61), 470.52 (M+, 25.19), 321.42(100). Anal. calcd for C21H16BrN3O3S (470.34): C, 53.63; H, 3.43; N 8.93; found: C, 53.88; H, 3.61; N, 9.12.
A suspension of 1,3,4-oxadiazole-2-thiol 9 (10 mmol, 3.84 g) and an appropriate secondary amine (10 mmol) was heated under reflux in ethanol (30 mL) with 36% formaldehyde (20 mmol) for 4–6 h. After cooling to room temperature, the resultant solid was crystallized from ethanol to get compound 17a–e.
Buff powder, yield 82%, m.p. 210–212 °C. IR (KBr, cm−1): 3059 (CH aromatic), 2974 (CH aliphatic), 1587 (CN), 1267 (CS). 1H NMR (400 MHz, DMSO-d6), δ ppm: 2.82–2.86 (m, 4H, CH2–N–CH2), 3.60–3.62 (m, 4H, CH2–O–CH2), 5.18 (s, 2H, CH2), 7.78–7.81 (m, 3H, Ar-H), 7.91 (t, 1H, Ar-H), 8.20 (d, 1H, J = 8.0 Hz. Ar-H), 8.28 (d, 2H, J = 8.8 Hz, Ar-H), 8.45 (s, 1H, Ar-H), 8.95 (d, 1H, J = 8.0 Hz, Ar-H), 13C NMR (100 MHz, DMSO-d6), δ ppm: 53.95(2C), 66.38(2C), 79.88, 122.55, 123.38, 124.87, 125.93, 126.61, 127.63, 128.61(2C), 129.93, 130.68, 131.75(2C), 137.82, 143.88, 155.15, 157.82, 179.34. MS m/z (%): 485.95 (M + 2, 17.04), 483.52 (M+, 14.99), 437.94 (100). Anal. calcd for C22H19BrN4O2S (483.38): C, 54.66; H, 3.96; N 11.59; found: C, 54.79; H, 4.12; N, 11.78.
Yellow powder, yield 77%, m.p. 192–194 °C, IR (KBr, cm−1): 3014 (CH aromatic), 2933 (CH aliphatic), 1587 (CN), 1246 (CS). 1H NMR (400 MHz, DMSO-d6), δ ppm: 1.35–1.64 (m, 6H, piperidine), 2.73–3.01 (m, 4H, piperidine), 5.17 (s, 2H, CH2), 7.76–7.88 (m, 3H, Ar-H), 7.87 (t, 1H, Ar-H), 8.16 (d, 1H, J = 8.0 Hz. Ar-H), 8.26 (d, 2H, J = 8.4 Hz, Ar-H), 8.36 (s, 1H, Ar-H), 9.15 (d, 1H, J = 8.0 Hz, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 22.23, 23.31(2C), 44.68(2C), 83.48, 116.21, 122.53, 124.69, 126.24, 126.60, 128.27, 129.25(2C), 130.93, 131.27, 132.25(2C), 137.60, 149.27, 155.00, 155.97, 181.05. MS m/z (%): 483.94 (M + 2, 36.44), 481.27 (M+, 36.79), 467.04 (100). Anal. calcd for C23H21BrN4OS (481.41): C, 57.38; H, 4.40; N 11.64; found: C, 57.45; H, 4.63; N, 11.90.
Yellow powder, yield 74%, m.p. 219–221 °C. IR (KBr, cm−1): 3423 (NH), 3064 (CH aromatic), 2931 (CH aliphatic), 1593 (CN), 1249 (CS). 1H NMR (400 MHz, DMSO-d6), δ ppm: 1.04 (s, 1H, NH, D2O exchangeable), 2.61 (s, 4H, piperazine), 2.91–3.07 (s, 4H, piperazine), 5.14 (s, 2H, CH2), 7.73–7.78 (m, 3H, Ar-H), 7.86 (t, 1H, Ar-H), 8.14 (d, 1H, J = 8.0 Hz, Ar-H), 8.26 (d, 2H, J = 7.6 Hz, Ar-H), 8.32 (s, 1H, Ar-H), 9.25 (d, 1H, J = 8.0 Hz, Ar-H). MS m/z (%): 484.08 (M + 2, 24.53), 481.98 (M+, 26.48), 284.45 (100). Anal. calcd for C22H20BrN5OS (482.40): C, 54.78; H, 4.18; N 14.52; found: C, 54.97; H, 4.40; N, 14.76.
Yellow powder, yield 80%, m.p. 156–158 °C. IR (KBr, cm−1): 3055 (CH aromatic), 2995 (CH aliphatic), 1589 (CN), 1265 (CS). 1H NMR (400 MHz, DMSO-d6), δ ppm: 2.83 (s, 3H, CH3), 3.18 (s, 4H, piperazine), 3.96 (s, 4H, piperazine), 5.23 (s, 2H, CH2), 7.65 (t, 1H, Ar-H), 7.71 (d, 2H, J = 8.4 Hz, Ar-H), 7.77 (t, 1H, Ar-H), 7.95 (d, 1H, J = 8.0 Hz, Ar-H), 8.07–8.11 (m, 3H, Ar-H), 8.85 (s, 1H, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 18.67, 53.39(2C), 56.43(2C), 69.34, 121.19, 121.87, 123.60, 125.24, 125.56, 128.30, 128.55, 129.42(2C), 130.23, 130.84, 132.18(2C), 137.21, 154.62, 154.72, 180.76. MS m/z (%): 498.15 (M + 2, 19.76), 496.72 (M+, 20.59), 323.33 (100). Anal. calcd for C23H22BrN5OS (496.43): C, 55.65; H, 4.47; N 14.11; found: C, 55.81; H, 4.63; N, 13.98.
Buff powder, yield 77%, m.p. 266–286 °C. IR (KBr, cm−1): 3055 (CH aromatic), 2997 (CH aliphatic), 1573 (CN), 1238 (CS). 1H NMR (400 MHz, DMSO-d6), δ ppm: 5.53 (s, 2H, CH2), 6.80 (t, 2H, Ar-H), 7.05 (d, 4H, J = 8 Hz, Ar-H), 7.21 (t, 4H, Ar-H), 7.71–7.76 (m, 3H, Ar-H), 7.87 (t, 1H, Ar-H), 8.13 (d, 1H, J = 8.4 Hz, Ar-H), 8.19 (d, 2H, J = 8.4 Hz, Ar-H), 8.33 (s, 1H, Ar-H), 8.74 (d, 1H, J = 8.4 Hz, Ar-H). MS m/z (%): 567.00 (M + 2, 15.43), 565.25 (M+, 17.63), 322.87 (100). Anal. calcd for C30H21BrN4OS (565.49): C, 63.72; H, 3.74; N, 9.91; found: C, 63.54; H, 3.89; N, 10.13.
4.2. Biological evaluation
See Appendix B.†4.3. In silico studies
Conflicts of interest
The authors declare that there are no potential conflicts of interest.References
- M. P. Curado, L. Voti and A. M. Sortino-Rachou, Cancer registration data and quality indicators in low and middle income countries: their interpretation and potential use for the improvement of cancer care, Cancer Causes & Control, 2009, 20, 751–756 CrossRef PubMed
.
- L. A. Torre, F. Bray, R. L. Siegel, J. Ferlay, J. Lortet-Tieulent and A. Jemal, Global cancer statistics, 2012, Ca-Cancer J. Clin., 2015, 65, 87–108 CrossRef PubMed
- M. Demaria, M. N. O'Leary, J. Chang, L. Shao, S. Liu, F. Alimirah, K. Koenig, C. Le, N. Mitin, A. M. Deal, S. Alston, E. C. Academia, S. Kilmarx, A. Valdovinos, B. Wang, A. de Bruin, B. K. Kennedy, S. Melov, D. Zhou, N. E. Sharpless, H. Muss and J. Campisi, Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse, Cancer Discovery, 2017, 7, 165–176 CrossRef CAS PubMed
- J. Chang, H. Ren, M. Zhao, Y. Chong, W. Zhao, Y. He, Y. Zhao, H. Zhang and C. Qi, Development of a series of novel 4-anlinoquinazoline derivatives possessing quinazoline skeleton: design, synthesis, EGFR kinase inhibitory efficacy, and evaluation of anticancer activities in vitro, Eur. J. Med. Chem., 2017, 138, 669–688 CrossRef CAS PubMed
- V. S. Kasture, D. S. Musmade, S. J. Aher and P. P. Patil, Tyrosine Kinases: promising targets for cancer chemotherapy, Drug Discov. Today, 2012, 2, 37–51 Search PubMed
- M. Huang, A. Shen, J. Ding and M. Geng, Molecularly targeted cancer therapy: some lessons from the past decade, Trends Pharmacol. Sci., 2014, 35, 41–50 CrossRef CAS PubMed
- A. A. Nasser, I. H. Eissa, M. R. Oun, M. A. El-Zahabi, M. S. Taghour, A. Belal, A. M. Saleh, A. B. M. Mehany, H. Luesch, A. E. Mostafa, W. M. Afifi, J. R. Rocca and H. A. Mahdy, Discovery of new pyrimidine-5-carbonitrile derivatives as anticancer agents targeting EGFRWT and EGFRT790M, Org. Biomol. Chem., 2020, 18, 7608–7634 RSC
- Y. Chen, J. Wu, A. Wang, Z. Qi, T. Jiang, C. Chen, F. Zou, C. Hu, W. Wang, H. Wu, Z. Hu, W. Wang, B. Wang, L. Wang, T. Ren, S. Zhang, Q. Liu and J. Liu, Discovery of N-(5-((5-chloro-4-((2-(isopropylsulfonyl)phenyl)amino)pyrimidin-2-yl)amino)-4-methoxy-2-(4-methyl-1,4-diazepan-1-yl)phenyl)acrylamide (CHMFL-ALK/EGFR-050) as a potent ALK/EGFR dual kinase inhibitor capable of overcoming a variety of ALK/EGFR associated drug resistant mutants in NSCLC, Eur. J. Med. Chem., 2017, 139, 674–697 CrossRef CAS PubMed
- Y. A. M. M. Elshaier, M. A. Shaaban, M. K. Abd El Hamid, M. H. Abdelrahman, M. A. Abou-Salim, S. M. Elgazwi and F. Halaweish, Design and synthesis of pyrazolo[3,4-d]pyrimidines: nitric oxide releasing compounds targeting hepatocellular carcinoma, Bioorg. Med. Chem., 2017, 25, 2956–2970 CrossRef CAS PubMed
- A. A. Gaber, A. H. Bayoumi, A. M. El-morsy, F. F. Sherbiny, A. B. M. Mehany and I. H. Eissa, Design, synthesis and anticancer evaluation of 1H-pyrazolo[3,4-d]pyrimidine derivatives as potent EGFRWT and EGFRT790M inhibitors and apoptosis inducers, Bioorg. Chem., 2018, 80, 375–395 CrossRef CAS PubMed
- T. Al-Warhi, A. A. Al-Karmalawy, A. A. Elmaaty, M. A. Alshubramy, M. Abdel-Motaal, T. A. Majrashi, M. Asem, A. Nabil, W. M. Eldehna and M. Sharaky, Biological evaluation, docking studies, and in silico ADME prediction of some pyrimidine and pyridine derivatives as potential EGFRWT and EGFRT790M inhibitors, J. Enzyme Inhib. Med. Chem., 2023, 38, 176–191 CrossRef CAS PubMed
- J. He, Z. Zhou, X. Sun, Z. Yang, P. Zheng, S. Xu and W. Zhu, The new opportunities in medicinal chemistry of fourth-generation EGFR inhibitors to overcome C797S mutation, Eur. J. Med. Chem., 2021, 210, 112995 CrossRef CAS PubMed
- M. Zheng, J. Huo, X. Gu, Y. Wang, C. Wu, Q. Zhang, W. Wang, Y. Liu, Y. Liu, X. Zhou, L. Chen, Y. Zhou and H. Li, Rational Design and Synthesis of Novel Dual PROTACs for Simultaneous Degradation of EGFR and PARP, J. Med. Chem., 2021, 64, 7839–7852 CrossRef CAS PubMed
- H. A. Allam, E. E. Aly, A. K. B. A. W. Farouk, A. M. El Kerdawy, E. Rashwan and S. E. S. Abbass, Design and Synthesis of some new 2,4,6-trisubstituted quinazoline EGFR inhibitors as targeted anticancer agents, Bioorg. Chem., 2020, 98, 103726 CrossRef CAS PubMed
- A. A. M. Eissa, K. F. M. Aljamal, H. S. Ibrahim and H. Abdelrasheed Allam, Design and synthesis of novel pyridopyrimidine derivatives with anchoring non-coplanar aromatic extensions of EGFR inhibitory activity, Bioorg. Chem., 2021, 116, 105318 CrossRef CAS PubMed
- A. M. Mohassab, H. A. Hassan, D. Abdelhamid, A. M. Gouda, B. G. M. Youssif, H. Tateishi, M. Fujita, M. Otsuka and M. Abdel-Aziz, Design and synthesis of novel quinoline/chalcone/1,2,4-triazole hybrids as potent antiproliferative agent targeting EGFR and BRAFV600E kinases, Bioorg. Chem., 2021, 106, 104510–104523 CrossRef CAS PubMed
- C.-H. Wu, M. S. Coumar, C.-Y. Chu, W.-H. Lin, Y.-R. Chen, C.-T. Chen, H.-Y. Shiao, S. Rafi, S.-Y. Wang, H. Hsu, C.-H. Chen, C.-Y. Chang, T.-Y. Chang, T.-W. Lien, M.-Y. Fang, K.-C. Yeh, C.-P. Chen, T.-K. Yeh, S.-H. Hsieh, J. T. A. Hsu, C.-C. Liao, Y.-S. Chao and H.-P. Hsieh, Design and Synthesis of Tetrahydropyridothieno[2,3-d]pyrimidine Scaffold Based Epidermal Growth Factor Receptor (EGFR) Kinase Inhibitors: The Role of Side Chain Chirality and Michael Acceptor Group for Maximal Potency, J. Med. Chem., 2010, 53, 7316–7326 CrossRef CAS PubMed
- H. Saito, T. Fukuhara, N. Furuya, K. Watanabe, S. Sugawara, S. Iwasawa, Y. Tsunezuka, O. Yamaguchi, M. Okada, K. Yoshimori, I. Nakachi, A. Gemma, K. Azuma, F. Kurimoto, Y. Tsubata, Y. Fujita, H. Nagashima, G. Asai, S. Watanabe, M. Miyazaki, K. Hagiwara, T. Nukiwa, S. Morita, K. Kobayashi and M. Maemondo, Erlotinib plus bevacizumab versus erlotinib alone in patients with EGFR-positive advanced non-squamous non-small-cell lung cancer (NEJ026): interim analysis of an open-label, randomised, multicentre, phase 3 trial, Lancet Oncol., 2019, 20, 625–635 CrossRef CAS PubMed
- K. Nakagawa, E. B. Garon, T. Seto, M. Nishio, S. Ponce Aix, L. Paz-Ares, C.-H. Chiu, K. Park, S. Novello, E. Nadal, F. Imamura, K. Yoh, J.-Y. Shih, K. H. Au, D. Moro-Sibilot, S. Enatsu, A. Zimmermann, B. Frimodt-Moller, C. Visseren-Grul, M. Reck, Q. Chu, A. Cortot, J.-L. Pujol, E. Fabre, C. Lamour, H. Bischoff, J. Kollmeier, M. Kimmich, W. Engel-Riedel, S. Hammerschmidt, W. Schütte, K. Syrigos, J. C. M. Ho, K.-H. Au, A. Ardizzoni, G. Pasello, V. Gregorc, A. Del Conte, D. Galetta, T. Takahashi, T. Kumagai, K. Hotta, Y. Goto, Y. Hosomi, H. Sakai, Y. Takiguchi, Y. H. Kim, T. Kurata, H. Yamaguchi, H. Daga, I. Okamoto, M. Satouchi, S. Ikeda, K. Kasahara, S. Atagi, K. Azuma, K. Aoe, Y. Horio, N. Yamamoto, H. Tanaka, S. Watanabe, N. Nogami, T. Ozaki, R. Koyama, T. Hirashima, H. Kaneda, K. Tomii, Y. Fujita, M. Seike, N. Nishimura, T. Kato, M. Ichiki, H. Saka, K. Hirano, Y. Nakahara, S. Sugawara, S.-W. Kim, Y. J. Min, H. W. Lee, J.-H. Kang, H. J. An, K. H. Lee, J.-S. Kim, G.-W. Lee, S. Y. Lee, A. Alexandru, A. A. Udrea, Ó. Juan-Vidal, E. Nadal-Alforja, I. Gil-Bazo, S. Ponce-Aix, B. Rubio-Viqueira, M. Alonso Garcia, E. Felip Font, J. Fuentes Pradera, J. Coves Sarto, M.-C. Lin, W.-C. Su, T.-C. Hsia, G.-C. Chang, Y.-F. Wei, J. Su, I. Cicin, T. Goksel, H. Harputluoglu, O. Ozyilkan, I. Henning, S. Popat, O. Hatcher, K. Mileham, J. Acoba, E. Garon, G. Jung, M. Raj, W. Martin and S. Dakhil, Ramucirumab plus erlotinib in patients with untreated, EGFR mutated, advanced non-small-cell lung cancer (RELAY): a randomised, double-blind, placebo-controlled, phase 3 trial, Lancet Oncol., 2019, 20, 1655–1669 CrossRef CAS PubMed
- V. Noronha, V. M. Patil, A. Joshi, N. Menon, A. Chougule, A. Mahajan, A. Janu, N. Purandare, R. Kumar, S. More, S. Goud, N. Kadam, N. Daware, A. Bhattacharjee, S. Shah, A. Yadav, V. Trivedi, V. Behel, A. Dutt, S. D. Banavali and K. Prabhash, Gefitinib Versus Gefitinib Plus Pemetrexed and Carboplatin Chemotherapy in EGFR-Mutated Lung Cancer, J. Clin. Oncol., 2020, 38, 124–136 CrossRef CAS PubMed
- W.-Z. Zhong, Q. Wang, W.-M. Mao, S.-T. Xu, L. Wu, Y.-C. Wei, Y.-Y. Liu, C. Chen, Y. Cheng, R. Yin, F. Yang, S.-X. Ren, X.-F. Li, J. Li, C. Huang, Z.-D. Liu, S. Xu, K.-N. Chen, S.-D. Xu, L.-X. Liu, P. Yu, B.-H. Wang, H.-T. Ma, J.-J. Yang, H.-H. Yan, X.-N. Yang, S.-Y. Liu, Q. Zhou and Y.-L. Wu, Gefitinib Versus Vinorelbine Plus Cisplatin as Adjuvant Treatment for Stage II-IIIA (N1-N2) EGFR-Mutant NSCLC: Final Overall Survival Analysis of CTONG1104 Phase III Trial, J. Clin. Oncol., 2021, 39, 713–722 CrossRef CAS PubMed
- Y. Kim, S.-H. Lee, J. S. Ahn, M.-J. Ahn, K. Park and J.-M. Sun, Efficacy and safety of afatinib for EGFR-mutant non-small cell lung cancer, compared with gefitinib or erlotinib, Cancer Res. Treat.: Off. J. Korean Cancer Assoc., 2019, 51, 502–509 CrossRef CAS PubMed
- A. Ayati, S. Moghimi, S. Salarinejad, M. Safavi, B. Pouramiri and A. Foroumadi, A review on progression of epidermal growth factor receptor (EGFR) inhibitors as an efficient approach in cancer targeted therapy, Bioorg. Chem., 2020, 99, 103811–103821 CrossRef CAS PubMed
- A. R. Lee, S. Lee, J. Y. Shin, J.-Y. Kim, K.-S. Moon and J. Jung, Biomarker LEPRE1 induces pelitinib-specific drug responsiveness by regulating ABCG2 expression and tumor transition states in human leukemia and lung cancer, Sci. Rep., 2022, 12, 2928–2942 CrossRef CAS PubMed
- P. Yadav and K. Shah, Quinolines, a perpetual, multipurpose scaffold in medicinal chemistry, Bioorg. Chem., 2021, 109, 104639–104657 CrossRef CAS PubMed
- J. Goldman, A. Martínez Bueno, C. Dooms, K. Jhaveri, M. J. de Miguel, S. A. Piha-Paul, N. Unni, A. Mahipal, J. M. Suga, C. Naltet, A. Zick, M. Antoñanzas Basa, J. Crown, Y. K. Chae, D. DiPrimeo, L. D. Eli, L. McCulloch and D. Mahalingam, Neratinib efficacy in patients with EGFR exon 18-mutant non-small-cell lung cancer: findings from the SUMMIT basket trial, Eur. J. Cancer, 2022, 174, S82 CrossRef
- K. D. Herman, C. G. Wright, H. M. Marriott, S. C. McCaughran, K. A. Bowden, M. O. Collins, S. A. Renshaw and L. R. Prince, The EGFR/ErbB inhibitor neratinib modifies the neutrophil phosphoproteome and promotes apoptosis and clearance by airway macrophages, Front. Immunol., 2022, 13, 1–19 Search PubMed
- M. Zou, J. Li, B. Jin, M. Wang, H. Chen, Z. Zhang, C. Zhang, Z. Zhao and L. Zheng, Design, synthesis and anticancer evaluation of new 4-anilinoquinoline-3-carbonitrile derivatives as dual EGFR/HER2 inhibitors and apoptosis inducers, Bioorg. Chem., 2021, 114, 105200–105214 CrossRef CAS PubMed
- H. Patel, A. Ansari, R. Pawara, I. Ansari, H. Jadhav and S. Surana, Design and synthesis of novel 2,4-disubstituted aminopyrimidines: reversible non-covalent T790M EGFR inhibitors, J. Recept. Signal Transduction, 2018, 38, 393–412 CrossRef CAS PubMed
- Y. Kim, J. Ko, Z. Cui, A. Abolhoda, J. S. Ahn, S.-H. Ou, M.-J. Ahn and K. Park, The EGFR T790M Mutation in Acquired Resistance to an Irreversible Second-Generation EGFR Inhibitor, Mol. Cancer Ther., 2012, 11, 784–791 CrossRef CAS PubMed
- L. V. Sequist, B. Besse, T. J. Lynch, V. A. Miller, K. K. Wong, B. Gitlitz, K. Eaton, C. Zacharchuk, A. Freyman, C. Powell, R. Ananthakrishnan, S. Quinn and J.-C. Soria, Neratinib, an Irreversible Pan-ErbB Receptor Tyrosine Kinase Inhibitor: Results of a Phase II Trial in Patients With Advanced Non-Small-Cell Lung Cancer, J. Clin. Oncol., 2010, 28, 3076–3083 CrossRef CAS PubMed
- S. Ding, Z. Gao, Z. Hu, R. Qi, X. Zheng, X. Dong, M. Zhang, J. Shen, T. Long, Y. Zhu, L. Tian, W. Song, R. Liu, Y. Li, J. Sun, W. Duan, J. Liu and Y. Chen, Design, synthesis and biological evaluation of novel osimertinib derivatives as reversible EGFR kinase inhibitors, Eur. J. Med. Chem., 2022, 238, 114492–114504 CrossRef CAS PubMed
- A. Ayati, S. Moghimi, M. Toolabi and A. Foroumadi, Pyrimidine-based EGFR TK inhibitors in targeted cancer therapy, Eur. J. Med. Chem., 2021, 221, 113523–113535 CrossRef CAS PubMed
- X. Hu, S. Tang, F. Yang, P. Zheng, S. Xu, Q. Pan and W. Zhu, Design, Synthesis, and Antitumor Activity of Olmutinib Derivatives Containing Acrylamide Moiety, Molecules, 2021, 26, 3041 CrossRef CAS PubMed
- E. B. Elkaeed, R. G. Yousef, H. Elkady, A. A. Alsfouk, D. Z. Husein, I. M. Ibrahim, A. M. Metwaly and I. H. Eissa, New Anticancer Theobromine Derivative Targeting EGFRWT and EGFRT790M: Design, Semi-Synthesis, In Silico, and In Vitro Anticancer Studies, Molecules, 2022, 27, 5859–5887 CrossRef CAS PubMed
- M. A. Mansour, A. M. AboulMagd, S. H. Abbas, H. M. Abdel-Rahman and M. Abdel-Aziz, Insights into fourth generation selective inhibitors of (C797S) EGFR mutation combating non-small cell lung cancer resistance: a critical review, RSC Adv., 2023, 13, 18825–18853 RSC
- Y. A. Ammar, A. A. Farag, A. M. Ali, S. A. Hessein, A. A. Askar, E. A. Fayed, D. M. Elsisi and A. Ragab, Antimicrobial evaluation of thiadiazino and thiazolo quinoxaline hybrids as potential DNA gyrase inhibitors; design, synthesis, characterization and morphological studies, Bioorg. Chem., 2020, 99, 103841–103854 CrossRef CAS PubMed
- J. M. A. Blair, M. A. Webber, A. J. Baylay, D. O. Ogbolu and L. J. V. Piddock, Molecular mechanisms of antibiotic resistance, Nat. Rev. Microbiol., 2015, 13, 42–51 CrossRef CAS PubMed
- K. U. Jansen, C. Knirsch and A. S. Anderson, The role of vaccines in preventing bacterial antimicrobial resistance, Nat. Med., 2018, 24, 10–19 CrossRef CAS PubMed
- Y. A. Ammar, A. A. Farag, A. M. Ali, A. Ragab, A. A. Askar, D. M. Elsisi and A. Belal, Design, synthesis, antimicrobial activity and molecular docking studies of some novel di-substituted sulfonylquinoxaline derivatives, Bioorg. Chem., 2020, 104, 104164–104176 CrossRef CAS PubMed
- A. Ragab, D. M. Elsisi, O. A. Abu Ali, M. S. Abusaif, A. A. Askar, A. A. Farag and Y. A. Ammar, Design, synthesis of new novel quinoxalin-2(1H)-one derivatives incorporating hydrazone, hydrazine, and pyrazole moieties as antimicrobial potential with in-silico ADME and molecular docking simulation, Arabian J. Chem., 2022, 15, 103497–103510 CrossRef CAS
- H. A. Hofny, M. F. A. Mohamed, H. A. M. Gomaa, S. A. Abdel-Aziz, B. G. M. Youssif, N. A. El-koussi and A. S. Aboraia, Design, synthesis, and antibacterial evaluation of new quinoline-1,3,4-oxadiazole and quinoline-1,2,4-triazole hybrids as potential inhibitors of DNA gyrase and topoisomerase IV, Bioorg. Chem., 2021, 112, 104920 CrossRef CAS PubMed
- M. Elagawany, L. Maram and B. Elgendy, Synthesis of 3-Aminoquinazolinones via a SnCl2-Mediated ANRORC-like Reductive Rearrangement of 1,3,4-Oxadiazoles, J. Org. Chem., 2023, 88, 17062–17068 CrossRef CAS PubMed
- S.-O. Zaraei, N. N. Al-Ach, H. S. Anbar, R. El-Gamal, H. Tarazi, R. T. Tokatly, R. R. Kalla, M. A. Munther, M. M. Wahba, A. M. Alshihabi, M. K. Shehata, R. M. Sbenati, A. I. Shahin, R. El-Awady, T. H. Al-Tel and M. I. El-Gamal, Design and synthesis of new quinoline derivatives as selective C-RAF kinase inhibitors with potent anticancer activity, Eur. J. Med. Chem., 2022, 238, 114434 CrossRef CAS PubMed
- D. I. A. Othman, K. B. Selim, M. A. A. El-Sayed, A. S. Tantawy, Y. Amen, K. Shimizu, T. Okauchi and M. Kitamura, Design, Synthesis and Anticancer Evaluation of New Substituted Thiophene-Quinoline Derivatives, Bioorg. Med. Chem., 2019, 27, 115026 CrossRef CAS PubMed
- H. M. Abd El-Lateef, D. Bafail, N. H. Y. Alhalees, E. E. Toson, A. H. A. Almaaty, E. H. Elsayed, I. Zaki and M. M. Youssef, Synthesis, characterization and biological research of novel 2-(quinoline-4-carbonyl) hydrazide-acrylamide hybrids as potential anticancer agents on MCF-7 breast carcinoma cells by targeting EGFR-TK, RSC Adv., 2024, 14, 23495–23504 RSC
- Z. Tang, Y. Peng and F. Liu, Design and synthesis of novel quinoline derivatives bearing oxadiazole, isoxazoline, triazolothiadiazole, triazolothiadiazine, and piperazine moieties, J. Heterocycl. Chem., 2020, 57, 2330–2338 CrossRef CAS
- M. D. Altıntop, B. Sever, G. Akalın Çiftçi, G. Turan-Zitouni, Z. A. Kaplancıklı and A. Özdemir, Design, synthesis, in vitro and in silico evaluation of a new series of oxadiazole-based anticancer agents as potential Akt and FAK inhibitors, Eur. J. Med. Chem., 2018, 155, 905–924 CrossRef PubMed
- S. Bajaj, M. S. Kumar, H. Tinwala and M. Yc, Design, synthesis, modelling studies and biological evaluation of 1,3,4-oxadiazole derivatives as potent anticancer agents targeting thymidine phosphorylase enzyme, Bioorg. Chem., 2021, 111, 104873 CrossRef CAS PubMed
- R. Kaur and K. Kumar, Synthetic and medicinal perspective of quinolines as antiviral agents, Eur. J. Med. Chem., 2021, 215, 113220 CrossRef CAS PubMed
- M. Wang, G. Zhang, J. Zhao, N. Cheng, Y. Wang, Y. Fu, Y. Zheng, J. Wang, M. Zhu, S. Cen, J. He and Y. Wang, Synthesis and antiviral activity of a series of novel quinoline derivatives as anti-RSV or anti-IAV agents, Eur. J. Med. Chem., 2021, 214, 113208 CrossRef CAS PubMed
- W. Yu, L. Ping Cheng, W. Pang and L. Ling Guo, Design, synthesis and biological evaluation of novel 1,3,4-oxadiazole derivatives as potent neuraminidase inhibitors, Bioorg. Med. Chem., 2022, 57, 116647 CrossRef CAS PubMed
- G. Jin, Z. Li, F. Xiao, X. Qi and X. Sun, Optimization of activity localization of quinoline derivatives: design, synthesis, and dual evaluation of biological activity for potential antitumor and antibacterial agents, Bioorg. Chem., 2020, 99, 103837 CrossRef CAS PubMed
- G. Jin, F. Xiao, Z. Li, X. Qi, L. Zhao and X. Sun, Design, Synthesis, and Dual Evaluation of Quinoline and Quinolinium Iodide Salt Derivatives as Potential Anticancer and Antibacterial Agents, ChemMedChem, 2020, 15, 600–609 CrossRef CAS PubMed
- H.-G. Fu, Z.-W. Li, X.-X. Hu, S.-Y. Si, X.-F. You, S. Tang, Y.-X. Wang and D.-Q. Song, Synthesis and Biological Evaluation of Quinoline Derivatives as a Novel Class of Broad-Spectrum Antibacterial Agents, Molecules, 2019, 24, 548–558 CrossRef PubMed
- S. M. Ghelani, H. R. Khunt and Y. T. Naliapara, Design, Synthesis, Characterization, and Antimicrobial Screening of Novel Indazole Bearing Oxadiazole Derivatives, J. Heterocycl. Chem., 2017, 54, 65–70 CrossRef CAS
- A. T. Bordei Telehoiu, D. C. Nuţă, M. T. Căproiu, F. Dumitrascu, I. Zarafu, P. Ioniţă, C. D. Bădiceanu, S. Avram, M. C. Chifiriuc, C. Bleotu and C. Limban, Design, Synthesis and In Vitro Characterization of Novel Antimicrobial Agents Based on 6-Chloro-9H-carbazol Derivatives and 1,3,4-Oxadiazole Scaffolds, Molecules, 2020, 25, 266–278 CrossRef PubMed
- H. Nikookar, M. Mohammadi-Khanaposhtani, S. Imanparast, M. A. Faramarzi, P. R. Ranjbar, M. Mahdavi and B. Larijani, Design, synthesis and in vitro α-glucosidase inhibition of novel dihydropyrano[3,2-c]quinoline derivatives as potential anti-diabetic agents, Bioorg. Chem., 2018, 77, 280–286 CrossRef CAS PubMed
- M. S. Ganesan, K. K. Raja, K. Narasimhan, S. Murugesan and B. K. Kumar, Design, synthesis, α-amylase inhibition and in silico docking study of novel quinoline bearing proline derivatives, J. Mol. Struct., 2020, 1208, 127873–127886 CrossRef CAS
- R. Hussain, W. Rehman, F. Rahim, S. Khan, M. Taha, Y. Khan, A. Sardar, I. Khan and S. A. A. Shah, Discovery of imidazopyridine derived oxadiazole-based thiourea derivatives as potential anti-diabetic agents: synthesis, in vitro antioxidant screening and in silico molecular modeling approaches, J. Mol. Struct., 2023, 1293, 136185–136199 CrossRef CAS
- A. M. Ghanim, A. S. Girgis, B. M. Kariuki, N. Samir, M. F. Said, A. Abdelnaser, S. Nasr, M. S. Bekheit, M. F. Abdelhameed, A. J. Almalki, T. S. Ibrahim and S. S. Panda, Design and synthesis of ibuprofen-quinoline conjugates as potential anti-inflammatory and analgesic drug candidates, Bioorg. Chem., 2022, 119, 105557 CrossRef CAS PubMed
- I. Chaaban, O. H. Rizk, T. M. Ibrahim, S. S. Henen, E.-S. M. El-Khawass, A. E. Bayad, I. M. El-Ashmawy and H. A. Nematalla, Synthesis, anti-inflammatory screening, molecular docking, and COX-1,2/-5-LOX inhibition profile of some novel quinoline derivatives, Bioorg. Chem., 2018, 78, 220–235 CrossRef CAS PubMed
- H. S. Abd-Ellah, M. Abdel-Aziz, M. E. Shoman, E. A. M. Beshr, T. Kaoud and A.-S. F. F. Ahmed, New 1,3,4-oxadiazole/oxime hybrids: design, synthesis, anti-inflammatory, COX inhibitory activities and ulcerogenic liability, Bioorg. Chem., 2017, 74, 15–29 CrossRef CAS PubMed
- S. S. Bharadwaj, B. Poojary, S. Madan Kumar, K. Byrappa, G. S. Nagananda, A. K. Chaitanya, K. Zaveri, N. S. Yarla, Y. Shiralgi, A. K. Kudva and B. L. Dhananjaya, Design, synthesis and pharmacological studies of some new quinoline Schiff bases and 2,5-(disubstituted-[1,3,4])-oxadiazoles, New J. Chem., 2017, 41, 8568–8585 RSC
- H. A. A. Ezelarab, H. A. Hassan, G. E.-D. A. Abuo-Rahma and S. H. Abbas, Design, synthesis, and biological investigation of quinoline/ciprofloxacin hybrids as antimicrobial and anti-proliferative agents, J. Iran. Chem. Soc., 2023, 20, 683–700 CrossRef CAS
- N. Ryad, A. A. Elmaaty, I. M Ibrahim, A. H. Ahmed Maghrabi, M. A. Yahya Alahdal, R. M. Saleem, I. Zaki and L. M. A. A. Ghany, Harnessing molecular hybridization approach to discover novel quinoline EGFR-TK inhibitors for cancer treatment, Future Med. Chem., 2024, 16, 1087–1107 CrossRef PubMed
- H. M. Abd El-Lateef, A. A. Elmaaty, L. M. A. Abdel Ghany, M. S. Abdel-Aziz, I. Zaki and N. Ryad, Design and Synthesis of 2-(4-Bromophenyl)Quinoline-4-Carbohydrazide Derivatives via Molecular Hybridization as Novel Microbial DNA-Gyrase Inhibitors, ACS Omega, 2023, 8, 17948–17965 CrossRef CAS PubMed
- D. S. Alshaya, R. M. Tawakul, I. Zaki, A. H. A. Almaaty, E. Fayad and Y. M. Abd El-Aziz, Design, synthesis and antiproliferative screening of newly synthesized acrylate derivatives as potential anticancer agents, RSC Adv., 2023, 13, 23538–23546 RSC
- H. M. Abd El-Lateef, L. M. A. Ghany, R. M. Saleem, A. H. A. Maghrabi, M. A. Y. Alahdal, E. H. K. Ali, B. Y. Beshay, I. Zaki and R. E. Masoud, Design, synthesis and antiproliferative screening of newly synthesized coumarin-acrylamide hybrids as potential cytotoxic and apoptosis inducing agents, RSC Adv., 2023, 13, 32547–32557 RSC
- D. N. Binjawhar, F. A. Al-Salmi, O. A. A. Ali, M. A. Alghamdi, E. Fayad, R. M. Saleem, I. Zaki and N. Farouk, Design, synthesis and cytotoxic activity of molecular hybrids based on quinolin-8-yloxy and cinnamide hybrids and their apoptosis inducing property, RSC Adv., 2024, 14, 11443–11451 RSC
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7, 42717–42729 CrossRef PubMed
- D. E. V. Pires, T. L. Blundell and D. B. Ascher, pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures, J. Med. Chem., 2015, 58, 4066–4072 CrossRef CAS PubMed
- A. Scalbert, C. Morand, C. Manach and C. Rémésy, Absorption and metabolism of polyphenols in the gut and impact on health, Biomed. Pharmacother., 2002, 56, 276–282 CrossRef CAS PubMed
- M. Thanou, J. C. Verhoef and H. E. Junginger, Oral drug absorption enhancement by chitosan and its derivatives, Adv. Drug Delivery Rev., 2001, 52, 117–126 CrossRef CAS PubMed
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS
- D. D. Levy, E. Zeiger, P. A. Escobar, A. Hakura, B.-j. M. van der Leede, M. Kato, M. M. Moore and K.-i. Sugiyama, Recommended criteria for the evaluation of bacterial mutagenicity data (Ames test), Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2019, 848, 403074–403084 CrossRef CAS PubMed
- S. Roy and M. K. Mathew, Fluid flow modulates electrical activity in cardiac hERG potassium channels, J. Biol. Chem., 2018, 293, 4289–4303 CrossRef CAS PubMed
- M. C. Sanguinetti, HERG1 channel agonists and cardiac arrhythmia, Curr. Opin. Pharmacol., 2014, 15, 22–27 CrossRef CAS PubMed
- E. Nicolaï, T. Güngör, J. Goyard, G. Cure, A. Fouquet, J. M. Teulon, C. Delchambre and A. Cloarec, Synthesis and aldose reductase inhibitory activity of N-(quinolinyl thiocarbonyl) glycine derivatives, Eur. J. Med. Chem., 1992, 27, 977–984 CrossRef
- K. Feist and M. Kuklinski, Synthetische Versuche in der 2-Phenyl-chinolin-Reihe I. (Synthese einiger brom-substituierter 2-Phenyl-chinolin-4-karbonsäuren, Untersuchungen über die Reaktionsfähigkeit des Bromatoms in ihnen sowie Abbau von 6- und 4′-Bromatophan nach Curtius), Arch. Pharm., 1936, 274, 244–255 CrossRef CAS
- R. Hamdy, S. A. Elseginy, N. I. Ziedan, A. T. Jones and A. D. Westwell, New Quinoline-Based Heterocycles as Anticancer Agents Targeting Bcl-2, Molecules, 2019, 24, 1274–1286 CrossRef PubMed
- H. M. Abd El-Lateef, E. E. M. Toson, A. H. Abu Almaaty, R. M. Saleem, A. H. A. Maghrabi, E. H. El-Sayed, I. Zaki and M. M. Youssef, Synthesis, Characterization and Biological Evaluation of New Enamide Fluorinated-Schiff Base Derivatives as Potential Cytotoxic and Apoptosis-Inducing Agents, ChemistrySelect, 2023, 8, e202303070 CrossRef CAS
- H. M. Abd El-Lateef, A. Gaafar, A. S. Alqahtani, A. A. Al-Mutairi, D. S. Alshaya, F. G. Elsaid, E. Fayad and N. Farouk, Design, synthesis, and antiproliferative screening of new quinoline derivatives bearing a cis-vinyl triamide motif as apoptosis activators and EGFR-TK inhibitors, RSC Adv., 2024, 14, 24781–24790 RSC
- L. Ma, S. Li, H. Zheng, J. Chen, L. Lin, X. Ye, Z. Chen, Q. Xu, T. Chen, J. Yang, N. Qiu, G. Wang, A. Peng, Y. Ding, Y. Wei and L. Chen, Synthesis and biological activity of novel barbituric and thiobarbituric acid derivatives against non-alcoholic fatty liver disease, Eur. J. Med. Chem., 2011, 46, 2003–2010 CrossRef CAS PubMed
- P. G. Baraldi, D. Preti, M. A. Tabrizi, F. Fruttarolo, G. Saponaro, S. Baraldi, R. Romagnoli, A. R. Moorman, S. Gessi, K. Varani and P. A. Borea, N6-[(Hetero)aryl/(cyclo)alkyl-carbamoyl-methoxy-phenyl]-(2-chloro)-5′'-N-ethylcarboxamido-adenosines: the first example of adenosine-related structures with potent agonist activity at the human A2B adenosine receptor, Bioorg. Med. Chem., 2007, 15, 2514–2527 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06712f |
| This journal is © The Royal Society of Chemistry 2024 |