
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design and synthesis of novel main protease inhibitors of COVID-19: quinoxalino[2,1-b]quinazolin-12-ones†
Atefeh Tirehdasta,
Seddigheh Sheikhi-Mohammareh a,
Hossein Sabet-Sarvestania,
Michael G. Organb,
Volodymyr Semeniuchenkob and
Ali Shiri*a
a,
Hossein Sabet-Sarvestania,
Michael G. Organb,
Volodymyr Semeniuchenkob and
Ali Shiri*a
aDepartment of Chemistry, Faculty of Science, Ferdowsi University of Mashhad, Mashhad, Iran. E-mail: alishiri@um.ac.irs
bDepartment of Chemistry and Biomolecular Sciences, Faculty of Science, University of Ottawa, Ottawa, Canada
First published on 13th September 2024
Abstract
The COVID-19 pandemic represents a substantial global challenge, being a significant cause of mortality in numerous countries. Thus, it is imperative to conduct research to develop effective therapies to combat COVID-19. The primary aim of this study is to employ a two-step tandem reaction involving 2,3-dichloroquinoxaline and 2-amino-N-substituted benzamides in alkaline media/DMF at an elevated temperature to design and synthesize a series of polycyclic derivatives endowed with quinoxalino[2,1-b]quinazolin-12-one framework. Following synthesis, the newly synthesized heterocycles were evaluated for their potential as inhibitors of the main protease of SARS-CoV-2 by means of molecular docking and dynamic simulation techniques. The in silico investigation demonstrated that all tested compounds effectively establish stable binding interactions, primarily through multiple hydrogen bonding and hydrophobic interactions, at the active site of the enzyme. These findings offer crucial structural insights that can be employed in future endeavors toward designing potent inhibitors targeting the main protease (Mpro). Among the investigated compounds, the p-tolylamino-substituted quinoxalino[2,1-b]quinazolinone derivative exhibited the most promise as an inhibitor of the main protease in COVID-19. Consequently, it warrants further investigation both in vitro and in vivo to identify it as a prospective candidate for anti-SARS-CoV-2 drug development.
Introduction
The global pandemic, instigated by Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), has resulted in significant consequences.1,2 Since its initial emergence in December 2019, SARS-CoV-2 (the causative agent of COVID-19) has impacted over 676 million individuals worldwide, leading to a mortality rate of 6.93 million, as cited by the World Health Organization on May 24th, 2023. Currently, individuals remain susceptible to recurrent SARS-CoV-2 infections. Despite the development of vaccines by Pfizer, Moderna, and AstraZeneca since 2020, these preventive measures have demonstrated insufficiency in countering the unforeseen mutations of SARS-CoV-2. Consequently, there is a pressing need to advance more effective antiviral therapeutics.3 SARS-CoV-2, a member of the β-coronavirus genus, is an RNA virus distinguished by its positive single-stranded genome. The replication process of this virus involves the participation of two proteolytic enzymes: the main protease (Mpro) and papain-like protease (PLpro).4–6 Mpro, also known as 3-chymotrypsin-like protease (3CLpro), shows significant promise as a target for therapeutic intervention against SARS-CoV-2 due to its noteworthy conservation across β-coronaviruses and limited similarity to human proteases.7,8 Moreover, studies have revealed substantial sequence similarity among Mpro proteins within the coronavirus group,9–12 implying that antivirals targeting SARS-CoV-2 Mpro may possess the potential to exhibit broad-spectrum effectiveness against various coronaviruses in the future.13,14 The catalytic dyad of 3CLpro, located within the active site between domains I (residues 8–101) and II (residues 102–184), comprises Cys145 and His41 amino acid residues. This dyad serves a crucial function in facilitating the replication of the viral genome.15–17 As a result, two separate classes of potent inhibitors, including both peptidomimetic and nonpeptidic inhibitors, have been identified as viable approaches in combating the swift spread of the COVID-19 pandemic.18–22As per FDA data covering the period from 2015 to 2020, it has been reported that approximately 88% of the small-molecule drugs approved contain heterocyclic skeletons.23 Quinazolinone, a nitrogen-containing heterocyclic building block that acts as a pharmacophore, exhibits a wide array of pharmacological activities spanning anti-inflammatory,24 antitumor,25 anticonvulsive,26 sedative,27 antihypertensive,28 vasodilatory,29 antimicrobial and antibacterial effects,30 thereby highlighting its potential for advancement within the domain of drugs and medicine. The literature survey revealed that 2-aminoquinazolin-4-(3H)-ones possess potent antiviral effects against both SARS-CoV-2 and MERS-CoV.31–33 In particular, 7-chloro-2-((3,5-dichlorophenyl)amino)quinazolin-4(3H)-one displayed significant efficacy (with an IC50 value of 0.23 μM) in combating SARS-CoV-2 when tested in a Vero cell assay. Additionally, the mentioned compound exhibited no toxicity and demonstrated favorable in vitro pharmacokinetic properties including high microsomal stability and minimal binding to hERG channels.31 A specific subset of quinazolinone compounds, namely the 2,3-fused quinazolinones, assumes pivotal roles in various pharmaceuticals and natural products, exemplified by Sclerotigenin, Benzomalvin A, Circumdatin H, Luotonin A, Rutaecarpine, Euxylophoricine B, Euxylophoricine E, Tryptanthrin, Mackinazolinone, Vasicinone, Isaindigotone, and Fumiquinazoline F (Fig. 1).34–36
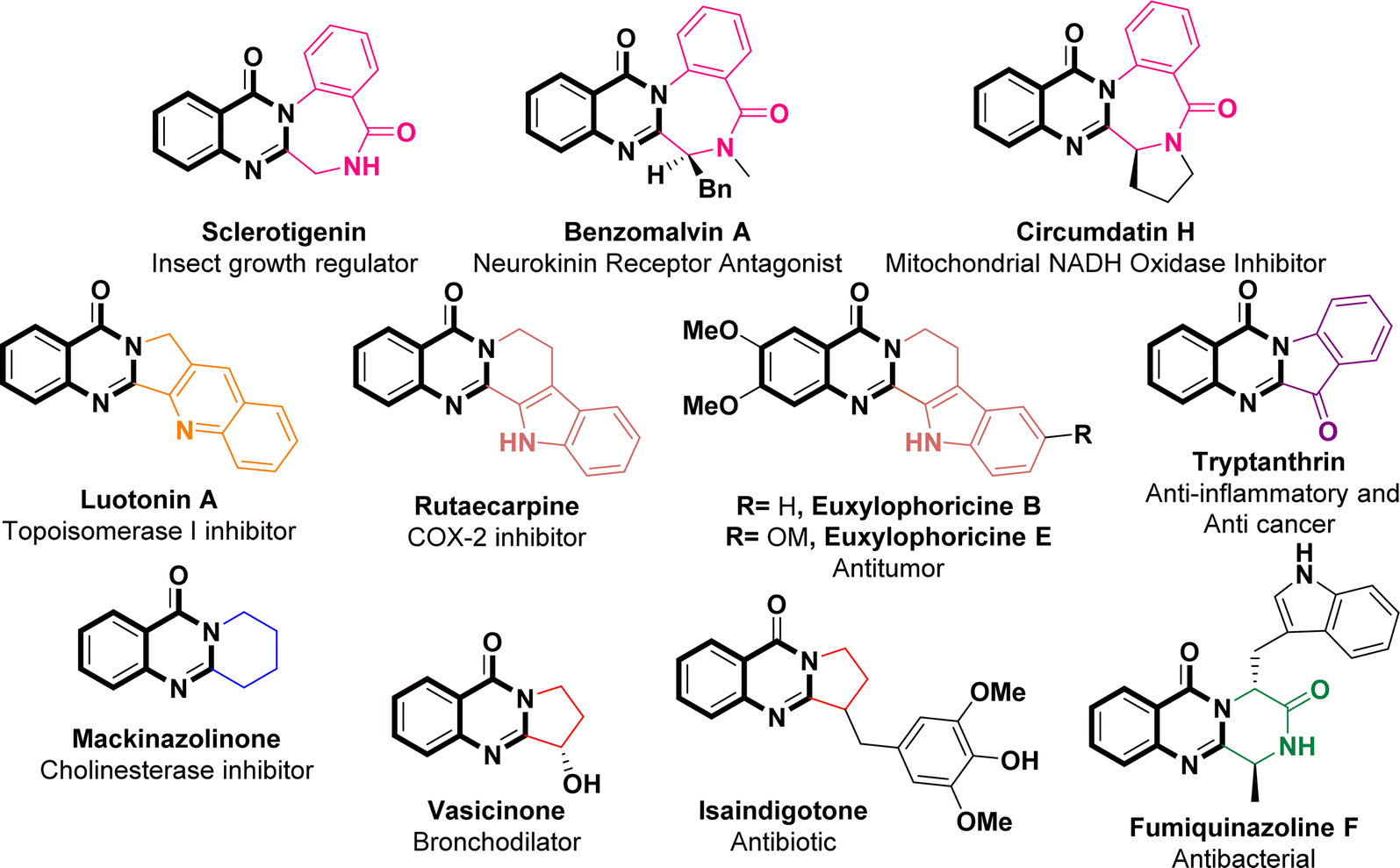 | ||
| Fig. 1 Representative natural products and drugs with 2,3-fused quinazolinone polycyclic skeletons. | ||
Despite the significant efforts made for the synthesis of novel valuable polycyclic quinazolinone-fused structures,37–39 there is still a demand for novel methodologies that can efficiently produce diverse quinoxalino[2,1-b]quinazolinones. This pursuit poses persistent challenges for organic chemists primarily because of the scarcity of literature on the synthesis of quinoxalino[2,1-b]quinazolinones, with only one study found thus far by Pan et al. in February 2024. The recently published procedure offers a solution to this gap by employing a cascade radical cyclization strategy, involving the reaction of 3-(2-isocyanophenyl)quinazolin-4(3H)-ones with ethers under photocatalytic and metal-free conditions.40
Soon after the outbreak of COVID-19, we designed and synthesized several novel heterocyclic architectures and theoretically evaluated their antiviral inhibitory against SARS-CoV-2 Mpro in order to contribute to the efforts in tackling the rising threat of fatal coronaviruses.41,42 With this background, stemming from our ongoing research interest in the synthesis of novel potentially bioactive heterocyclic scaffolds,43–52 and considering the potent applications of quinazolinone-based heterocycles against both SARS-CoV-2 and MERS-CoV,31–33 we have developed a novel methodology for synthesizing derivatives of quinoxalino[2,1-b]quinazolinones. Computational assessments were carried out to explore the interaction of the synthetic N-heterocyclic compounds with the SARS-CoV-2 Mpro, utilizing docking and molecular dynamics approaches. These assessments aimed to describe novel nonpeptidic and non-covalent inhibitors of COVID-19 Mpro derived from a two-component reaction involving 2,3-dichloroquinoxaline and 2-amino-N-substituted benzamides in DMF.
Results and discussion
Initially, the ring opening of isatoic anhydride through the reaction of several primary amines in aqueous media at room temperature resulted in the formation of off-white anthranilamide derivatives (1a–h) (Scheme 1).53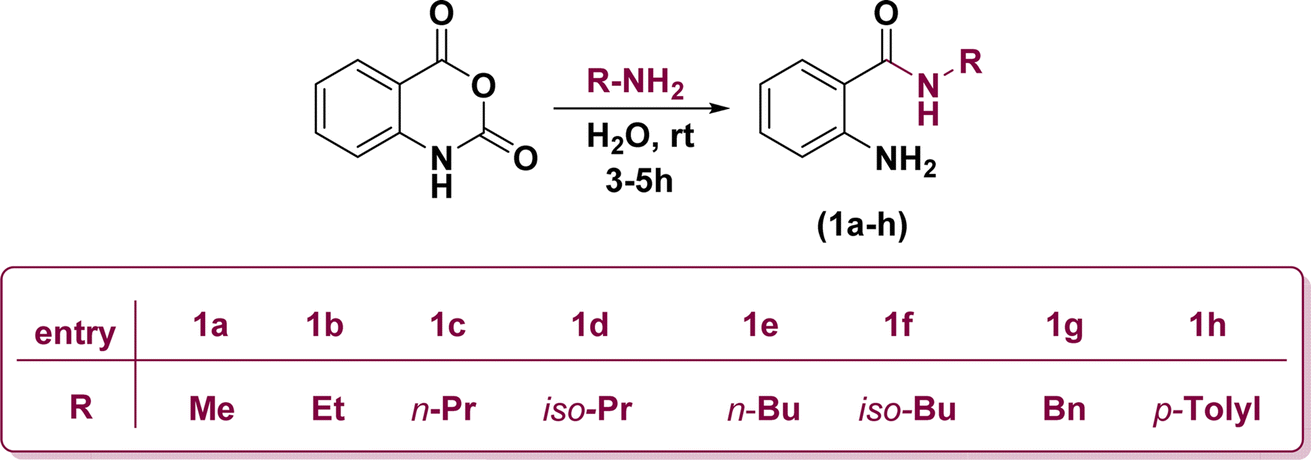 | ||
| Scheme 1 Construction of anthranilamide derivatives. | ||
On the other hand, 2,3-dichloroquinoxaline (2) was also synthetically prepared through the treatment of equimolar amounts of ortho-phenylenediamine and diethyloxalate in refluxing ethanol for 6 h followed by chlorination of the obtained quinoxaline-2,3-diol (Scheme 2).54
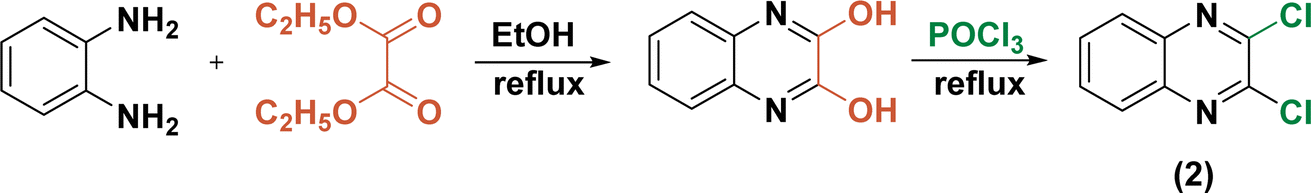 | ||
| Scheme 2 Construction of 2,3-dicholoroquinoxaline. | ||
Replacement of one of the chlorine atoms of the substrate (2) with –NH2 group of compounds (1a–h) through SNAr amination afforded quinoxaline derivatives (3a–h) endowed with ortho-amino-N-alkyl/phenyl benzamide substituents appended in C-2 position. These substitution reactions proceeded at room temperature in the presence of potassium hydroxide dissolved in dimethylformamide (Scheme 3).
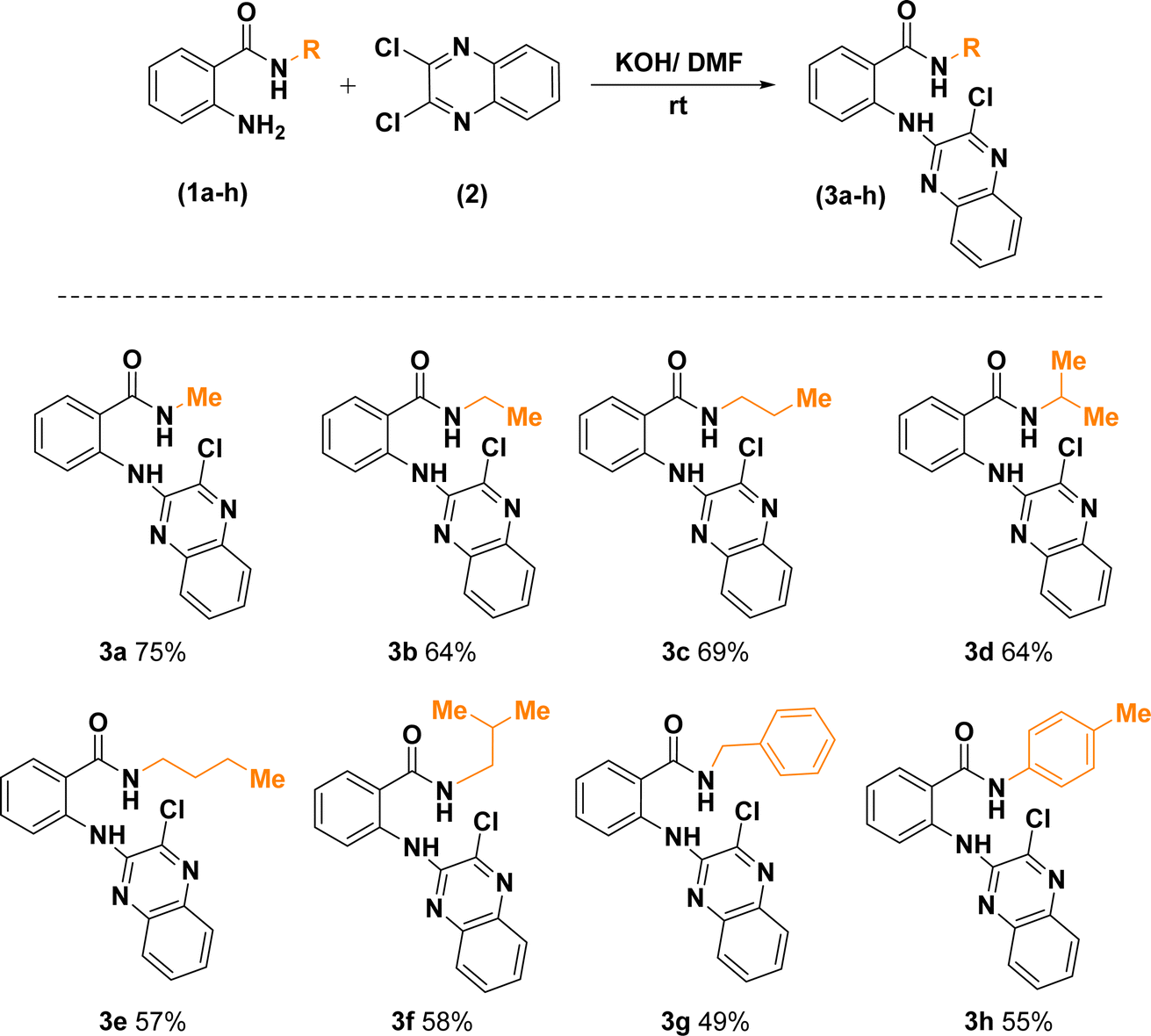 | ||
| Scheme 3 Synthetic route for the construction of 2-((3-chloroquinoxalin-2-yl)amino)-N-substituted benzamides. | ||
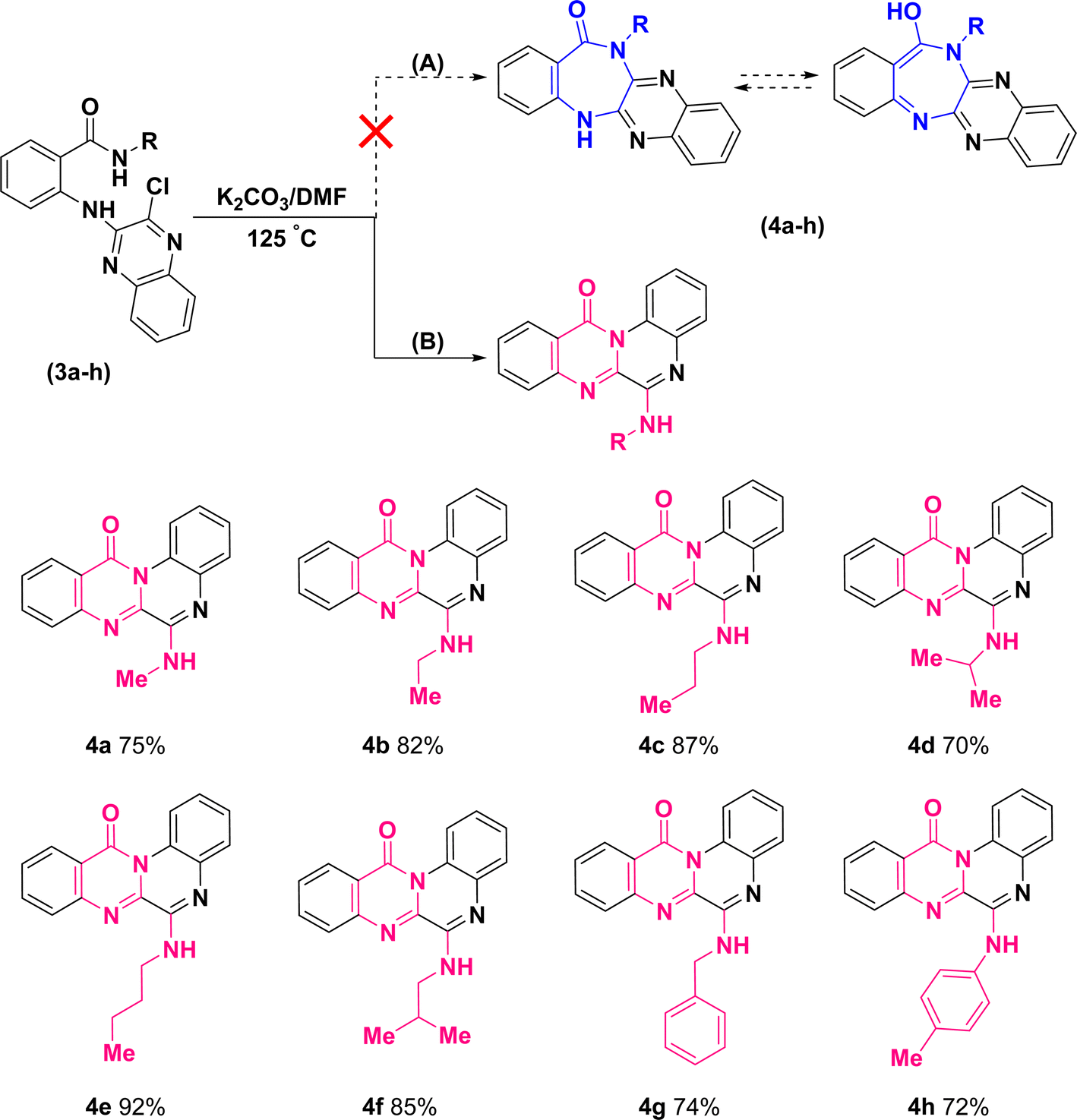
Further treatment of di-hetero(aryl) amine compounds (3a–h) with K2CO3 in DMF medium at elevated temperature facilitated the synthesis of products (4a–h) that displayed a blue shift for amidic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band (based on the infrared spectra) as well as chlorine atom removal (according to the mass spectra). These findings which uniformly observed across all the newly synthesized compounds, provided unequivocal establishment for the definite occurrence of heterocyclization. However, direct intramolecular nucleophilic substitution of amidic moiety on C(sp2)–Cl of quinoxaline scaffold (pathway (A)) or tandem multistep pathway (B) leading to annulation of diazepinones or pyrimidinones, respectively, is open to dispute (Scheme 4).
O stretching band (based on the infrared spectra) as well as chlorine atom removal (according to the mass spectra). These findings which uniformly observed across all the newly synthesized compounds, provided unequivocal establishment for the definite occurrence of heterocyclization. However, direct intramolecular nucleophilic substitution of amidic moiety on C(sp2)–Cl of quinoxaline scaffold (pathway (A)) or tandem multistep pathway (B) leading to annulation of diazepinones or pyrimidinones, respectively, is open to dispute (Scheme 4).
 | ||
| Scheme 4 Synthetic route for the construction of 6-(alkylamino)-12H-quinoxalino[2,1-b]quinazolin-12-ones. | ||
The structural assignments of all the newly synthesized compounds (4a–h) were corroborated by spectroscopic and microanalytical data. Although the formation of both structural isomers (A) and (B) was predicted, the experimental results revealed the formation of only one isomer, either (A) or (B). In the 1H-NMR spectra of compounds (4a–h), a broad signal (with slight multiplicity) around δ 7.10–7.25 ppm and a singlet signal at δ 9.77 ppm, belonging to a D2O-exchangeable proton, were observed for compounds (4a–g) and (4h), respectively. Regarding compound (4a), for instance, upon exchange of XH to XD (X: O in isomer (A) or N in isomer (B)) in the presence of D2O, the corresponding broad signal at δ 7.12 ppm vanished and the doublet signal of methyl protons was transformed into a singlet signal at δ 3.21 ppm (S32, ESI†).
This phenomenon can be justified only in the case of a correlation between the exchangeable proton and methyl protons, either in terms of bonding or space. Consequently, 1H–1H correlation spectroscopy (COSY) (S33, ESI†) and nuclear Overhauser effect spectroscopy (NOESY) (S34, ESI†) were conducted on compound (4a). Resembling the 1H-NMR, both of the two-dimensional spectra supported that the aforementioned hydrogen groups were coupled to each other due to the existence of cross-peaks.
As 1D and 2D-NMR evidences did not succeed to differentiate between frameworks (A) and (B), the following structures could still be the robust isomeric candidates for cyclization of (3a) into (4a) (Fig. 2).
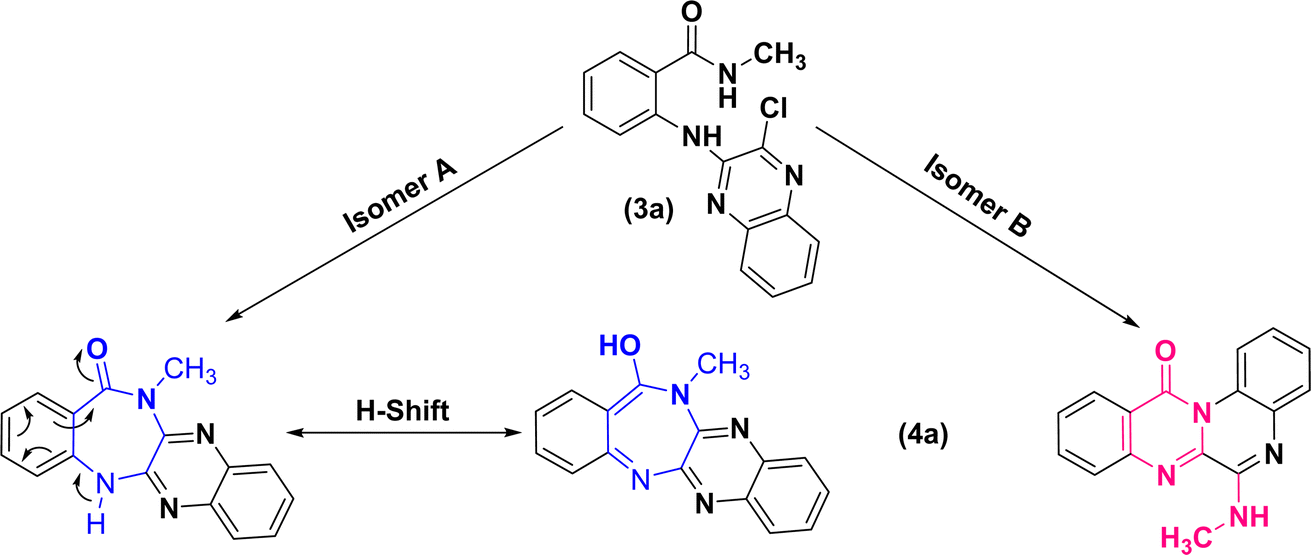 | ||
| Fig. 2 Structural isomeric candidates for annulation of (3a) into (4a). | ||
Eventually, a single crystal X-ray diffraction analysis should be employed to ascertain how cyclization has occurred. In this regard, slow evaporation of (4e)–ethanol solution yielded thin poorly diffracting needles, which allowed solving atoms connectivity, and confirmed the structure (4e). High quality crystals of (4e)–perchlorate salt were grown by slow evaporation from a binary mixture of ethanol–aqueous perchloric acid. Fig. 3 depicts the molecular structure of 4e perchlorate.
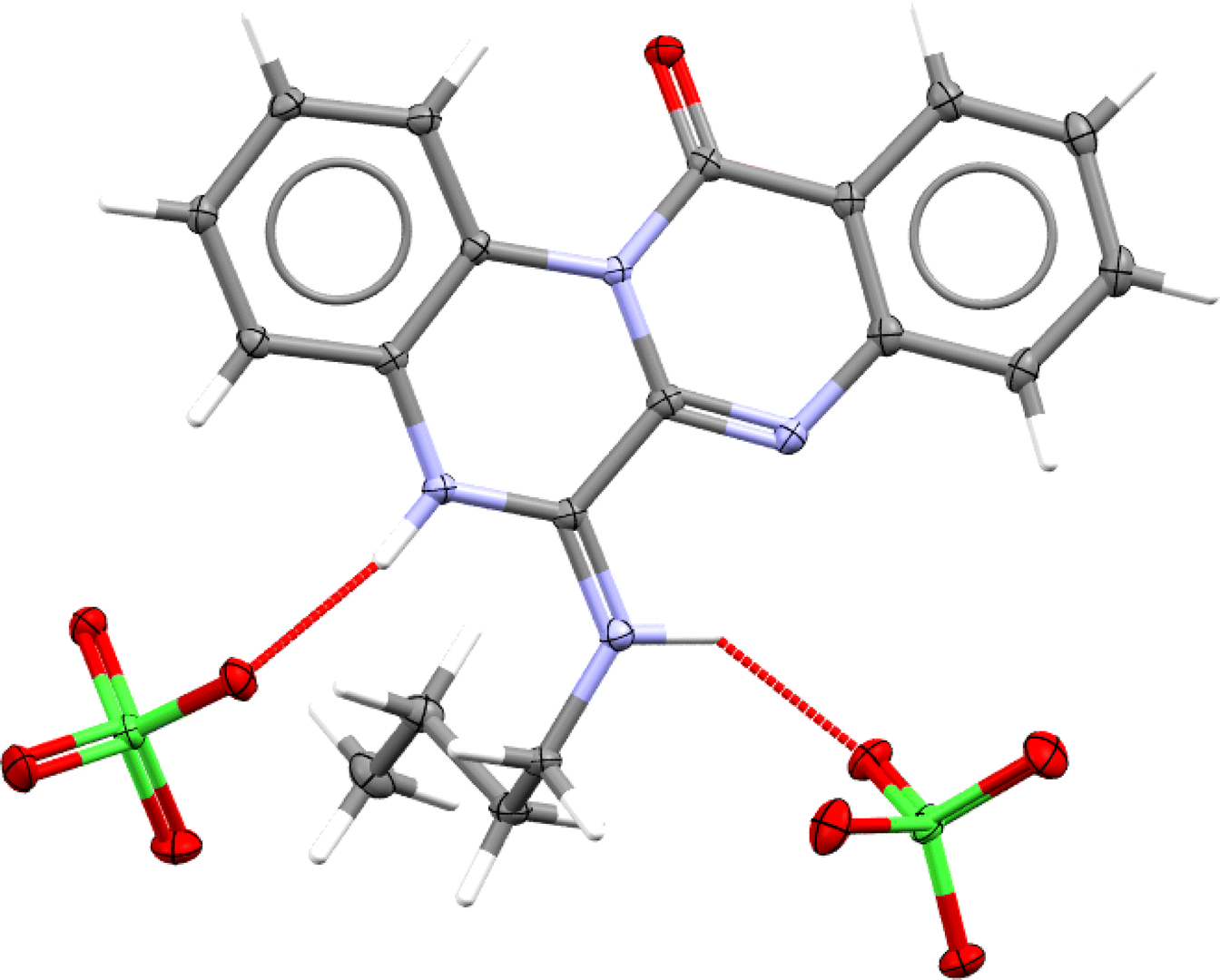 | ||
| Fig. 3 Molecular structure of 4e–perchlorate (CCDC 2357348). Asymmetric unit contains one 4e–H+ cation and one ClO4− anion, however, hydrogen bonding connects one cation to two anions (as shown here) and vice versa. | ||
According to these results, the plausible mechanism for the synthesis of quinoxalino[2,1-b]quinazolinone derivatives (4a–h) from (3a–h) has been proposed as a self-explanatory domino pathway. This pathway includes an intramolecular nucleophilic substitution of amidic carbonyl followed by an in situ SNAr amination. As depicted in Fig. 4, first, the endocyclic N of quinoxaline moiety of (3a–h) performs a nucleophilic attack on the carbonyl group leading to the cyclization of pyrimidinone. Then, the replacement of chlorine atom with non-isolated nucleophiles (–NH2R) accompanied by the elimination of HCl leads to the formation of the corresponding derivatives of (4a–h).
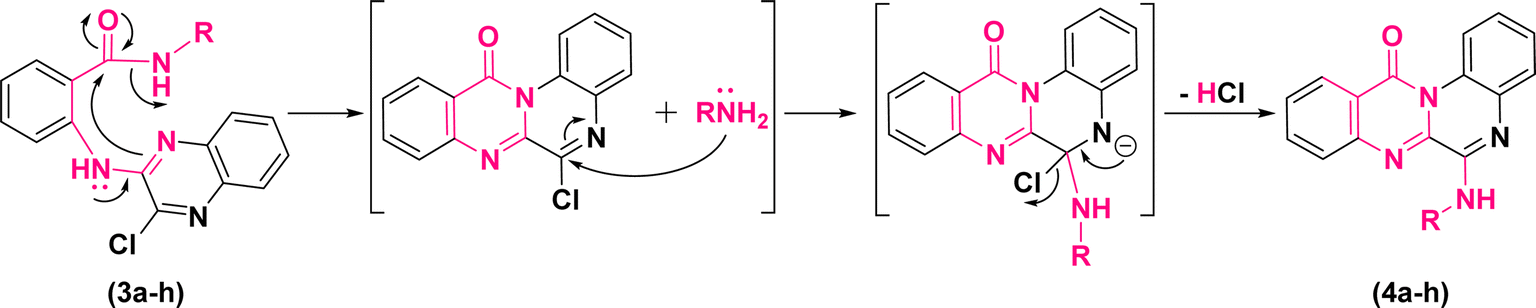 | ||
| Fig. 4 Plausible mechanism for the synthesis of 6-(alkylamino)-12H-quinoxalino[2,1-b]quinazolin-12-ones. | ||
Computational analyses
Computer-aided drug design (CADD) is a powerful approach that utilizes computational methods and algorithms to expedite and improve the drug discovery and development process. CADD techniques aid in identifying and validating potential drug targets by analyzing biological data and understanding the interactions between proteins, enzymes, receptors, and other molecules involved in disease pathways. This helps researchers pinpoint the most promising targets for therapeutic intervention.Methodology
| Compound | Interacting residues | Binding affinity (kcal mol−1) |
|---|---|---|
| (4a) | His163, Cys145, Met49 | −7.7 |
| (4b) | His41, Cys145, Met49, Leu27, Met165 | −7.6 |
| (4c) | His163, Cys145, Met49, Leu27, Met165 | −7.7 |
| (4d) | Met165, His41, Cys145, Leu27 | −7.9 |
| (4e) | His41, Cys145, Leu27, Gln189, Ser144, Gly143, Asn142 | −7.8 |
| (4f) | Met165, His41, Cys145, Gln189, Asn142, Gly143, Ser144, Leu27 | −8.1 |
| (4g) | Met165, His163, Cys145, Met49, Thr25 | −8.7 |
| (4h) | Met165, His41, Cys145 | −9.2 |
| N3(Ref) | Leu287, Leu272, Tyr237, Ala193, Thr169 | −6.4 |
Fig. 5 illustrates the interactions of amino acids and the binding modes of compounds (4g) and (4h).
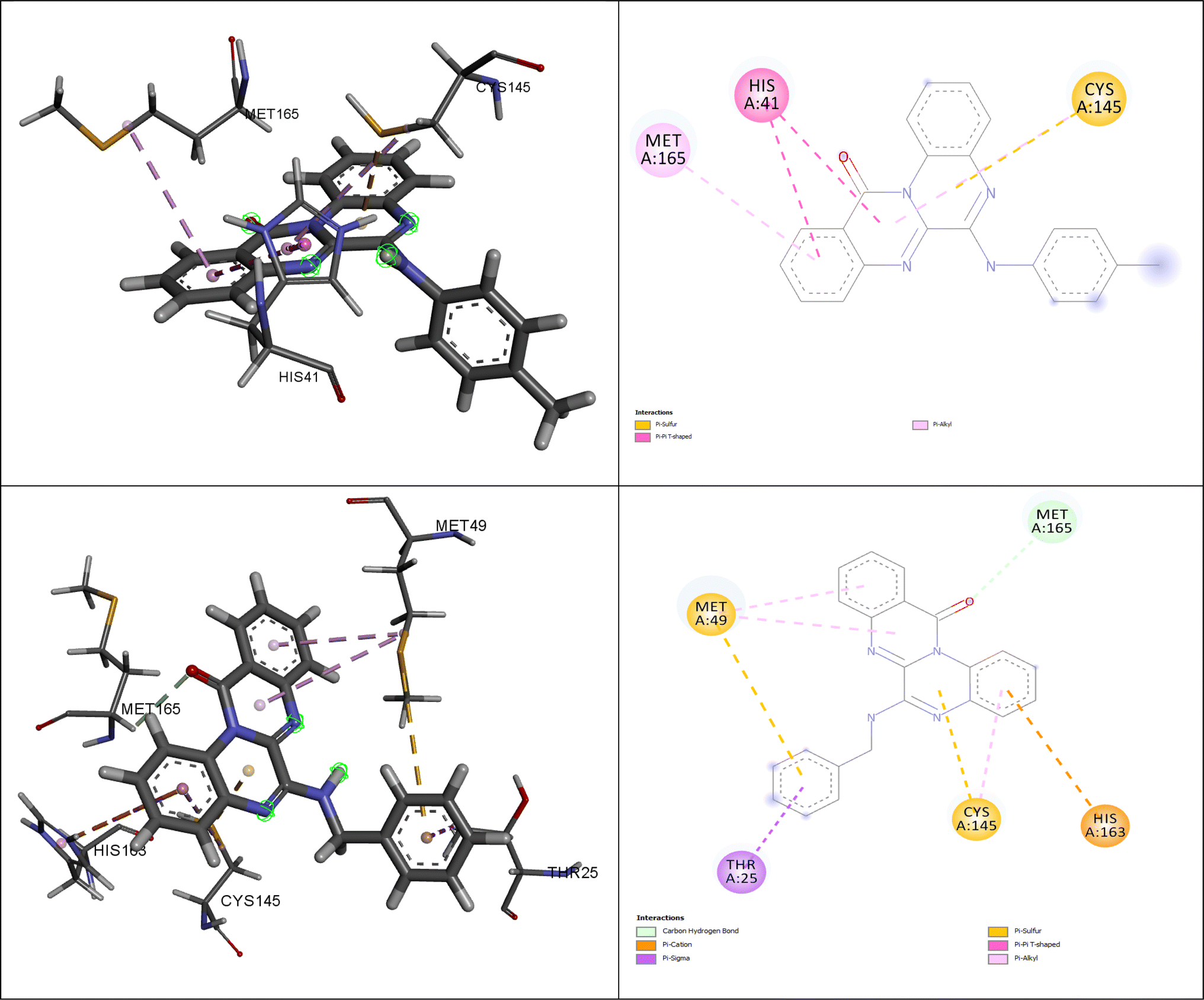 | ||
| Fig. 5 The interacting amino acids for the best-docked synthesized compounds: (4g) (down) and (4h) (up). | ||
The entire system underwent energy minimization without restraints using the steepest descent algorithm65 for 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 iteration steps to attain equilibrium. Equilibration was achieved through two distinct simulation steps. Initially, the systems were equilibrated under a constant number of particles, volume, and temperature (NVT ensemble)66 for 200 ps at 310 K, employing the Verlet scheme to regulate temperature. Subsequently, equilibration continued under a constant number of particles, pressure, and temperature (NPT ensemble)67 at 300 K for 200 ps, with Berendsen barostat and Verlet scheme68 used to control pressure (set to 1 bar) and temperature, respectively. The LINCS algorithm was utilized to manage covalent bond constraints during equilibration.69
000 iteration steps to attain equilibrium. Equilibration was achieved through two distinct simulation steps. Initially, the systems were equilibrated under a constant number of particles, volume, and temperature (NVT ensemble)66 for 200 ps at 310 K, employing the Verlet scheme to regulate temperature. Subsequently, equilibration continued under a constant number of particles, pressure, and temperature (NPT ensemble)67 at 300 K for 200 ps, with Berendsen barostat and Verlet scheme68 used to control pressure (set to 1 bar) and temperature, respectively. The LINCS algorithm was utilized to manage covalent bond constraints during equilibration.69
Following equilibration, production MD simulations were conducted for 100 ns at a constant temperature of 300 K, with simulation trajectories saved every 1 fs. Each MD simulation was performed in triplicate for every complex, and the mean values of obtained parameters were used for structural analysis. The results of MD simulation were illustrated in Fig. 6(a–d) which a–d corresponds to the Root Mean Square Deviation (RMSD), the Root Mean Square Fluctuation (RMSF), the Radius of Gyration (Rg) and the numbers of hydrogen bonds plots, respectively.
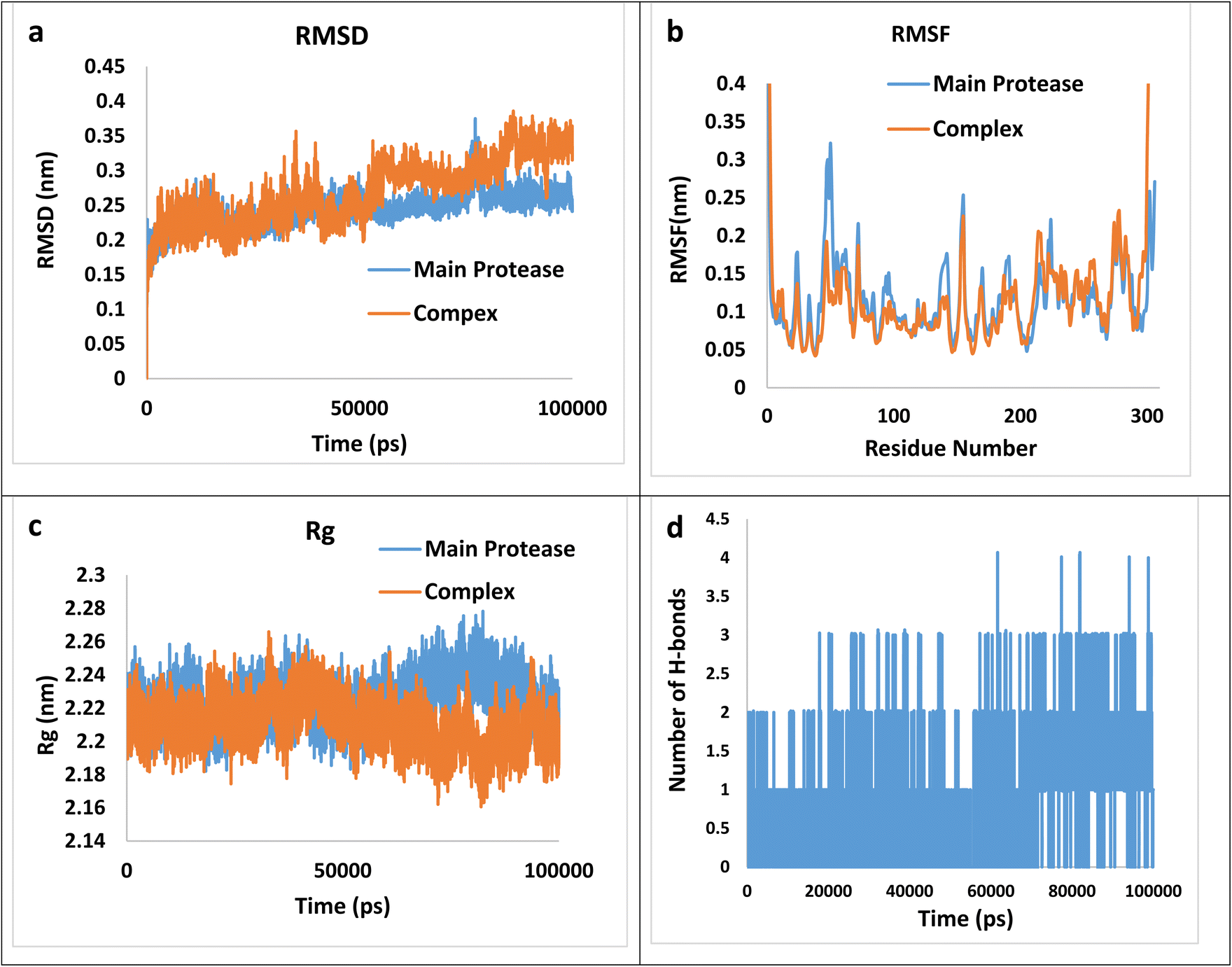 | ||
| Fig. 6 The RMSD (a), RMSF (b), Rg (c), and the numbers of hydrogen bonds plots (d) for the (4h)–main protease complex during 100 ns simulation. | ||
RMSD graphs illustrate the stability and conformational alterations observed in both the main protease and its associated complex with (4h) throughout the simulation. Mean RMSD values ranging from 0.1 to 0.3 nm are generally deemed satisfactory and indicative of stability. However, values exceeding 0.3 nm are considered unfavorable and are typically disregarded.70 The average calculated RMSD value stands at 0.27 nm, falling within an acceptable range. Furthermore, RMSF calculation proves valuable for identifying local fluctuations over the course of the simulation. The RMSF index provides insight into the flexibility of the protein chain relative to a reference frame. It measures the deviation of Cα atom coordinates from their average position, serving as a criterion for assessing these effects. The peaks observed in the RMSF plot indicate that certain residues exhibit greater fluctuations compared to others within the studied complex. Specifically, RMSF values for the complexes reveal significant fluctuations at the N- and C-termini in contrast to other regions. This phenomenon is commonly observed and does not require further investigation. Regarding the average values 0.117 and 0.120 nm for the main protease and the complex, respectively, can be concluded that the main protease does not involve abnormal fluctuations. The radius of gyration (Rg) factor serves as a crucial metric for assessing the compactness of the ligand–protein complex throughout molecular simulations. It quantifies the extent of variation in distance between the center of mass of the protein atoms and its terminal. A lower Rg value in the ligand–protein complex indicates higher compactness, implying more effective interactions between the ligand and the protein. The mean Rg values for the complex and the main protease are 2.21 and 2.23 nm, suggesting more compactness of the complex. Finally, the number of hydrogen bonds during the simulation is an effective factor in the stability of the complex, the calculated average value of hydrogen bonds during the time of simulation is 0.81. Thus, besides the hydrogen bonding, other intermolecular forces may exert a greater effect on the total bending energy.
Conclusion
In summary, with the aim of discovering potent nonpeptidic and non-covalent inhibitors targeting 3CLpro, a key enzyme implicated in the replication of the coronavirus, an efficient two-step procedure was established for the synthesis of quinoxalino[2,1-b]quinazolin-12-one derivatives from synthetically accessible starting materials 2,3-dichloroquinoxaline and anthranilamides. Following that, molecular docking and molecular dynamics simulations were employed to assess the inhibitory potential of synthesized products against the main protease of COVID-19. According to molecular docking results, 6-(p-tolylamino)-12H-quinoxalino[2,1-b]quinazolin-12-one (4h) displayed the lowest binding affinity (−9.0 kcal mol−1), indicating their favorable orientation within the active site of the chymotrypsin-like cysteine protease (3CLpro). To gain deeper insights into ligand–protein interactions, molecular docking outcomes served as inputs for molecular dynamics simulations. The simulations revealed that the binding energy value of the (4h)–main protease complex is −131.96 kJ mol−1, also van der Waals and polar solvation energies are the most effective parameters on the total binding energy. Thus, the findings suggest that compound (4h) bearing p-tolylamino substitution on C-6 of quinoxalino[2,1-b]quinazolinone is a promising inhibitor against the main protease of COVID-19.Experimental
Melting points were measured by an Electrothermal type 9200 melting point apparatus. The 1H NMR (300 MHz) and the 13C NMR (75 MHz) spectra were obtained on a Bruker Avance DRX-300 Fourier transform spectrometer using tetramethylsilane as an internal standard. An Avatar 370 FT-IR Thermo Nicolet spectrometer was employed to record the IR spectra and a Varian Mat CH-7 instrument for scanning mass spectra at 70 eV. Micro analytical data were obtained on a Thermo Finnigan Flash EA 1112 microanalyzer.General procedure for the synthesis of 2-((3-chloroquinoxalin-2-yl)amino)-N-substituted benzamide derivatives (3a–h)
A mixture of appropriate 2-amino-N-substituted benzamide derivatives (1a–h) (1 mmol), 2,3-dichloroquinoxaline (2) (1 mmol, 0.199 g) and excess amount of KOH (6–8 granular pellets) in DMF (3 ml) was stirred at room temperature for 16 h. The progress of the reaction was monitored by TLC using chloroform:methanol 20:1 eluent. After the completion of the reaction, the mixture was cooled, poured into an ice/water bath and neutralized with aqueous 5% HCl solution. The resulting solid product was collected by filtration, washed with water (2 × 20 ml) and recrystallized from EtOH.
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) –CO). 13C NMR (75 MHz, DMSO-d6, ppm): δC 26.9, 120.0, 120.4, 122.3, 126.7, 126.8, 127.9, 128.5, 130.9, 132.4, 136.8, 139.2, 139.9, 140.4, 145.4, 169.5. MS (m/z) = 312 (M+). Anal. calcd for C16H13ClN4O (%): C, 61.45; H, 4.19; N, 17.91. Found: C, 61.43; H, 4.14; N, 17.80.–CO). 13C NMR (75 MHz, DMSO-d6, ppm): δC 15.0, 34.6, 120.0, 120.7, 122.2, 126.6, 126.7, 127.9, 128.6, 130.8, 132.3, 136.8, 139.1, 139.9, 140.4, 145.4, 168.8; MS (m/z) = 326 (M+). Anal. calcd for C17H15ClN4O (%): C, 62.48; H, 4.63; N, 17.15. Found: C, 62.43; H, 4.60; N, 17.13.–CO). 13C NMR (75 MHz, DMSO-d6, ppm): δC 11.9, 22.6, 41.5, 120.0, 120.9, 122.3, 126.7, 126.8, 127.9, 128.7, 131.0, 132.3, 136.8, 139.2, 139.9, 140.3, 145.5, 169.0. MS (m/z) = 340 (M+). Anal. calcd for C18H17ClN4O (%): C, 63.44; H, 5.03; N, 16.44. Found: C, 63.40; H, 5.01; N, 16.42.–CO). 13C NMR (75 MHz, DMSO-d6, ppm): δC 22.6, 41.6, 120.0, 121.1, 122.3, 126.7, 126.8, 128.0, 128.9, 131.0, 132.3, 136.8, 139.2, 140.0, 140.2, 145.5, 168.2. MS (m/z) = 340 (M+). Anal. calcd for C18H17ClN4O (%): C, 63.44; H, 5.03; N, 16.44. Found: C, 63.42; H, 5.02; N, 16.42.–CO). 13C NMR (75 MHz, DMSO-d6, ppm): δC 14.2, 20.1, 31.5, 39.4, 120.0, 121.0, 122.4, 126.7, 126.9, 128.0, 128.7, 131.1, 132.4, 136.9, 139.2, 140.0, 140.2, 145.5, 169.0. MS (m/z) = 354 (M+). Anal. calcd for C19H19ClN4O (%): C, 64.31; H, 5.40; N, 15.79. Found: C, 64.28; H, 5.38; N, 15.75.–CO). 13C NMR (75 MHz, DMSO-d6, ppm): δC 20.7, 28.5, 47.1, 120.0, 121.1, 122.3, 126.7, 126.8, 128.0, 128.7, 131.0, 132.3, 136.8, 139.1, 139.9, 140.2, 145.4, 169.1. MS (m/z) = 354 (M+). Anal. calcd for C19H19ClN4O (%): C, 64.31; H, 5.40; N, 15.79. Found: C, 64.28; H, 5.38; N, 15.77.–CO). 13C NMR (75 MHz, DMSO-d6, ppm): δC 43.1, 120.1, 120.5, 122.4, 126.7, 126.9, 127.4, 127.7, 128.0, 128.7, 128.8, 131.0, 132.6, 136.9, 139.2, 139.6, 139.9, 140.4, 145.4, 169.1. MS (m/z) = 388 (M+). Anal. calcd for C22H17ClN4O (%): C, 67.95; H, 4.41; N, 14.41. Found: C, 67.91; H, 4.40; N, 14.39.
–CO). 13C NMR (75 MHz, DMSO-d6, ppm): δC 26.9, 120.0, 120.4, 122.3, 126.7, 126.8, 127.9, 128.5, 130.9, 132.4, 136.8, 139.2, 139.9, 140.4, 145.4, 169.5. MS (m/z) = 312 (M+). Anal. calcd for C16H13ClN4O (%): C, 61.45; H, 4.19; N, 17.91. Found: C, 61.43; H, 4.14; N, 17.80.–CO). 13C NMR (75 MHz, DMSO-d6, ppm): δC 15.0, 34.6, 120.0, 120.7, 122.2, 126.6, 126.7, 127.9, 128.6, 130.8, 132.3, 136.8, 139.1, 139.9, 140.4, 145.4, 168.8; MS (m/z) = 326 (M+). Anal. calcd for C17H15ClN4O (%): C, 62.48; H, 4.63; N, 17.15. Found: C, 62.43; H, 4.60; N, 17.13.–CO). 13C NMR (75 MHz, DMSO-d6, ppm): δC 11.9, 22.6, 41.5, 120.0, 120.9, 122.3, 126.7, 126.8, 127.9, 128.7, 131.0, 132.3, 136.8, 139.2, 139.9, 140.3, 145.5, 169.0. MS (m/z) = 340 (M+). Anal. calcd for C18H17ClN4O (%): C, 63.44; H, 5.03; N, 16.44. Found: C, 63.40; H, 5.01; N, 16.42.–CO). 13C NMR (75 MHz, DMSO-d6, ppm): δC 22.6, 41.6, 120.0, 121.1, 122.3, 126.7, 126.8, 128.0, 128.9, 131.0, 132.3, 136.8, 139.2, 140.0, 140.2, 145.5, 168.2. MS (m/z) = 340 (M+). Anal. calcd for C18H17ClN4O (%): C, 63.44; H, 5.03; N, 16.44. Found: C, 63.42; H, 5.02; N, 16.42.–CO). 13C NMR (75 MHz, DMSO-d6, ppm): δC 14.2, 20.1, 31.5, 39.4, 120.0, 121.0, 122.4, 126.7, 126.9, 128.0, 128.7, 131.1, 132.4, 136.9, 139.2, 140.0, 140.2, 145.5, 169.0. MS (m/z) = 354 (M+). Anal. calcd for C19H19ClN4O (%): C, 64.31; H, 5.40; N, 15.79. Found: C, 64.28; H, 5.38; N, 15.75.–CO). 13C NMR (75 MHz, DMSO-d6, ppm): δC 20.7, 28.5, 47.1, 120.0, 121.1, 122.3, 126.7, 126.8, 128.0, 128.7, 131.0, 132.3, 136.8, 139.1, 139.9, 140.2, 145.4, 169.1. MS (m/z) = 354 (M+). Anal. calcd for C19H19ClN4O (%): C, 64.31; H, 5.40; N, 15.79. Found: C, 64.28; H, 5.38; N, 15.77.–CO). 13C NMR (75 MHz, DMSO-d6, ppm): δC 43.1, 120.1, 120.5, 122.4, 126.7, 126.9, 127.4, 127.7, 128.0, 128.7, 128.8, 131.0, 132.6, 136.9, 139.2, 139.6, 139.9, 140.4, 145.4, 169.1. MS (m/z) = 388 (M+). Anal. calcd for C22H17ClN4O (%): C, 67.95; H, 4.41; N, 14.41. Found: C, 67.91; H, 4.40; N, 14.39.General procedure for the synthesis of (4a–h)
A mixture of the appropriate 2-((3-chloroquinoxalin-2-yl)amino)-N-substituted benzamide derivatives (3a–h) (1 mmol) and K2CO3 (4 mmol, 0.552 g) in DMF (3 ml) was heated at 125 °C for 5 h. After the completion of the reaction (monitored by TLC, CHCl3:MeOH, 20:1), the mixture was cooled, poured onto ice water and neutralized with aqueous 5% HCl solution. The resulting solid product was collected by filtration and recrystallized from acetone to form crystalline yellow needle precipitates.
Single crystal (thin needle, 0.055 × 0.092 × 0.174 mm) of compound (4e) was grown by slow evaporation of ethanol solution. Diffraction (crystal was cooled to 100 K) to 1.5 Å resolution with reasonable statistic (R-factor 0.4) allowed to determine approximate unit cell (a = 7.01, b = 10.24, c = 20.92, α = 90.11, β = 90.03, γ = 90.15) and solve the structure in P1 space group (Z = 4), as weak reflections did not allow to refine unit cell and assign correct space group. Nevertheless, atoms connectivity confirmed the structure of compound (4e). Then it was subjected to slow evaporation from aqueous perchloric acid–ethanol mixture, which resulted in large crystals of (4e)–perchlorate salt, suitable for high resolution X-ray diffraction experiment. Full crystallographic report is available in ESI† (CCDC deposition number 2357348).
Data availability
Crystallographic data for compound (4e) has been deposited at the Cambridge Crystallographic Data Centre under deposition no. CCDC 2357348. The other data sets used and analyzed during the current study are available in ESI.†Conflicts of interest
The authors declares that they have no conflicts of interest.Acknowledgements
The authors gratefully acknowledge the Research Council of Ferdowsi University of Mashhad for financial support of this project (3/47643) and the University of Ottawa for accepting Ali Shiri as a visiting professor and completing the research related to this project.References
- M.-K. Looi, BMJ, 2020, 371, m4113 CrossRef PubMed.
- P. Smith, BMJ, 2020, m3350 CrossRef PubMed.
- K. Al Adem, J. C. Ferreira, S. Fadl and W. M. Rabeh, J. Biol. Chem., 2023, 299, 102790 CrossRef CAS PubMed.
- A. E. Gorbalenya, S. C. Baker, R. S. Baric, R. J. de Groot, C. Drosten, A. A. Gulyaeva, B. L. Haagmans, C. Lauber, A. M. Leontovich and B. W. Neuman, Nat. Microbiol., 2020, 5, 536–544 CrossRef PubMed.
- P. Zhou, X.-L. Yang, X.-G. Wang, B. Hu, L. Zhang, W. Zhang, H.-R. Si, Y. Zhu, B. Li and C.-L. Huang, Nature, 2020, 579, 270–273 CrossRef CAS PubMed.
- F. Wu, S. Zhao, B. Yu, Y.-M. Chen, W. Wang, Z.-G. Song, Y. Hu, Z.-W. Tao, J.-H. Tian and Y.-Y. Pei, Nature, 2020, 579, 265–269 CrossRef CAS PubMed.
- A. Citarella, A. Scala, A. Piperno and N. Micale, Biomolecules, 2021, 11, 607 CrossRef CAS PubMed.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang and C. Peng, Nature, 2020, 582, 289–293 CrossRef CAS PubMed.
- S. Su, G. Wong, W. Shi, J. Liu, A. C. K. Lai, J. Zhou, W. Liu, Y. Bi and G. F. Gao, Trends Microbiol., 2016, 24, 490–502 CrossRef CAS PubMed.
- M. D. Sacco, C. Ma, P. Lagarias, A. Gao, J. A. Townsend, X. Meng, P. Dube, X. Zhang, Y. Hu and N. Kitamura, Sci. Adv., 2020, 6, eabe0751 CrossRef CAS PubMed.
- L. Fu, F. Ye, Y. Feng, F. Yu, Q. Wang, Y. Wu, C. Zhao, H. Sun, B. Huang and P. Niu, Nat. Commun., 2020, 11, 1–8 CrossRef PubMed.
- C. Ma, M. D. Sacco, B. Hurst, J. A. Townsend, Y. Hu, T. Szeto, X. Zhang, B. Tarbet, M. T. Marty and Y. Chen, Cell Res., 2020, 30, 678–692 CrossRef CAS PubMed.
- C. Ma, M. D. Sacco, Z. Xia, G. Lambrinidis, J. A. Townsend, Y. Hu, X. Meng, T. Szeto, M. Ba and X. Zhang, ACS Cent. Sci., 2021, 7, 1245–1260 CrossRef CAS PubMed.
- Y. M. Bar-On, A. Flamholz, R. Phillips and R. Milo, Elife, 2020, 9, e57309 CrossRef PubMed.
- F. Wang, C. Chen, W. Tan, K. Yang and H. Yang, Sci. Rep., 2016, 6, 22677 CrossRef CAS PubMed.
- L. Zhang, D. Lin, X. Sun, U. Curth, C. Drosten, L. Sauerhering, S. Becker, K. Rox and R. Hilgenfeld, Science, 2020, 368, 409–412 CrossRef CAS PubMed.
- Z. Ren, L. Yan, N. Zhang, Y. Guo, C. Yang, Z. Lou and Z. Rao, Protein Cell, 2013, 4, 248 CrossRef PubMed.
- L. Zhang, D. Lin, Y. Kusov, Y. Nian, Q. Ma, J. Wang, A. Von Brunn, P. Leyssen, K. Lanko and J. Neyts, J. Med. Chem., 2020, 63, 4562–4578 CrossRef CAS PubMed.
- N. Verma, J. A. Henderson and J. Shen, J. Am. Chem. Soc., 2020, 142, 21883–21890 CrossRef CAS PubMed.
- P. Thanigaimalai, S. Konno, T. Yamamoto, Y. Koiwai, A. Taguchi, K. Takayama, F. Yakushiji, K. Akaji, S.-E. Chen and A. Naser-Tavakolian, Eur. J. Med. Chem., 2013, 68, 372–384 CrossRef CAS PubMed.
- V. Frecer and S. Miertus, RSC Adv., 2020, 10, 40244–40263 RSC.
- C.-H. Zhang, K. A. Spasov, R. A. Reilly, K. Hollander, E. A. Stone, J. A. Ippolito, M.-E. Liosi, M. G. Deshmukh, J. Tirado-Rives and S. Zhang, ACS Med. Chem. Lett., 2021, 12, 1325–1332 CrossRef CAS PubMed.
- P. Bhutani, G. Joshi, N. Raja, N. Bachhav, P. K. Rajanna, H. Bhutani, A. T. Paul and R. Kumar, J. Med. Chem., 2021, 64, 2339–2381 CrossRef CAS PubMed.
- J. Qiu, Q. Zhou, Y. Zou, S. Li, L. Yang, W. Chen, J. Gao and X. Gu, Eur. J. Med. Chem., 2022, 231, 114159 CrossRef CAS PubMed.
- J. Hricovíniová, Z. Hricovíniová and K. Kozics, Int. J. Mol. Sci., 2021, 22, 610 CrossRef PubMed.
- P. S. Auti, G. George and A. T. Paul, RSC Adv., 2020, 10, 41353–41392 RSC.
- A. M. Soliman, H. M. Karam, M. H. Mekkawy, M. Higgins, A. T. Dinkova-Kostova and M. M. Ghorab, Eur. J. Med. Chem., 2020, 200, 112467 CrossRef CAS PubMed.
- S. M. Elfeky, T. R. Sobahi, M. M. Gineinah and N. S. Ahmed, Arch. Pharm., 2020, 353, 1900211 CrossRef CAS PubMed.
- L. Hudson, J. Mui, S. Vázquez, D. M. Carvalho, E. Williams, C. Jones, A. N. Bullock and S. Hoelder, J. Med. Chem., 2018, 61, 7261–7272 CrossRef CAS PubMed.
- R. Bouley, D. Ding, Z. Peng, M. Bastian, E. Lastochkin, W. Song, M. A. Suckow, V. A. Schroeder, W. R. Wolter and S. Mobashery, J. Med. Chem., 2016, 59, 5011–5021 CrossRef CAS PubMed.
- J. Y. Lee, Y. S. Shin, S. Jeon, S. I. Lee, S. Noh, J.-E. Cho, M. S. Jang, S. Kim, J. H. Song and H. R. Kim, Bioorg. Med. Chem. Lett., 2021, 39, 127885 CrossRef CAS PubMed.
- J. Y. Lee, Y. S. Shin, S. Jeon, S. I. Lee, J. Cho, S. Myung, M. S. Jang, S. Kim, J. H. Song and H. R. Kim, Bull. Korean Chem. Soc., 2022, 43, 412–416 CrossRef CAS PubMed.
- Y. S. Shin, J. Y. Lee, S. Jeon, J.-E. Cho, S. Myung, M. S. Jang, S. Kim, J. H. Song, H. R. Kim and H. Park, Pharmaceuticals, 2022, 15, 831 CrossRef CAS PubMed.
- U. A. Kshirsagar, Org. Biomol. Chem., 2015, 13, 9336–9352 RSC.
- D. He, M. Wang, S. Zhao, Y. Shu, H. Zeng, C. Xiao, C. Lu and Y. Liu, Fitoterapia, 2017, 119, 136–149 CrossRef CAS PubMed.
- S. Eguchi, Bioact. Heterocycl., 2006, I, 113–156 Search PubMed.
- P. Ghosh, B. Ganguly and S. Das, Org. Biomol. Chem., 2020, 18, 4497–4518 RSC.
- R. S. Rohokale and U. A. Kshirsagar, Synthesis, 2016, 48, 1253–1268 CrossRef CAS.
- W. Wang, P.-S. Zou, L. Pang, C.-X. Pan, D.-L. Mo and G.-F. Su, Org. Biomol. Chem., 2022, 20, 6293–6313 RSC.
- X. Wu, L. Liu, C. Xiang, J.-T. Yu and C. Pan, Chem. Commun., 2024, 60, 2556–2559 RSC.
- P. Moghimi, H. Sabet-Sarvestani, O. Kohandel and A. Shiri, J. Org. Chem., 2022, 87, 3922–3933 CrossRef CAS PubMed.
- P. Moghimi, H. Sabet-Sarvestani and A. Shiri, J. Biomol. Struct. Dyn., 2023, 41, 13198–13210 CrossRef CAS PubMed.
- S. Sheikhi-Mohammareh, F. Oroojalian, H. Beyzaei, M. Moghaddam-Manesh, A. Salimi, F. Azizollahi and A. Shiri, Talanta, 2023, 262, 124723 CrossRef CAS PubMed.
- I. Farzamnezhad, S. Sheikhi-Mohammareh, H. Beyzaei, E. Yarmohammadi and A. Shiri, Polycyclic Aromat. Compd., 2024, 44, 807–817 CrossRef CAS.
- R. Maghamifar, S. Sheikhi-Mohammareh, H. Beyzaei, Z. Ebrahimnezhad and A. Shiri, J. Iran. Chem. Soc., 2023, 20, 2297–2305 CrossRef CAS.
- S. Bigonah-Rasti, S. Sheikhi-Mohammareh, K. Saadat and A. Shiri, Polycyclic Aromat. Compd., 2022, 42, 2644–2654 CrossRef CAS.
- O. Kohandel, S. Sheikhi-Mohammareh, F. Oroojalian, T. Memariani, J. Mague and A. Shiri, Mol. Diversity, 2022, 26, 1621–1633 CrossRef CAS PubMed.
- S. Sheikhi-Mohammareh, A. Shiri and J. Mague, Mol. Diversity, 2022, 26, 923–937 CrossRef CAS PubMed.
- S. Sheikhi-Mohammareh, A. Shiri, E. H. Maleki, M. M. Matin, H. Beyzaei, P. Baranipour, F. Oroojalian and T. Memariani, ChemistrySelect, 2020, 5, 10060–10066 CrossRef CAS.
- S. Sheikhi-Mohammareh, A. Shiri, H. Beyzaei and E. Yarmohammadi, Monatsh. Chem., 2020, 151, 963–969 CrossRef CAS.
- S. Sheikhi-Mohammareh, M. Mashreghi and A. Shiri, J. Iran. Chem. Soc., 2020, 17, 1555–1566 CrossRef CAS.
- S. Sheikhi-Mohammareh and A. Shiri, J. Heterocycl. Chem., 2018, 55, 2055–2060 CrossRef CAS.
- M. Mahdavi, N. Foroughi, M. Saeedi, M. Karimi, H. Alinezhad, A. Foroumadi, A. Shafiee and T. Akbarzadeh, Synlett, 2014, 25, 385–388 CAS.
- K. V. G. Chandra Sekhar, V. S. Rao, W. Deuther-Conrad, D. Sridhar, H. N. Nagesh, V. S. Kumar, P. Brust and M. M. K. Kumar, Med. Chem. Res., 2013, 22, 1660–1673 CrossRef CAS.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- D. S. Biovia, Discovery Studio Modeling Environment, Dassault System Release, 2015, vol. 4 Search PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CrossRef CAS PubMed.
- M. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Petersson, Uranyl extraction by N,N-dialkylamide ligands studied by static and dynamic DFT simulations, in Gaussian 09, Inc., Wallingford CT, 2009 Search PubMed.
- J. M. Bofill, Mol. Model. Annu., 1995, 1, 11–17 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1, 19–25 CrossRef.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren and J. Hermans, in Intermolecular Forces: Proceedings of the fourteenth Jerusalem symposium on Quantum Chemistry and Biochemistry Held in jerusalem, israel, April 13–16, 1981, Springer, 1981, pp. 331–342 Search PubMed.
- X. Yuan, Y. Wang, Z. Qu, J. Ren, X. Wu and J. Wang, Chem. Res. Chin. Univ., 2013, 29, 934–940 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 1–8 CrossRef PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- S. S. Petrova and A. D. Solovev, Hist. Math., 1997, 24, 361–375 CrossRef.
- S. Thanna, C. M. Goins, S. E. Knudson, R. A. Slayden, D. R. Ronning and S. J. Sucheck, J. Org. Chem., 2017, 82, 3844–3854 CrossRef CAS PubMed.
- D. Zhang, C.-F. Chen, B.-B. Zhao, L.-L. Gong, W.-J. Jin, J.-J. Liu, J.-F. Wang, T.-T. Wang, X.-H. Yuan and Y.-W. He, PLoS One, 2013, 8, e80636 CrossRef PubMed.
- D. E. Elmore and D. A. Dougherty, Biophys. J., 2001, 81, 1345–1359 CrossRef CAS PubMed.
- X.-H. Yuan, Y.-C. Wang, W.-J. Jin, B.-B. Zhao, C.-F. Chen, J. Yang, J.-F. Wang, Y.-Y. Guo, J.-J. Liu and D. Zhang, PLoS One, 2012, 7, e32938 CrossRef CAS PubMed.
- B. N. Diallo, T. Swart, H. C. Hoppe, Ö. Tastan Bishop and K. Lobb, Sci. Rep., 2021, 11, 1413 CrossRef CAS PubMed.
- N. A. Baker, D. Sept, S. Joseph, M. J. Holst and J. A. McCammon, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10037–10041 CrossRef CAS PubMed.
- R. Kumari, R. Kumar, O. S. D. D. Consortium and A. Lynn, J. Chem. Inf. Model., 2014, 54, 1951–1962 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2357348. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra06025c |
| This journal is © The Royal Society of Chemistry 2024 |