
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of vanillin via oxidation of kenaf stalks in the presence of CeO2: tuning the catalytic behaviour of CeO2 via nanostructure morphology†
Anita Ramli *a,
Nur Akila Syakida Idayu Khairul Anuara,
Normawati Mohamad Yunusb and
Alina Rahayu Mohamedc
*a,
Nur Akila Syakida Idayu Khairul Anuara,
Normawati Mohamad Yunusb and
Alina Rahayu Mohamedc
aHICoE Centre of Biofuel and Biochemical Research (CBBR), Institute of Sustainable Energy & Resources (ISER), Department of Fundamental & Applied Sciences, Universiti Teknologi PETRONAS, Seri Iskandar 32610, Perak, Malaysia. E-mail: anita_ramli@utp.edu.my
bCentre of Research in Ionic Liquids (CORIL), Institute of Sustainable Energy & Resources (ISER), Department of Fundamental and Applied Sciences, Universiti Teknologi PETRONAS, Seri Iskandar 32610, Perak, Malaysia. E-mail: normaw@utp.edu.my
cFaculty of Chemical Engineering & Technology, UniMAP, Complex of Academics Jejawi 3, Jejawi, Arau 02600, Perlis, Malaysia. E-mail: alina@unimap.edu.my
First published on 13th November 2024
Abstract
Different CeO2 nanostructures were synthesized using a hydrothermal method and treated with alkaline NaOH, followed by drying at 120 °C for 16 h and calcined at 400 °C for the direct oxidation of kenaf stalks to vanillin under microwave irradiation. The catalysts were characterized for their physicochemical properties using XRD, BET, Raman spectroscopy, TPR, TPO, and XPS. All synthesized CeO2 nanostructures show diffraction peaks corresponding to the formation of cubic fluorite, which agrees with Raman spectra of the F2g mode. The N2 adsorption–desorption isotherms showed that all catalysts possess a type IV isotherm, indicating a mesoporous structure. TPR and TPO analyses display formation peaks corresponding to surface-to-bulk reducibility and the oxidized oxygen ratio, which is responsible for the redox properties of ceria nanostructures. The XPS analysis of CeO2 nanostructures proved that Ce exists in the Ce3+ and Ce4+ oxidation states. All catalysts were tested for direct oxidation of kenaf stalks under microwave irradiation with the highest vanillin yield obtained by the CeO2-Nps-400 heterogeneous catalyst at 3.84%, whereas 4.66% vanillin was produced using 2 N NaOH as a homogeneous catalyst.
Introduction
Vanillin (4-hydroxy-3-methoxy benzaldehyde) is a vital commercial compound with a wide range of applications in the food and perfumery industries, and it is naturally extracted from dried pods of the vanilla plant.1 The commercial vanillin market is served by three major sources: natural vanilla from vanilla beans (approximately 0.2%); vanillin produced from petroleum-based intermediates, especially guaiacol, which accounts for 85% of the world supply; while the remaining 15% is produced from lignin derived from the wood pulping process.2 Synthetic vanillin cannot be labelled as natural but rather as a “synthetic” or “artificial” vanilla flavor.3 However, according to EU regulations, if the process and base material are natural, such as lignin, phenolic stilbenes, isoeugenol, eugenol, ferulic acid and aromatic acids, the vanillin produced can be considered ‘natural vanilla aroma’.4 Therefore, lignin could be used as a natural precursor to meet the growing demand for natural vanillin.Various studies have been conducted to produce vanillin from lignocellulosic biomass under a catalyzed medium. Deng et al.5 reported 5.3% vanillin yield obtained from wet aerobic oxidation of lignin from cornstalks under 2 M NaOH as a solution with 5 wt% of LaCo0.8Cu0.2O3 as a catalyst. Another study by Qu et al.6 reported that 5% vanillin yield was obtained through direct oxidation of Japanese cedar biomass using microwave irradiation in a 2 N NaOH alkaline medium with peroxide as an oxidant and CuO as a catalyst. Ceria is a well-known catalyst or catalyst promoter for numerous industrial redox processes. The interconversion between the +4 and +3 oxidation states of cerium makes CeO2 a suitable candidate for redox catalysts.7 Both surface and bulk oxygen vacancies in CeO2 are suitable sites for adsorption. Hence, besides being useful as a support, CeO2 can actively participate in chemical reactions and provide an active site as an oxygen carrier.8
The oxidation of commercial organosolv lignin was carried out using a batch-type high-pressure autoclave reactor at 170–185 °C for 24 h in a MeOH solution medium under continuous stirring with 1–10 bars purged of oxygen and Pd/CeO2 as a catalyst. The highest vanillin yield of 5.2% was achieved.9 However, Rawat et al.10 reported that selective production of vanillin from oxidative depolymerization of alkali lignin catalyzed using MoPO/CeO2 was carried out with a batch reactor under continuous stirring at 150 °C for 3 h in 2 M NaOH solution with 5 bars of oxygen as an oxidant yielding 9% of vanillin. Our initial work employing Ce and Zr catalysts supported by MgO to oxidize kenaf stalks to produce vanillin directly indicates that CeO2 is active in vanillin production. The oxidation of lignin from the kenaf stalk using Ce/MgO catalyst under microwave gives a 3.70 yield of vanillin, while bare MgO and Zr/MgO do not give any vanillin product.11 These facts provoked our interest in conducting the present investigation on ceria.
Different shapes of CeO2 nanostructures give a prevalence of specific crystal facets that display different reactivities in various catalytic processes.12 By tailoring the shape and size of the particles, certain crystal facets can be exposed, leading to different structural and redox properties, as reported by researchers.13–15 Thus, methods that control crystal shape may provide a course to ascertain an insight aspect required to enhance catalytic activity.16 Theoretically, the presence of particular active facets of the ceria surface affected oxygen vacancy formation and its ability to act as an oxygen donor–acceptor.17 The oxygen storage/release capacity is capable of explaining the quantitative amount of oxygen that can be substituted by a reducible oxide in the gas phase or with an active metal component. Konsolakis et al.17 and Qiao et al.14 found that the rod-shaped ceria nanostructures prevailing [110] and [100] facets possess a high catalytic conversion of CO oxidation, where they show a consistent trend with the highest oxygen storage capacity (OSC) and surface to bulk ratio compared to nanocubes and nanopolyhedral. Altering the ceria structure generates differences in the surface atomic arrangement and the electronic properties, which also significantly influence the oxygen vacancy formation energy.18 This research investigated catalytic activity and correlated it with the number of oxygen vacancies and OSC, especially for the oxidation of kenaf stalks to vanillin production. Nevertheless, the reactivity of oxygen vacancies on different crystal facets for vanillin production has rarely been discussed.
The kenaf plant was used as a biomass feedstock. Kenaf is an annual fiber crop plant that is composed of inner core fiber (75–60%) and bast fiber (24–40%)19 and has a high syringyl to guaiacyl ratio in aromatic composition owing to its lignin characteristic. It is reported that kenaf stalks contain 13–15% lignin.20,21 This paper is based on the use of CeO2 in the oxidation of the lignocellulosic biomass of kenaf to vanillin reported recently. In a previous paper, CeO2 nanostructures were prepared by varying the calcination temperature from 400 to 600 °C. Catalyst screening was done to choose the ideal calcination temperature based on the performance of vanillin production. It was found that the CeO2 nanostructure calcined at 400 °C showed a higher production of vanillin due to its high characteristics in textural properties compared to other catalysts.22 The conditions for catalyst screening were 180 °C of microwave heating for 20 min with 10 wt% of a catalyst under 200 W microwave irradiation, and it was found that a calcination temperature of 400 °C gave the highest yield. The chosen catalyst was further utilized to determine the optimum reaction conditions.22 Our recently published paper shows different active facets on different nanostructures of CeO2 that have been successfully proven and discussed using TEM and HRTEM. The uniform shape of the CeO2 nanostructures (nanoparticles, nanorods and nanocubes) from the TEM image was found, while HRTEM images show that the CeO2 nanostructures are single crystalline in nature.22 Thus, this research discussed the correlation of different CeO2 nanostructure morphologies for renewable vanillin produced directly from biomass under microwave heating and was further tested for optimized reaction conditions.
Experimental
Materials
Dried kenaf stalks were obtained from the National Kenaf and Tobacco Board, Malaysia. Methanol (CH3OH), sodium hydroxide pallet (NaOH), ethanol, ethyl acetate, hydrochloric acid (HCl), hydrogen peroxide and vanillin laboratory standard (as standard) were obtained from Aldrich, USA. Sodium hydroxide and cerium hexahydrate (Ce(NO3)3·6H2O) were obtained from Merck, Germany.Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ. Crystallite size was analyzed using HighScore Plus software (version 3.0), Malvern, UK, with an X-ray wavelength of Cu Kα radiation at λ = 1.54 A, where θ is the Bragg angle. β is the full width at half maximum in radians, corresponding to the 2θ value at the (111) plane. The unknown shape factor, k, was assumed to be 0.89, and the reflecting peak at 2θ was chosen for the entire sample.
cosθ. Crystallite size was analyzed using HighScore Plus software (version 3.0), Malvern, UK, with an X-ray wavelength of Cu Kα radiation at λ = 1.54 A, where θ is the Bragg angle. β is the full width at half maximum in radians, corresponding to the 2θ value at the (111) plane. The unknown shape factor, k, was assumed to be 0.89, and the reflecting peak at 2θ was chosen for the entire sample.The surface area and pore size of the catalysts were analyzed using Brunauer–Emmett–Teller (BET) (Micromeritics ASAP 2020, Norcross, GA, USA). The catalysts were degassed at 200 °C for 24 h prior to N2 adsorption measurement at −77 °C.
The TPR experiment on H2 consumption and oxygen capacity storage was conducted using a Thermo Scientific TPDRO 1100 with a TCD detector. The samples (0.5 g) were treated by heating from 298 to 1073 K at a rate of 10 K min−1 in a flow of 5 vol% H2 in He. The total gas flow rate was 10 mL (NTP) per min. The TPO experiment was conducted on a Thermo Scientific TPDRO 1100 using a TCD detector. The samples (0.5 g) were treated by heating from 298 to 1073 K at a rate of 10 K min−1 in a flow of 5 vol% O2 in He. The total gas flow rate was 10 mL (NTP) per min.
The Raman spectra of the glasses were collected at room temperature using a Jasco NRS-3300. Raman spectrometer was equipped with a CCD detector (−69 °C) using the 514 nm line of an air-cooled Ar ion laser (Melles Griot), 600 lines per mm grating, 0.1 × 6 mm slit and 100× Olympus objective lens.
Thermo Scientific X-ray photoelectron spectroscopy (XPS) equipment with MgKα X-ray radiation source (E = 1253.6 eV) was performed to analyze each element's surface composition and chemical state present in the catalyst structure. The C 1s peak set at 284.6 eV was used as the charge referencing. The atomic concentration ratios in the outer layers of the samples were evaluated from the corresponding XPS area ratios using an effective ionization cross-section of the ejected electrons.24 According to the ratio of the Ce3+ ion peak area to that of the total Ce3+ and Ce4+ ion peak areas, the following equations were used to calculate the relative content of Ce3+ in the CeO2-Nps-400 catalyst:25
| ACe3+ = Av′ + Au′, |
| ACe4+ = Av + Av′′ + Av′′′ + Au + Au′′ + Au′′′. |
| Relative content of Ce3+ = ACe3+/(ACe3+ + ACe4+). | (1) |
The surface chemisorbed oxygen ratio [R(Os)] can be obtained as follows:26
| R(Os) = A(Os)/[A(Os) + A(Ob)]. | (2) |
:2, agitated for 15 seconds with a vortex agitator at 5000 rpm, and then centrifuged for 15 minutes at 1000 rpm. The supernatant was then treated with 1:1 ethyl acetate. Vanillin and other low-molecular weight molecules were extracted into the organic phase. The mixture was agitated for 60 seconds at 5000 rpm and then centrifuged for 5 minutes at 1000 rpm, separating the mixture into two phases. The upper phase was placed in a vial. At 50 °C and 400 mbar, excess solvent was evaporated using a rotary evaporator.11,16:10, and the mass range (m/z) was in the range of 40–600 m/z.6Results and discussions
Catalyst characterization
Fig. 1 shows the XRD patterns of synthesized CeO2 nanoparticles (CeO2-Nps-400), nanorods (CeO2-Ncs-400) and nanocubes (CeO2-Nrs-400) calcined at 400 °C for 2 h. Multiple sharp peaks were observed for all catalyst samples, indicating the nanocrystalline nature of CeO2. The XRD results demonstrated that all prepared CeO2 nanostructures show the formation of cubic fluorite that is perfectly indexed to ICSD: 81-0792 with different intensities. The diffraction peaks at 2θ = 28.53, 33.07, 47.46, 56.31, 59.06, 69.38, 76.66, and 79.03° correspond to the (111), (200), (220), (311), (222), (400), (331) and (420) planes, respectively. These observations agree with those of Lykaki et al.,27 who reported that the presence of all diffraction peaks of CeO2 nanostructures indicates the prominent peak of a pure ceria face-centered cubic fluorite structure (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m symmetry). In general, the results also indicate that the peak intensity for CeO2-Ncs-400 is higher and has a well-defined sharp peak compared to CeO2-Nrs-400 and CeO2-Nps-400, which means an increase in the size and crystallinity of the products.28
m symmetry). In general, the results also indicate that the peak intensity for CeO2-Ncs-400 is higher and has a well-defined sharp peak compared to CeO2-Nrs-400 and CeO2-Nps-400, which means an increase in the size and crystallinity of the products.28
 | ||
| Fig. 1 XRD patterns of CeO2 nanostructures calcined at 400 °C. | ||
Based on the full width at half maximum (FWHM) of the diffraction peak (111), crystallite sizes of CeO2 nanostructures calcined at 400 °C were estimated via the Scherrer equation and are summarized along with textural properties of catalysts (surface area, pore volume and average pore size), as illustrated in Table 1. The obtained nanostructures of CeO2 in the forms of nanoparticles, nanorods and nanocubes were prepared by adjusting the concentration of sodium hydroxide and hydrothermal reaction temperature.
| Catalyst | SBET (m2 g−1) | SV (cm3 g−1) | SP (nm) | Ssize (nm) | FWHM (cm−1) | AD/AF2g |
|---|---|---|---|---|---|---|
| CeO2-Nps-400 | 66.1 | 0.275 | 27.5 | 11.3 | 50 | 0.136 |
| CeO2-Nrs-400 | 57.8 | 0.145 | 24.0 | 15.0 | 46 | 0.113 |
| CeO2-Ncs-400 | 22.5 | 0.134 | 17.1 | 28.7 | 19 | 0.065 |
In particular, the crystallite sizes of CeO2 are 11.3, 15.0 and 28.7 nm for CeO2-Nps-400, CeO2-Nrs-400 and CeO2-Ncs-400, respectively, while the crystallite size of CeO2 nanostructures increases in the order of CeO2-Ncs-400 < CeO2-Nrs-400 < CeO2-Nps-400. The large crystallite size of CeO2-Ncs-400 might be due to the expansion of lattice between nanocubes crystals, which agrees with the report by Hailstone et al.29 They also found that the increase in the cluster size was due to lattice expansion ascribed to the decreased amounts of Ce3+ at the surface of smaller clusters, which directed the reduced Ce–O electrostatic attraction.
The results showed that CeO2 nanoparticles have the highest BET surface area, pore volume and average pore size with increasing order of CeO2-Ncs-400 < CeO2-Nrs-400 < CeO2-Nps-400. In addition, the surface areas found in the present study correlate with the crystallinity of the catalysts, as shown in the XRD results. According to Lykaki et al.,27 these differences in the distribution of textural properties are linked to different ceria morphologies and can be mainly accounted for by the observed variations in the BET surface area. Moreover, it is noteworthy that the BET surface area follows the reverse order of the crystallite size of CeO2 (111) phases (Table 1). This shows that the larger the crystallite size, the lower the BET surface area. Fig. 2 shows the N2 adsorption–desorption isotherms of the nanostructures of CeO. According to the International Union of Pure and Applied Chemistry (IUPAC) classification, all the synthesized CeO2 nanostructures resemble type IV isotherm with type H1 desorption hysteresis according to IUPAC classification, which indicates the formation of a well-developed mesoporous structure.30 The hysteresis loop of type H1 is commonly found in highly ordered mesoporous materials, such as MCM-41, MCM-48 and SBA 15.31
 | ||
| Fig. 2 N2 adsorption–desorption of CeO2 nanostructures. | ||
Raman spectroscopy was used to characterize the oxygen vacancy abundances of ceria's exposed planes on the structural defects. Fig. 3 shows the Raman spectra of the CeO2 nanostructures catalyst. The spectra are dominated by a strong peak at 463 cm−1 from the F2g mode of the CeO2 Fmm cubic fluorite, which agrees with the XRD patterns (Fig. 2). Kainbayev et al.32 reported that the observed peak inherent to cerium oxide ranges from around 460 to 463 cm−1, which is in accordance with the F2g mode corresponding to the symmetric vibrations of oxygen ions around Ce4+ ions in cerium oxide, while the D band observed at 598–615 cm−1 is assigned to structural defects caused by the perturbations in cerium lattice.27 For CeO2-Nps-400, the Raman band is detected at 610 cm−1 with high intensity compared to CeO2-Nrs-400 and CeO2-Ncs-400 catalyst, which is also assigned to the presence of oxygen atom detachment, resulting from Ce4+ transforming to Ce3+ partial reduction.33 As presented in Table 1, the intensity of the peak and FWHM values is the highest for CeO2-Nps-400, followed by CeO2-Nrs-400 and CeO2-Ncs-400. According to Ndifor et al.,34 the FWHM of CeO2 increased with a decreasing crystallite size and/or a greater concentration of oxygen vacancies. The Raman shifts in the mode corresponded to the particle size effect, lattice constant, and crystallite size of the catalyst.27 Lattice deformation and oxygen vacancies of CeO2 nanostructures were measured using the AD/AF2g ratio, following the order of CeO2-Nps-400 > CeO2-Nrs-400 > CeO2-Ncs-400, which interestingly agrees with the reducibility order of TPR analysis for CeO2 nanostructures.
 | ||
| Fig. 3 Raman spectra corresponding to different CeO2 nanostructures. | ||
As depicted in Fig. 4, hydrogen temperature programmed reduction (H2-TPR) was further used to study the reducibility of these ceria nanostructures. However, temperature programmed oxidation (O2-TPO) was chosen for further characterization to determine the oxidation behaviors of ceria nanostructures, as shown in Fig. 5. TP profiles of ceria particles reveal a reduction peak at about 350–500 °C assigned to consumption of surface oxygen (Os),35 where the total consumption in this region is well correlated with the polycrystalline of CeO2 surface area.36 On the contrary, the reduction peaks above 800 °C are attributed to the reduction of oxygen species in bulk CeO2 (Ob),37 resulting from diffusion of O out of the CeO2 bulk to the CeO2 surface, where it can react with adsorbed or gas phase H species.38 Consequently, measurements of OSC were determined from lower temperature reduction regions, as it mainly reflects on surface oxygen.
 | ||
| Fig. 4 H2-TPR profiles of CeO2 nanostructures. | ||
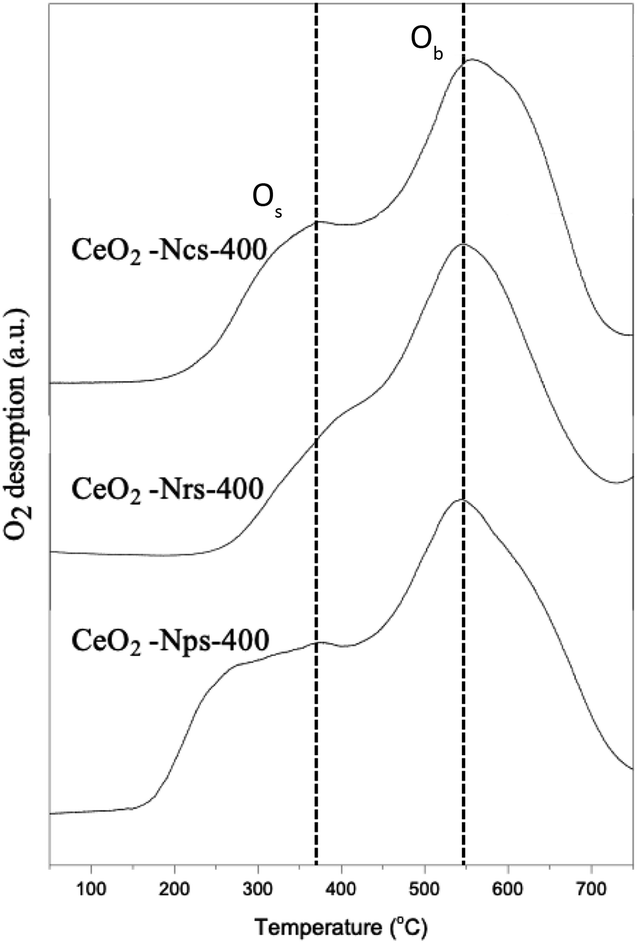 | ||
| Fig. 5 O2-TPO profiles of CeO2 nanostructures. | ||
As observed in Fig. 4, the Os peak is smaller for ceria nanocubes compared to nanorods and nanoparticles. This is attributed to the smaller amount of easily reduced oxygen species available in the cubic samples, which can also be closely related to the lowest lattice deformation and oxygen vacancies (AD/AF2g) and exposed surface facets. A few studies have also found that exploring the role of exposed surface facets using ceria nanostructures of different morphologies revealed differences in reduction behavior.39,40 These results agree with a study by Lykaki et al.,27 which reported that ceria nanocubes exhibit a smaller Os peak compared with nanorods and nanopolyhedra, indicating that the lower reducibility of the cube-shaped sample possesses the smallest population of weakly bound oxygen species.39,40 The reduction of ceria through a temperature-programmed reduction in hydrogen (H2-TPR) also indicates a superior behavior of CeO2-Nps-400 compared to that of CeO2-Nrs-400 and CeO2-Ncs-400, as shown in Table 2. This is evidenced by the anticipation of the onset of surface Ce4+ reduction and by the increase in reduction degree at low temperatures, which can be associated with easier oxygen removal from exposed [100] and [111] surfaces and the higher density of surface defects present in CeO2-Nps-400.41 The results in Table 2 demonstrate that H2 at lower temperatures removed surface oxygen as the lattice strain increased, suggesting that catalytic activity was associated with the reversible oxygen storage capacity of the catalysts.38 Consumption of H2 resulting from surface reduction occurred at lower temperatures over nanoparticles and nanorods than over the nanocubes, corroborating the ease of reducibility of CeO2 surface facets exhibited by each nanostructure.
| Catalyst | H2 consumption (μmol H2 per g) | Os/Ob | Peak temperature (°C) | |||||
|---|---|---|---|---|---|---|---|---|
| Os | Ob | Total | Os | Ob | ||||
| Peak 1 | Peak 2 | Peak 3 | ||||||
| CeO2-Nps-400 | 7219.7 | 6262.0 | 6400.8 | 11112.3 |
30994.8 |
1.79 | 418–484 | 847 |
| CeO2-Nrs-400 | 5348.8 | 4603.0 | 4233.1 | 9293.8 | 23478.7 |
1.53 | 401–492 | 842 |
| CeO2-Ncs-400 | 6535.0 | 6101.2 | — | 20791.2 |
835.3 | 0.61 | 386–509 | 863 |
| Catalyst | O2 consumption (μmol O2 per g) | Os/Ob | Peak temperature (°C) | |||
|---|---|---|---|---|---|---|
| Os | Ob | Total | Os | Ob | ||
| CeO2-Nps-400 | 4035.0 | 4453.0 | 8488.00 | 0.91 | 378 | 546 |
| CeO2-Nrs-400 | 4248.5 | 4882.5 | 9131.0 | 0.87 | 401 | 553 |
| CeO2-Ncs-400 | 2362.4 | 3676.8 | 6039.2 | 0.64 | 372 | 556 |
Differences in the surface area of ceria can also influence the overall H2-TPR profiles by changing the order of reactivity between the CeO2 nanostructures. During H2-TPR experiments, the reduction of Ce4+ to Ce3+ indicated oxygen removal and oxygen vacancy formation. In contrast, O2-TPO experiments were thus conducted to present the oxygen uptake and storage expedition charge transfer of ligand (O 2p) to metal (Ce 4f) by the primary photoionization process. The lower binding energy of v° and u° spin–orbit doublet located at 889.7 and 908.6 eV associated capacity.42 The TPO profile exhibits two main O2 consumption areas with broad peaks that contributed to the oxidation of CeO2 species in sequence from low to high temperatures at around 350 and 540 °C for all CeO2 nanostructured catalysts. As shown in Fig. 5, it is proposed that the first stage (200–450 °C) might be attributed to oxygen uptake as a surface lattice to oxygen (Os) and the second (450–700 °C) could be attributed to oxygen uptake as bulk oxygen (Ob).43 From the result, it can be observed clearly that the intensity of O2 consumption was increased in the order of CeO2-Ncs-400 < CeO2-Nrs-400 < CeO2-Nps-400, which revealed a good agreement with the measured reduction of CeO2 nanostructures in TPR analysis. Due to the highly oxidized particle-shaped surface of CeO2, CeO2-Nps-400 exhibits the highest population of weakly bound oxygen.44,45
Subsequently, the samples were further characterized using XPS analysis for a comparative assessment of each CeO2 nanostructure, which gives insight into the impact of morphology on elemental chemical states and surface composition, as depicted in Fig. 6. The XPS analysis on CeO2 nanostructure was performed to obtain more profound insight into the valence state of the chemical compound and chemical composition contained in the sample. Fig. 6 shows the Ce 3d core level spectra of different CeO2 nanostructure catalysts. The XPS survey spectra indicated that the samples included no components besides C, Ce, and O, confirming the exceptional chemical purity of the CeO2 nanoparticles (Fig. 10), which is consistent with the conclusion made by Bortamuly et al.43 More specifically, the spectra of Ce 3d revealed that Ce exists in the +4 and +3 oxidation states for all samples. This could be determined as Ce 3d5/2,3/2 spin–orbit doublet peaks, splitting at around 27 eV. Based on Burroughs et al.44 studies, the Ce 3d spectrum can be represented as u and v doublet peaks, indicating the 3d3/2 and 3d5/2 spin–orbit states, respectively. The v′′′ and u′′′ doublets at 906.2 and 925.2 eV with 19 eV separation correspond to the primary photoemission O 2p orbital to an empty core level of Ce 4f orbital with Ce 3d94f1 O 2p5 and Ce 3d94f2 O 2p4 final state shake-down satellite features, respectively.23,39,45 According to Anandan et al.,46 the satellite features are due to the photoionization from Ce3+ with Ce 3d94f2 O 2p5 final state. However, the v′ and u′ spin–orbits at 898.2 and 915.2 eV indicate the photoemission of Ce3+ from Ce 3d94f1 O 2p6 final state.47,48 Stetsovych et al.49 reported that the Ce3+ phase is buried under the CeO2 layer and the fully oxidized CeO2 layer is grown on top of the interfacial phase.
 | ||
| Fig. 6 Ce 3d core level XPS spectra of the CeO2 nanostructure catalyst. | ||
The XPS scan of the O 1s core level for different CeO2 nanostructure catalysts is depicted in Fig. 7. The spectra of the samples were deconvoluted into three peaks at binding energies of around 536.2, 538.1, and 540.3 eV. According to ref. 50, the lowest binding energy peak at 536.2 eV is attributed to the lattice oxygen in CeO2; the binding energy peak appearing at 538.1 eV is ascribed to the surface oxygen, while the observed binding energy at 540.3 eV is due to the hydroxyl on the surface of CeO2. According to Zhang et al.,51 surface oxygen content influences the catalytic performance of CeO2 materials. Moreover, the surface oxygen vacancy adsorbs and activates oxygen molecules to produce adsorbed oxygen species. This mechanism promotes the redox properties of CeO2.52 The obtained deconvoluted peaks differ in binding energy values around 4–5 eV compared to the literature findings. This result is consistent with the findings reported by Sudarsanam et al.53 and Hillary et al.,54 which state that the formation peak at a lower binding energy corresponds to the lattice oxygen of the metal oxide phases (O2−). However, the peak occurring at a higher binding energy is ascribed to surface chemisorbed oxygen, including adsorbed oxygen (O−/O22−), adsorbed water, hydroxyl (OH−), and carbonate (CO32−) species.
 | ||
| Fig. 7 O 1s core level XPS spectra of the CeO2 nanostructure catalyst. | ||
The calculated values of relative Ce3+ content and surface chemisorbed oxygen ratio of the CeO2 nanostructure are shown in Table 3. Because oxygen vacancy is generated when Ce4+ is reduced to Ce3+, it is crucial to calculate the relative Ce3+ content and chemisorption oxygen ratio to evaluate the generation of oxygen vacancy on the catalyst surface. Both the relative contents of Ce3+ and the surface chemisorbed oxygen ratio of CeO2 display an increasing pattern in the order of CeO2-Nps-400 > CeO2-Nrs-400 > CeO2-Ncs-400. Overall, the nanostructured ceria's direct oxidation intrinsic catalytic activity increases as the amount of Ce3+ increases, and the presence of Ce3+ ions demonstrates the formation on the surface of the non-stoichiometric CeO2.55 The Ce3+ ions accompanying the oxygen vacancies play a crucial role in the oxidation activity, which is also associated with the surface chemisorption of oxygen and activation of oxygen surface vacancies on the material and the migration of oxygen toward the surface material, as confirmed by observing the catalytic performance of the CeO2-Nps-400 catalyst that recorded high production of vanillin compared to CeO2-Nrs-400 and CeO2-Ncs-400 catalysts.56 The Ce3+ and Os species on the CeO2 surface play major roles in controlling catalytic oxidation for vanillin production, which are directly related to oxygen vacancies and the oxygen storage capacity of the catalyst. Based on the summarized data in Table 3, the results regarding the chemisorption oxygen of CeO2-Nps-400 agree with those of the relative Ce3+ content.
| Sample | Relative to Ce3+ content | Surface chemisorption oxygen |
|---|---|---|
| CeO2-Nps-400 | 0.197 | 0.139 |
| CeO2-Nrs-400 | 0.126 | 0.111 |
| CeO2-Ncs-400 | 0.119 | 0.098 |
Catalytic oxidation of raw kenaf
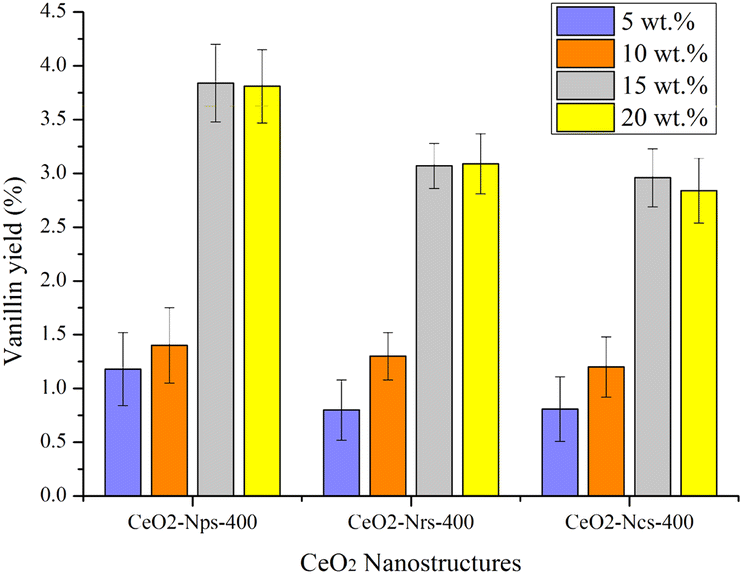 | ||
| Fig. 8 Reaction conditions: 2 g of dried kenaf stalks, 20 mL of 0.01 N NaOH solution, and 1 mL of H2O2 under 300 W microwave heating at 170 °C for 20 min. | ||
The outcome in Fig. 8 demonstrates the increased number of yields with the increased amount of catalyst loading for 15 wt% with a vanillin yield of 3.84% (CeO2-Nps-400), 3.07% (CeO2-Nrs-400) and 2.96% (CeO2-Ncs-400). This result agrees with the study conducted by Prado et al.,58 which states that vanillin yield increases from 0.015 to 0.22% when catalyst loading increases from 1 to 20%. This is attributed to the higher total contact frequency of catalysts with lignin, which is also consistent with the study conducted by Qu et al.6 They stated that the higher the catalyst loading, the higher the conversion of kenaf stalks. According to Chen et al.,18 it was found that the catalysts had a greater overall contact frequency with lignin. It is also believed that higher catalyst loading made more active sites accessible to break down lignin into aromatic aldehyde compounds. Based on the surface area of the CeO2 nanostructures catalyst, CeO2-Nps-400 possesses a high surface area among all CeO2 nanostructures. Due to its high surface area, CeO2-Nps-400 provided many active surfaces for the oxidation reaction of lignin to occur compared to other CeO2 nanostructured catalysts. Furthermore, the surface oxygen provided by CeO2-Nps-400 promoted oxidation as the catalyst loading increased. This showed a considerable effect on the vanillin yield, greatly improving the selectivity of the conversion of lignin to vanillin. According to Zhang et al.,59 materials in the nanoscale possess a surfeit of oxygen defects and improved catalytic performance as the crystallite decreases. In fact, the fine-tuning of the ceria with textural and defect structural characteristics exhibited by nanoparticles ceria as a catalyst proves its strong dependence between support morphology and catalytic activity, as reported in previous studies.60,61 However, as the catalyst loading increased to 20 wt%, the vanillin produced a slight decrease. These findings agreed with those of Behling et al., who found that increased loading of Co3O4 from 1 to 2 wt% led to increased yield of vanillin from around 13% to 19%. However, the vanillin yield shows a slightly decreased pattern when the catalyst loading further increases to 20 wt%, which might be due to the overoxidation of vanillin that is completely formed with available oxidizing species in the media. Excessive loading of the catalyst might also result in increased side reactions due to the overcrowding of the active sites.62
Conversion through a chemical transformation in heterogeneous catalysts is usually generated on specific sites on the catalyst surface, and it is generally referred to as active sites and intrinsic activity on a per site basis. The intrinsic activity of catalysts can normalize with different parameters that normally provide information on certain features of a catalyst, such as overall mass (catalyst loading), the mass of a specific component or surface area. In this study, various masses of catalyst loadings were used to show the normalized rate of CeO2 nanostructure on the production of vanillin. Direct oxidation tests from kenaf stalks for each CeO2 nanostructure catalyst were performed from 5 wt% to 20 wt% of catalyst loading with a constant 20 min reaction duration, 170 °C temperature, and 300 W microwave power output. Different catalyst loadings were used as parameters to observe the normalized pattern for each catalyst to allow for a direct comparison of the reaction rate and its effect on vanillin production on CeO2 nanostructures. The slope trends of CeO2-Nps-400 depicted in Fig. 9 are 0.256, 0.094, 0.246 and 0.178 for 5, 10, 15 and 20 wt%, respectively, where it shows a decreasing pattern from 5 to 10 wt% and an increasing pattern at loading 15 wt%, followed by a decreasing normalized value for loading greater than 15 wt%. This indicates that the production of vanillin is independent of 10 and 20 wt% of catalyst loading, which can be explained by the mass transfer limited of the catalyst, where it showed a decreasing order to the factor of three and two, respectively.62 In contrast, normalized for 15 wt% shows an increasing normalized rate approximately to the factor of three. Meanwhile, CeO2-Nrs-400 and CeO2-Ncs-400 displayed the same pattern as CeO2-Nps-400.63 It could be concluded that catalyst loadings of 5 and 15 wt% show a significant normalized value to the production of vanillin through the direct oxidation test and the least significant for catalyst loadings at 10 and 20 wt%, respectively, as illustrated by the transition slopes in Fig. 10.
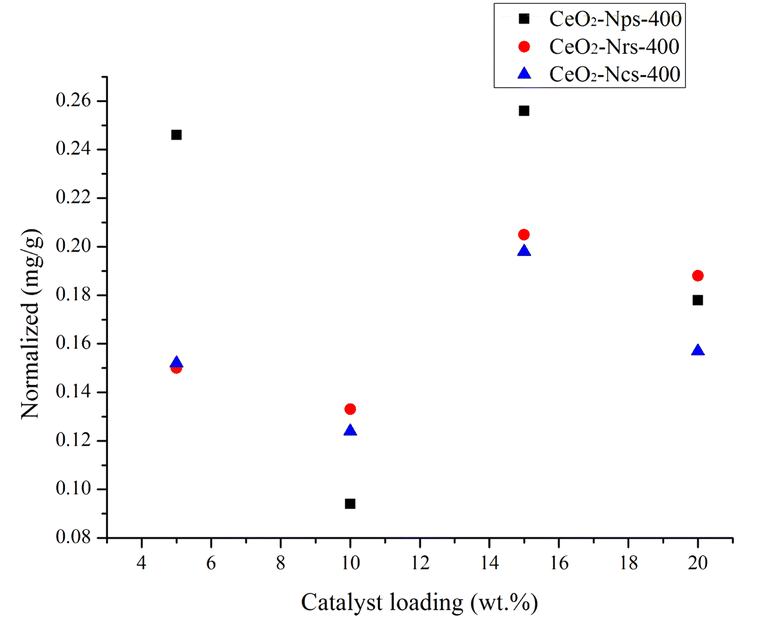 | ||
| Fig. 9 Comparison of the normalized rate of the production of vanillin to the loading of each type of catalyst from 5–20 wt%. | ||
 | ||
| Fig. 10 Reaction conditions: 2 g of dried kenaf stalks, 20 mL of 0.01 N NaOH solution, 15 wt% of catalyst, and 1 mL of H2O2 under 300 W of microwave heating at 170 °C for 20 min. | ||
The yield of vanillin obtained for the CeO2-Nps-400 heterogeneous catalyst was 3.84%, while 4.66% vanillin was produced using 2 N (normality) NaOH as a homogeneous catalyst. This is noteworthy because vanillin production was directly from biomass without extracting the lignin from the hemicellulose and cellulose components. This result agrees with a study conducted by Khairul et al.11 in which a 4.45% vanillin yield was obtained utilizing 2 N of NaOH as a homogeneous catalyst. In comparison, 2.90% vanillin was produced using 30Ce/MgO-48 as a heterogeneous catalyst at 170 °C for 20 min. They also reported that although heterogeneous catalysts possess high selectivity and conversion percentages for vanillin production, there are drawbacks throughout the reaction process due to surface saturation/deactivation in the formation of alcohols and aromatic compounds. On the contrary, the homogeneous catalyst is considered a flexible catalyst that allows it to freely contact the CAr–O bonds without high steric limitations because the reaction occurs solely in a liquid phase that also reacts as a reaction medium simultaneously, as reported in previous studies.11,64
 | ||
| Fig. 11 Catalyst reusability of CeO2-Nps-400. | ||
The catalyst's physicochemical properties for pre-reaction and post-reaction were compared to identify the structural integrity of the catalyst as the active oxygen sites on the catalyst's surface are exposed and play the primary role during the direct oxidation reaction. The CeO2-Nps-400 was used for up to three cycles under 170 °C microwave heating for 20 min at 300 W power output. The catalyst was treated for reusability studies, and the third cycle catalyst was used for further characterization using XRD and Raman analyses. Fig. 12 displays the (a) XRD and Raman (b) analyses of CeO2-Nps-400 for pre-reaction and post-reaction after the three cycles used. As observed in the XRD figure of CeO2-Nps-400, there are no significant changes in the formation of peaks with identical peaks of the ceria before and after the use of the catalyst. However, there are slight differences in diffraction intensity as the post-reaction catalyst showed a slight decrease in diffraction intensity peak compared to a pre-reaction catalyst for diffraction peak at higher 2θ = 30°. This was proved by comparing the crystallite size, where the post-reaction catalyst was 10.1 nm with a 1.2 nm difference from the pre-reaction catalyst (11.3 nm). There is a slight peak shift to the right with a shifted value of 2θ around 0.1 to 0.01° for the post-reaction catalyst compared to the original diffraction peaks before the reaction. The different intensities of the diffraction peaks indicate that there are structural changes between the crystal lattices. This might be caused by the agglomeration of the particles at a higher product composition owing to uneven particle distribution, which is evident by the differences in the peak intensity and crystallite size of the catalysts.68
 | ||
| Fig. 12 Characterization of CeO2-Nps-400 using XRD and Raman analyses for the post-reaction. | ||
The same goes for Raman analysis, where the absorption peak intensity and peak shift for post-reaction can be detected at the F2g and D modes with slightly different pre-reaction catalysts. The defect oxygen (D) Raman for post-reaction demonstrated an intensity decrease compared to pre-reaction CeO2-Nps-400 at 601 cm−1. In contrast, there are no significant differences in Raman intensity for the observed peak inherent to cerium oxide at 463 cm−1. The Ce oxidation state reduces as oxygen is released and oxygen vacancies are formed. As the concentration of oxygen vacancies increased, the non-stoichiometry F2g in the lattice becomes asymmetric and the shoulder at band ∼600 cm−1, which is associated with defect species, broadens and shades at high temperatures as the lattice becomes more mobile for the post-reaction catalyst due to the regeneration of lattice Ce–O species to replenish oxygen vacancies simultaneously in the reduced state of metal oxide.69,70
The CeO2 nanostructures also showed a clear contradiction pattern between XRD and BET analyses, where it showed an increasing crystallite size in the order of CeO2-Nps-400 > CeO2-Nrs-400 > CeO2-Ncs-400.72 Simultaneously, the surface area of the catalyst follows the reverse order. It was found that by decreasing crystallite size, the CeO2 nanostructures were exposed to the high OSC.73 Thus, the large surface area to volume ratio existing in a catalyst permits CeO2 to react differently, resulting in unique properties. A larger particle size results in lattice expansion in the crystal structure. The expansion of the lattice decreases its oxygen release and reabsorption capabilities, as reported by Hailstone et al.29 This is proved by observing the catalytic performance of the CeO2-Ncs-400 catalyst, which produced the least amount of vanillin yield compared to the other catalysts. In particular, high surface area-engaged nanostructures are considered perfect for the oxidation reaction by providing a greater tendency for active species to come into contact with reactants and improve catalytic performance.74 The relationship between the specific area and OSC was investigated by several authors,73,75,76 considering that materials with high specific areas display more active sites to adsorb oxygen from external sources, such as oxidants that supply more active oxygen species.77 The summarized results in Table 1 show that the CeO2-Nps-400, CeO2-Nrs-400 and CeO2-Ncs-400 whose areas correspond to 66.1, 57.8 and 22.5 m2 g−1, respectively, while the OSC values are 1664.6, 1340.4 and 835.3 μmol O2 per g, respectively. As the BET surface area of the materials increased, the OSC value also increased. The comparable OSC values in different nanostructures of CeO2 indicate that specific surface area and OSC have a high relation and depend on each other as the vanillin yield also shows an increasing pattern with increasing BET surface area and lower temperature OSC.
Different structural characteristics exposed to different active facets established a direct correlation between lower-temperature OSC and the presence of defects.78 Generally, there are three low-index lattice planes on the surface of CeO2 nanostructures that are facet (100), (110) and (111). The stability and excess charge localization of all three facets follow the sequence (111) > (110) > (100), whereas the activity follows the reverse order.78–81 According to initial research conducted by Ramli et al.,22 the reported TEM and HRTEM results show that CeO2 nanoparticles promoted the exposure of Ce(111) and Ce(100) facets, while CeO2 nanorods preferentially enclosed (110) facets,82–84 followed by ceria nanocubes promoting the exposure of Ce(100) facets. The (110) and (100) facets are considered more active than the (111) facets in terms of catalytic activity. However, in this study, it was found that nanoparticles promote (100) and (111), which contradicts the activity order of the facets for ceria nanostructures. Moreover, a nanocube was found to promote (100), but its reactivity on the conversion of vanillin shows a poorer yield among other ceria nanostructures. Konsolakis and Lykaki et al.27 reported that in terms of the relation to oxygen storage capacity (OSC) obtained for bare ceria nanostructures in their study, the order shows (111) > (100) > (110), which also can correlate with the energy formation of anionic vacancies. In this study, the facets (100) and (111) enclosed by ceria nanoparticles are found to express significantly enhanced redox properties required for higher catalytic activities in converting vanillin from direct oxidation of kenaf stalks,22 which also reconfirms the involvement of the active crystal facets in chemical reactions. They also said that both the (100) and (111) facets facilitate the regeneration of active sites due to their improved oxygen exchange kinetics, thus favoring the regeneration of active sites27 for the oxidation reaction.
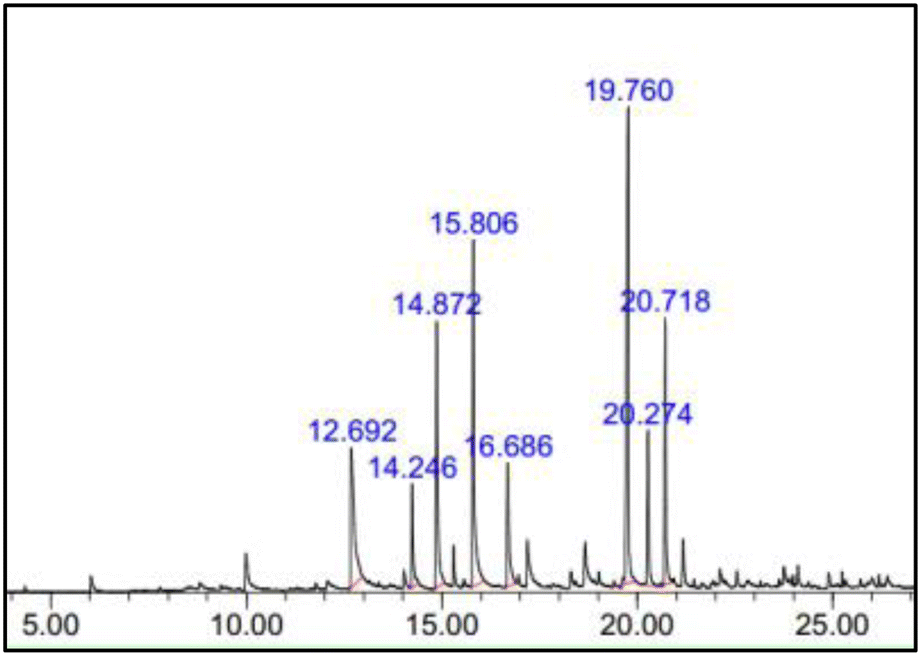 | ||
| Fig. 13 Gas chromatogram of degradation wood products. | ||
Despite the inclusion of additional aromatic and non-aromatic monomer chemicals in the products, this is remarkable because vanillin is extracted without even isolating the lignin components from cellulose and hemicellulose in kenaf stalks. According to Qu et al.,6 direct vanillin production from Japanese cedar wood yielded 5% vanillin when CuO is present as a catalyst, and degradation products detected the presence of other aromatic monomer compounds, such as guaiacol, acetovanillone, vanillic acid, syringaldehyde, and syringic acid. This showed that syringaldehyde was identified with a higher peak area than vanillin. This is because hardwood lignin contains both guaiacylpropane and syringylpropane units. Thus, the alkaline oxidation of angiosperm lignin generates large amounts of syringaldehyde. Syringaldehyde has the most similar applications as vanillin because both possess almost the same chemical structure and properties, which is also one reason for the difficulty of separating syringaldehyde from pure vanillin. Vanillin purification is also challenging due to contaminants, such as carbohydrates, various side products, and unreacted lignin.84
Conclusions
Vanillin was successfully produced from the direct oxidation of kenaf stalks under microwave heating using H2O2 as an oxidizing agent in the presence of CeO2-Nps-400, CeO2-Nrs-400 and CeO2-Nps-400 catalysts at pH 11.5. A vanillin yield of 3.84% was achieved using CeO2-Nps-400 as a heterogeneous catalyst, while 4.66% was obtained when NaOH was used as a homogeneous catalyst. The heterogeneous CeO2-Nps-400 catalyst is chemically stable and can be repeated use for 3 times. Based on the catalytic evaluation, vanillin production showed increasing yield in order of CeO2-Ncs-400 < CeO2-Nrs-400 < CeO2-Nps-400, which also follows the same trend as characteristic morphology in textural and redox properties of CeO2 nanostructures. The synthesized CeO2 nanoparticles proved that the highest vanillin yield was affected by its morphology, where it was recorded with high- and low-temperature OSC value, easily weakly bound of oxygen species through TPR and TPO and exceptional high oxygen vacancies from Raman and XPS analyses.Data availability
Data are present within the article and its ESI.†Author contributions
Conceptualization, A. R.; methodology, N. A. S. I. K. A.; software, N. A. S. I. K. A.; validation, A. R., N. M. Y. and A. R. M.; formal analysis, N. A. S. I. K. A.; investigation, N. A. S. I. K. A.; resources, A. R., N. M. Y. and A. R. M.; data curation, N. A. S. I. K. A.; writing—original draft preparation, N. A. S. I. K. A.; writing—review and editing, A. R.; visualization, A. R., N. M. Y. and A. R. M.; supervision, A. R., N. M. Y. and A. R. M.; project administration, A. R.; funding acquisition, A. R., N. M. Y. and A. R. M. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors would like to thank Malaysian Ministry of Higher Education under the Fundamental Research Grant Scheme (FRGS/1/2020/STG04/UTP/02/2, cost centre 015MA0-125, funder ID 10.13039/501100003093) for the research grants awarded to conduct the research.Notes and references
- M. Fache, B. Boutevin and S. Caillol, ACS Sustain. Chem. Eng., 2015, 4, 35–46 CrossRef.
- N. J. Walton, M. J. Mayer and A. Narbad, Phytochemistry, 2003, 63, 505–515 CrossRef CAS PubMed.
- E. B. da Silva, M. Zabkova, J. D. Araújo, C. A. Cateto, M. F. Barreiro, M. N. Belgacem and A. E. Rodrigues, Chem. Eng. Res. Des., 2009, 87, 1276–1292 CrossRef.
- F. S. Chakar and A. J. Ragauskas, Ind. Crops Prod., 2004, 20, 131–141 CrossRef CAS.
- H. Deng, L. Lin and S. Liu, Energy Fuels, 2010, 24, 4797–4802 CrossRef CAS.
- C. Qu, M. Kaneko, K. Kashimura, K. Tanaka, S. Ozawa and T. Watanabe, ACS Sustain. Chem. Eng., 2017, 5, 11551–11557 CrossRef CAS.
- D. Mukherjee, R. Singuru, P. Venkataswamy, D. Damma and B. M. Reddy, ACS Omega, 2019, 4, 4770–4778 CrossRef CAS PubMed.
- F. Liu, L. Chen, J. K. Neathery, K. Saito and K. Liu, Ind. Eng. Chem. Res., 2014, 53, 16341–16348 CrossRef CAS.
- W. Deng, H. Zhang, X. Wu, R. Li, Q. Zhang and Y. Wang, Green Chem., 2015, 17, 5009–5018 RSC.
- S. Rawat, P. Gupta, B. Singh, T. Bhaskar, K. Natte and A. Narani, Appl. Catal., A, 2020, 598, 117567 CrossRef CAS.
- N. A. S. I. Khairul Anuar, A. Ramli and L. Jun Wei, Catalysts, 2021, 11, 1449 CrossRef CAS.
- W. I. Hsiao, Y. S. Lin, Y. C. Chen and C. S. Lee, Chem. Phys. Lett., 2007, 441, 294–299 CrossRef CAS.
- M. Melchionna and P. Fornasiero, Mater. Today, 2014, 17, 349–357 CrossRef CAS.
- Z. A. Qiao, Z. Wu and S. Dai, ChemSusChem, 2013, 6, 1821–1833 CrossRef CAS PubMed.
- Z. Wu, M. Li and S. H. Overbury, J. Catal., 2012, 285, 61–73 CrossRef CAS.
- W. Huang, Top. Catal., 2013, 56, 1363–1376 CrossRef CAS.
- M. Konsolakis and M. Lykaki, Catalysts, 2021, 11, 452 CrossRef CAS.
- D. Chen, D. He, J. Lu, L. Zhong, F. Liu, J. Liu, J. Yu, G. Wan, S. He and Y. Luo, Appl. Catal., B, 2017, 218, 249–259 CrossRef CAS.
- Y. Ohtani, B. B. Mazumder and K. Sameshima, J. Wood Sci., 2001, 47, 30–35 CrossRef CAS.
- G. Al-Naqeb, M. Ismail, G. Bagalkotkar and H. A. Adamu, Food Res. Int., 2010, 43, 2437–2443 CrossRef CAS.
- N. Saba, M. T. Paridah and M. Jawaid, Constr. Build. Mater., 2015, 76, 87–96 CrossRef.
- A. Ramli, N. A. S. I. K. Anuar, N. A. A. Bakhtiar, N. M. Yunus and A. R. Mohamed, Molecules, 2023, 28, 4963 CrossRef CAS PubMed.
- L. Torrente-Murciano and R. S. Chapman, Phys. Chem. Chem. Phys., 2016, 18, 15496–15500 RSC.
- M. Kovacevic, B. L. Mojet, J. G. van Ommen and L. Lefferts, Catal. Lett., 2016, 146, 770–777 CrossRef CAS.
- J. Hierso, Ö. Sel, A. Ringuedé, C. Laberty-Robert, L. Bianchi, D. Grosso and C. Sánchez, Chem. Mater., 2009, 21, 2184–2192 CrossRef CAS.
- L. Ma, D. Wang, J. Li, B. Bai, L. Fu and Y. Li, Appl. Catal., B, 2014, 148–149, 36–43 CAS.
- M. Lykaki, S. Stefa, S. A. Carabineiro, P. K. Pandis, V. N. Stathopoulos and M. Konsolakis, Catalysts, 2019, 9, 371 CrossRef CAS.
- F. Gao, Q. Lu and S. Komarneni, J. Nanosci. Nanotechnol., 2006, 6, 3812–3819 CrossRef CAS PubMed.
- R. K. Hailstone, A. G. DiFrancesco, J. G. Leong, T. D. Allston and K. J. Reed, J. Phys. Chem. C, 2009, 113, 15155–15159 CrossRef CAS.
- M. Thommes, K. Kaneko, V. Neimark, P. Alexander, J. R. F. Olivier, J. Rouquerol and S. W. K. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- J. Arnaldo, Microporous Mesoporous Mater., 2020, 291, 109698 CrossRef.
- N. Kainbayev, M. Sriubas, D. Virbukas, Z. Rutkuniene, K. Bockute, S. Bolegenova and G. Laukaitis, Coatings, 2020, 10, 432 CrossRef CAS.
- M. Yang, G. Shen, Q. Wang, K. Deng, M. Liu, Y. Chen and Z. Wang, Molecules, 2021, 26, 6363 CrossRef CAS PubMed.
- E. N. Ndifor, T. Garcia, B. Solsona and S. H. Taylor, Appl. Catal., B, 2007, 76, 248–256 CrossRef CAS.
- J. Shyu, J. Catal., 1989, 115, 16–23 CrossRef CAS.
- J. D. Kammert, J. Moon and Z. Wu, Chin. J. Catal., 2020, 41, 901–914 CrossRef CAS.
- G. Ranga Rao and B. G. Mishra, Bulletin of Catalysis Society of India, 2003, 2, 122–134 Search PubMed.
- Tana, M. Zhang, J. Li, H. Li, Y. Li and W. Shen, Catal. Today, 2009, 148, 179–183 CrossRef CAS.
- L. Liu, Y. Cao, W. Sun, Z. Yao, B. Liu and F. Gao, Catal. Today, 2011, 175, 48–54 CrossRef CAS.
- T. Désaunay, G. Bonura, V. Chiodo, S. Freni, J. P. Couzinié, J. Bourgon and M. Cassir, J. Catal., 2013, 297, 193–201 CrossRef.
- B. Murugan and A. V. Ramaswamy, J. Am. Chem. Soc., 2007, 129, 3062–3063 CrossRef CAS PubMed.
- S. Hamoudi, F. Larachi, G. Cerrella and M. Cassanello, Ind. Eng. Chem. Res., 1998, 37, 3561–3566 CrossRef CAS.
- R. Bortamuly, G. Konwar, P. K. Boruah, M. R. Das, D. Mahanta and P. Saikia, Ionics, 2020, 26, 5747–5756 CrossRef CAS.
- P. Burroughs, A. Hamnett, A. F. Orchard and G. Thornton, Dalton Trans., 1976, 17, 1686 RSC.
- M. Pan, S. Zhang, Y. Xu and R. Li, Appl. Surf. Sci., 2018, 448, 435–443 CrossRef.
- C. Anandan and P. Bera, Appl. Surf. Sci., 2013, 283, 297–303 CrossRef CAS.
- E. Bêche, P. Charvin, D. Perarnau, S. Abanades and G. Flamant, Surf. Interface Anal., 2008, 40, 264–267 CrossRef.
- B. M. Reddy, A. Khan, Y. Yamada, T. Kobayashi, S. Loridant and J. C. Volta, J. Phys. Chem., 2003, 107, 5162–5167 CrossRef CAS.
- V. Stetsovych, F. Pagliuca, F. Dvořák, T. Duchoň, M. Vorokhta, M. Aulická and V. Matolín, J. Phys. Chem. Lett., 2013, 4, 866–871 CrossRef CAS.
- S. Niroumandrad, M. Rostami and B. Ramezanzadeh, Appl. Surf. Sci., 2015, 357, 2121–2130 CrossRef CAS.
- B. Zhang, Y. Huyan, J. Wang, W. Wang, Q. Zhang and H. Zhang, J. Am. Ceram. Soc., 2018, 102, 2218–2227 CrossRef.
- Z. Su, W. Yang, C. Wang, S. Xiong, X. Cao, Y. Peng, W. Si, Y. Weng, M. Xue and J. Li, Environ. Sci. Technol., 2020, 54, 12684–12692 CrossRef CAS PubMed.
- P. Sudarsanam, B. Hillary, M. H. Amin, N. Rockstroh, U. Bentrup, A. Brückner and S. K. Bhargava, Langmuir, 2018, 34, 2663–2673 CrossRef CAS PubMed.
- B. Hillary, P. Sudarsanam, M. H. Amin and S. K. Bhargava, Langmuir, 2017, 33, 1743–1750 CrossRef CAS PubMed.
- J. M. López, A. Gilbank, T. García, B. Solsona, S. Agouram and L. Torrente-Murciano, Appl. Catal., B, 2015, 174, 403–412 CrossRef.
- A. I. Y. Tok, S. Du, F. Boey and W. K. Chong, Mater. Sci. Eng., A, 2007, 466, 223–229 CrossRef.
- X. Xu, P. Li, Y. Zhong, J. Yu, C. Miao and G. Tong, J. Biol. Macromol., 2023, 243, 125203 CrossRef CAS PubMed.
- R. Prado, X. Erdocia, G. F. de Gregorio, J. Labidi and T. Welton, ACS Sustain. Chem. Eng., 2016, 4, 5277–5288 CrossRef CAS.
- D. Zhang, X. Du, L. Shi and R. Gao, Dalton Trans., 2012, 41, 14455–14475 RSC.
- M. Zabilskiy, P. Djinović, E. Tchernychova, O. P. Tkachenko, L. M. Kustov and A. Pintar, ACS Catal., 2015, 5, 5357–5365 CrossRef CAS.
- M. Lykaki, E. Pachatouridou, S. A. C. Carabineiro, E. Iliopoulou, C. Andriopoulou, N. Kallithrakas-Kontos, S. Boghosian and M. Konsolakis, Appl. Catal., B, 2018, 230, 18–28 CrossRef CAS.
- R. Behling, G. Chatel and S. Valange, Ultrason, 2017, 36, 27–35 CAS.
- J. C. Walter, A. Zurawski, D. M. Montgomery, M. Thornburg and S. T. Revankar, J. Power Sources, 2008, 179, 335–339 CrossRef CAS.
- A. M. da Costa Lopes, J. R. Gomes, J. A. Coutinho and A. J. Silvestre, Green Chem., 2020, 22, 2474–2487 RSC.
- B. S. Takale, M. Bao and Y. Yamamoto, Org. Biomol. Chem., 2014, 12, 2005 RSC.
- S. Santra, A. K. Bagdi, A. Majee and A. Hajra, RSC Adv., 2013, 47, 24931 RSC.
- M. J. Sampaio, A. Benyounes, P. Serp, J. L. Faria and C. G. Silva, Appl. Catal., A, 2018, 551, 71–78 CrossRef CAS.
- N. J. Abd Rahman, PhD thesis, Universiti Teknologi PETRONAS, Perak, Malaysia, 2020.
- A. Younis, D. Chu and S. Li, Funct. Nanomater., 2016, 3, 53–68 Search PubMed.
- G. Sharma, D. D. Dionysiou, S. Sharma, A. Kumar, A. H. Al-Muhtaseb, M. Naushad and F. J. Stadler, Catal. Today, 2019, 335, 437–451 CrossRef CAS.
- D. Wei, M. Yue, S. Qin, S. Zhang, Y. Wu, G. Xu, H. Zhang, T. Zhang and J. Li, J. Am. Chem. Soc., 2021, 143, 15635–15643 CrossRef CAS PubMed.
- Y. Xiao, H. Li and K. Xie, Angew. Chem., Int. Ed., 2021, 60, 5240–5244 CrossRef CAS PubMed.
- J. Wang, M. Shen, J. Wang, J. Gao, J. Ma and S. Liu, Catal. Today, 2011, 175, 65–71 CrossRef CAS.
- V. P. Santos, M. Pereira and J. Órfão, Appl. Catal., B, 2010, 99, 353–363 CrossRef CAS.
- A. Migani, G. N. Vayssilov, S. T. Bromley, F. Illas and K. M. Neyman, J. Mater. Chem., 2010, 20, 10535–10546 RSC.
- Z. Minwei, M. Shen and J. Wang, J. Catal., 2007, 248, 258–267 CrossRef.
- N. Kamiuchi, M. Haneda and M. Ozawa, Catal. Today, 2014, 232, 179–184 CrossRef CAS.
- M. Cui, Y. Li, X. Wang, J. Wang and M. Shen, J. Rare Earths, 2013, 31, 572–576 CrossRef CAS.
- E. Mamontov, T. Egami, R. Brezny, M. Koranne and S. Tyagi, J. Phys. Chem. B, 2000, 104, 11110–11116 CrossRef CAS.
- D. C. Sayle, S. A. Maicaneanu and G. W. Watson, J. Am. Chem. Soc., 2002, 124, 11429–11439 CrossRef CAS PubMed.
- Y. Jiang, J. B. Adams and M. van Schilfgaarde, J. Chem. Phys., 2005, 123, 64701 CrossRef PubMed.
- Y. Chen, P. Hu, M. H. Lee and H. Wang, Surf. Sci., 2008, 602, 1736–1741 CrossRef CAS.
- T. X. T. Sayle, M. Cantoni, U. M. Bhatta, S. C. Parker, S. R. Hall, G. Möbus, M. Molinari, D. Reid, S. Seal and D. C. Sayle, Chem. Mater., 2012, 24, 1811–1821 CrossRef CAS.
- R. Zhang, R. Maltari, M. Guo, J. Kontro, A. Eronen and T. Repo, Ind. Crops Prod., 2020, 145, 112095 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra05833j |
| This journal is © The Royal Society of Chemistry 2024 |