
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanism and toxicity assessment of carbofuran degradation by persulfate-based advanced oxidation process†
Chenxi Zhang ab,
Youxin Xuab,
Bingbing Chua and
Xiaomin Sun*b
ab,
Youxin Xuab,
Bingbing Chua and
Xiaomin Sun*b
aShandong Provincial University Laboratory for Protected Horticulture, Weifang University of Science and Technology, Weifang 262700, China
bEnvironment Research Institute, Shandong University, Qingdao 266200, P. R. China. E-mail: sxmwch@sdu.edu.cn
First published on 24th September 2024
Abstract
The advanced oxidation process based on persulfate has been proven to be a promising method for degrading the highly toxic carbamate pesticide carbofuran (CBF). However, the mechanism of CBF degradation by sulfate radicals (SO4·−) and hydroxyl radicals (·OH) is still unclear and requires further research and discussion. This study investigated the mechanism and toxicity assessment of CBF degradation using density functional theory (DFT) theory calculation methods. The results indicated that SO4·− and ·OH can undergo addition and abstraction reactions with CBF. Thermodynamic and kinetic analysis showed that the abstraction reaction between SO4·− and the secondary H atom is the optimal reaction pathway, exhibiting the highest branching ratio (Γ = 41.84%). The rate constants for the reactions of CBF with SO4·− and ·OH at room temperature were found to be 3.66 × 109 and 8.96 × 108 M−1 s−1, respectively, which are consistent with experimental data reported in previous studies. The acute and chronic toxicity of CBF and its degradation products to aquatic organisms was predicted through an ecological toxicity assessment model. The toxicity of the degradation products was lower than that of the parent CBF, confirming the viability of using persulfate-based advanced oxidation processes for water treatment.
1 Introduction
Carbofuran (CBF) is a broad-spectrum systemic N-methyl carbamate pesticide widely used as an insecticide, nematicide, and acaricide in agriculture, household, and industrial sectors.1,2 It belongs to the highly toxic pesticides among all n-methyl carbamate insecticides, posing significant acute toxicity to humans.3,4 CBF is particularly hazardous to birds among various environmental organisms.5,6 When raptors, small mammals, or reptiles feed on small birds that have been poisoned to death by carbofuran, it can cause secondary poisoning and lead to death. CBF is soluble in water and highly mobile in soil, with a long half-life of up to 50 ∼ 110 days.7,8 And it can contaminate groundwater and nearby water bodies through precipitation runoff.9 Research indicates that exposure to CBF concentrations of 0.0044 mg L−1 or higher for more than 7 days in aquatic environments can alter the histology of gills, liver, and kidneys in Nile tilapia, thereby compromising fish health.10 Moreover, studies suggest serious neurological damage in humans associated with CBF, potentially linking it to cancer.11 Therefore, due to its endocrine-disrupting effects and neurotoxic inhibition, CBF poses high risks to aquatic ecosystems and public health, emphasizing the critical need to control its presence in aquatic environments.Currently, methods for removing CBF from drinking water and wastewater mainly include adsorption, bioremediation, and chemical degradation.12–16 Sugarcane bagasse-peanut shell magnetic composites, bamboo shoot shell-based activated carbon, and coated polystyrene beads have been studied for their adsorption capabilities in water containing CBF.17–19 While these methods show promising results, effective treatment of the waste and pollutants generated during the adsorption process is currently lacking, posing environmental risks.20 Additionally, bioremediation is considered a time-consuming process.21,22 Therefore, chemical degradation is seen as a viable approach for treating CBF-contaminated water bodies, particularly through persulfate-based advanced oxidation processes (AOPs).23 Many laboratory and field studies have shown that persulfate-based AOPs has been widely applied to pesticide pollutants in the environment, such as dimethoate, ethyl-parathion, atrazine, and imidacloprid.24–27 This technology generates reactive oxygen species (ROS) such as sulfate radicals (SO4·−, E0 = 2.5–3.1 V), which have a higher redox potential compared to hydroxyl radicals (·OH, E0 = 2.8 V).28 Additionally, SO4·− has a longer half-life, faster kinetics, greater stability, and a larger transport distance, can operate over a broader pH range.29 Therefore, it represents a promising technology for the treatment of organic pollutants.
In general, the ROS produced during the activation process of persulfates vary depending on the activation method used. These species primarily include SO4·−, ·OH, singlet oxygen (1O2), and superoxide anions (O2·−).30 Accurately analyzing the ROS in AOPs is crucial for understanding the degradation mechanisms of pollutants. Identification methods include electron paramagnetic resonance (EPR), high performance liquid chromatography (HPLC), fluorescence probes, and quenching experiments.31 EPR is effective in detecting reactive species in water but cannot detect surface-bound radicals.32 HPLC is affected by various factors in complex systems, and its accuracy needs further improvement.31 Fluorescence probe methods can be interfered with by background light sources when detecting 1O2.33 Quenching experiments, as a simple and effective ROS detection method, not only identify the types of ROS but also calculate and analyze their contribution rates and concentrations, and have been widely used in ROS detection.34 Samy et al. synthesized a novel nanocomposite of magnetite for the first time, combining photocatalysis with persulfate activation to synergistically generate more radicals, achieving a degradation efficiency of 92.8% for CBF in a short time.23 Through quenching experiments, with the addition of the ethanol (SO4·− scavenger), isopropanol (·OH scavenger), sodium azide (1O2 scavenger) and benzoquinone (O2·− scavenger), the degradation efficiency decreased to 28.8%, 46%, 90.3% and 80.4%, respectively, confirming the crucial roles of ·OH and SO4·−. Jiang et al. developed cobalt-based catalysts to activate persulfate for CBF degradation, determining the contributions of ·OH and SO4·− through quenching experiments and EPR tests.35 Persulfate technology rapidly oxidizes and decomposes recalcitrant CBF, achieving effective treatment with better environmental and safety profiles compared to other chemical degradation methods. However, the precise mechanism of persulfate technology for CBF degradation remains unclear and requires further research and discussion.
Density functional theory (DFT) calculations can assist in studying the mechanism of radical reactions and in researching the kinetic processes of reactions.36,37 Atifi et al. studied the photodegradation of CBF in aqueous medium using DFT method and explained the dissociation behavior after irradiation based on thermodynamic analysis of the bond dissociation energy of CBF.38 According to the calculations, the photodegradation of CBF is predicted to occur through the C–O bond cleavage in the carbamate moiety. The results of the Laser Flash Photolysis (LFP) experiment showed the generation of phenoxyl groups, which is consistent with the theoretical calculations. Ćwieląg-Piasecka et al. applied DFT method to study the oxidation reaction of CBF with ·OH, where hydrogen atom transfer is the preferred mechanism.39 Currently, there is no theoretical study on CBF with SO4·−, and research on the interaction with ·OH is limited to initiation steps, with the degradation mechanism remaining unclear. Therefore, this study proposes to use DFT calculations to investigate the degradation mechanisms of CBF with SO4·− and ·OH. In addition, in many cases, products formed during degradation often exhibit higher toxicity than the parent compounds.40,41 Thus, ecological toxicity assessments also explore the potential environmental risks of CBF and its products. These findings will provide theoretical support for the treatment of CBF wastewater using persulfate oxidation.
2 Computational methods
All geometrical optimizations involved in the degradation process were performed using Gaussian 16 program.42 The geometric configurations of reactants (R), intermediates (IM), transition states (TS), and products (P) were optimized at the M06-2X/6-31+G(d,p) level of theory. Previous studies have shown that M06-2X is one of the best functionals for a combination of main-group thermochemistry and kinetics.43,44 For more accurate energy evaluations, single-point energy calculations were performed at the M06-2X/6-311++G(3df,3pd) level on all structures. To account for solvent effects in the entire system, the solvation model density (SMD) model based on self-consistent reaction field theory was employed.45KisThelP software, based on transition state theory (TST) and Wigner tunneling correction, was used for the kinetic calculations.46,47 The thermodynamic equivalent of eqn (1) is employed in KiSThelP.46
 | (1) |
Lastly, toxicity assessments of CBF and its degradation products were conducted using the Ecological Structure Activity Relationship (ECOSAR) predictive model.48 In the model, fish, water fleas, and green algae were selected as aquatic organisms to assess acute and chronic toxicity risks. Acute toxicity to fish and water fleas was evaluated using the median lethal concentration (LC50), indicating the concentration at which 50% of fish and water fleas die after 96 hours and 48 hours of exposure, respectively. The acute toxicity to green algae was determined using the median effective concentration (EC50), representing the concentration at which 50% of the algae show adverse effects after 96 hours of exposure.
3 Results and discussion
3.1 Structural analysis of CBF
The structure of CBF is depicted in Fig. 1(a), with each atom numbered for clarity. In the structure of CBF, there is a phenyl ring, a furan ring, an amino formate group, and three methyl groups. To elucidate the sensitivity of its potential reaction sites, we plotted the electrostatic potential (ESP) distribution of CBF in Fig. 1(b) and highlighted the Fukui function (f0) and dual descriptor (CDD) values of CBF in Table 1. | ||
| Fig. 1 (a) The structure of CBF. (b) Molecular surface ESP distribution diagram of CBF. | ||
| Atom | f0 | CDD | Atom | f0 | CDD |
|---|---|---|---|---|---|
| 1(C) | 0.0341 | −0.0513 | 17(H) | 0.0269 | −0.0199 |
| 2(C) | 0.0312 | −0.0267 | 18(H) | 0.0324 | −0.0370 |
| 3(C) | 0.0674 | −0.1068 | 19(H) | 0.0354 | −0.0046 |
| 4(C) | 0.0281 | −0.0385 | 20(H) | 0.1086 | 0.1935 |
| 5(C) | 0.0319 | −0.0626 | 21(H) | 0.0983 | 0.1685 |
| 6(C) | 0.0400 | −0.0760 | 22(H) | 0.0283 | 0.0466 |
| 7(C) | 0.0279 | 0.0498 | 23(H) | 0.0615 | 0.0946 |
| 8(C) | 0.0494 | 0.0797 | 24(H) | 0.0050 | −0.0037 |
| 9(C) | 0.0109 | −0.0121 | 25(H) | 0.0149 | −0.0085 |
| 10(C) | 0.0097 | −0.0049 | 26(H) | 0.0216 | −0.0130 |
| 11(C) | 0.0122 | −0.0116 | 27(H) | 0.0183 | −0.0101 |
| 12(C) | 0.0054 | −0.0087 | 28(H) | 0.0179 | 0.0001 |
| 13(O) | 0.0189 | −0.0138 | 29(H) | 0.0111 | −0.0033 |
| 14(O) | 0.0198 | 0.0249 | 30(H) | 0.0222 | −0.0159 |
| 15(O) | 0.0622 | −0.1301 | 31(H) | 0.0215 | −0.0212 |
| 16(N) | 0.0232 | 0.0150 |
In the ESP distribution map, red and blue colors represent electron-deficient and electron-rich regions of CBF, respectively. It can be observed that the phenyl ring and the carbamate group have lower electrostatic potential values, making them susceptible to attack by electrophilic reactive oxygen species SO4·− and ·OH. According to the values of f0 and CDD for CBF, higher f0 values indicate greater susceptibility to attack by reactive radicals, while more negative CDD values suggest greater susceptibility of that region to attack by electrophilic reagents.49 The f0 values at C3 and C6 positions on the phenyl ring are highest, and the CDD values at C3 and C6 on the phenyl ring are most negative, indicating that these are the sites most susceptible to attack by SO4·− and ·OH. Furthermore, the f0 value at O15 position is relatively high, and its CDD value is the lowest, suggesting it is susceptible to attack by reactive radicals or electrophilic reagents. However, being a saturated site, it makes it difficult for it to react with radicals.
In addition, SO4·− and ·OH are highly oxidative and readily undergo hydrogen atom abstraction reactions.36 Therefore, the next focus will primarily be on the addition reactions of SO4·− and ·OH with the phenyl ring and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in the formate group, as well as their hydrogen atom abstraction reactions.
O bond in the formate group, as well as their hydrogen atom abstraction reactions.
3.2 The addition reaction of CBF with SO4·− and ·OH
Fig. 2 and 3 illustrate the addition reactions of CBF with SO4·− and ·OH, respectively. In the figures, ΔG≠ represents the Gibbs free energy barrier, and ΔrG denotes the change in Gibbs free energy of the reaction, measured in kcal mol−1. The transition state configurations of CBF reacting with SO4·− and ·OH are depicted in Fig. S1 and S2,† respectively.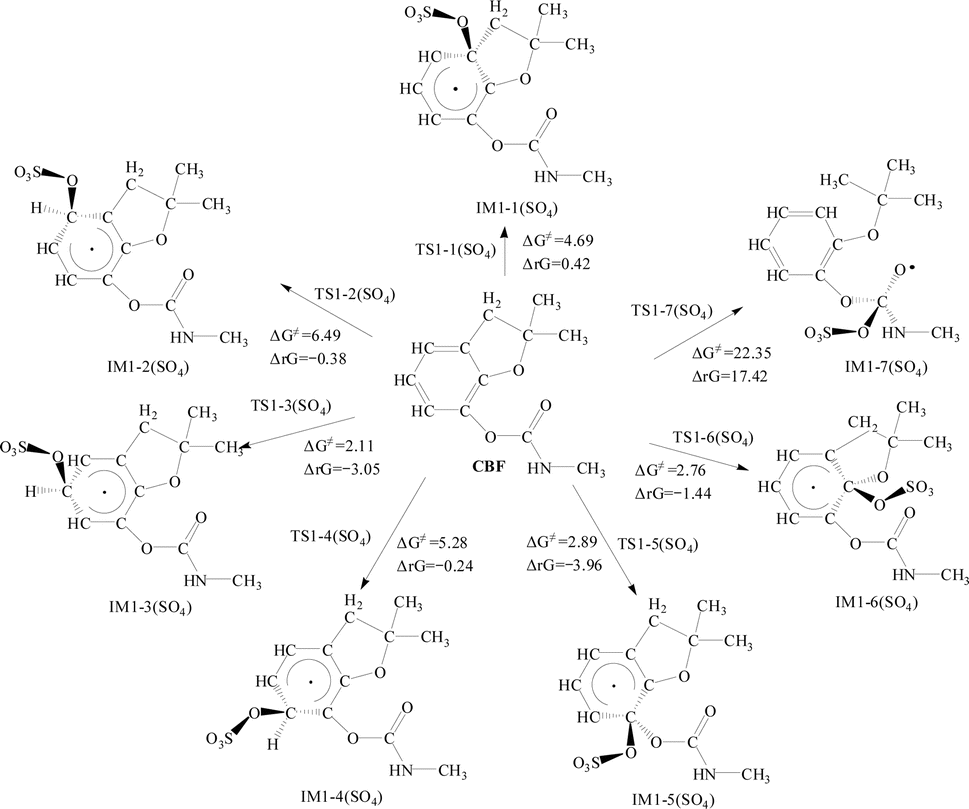 | ||
| Fig. 2 The Gibbs free energy barrier ΔG≠ (kcal mol−1) and the Gibbs free energy change ΔrG (kcal mol−1) at 298 K for the addition pathways of CBF with SO4·−. | ||
 | ||
| Fig. 3 The Gibbs free energy barrier ΔG≠ (kcal mol−1) and the Gibbs free energy change ΔrG (kcal mol−1) at 298 K for the addition pathways of CBF with ·OH. | ||
For the addition reaction of CBF with SO4·−, the analysis of ΔG≠ and ΔrG for all C positions on the phenyl ring was conducted. It is observed that ΔG≠ ranges between 2.11 and 6.49 kcal mol−1. Notably, the ΔG≠ values at the C3 and C6 positions are the lowest, at 2.11 and 2.76 kcal mol−1, respectively. This is consistent with the results obtained from the analysis using f0 and CDD values.
Apart from the ΔrG value being positive at the C1 position, all others are negative, indicating that the addition reaction of SO4·− with the phenyl ring is spontaneous. The addition reaction of SO4·− with the CO bond in the formate group shows a ΔG≠ of 22.35 kcal mol−1, indicating that the potential barrier is much higher than that of the addition reaction with the phenyl ring. The ΔrG is 17.42 kcal mol−1, which is positive, indicating that the reaction cannot proceed spontaneously.
The addition reaction of CBF with ·OH, as shown in Fig. 3, follows a pathway similar to the addition reaction with SO4·−. The ΔG≠ for the addition to the phenyl ring ranges from 5.39 to 8.98 kcal mol−1, with the lowest ΔG≠ still observed at the C3 and C6 positions. All reactions exhibit negative ΔrG values, indicating that the addition of ·OH to the phenyl ring of CBF is spontaneous. Through the analysis of ΔG≠ and ΔrG, it is evident that the addition reaction of ·OH with the CO bond in the formate group presents a higher energy barrier (ΔG≠ = 21.72 kcal mol−1) and is non-spontaneous (ΔrG = 6.88 kcal mol−1).
3.3 The abstraction reaction of CBF with SO4·− and ·OH
Due to the strong oxidizing properties of SO4·− and ·OH, they are prone to undergo hydrogen (H) atom abstraction reactions. There are four types of H atoms on CBF: H atoms on the phenyl ring (H17, H18, and H19), H atom on the amino group (H20), primary H atoms (H21 ∼ H29), and secondary H atoms (H30 and H31). The specific reaction pathways are shown in Fig. 4 and 5. | ||
| Fig. 4 The Gibbs free energy barrier ΔG≠ (kcal mol−1) and the Gibbs free energy change ΔrG (kcal mol−1) at 298 K for the abstraction reactions of CBF with SO4·−. | ||
 | ||
| Fig. 5 The Gibbs free energy barrier ΔG≠ (kcal mol−1) and the Gibbs free energy change ΔrG (kcal mol−1) at 298 K for the abstraction reactions of CBF with ·OH. | ||
For the H atom abstraction reactions between CBF and SO4·−, the ΔG≠ for H atom abstract from the phenyl ring is between 19.05 to 21.99 kcal mol−1, with ΔrG being negative or close to negative; for H atom abstract from the amino group, the ΔG≠ is 11.20 kcal mol−1, with ΔrG of −4.75 kcal mol; for the abstraction reactions of primary H atoms, the ΔG≠ ranges from 11.37 to 14.24 kcal mol−1, while the ΔrG ranges from −10.40 to −15.86 kcal mol; for the abstraction reactions of secondary H atoms, ΔG≠ is 1.54 kcal mol−1, with ΔrG of −24.99 kcal mol−1. From a thermodynamic perspective, the activation barrier for secondary H atoms abstraction has the lowest potential barrier and is most likely to occur spontaneously.
Similar results are observed for the H atom abstraction reactions between CBF and ·OH, with the order of ΔG≠ being the abstraction reactions of secondary H atoms < the abstraction reactions of primary H atoms < H atom abstract from the phenyl ring < H atom abstract from the amino group. This research result is consistent with the findings on ·OH and the carbamate insecticide isoprocarb, where H atoms are more easily abstracted from the –CH(CH3)2 and –CH3 groups than from the –CONH– group and the aromatic ring.50 Additionally, Xu et al. studied the reactions of N,N-diethyl-m-toluamide with SO4·− and ·OH at the M06-2X/6-31 + G(d,p) level, found that hydrogen abstraction from –CH2– is easier than from –CH3–, which is consistent with our findings.20 For the abstraction reactions of secondary H atoms, ΔG≠ is 7.66 kcal mol−1, which is higher than the reaction with SO4·−. Therefore, from a thermodynamic perspective, the abstraction reaction of CBF with SO4·− is slightly easier compared to that of ·OH.
3.4 Rate constants analysis
At 298 K, the rate constants (k) for the reactions of CBF with SO4·− and ·OH were calculated, including all possible addition and abstraction reactions. The calculation results are shown in Table 2. In the table, kadd represents the rate constant for addition reactions of SO4·− and ·OH, kabs represents the rate constant for abstraction reactions of SO4·− and ·OH, and Γ represents the branching ratio. The calculation formula for Γ is Γ = ki/ktotal, where ktotal is the sum of the rate constants for addition and abstraction reactions of SO4·− and ·OH with CBF.| Reaction | k298K (M−1 s−1) | Γ (%) | Reaction | k298K (M−1 s−1) | Γ (%) |
|---|---|---|---|---|---|
| CBF + SO4·− → IM1-1(SO4) | 2.46 × 107 | 0.54 | CBF + ·OH → IM1-1(OH) | 1.49 × 107 | 0.33 |
| CBF + SO4·− → IM1-2(SO4) | 2.88 × 106 | 0.06 | CBF + ·OH → IM1-2(OH) | 1.89 × 106 | 0.04 |
| CBF + SO4·− → IM1-3(SO4) | 1.09 × 109 | 23.86 | CBF + ·OH → IM1-3(OH) | 1.03 × 108 | 2.26 |
| CBF + SO4·− → IM1-4(SO4) | 1.99 × 107 | 0.44 | CBF + ·OH → IM1-4(OH) | 1.81 × 106 | 0.04 |
| CBF + SO4·− → IM1-5(SO4) | 1.47 × 108 | 3.22 | CBF + ·OH → IM1-5(OH) | 5.54 × 106 | 0.12 |
| CBF + SO4·− → IM1-6(SO4) | 4.73 × 108 | 10.38 | CBF + ·OH → IM1-6(OH) | 7.93 × 108 | 16.20 |
| CBF + SO4·− → IM1-7(SO4) | 2.77 × 10−4 | 0 | CBF + ·OH → IM1-7(OH) | 9.13 × 10−4 | 0 |
| kaddSO4·− | 1.75 × 109 | 38.51 | kadd·OH | 8.66 × 108 | 18.99 |
| CBF + SO4·− → IM1-8 + HSO4·− | 1.42 | 0 | CBF + ·OH → IM1-8 + H2O | 3.95 × 104 | 0 |
| CBF + SO4·− → IM1-9 + HSO4·− | 1.07 × 101 | 0 | CBF + ·OH → IM1-9 + H2O | 5.36 × 103 | 0 |
| CBF + SO4·− → IM1-10 + HSO4·− | 1.89 × 10−2 | 0 | CBF + ·OH → IM1-10 + H2O | 9.48 × 103 | 0 |
| CBF + SO4·− → IM1-11 + HSO4·− | 8.49 × 103 | 0 | CBF + ·OH → IM1-11 + H2O | 1.87 × 102 | 0 |
| CBF + SO4·− → IM1-12 + HSO4·− | 4.46 × 104 | 0 | CBF + ·OH → IM1-12 + H2O | 1.65 × 106 | 0.04 |
| CBF + SO4·− → IM1-13 + HSO4·− | 3.61 × 102 | 0 | CBF + ·OH → IM1-13 + H2O | 4.31 × 106 | 0.10 |
| CBF + SO4·− → IM1-14 + HSO4·− | 1.24 × 103 | 0 | CBF + ·OH → IM1-14 + H2O | 5.82 × 105 | 0.01 |
| CBF + SO4·− → IM1-15 + HSO4·− | 1.91 × 109 | 41.84 | CBF + ·OH → IM1-15 + H2O | 2.32 × 107 | 0.51 |
| kabsSO4·− | 1.91 × 109 | 41.84 | kabs·OH | 2.98 × 107 | 0.66 |
Analysis of the calculation results reveals that the abstraction reaction between SO4·− and secondary H atom has the highest Γ (41.84%). Next are the addition reactions of SO4·− at the C3 position of the benzene ring (Γ = 23.86%), the addition reactions of ·OH at the C6 position of the benzene ring (Γ = 16.20%), the addition reactions of SO4·− at the C6 position of the benzene ring (Γ = 10.38%), the addition reactions of SO4·− at the C5 position of the benzene ring (Γ = 3.22%), the addition reactions of ·OH at the C3 position of the benzene ring (Γ = 2.26%), and the abstraction reaction between ·OH and secondary H atom (Γ = 0.51%). The other reaction pathways can be considered negligible. At 298 K, the calculated total rate constants for the reaction of CBF with SO4·− and ·OH are 3.66 × 109 and 8.96 × 108 M−1 s−1, respectively. Zhang et al. experimentally measured the rate constant of CBF and ·OH to be 9.4 × 107 M−1 s−1, which is an order of magnitude lower than the theoretical value.51 This may be due to the fact that theoretical calculations are typically based on simplified reaction mechanism models, while actual reactions may involve more complex intermediate steps or parallel reactions. Due to the lack of experimental data for CBF with SO4·−, we compared it with the experimental rate constant of phenylurea with SO4·−, approximately 5 × 109 M−1 s−1, which is consistent with our calculated value.52,53 These results indicate that the calculated results and subsequent theoretical analysis are reliable and valuable.
In summary, based on the analysis of thermodynamic and kinetic results, it is evident that the abstraction reaction between SO4·− and secondary H atom is the optimal reaction pathway. Additionally, the addition reactions of SO4·− at the C3 and C6 positions of the benzene ring, as well as the addition reactions of SO4·− at the C5 position, cannot be ignored. Therefore, we will focus on studying the subsequent reactions of IM1-3(SO4), IM1-6(SO4), IM1-3(OH), IM1-6(OH), IM1-5(SO4), and IM1-15.
3.5 Subsequent reactions of main intermediates
The reaction pathways of the addition products IM1-3(OH) and IM1-3(SO4) are similar, both of which can be abstracted by SO4·− and ·OH to form stable structures. The specific path is shown in Fig. 6. | ||
| Fig. 6 Subsequent reactions of IM1-3(OH) and IM1-3(SO4). | ||
IM1-3(OH) and IM1-3(SO4) can proceed with very low barriers to form products P1 and P2. P1 was detected during the activation of persulfate degradation experiments using nano-magnetite/ZnO/activated carbon nanohybrid.23 However, the sulfate-originated product P2 was not detected in the experiment, which may be due to its weak stability and rapid evolution into hydroxylated compounds through substitution.54 The ΔrG of this process is negative, indicating that the reaction will proceed spontaneously.
IM1-6(OH) and IM1-6(SO4) undergo bond cleavage reactions by breaking the C6–O15 bond, thereby opening the furan ring. As shown in Fig. 7, the ΔG≠ of these two reaction processes is relatively high, measured at 23.96 and 24.01 kcal mol−1 respectively. The ΔrG is positive, indicating that the reaction will not proceed spontaneously. P3 and P4 were not detected in the experiment, which also confirms that the reaction is difficult to proceed.23
 | ||
| Fig. 7 Subsequent reactions of IM1-6(OH) and IM1-6(SO4). | ||
The subsequent reaction of IM1-5(SO4) is depicted in Fig. 8. This process involves initially overcoming a barrier of 10.35 kcal mol−1 and releases a significant amount of heat, making it easily spontaneous. This process yields products P5 and intermediate IM4, which can abstract an H atom from the water in the system to obtain stable product P6. The sulfate-originated product P5 has weak stability, will rapidly convert into hydroxylated compounds.23 The P6 is carbamic acid, which has been proven to decompose into carbon dioxide and methylamine.55
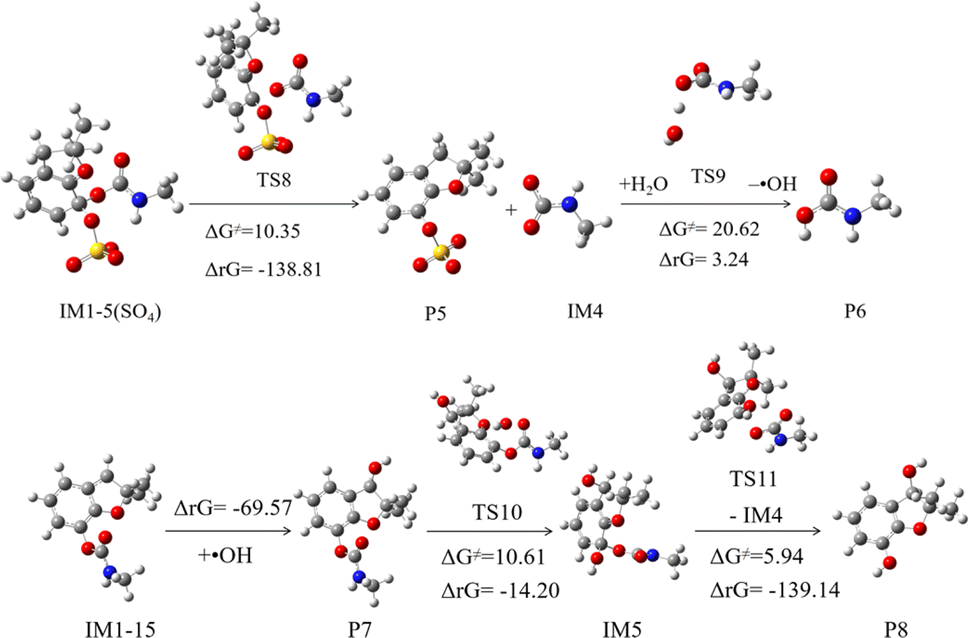 | ||
| Fig. 8 Subsequent reactions of IM1-5(SO4) and IM1-15. | ||
The abstraction product IM1-15, with the highest branching ratio, can rapidly undergo barrier-free addition reactions with ·OH present in the system to form stable product P7, releasing a substantial amount of heat. Subsequently, as depicted in Fig. 7, P7 can continue to undergo addition reactions with ·OH. The addition at the C5 position facilitates the formation of small molecule products through bond cleavage. The ΔG≠ for this addition process is low, at 10.61 kcal mol−1, and the ΔrG is negative, indicating that the reaction will readily proceed spontaneously. After bond cleavage, product P8 will be generated, which corresponds to the product with m/z of 181 detected by liquid chromatography-tandem mass spectroscopy (LC-MS/MS) in the experiment.23,55
3.6 Ecotoxicity assessment
Studies have shown that during the degradation of pesticides using persulfate based AOPs, a large amount of more toxic parent pesticide by-products are generated.56 Therefore, it crucial to estimate the toxicity of intermediates during pesticide degradation. ECOSAR is widely used to test the toxicity of by-products of pesticides.57 Xu et al. used ECOSAR software to predict the toxicity of chlorpyrifos products after treatment with persulfate-based AOPs.20 The results indicated that the acute and chronic toxicity of eight major degradation products to fish, aquatic invertebrates, and green algae were lower than that of the parent. Kan et al. predicted the acute and chronic toxicity of imidacloprid degradation products using ECOSAR software. The study found that products P1, P2, and P8 were more hazardous than the parent chemical.58 The ECOSAR ecological toxicity model was used to predict the acute and chronic toxicity of CBF and its products to fish, water fleas, and green algae.Table S1† lists the acute and chronic toxicity values of CBF and its degradation products. From the table, it can be seen that the chronic toxicity value of CBF to fish is 0.408 mg L−1, which is consistent with the results of 0.44 mg L−1 for tilapia obtained by Américo-Pinheiro et al.59 Fig. 9 categorizes the toxicity of CBF and its degradation products. As shown in Fig. 9(a), CBF, P1, P3, and P7 are highly toxic compounds in terms of acute toxicity to water fleas. For fish and green algae, only CBF and P1 are toxic compounds, while most of the other degradation products are harmless. In Fig. 9(b), from the perspective of chronic toxicity, only CBF and P1 are highly toxic to water fleas, while P3 and P7 are toxic compounds. For fish and green algae, most degradation products are harmless. The changes in acute and chronic toxicity indicate that the degradation products have lower acute and chronic toxicity to fish, water fleas, and green algae compared to the parent compound CBF. The main product P8 is harmless to all organisms. This result is consistent with the toxicity assessment of degradation products of CBF using persulfate-based AOPs by Samy et al.23 Additionally, Singh et al. evaluated the ecological toxicity of CBF wastewater treated with OH radicals using algal cell vitality assays and found that the toxicity gradually decreased, with minimum toxicities of 3.04%, 4.43%, and 7.30% at 24, 48, and 72 hours, respectively.41 This suggests that persulfate-based AOP is an effective method for remediating CBF pollution in water bodies.
 | ||
| Fig. 9 Classification of acute toxicity of CBF and transformation products to aquatic organisms. (a) The acute toxicity index: LC50/EC50 and (b) the chronic toxicity index: ChV (unit: mg L−1). | ||
4 Conclusions
This study investigated the mechanism of CBF degradation based on persulfate oxidation using DFT theory calculation methods. The optimal pathways initiated by SO4·− and ·OH were identified for degradation. Further evaluation of the toxicity of CBF degradation products was conducted to assess the feasibility of using persulfate-based AOP to degrade CBF. The results indicated that SO4·− and ·OH can undergo addition and abstraction reactions with CBF. Thermodynamic and kinetic calculations have confirmed that SO4·− and ·OH readily adds to the benzene ring, while the abstraction reaction primarily occurs on the furan ring. The initiating effect of SO4·− is greater than that of ·OH, which is consistent with experimental results. And the abstraction reaction between SO4·− and the secondary H atom on the furan ring is the optimal reaction pathway. The acute and chronic toxicity changes indicate that the degradation products exhibit lower toxicity compared to the CBF. In the products, P1, P3 and P7 still exhibits toxic or highly toxic levels, potentially posing a risk to human health. However, the primary product P8 is harmless to all organisms. The significant reduction in acute toxicity confirms that this system is a promising technology for treating CBF-contaminated water.Data availability
The datasets supporting this article have been provided below. All the data presented in this article is present in the form of figures and tables in the manuscript itself.Author contributions
Chenxi Zhang: conceptualization; software; data curation; writing-original draft. Youxin Xu: visualization; investigation, supervision. Bingbing Chu: software, investigation. Xiaomin Sun: methodology, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Science and Technology Development Program of Weifang City (grant number 2022GX036, 2023GX054), the Science and Technology Development Program of Shouguang City (grant number 2022JH08) and the Fundamental Research Funds of Weifang University of Science and Technology (grant number KJRC2022013).Notes and references
- J. Lan, W. Sun, L. Chen, H. Zhou, Y. Fan, X. Diao, B. Wang and H. Zhao, Food Agric. Immunol., 2020, 31, 165–175 CrossRef CAS.
- J. Gupta, R. Rathour, R. Singh and I. S. Thakur, Bioresour. Technol., 2019, 282, 417–424 CrossRef CAS PubMed.
- D. W. Gammon, Z. Liu and J. M. Becker, Pest Manage. Sci., 2012, 68, 362–370 CrossRef CAS PubMed.
- A. Abad, M. J. Moreno and A. Montoya, J. Agric. Food Chem., 1999, 47, 2475–2485 CrossRef CAS PubMed.
- P. O. Otieno, J. O. Lalah, M. Virani, I. O. Jondiko and K. W. Schramm, J. Environ. Sci. Health, Part B, 2010, 45, 137–144 CrossRef CAS PubMed.
- S. Campbell, M. D. David, L. A. Woodward and Q. X. Li, Chemosphere, 2004, 54, 1155–1161 CrossRef CAS PubMed.
- M. Umar Mustapha, N. Halimoon, W. L. Wan Johari and M. Y. Abd Shukor, Molecules, 2020, 25, 2771 CrossRef.
- T. P. O. Nguyen, D. E. Helbling, K. Bers, T. T. Fida, R. Wattiez, H. P. E. Kohler, D. Springael and R. De Mot, Appl. Microbiol. Biotechnol., 2014, 98, 8235–8252 CrossRef CAS.
- A. Hmimou, A. Maslouhi, K. Tamoh and L. Candela, C. R. Geosci., 2014, 346, 255–261 CrossRef.
- J. H. P. Américo-Pinheiro, A. A. Machado, C. da Cruz, M. M. Aguiar, L. F. R. Ferreira, N. H. Torres and J. G. Machado-Neto, Water, Air, Soil Pollut., 2020, 231, 228 CrossRef.
- S. Mishra, W. Zhang, Z. Lin, S. Pang, Y. Huang, P. Bhatt and S. Chen, Chemosphere, 2020, 259, 127419 CrossRef CAS PubMed.
- N. Cao, X. Zong, X. Guo, X. Chen, D. Nie, L. Huang, L. Li, Y. Ma, C. Wang and S. Pang, Chemosphere, 2024, 350, 140992 CrossRef CAS PubMed.
- K. Stoyanova, M. Gerginova, N. Peneva, I. Dincheva and Z. Alexieva, Processes, 2023, 11, 3343 CrossRef CAS.
- H. Park, S. I. Seo, J.-H. Lim, J. Song, J.-H. Seo and P. I. Kim, Metabolites, 2022, 12, 219 CrossRef CAS PubMed.
- R. Patowary, P. Jain, C. Malakar and A. Devi, Environ. Sci. Pollut. Res., 2023, 30, 115185–115198 CrossRef CAS.
- S. H. Alrefaee, A. M. Al-bonayan, H. H. Alsharief, M. Aljohani, K. F. Alshammari, F. A. Saad, H. M. Abumelha and N. M. El-Metwaly, Surf. Interfaces, 2023, 40, 103133 CrossRef CAS.
- H. P. Toledo-Jaldin, V. Sánchez-Mendieta, A. Blanco-Flores, G. López-Téllez, A. R. Vilchis-Nestor and O. Martín-Hernández, Environ. Sci. Pollut. Res., 2020, 27, 7872–7885 CrossRef CAS PubMed.
- Z. N. Garba, A. Tanimu and Z. U. Zango, Bull. Chem. Soc. Ethiop., 2019, 33, 425–436 CrossRef CAS.
- J. M. Schöntag, A. A. Alves, L. G. Romero Esquivel and M. L. Sens, Environ. Technol., 2019, 40, 2833–2839 CrossRef.
- Y. Xu, C. Zhang, H. Zou, G. Chen, X. Sun, S. Wang and H. Tian, Toxics, 2024, 12, 207 CrossRef CAS.
- K. Ruíz-Hidalgo, M. Masís-Mora, E. Barbieri, E. Carazo-Rojas and C. E. Rodríguez-Rodríguez, Chemosphere, 2016, 144, 864–871 CrossRef PubMed.
- S. Li, Z. Liu, Z. Qu, C. Piao, J. Liu, D. Xu, X. Li, J. Wang and Y. Song, J. Photochem. Photobiol., A, 2020, 389, 112246 CrossRef CAS.
- M. Samy, A. G. Kumi, E. Salama, M. ElKady, K. Mensah and H. Shokry, Process Saf. Environ. Prot., 2023, 169, 337–351 CrossRef CAS.
- Y. Aimer, O. Benali and K. G. Serrano, Sep. Purif. Technol., 2019, 208, 27–33 CrossRef CAS.
- D. Miao, S. Zhao, K. Zhu, P. Zhang, T. Wang, H. Jia and H. Sun, Chemosphere, 2020, 253, 126679 CrossRef CAS PubMed.
- Z. Shen, H. Zhou, P. Zhou, H. Zhang, Z. Xiong, Y. Yu, G. Yao and B. Lai, J. Hazard. Mater., 2022, 425, 127781 CrossRef CAS.
- Y. J. Zou, X. Huang, X. L. Guo, C. H. Jia, B. T. Li, E. C. Zhao and J. X. Wu, Ecotoxicol. Environ. Saf., 2022, 241, 113815 CrossRef PubMed.
- Z. Liu, X. Ren, X. Duan, A. K. Sarmah and X. Zhao, Sci. Total Environ., 2023, 863, 160818 CrossRef CAS.
- A. Romero, A. Santos, F. Vicente and C. González, Chem. Eng. J., 2010, 162, 257–265 CrossRef CAS.
- J. L. Wang and S. Z. Wang, Chem. Eng. J., 2018, 334, 1502–1517 CrossRef CAS.
- J. L. Wang and S. Z. Wang, Chem. Eng. J., 2020, 401, 126158 CrossRef CAS.
- C. Zhu, F. Liu, C. Ling, H. Jiang, H. Wu and A. Li, Appl. Catal., B, 2019, 242, 238–248 CrossRef CAS.
- Z. H. Xie, C. S. He, D. N. Pei, Y. Dong, S. R. Yang, Z. Xiong, P. Zhou, Z. C. Pan, G. Yao and B. Lai, Chem. Eng. J., 2023, 468, 143778 CrossRef CAS.
- L. Wang, X. Lan, W. Peng and Z. Wang, J. Hazard. Mater., 2021, 408, 124436 CrossRef CAS.
- X. Y. Jiang, E. Kwon, H. C. Chang, N. N. Huy, X. Duan, S. Ghotekar, Y. C. Tsai, A. Ebrahimi, F. Ghanbari and K. Y. A. Lin, Sep. Purif. Technol., 2023, 308, 122789 CrossRef CAS.
- C. Zhang, Y. Xu, W. Liu, H. Zhou, N. Zhang, Z. Fang, J. Gao, X. Sun, D. Feng and X. Sun, Ecotoxicol. Environ. Saf., 2023, 263, 115298 CrossRef CAS.
- J. Yang, Y. Xu, G. Lv, T. Li and X. Sun, J. Cleaner Prod., 2024, 434, 140078 CrossRef CAS.
- A. Atifi, M. Talipov, H. Mountacer, M. D. Ryan and M. Sarakha, J. Photochem. Photobiol., A, 2012, 235, 1–6 CrossRef CAS.
- I. Cwielag-Piasecka, M. Witwicki, M. Jerzykiewicz and J. Jezierska, Environ. Sci. Technol., 2017, 51, 14124–14134 CrossRef CAS PubMed.
- M. Lapertot, S. Ebrahimi, I. Oller, M. I. Maldonado, W. Gernjak, S. Malato and C. ulgarín, Ecotoxicol. Environ. Saf., 2008, 69, 546–555 CrossRef CAS.
- R. K. Singh, L. Philip and S. Ramanujam, Ind. Eng. Chem. Res., 2016, 55, 7201–7209 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel and G. E. Scuseria, et al., Gaussian 16 Rev. A.03, Gaussian: Wallingford, CT, USA, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- J. Zheng, Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2009, 5, 808–821 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS.
- S. Canneaux, F. Bohr and E. Henon, J. Comput. Chem., 2014, 35, 82–93 CrossRef CAS PubMed.
- K. J. Laidler, Theories of chemical reaction rates, McGraw-Hill Book Company, New York, NY, USA, 1969 Search PubMed.
- U. S. EPA. Ecological Structure—Activity Relationships Program (ECOSAR) Operation Manual, vol. 2.2; U. S. EPA: Washington, DC, USA, 2022 Search PubMed.
- X. Y. Ding, X. Song, X. Chen, D. Ding, C. Xu and H. Chen, Chemosphere, 2022, 286, 131720 CrossRef CAS.
- C. Zhang, W. Yang, J. Bai, Y. Zhao, C. Gong, X. Sun, Q. Zhang and W. Wang, Atmos. Environ., 2012, 60, 460–466 CrossRef CAS.
- B. Zhang, T. Hiramatsu, S. Hamano, M. Fujii, M. G. Alalm, S. Yoshikawa, H. Matsumoto and S. Ookawara, J. Environ. Chem. Eng., 2022, 10, 108801 CrossRef CAS.
- M. Canle Lopez, S. Rodrígez, L. F. R. Vazques, J. A. Santaballa and S. Steenken, J. Mol. Struct., 2001, 565–566, 133–139 CrossRef.
- M. Canle Lopez, M. I. Fernández, S. Rodrígez, J. A. Santaballa, S. Steenken and E. Vulliet, Phys. Chem. Chem. Phys., 2005, 6, 2064–2074 CrossRef CAS.
- A. Abdelhaleem and W. Chu, Chemosphere, 2019, 237, 124487 CrossRef CAS PubMed.
- K. Fouad, M. G. Alalm, M. Bassyouni and M. Y. Saleh, Environ. Technol. Innovation, 2021, 23, 101778 CrossRef CAS.
- S. Khan, M. Sohail, C. Han, J. A. Khan, H. M. Khan and D. D. Dionysiou, J. Hazard. Mater., 2021, 402, 123558 CrossRef CAS.
- A. Derbalah and H. Sakugawa, Int. J. Environ. Res., 2024, 18, 11 CrossRef CAS.
- Q. Kan, K. Lu, S. Dong, D. Shen, Q. Huang, Y. Tong, W. Wu, S. Gao and L. Mao, Environ. Pollut., 2020, 267, 115438 CrossRef CAS PubMed.
- J. H. P. Américo-Pinheiro, A. A. Machado, C. da Cruz, M. M. Aguiar, L. F. R. Ferreira, N. H. Torres and J. G. Machado-Neto, Water, Air, Soil Pollut., 2020, 231, 228 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra05365f |
| This journal is © The Royal Society of Chemistry 2024 |