
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
FeBr3-catalysed synthesis of 3-aroylimidazo[1,2-a]pyridine and 3,3′-(arylmethylene)bis(2-phenylimidazo[1,2-a]pyridines) derivatives from 2-arylimidazo[1,2-a]pyridines and aromatic aldehydes: an investigation about mechanistic insights†
Tran Quang Hung*ad,
Ban Van Phuc‡
a,
Mai Phuong Nguyen‡b,
Tuan Linh Tran‡b,
Dang Van Dob,
Ha Thanh Nguyen ad,
Van Tuyen Nguyenad,
Hien Nguyenc and
Tuan Thanh Dang*b
ad,
Van Tuyen Nguyenad,
Hien Nguyenc and
Tuan Thanh Dang*b
aInstitute of Chemistry, Vietnam Academy of Science and Technology, Vietnam. E-mail: tqhung@ich.vast.vn
bFaculty of Chemistry, Hanoi University of Science, Vietnam National University (VNU), Vietnam. E-mail: dangthanhtuan@hus.edu.vn
cFaculty of Chemistry, Hanoi National University of Education (HNUE), Vietnam
dGraduate University of Science and Technology, Vietnam Academy of Science and Technology, Vietnam
First published on 18th September 2024
Abstract
In a new approach, a series of 3-aroylimidazo[1,2-a]pyridine derivatives were prepared in high yields. This approach revealed the direct Fe-catalyzed functionalization of imidazo[1,2-a]pyridine derivatives with aryl aldehydes via an aerobic oxidative cross-dehydrogenative coupling process. This transformation occurred in the presence of air, and FeBr3 served as a homogeneous Lewis catalyst. O2 was found to be the principal oxidant responsible for the method's success. Interestingly, when these reactions were carried out under an argon atmosphere, 3,3′-(arylmethylene)bis(2-phenylimidazo[1,2-a]pyridines) derivatives were prepared in good yields.
Introduction
In the field of drug discovery, fused N-heterocycles have long been recognized as essential targets.1–4 Specifically, imidazo[1,2-a]pyridine derivatives are crucial building blocks in several important drugs and bioactive molecules due to their wide bioactivities.5 Several commercial pharmaceutics contain an imidazo[1,2-a]pyridine core, including anxiolytics Alpidem, Necopidem, Soraprazan, Saripidem and Zolimidine, as well as Zolpidem, which is used to treat brain abnormalities (Fig. 1).5 Furthermore, the antibacterial, anticancer, anti-inflammatory, antiprotozoal, antiviral, antiparasitic, analgesic, and antipyretic activities of a number of new compounds with an imidazo[1,2-a]pyridine core have been convincingly demonstrated.6 In addition to pharmacological research, imidazo[1,2-a]pyridine derivatives have also shown highly fluorescent characteristics with high quantum yields.7 Therefore, their synthetic approaches have gained substantial attention in recent years.5,8 | ||
| Fig. 1 Imidazo[1,2-a]pyridine derivatives with various biological activities. | ||
Recently, 3-aroylimidazo[1,2-a]pyridine derivatives have found promising applications in the development of pharmaceutical chemistry research such as anticancer and antimitotic agents (Fig. 1, compounds A, B, C).9 In fact, various approaches to prepare 3-aroylimidazo[1,2-a]pyridine derivatives have been reported.9 The first effort for direct coupling reaction of 2-aminopyridines with chalcones by Cu-catalysed aerobic oxidation was disclosed by Monir and coworkers.10 In 2014 Kaswan et al. reported a similar research on the Cu-catalysed synthesis of 3-aroylimidazo[1,2-a]pyridines from 2-aminopyridines with chalcones under air.11 Then, Nguyen et al. demonstrated a protocol for the aerobic coupling of 2-aminopyridines with chalcones using CuFe2O4 nanoparticles catalyst to give 3-aroylimidazo[1,2-a]pyridines in good yields.12 In this procedure, both iodine and oxygen must be used as oxidants for the success of this transformation. In 2015, Kaswan et al. also reported a method to prepare 3-aroylimidazo[1,2-a]pyridines by one-pot, three-component reaction of 2-aminopyridines, acetophenones, and benzaldehyde derivatives in the presence of CuCl2 catalyst under air.13 One year later, Xing and coworkers described a practical synthesis of 3-aroylimidazo[1,2-a]pyridines by Iodine-promoted oxidative coupling reaction of 2-aminopyridines and chalcones.14 From 2015, several reports in the synthesis of 3-aroylimidazo[1,2-a]pyridines based on the tandem Cu(I)-catalysed oxidative cyclization reactions of 2-aminopyridines and 1-phenyl-3-(aryl)prop-2-yn-1-ones via N-(2-pyridinyl) enaminone intermediates were also disclosed.15
Recently, direct functionalization of imidazo[1,2-a]pyridines have been identified as an efficient approach to prepare bioactive imidazo[1,2-a]pyridine derivatives.16 In fact, the functionalization of imidazo[1,2-a]pyridines at the C3 position could not be directly made via the classical Friedel–Crafts acylation using aroyl chlorides and Lewis acid catalysts.9 Formal synthesis of 3-aroylimidazo[1,2-a]pyridine required a three-step procedure involving: (i) formylation of imidazo[1,2-a]pyridines, (ii) reaction of imidazo[1,2-a]pyridine-3-carbaldehydes with Grignard reagent to form secondary alcohols and (iii) oxidation of secondary alcohols to give 3-aroylimidazo[1,2-a]pyridines.17 In continuation of our efforts on the synthesis of imidazoheterocycles as well as indole-fused N-heterocycles,18 herein, we wish to report two practical procedures: (i) a direct FeBr3-catalysed functionalization of 2-arylimidazo[1,2-a]pyridine derivatives using aromatic aldehydes via aerobic oxidative cross-dehydrogenative coupling reaction; (ii) the FeBr3-catalysed alkylation of 2-arylimidazo[1,2-a]pyridine derivatives using aromatic aldehydes to give 3,3′-(arylmethylene)bis(2-phenylimidazo[1,2-a]pyridines) under argon atmosphere (Scheme 1).
 | ||
| Scheme 1 Several approaches to prepare 3-aroylimidazo[1,2-a]pyridines and 3,3′-(arylmethylene)bis(2-phenylimidazo[1,2-a]pyridines). | ||
Results and discussion
The first step of this research was initiated by the preparation of 2-arylimidazo[1,2-a]pyridines as the key starting materials following a well-established method.19 In order to optimise this transformation, we choose the reaction between compound 1a and picolinaldehyde 2a as the representative reaction, as demonstrated in Table 1. With the purpose of regioselective functionalization of imidazo[1,2-a]pyridines at the C3 position, our attention was solely directed towards the investigation of various Lewis acid catalysts with the aim of identifying the optimal conditions for this transformation. Initially, the typical Lewis acids for Friedel–Crafts acylation was employed, resulting in a promising 60% yield of the desired product in the presence of FeCl3 catalyst (entries 1–4). The employment of several commonly used Fe(III) salts as catalysts was examined which showed that FeBr3 could be the most suitable catalyst for this transformation (entries 5–9). Then, Fe(II) salts were also examined as Lewis acid catalysts. However, we only obtained the desired product 3a in low yields (entries 10–12). Finally, further optimisations using different solvents, reaction time and temperatures did not give us better results (entries 13–19). In order to understand the real role of FeBr3 catalyst and the oxidant in the present study, two control experiments were carried out in the absence of catalyst and oxygen. In the absence of FeBr3 catalyst, only 9% of product 3a was obtained (entry 20). Notably, only 12% yield of product 3a was isolated from reaction mixture when this reaction was performed under argon atmosphere (entry 21). Hence, oxygen in air should be involved in the catalytic cycle as the oxidant.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Time (h) | Temp. (°C) | Yieldb (%) |
| a Condition: 1a (0.3 mmol), 2a (1.5 equiv.), [Fe] catalyst (20 mol%), 110 °C, 24 h.b Yield of isolated products are given.c Reaction was performed in argon atmosphere.d Reaction was performed in oxygen atmosphere. | |||||
| 1 | AlCl3 | Toluene | 16 | 110 | — |
| 2 | ZrCl4 | Toluene | 16 | 110 | — |
| 3 | CuCl2 | Toluene | 16 | 110 | 23 |
| 4 | FeCl3 | Toluene | 16 | 110 | 60 |
| 5 | FeBr3 | Toluene | 16 | 110 | 70 |
| 6 | Fe2(SO4)3·9H2O | Toluene | 16 | 110 | — |
| 7 | Fe(NO3)3·9H2O | Toluene | 16 | 110 | — |
| 8 | Fe(acac)3 | Toluene | 16 | 110 | — |
| 9 | Fe(OTf)3 | Toluene | 16 | 110 | 65 |
| 10 | Fe(OAc)2·4H2O | Toluene | 16 | 110 | 20 |
| 11 | FeCl2 | Toluene | 16 | 110 | 15 |
| 12 | FeSO4·7H2O | Toluene | 16 | 110 | 2 |
| 13 | FeBr3 | DMF | 16 | 110 | 10 |
| 14 | FeBr3 | 1,4-Dioxane | 16 | 110 | 5 |
| 15 | FeBr3 | Xylene | 16 | 110 | 30 |
| 16 | FeBr3 | Toluene | 16 | 120 | 65 |
| 17 | FeBr3 | Toluene | 16 | 100 | 56 |
| 18 | FeBr3 | Toluene | 24 | 110 | 72 |
| 19 | FeBr3 | Toluene | 8 | 110 | 45 |
| 20 | — | Toluene | 16 | 110 | 9 |
| 21 | FeBr3 | Toluene | 16 | 110 | 12c |
| 22 | FeBr3 | Toluene | 16 | 110 | 73d |

With the optimised condition in hand, we proceeded to investigate the potential application of this reaction of 2-arylimidazo[1,2-a]pyridines 1a–d with various aldehydes 2a–g, as described in Table 2. In general, the desired products were successfully prepared, resulting in isolated yields of up to 89%. Typically, the functionalization of 2-arylimidazo[1,2-a]pyridines with picolinaldehyde and isopicolinaldehydes frequently afforded to corresponding products 3a–h and 3o–r in high yields, reaching up to 89%. Interestingly, benzaldehyde and its derivatives could be successfully employed in the reaction with 2-phenylimidazo[1,2-a]pyridine 1a which only afforded to the corresponding products 3k–n in moderate yields.
| a Condition: 1a–d (0.5 mmol), 2a–g (1.5 equiv.), FeBr3 catalyst (20 mol%), 110 °C, 16 h; yields of isolated products are given. |
|---|
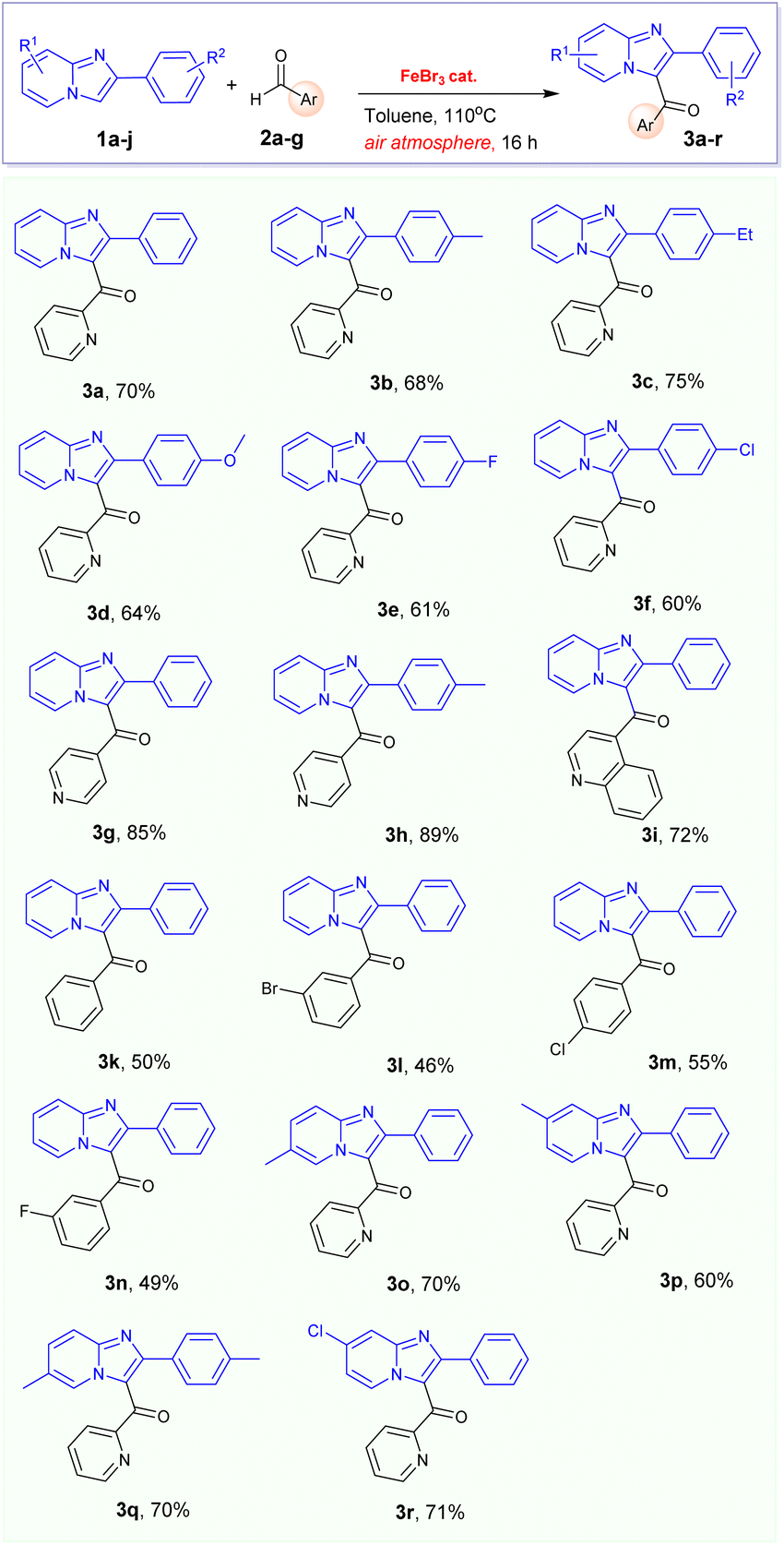 |
Especially, the oxidative coupling reaction of phenylimidazo[1,2-a]pyridine 1a with 4-methylbenzaldehyde did not give the desired 3-aroylimidazo[1,2-a]pyridine under optimized condition. It is interesting to report that a new 3,3′-(p-tolylmethylene)bis(2-phenylimidazo[1,2-a]pyridine) 4b (Table 3) was formed in this reaction as the main product in 55% yield. This observation led us to conclude that the electrophilic activity of the aldehyde substrates plays a major role in the formation of products. In previous literature, only one example for the FeCl3-catalysed formation of product 4b was described in 2016 by Hajra's group.16 However, the authors did not clarify the reaction mechanism and the real role of oxygen. We consequently came to the conclusion that oxygen in air might be crucial to this transformation. In our effort to prepare the desired 3-aroylimidazo[1,2-a]pyridine product from 4-methylbenzaldehyde, this reaction was carried out under oxygen atmosphere. However, we also failed to obtain this product in reasonable yield. Remarkably, 3,3′-(p-tolylmethylene)bis(2-phenylimidazo[1,2-a]pyridine) product 4b can be prepared in improved yield (65%) when this reaction was carried out under argon atmosphere in the presence of FeBr3 catalyst. Consequently, we are interested in extending the scope of this transformation with other benzaldehyde derivatives. A series of 3,3′-(arylmethylene)bis(2-phenylimidazo[1,2-a]pyridines) 4a–e were prepared in good yields from the FeBr3-catalysed alkylation of phenylimidazo[1,2-a]pyridine 1a with benzaldehyde derivatives (Table 3).
| a Condition: 1a (0.515 mmol), 2a–g (1.0 equiv.), FeBr3 catalyst (20 mol%), 110 °C, under argon, 16 h; yields of isolated products are given. |
|---|
 |
Surprisingly, FeBr3-catalysed alkylation of phenylimidazo[1,2-a]pyridine 1a with hexanal did not give either 1-(2-phenylimidazo[1,2-a]pyridin-3-yl)hexan-1-one or 3,3′-(hexane-1,1-diyl)bis(2-phenylimidazo[1,2-a]pyridine) products under air or argon atmosphere conditions. Interestingly, only (E)-3-(hex-1-en-1-yl)-2-phenylimidazo[1,2-a]pyridine product 5a was obtained in 75% isolated yield (Scheme 5).
We conducted several control experiments to better understand the mechanism underlying this transformation (Scheme 2). First, we discovered that the FeBr3-catalysed aerobic oxidative coupling reactions of 2-phenylimidazo[1,2-a]pyridine 1a with benzaldehyde and benzoic acid yielded the same product 3a in 50% and 65%, respectively (reactions [1], [2], Scheme 2). Based on these findings, we hypothesized that this transformation may happen in two steps. The first step would be the oxidation of benzaldehyde to produce benzoic acid in the presence of oxygen in air, followed by a second Friedel–Crafts-type alkylation reaction of 1a with in situ-formed benzoic acid to produce 3a. In order to confirm this hypothesis, the oxidation of benzaldehyde under optimized condition was performed which gave benzoic acid in 72% yield. This oxidation reaction was well established in literature.20 A very similar Fe(III)-catalysed transformation of benzaldehyde to form benzoic acid using oxygen as oxidant was also reported by Wang and coworkers.20 Notably, the FeBr3-catalysed alkylation reaction of 2-phenylimidazo[1,2-a]pyridine 1a with benzaldehyde did not result in the formation of product 3a when this reaction was carried out under argon atmosphere (reaction [4]). Indeed, we obtained 3,3′-(phenylmethylene)bis(2-phenylimidazo[1,2-a]pyridine) product 4a in 45% isolated yield when was carried out under argon atmosphere in the employment of FeBr3 catalyst.
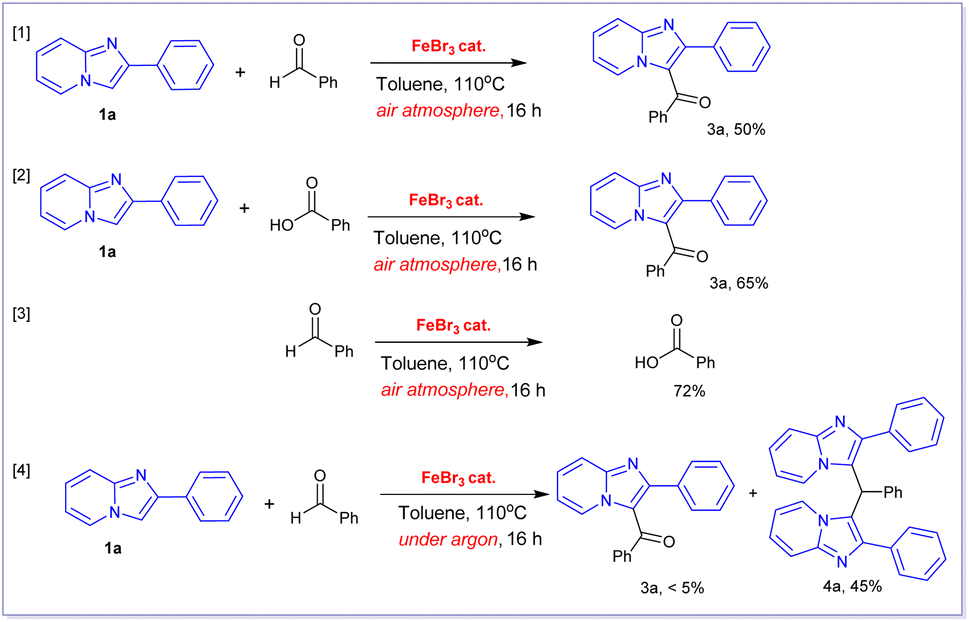 | ||
| Scheme 2 Control experiments. | ||
Based on the observed results in control experiments, a plausible mechanism for the FeBr3-catalysed synthesis of 3-aroylimidazo[1,2-a]pyridines from 2-arylimidazo[1,2-a]pyridine and aromatic aldehydes is proposed (Scheme 3). Firstly, the FeBr3-activated benzaldehyde may well react with oxygen in air to form a benzoic acid intermediate which subsequently react with 2-phenylimidazo[1,2-a]pyridine 1a via a Friedel–Crafts-type acylation reaction in the presence of FeBr3 catalyst. The in situ-formed intermediate A would be converted to 3-aroylimidazo[1,2-a]pyridine product 3 and regenerate the FeBr3 catalyst for the next catalytic cycle.
 | ||
| Scheme 3 Plausible mechanism for the FeBr3-catalysed formation of 3-aroylimidazo[1,2-a]pyridines 3 in the presence of oxygen. | ||
Scheme 4 illustrates a possible process for the FeBr3 catalysed synthesis of 3,3′-(phenylmethylene)bis(2-phenylimidazo[1,2-a]pyridines) product 4. First, the nucleophilic attack of 2-phenylimidazo[1,2-a]pyridine 1a on FeBr3-activated benzaldehyde yielded the intermediate A, which was subsequently converted to intermediates B and C. Then, a second nucleophilic addition of 2-phenylimidazo[1,2-a]pyridine 1a to the intermediate C occurred to afford to the intermediate D. In order to produce 3,3′-(phenylmethylene)bis(2-phenylimidazo[1,2-a]pyridines) product 4a, the intermediate D may finally be deprotonated by a bromide anion. This will also remove a Fe(OH)Br2 molecule, which will then react with the in situ-formed HBr to regenerate FeBr3 catalyst for the subsequent catalytic cycle.
 | ||
| Scheme 4 Possible mechanism for the FeBr3-catalysed formation of 3,3′-(arylmethylene)bis(2-phenylimidazo[1,2-a]pyridines) products under argon atmosphere. | ||
As described above, FeBr3-catalysed alkylation of 2-phenylimidazo[1,2-a]pyridine 1a with hexanal resulted in the formation of only (E)-3-(hex-1-en-1-yl)-2-phenylimidazo[1,2-a]pyridine product 5a in 75% isolated yield. A plausible mechanism for the FeBr3-catalysed synthesis of (E)-3-(hex-1-en-1-yl)-2-phenylimidazo[1,2-a]pyridine product 5a from 2-phenylimidazo[1,2-a]pyridine and hexanal was proposed (Scheme 5). Firstly, 2-phenylimidazo[1,2-a]pyridine 1a react with FeBr3-activated hexanal to form similar proposed intermediates A, B. However, the in situ-formed bromide anion took β-proton from intermediate B to produce the alkene product 5a in 75% isolated yield.
 | ||
| Scheme 5 Plausible mechanism for the FeBr3-catalysed formation of (E)-3-(hex-1-en-1-yl)-2-phenylimidazo[1,2-a]pyridine product 5a. | ||
Conclusions
In conclusion, we are reporting a practical and convenient method for the synthesis of either 3-aroylimidazo[1,2-a]pyridines or 3,3′-(arylmethylene)bis(2-phenylimidazo[1,2-a]pyridines) derivatives from the same starting materials such as 2-arylimidazo[1,2-a]pyridines and aromatic aldehydes. To the best of our knowledge, this direct FeBr3-catalysed functionalization of 2-arylimidazo[1,2-a]pyridine derivatives using aryl aldehydes via aerobic oxidative cross-dehydrogenative coupling strategy has not been reported in the literature before. In addition, when this reaction was carried out under argon atmosphere, 3,3′-(arylmethylene)bis(2-phenylimidazo[1,2-a]pyridines) derivatives were chemoselectively formed in good yields. Plausible mechanisms for the selective formation of 3-aroylimidazo[1,2-a]pyridines or 3,3′-(arylmethylene)bis(2-phenylimidazo[1,2-a]pyridines) involving FeBr3 as a homogeneous catalyst have been proposed relying on observed experimental results. Remarkably, the role of oxygen was figured out to be the key oxidant for the chemoselective formation of 3-aroylimidazo[1,2-a]pyridines. The findings reported herein could be highly beneficial for the development of practical synthetic applications in materials science and medicinal chemistry. Currently, several biological activities investigations on 3-aroylimidazo[1,2-a]pyridine and 3,3′-(arylmethylene)bis(2-phenylimidazo[1,2-a]pyridines) derivatives are currently carried out in our laboratory.Experimental
General procedure A for synthesis of compound 1a
In a round-bottom flask, 2-bromo-1-phenylethan-1-one (0.318 g, 1.6 mmol), 2-aminopyridine (0.181 g, 1.920 mmol), and sodium bicarbonate (0.134 g, 1.6 mmol) were dissolved in ethanol (3 mL). The reaction mixture was stirred at 70 °C for 6 hours. After cooling to room temperature, the solvent was removed under reduced pressure. The residue was extracted with ethyl acetate and water, and the organic layer was dried over anhydrous sodium sulfate. Evaporation of the solvent afforded the crude product, which was purified by column chromatography on silica gel using a hexane/ethyl acetate (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solvent system to afford 2-phenylimidazo[1,2-a]pyridine 1a as a white solid (0.280 g, 90%).
:1) solvent system to afford 2-phenylimidazo[1,2-a]pyridine 1a as a white solid (0.280 g, 90%).
1H NMR (600 MHz, CDCl3) δ 8.07 (dt, J = 6.7, 1.2 Hz, 1H), 7.97–7.93 (m, 2H), 7.82 (d, J = 0.8 Hz, 1H), 7.62 (dq, J = 9.0, 1.0 Hz, 1H), 7.45–7.40 (m, 2H), 7.34–7.30 (m, 1H), 7.14 (ddd, J = 9.1, 6.7, 1.3 Hz, 1H), 6.74 (td, J = 6.7, 1.2 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 145.8, 145.7, 133.8, 128.7, 127.9, 126.1, 125.6, 124.6, 117.6, 112.4, 108.1.
General procedure B for synthesis of compound 3a
In a reaction tube, 2-phenylimidazo[1,2-a]pyridine 1a (100 mg, 0.515 mmol), pyridine-2-carbaldehyde 2a (83 mg, 0.772 mmol), and iron(III) bromide (FeBr3) (30.4 mg, 0.103 mmol) were dissolved in toluene (0.6 mL). The reaction mixture was stirred at 110 °C for 16 hours. After this reaction completed, the reaction mixture was extracted with water and ethyl acetate. The organic layer was dried over anhydrous sodium sulfate, and then the solvent was removed under reduced pressure. The crude residue was purified by column chromatography on silica gel using a hexane/ethyl acetate (2:1) solvent system to afford (2-phenylimidazo[1,2-a]pyridin-3-yl)(pyridin-2-yl)methanone 3a as a white solid (108 mg, 70%).
1H NMR (600 MHz, CDCl3) δ 9.65 (dt, J = 6.9, 1.2 Hz, 1H), 8.09 (ddd, J = 4.8, 1.7, 0.9 Hz, 1H), 7.82 (dt, J = 8.9, 1.2 Hz, 1H), 7.69 (dt, J = 7.8, 1.1 Hz, 1H), 7.60 (td, J = 7.7, 1.7 Hz, 1H), 7.55 (ddd, J = 8.9, 6.9, 1.3 Hz, 1H), 7.32–7.27 (m, 2H), 7.16–7.04 (m, 5H). 13C NMR (151 MHz, CDCl3) δ 185.5, 156.8, 155.6, 148.4, 147.6, 136.2, 134.7, 129.7, 129.6, 128.5, 127.9, 127.7, 125.4, 123.8, 119.9, 117.5, 114.9.
General procedure C for synthesis of compound 4a
In a reaction tube, 2-phenylimidazo[1,2-a]pyridine 1a (100 mg, 0.515 mmol), pyridine-2-carbaldehyde 2a (83 mg, 0.772 mmol), and iron(III) bromide (FeBr3) (30.4 mg, 0.103 mmol) were dissolved in toluene (0.6 mL). Then, the reaction tube was backfilled with argon gas 3 times. The reaction mixture was stirred at 110 °C for 16 hours. After this reaction completed, the reaction mixture was extracted with water and ethyl acetate. The organic layer was dried over anhydrous sodium sulfate, and then the solvent was removed under reduced pressure. The crude residue was purified by column chromatography on silica gel using a hexane/ethyl acetate (2:1) solvent system to obtain 3,3′-(phenylmethylene)bis(2-phenylimidazo[1,2-a]pyridine) 4a as a brown solid (56 mg, 45%).
1H NMR (600 MHz, CDCl3) δ 7.60 (dt, J = 9.0, 1.1 Hz, 2H), 7.35–7.28 (m, 4H), 7.23 (dt, J = 6.9, 1.2 Hz, 2H), 7.21–7.15 (m, 3H), 7.15–7.05 (m, 7H), 6.86–6.76 (m, 2H), 6.53 (s, 1H), 6.41 (td, J = 6.8, 1.2 Hz, 2H). 13C NMR (151 MHz, CDCl3) δ 145.3, 144.7, 135.7, 133.8, 129.2, 128.7, 128.0, 127.9, 127.7, 127.7, 124.5, 124.1, 118.0, 117.3, 112.2, 38.5.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.† Data which are reported in this manuscript are available from the authors upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Vietnam Academy of Science and Technology has provided funding for this research under Grant Number ĐL0000.04/22-24.Notes and references
- (a) R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845 CrossRef CAS PubMed; (b) A. Gomtsyan, Chem. Heterocycl. Compd., 2012, 48, 7 CrossRef CAS.
- (a) R. Vardanyan and V. Hruby, Synthesis of Best-Seller Drugs, Elsevier Science, 2016, p. 868 Search PubMed; (b) J. A. Joule and K. Mills, Heterocyclic Chemistry, Wiley, Chichester, UK, 5th edn, 2010 Search PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed.
- M. M. Heravi and V. Zadsirjan, RSC Adv., 2020, 10, 44247 RSC.
- Z. Tashrifi, M. M. Khanaposhtani, B. Larijani and M. Mahdavi, Eur. J. Org Chem., 2020, 269 CrossRef CAS.
- (a) A. Anaflous, N. Benchat, M. Mimouni, S. Abouricha, T. Ben-Hadda, B. El-Bali, A. Hakkou and B. Hacht, Lett. Drug Des. Discov., 2004, 1, 224 CrossRef CAS; (b) T. H. Al-Tel and R. A. Al-Qawasmeh, Eur. J. Med. Chem., 2010, 45, 5848 CrossRef CAS PubMed; (c) Y. Rival, G. Grassy and G. Michel, Chem. Pharm. Bull., 1992, 40, 1170 CrossRef CAS PubMed; (d) Y. K. Marquez-Flores, M. E. Campos-Aldrete, H. Salgado-Zamora, J. Correa-Basurto and M. E. Melendez-Camargo, Med. Chem. Res., 2011, 21, 3491 CrossRef; (e) N. Devi, D. Singh, R. K. Rawal, J. Bariwal and V. Singh, Curr. Top. Med. Chem., 2016, 16, 2963 CrossRef CAS PubMed.
- (a) V. Tyagi, S. Khan, V. Bajpai, H. M. Gauniyal, B. Kumar and P. M. S. Chauhan, J. Org. Chem., 2012, 77, 1414 CrossRef CAS PubMed; (b) A. El Akkaoui, M. A. Hiebel, A. Mouaddib, S. Berteina-Raboin and G. Guillaumet, Tetrahedron, 2012, 68, 9131 CrossRef CAS; (c) L. Kielesinski, M. Tasior and D. T. Gryko, Org. Chem. Front., 2015, 2, 21 RSC; (d) B. Banerji, S. Chatterjee, K. Chandrasekhar, S. Ghosh, K. Mukherjee and C. Mandal, J. Org. Chem., 2018, 83, 13011 CrossRef CAS PubMed; (e) Y. Prostota, O. D. Kachkovsky, L. V. Reis and P. F. Santos, Dyes Pigm., 2013, 96, 554 CrossRef CAS; (f) C.-H. Ke, B.-C. Kuo, D. Nandi and H. M. Lee, Organometallics, 2013, 32, 4775 CrossRef CAS; (g) S. Takizawa, J. Nishida, T. Tsuzuki, S. Tokito and Y. Yamashita, Chem. Lett., 2005, 34, 1222 CrossRef CAS.
- V. Kurteva, ACS Omega, 2021, 6, 35173 CrossRef CAS PubMed.
- R. Rawat and S. M. Verma, Synth. Commun., 2020, 23, 3507 CrossRef.
- K. Monir, A. K. Bagdi, S. Mishra, A. Majee and A. Hajra, Adv. Synth. Catal., 2014, 356, 1105 CrossRef CAS.
- P. Kaswan, K. Pericherla and A. R. Kumar, Tetrahedron, 2014, 70, 8539 CrossRef CAS.
- O. T. K. Nguyen, P. T. Ha, H. V. Dang, Y. H. Vo, T. T. Nguyen, N. T. H. Le and N. T. S. Phan, RSC Adv., 2019, 9, 5501 RSC.
- P. Kaswan, K. Pericherla, H. K. Saini and A. Kumar, RSC Adv., 2015, 5, 3670 RSC.
- M. M. Xing, M. Xin, C. Shen, J. R. Gao, J. H. Jia and Y. J. Li, Tetrahedron, 2014, 70, 8539 CrossRef.
- (a) S. Cacchi, A. Ciogli, N. Demitri, G. Fabrizi, F. Ghirga, A. Goggiamani, A. Iazzetti and D. Lamba, Synthesis, 2018, 50, 3513 CrossRef CAS; (b) K. R. Reddy, A. S. Reddy, R. Shankar, R. Kant and P. Das, Asian J. Org. Chem., 2015, 4, 573 CrossRef CAS.
- S. Samanta, S. Moldal, S. Santra, G. Kibriya and A. Hajra, J. Org. Chem., 2016, 81, 10088 CrossRef CAS PubMed.
- Y.-S. Tung, et al., J. Med. Chem., 2011, 54, 3076 CrossRef CAS PubMed.
- (a) T. Q. Hung, N. M. Quan, H. V. Dong, T. D. Nguyen, H. L. T. Anh, T. Q. Hung, N. V. Tuyen, N. T. Thuan, T. T. Dang and P. Langer, Synlett, 2019, 30, 303 CrossRef CAS; (b) B. V. Phuc, Q. H. Dinh, N. L. Chi, Q. T. Nguyen, T. T. N. Truong, N. V. Tuyen, H. Nguyen, P. Langer, T. T. Dang and T. Q. Hung, Tetrahedron, 2023, 136, 133360 CrossRef; (c) D. V. Do, H. N. Do, N. K. Nguyen, T. A. Le, T. M. Le, B. V. Phuc, T. S. Le, Q. A. Ngo, H. Nguyen, T. Q. Hung and T. T. Dang, Tetrahedron Lett., 2023, 122, 154504 CrossRef; (d) T. N. Ngo, P. Ehlers, T. T. Dang and P. Langer, Org. Biomol. Chem., 2015, 13, 3321 RSC; (e) T. Q. Hung, T. T. Dang, J. Janke, A. Villinger and P. Langer, Org. Biomol. Chem., 2015, 13, 1375 RSC; (f) T. N. Ngoc, O. A. Akrawi, T. T. Dang, A. Villinger and P. Langer, Tetrahedron Lett., 2015, 56, 86 CrossRef; (g) T. Q. Hung, A. Villinger, T. T. Dang and P. Langer, Org. Biomol. Chem., 2015, 13, 583 RSC; (h) N. N. Pham, T. T. Dang, T. N. Ngo, P. Ehlers and P. Langer, Org. Biomol. Chem., 2015, 13, 6047 RSC; (i) T. Q. Hung, N. N. Thang, D. H. Hoang, T. T. Dang, A. Villinger and P. Langer, Org. Biomol. Chem., 2014, 12, 2596 RSC; (j) T. Q. Hung, N. N. Thang, D. H. Hoang, T. T. Dang, A. Villinger, K. Ayub, S. Lochbrunner, G. Flechsig and P. Langer, Eur. J. Org Chem., 2015, 1007 CrossRef CAS; (k) H. N. Do, N. M. Quan, B. V. Phuc, D. V. Tinh, N. Q. Tien, T. T. T. Nga, T. Q. Hung, T. T. Dang and P. Langer, Synlett, 2021, 32, 611 CrossRef CAS; (l) B. V. Phuc, N. T. Nguyen, N. T. H. Van, T. L. Nguyen, V. H. Nguyen, C. M. Tran, H. Nguyen, M. T. Nguyen, T. Q. Hung and T. T. Dang, Chem. Commun., 2023, 59, 1947 RSC; (m) T. Q. Hung, B. C. Q. Nguyen, B. V. Phuc, T. D. Dang-Van, C. M. Trang, Q. T. K. Anh, D. V. Do, H. Nguyen, Q. A. Ngo and T. T. Dang, Org. Biomol. Chem., 2023, 21, 8813 RSC.
- K. Pericherla, A. Jha, B. Khungar and A. Kumar, Org. Lett., 2013, 15, 4304 CrossRef CAS PubMed.
- Y. Zhang, Y. Cheng, H. Cai, S. He, Q. Shan, H. Zhao, Y. Chen and B. Wang, Green Chem., 2017, 19, 5708 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra05198j |
| ‡ All authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2024 |