
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of novel piperazine-based bis(thiazole)(1,3,4-thiadiazole) hybrids as anti-cancer agents through caspase-dependent apoptosis†
Doaa M. Mohameda,
Nabila A. Khedera,
Marwa Sharakyb,
Mohamed S. Nafie cd,
Kamal M. Dawood*a and
Ashraf A. Abbas*a
cd,
Kamal M. Dawood*a and
Ashraf A. Abbas*a
aDepartment of Chemistry, Faculty of Science, Cairo University, Giza 12613, Egypt. E-mail: kmdawood@sci.cu.edu.eg; ashrafabbas@cu.edu.eg; Fax: +202 35727556; Tel: +202 35676602
bPharmacology Unit, Cancer Biology Department, National Cancer Institute, Cairo University, Cairo, Egypt
cDepartment of Chemistry, College of Sciences, University of Sharjah, P. O. 27272, Sharjah, United Arab Emirates
dDepartment of Chemistry, Faculty of Science, Suez Canal University, Ismailia, 41522, Egypt
First published on 9th August 2024
Abstract
A series of novel piperazine-based bis(thiazoles) 13a–d were synthesized in moderate to good yields via reaction of the bis(thiosemicarbazones) 7a, b with an assortment of C-acetyl-N-aryl-hydrazonoyl chlorides 8a–f. Similar treatment of the bis(thiosemicarbazone) 7a, b with C-aryl-N-phenylhydrazonoyl chlorides 10a, b afforded the expected bis(thiadiazole) based piperazine products 13b–d in reasonable yields. Cyclization of 7a, b with two equivalents of α-haloketones 14a–d led to the production of the corresponding bis(4-arylthiazol)piperazine derivatives 15a–h in good yields. The structures of the synthesized compounds were confirmed from elemental and spectral data (FTIR, MALDI-TOF, 1H, and 13C NMR). The cytotoxicity of the new compounds was screened against hepatoblastoma (HepG2), human colorectal carcinoma (HCT 116), breast cancer (MCF-7), and Human Dermal Fibroblasts (HDF). Interestingly, all compounds showed promising cytotoxicity against most of the cell lines. Interestingly, compounds 7b, 9a, and 9i exhibited IC50 values of 3.5, 12.1, and 1.2 nM, respectively, causing inhibition of 89.7%, 83.7%, and 97.5%, compared to Erlotinib (IC50 = 1.3 nM, 97.8% inhibition). Compound 9i dramatically induced apoptotic cell death by 4.16-fold and necrosis cell death by 4.79-fold. Compound 9i upregulated the apoptosis-related genes and downregulated the Bcl-2 as an anti-apoptotic gene. Accordingly, the most promising EGFR-targeted chemotherapeutic agent to treat colon cancer was found to be compound 9i.
1. Introduction
Piperazine has diverse synthetic potential due to the participation of its N1 and N4 atoms in the construction of a huge number of bis-functionalized organic molecules with broad pharmacological applications. Based on statistical analysis, piperazine is the most abundant N-heterocycle after piperidine and pyridine used in developing pharmaceutical small molecule drugs and biomedical materials.1–6 Piperazine derivatives have emerged as privileged pharmacophores with several therapeutic versatilities.7–11 In addition, some piperazine-based commercial drugs were approved for cancer treatment, such as Olaparib, Abemaciclib, Palbociclib (breast cancer drugs), Rociletinib (lung cancer drug), and Imatinib (acute lymphoblastic leukemia drug)12,13 (Fig. 1), and some other approved commercial drugs also employed piperazine moieties such as lorpiprazole (anxiolytic drug), dapiprazole (ophthalmic solutions)14,15 and piperaquine (antiparasitic drug)16 (Fig. 1). The piperazine motif has also been found to be an essential part of numerous bioactive natural products.17,18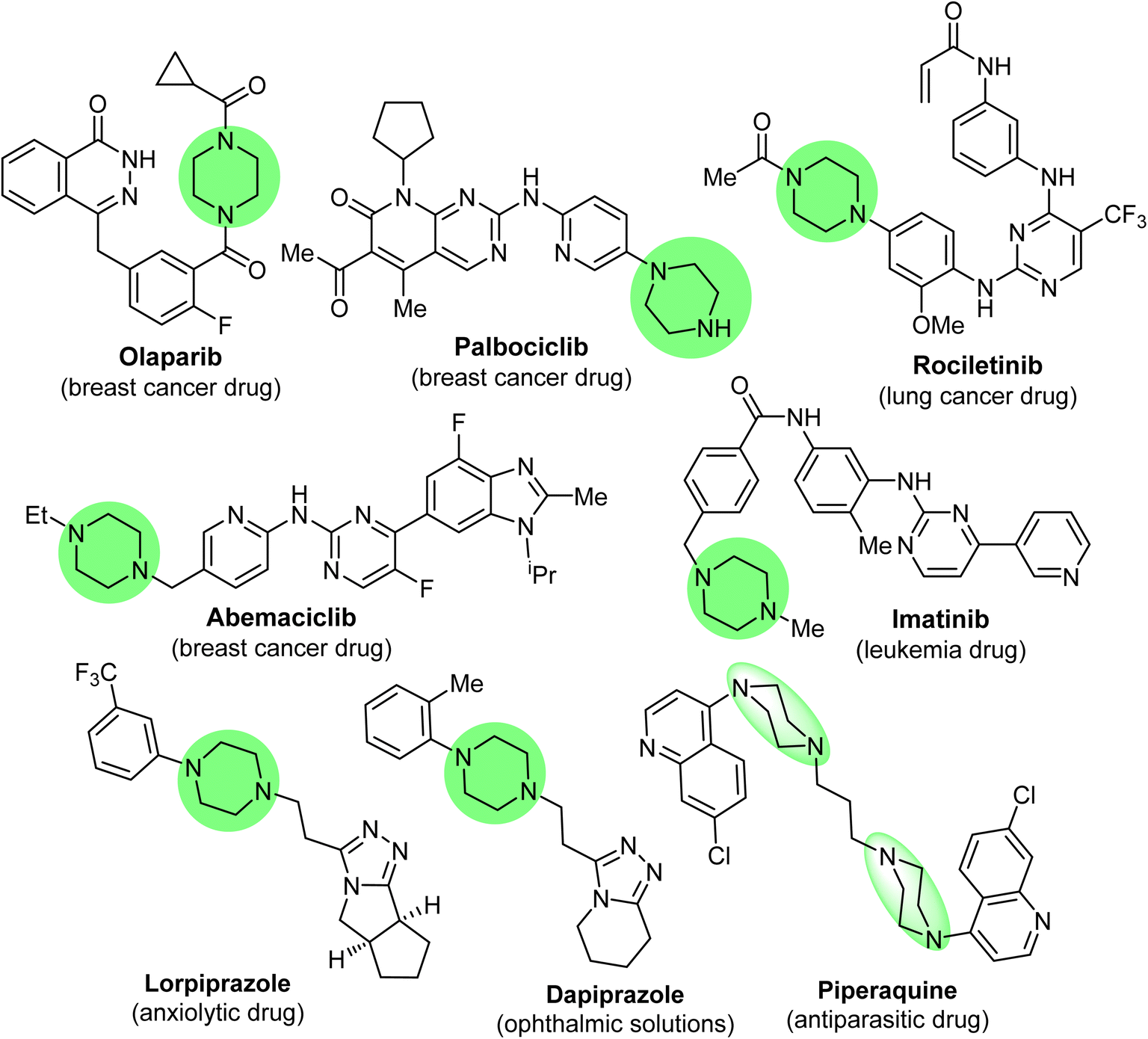 | ||
| Fig. 1 Examples of some piperazine-based marketed drugs. | ||
Furthermore, several scientific reports discussed the biological potencies of thiazole-based heterocyclic compounds.19–23 It was reported that the 1,3-thiazole nucleus had been an integral part of tiazofurin,24 dasatinib,25 and dabrafenib26 (Fig. 2), clinically useful anti-cancer drugs. Furthermore, 1,3,4-thiadiazole derivatives have outstanding pharmacological applications due to their broad spectrum of inhibitory activities, particularly 1,3,4-thiadiazoles, which are of immense significance as potent anti-cancer agents.27–33 Some marketed anti-cancer FDA-approved drugs were also found to employ the 1,3,4-thiadiazole moiety, such as azeteta, litronesib, and filanesib drugs (Fig. 2).
 | ||
| Fig. 2 Marketed anti-cancer drugs employing thiazole and 1,3,4-thiadiazole motifs. | ||
In addition, organic compounds having bis-carboxamide functionality were found to be components of remarkable biologically active natural products.34–37 Synthetic bis-carboxamide-based compounds exhibited wide-array inhibitory effects,38–42 including anti-cancer potencies.43–45 Bis-carboxamide derivatives were employed as essential constituents of the two marketed drugs, lacosamide and batimastat, and the two anti-cancer agents bis-carboxamides46 I and II (Fig. 3).
 | ||
| Fig. 3 Marketed bis-carboxamide anti-cancer medication. | ||
Recently, our research target has been aimed at building up a wide array of simple- and bis-organic molecules incorporating various heterocyclic nuclei and different functionalities with promising anti-cancer inhibitory activities against numerous human cell lines,47–57 the reported anti-cancer potencies of compounds employing piperazine, thiazole and/or 1,3,4-thiadiazole, and carboxamide pharmacophores inspired us to synthesize a new library of novel piperazine-based bis-thiazoles and/or thiadiazole hybrids (Fig. 4) having bis-amide linkers. Building on the bis(2-chloroacetamide) derivative 3, the target compounds were tested for their inhibitory effectiveness as anti-cancer medicines against various cancer cell lines. Additionally, the effective molecular target and apoptotic cell death were investigated.
 | ||
| Fig. 4 Rationale design for molecular hybridization of our targets toward new anti-cancer agents. | ||
2. Results and discussion
2.1. Chemistry
At first, 1,4-bis(chloroacetyl)piperazine 3 was selected as a key starting material to achieve our goals, as shown in Scheme 1. Thus, reaction of 2 equivalents of chloroacetyl chloride (1) with piperazine (2) in chloroform at 0 °C afforded compound 3 in a good yield. The reaction of the bis(chloroacetyl) compound 3 with the potassium salts of ortho- and para-hydoxybenzaldehydes 4a, b [isolated from treatment of o- and p-hydoxybenzaldehydes with methanolic KOH solution] in DMF under boiling conditions for 10 min afforded the corresponding 1,4-bis(formylphenoxyacetamido)piperazines 5a, b in ∼60% yields. In anticipation of the low yield of the previous reaction, the same reaction was repeated using sodium ethoxide as a basic medium instead of KOH/DMF, where the bis-aldehydes 5a, b were isolated in 71–73% yields.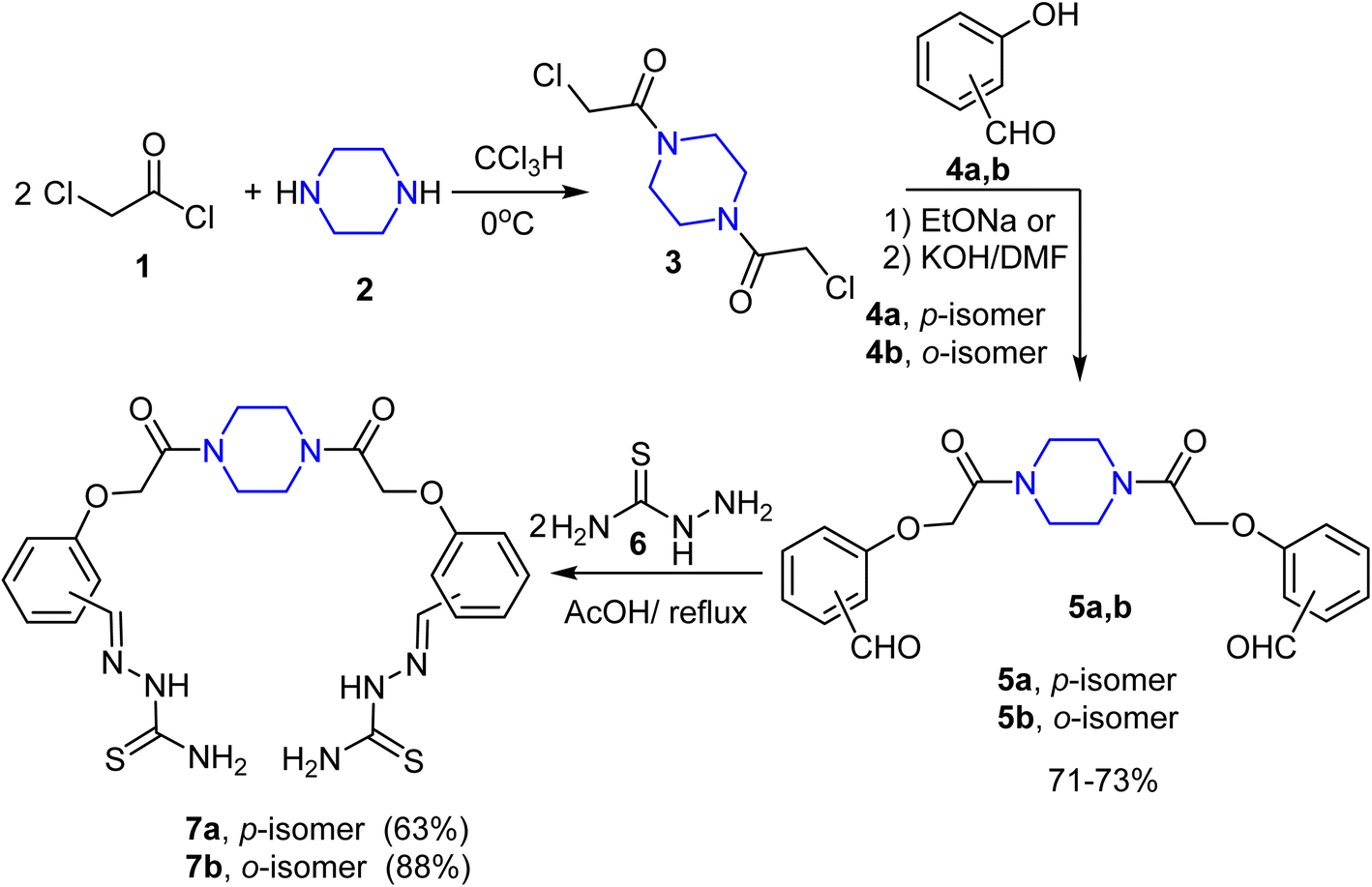 | ||
| Scheme 1 Synthetic route to the bis-thiosemicarbazones 7a, b. | ||
The bis-aldehydes 5a, b were then used to construct the novel key starting materials (piperazine-1,4-diyl)bis(hydrazine-1-carbothioamides) 7a, b as depicted in Scheme 1. Therefore, reaction of the bis-aldehydes 5a, b with two equivalents of thiosemicarbazide (6) in acetic acid at reflux temperature furnished the novel bis-thiosemicarbazones 7a and 7b in 63% and 88% yields, respectively. The structure of the obtained products 7a, b were fully confirmed by both elemental and spectral (IR, 1H NMR, and MALDI-TOF) analyses. The IR spectrum of the thiosemicarbazone 7a showed sharp peaks at v 3270, 3439, and 1652 cm−1 for NH, NH2, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively. 1H NMR spectrum of 7a showed characteristic signals for methylene protons of piperazine moiety at δ 3.48–3.54, and a sharp singlet at δ 4.93 assignable to two OCH2 protons, two doublets at δ 6.96 and 7.72 for the para-phenylene protons and two singlet signals at δ 7.99 and 11.30 assigned to CHN, and NH protons, and finally two singlets at δ 7.89 and 8.09 due to the protons of NH2. This is due to the partial double bond character induced by the resonance of the thioamide group as outlined in Fig. 5, which made the germinal protons of amino function electronically non-equivalent. This result is similar to related published studies characteristic of thiosemicarbazone derivatives.58,59
O, respectively. 1H NMR spectrum of 7a showed characteristic signals for methylene protons of piperazine moiety at δ 3.48–3.54, and a sharp singlet at δ 4.93 assignable to two OCH2 protons, two doublets at δ 6.96 and 7.72 for the para-phenylene protons and two singlet signals at δ 7.99 and 11.30 assigned to CHN, and NH protons, and finally two singlets at δ 7.89 and 8.09 due to the protons of NH2. This is due to the partial double bond character induced by the resonance of the thioamide group as outlined in Fig. 5, which made the germinal protons of amino function electronically non-equivalent. This result is similar to related published studies characteristic of thiosemicarbazone derivatives.58,59
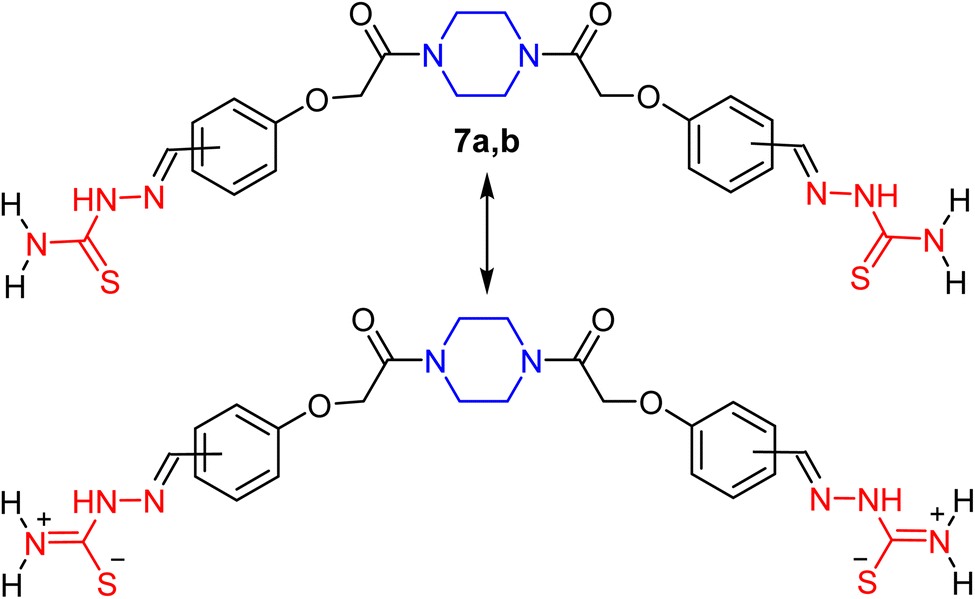 | ||
| Fig. 5 The tautomeric form of compounds 7a, b with restricted rotation of thioamide group. | ||
The formation of the novel bis(thiosemicarbazones) 7a, b in good yields encouraged us to continue our ongoing interest in the synthesis of some potent biologically active bis(heterocycles). Thus, treatment of the bis-thiosemicarbazone 7a with 2-oxo-N-phenylpropanehydrazonoyl chloride (8a)60 in DMF/ethanol (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), at reflux temperature, containing two equivalents of triethylamine as a base, afforded the corresponding bis-thiazole derivative 9a as a single product in 36% yield. The structure of 9a was fully elucidated by both spectral (IR, 1HNMR, and 13CNMR) as well as elemental analyses and mass spectrometry (cf. Experimental section). The IR spectrum of the newly synthesized compound 9a showed two peaks characteristic for NH and CO at ν 3415 and 1661 cm−1, respectively, and its 1H NMR spectrum showed the expected features with the appropriate signals, where it revealed three signals at δ 2.59, 3.51–3.57 and 4.99 for the aliphatic protons; two thiazole-methyl groups, four NCH2 and two OCH2 groups, respectively, and two singlet signals at δ 8.61 and 10.54 for CHN, and NH, respectively, in addition to a multiplet in the region δ 7.09–7.83 for the aromatic protons. The 13C NMR spectrum of 9a showed a total of 17 peaks identified as four peaks at δ 16.43, 40.99, 41.22, 43.80, 44.05 and 65.73 due to aliphatic carbons,61 and ten peaks at δ 114.12, 115.14, 122.09, 126.68, 129.21, 129.91, 137.87, 143.35, 159.55, 160.81 for the aromatic carbons, besides three peaks at δ 165.64, 171.96, 177.95 for 2CN and CO carbons which are compatible with the suggested structure. The positive ion mode MALDI-TOF mass spectrum of 9a exhibited a molecular ion peak of the form [M + H]+ at m/z = 841.09.
:1), at reflux temperature, containing two equivalents of triethylamine as a base, afforded the corresponding bis-thiazole derivative 9a as a single product in 36% yield. The structure of 9a was fully elucidated by both spectral (IR, 1HNMR, and 13CNMR) as well as elemental analyses and mass spectrometry (cf. Experimental section). The IR spectrum of the newly synthesized compound 9a showed two peaks characteristic for NH and CO at ν 3415 and 1661 cm−1, respectively, and its 1H NMR spectrum showed the expected features with the appropriate signals, where it revealed three signals at δ 2.59, 3.51–3.57 and 4.99 for the aliphatic protons; two thiazole-methyl groups, four NCH2 and two OCH2 groups, respectively, and two singlet signals at δ 8.61 and 10.54 for CHN, and NH, respectively, in addition to a multiplet in the region δ 7.09–7.83 for the aromatic protons. The 13C NMR spectrum of 9a showed a total of 17 peaks identified as four peaks at δ 16.43, 40.99, 41.22, 43.80, 44.05 and 65.73 due to aliphatic carbons,61 and ten peaks at δ 114.12, 115.14, 122.09, 126.68, 129.21, 129.91, 137.87, 143.35, 159.55, 160.81 for the aromatic carbons, besides three peaks at δ 165.64, 171.96, 177.95 for 2CN and CO carbons which are compatible with the suggested structure. The positive ion mode MALDI-TOF mass spectrum of 9a exhibited a molecular ion peak of the form [M + H]+ at m/z = 841.09.
Now, the scope of the last reaction was extended to the bis-thiosemicarbazone 5b to generate the corresponding bis-thiazole derivatives. Therefore, under typical reaction conditions, the bis-thiosemicarbazones 7a, b reacted with various hydrazonoyl chlorides 8a–f (ref. 60, 62 and 63) to afford the corresponding piperazine-based bis(thiazoles) 9b–j in 33–65% yields. The structures of 9b–j were enlightened by both elemental and spectral (IR, MALDI-TOF, 1H NMR, and 13C NMR) analyses (cf. Experimental section) (Scheme 2).
 | ||
| Scheme 2 Synthetic route to piperazine-based bis(thiazole) derivatives 9a–j. | ||
The next target was aimed to explore the synthetic potential of the piperazine-based bis(thiosemicarbazones) 7a, b in the construction of the bis(1,3,4-thiadiazole) derivatives 13a–d. Thus, reaction of the reactive substrate 7a with C,N-diphenylhydrazonoyl chloride (10a) (ref. 64) in DMF/ethanol mixture containing triethylamine at reflux temperature gave the corresponding bis(1,3,4-thiadiazole) 13a in 63% yield, as shown in Scheme 3. The structure of compound 13a was confirmed using spectral data. The absorption band of the imine (CN) group appeared in the IR spectrum of compound 13a at ν 1604 cm−1, and the CO starching band appeared also at ν 1658 cm−1. The 1H NMR spectrum of 13a showed the existence of multiple signals for the protons of four CH2N of piperazine moiety at δ 3.50–3.55 as well as a singlet signal for the protons of two OCH2 linkers at δ 4.95, and a singlet signal at δ 8.43 due to the CHN function. Furthermore, multiple signals corresponding to the aromatic protons appeared at δ 7.03–8.10. Finally, the mass spectrum of compound 13a revealed a correct molecular ion peak for [M + H+] at m/z = 912.32. A similar reaction of the bis(thiosemicarbazone) 7a with the hydrazonoyl chloride 10b (ref. 65) gave the corresponding bis(1,3,4-thiadiazole) 13b in a moderate yield. Under the same reaction conditions, heteroannulation of the ortho-isomeric bis(thiosemicarbazone) 7b with N-arylbenzohydrazonoyl chlorides 10a, b gave the anticipated novel piperazine-based bis(1,3,4-thiadiazoles) 13c, d in reasonable yields as depicted in Scheme 3. Formation of the target compounds 13a–d took place via the proposed mechanistic pathway displayed in Scheme 3. Due to the basic condition, the sequential removal of two molecules of hydrochloric acid gave the intermediate 11, which underwent an intramolecular cyclization to give 12. Extrusion of two ammonia molecules from the intermediate 12 gave the final product 13.
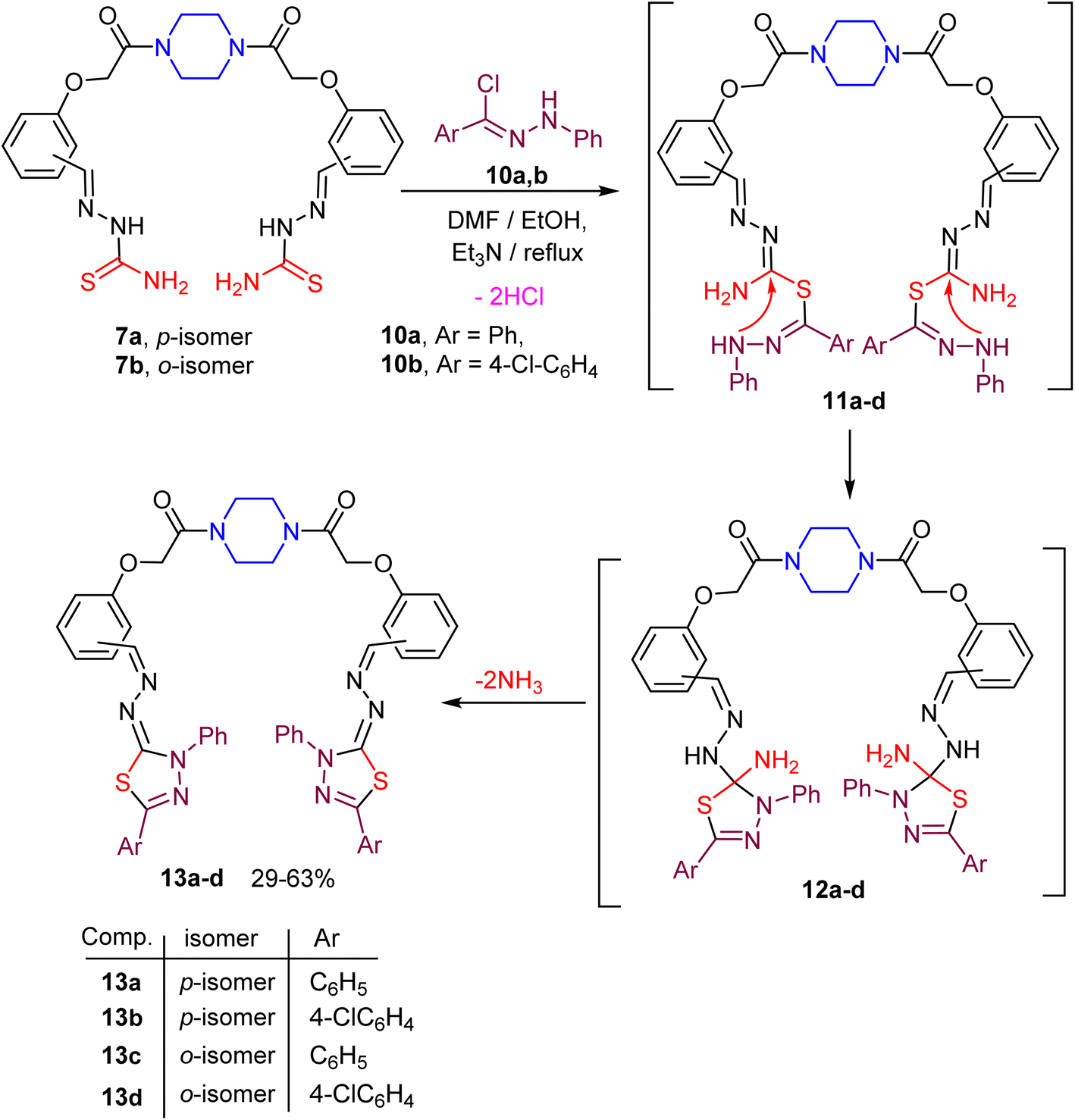 | ||
| Scheme 3 Synthetic route to piperazine-based bis(1,3,4-thiadiazole) hybrids 13a–d. | ||
Finally, the scope of our approach to synthesize a new series of functionalized piperazine-based bis(thiazol-2-ylidene) derivatives 15a–h is achieved as shown in Scheme 4. Therefore, under the above reaction conditions, treatment of the bis-thiosemicarbazone 7a with phenacyl bromide (14a)66 in a 1:2 molar ratio led to the production of the corresponding 1,1′-(piperazine-1,4-diyl)bis(2-(4-phenylthiazol-2(3H)ylidene-hydrazineylidene)methyl)phenoxy-ethan-1-one (15a) as a single product in 48% yield. The 1H-NMR spectrum of compound 15a exhibited multiplet peaks in the region δ 3.49–3.56 corresponding to the piperazine protons, and a singlet signal at δ 4.92 for the OCH2 protons, in addition to three singlet peaks at δ 7.3, 7.86, and 11.99 assigned for the protons of thiazole-5-CH, imine (CHN), and NH, respectively. The 13C NMR spectrum displayed five aliphatic carbon atoms at δ 41.18, 41.40, 44.03, 44.53, and 66.03 assigned to the CH2's of piperidine moiety and OCH2 linker, ten aromatic carbon atoms at δ 94.41, 103.49, 115.18, 125.61, 127.53, 127.75, 128.71, 134.83, 141.25, and 150.60 in addition to three signals at δ 159.12, 166.02 and 168.45 assigned to two CN and CO carbons.
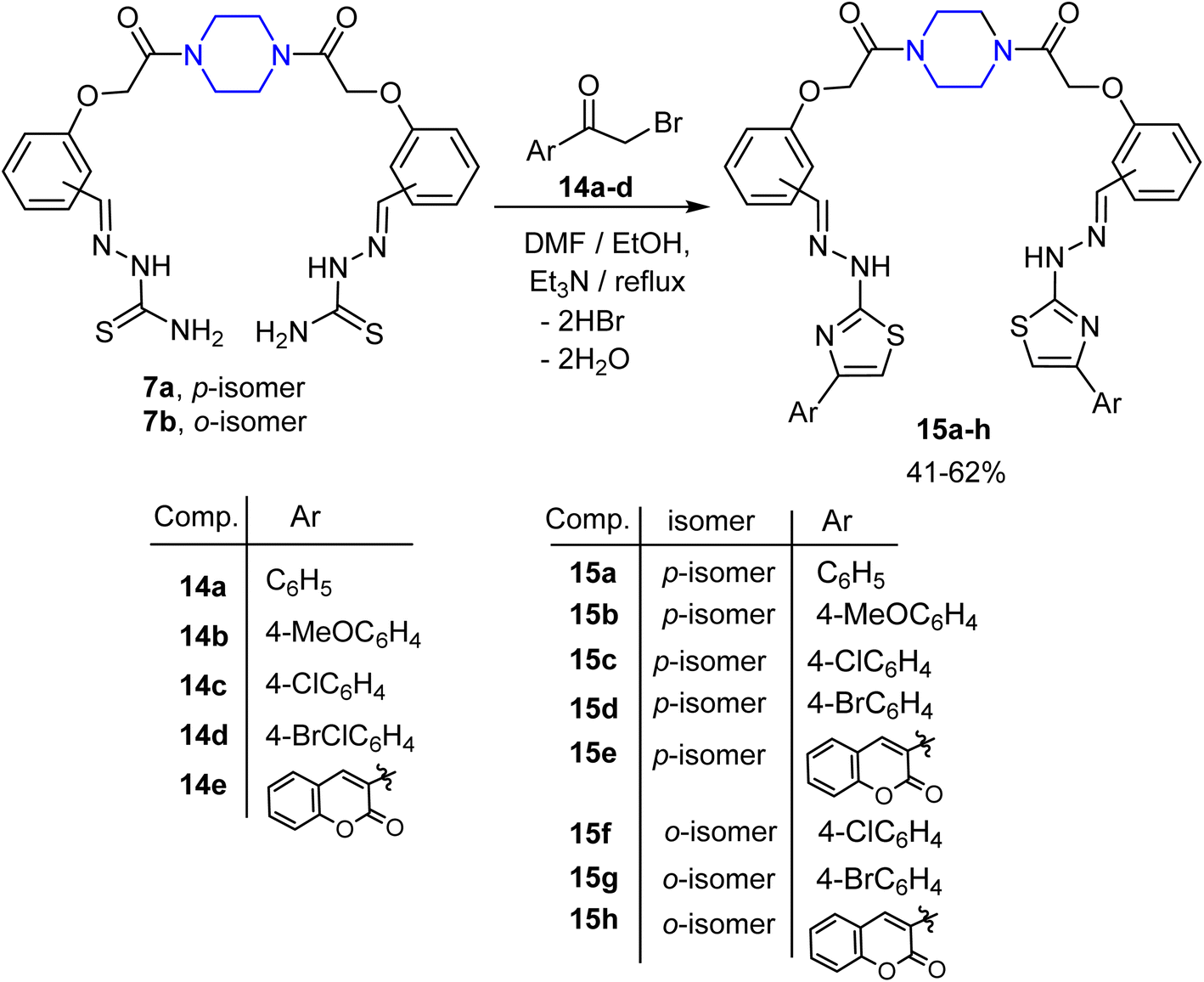 | ||
| Scheme 4 Synthetic route to the piperazine-based bis(thiazol-2-ylidene) derivatives 15a–h. | ||
The above reaction was generalized by applying a typical procedure for the reaction of bis-thiosemicarbazones 7a, b with different reactive synthons of the α-bromoketones 14b–e (ref. 67 and 68). The reaction proceeded straightforwardly and produced the corresponding piperazine-based bis((4-arylthiazol-2-ylidene)hydrazono) derivatives 15b–h in moderate to good yields, as shown in Scheme 4. Spectroscopic data and elemental analyses were used to establish the constitution of the targeted compounds 15a–h (see experimental data).
2.2. Biological evaluation
The effect of the addition of different concentrations of the newly synthesized compounds on HCT-116, HEPG2, MCF7, and HDF cell lines was examined. All the synthesized compounds were used in single-dose 100 μM for 48 h on HCT116, HEPG2, MCF7, and HDF cell lines as described in Table 1. Interestingly, compounds 7b, 9a, 9b, 9e, 9i, and 13b showed potent percentage of cell growth inhibition on the cancer cell lines exceeding 80% with low percentage against normal cells, and hence, they were employed in measuring their IC50 values, where the extent of cytotoxicity of the selected compounds on cancer cell lines was significantly prominent. This resulted in a marked inhibition in the cellular proliferation of cancer cell lines in 100 μM concentration, as presented in Table 1 and Fig. 6. Compound 9i exhibited the most potent inhibitory activity against HCT116, HEPG2, and MCF7 cancer cells with IC50 values of 8.51 ± 2.5, 22.02 ± 2.9, and 13.01 ± 2.8 μM, respectively. Additionally, compound 9a exhibited potent cytotoxicity against HCT116, HEPG2 and MCF7 with IC50 values of 9.98 ± 2, 23.78 ± 4 and 13.67 ± 2.3 μM, respectively (Table 2).
| Comp. | Percentage of cell growth inhibition at [100 μM] | |||
|---|---|---|---|---|
| HCT116 | HEPG2 | MCF7 | HDF | |
| 7a | 86.23 ± 5.33 | 83.30 ± 7.47 | 85.29 ± 5.20 | 45.09 ± 30.14 |
| 7b | 90.07 ± 3.41 | 89.13 ± 2.22 | 88.90 ± 5.46 | 66.45 ± 11.48 |
| 9a | 81.00 ± 3.91 | 74.72 ± 4.49 | 85.88 ± 5.94 | 18.16 ± 11.54 |
| 9b | 85.16 ± 2.11 | 56.50 ± 4.93 | 89.48 ± 2.34 | 27.57 ± 25.06 |
| 9c | 87.20 ± 4.53 | 77.19 ± 3.10 | 90.27 ± 1.30 | 67.68 ± 11.53 |
| 9d | 89.11 ± 2.04 | 73.11 ± 5.28 | 90.45 ± 1.99 | 57.72 ± 17.99 |
| 9e | 86.85 ± 2.09 | 77.52 ± 7.73 | 84.60 ± 2.019 | 18.59 ± 32.06 |
| 9f | 85.36 ± 6.41 | 93.45 ± 2.072 | 85.53 ± 2.11 | 78.32 ± 3.64 |
| 9g | 80.64 ± 7.04 | 91.80 ± 3.29 | 88.68 ± 4.11 | 71.06 ± 21.58 |
| 9h | 77.71 ± 8.59 | 83.97 ± 3.07 | 81.05 ± 7.44 | 55.95 ± 23.63 |
| 9i | 89.61 ± 5.30 | 90.95 ± 3.56 | 89.87 ± 2.30 | 71.60 ± 17.26 |
| 9j | 87.68 ± 4.30 | 83.09 ± 7.22 | 71.40 ± 20.50 | 12.01 ± 6.04 |
| 13a | 78.07 ± 5.78 | 71.42 ± 4.92 | 88.25 ± 1.88 | 52.49 ± 14.75 |
| 13b | 89.34 ± 2.83 | 85.14 ± 3.16 | 90.81 ± 1.39 | 79.15 ± 3.71 |
| 13c | 76.41 ± 5.38 | 68.83 ± 12.69 | 86.16 ± 1.83 | 60.79 ± 3.0939 |
| 13d | 88.87 ± 1.38 | 80.02 ± 3.39 | 86.37 ± 2.56 | 64.02 ± 10.80 |
| 15a | 78.09 ± 6.50 | 67.93 ± 7.03 | 87.26 ± 1.63 | 50.19 ± 21.51 |
| 15b | 21.75 ± 16.49 | 83.39 ± 9.48 | 50.64 ± 10.22 | 48.00 ± 48.46 |
| 15c | 53.11 ± 19.55 | 42.58 ± 11.48 | 82.42 ± 1.15 | 40.60 ± 13.65 |
| 15d | 74.82 ± 0.38 | 62.26 ± 18.32 | 89.20 ± 1.59 | 58.44 ± 25.12 |
| 15e | 73.88 ± 4.08 | 74.44 ± 4.88 | 68.37 ± 7.33 | 68.13 ± 10.09 |
| 15f | 63.59 ± 5.34 | 71.19 ± 3.61 | 75.98 ± 10.50 | 66.86 ± 12.10 |
| 15g | 61.76 ± 8.01 | 74.828 ± 2.18 | 71.29 ± 11.94 | 54.76 ± 21.19 |
| 15h | 36.44 ± 13.81 | 70.12 ± 8.68 | 46.72 ± 11.33 | 44.00 ± 51.49 |
| DOX | 82.77 ± 2.98 | 89.09 ± 0.63 | 86.37 ± 1.82 | 30.53 ± 9.75 |
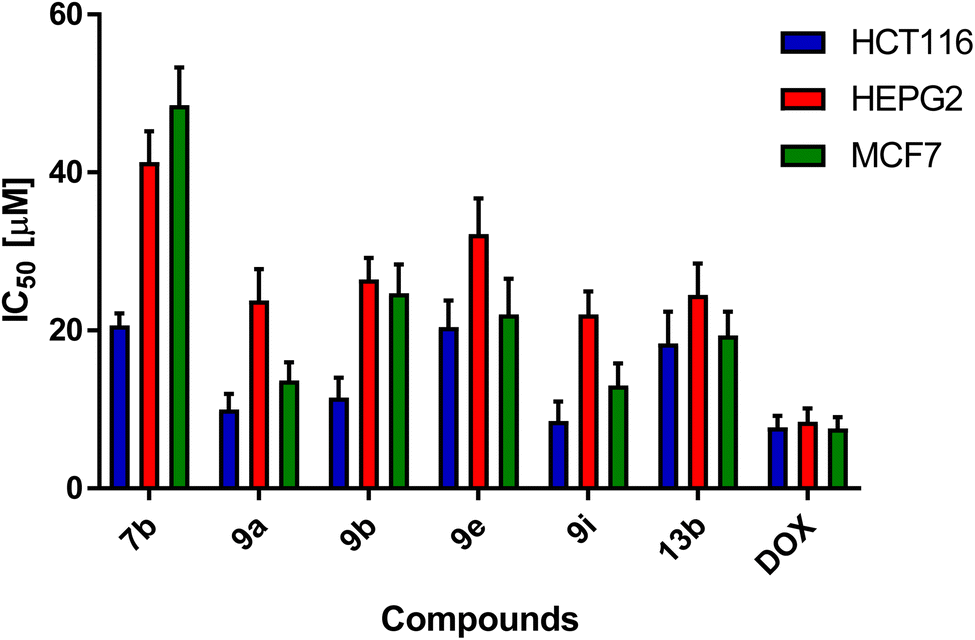 | ||
| Fig. 6 Cytotoxicity of selected compounds on HCT116, HEPG2, and MCF7 cell lines after 48 h treatment by sulphorhodamine assay. Graphs and data analysis were performed using GraphPad InStat, version 8. | ||
| Comp. | IC50 (μM) ± SD | ||
|---|---|---|---|
| HCT116 | HEPG2 | MCF7 | |
| 7b | 20.66 ± 1.5 | 41.3 ± 3.9 | 48.5 ± 4.8 |
| 9a | 9.98 ± 2 | 23.78 ± 4 | 13.67 ± 2.3 |
| 9b | 11.5 ± 2.5 | 26.46 ± 2.7 | 24.73 ± 3.6 |
| 9e | 20.40 ± 3.4 | 32.2 ± 4.5 | 22.01 ± 4.5 |
| 9i | 8.51 ± 2.5 | 22.02 ± 2.9 | 13.01 ± 2.8 |
| 13b | 18.36 ± 4 | 24.49 ± 4 | 19.38 ± 3 |
| DOX | 7.7 ± 1.5 | 8.42 ± 1.7 | 7.59 ± 1.4 |
| Compound | % of EGFR inhibition | IC50 ± SDa (nM) |
|---|---|---|
| a “Values are expressed as an average of three independent replicates”. “IC50 values were calculated using sigmoidal non-linear regression curve fit of percentage inhibition against five concentrations of each compound”. | ||
| 7b | 89.7 ± 2.8 | 3.5 ± 0.1 |
| 9a | 83.7 ± 1.4 | 12.1 ± 0.04 |
| 9b | 74.8 ± 1.6 | 54.6 ± 0.9 |
| 9i | 97.5 ± 2.1 | 1.2 ± 0.05 |
| Erlotinib | 97.8 ± 2.8 | 1.3 ± 0.01 |
2.2.3.1. Annexin V/PI staining with cell cycle analysis. Using flow cytometric examination of Annexin V/PI staining, the apoptotic cell death in both untreated and treated HCT-116 cells was investigated to assess the apoptotic activity of compound 9i (IC50 = 8.5 μM, 48 h). Fig. 7A showed that compound 9i significantly induced apoptotic cell death; it induced total apoptosis by 16.85% (16.31% for late apoptosis, 0.54% for early apoptosis) compared to the untreated control group (2.36% for late apoptosis, 1.69% for early apoptosis). Additionally, it induced necrosis cell death by 4.79-fold, and it induced necrosis by 35.74% compared to 7.45% in the untreated control.
 | ||
| Fig. 7 (A) Cryptographs of Annexin-V/propidium iodide staining of untreated and 9i-treated HCT-116 cells with (IC50 = 8.5 μM, 48 h). (B) Percentage of cell population at each cell cycle “G1, and G2” using DNA content-flow cytometry aided cell cycle analysis. Cell arrest happened at the P2 phase (arrow indicated). | ||
Next, DNA flow cytometry was used to estimate the cell population in each cell phase following treatment with cytotoxic agents. As seen in Fig. 7B, compound 9i treatment increased the cell population at the P2 phase by 51.89% compared to control 3.77%. Compound 9i caused cell death in HCT-116 cells, stopping their growth in the P2 phase which represent the apoptosis cell death.
2.2.3.2. RT-PCR. It was determined by comparing the RT-PCR levels of apoptosis-mediated genes P53, bax, caspase-3,8,9, and Bcl-2 in the untreated and treated HCT-116 cells, compound 9i induced apoptotic cell death in these cells. As seen in Fig. 8, compound 9i upregulated caspase-3,8,9 levels by 9.87, 7.6, and 11.6-fold change, PUMA by 8.5-fold change, and P53 by 10.87-fold change, while it downregulated the Bcl-2 expression by 0.76-fold change. The results showed that compound 9i administration triggered cell death by apoptosis through intrinsic and extrinsic pathways.
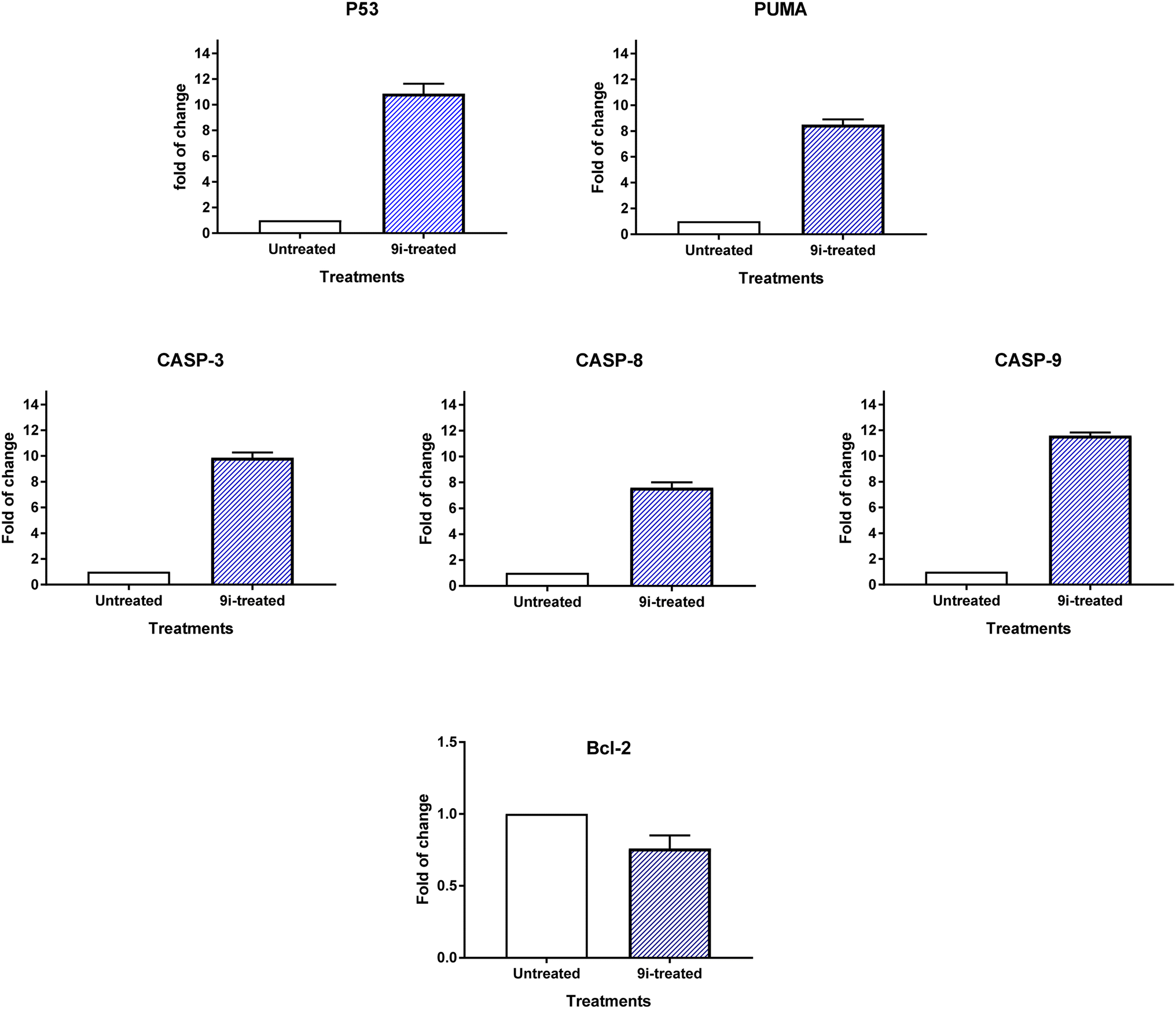 | ||
| Fig. 8 Quantitative RT-PCR results analysis of the apoptosis-related genes; P53, bax, caspases 3, 8, 9, and Bcl-2, respectively in HCT-116 cells treated with compound 9i with (IC50 = 8.5 μM, 48 h). The data illustrated is the average of 3 independent experimental runs (mean ± SD). Fold change of untreated control = 1. | ||
3. Experimental
3.1. Chemistry
3.1.1.1 Molecular weight determination using MALDI-TOF/MS. Matrix-assisted Laser Desorption Ionization Time-of-flight mass spectrometry (MALDI-TOF/MS) (Bruker, model ultraFlexXtreme) was used to determine the molecular weight of the new products. The measurements were examined in positive mode using a reflectron time-of-flight mass spectrometer. The instrument was calibrated with a peptide calibration standard. The sample was prepared as follows: the matrix DCTB dissolved in tetrahydrofuran (THF) (DCTB = trans-2-3-(4-tert-butylphenyl)-2-(methyl-2-propenylidene)malononitrile ≥99.0%) was purchased from Sigma. All samples were dissolved in THF. The samples were deposited on the target plate as follows: 2 μL of the sample was mixed with 2 μL of the matrix solution in a 1
:1 ratio using an Eppendorf Research Plus micropipette. Using a micropipette, a single drop of the solution mixture was placed on a specific labeled location in the target plate and then allowed to dry at room temperature.
3.1.2.1 Method A. Potassium hydroxide (1.14 g, 20 mmol) was dissolved in methanol (10 mL), then the resulting solution was mixed with p- or o-hydroxybenzaldehyde derivatives 4a, b (2.44 g, 20 mmol). The resulting mixtures were stirred at ambient temperature for 10 min. Then the solvent was evaporated under reduced pressure. The obtained potassium salts were treated with dry ether and involved in the next step as crude materials. Dissolving potassium salts (20 mmol) in dimethylformamide (15 mL), then adding the bis(dichloro) compound 3 (10 mmol) and leaving the reaction mixture to boil at reflux for 10 min, potassium chloride was extruded. After that, the reaction solvent was evaporated and the precipitated materials were collected and washed with water. The isolated solid products obtained were purified by recrystallization from the ethanol/acetic acid to produce the corresponding bis(aldehyde) derivatives 5a, b in about 60% yield.71
3.1.2.2 Method B. p- or o-hydroxybenzaldehydes 4a or 4b (2.44 g, 20 mmol) was added to sodium ethoxide solution [prepared from sodium (0.5 g, 20 mmol) in dry ethyl alcohol (30 mL)]. The product mixture was heated at reflux temperature for 10 min., then the bis(dichloro) derivative 3 (10 mmol) was added. The reaction mixture was heated at reflux condition for 4 h. The solvent was then evaporated under reduced pressure, and the remaining solid products were washed with cold water. The solid obtained was collected and purified by recrystallization from ethanol/acetic acid to give the bis(aldehyde) derivatives 5a, b in about 72% yield.
3.1.3.1 General procedure. The appropriate bis-aldehydes 5a or 5b (2.05 g, 5 mmol) were dissolved in acetic acid (20 mL), then thiosemicarbazide (6) (0.91 g, 10 mmol) was added and the produced mixture was refluxed for 1 h with stirring. The reaction contents were left to cool to ambient temperature and the produced solid material was collected by filtration, washed with ethanol, and dried. Purification of the products by recrystallization from DMF/EtOH (1
:1 v/v) gave the corresponding bis-thiosemicarbazone derivatives 7a, b.
3.1.3.2 1,1′-(Piperazine-1,4-diyl)bis(2-(4-(2-carbamothioylhydrazono)methylphenoxy)ethanone) (7a). Pale yellow crystals, 0.85 g (yield 63%). Mp 236–238 °C; IR (KBr) v 3270, 3439 (NH, NH2), 1652 (C
O), 1243 (CS) cm−1; MS: MALDI-TOF: calcd for [M + H]+ m/z = 557.66, found 557.33; 1H NMR (DMSO-d6) δ 3.48–3.54 (m, 8H, 4N–CH2), 4.93 (s, 4H, 2OCH2), 6.96 (d, J = 8.7 Hz, 4H, ArH's), 7.72 (d, J = 8.4 Hz, 4H, ArH's), 7.89, 8.09 (2 s, 4H, 2NH2), 7.99 (s, 2H, CHN), 11.30 (s, 2H, 2NH); Anal. calcd: for C24H28N8O4S2 (556.66): C, 51.78; H, 5.07; N, 20.13%. Found: C, 51.60; H, 4.78; N, 20.30%.
3.1.3.3 1,1′-(Piperazine-1,4-diyl)bis(2-(2-(2-carbamothioylhydrazono)methylphenoxy)ethanone) (7b). Pale yellow crystals, 1.2 g (yield 88%). Mp 230–232 °C; IR (KBr) v 3286, 3387 (NH, NH2), 1670 (C
O), 1253 (CS) cm−1; MS: MALDI-TOF: calcd for [M + H]+ m/z = 557.66, found 557.43; 1H NMR (DMSO-d6) δ 3.52–3.55 (m, 8H, 4 NCH2), 4.96 (s, 4H, 2OCH2), 6.94–7.0 (m, 4H, ArH's), 7.34 (t, J = 8.4 Hz, 2H, ArH's), 7.94 (s, 2H, 2 NH), 8.07–8.01 (m, 4H, 2 NH & 2 ArH's), 8.45 (s, 2H, 2CHN), 11.47 (s, 2H, 2 NH) ppm; 13C NMR (DMSO-d6) δ 41.06, 41.45, 44.08, 44.47, 66.31, 112.79, 120.99, 122.46, 126.33, 131.05, 138.04, 156.52, 165.91, 177.87 ppm; Anal. calcd: for C24H28N8O4S2 (556.66): C, 51.78; H, 5.07; N, 20.13%. Found: C, 51.84; H, 5.12; N, 20.02%.
3.1.4.1 General procedure. A mixture of the appropriate bis-thiosemicarbazones 7a, b (0.5 mmol, 0.265 g) and the appropriate N-aryl-hydrazonoyl chlorides 8a–f or 12a, b (1 mmol) in absolute ethanol/DMF (1
:3, 10 mL), was refluxed for 6–8 hours in the presence of triethylamine (0.1 mL). The reaction vessel was left to cool to ambient temperature and the isolated solid product was collected by filtration, washed with ethanol, and dried. The dried products were then purified by recrystallization from DMF/EtOH mixture to give the targeted bis-(1,3-thiazoles) 9a–j or bis-(1,3,4-thiadiazoles) 13a–d, respectively.
3.1.4.2 1,1′-(Piperazine-1,4-diyl)bis(2-(4-(((4-methyl-5-(phenylazo)thiazol-2-yl)hydrazono)methyl)-phenoxy)ethanone) (9a). Orange crystals, yield (0.11 g, 36%), mp 180–182 °C: IR (KBr) v 3415 (NH), 1661 (C
O) cm−1; MS: MALDI-TOF: calcd for [M + H]+ m/z = 841.98, found 841.09; 1H NMR (DMSO-d6) δ 2.59 (s, 6H, 2CH3), 3.51–3.57 (m, 8H, 4 NCH2), 4.99 (s, 4H, 2OCH2), 6.98 (t, J = 6.9 Hz, 4H, ArH's), 7.09 (d, J = 8.4 Hz, 4H, ArH's), 7.30–7.38 (m, 6H, ArH's), 7.83 (d, J = 8.4 Hz, 4H, ArH's), 8.61 (s, 2H, 2CHN), 10.54 (s, 2H, 2 NH); 13C NMR (DMSO-d6) δ 16.43, 40.99, 41.22, 43.80, 44.05, 65.73, 114.12, 115.14, 122.09, 126.68, 129.21, 129.91, 137.87, 143.35, 159.55, 160.81, 165.64, 171.96, 177.95. Anal. calcd: for C42H40N12O4S2 (840.98): C, 59.98; H, 4.79; N, 19.99%. Found: C, 59.75; H, 4.90; N, 20.03%.
3.1.4.3 1,1′-(Piperazine-1,4-diyl)bis(2-(4-(((4-methyl-5-(4-tolylazo)thiazol-2-yl)hydrazono)methyl)-phenoxy)ethanone) (9b). Orange crystals, yield (0.2 g, 65%), mp 192–194 °C: IR (KBr) v 3421 (NH), 1652 (C
O) cm−1; MS: MALDI-TOF: calcd for [M + H]+ m/z = 870.04, found 871.25; 1H NMR (DMSO-d6) δ 2.26 (s, 6H, 2CH3Ar), 2.57 (s, 6H, 2CH3), 3.51–3.57 (m, 8H, 4CH2N), 4.99 (s, 4H, 2OCH2), 7.08–7.15 (m, 8H, ArH's), 7.26 (d, J = 7.5 Hz, 4H, ArH's), 7.82 (d, J = 7.2 Hz, 4H, ArH's), 8.59 (s, 2H, 2CHN), 10.49 (s, 2H, 2 NH) ppm; 13C NMR (DMSO-d6) δ 16.57, 20.48, 41.10, 41.60, 43.89, 44.15, 65.85, 114.28, 115.28, 126.85, 129.59, 130.02, 131.30, 137.36, 141.17, 159.52, 160.91, 165.80, 172.19, 177.93 ppm. Anal. calcd: for C44H44N12O4S2 (869.04): C, 60.81; H, 5.10; N, 19.34%. Found: C, 60.75; H, 4.95; N, 19.23%.
3.1.4.4 1,1′-(Piperazine-1,4-diyl)bis(2-(4-(((4-methyl-5-(4-fluorophenylazo)thiazol-2-yl)hydrazono)-methyl)phenoxy)ethanone) (9c). Orange crystals, yield (0.16 g, 52%), mp 210–212 °C; IR (KBr) δ 3422 (NH), 1662 (C
O) cm−1; MS: MALDI-TOF: calcd for [M + K]+ m/z = 915.96, found 915.24; 1H NMR (DMSO-d6) δ 2.58 (s, 6H, 2CH3), 3.51–3.57 (m, 8H, 4 CH2 N), 4.99 (s, 4H, 2OCH2), 7.09 (d, J = 8.4 Hz, 4H, ArH's), 7.17 (t, J = 8.4, 4H, ArH's) 7.34–7.38 (m, 4H, ArH's), 7.82 (d, J = 8.4 Hz, 4H, ArH's), 8.61 (s, 2H, 2CHN), 10.55 (s, 2H, 2 NH) ppm; 13C NMR (DMSO-d6) δ 16.40, 40.21, 40.98, 43.54, 44.04, 65.76, 115.40, 115.51, 115.75, 116.05, 126.66, 129.92, 137.89, 139.95, 159.59, 160.82, 165.65, 171.86, 177.94 ppm. Anal. calcd: for C42H38F2N12O4S2 (876.96): C, 57.52; H, 4.37; N, 19.17%. Found: C, 57.65; H, 4.15; N, 19.25%.
3.1.4.5 1,1′-(Piperazine-1,4-diyl)bis(2-(4-(((4-methyl-5-(4-chlorophenylazo)thiazol-2-yl)hydrazono)-methyl)phenoxy)ethanone) (9d). Pale brown crystals, yield (0.21 g, 63%), mp 220–222 °C; IR (KBr) ν 1662 (C
O) cm−1; MS: MALDI-TOF: calcd for [M + H]+ m/z = 910.87, found 910.90; 1H NMR (DMSO-d6) δ 2.58 (s, 6H, 2CH3), 3.50–3.56 (m, 8H, 4CH2N), 4.99 (s, 4H, 2OCH2), 7.09 (d, J = 8.1 Hz, 4H, ArH's), 7.36 (s, 8H, ArH's), 7.81 (d, J = 8.7 Hz, 4H, ArH's), 8.61 (s, 2H, 2CHN), 10.61 (s, 2H, 2 NH). Anal. calcd: for C42H38Cl12N12O4S2 (909.87): C, 55.44; H, 4.21; N, 18.47%. Found: C, 55.62; H, 4.25; N, 18.22%.
3.1.4.6 1,1′-(Piperazine-1,4-diyl)bis(2-(4-(((4-methyl-5-(4-nitrophenylazo)thiazol-2-yl)hydrazono)methyl)-phenoxy)ethanone) (9e). Deep brown crystals, yield (0.15 g, 45%), mp <300 °C; IR (KBr) v 1644 (C
O), 1324, 1498 (NO2) cm−1; MS: MALDI-TOF: calcd for [M + H]+ m/z = 931.89, found 931.04; 1H NMR (DMSO-d6) δ 2.61 (s, 6H, 2CH3), 3.51–3.57 (m, 8H, 4CH2N), 4.50 (s, 4H, 2OCH2), 7.09 (d, J = 7.5 Hz, 4H, ArH's), 7.46 (d, J = 8.1 Hz, 4H, ArH's), 7.83 (d, J = 7.8 Hz, 4H, ArH's), 8.19 (d, J = 7.8 Hz, 4H, ArH's), 8.64 (s, 2H, 2CHN), 11.06 (s, 2H, 2 NH) ppm; 13C NMR (DMSO-d6) δ 16.65, 40.93, 41.43, 43.74, 44.24, 65.92, 113.82, 115.29, 125.78, 126.62, 130.27, 141.03, 142.31, 149.20, 160.78, 161.16, 165.79, 171.40, 178.53. Anal. calcd: for C42H38N14O8S2 (930.98): C, 54.19; H, 4.11; N, 21.06%. Found: C, 54.16; H, 4.26; N, 21.31%.
3.1.4.7 1,1′-(Piperazine-1,4-diyl)bis(2-(2-(((4-methyl-5-(4-tolylazo)thiazol-2-yl)hydrazono)methyl)-phenoxy)ethanone) (9f). Orange crystals, yield (0.1 g, 33%), mp 264–266 °C; IR (KBr) v 3219 (NH), 1629 (C
O), cm−1; MS: MALDI-TOF: calcd for [M + H]+ m/z = 870.04, found 870.37; 1H NMR (DMSO-d6) δ 2.26 (s, 6H, 2CH3Ar), 2.58 (s, 6H, 2CH3), 3.51–3.56 (m, 8H, 4CH2N), 5.06 (s, 4H, 2OCH2), 7.06–7.15 (m, 8H, ArH's), 7.26 (d, 4H, J = 8.1 Hz, 4H, ArH's), 7.44–7.65 (m, 2H, ArH's), 7.99 (d, J = 7.5 Hz, 2H, ArH's), 8.95 (s, 2H, 2CHN), 10.54 (s, 2H, 2 NH); 13C NMR (DMSO-d6) δ 16.65, 20.54, 40.99, 41.39, 43.85, 44.25, 66.23, 113.72, 114.42, 121.32, 122.18, 126.49, 129.91, 131.51, 133.23, 137.29, 141.18, 155.21, 157.79, 166.01, 173.19, 178.38 ppm. Anal. calcd: for C44H44N12O4S2 (869.04): C, 60.81; H, 5.10; N, 19.34%. Found: C, 60.62; H, 4.88; N, 19.54%.
3.1.4.8 1,1′-(Piperazine-1,4-diyl)bis(2-(2-(((4-methyl-5-(4-fluorophenylazo)thiazol-2-yl)hydrazono)-methyl)phenoxy)ethanone) (9g). Orange crystals, yield (0.15 g, 48%), mp 266–268 °C; IR (KBr) v 3213 (NH), 1648 (C
O) cm−1; MS: MALDI-TOF: calcd for [M + Na]+ m/z = 899.96, found 899.05; 1H NMR (DMSO-d6) δ 2.58 (s, 6H, 2CH3), 3.51–3.57 (m, 8H, 4 CH2N), 5.06 (s, 4H, 2OCH2), 7.06–7.19 (m, 8H, ArH's), 7.35 (s, 2H, ArH's), 7.46–4.48 (m, 2H, ArH's), 7.65–7.9 (m, 2H, ArH's), 7.98 (d, J = 7.5 Hz, 2H, ArH's), 8.96 (s, 2H, 2CHN), 10.59 (s, 2H, 2 NH); 13C NMR (DMSO-d6) δ 16.66, 41.19, 41.47, 43.92, 44.19, 66.26, 113.70, 115.81, 116.30, 121.35, 122.18, 126.56, 133.31, 137.96, 140.09, 155.46, 156.44, 157.83, 166.05, 173.07, 178.58 ppm. Anal. calcd: for C42H38F2N12O4S2 (876.96): C, 57.52; H, 4.37; N, 19.17%. Found: 57.38; H, 4.44; N, 19.03%.
3.1.4.9 1,1′-(Piperazine-1,4-diyl)bis(2-(2-(((4-methyl-5-(4-chlorophenylazo)thiazol-2-yl)hydrazono)-methyl)phenoxy)ethanone) (9h). Deep red crystals, yield (0.13 g, 41%), mp 280–282 °C; IR (KBr) v 3217 (NH), 1628 (C
O), cm−1; MS: MALDI-TOF: calcd for [M + H]+ m/z = 910.87, found 910.06; 909.01; 1H NMR (DMSO-d6) δ 2.58 (s, 6H, 2CH3), 3.50–3.56 (m, 8H, 4CH2N), 5.06 (s, 4H, 2OCH2), 7.06–7.14 (m, 3H, ArH's), 7.35 (s, 9H, ArH's), 7.46 (t, J = 7.2 Hz, 2H, ArH's), 7.98 (d, J = 7.8 Hz, 2H, ArH's), 8.96 (s, 2H, 2CHN), 10.64 (s, 2H, 2NH); 13C NMR (DMSO-d6) δ 16.52, 41.40, 41.67, 43.73, 44.05, 66.22, 113.59, 115.70, 121.12, 122.02, 125.74, 126.39, 129.13, 133.12, 138.61, 142.36, 155.44, 157.72, 165.82, 172.76, 178.40. Anal. calcd: for C42H38Cl12N12O4S2 (909.87): C, 55.44; H, 4.21; N, 18.47%. Found: C, 55.32; H, 4.08; N, 18.53%.
3.1.4.10 1,1′-(Piperazine-1,4-diyl)bis(2-(2-(((4-methyl-5-(4-bromophenylazo)thiazol-2-yl)hydrazono)-methyl)phenoxy)ethanone) (9i). Orange crystals, yield (0.13 g, 37%), mp 282–284 °C; IR (KBr) v 3212 (NH), 1629 (C
O) cm−1; MS: MALDI-TOF: calcd for [M + H]+ m/z = 999.77, found 999.04; 1H NMR (DMSO-d6) δ 2.59 (s, 6H, 2CH3), 3.50–3.56 (m, 8H, 4 CH2N), 5.06 (s, 4H, 2OCH2), 7.0–7.14 (m, 4H, ArH's), 7.28–7.31 (m, 4H, ArH's), 7.48 (brs, 6H, ArH's), 7.98 (d, J = 7.2 Hz, 2H, ArH's), 8.96 (s, 2H, 2CHN), 10.65 (s, 2H, 2NH) ppm; 13C NMR (DMSO-d6) δ 16.77, 41.19, 41.44, 43.95, 44.21, 66.40, 113.88, 116.36, 121.39, 122.17, 126.58, 132.25, 133.43, 138.92, 142.96, 155.73, 157.96, 166.04, 172.93, 178.74. Anal. calcd: for C42H38Br2N12O4S2 (998.77): C, 50.51; H, 3.84; N, 16.83%. Found: C, 50.62; H, 3.73; N, 16.92%.
3.1.4.11 1,1′-(Piperazine-1,4-diyl)bis(2-(2-(((4-methyl-5-(4-nitrophenylazo)thiazol-2-yl)hydrazono)-methyl)phenoxy)ethanone) (9j). Dark brown crystals, yield (0.13 g, 40%), mp 240–242 °C; IR (KBr) δ 3196 (NH), 1648 (C
O), 1324, 1485 (NO2) cm−1; MS: MALDI-TOF: calcd for [M + H]+ m/z = 931.98, found 931.91; 1H NMR (DMSO-d6) δ 2.62 (s, 6H, 2CH3), 3.50–3.56 (m, 8H, 4 CH2N), 5.06 (s, 4H, 2OCH2), 7.07–7.15 (m, 6H, ArH's), 7.48 (m, 4H, ArH's), 7.98 (d, J = 7.8 Hz, 2H, ArH's), 8.19 (brs, 4H, ArH's), 8.98 (s, 2H, 2CHN), 11.09 (s, 2H, 2 NH) ppm; 13C NMR (DMSO-d6) δ 16.86, 41.29, 41.75, 43.98, 44.50, 66.36, 112.64, 114.04, 121.39, 122.09, 125.91, 126.67, 133.62, 141.25, 142.30, 149.28, 156.56, 158.04, 166.06, 172.53, 179.14. Anal. calcd: for C42H38N14O8S2 (930.98): C, 54.19; H, 4.11; N, 21.06%. Found: C, 54.31; H, 4.23; N, 21.15%.
3.1.4.12 1,1′-(Piperazine-1,4-diyl)bis(2-(4-(((3,5-diphenyl-1,3,4-thiadiazol-2(3H)-ylidene)hydrazono)-methyl)phenoxy)ethanone) (13a). Pale yellow crystals, yield (0.2 g, 63%), mp 214–216 °C; IR (KBr) v 1604 (C
N), 1658 (CO), cm−1; MS: MALDI-TOF: calcd for [M + H]+ m/z = 912.07, found 912.32; 1H NMR (DMSO-d6) δ 3.50–3.55 (m, 8H, 4 CH2N), 4.95 (s, 4H, 2OCH2), 7.03 (d, J = 7.5 Hz, 4H, ArH's), 7.34 (t, J = 7.8 Hz, 4H, ArH's), 7.51–7.56 (m, 8H, ArH's), 7.74 (d, J = 7.5 Hz, 4H, ArH's), 7.86 (s, 4H, ArH's), 8.10 (d, J = 7.8 Hz, 4H, ArH's), 8.43 (s, 2H, 2CHN). Anal. calcd: for C50H42N10O4S2 (911.07): C, 65.92; H, 4.65; N, 15.37%. Found: C, 65.78; H, 4.48; N, 15.42%.
3.1.4.13 1,1′-(Piperazine-1,4-diyl)bis(2-(4-(((5-(4-chlorophenyl)-3-phenyl-1,3,4-thiadiazol-2(3H)-ylidene)-hydrazono)methyl)phenoxy)ethanone) (13b). Yellow crystals, yield (0.15 g, 43%), mp 260–262 °C; IR (KBr) v 1607 (C
N), 1660 (CO) cm−1; MS: MALDI-TOF: calcd for [M + H]+ m/z = 980.96, found 980.04; 1H NMR (DMSO-d6) δ 3.50–3.57 (m, 8H, 4CH2N), 4.96 (s, 4H, 2OCH2), 7.03 (d, J = 8.4 Hz, 4H, ArH's), 7.34 (t, J = 7.5 Hz, 2H, ArH's), 7.34 (t, J = 7.5 Hz, 4H, ArH's), 7.61 (d, J = 8.4 Hz, 4H, ArH's), 7.74 (d, J = 8.4 Hz, 4H, ArH's), 7.88 (d, J = 8.4 Hz, 4H, ArH's), 8.09 (d, J = 8.4 Hz, 4H, ArH's), 8.44 (s, 2H, 2CHN); 13C NMR (DMSO-d6) δ 40.99, 41.41, 43.80, 44.05, 66.06, 115.32, 121.93, 126.54, 127.53, 128.17, 128.66, 129.25, 129.66, 135.89, 139.48, 149.30, 154.75, 160.19, 164.28, 166.13 ppm. Anal. calcd: for C50H40Cl2N10O4S2 (979.96): C, 61.28; H, 4.11; N, 14.29%. Found: C, 61.35; H, 4.23; N, 14.04%.
3.1.4.14 1,1′-(Piperazine-1,4-diyl)bis(2-(2-(((3,5-diphenyl-1,3,4-thiadiazol-2(3H)-ylidene)hydrazono)-methyl)phenoxy)ethanone) (13c). Yellow crystals, yield (0.14 g, 44%), mp 144–146 °C; IR (KBr) v 1602 (C
N), 1659 (CO) cm−1 MS: MALDI-TOF: calcd for [M + H]+ m/z = 912.07, found 912.21; 1H NMR (DMSO-d6) δ 3.50–3.56 (m, 8H, 4CH2N), 5.01 (s, 4H, 2OCH2), 7.03–7.09 (m, 6H, ArH's), 7.31–7.42 (m, 6H, ArH's), 7.42–7.57 (m, 6H, ArH's), 7.88 (brs, 4H, ArH's), 7.94 (d, J = 7.8 Hz, 2H, ArH's), 8.10 (d, J = 8.1 Hz, 4H, ArH's), 8.77 (s, 2H, 2CHN). Anal. calcd: for C50H42N10O4S2 (911.07): C, 65.92; H, 4.65; N, 15.37%. Found: C, 65.78; H, 4.44; N, 15.45%.
3.1.4.15 1,1′-(Piperazine-1,4-diyl)bis(2-(2-(((5-(4-chlorophenyl)-3-phenyl-1,3,4-thiadiazol-2(3H)-ylidene)-hydrazono)methyl)phenoxy)ethanone) (13d). Yellow crystals, yield (0.1 g, 29%), mp 278–280 °C; IR (KBr) ν 1604 (C
N), 1667 (CO) cm−1; MS: MALDI-TOF: calcd for [M + H1]+ m/z = 980.96, found 980.96; 1H NMR (DMSO-d6) δ 3.49–3.55 (m, 8H, 4CH2N), 5.0 (s, 4H, 2OCH2), 7.02–7.09 (m, 4H, ArH's), 7.31–7.42 (m, 4H, ArH's), 7.53 (t, J = 7.2 Hz, 4H, ArH's), 7.60 (d, J = 7.8 Hz, 4H, ArH's), 7.87–7.93 (m, 6H, ArH's), 8.08 (d, J = 7.8 Hz, 4H, ArH's), 8.76 (s, 2H, 2CHN). Anal. calcd for C50H40Cl12N10O4S2 (979.96): C, 61.28; H, 4.11; N, 14.29%. Found: C, 61.12; H, 4.23; N, 14.11%.
3.1.5.1 General procedure. The bis-thiosemicarbazone 7a or 7b (0.278 g, 0.5 mmol), were mixed with the appropriate α-bromocarbonyl compounds 14a–e (1.0 mmol) in dry EtOH/DMF (1
:3, 10 mL), then heated at reflux condition for 8 hours, during which triethylamine (0.1 mL) was added. The reaction vessel was left to cool to ambient temperature, and the isolated solid product was collected by filtration, washed with ethanol, and dried. Purification of the crude products by recrystallization from EtOH/DMF mixed solvents produced the corresponding bis-thiazole derivatives 15a–h, respectively.
3.1.5.2 1,1′-(Piperazine-1,4-diyl)bis(2-(4-(((4-phenylthiazol-2(3H)-ylidene)hydrazineylidene)methyl)-phenoxy)ethanone) (15a). Yellow crystals, yield (0.13 g, 48%), mp 262–264 °C; IR (KBr) v 3292 (NH), 1670 (C
O), 1605 (CN) cm−1; MS: MALDI-TOF: calcd for [M + H]+ m/z = 757.90, found 757.14; 1H NMR (DMSO-d6) δ 3.49–3.56 (m, 8H, 4NCH2), 4.92 (s, 4H, 2OCH2), 7.0 (d, J = 8.7 Hz, 4H, ArH's), 7.28–7.31 (m, 4H, thiazole-2-CH, 2ArH's), 7.39 (t, J = 7.8 Hz, 4H, ArH's), 7.59 (d, J = 8.4 Hz, 4H, ArH's), 7.84 (d, J = 8.1 Hz, 4H, ArH's), 7.86 (s, 2H, 2CHN), 11.99 (s, 2H, 2NH) ppm; 13C NMR (DMSO-d6) δ 41.18, 41.40, 44.03, 44.53, 66.03, 94.41, 103.49, 115.18, 125.61, 127.53, 127.75, 128.71, 134.83, 141.25, 150.60, 159.12, 166.02, 168.45 ppm. Anal. calcd: for C40H36N8O4S2 (756.90): C, 63.47; H, 4.79; N, 14.80%. Found: C, 63.72; H, 4.76; N, 14.69%.
3.1.5.3 1,1′-(Piperazine-1,4-diyl)bis(2-(4-(((4-(4-methoxyphenyl)thiazol-2(3H)-ylidene)hydrazineylidene)-methyl)phenoxy)ethanone) (15b). Dark brown crystals, yield (0.13 g, 44%), mp 260–262 °C; IR (KBr) v 3297 (NH), 1653 (C
O), 1604 (CN) cm−1; MS: MALDI-TOF: calcd for [M + H]+ m/z = 817.95, found 817.22; 1H NMR (DMSO-d6) δ 3.50–3.56 (m, 8H, 4CH2N), 3.75 (s, 6H, 2CH3O), 4.92 (s, 4H, 2OCH2), 6.96 (d, J = 8.7 Hz, 4H, ArH's), 7.0 (d, J = 8.7 Hz, 4H, ArH's), 7.1 (s, 2H, thiazole-2-CH), 7.59 (d, J = 8.7 Hz, 4H, ArH's), 7.77 (d, J = 8.4 Hz, 4H, ArH's), 7.98 (s, 2H, 2CHN), 11.95 (br, 2H, 2NH); 13C NMR (DMSO-d6) δ 41.06, 41.31, 43.87, 44.37, 55.09, 65.83, 101.22, 113.94, 115.05, 126.84, 127.45, 127.62, 141.03, 150.31, 154.34, 158.74, 158.97, 165.93, 168.24. Anal. calcd: for C42H40N8O6S2 (816.95): C, 61.75; H, 4.94; N, 13.72%. Found: C, 61.88; H, 4.78; N, 13.55.
3.1.5.4 1,1′-(Piperazine-1,4-diyl)bis(2-(4-(((4-(4-chloroyphenyl)thiazol-2(3H)-ylidene)hydrazineylidene)-methyl)phenoxy)ethanone) (15c). Pale yellow crystals, yield (0.12 g, 41%), mp 278–280 °C; IR (KBr) v 3181 (NH), 1660 (C
O), 1607 (CN) cm−1; MS: MALDI-TOF: calcd for [M + H]+ m/z = 826.78, found 826.20; 1H NMR (DMSO-d6) δ 3.50–3.56 (m, 8H, 4CH2N), 4.93 (s, 4H, 2OCH2), 7.01 (d, J = 7.8 Hz, 4H, ArH's), 7.35 (s, 2H, thiazole-2-CH), 7.45 (d, J = 8.1 Hz, 4H, ArH's), 7.59 (d, J = 8.1 Hz, 4H, ArH's), 7.86 (d, J = 7.5 Hz, 4H, ArH's), 7.99 (s, 2H, 2CHN), 12.0 (s, 2H, 2 NH); 13C NMR (DMSO-d6) δ 41.05, 41.35, 43.87, 44.20, 65.88, 104.18, 115.05, 127.19, 127.34, 127.68, 128.61, 131.88, 133.57, 141.31, 149.27, 159.03, 165.89, 168.47. Anal. calcd for C40H34Cl2N8O4S2 (825.78): C, 58.18; H, 4.15; N, 13.57%. Found: C, 58.34; H, 4.27; N, 13.27%.
3.1.5.5 1,1′-(Piperazine-1,4-diyl)bis(2-(4-(((4-(4-bromoyphenyl)thiazol-2(3H)-ylidene)hydrazineylidene)-methyl)phenoxy)ethanone) (15d). Pale yellow solid, yield (0.16 g, 50%), mp 288–290 °C; IR (KBr) v 3292 (NH), 1660 (C
O), 1606 (CN) cm−1; 1H NMR (DMSO-d6) δ 3.49–3.55 (m, 8H, 4 CH2N), 4.92 (s, 4H, 2OCH2), 7.0 (d, J = 7.2 Hz, 4H, ArH's), 7.35 (s, 2H, thiazole-2-CH), 7.45 (d, J = 8.1 Hz, 4H, ArH's), 7.59 (d, J = 7.8 Hz, 4H, ArH's), 7.85 (d, J = 8.1 Hz, 4H, ArH's), 7.99 (s, 2H, 2CHN), 11.99 (s, 2H, 2 NH). 13C NMR (DMSO-d6) δ 40.81, 41.41, 43.98, 44.58, 65.91, 104.31, 115.16, 127.31, 127.78, 128.72, 131.98, 133.68, 141.44, 149.38, 159.14, 166.01, 168.57. Anal. calcd: for C40H34Br2N8O4S2 (914.69): C, 52.52; H, 3.75; N, 12.25%. Found: C, 52.41; H, 3.88; N, 12.11%.
3.1.5.6 1,1′-(Piperazine-1,4-diyl)bis(2-(4-(((4-(coumarin-3-yl)thiazol-2(3H)-ylidene)hydrazineylidene)-methyl)phenoxy)ethanone) (15e). Yellow solid, yield (0.18 g, 56%), mp 320–322 °C; IR (KBr) v 3230 (NH), 1713, 1662 (2C
O), 1605 (CN) cm−1; MS: MALDI-TOF: calcd for [M + H]+ m/z = 893.96, found 893.00; 1H NMR (DMSO-d6) δ 3.49–3.56 (m, 8H, 4CH2N), 4.93 (s, 4H, 2OCH2), 7.01 (d, J = 8.4 Hz, 4H, ArH's), 7.36–7.46 (m, 4H, ArH's), 7.59–7.65 (m, 6H, ArH's), 7.75 (s, 2H, thiazole-2-CH), 7.85 (d, J = 7.8 Hz, 2H, ArH's), 8.01 (s, 2H, ArH's), 8.54 (s, 2H, 2CHN), 12.03 (s, 2H, 2NH) ppm. Anal. calcd: for C46H36N8O8S2 (892.96): C, 61.87; H, 4.06; N, 12.55%. Found: C, 61.95; H, 4.21; N, 12.38%.
3.1.5.7 1,1′-(Piperazine-1,4-diyl)bis(2-(2-(((4-(4-chloroyphenyl)thiazol-2(3H)-ylidene)hydrazineylidene)-methyl)phenoxy)ethanone) (15f). Yellow crystals, yield (0.18 g, 62%), mp 208–210 °C; IR (KBr) v 3191 (NH), 1659 (C
O), 1602(CN) cm−1; MS: MALDI-TOF: calcd for [M + H]+ m/z = 826.78, found 826.18; 1H NMR (DMSO-d6) δ 3.53–3.57 (m, 8H, 4 NCH2), 4.99 (s, 4H, 2OCH2), 6.70–7.04 (m, 4H, ArH's), 7.31 (d, J = 7.8 Hz, 2H, ArH's), 7.36 (s, 2H, thiazole-2-CH), 7.45 (d, J = 8.7 Hz, 4H, ArH's), 7.80 (d, J = 8.1 Hz, 2H, ArH's), 7.85 (d, J = 8.4 Hz, 4H, ArH's), 8.42 (s, 2H, 2CHN), 12.18 (s, 2H, 2NH); 13C NMR (DMSO-d6) δ 41.13, 41.41, 44.02, 44.27, 66.20, 104.39, 112.96, 121.20, 122.72, 125.01, 127.25, 128.66, 130.49, 131.97, 133.58, 137.04, 149.38, 155.90, 165.96, 168.48. Anal. calcd: for C40H34Cl2N8O4S2 (825.78): C, 58.18; H, 4.15; N, 13.57%. Found: C, 58.32; H, 4.22; N, 13.48%.
3.1.5.8 1,1′-(Piperazine-1,4-diyl)bis(2-(2-(((4-(4-bromoyphenyl)thiazol-2(3H)-ylidene)hydrazineylidene)-methyl)phenoxy)ethanone) (15g). Yellow crystals, yield (0.14 g, 44%), mp 218–220 °C; IR (KBr) v 3196 (NH), 1642 (C
O), 1601 (CN) cm−1; 1H NMR (DMSO-d6) δ 3.53–3.56 (m, 8H, 4CH2N), 4.99 (s, 4H, 2OCH2), 6.99–7.02 (m, 4H, ArH's), 7.31 (d, J = 7.8 Hz, 2H, ArH's), 7.37 (s, 2H, thiazole-2-CH), 7.45 (d, J = 7.5 Hz, 4H, ArH's), 7.79 (d, J = 7.8 Hz, 2H, ArH's), 7.86 (d, J = 7.5 Hz, 4H, ArH's), 8.42 (s, 2H, 2CHN), 12.18 (s, 2H, 2 NH); 13C NMR (DMSO-d6) δ 41.14, 41.44, 44.05, 44.29, 66.36, 104.39, 112.96, 121.18, 122.71, 124.99, 127.24, 128.63, 130.46, 131.93, 133.57, 137.04, 149.34, 155.88, 165.95, 168.43. Anal. calcd: for C40H34Br2N8O4S2 (914.69): C, 52.52; H, 3.75; N, 12.25%. Found: C, 52.45; H, 3.88; N, 12.12%.
3.1.5.9 1,1′-(Piperazine-1,4-diyl)bis(2-(2-(((4-(coumarin-3-yl)thiazol-2(3H)-ylidene)hydrazineylidene)-methyl)phenoxy)ethanone) (15h). Brown crystals, yield (0.16 g, 50%), mp 198–200 °C; IR (KBr) v 3200 (NH), 1650, 1713 (2C
O), 1603 (CN) cm−1; MS: MALDI-TOF: calcd for [M + H]+ m/z = 893.96, found 893.17; 1H NMR (DMSO-d6) δ 3.44–3.54 (m, 8H, 4 NCH2), 5.01 (s, 4H, 2OCH2), 7.02–7.04 (m, 4H, ArH's), 7.31–7.45 (m, 6H, ArH's), 7.59–7.64 (m, 2H, ArH's), 7.73 (s, 2H, thiazole-2-CH), 7.75–7.84 (m, 4H, ArH's), 8.45 (s, 2H, ArH's), 8.51 (s, 2H, 2CHN), 12.21 (s, 2H, 2 NH). Anal. calcd: for C46H36N8O8S2 (892.96): C, 61.87; H, 4.06; N, 12.55%. Found: 61.72; H, 4.21; N, 12.32%.
3.2. Biological evaluation
3.2.1.1 Drugs and chemicals. The samples were dissolved in DMSO to yield a stock solution of 2 mg mL−1 and serially diluted in DMEM (Dulbecco's Modified Eagle Medium) supplemented medium immediately before use to yield concentrations ranging between 0–50 μg mL−1 for the human colon carcinoma (HCT116), human liver carcinoma (HEPG2), human breast carcinoma (MCF7) and human dermal normal (HDF) cell lines. The final concentration of DMSO never exceeded 0.1% (v/v) in control and treated samples to avoid potential toxicity to the cell lines that could lead to cell death.
3.2.1.2 Human cancer cell lines. Human carcinoma (HCT116, HEPG2, and MCF7) cell lines and human normal dermal cell lines (HDF) were obtained from the National Cancer Institute, Cairo University, Egypt. The tumor cell line was maintained as monolayer cultures in DMEM supplemented with 10% FBS and 1% penicillin–streptomycin.
3.2.1.3 Cytotoxicity using SRB assay. The percentage of cell growth inhibition at the single dose [100 μM] and cytotoxicity (IC50) were determined using the sulforhodamine-B (SRB) method69,70 on both normal skin (HDF) cell line and the cancer cell lines: HCT116, HEPG2, and MCF7. This was done at the beginning of the study to determine a single dose % inhibition at 100 μM for the compounds to select the most effective ones among them as anti-cancer agents. Then, the cytotoxicity of the drugs on the malignant cell line compared to the normal cell line was evaluated. Cells were treated for 48 h with different concentrations (0, 6.25, 12.5, 25, and 50 μM) of compounds selected. The optical density (OD) was measured spectrophotometrically at 570 nm using an ELISA microplate reader (Sunrise TM, TECAN, Germany). The mean values were estimated as the percentage of cell viability as follows:
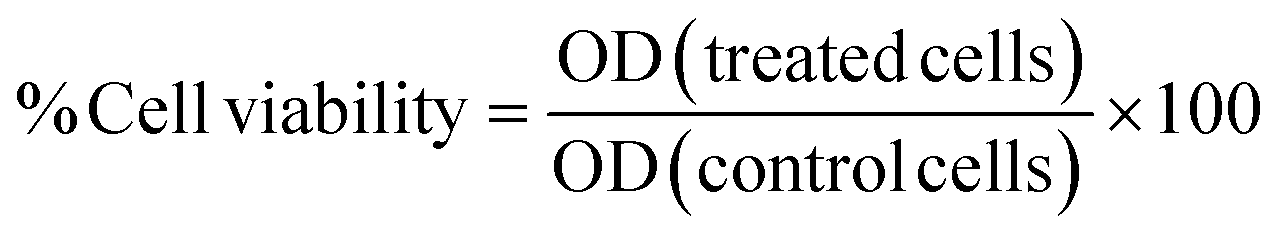
The IC50 value of each drug was calculated using dose–response curve-fitting models (Graph-Pad Prism software, version, 8). The concentration of IC50 was used for further treatment to do other molecular pathways.
3.2.3.1 Annexin V/PI staining and cell cycle analysis. HCT-116 cells were seeded into 6-well culture plates (3–5 × 105 cells per well) and incubated overnight. Cells were treated with compound 9i at their IC50 values for 48 h. Next, media supernatants and cells were collected and rinsed with ice-cold PBS. Then, cells were suspended the cells in 100 μL of annexin binding buffer solution “1.4 M NaCl, 25 mM CaCl2, and 0.1 M Hepes/NaOH, pH 7.4” and incubated with “Annexin V-FITC solution (1
:100) and propidium iodide (PI)” at a concentration equals 10 μM in the dark for 30 min. Stained cells were then acquired by Cytoflex from Beckham Coulter Flow Cytometer with cytexpert software.73–76
3.2.3.2 Real time-polymerase chain reaction for the selected genes. Gene expression of Bcl-2, the anti-apoptotic gene, and the pro-apoptotic genes P53, PUMA, and caspases-3,8,9 were evaluated to delve deeper into the apoptotic pathway. HCT-116 cells were treated with compound 9i at their IC50 values for 48 h. After treatment, the RT-PCR reaction was carried out following routine work. Then, the Ct values were collected to calculate the relative genes' expression in all samples by normalization to the β-actin housekeeping gene.73,77
4. Conclusion
Convenient synthetic routes for constructing some novel bis-thiazoles and bis-1,3,4-thiadiazoles linked to piperazine core were reported in good outcomes. The easy synthesis of the target compounds in good yields under mild reaction conditions in a short reaction time utilizing affordable starting materials was one of these reactions' benefits. Another advantage was that simple crystallization could easily isolate the desired compounds without chromatographic purification. The anti-cancer activity of synthesized compounds was assessed, and the results showed promising results of bis-thiazoles 9a and 9i against HCT116 cell lines. Interestingly, compound 9i had potent EGFR kinase inhibition with an IC50 value of 1.2 nM with inhibition of 97.5% compared to Erlotinib (IC50 = 1.3 nM, 97.8% inhibition). Moreover, compound 9i significantly induced apoptosis in HCT-16 cells by 4.16-fold by having 16.85% total apoptosis in treated cells compared to 4.05% for control moreover, arresting the cell cycle at the P2 phase. A promising EGFR-targeted chemotherapeutic drug for the treatment of colon cancer, compound 9i was thus verified.Data availability
All data associated with this manuscript will be available upon reasonable request from the corresponding authorConflicts of interest
There are no conflicts to declare.References
- K. E. Gettys, Z. Ye and M. Dai, Synthesis, 2017, 49, 2589–2604 CrossRef CAS.
- C. C. Guo, H.-P. Li and X.-B. Zhang, Bioorg. Med. Chem., 2003, 11, 1745–1751 CrossRef CAS PubMed.
- C. C. Guo, R.-B. Tong and K.-L. Li, Bioorg. Med. Chem., 2004, 12, 2469–2475 CrossRef CAS PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS.
- M. Rizwan, S. Noreen, S. Asim, Z. Liaqat, M. Shaheen and H. Ibrahim, Chem. Inorg. Mater., 2024, 2, 100041 Search PubMed.
- M. Shaquiquzzaman, G. Verma, A. Marella, M. Akhter, W. Akhtar, M. F. Khan, S. Tasneem and M. M. Alam, Eur. J. Med. Chem., 2015, 102, 487–529 CrossRef CAS.
- N. A. Meanwell and O. Loiseleur, J. Agric. Food Chem., 2022, 70, 10942–10971 CrossRef CAS.
- P. Meena, V. Nemaysh, M. Khatri, A. Manral, P. M. Luthra and M. Tiwari, Synthesis, Bioorg. Med. Chem., 2015, 23, 1135–1148 CrossRef CAS.
- L. Yurtta, Z. A. Kaplancikli and Y. Ozkay, J. Enzyme Inhib. Med. Chem., 2013, 28, 1040–1047 CrossRef.
- R. S. Upadhayaya, N. Sinha, S. Jain, N. Kishore, R. Chandra and S. K. Arora, Bioorg. Med. Chem., 2004, 12, 2225–2238 CrossRef CAS.
- P. Chaudhary, R. Kumar and A. K. Verma, Bioorg. Med. Chem., 2006, 14, 1819–1826 CrossRef CAS PubMed.
- F. Stegmeier, M. Warmuth, W. R. Sellers and M. Dorsch, Clin. Pharmacol. Ther., 2010, 87, 543–552 CrossRef CAS PubMed.
- P. W. Manley, F. Blasco, J. Mestan and R. Aichholz, Bioorg. Med. Chem., 2013, 21, 3231–3239 CrossRef CAS PubMed.
- W. Sneader, C. Hansch, P. Sammnes and J. Taylor, Comprehensive Medicinal Chemistry, Pergamon Press, London, 1990, 1, pp. 65–70 Search PubMed.
- R. Aggarwal and G. Sumran, Eur. J. Med. Chem., 2020, 205, 112652 CrossRef CAS PubMed.
- T. M. Davis, T.-Y. Hung, I.-K. Sim, H. A. Karunajeewa and K. F. Ilett, Drugs, 2005, 65, 75–87 CrossRef CAS PubMed.
- R.-H. Zhang, H.-Y. Guo, H. Deng, J. Li, Z.-S. Quan and J. Enzyme Inhib, Med. Chem., 2021, 36, 1165–1197 CAS.
- R. Wang, A. M. Piggott, Y.-H. Chooi and H. Li, Nat. Prod. Rep., 2023, 40, 387–411 RSC.
- P. C. Sharma, K. K. Bansal, A. Sharma, D. Sharma and A. Deep, Eur. J. Med. Chem., 2020, 188, 112016 CrossRef CAS.
- A. Petrou, M. Fesatidou and A. Geronikaki, Molecules, 2021, 26, 3166 CrossRef CAS.
- A. Ayati, S. Emami, S. Moghimi and A. Foroumadi, Future Med. Chem., 2019, 11, 1929 CrossRef CAS PubMed.
- M. T. Chhabria, S. Patel, P. Modi and P. S. Brahmkshatriya, Curr. Top. Med. Chem., 2016, 16, 2841 CrossRef CAS.
- A. B. Muhsinah, M. M. Alharbi, N. A. Kheder, S. M. Soliman, H. A. Ghabbour and Y. N. Mabkhot, J. Mol. Struct., 2024, 1316, 139083 CrossRef CAS.
- P. Franchetti, L. Cappellacci, M. Grifantini, A. Barzi, G. Nocentini, H. Yang, A. O'Connor, H. N. Jayaram, C. Carrell and B. M. Goldstein, J. Med. Chem., 1995, 38, 3829 CrossRef CAS.
- X. Li, Y. He, C. H. Ruiz, M. Koenig and M. D. Cameron, Drug Metab. Dispos., 2009, 37, 1242 CrossRef CAS.
- S. Hu-Lieskovan, S. Mok, B. Homet Moreno, J. Tsoi, L. Robert, L. Goedert, E. M. Pinheiro, R. C. Koya, T. G. Graeber, B. Comin-Anduix and A. Ribas, Sci. Transl. Med., 2015, 18, 279 Search PubMed.
- K. M. Dawood, T. M. Eldebss, H. S. El-Zahabi, M. H. Yousef and P. Metz, Eur. J. Med. Chem., 2013, 70, 740–749 CrossRef CAS PubMed.
- K. M. Dawood and S. M. Gomha, J. Heterocycl. Chem., 2015, 52, 1400–1405 CrossRef CAS.
- K. M. Dawood and T. A. Farghaly, Expert Opin. Ther. Pat., 2017, 27, 477–505 CrossRef CAS PubMed.
- S. M. Gomha, M. R. Abdelaziz, N. A. Kheder, H. M. Abdel-Aziz, S. Alterary and Y. N. Mabkhot, Chem. Cent. J., 2017, 11, 105 CrossRef PubMed.
- S. M. Gomha, N. A. Kheder, M. R. Abdelaziz, Y. N. Mabkhot and A. M. Alhajoi, Chem. Cent. J., 2017, 11, 25 CrossRef.
- X. H. Yang, Q. Wen, T. T. Zhao, J. Sun, X. Li, M. Xing, X. Lu and H. L. Zhu, Bioorg. Med. Chem., 2012, 20, 1181–1187 CrossRef CAS PubMed.
- H. Rajak, A. Agarawal, P. Parmar, B. S. Thakur, R. Veerasamy, P. C. Sharma and M. D. Kharya, Bioorg. Med. Chem. Lett., 2011, 21, 5735–5738 CrossRef CAS PubMed.
- Q. Ma, W. Ding, Z. Chen and Z. Ma, Nat. Prod. Res., 2018, 32, 761–766 CrossRef CAS PubMed.
- B. Li, G. Chen, J. Bai, Y. K. Jing and Y. H. Pei, J. Asian Nat. Prod. Res., 2011, 13, 1146–1150 CrossRef CAS.
- L. Zhang, L. H. Wang, Y. F. Yang, S. M. Yang, J. H. Zhang and C. H. Tan, Nat. Prod. Res., 2011, 25, 1676–1679 CrossRef CAS PubMed.
- M. S. Abdelfattah, K. Toume, F. Ahmed, S. K. Sadhu and M. Ishibashi, Chem. Pharm. Bull., 2010, 58, 1116–1118 CrossRef CAS.
- A. V. Pakhomova, V. E. Nebolsin, O. V. Pershina, V. A. Krupin, L. A. Sandrikina, E. S. Pan, N. N. Ermakova, O. E. Vaizova, D. Widera, W. D. Grimm and V. Y. E. Kravtsov, Int. J. Mol. Sci., 2020, 21, 991 CrossRef CAS.
- J. Han, H. W. Lee, Y. Jin, D. B. Khadka, S. Yang, X. Li, M. Kim and W. J. Cho, Eur. J. Med. Chem., 2020, 188, 112031 CrossRef CAS PubMed.
- S. Yang, K. R. Jyothi, S. Lim, T. G. Choi, J. H. Kim, S. Akter, M. Jang, H. J. Ahn, H. Y. Kim, M. P. Windisch, D. B. Khadka, C. Zhao, Y. Jin, I. Kang, J. Ha, B. C. Oh, M. Kim, S. S. Kim and W. J. Cho, J. Med. Chem., 2015, 58, 9546–9561 CrossRef CAS PubMed.
- A. H. Rezayan, S. Hariri, P. Azerang, G. Ghavami, I. Portugal and S. Sardari, Iran. J. Pharm. Res., 2017, 16, 745–755 Search PubMed.
- K. Singh, S. Banerjee and A. K. Patra, RSC Adv., 2015, 5, 107503–107513 RSC.
- M. I. El-Gamal, M. S. Abdel-Maksoud, M. M. G. El-Din, K. H. Yoo, D. Baek and C. H. Oh, Arch. Pharm., 2014, 347, 635–641 CrossRef CAS PubMed.
- M. S. Ayoup, M. A. Fouad, H. Abdel-Hamid, E.-S. Ramadan, M. M. Abu-Serie, A. Noby and M. Teleb, Eur. J. Med. Chem., 2020, 186, 111875 CrossRef CAS PubMed.
- M. P. Gajewski, H. Beall, M. Schnieder, S. M. Stranahan, M. D. Mosher, K. C. Rider and N. R. Natale, Bioorg. Med. Chem. Lett., 2009, 19, 4067–4069 CrossRef CAS PubMed.
- Y. Ling, Z. Wang, H. Zhu, X. Wang, W. Zhang, X. Wang, L. Chen, Z. Huang and Y. Zhang, Bioorg. Med. Chem., 2014, 22, 374–380 CrossRef CAS PubMed.
- H. A. Abdel-Aziz, H. S. El-Zahabi and K. M. Dawood, Eur. J. Med. Chem., 2010, 45, 2427–2432 CrossRef CAS PubMed.
- K. M. Dawood, H. Abdel-Gawad, H. A. Mohamed and F. A. Badria, Med. Chem. Res., 2011, 20, 912–919 CrossRef CAS.
- K. M. Dawood, M. A. Raslan, A. A. Abbas, B. E. Mohamed, M. H. Abdellattif, M. S. Nafie and M. K. Hassan, Front. Chem., 2021, 9, 694870 CrossRef CAS PubMed.
- F. M Thabet, K. M. Dawood, E. A. Ragab, M. S. Nafie and A. A. Abbas, RSC Adv., 2022, 12, 23644–23660 RSC.
- K. M. Dawood, M. A. Raslan, A. A. Abbas, B. E. Mohamed and M. S. Nafie, Anti-Cancer Agents Med. Chem., 2023, 23, 328–345 CrossRef CAS PubMed.
- A. A. Abbas and K. M. Dawood, Expert Opin. Drug Discovery, 2022, 17, 1357–1376 CrossRef CAS.
- M. E. Salem, E. M. Mahrous, E. A. Ragab, M. S. Nafie and K. M. Dawood, MBC Chem., 2023, 17(51), 1–17 Search PubMed.
- M. E. Salem, E. M. Mahrous, E. A. Ragab, M. S. Nafie and K. M. Dawood, ACS Omega, 2023, 8, 35359–35369 CrossRef CAS PubMed.
- M. S. Nafie, S. H. Kahwash, M. M. Youssef and K. M. Dawood, Arch. Pharm., 2024, e2400225 CrossRef PubMed.
- A. A. Abbas, T. A. Farghaly and K. M. Dawood, RSC Adv., 2024, 14, 19752–19779 RSC.
- K. M. Dawod and A. A. Abbas, ChemistrySelect, 2021, 6, 279–305 CrossRef.
- M. A. El-Atawy, A. Z. Omar, M. Hagar and E. M. Shashira, Green Chem. Lett. Rev., 2019, 12, 364–376 CrossRef CAS.
- S. Bondock, T. Albarqi, M. Abboud, T. Nasr, N. M. Mohamed and M. M. Abdou, RSC Adv., 2023, 13, 24003–24022 RSC.
- W. Dieckmann and O. Platz, Chem. Ber., 1906, 38, 2989–2995 Search PubMed.
- R. Wodtke, J. Steinberg, M. Köckerling, R. Löser and C. Mamat, RSC Adv., 2018, 8, 40921–40933 RSC.
- N. F. Eweiss and A. Osman, J. Heterocycl. Chem., 1980, 17, 1713–1717 CrossRef CAS.
- M. M. El-Abadelah, A. Q. Hussein and B. A. Thaher, Heterocycles, 1991, 32, 1879–1895 CrossRef CAS.
- P. Wolkoff, Can. J. Chem., 1975, 53, 1333–1335 CrossRef CAS.
- Y. C. Tian, J. K. Li, F. G. Zhang and J. A. Ma, Adv. Synth. Catal., 2021, 363, 2093–2097 CrossRef CAS.
- R. M. Cowper and L. H. Davidson, Org. Synth., 1943, 2, 480 Search PubMed.
- R. B. Mohan and N. G. Reddy, Synth. Commun., 2013, 43, 2603–2614 CrossRef CAS.
- C. F. Koelsch, J. Am. Chem. Soc., 1950, 72, 2993–2995 CrossRef CAS.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, V. D. McMahon, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107 CrossRef CAS PubMed.
- M. Sharaky, M. Kamel, M. A. Aziz, M. Omran, M. M. Rageh, K. A. Abouzid and S. A. Shouman, J. Enzyme Inhib. Med. Chem., 2020, 35, 1641–1656 CrossRef CAS PubMed.
- M. E. Salem, I. M. Fares, S. A. Ghozlan, M. M. Abdel-Aziz, I. A. Abdelhamid and A. H. M. Elwahy, J. Heterocycl. Chem., 2022, 59, 1907–1926 CrossRef CAS.
- J. L. Nakamura, Expert Opin. Ther. Targets, 2007, 11, 463–472 CrossRef CAS PubMed.
- M. S. Nafie, K. Arafa, N. K. Sedky, A. A. Alakhdar and R. K. Arafa, Chem.-Biol. Interact., 2020, 324, 109087 CrossRef CAS.
- M. S. Nafie, A. M. Amer, A. K. Mohamed and E. S. Tantawy, Bioorg. Med. Chem., 2020, 28, 115828 CrossRef CAS PubMed.
- E. M. Gad, M. S. Nafie, E. H. Eltamany, M. S. A. G. Hammad, A. Barakat and A. T. A. Boraei, Molecules, 2020, 25, 2523 CrossRef CAS PubMed.
- M. S. Nafie, S. M. Kishk, S. Mahgoub and A. M. Amer, Chem. Biol. Drug Des., 2022, 99, 547–560 CrossRef CAS.
- M. S. Nafie, S. Mahgoub and A. M. Amer, Chem. Biol. Drug Des., 2021, 97, 553–564 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra05091f |
| This journal is © The Royal Society of Chemistry 2024 |