
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of a new experimental NMR strategy for covalent cysteine protease inhibitors screening: toward enhanced drug discovery†
Abdelali Chihaba,
Nabil El Brahmia,
Ghanem Hamdouna,
Abdelmoula El Abbouchia,
Hamza Ghammazb,
Nadia Touilc,
Mostafa Bousminaa,
Elmostafa El Fahimeb and
Saïd El Kazzouli *a
*a
aEuromed University of Fes, UEMF, Morocco. E-mail: s.elkazzouli@ueuromed.org
bCentre National de la Recherche Scientifique et Technique (CNRST), Angle avenues des FAR et Allal El Fassi, Hay Ryad, 10102 Rabat, Morocco
cCell Culture Unit, Center of Virology, Infectious, and Tropical Diseases, Mohammed V Military Hospital, Rabat, Morocco
First published on 23rd August 2024
Abstract
In the development of antiviral drugs, proteases and polymerases are among the most important targets. Cysteine proteases, also known as thiol proteases, catalyze the degradation of proteins by cleaving peptide bonds using the nucleophilic thiol group of cysteine. As part of our research, we are examining how cysteine, an essential amino acid found in the active site of the main protease (Mpro) enzyme in SARS-CoV-2, interacts with electrophilic groups present in ethacrynic acid (EA) and compounds 4, 6, and 8 to form sulfur–carbon bonds. Nuclear magnetic resonance (NMR) spectroscopy was used to monitor the reaction rate between cysteine and Michael acceptors. We found that the inhibitory activity of these compounds towards Mpro is correlated to their chemical reactivity toward cysteine. This approach may serve as a valuable tool in drug development for detecting potential covalent inhibitors of Mpro and other cysteine proteases.
Introduction
Proteases, also known as peptidases, proteinases, or proteolytic enzymes, play an important role in many biological processes of bacteria, viruses, and mammals.1 Through catalysing scissile bond hydrolysis, these enzymes cleave proteins into smaller fragments.2 According to their catalytic site, proteases are classified into four major groups; namely serine proteases, aspartic proteases, metalloproteases and cysteine proteases.3 Cysteine proteases, or thiol proteases, are characterized by an active site comprising a cysteine residue and another critical amino acid, typically histidine. In this last case, histidine functions as a base, facilitating the deprotonation of cysteine's thiol group. This process enhances the nucleophilicity of cysteine, defining a key aspect of the enzymatic activity of cysteine proteases.4 Papain, discovered in 1873, stands as the pioneering exemplar of cysteine proteases.4 Various thiol proteases have been isolated and completely characterized since then, including cathepsins,5 calpain,6 and 3C-like proteases, known as main proteases Mpro.7 Due to its vital role in coronavirus replication cycle, the Mpro (3CLpro) has been the subject of extensive research around the world.8,9 Its conservation among several coronaviruses including SARS-CoV and SARS-CoV-2, along with its distinct and unique cleavage recognition site, make it an attractive target for drug development.8Since the apparition of the COVID-19 pandemic in late 2019, many research teams have focused on developing inhibitors that block the activity of the Mpro and thereby, inhibit the SARS-CoV-2 virus, the primary cause of this global health crisis.10,11 Thus, a multitude of potential inhibitors have been identified, several of which are currently undergoing clinical evaluation.12 Furthermore, Pfizer had successfully brought Paxlovid, a combination of a Mpro inhibitor (nirmatrelvir) and an HIV protease inhibitor (ritonavir), to the market as an approved treatment for COVID-19.13 Overall, the majority of existing Mpro inhibitors are competitive inhibitors that target the active cysteine 145 and form a covalent bond with it.8,14–17 The strategy of forming a covalent linkage involves the use of electrophilic warheads such as aldehyde,14 active esters,18 α-ketoamide19 and Michael acceptors20 etc., to form a sulfur–carbon (S–C) covalent bond with the cysteine's thiol as depicted in Fig. 1.
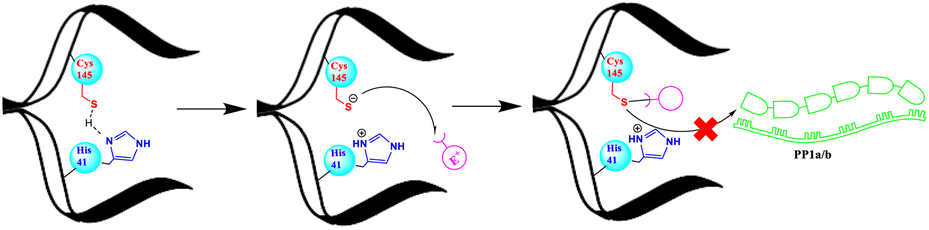 | ||
| Fig. 1 Schematic representation of the action mode of covalent inhibitors. | ||
The formation of the S–C covalent bond with cysteine inhibits its reactivity towards other electrophilic groups, including the scissile bonds within the polyproteins PP1a and PP1b. This inhibition effectively blocks the activity of Mpro.14,16
Compounds bearing Michael acceptors have been extensively used as potent bioactive molecules in wide range of pharmaceutical applications including anti-inflammatory, antiproliferative and antivirals.21–23 By strengthening their electrophilicity, their biological and pharmacological properties were improved.22,24 Moreover, their stability in forming thiol-Michael adducts was found to have a significance impact in their activity of inhibiting kinases.25
Presently, bioinformatics plays a crucial role in the advancement of novel drugs, having emerged as an integral element in drug development. Beyond its significance in the examination of various biological components such as genomes, genes, proteins, and enzymes, it serves as a robust screening method facilitating the identification of promising leads exhibiting potent activities.26,27 There are, however, still some limitations, notably the generation of false positives hits.28 Therefore, new screening methods are needed to overcome this issue and enhance drug discovery tools.
The present study presents three novel derivatives of EA, specifically compounds 4, 6 and 8, which exhibit significant inhibitory properties against the SARS-CoV-2 virus. Additionally, the biological evaluation and chemical reactivity analysis of compound 9, a reduced analogue of compound 8 that lacks the double bond, indicates that the Michael acceptor plays a crucial role in the inhibitory activity within this compound's family. In a subsequent NMR study, we investigate the formation of the S–C covalent bond between the inhibitor and the thiol group of cysteine, a crucial amino acid within the active site of Mpro of SARS-CoV and SARS-CoV-2. This study revealed a direct correlation between the inhibitory activity and the chemical reactivity of the corresponding inhibitor towards cysteine. These findings indicate the potential utility of this method in drug development for identifying potent inhibitors of Mpro and other cysteine proteases. Beyond its ease of use, this method offers a cost-effective and time-efficient alternative to other screening tools, such as high-throughput screening (HTS), in vitro screening, or in vivo screening. These conventional methods frequently necessitate expensive equipment, large compound libraries, and extended processing times, as well as complex data interpretation and ethical concerns.29,30
Results and discussion
Synthesis
The EA derivatives 4, 6 and 8 were prepared as described in Scheme 1. First, the N1-alkylation of 7-nitro-1H-idazole 1 was achieved as reported in the literature.31 Methyl iodide was added to a mixture of 1 with sodium hydride (NaH) in dimethylformamide (DMF) to afford intermediate 2a in a yield of 86% after stirring at room temperature for 12 h. The analogue 2b was obtained using the procedure reported by M. Cheung et al.32 1 reacted with trimethyloxonium tetrafluoroborate ((CH3)3O(BF4)) in ethyl acetate (EtOAc) at room temperature to provide 2b in a yield of 92%. The resulting intermediates 2a and 2b were treated with ammonium tetra chloride (NH4Cl) in a mixture of ethanol/water as solvent to give their corresponding amine intermediates 3 and 5 in quantitative yields (Scheme 1A). This intermediates were reacted in the next step, without any further purification, with EA in the presence of N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl) and 1-hydroxybenzotriazole hydrate (HOBt) as activator agents in dichloromethane (DCM) at room temperature overnight to produce the desired products 4 and 6 in 44 and 46% yields, respectively. Reaction of EA with the amine 7 under the same conditions afforded compound 8 in a yield of 35% (Scheme 1B). | ||
| Scheme 1 Synthesis pathway of compounds 4, 6, 8 and 9. | ||
Compound 9, the reduced analogue of 8 was prepared as described in Scheme 1C, first, a Friedel–Crafts acylation of the anisole 1a using butyryl chloride was performed to give intermediate 1b′ which was used in the next step without purification. This step was followed by deprotection of the methoxy group by the action of aluminum chloride (AlCl3) to afford intermediate 1b. This intermediate underwent a nucleophilic substitution using ethyl-2-bromoacetate in the presence of potassium tert-butoxide (tBuOK) as base in tetrahydro furane (THF) to afford intermediate 1c which after basic hydrolysis provided the EA reduced analogue 1d. The same coupling reaction between 1d and 7 gave compound 9 in a yield of 20%.
Compounds 4, 6, 8 and 9 were identified and characterized via different characterization techniques, including 1H and 13C NMR, Fourier-transform infrared spectroscopy (FT-IR), High-resolution mass spectrometry (HRMS), and melting point (MP).
ADMET analysis
The absorption, distribution, metabolism, excretion and toxicity (ADMET) are fundamental parameters that provide valuable information about the drug likeness and the pharmacokinetic properties of a specific lead. They often exhibit as the most challenging part in the drug discovery process. Therefore, we used SwissADME (http://www.swissadme.ch/) online tool to predict the ADMET properties of our lead compounds and compare them to EA ones which is an already approved drug (Table 1 and Fig. 2).| Parameters | EA | Comp. 4 | Comp. 6 | Comp. 8 |
|---|---|---|---|---|
| Molecular weight (g mol−1) | 303.18 | 432.30 | 432.30 | 431.31 |
| H-bond donors | 1 | 1 | 1 | 2 |
| H-bond acceptors | 4 | 4 | 4 | 3 |
| Rotatable bonds | 6 | 8 | 8 | 9 |
| Molar refractivity | 73.67 | 115.48 | 115.48 | 116.05 |
| TPSA (Å2) | 63.60 | 73.22 | 73.22 | 71.19 |
| Bioavailability score | 0.85 | 0.55 | 0.55 | 0.55 |
Lipophilicity (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) P) |
3.61 | 4.86 | 4.12 | 4.49 |
| Solubility (logS) |
−3.96 | −5.39 | −5.39 | −5.58 |
| GI | High | High | High | High |
| LogBB |
−0.313 | −0.704 | −0.715 | −0.378 |
| P-gp substrate | No | No | No | No |
| CYP1A2 inhibitor | No | No | No | Yes |
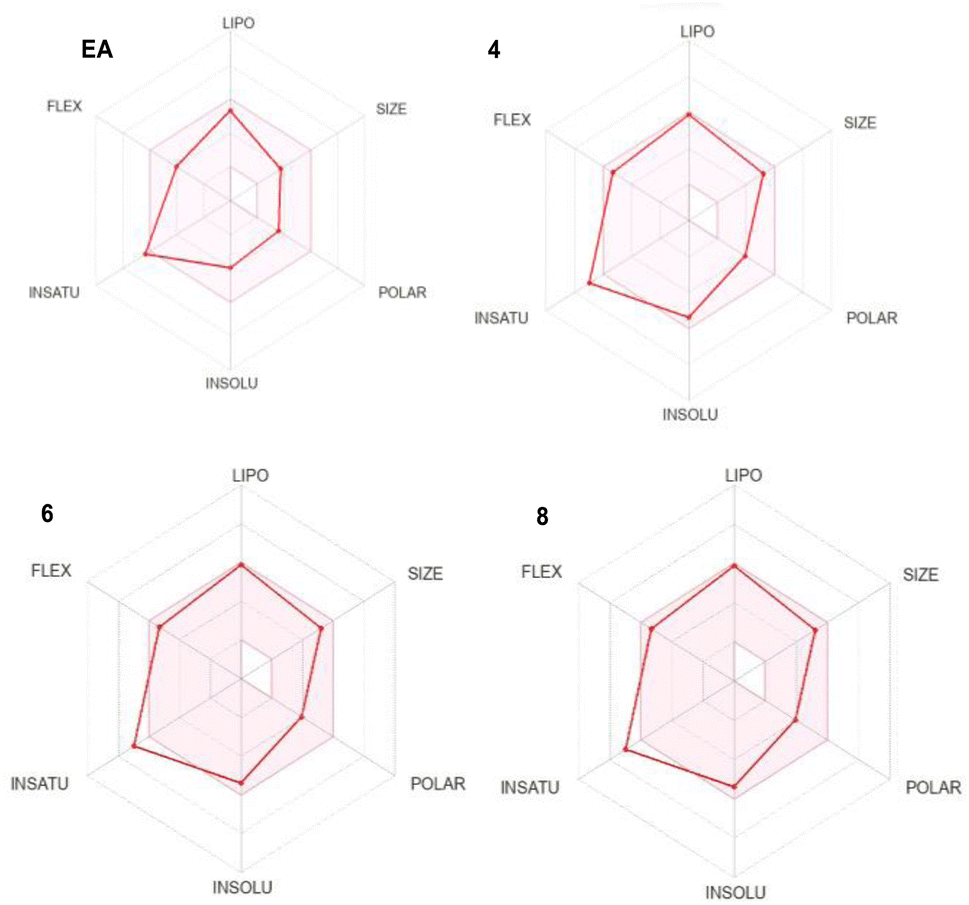 | ||
| Fig. 2 Bioavailability radar of compound EA, 4, 6 and 8. | ||
The investigation of the ADMET properties of our compounds started with an assessment against key drug-likeness criteria derived from Lipinski, Ghose, Veber, and Egan's rules. Notably, all the compounds exhibit clear adherence to all evaluated criteria, suggesting highly favorable pharmacokinetic properties. According to Lipinski's Rule of Five, all the compounds 4, 6 and 8 demonstrate molecular weight below 500 daltons, a logP value under 5, and zero violations for both hydrogen bond donors and acceptors. Ghose's Rule is satisfied, as the compounds falls within the acceptable range for both lipophilicity (logP) and molar refractivity. In accordance with Veber's Rule, the compounds display limited number of rotatable bonds and a topological polar surface area (TPSA) of 73.22, 73.22 and 71.19 Å2 for 4, 6 and 8, respectively, which is below the stipulated threshold of TPSA of 140 Å2. Furthermore, compounds 4, 6 and 8 fall within the specified range outlined by Egan's Rule, indicating a favorable likelihood of oral bioavailability. Regarding the pharmacokinetic properties, all the studied compounds showed good gastrointestinal (GI) absorption, and no CYP1A2 inhibition except compound 8. The blood–brain barrier permeability logBB values of −0.704, −0.715, −0.378 for 4, 6 and 8, respectively, fall within the range associated with compounds capable of accessing the central nervous system (CNS), in accordance with findings by Santiago Vilar et al.33 In summary the overall ADME profile of the three compounds suggests their potential as viable drug candidates.
Antiviral activity
The SARS-CoV-2 inhibitory activities of compounds 4 and 6 have been evaluated in our previous work.34 Both compounds exhibited potent activities with EC50 value of 3.9 μM for compound 4 and EC50 value of 4.8 μM for compound 6. In our continuous efforts to discover new SARS-CoV-2 Mpro inhibitors, we opted to evaluate the potency of compound 8 and its reduced analogue 9 to inhibit the replication of SARS-CoV-2 in vitro. For this purpose, we first conducted cytotoxicity experiments in Vero E6 cells to determine the compound's cytotoxicity. As indicated in Table 2 and Fig. S8,† compound 8 showed no apparent cytotoxicity (CC50 > 100 μM). This value is very far from the cytotoxicity concentration as compared to that of the analogue 9 which presented a CC50 ≥2 μM. Moreover, As assessed using the quantitative VeroE6 cell-based assay with RNA-qPCR, the medians EC50 value of compound 8 is 7.8 μM while that of the analogue 9 exceeds 100 μM (Table 2 and Fig. S8†). This finding highlights the crucial role of the Micheal acceptor moiety in biological activity of this family of compounds.NMR study
In order to check the ability of EA, and its analogues 4, 6 and 8 to form a S–C covalent bond with cysteine 145 (Cys145), a nucleophilic substitution reaction of these compounds with cysteine in the presence of histidine was carried out, as illustrated in Scheme 2. The formation of the corresponding products 10, 11, 12 or 13 in which the sulfur atom of cysteine is covalently linked to the corresponding starting material was monitored and confirmed by NMR spectroscopy.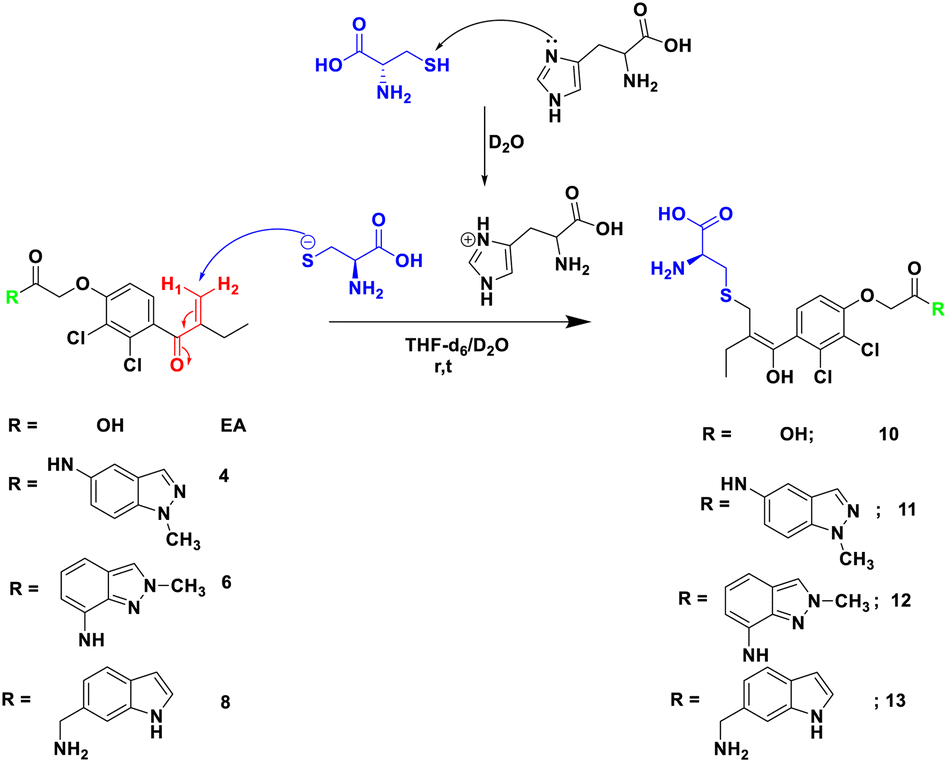 | ||
| Scheme 2 Reaction of Michael acceptor EA, and its analogues 4, 6 and 8 with cysteine in the presence of histidine as base in THF-d6/D2O. | ||
To mimic the chemical reaction that leads to the covalent bond between Cys145 and the inhibitor, we initially mixed cysteine and histidine, histidine serves as a base to remove the proton from the thiol group, which activated the cysteine, followed by the addition of either EA, 4, 6 or 8. The reaction was carried out at room temperature in NMR tubes and monitored using a 1H NMR experiment.
As depicted in Fig. 3, before mixing, the β-carbonyl CH2 of EA appears as two separate singlets at 5.56 and 5.94 ppm. These two characteristic singlets of the Michael acceptor were used to follow the evolution of the rection between EA and cysteine. After 20 min of reaction, the signals intensity decreases by about 70% due to S–C bond formation and after 30 min all signals of the β-carbonyl CH2 of EA completely disappeared (Fig. 3).
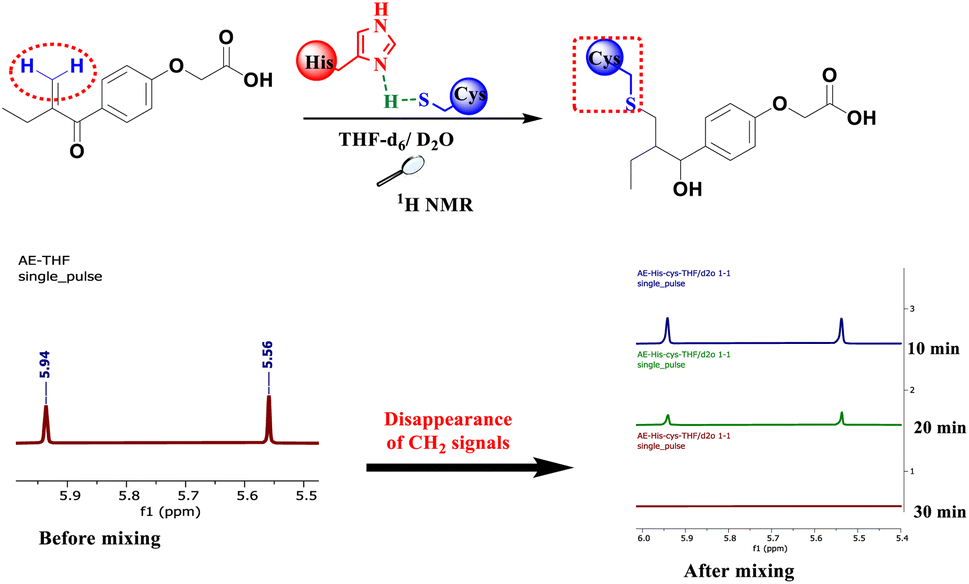 | ||
| Fig. 3 Evolution of the reaction of EA with cysteine in the presence of histidine time. | ||
However, when the same process was applied to compounds 4, 6 and 8, the reaction with cysteine occurs instantaneously as demonstrated by the immediate disappearance of their characteristic CH2 signals at 5.48 and 5.94 ppm for compound 4, 5.58 and 5.96 ppm for compound 6 and 5.51 and 5.93 ppm for compound 8 as depicted in Fig. 4. These reactions were repeated a number of times with varying number of equivalents of the starting materials. The mixtures were analyzed using 1H NMR spectra with 2 equivalents of the related Michael acceptor. The aliphatic zone in Fig. 4 shows both the initial material and its S–C bonded products. Once reaching stoichiometric conditions, the corresponding starting Micheal acceptor disappears immediately.
 | ||
| Fig. 4 Instantaneous reactions of 4, 6 and 8 with cysteine in the presence of histidine. | ||
Compound 9 showed no reaction under the described conditions, confirming that its electrophilic carbonyl center does not react with cysteine to form an S–C covalent bond (Fig. 5).
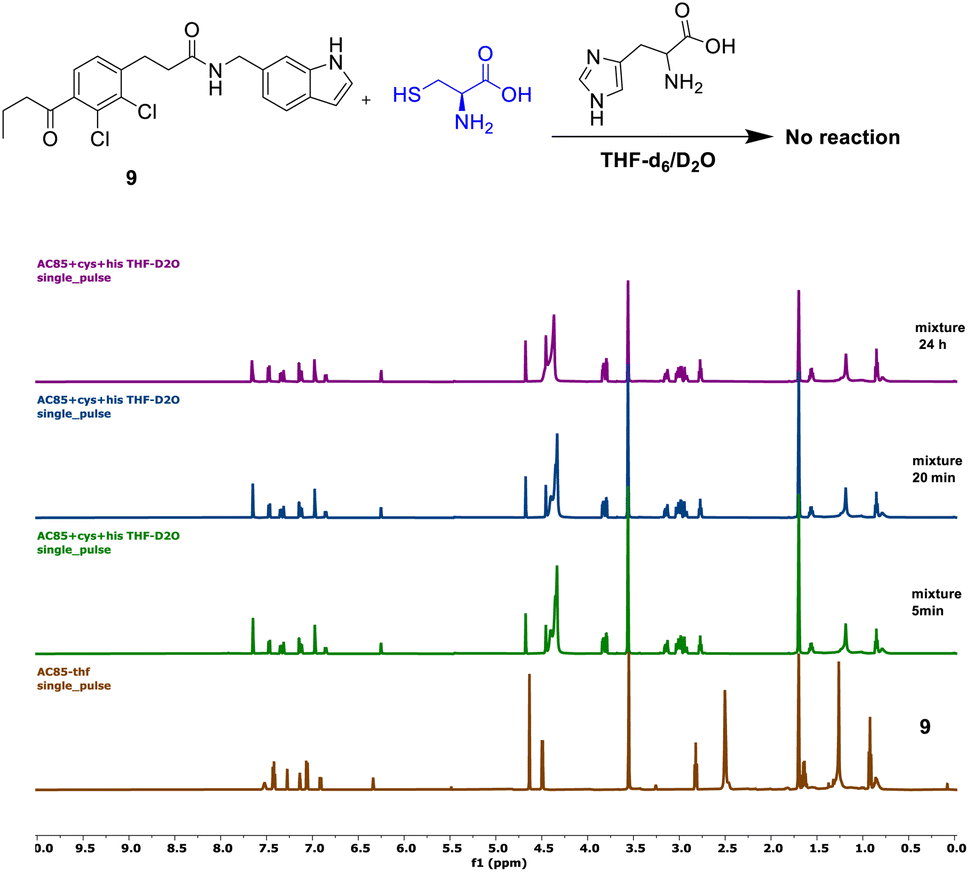 | ||
| Fig. 5 Investigation of the reactivity of 9 towards cysteine. | ||
Carmofur is recognized for its ability to inhibit the SARS-CoV-2 main protease. It covalently alters the Cys145 residue through the formation of a C–S bond.36 Our investigation, utilizing NMR monitoring, examinates the reactivity of carmofur towards cysteine and its effectiveness in forming a carbon-sulfur covalent bond. As shown by 1H NMR, after 2 h, 75% of the starting carmofur was consumed by cysteine. Carmofur's aromatic doublet at 8.30 ppm totally disappeared after 6 hours, being replaced by the proton signal of the new product 14, at 7.05 ppm. The reaction rate is, however, slow compared to reactions with EA analogues, taking 6 hours to fully consume the starting material, perhaps due to carmofur's reactivity, which could be addressed by further optimization of the reaction parameters (Fig. 6).
 | ||
| Fig. 6 Reaction of carmofur with cysteine in the presence of histidine. | ||
Molecular docking
In order to check the ability of the studied compounds to fit into the active site of the Mpro and to create favorable interactions with the catalytic residues, we performed a molecular docking using the protein Mpro (6LU7). Fig. 7 presents the docking score of each compound docked into the enzyme active site as well as, the 3D and 2D visualization of protein-ligand interactions. All the compounds showed good binding energies ranging from −6.5 to −7.3 kcal mol−1. In addition, several interactions have been established with numerous amino acids within the enzyme active site. | ||
| Fig. 7 Molecular docking of compounds 4, 6, 8 and 9 with Mpro (pdb: 6LU7). | ||
Correlation between the chemical reactivity and the biological activity
EA was previously identified as a weak inhibitor of Mpro, displaying only approximately 20% inhibition at 100 μM.37 In contrast, compounds 4 and 6 exhibited potent inhibitory activity against SARS-CoV-2 Mpro.34 Additionally, compound 8 proved to be an effective SARS-CoV-2 inhibitor, as indicated by its EC50 value of 7.8 μM. In addition, compound 9 did not show any inhibitory activity against the virus even at a concentration of 100 μM. Despite these differences in activity, our docking study reveals that all compounds 4, 6 and 8 displayed the ability to interact with the Mpro active site, presenting comparable binding energy values, as illustrated in Fig. 7 and S9.† This highlights that using molecular docking alone can lead to erroneous conclusions by generating false-positive results. However, our new NMR-based approach has demonstrated its ability to rank these compounds according to their reactivity towards cysteine. Indeed, compounds 4, 6, and 8 show a remarkably rapid reaction kinetic compared to EA, which requires about 30 min for complete conversion of the starting material, and compound 9, which did not produce any S–C linked derivative. These results indicate that compounds 4, 6, and 8 are highly reactive and capable of forming the expected S–C bond with the catalytic cysteine of Mpro, which may explain their significant inhibitory activities. These findings provide further evidence that the biological activity of these compounds is directly related to their chemical reactivity.Since the majority of Mpro inhibitors, as mentioned earlier, are competitive inhibitors that interact with Cys145 through covalent bonding, this new screening method was evaluated using a deferent electrophilic warhead such as the amide group present within the structure of carmofur. As expected, a S–C covalent bond has been formed between carmofur and cysteine which is in accordance with the covalent inhibition mode reported by Z. Jin et al.36
In light of the findings in this study, an effective screening tool has been formulated. According to this new tool, a good inhibitor candidate should possess at least one of these two properties: (1) forming a sulfur–carbon bond with cysteine or (2) based on the nature of the electrophilic center, the reaction rate can serve as a benchmark for ranking compounds belonging to the same family based on their respective reactivity rates.
Conclusions
This work introduces an innovative screening approach employing NMR spectroscopy to identify a prospective inhibitor for Mpro and other cysteine proteases. The method focuses on revealing the formation of S–C bonds between the thiol group of cysteine and the Michael acceptors found in EA and its analogues 4, 6, and 8. A reduced analogue of 8, compound 9, failed to afford a S–C bond, explaining its poor in vitro activity. Accordingly, our new method can be used to identify Mpro inhibitors based on their chemical reactivity with cysteine, revealing a direct relationship between inhibitory activity and chemical reactivity. The mechanistic similarity between almost all the cysteine proteases makes our approach applicable to many other protases.Experimental section
NMR experiments
All 1H NMR measurements were performed using a JEOL 600 MHz spectrometer (with JEOL ROYAL HFX Probe). All spectra were recorded at 29815 K after 5 min for thermal equilibration.
• Activation of cysteine (mimic of Mpro's active site)
A mixture of histidine (4.5 mg; 0.02 mmol) and cysteine (3.5 mg; 0.02 mmol) was prepared in NMR tube dissolved in 0.7 mL of D2O to give a neutral solution (pH = 7) (S1).
• Addition of lead inhibitors
✓ Case of EA
To the solution S1, a stoichiometric amount of EA (8.4 mg; 0.02 mmol), in THF-d6 was added. The resulting mixture in THF-d6/D2O (2/1) was then analyzed using 1H NMR experiment at room temperature.
✓ Case of lead compounds 4, 6 and 8.
Because of its instantaneous reaction with cysteine (see result and discussion for more Detaille) first 2 equivalents of 4, 6 or 8 were added to a new S1 solution in NMR tube, a first NMR spectra was obtained. Then, the remaining amount of cysteine and histidine were added to get stoichiometric conditions, this last mixture was analyzed using 1H NMR experiment in the same conditions.
Biological assays
The protocol used follows a prophylactic approach (4 h incubation with 300 TCID50 before in vitro infection). Indeed, cells are incubated in the presence or absence of the tested compounds for 4 h and then infected at an MOI (Multiplicity of Infection) of 0.01 for duration of 48 h in 37 °C under 5% CO2.
The effect on viral production (antiviral effect) in vitro is measured by RT-qPCR and the determination of infectious titers (logTCID50 mL−1) is performed on Vero-E6 cells after 48 h of incubation as described by Reed and Muench method (1938). The ratio of the infectious titer in each condition was expressed as a function of the titer measured in the control condition (without treatment).
Docking studies
Data availability
Source data: crystal structure of SARS-CoV-2 main protease from the protein data bank (https://www.rcsb.org/) using the ID 6LU7 (Mpro in complex with inhibitor N3). Software availability: MGLTools: https://ccsb.scripps.edu/mgltools/downloads/, AutoDock: https://autodock.scripps.edu/download-autodock4/, BIOVIA discovery studio: https://discover.3ds.com/discovery-studio-visualizer-download. Underlying data: All the data underlying the results of this study are available as part of this article and ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was carried out with the assistance of the Hassan 2 Academy of Sciences and Technologies. The authors are grateful to the Hassan II Academy of Sciences and Techniques and the Euromed University of Fes for funding. The authors are also grateful to the Euromed University of Fes for providing their facilities. AC is grateful for the Euromed University for the scholarship.References
- S. Verma, R. Dixit and K. C. Pandey, Front. Pharmacol, 2016, 7, 193290 Search PubMed.
- C. López-Otín and J. S. Bond, J. Biol. Chem., 2008, 283, 30433 Search PubMed.
- M. B. Rao, A. M. Tanksale, M. S. Ghatge and V. V. Deshpande, Microbiol. Mol. Biol. Rev., 1998, 62, 597 CAS.
- A. Rawat, M. Roy, A. Jyoti, S. Kaushik, K. Verma and V. K. Srivastava, Microbiol. Res., 2021, 249, 126784 CrossRef CAS PubMed.
- S. P. M. Lutgens, K. B. J. M. Cleutjens, M. J. A. P. Daemen and S. Heeneman, FASEB J., 2007, 21, 3029–3041 CAS.
- L. Yu, M. Yin, X. Yang, M. Lu, F. Tang and H. Wang, Can. J. Physiol. Pharmacol., 2018, 96, 60–67 CrossRef CAS PubMed.
- K. Anand, J. Ziebuhr, P. Wadhwani, J. R. Mesters and R. Hilgenfeld, Science, 2003, 300, 1763–1767 CrossRef CAS PubMed.
- Z. Jin, et al., Nature, 2020, 582, 289–293 CAS.
- L. Zhang, D. Lin, X. Sun, U. Curth, C. Drosten, L. Sauerhering, S. Becker, K. Rox and R. Hilgenfeld, Science, 2020, 368, 409–412 CAS.
- H. Yang and J. Yang, RSC Med. Chem., 2021, 12, 1026–1036 CAS.
- L. A. Beltrán, S. De La Hoz-Rodríguez, L. B. Iserte, S. Rodríguez, A. Fernández-De-La-pradilla and F. V. González, Molecules, 2022, 27, 2523 Search PubMed.
- B. X. Quan, et al., Nat. Microbiol., 2022, 7, 716–725 CrossRef CAS PubMed.
- M. Marzi, M. K. Vakil, M. Bahmanyar and E. Zarenezhad, Biomed Res. Int., 2022, 7, 7341493 Search PubMed.
- W. Dai, et al., Science, 2020, 368, 1331–1335 CAS.
- J. Qiao, et al., Science, 2021, 371, 1374–1378 CrossRef CAS PubMed.
- X. Chen, et al., Nat. Microbiol., 2024, 9, 1075–1088 CAS.
- G. La Monica, A. Bono, A. Lauria and A. Martorana, J. Med. Chem., 2022, 65, 12500–12534 CrossRef CAS PubMed.
- S. I. Hattori, et al., MBio, 2020, 11, 1–16 Search PubMed.
- C. Ma, et al., Cell Res., 2020, 30, 678–692 CrossRef CAS PubMed.
- G. Amendola, et al., J. Chem. Inf. Model., 2021, 61, 2062–2073 CAS.
- G. Yang, X. Mi, Y. Wang, S. Li, L. Yu, X. huang, S. Tan and H. Yu, Bioorg. Chem., 2023, 134, 106467 CrossRef CAS PubMed.
- A. El Abbouchi, N. El Brahmi, M. A. Hiebel, H. Ghammaz, E. El Fahime, J. Bignon, G. Guillaumet, F. Suzenet and S. El Kazzouli, Molecules, 2023, 28, 910 CAS.
- M. Liu, B. Xu, Y. Ma, L. Shang, S. Ye and Y. Wang, Antiviral Res., 2021, 192, 105102 CrossRef CAS PubMed.
- S. T. Liang, C. Chen, R. X. Chen, R. Li, W. L. Chen, G. H. Jiang and L. L. Du, Front. Pharmacol, 2022, 13, 1033003 CAS.
- S. Krishnan, R. M. Miller, B. Tian, R. D. Mullins, M. P. Jacobson and J. Taunton, J. Am. Chem. Soc., 2014, 136, 12624–12630 CAS.
- O. M. Lage, M. C. Ramos, R. Calisto, E. Almeida, V. Vasconcelos and F. Vicente, Mar. Drugs, 2018, 16, 279 Search PubMed.
- J. P. Hughes, S. S. Rees, S. B. Kalindjian and K. L. Philpott, Br. J. Pharmacol., 2011, 162, 1239–1249 CAS.
- P. Szymański, M. Markowicz and E. Mikiciuk-Olasik, Int. J. Mol. Sci., 2012, 13, 427 Search PubMed.
- F. Ver Donck, K. Downes and K. Freson, J. Thromb. Haemostasis, 2020, 18, 1839–1845 CrossRef PubMed.
- X. X. Yang, W. Gu, L. Liang, H. L. Yan, Y. F. Wang, Q. Bi, T. Zhang, J. Yu and G. X. Rao, RSC Adv., 2017, 7, 3089–3100 CAS.
- M. Naas, S. El Kazzouli, E. M. Essassi, M. Bousmina and G. Guillaumet, J. Org. Chem., 2014, 79, 7286–7293 CAS.
- M. Cheung, A. Boloor and J. A. Stafford, J. Org. Chem., 2003, 68, 4093–4095 CAS.
- S. Vilar, M. Chakrabarti and S. Costanzi, J. Mol. Graphics Modell., 2010, 28, 899–903 CAS.
- S. El Kazzouli, N. Touil, E. El Fahime, A. El Abbouchi, M. Hemlali, N. El Brahmi, A. El Alaoui, S. Bounou and M. Bousmina, Chinese Pat., CN 117730074A, 2024.
- C. Isgrò, A. M. Sardanelli and L. L. Palese, Viruses, 2021, 13, 106 Search PubMed.
- Z. Jin, et al., Nat. Struct. Mol. Biol., 2020, 27, 529–532 CrossRef CAS PubMed.
- U. Kaeppler, et al., J. Med. Chem., 2005, 48, 6832–6842 CAS.
- F. Amanat, et al., Curr. Protoc. Microbiol., 2020, 58, e108 CAS.
- D. E. Gordon, et al., Nature, 2020, 583, 459–468 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04938a |
| This journal is © The Royal Society of Chemistry 2024 |