
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface chemistry and catalytic activity in H2O2 decomposition of pyrolytically fluoralkylated activated carbons†
Gauhar Mussabek‡
 ab,
Saule Baktygereyab,
Yerzhan Taurbayeva,
Dana Yermukhamedab,
Nazym Zhylkybayevaab,
Alexander N. Zaderkoc,
Vitaliy E. Diyuk‡d,
Sergii Afonine,
Gulmira Yar-Mukhamedovaa,
Ruslan T. Mariychukf,
Liudmyla M. Grishchenkod,
Mária Kaňuchovág and
Vladyslav V. Lisnyak*adhi
ab,
Saule Baktygereyab,
Yerzhan Taurbayeva,
Dana Yermukhamedab,
Nazym Zhylkybayevaab,
Alexander N. Zaderkoc,
Vitaliy E. Diyuk‡d,
Sergii Afonine,
Gulmira Yar-Mukhamedovaa,
Ruslan T. Mariychukf,
Liudmyla M. Grishchenkod,
Mária Kaňuchovág and
Vladyslav V. Lisnyak*adhi
aNanotechnological Laboratory of Open Type, Al-Farabi Kazakh National University, 050040 Almaty, Kazakhstan
bInstitute of Information and Computational Technologies, 050012 Almaty, Kazakhstan
cLight Matter Institute, UMR-5306, Claude Bernard University of Lyon/CNRS, Université de Lyon, 69622 Villeurbanne Cedex, France
dChemical Faculty, Taras Shevchenko National University of Kyiv, 01033 Kyiv, Ukraine. E-mail: lisnyak@univ.kiev.ua
eInstitute of Biological Interfaces (IBG-2), Karlsruhe Institute of Technology, POB 3640, 76021 Karlsruhe, Germany
fDepartment of Ecology, Faculty of Humanities and Natural Sciences, University of Presov, 08001, Presov, Slovakia
gInstitute of Earth Resources, Faculty of Mining, Ecology, Process Control and Geotechnology, Technical University of Kosice, 042 00 Kosice, Slovakia
hWestern Caspian University, AZ 1001 Baku, Republic of Azerbaijan
iInstitute of Macromolecular Chemistry, The National Academy of Sciences of Ukraine, 02160, Kyiv, Ukraine
First published on 12th September 2024
Abstract
According to the proposed pyrolytic method, granular activated carbon (AC) Norit 830 W was functionalized by thermal treatment of AC in hydrofluorocarbon (HFC) gases, pentafluoroethane and 1,1,1,2-tetrafluoroethane, at 400–800 °C. This method does not require activation by plasma and photons. Chemical and elemental analysis showed that the pyrolytic treatment provides a loading of 2.95 mmol (5.6 wt%) of fluorine per gram of AC. Nitrogen adsorption measurements indicated that the microporous structure contracted when AC was treated with HFC at temperatures above 400 °C. Thermogravimetry, Fourier transform infrared spectroscopy (FTIR) with attenuated total reflectance (ATR), and X-ray photoelectron spectroscopy (XPS) demonstrated the evolution of oxygen-containing and fluorine-containing groups to more thermostable groups with treatment temperature. The fluorine-containing groups grafted at high temperature, above 600 °C exhibited the highest thermal stability up to 1250 °C in dry argon. From the data of XPS and solid-state 19F nuclear magnetic resonance spectroscopy data, the grafted fluorine exists in several types of grafted F-containing groups, the HFC residues. By changing the thermal regime of fluorination, the composition of fluorine-containing groups on a carbon surface can be regulated. Isolated fluoroalkyl groups can be grafted at temperatures of 400–500 °C, while at 600 °C and above, the semi-ionic fluorine groups increase significantly. The hydrophobized surface demonstrated the ability to effectively decompose H2O2 in methanol solutions.
1. Introduction
In recent years, fluorinated carbon materials have attracted considerable interest from researchers in various fields.1 The valuable properties that can be obtained by fluorination, such as unsurpassed superhydrophobicity, enhanced conductivity, and specific adsorption, are significant additions to the unique properties of conventional carbons.2 One potential area for their use is in energy technologies, including power generation and energy harvesting.3 Currently, the most common of several methods4 for obtaining fluorinated carbon materials is the use of chemical reactions with elemental fluorine, which effectively modifies the physicochemical parameters of carbon materials.5 Plasma methods, such as plasma deposition of fluorocarbon films obtained from CH2F2/Ar and C2H2F4/Ar gas mixtures as fluorocarbon plasma sources, and plasma fluorination with CF4, C4F8, and C6F6 at ambient temperature, are the methods occupying second place.6 An alternative approach is to use anhydrous fluorinating systems, such as KF/18-crown-6/acetonitrile, or even HF for substitutive fluorination.7In the paradigm of gas-phase reactions, hydrofluorocarbons (HFCs) deserve special attention. Today, HFCs are known to be used, after activation, as reagents in organic synthesis, but their use is limited to the production of low-molecular-weight fluorinated organic compounds; most typically, they serve as intermediates in the so-called “building block” approach to obtaining effective pharmaceutical and agrochemical active ingredients.8 On the other hand, various strategies are currently used to reduce HFC emissions, including recovery methods, incineration, plasma treatment, catalytic decomposition, and pyrolytic and hydrolytic treatment technologies.9 Previously, some works10 have reported the results of gas-phase and plasma pyrolysis of tetrafluoroethane C2H2F4 (HFC-134a) and pentafluoroethane C2HF5 (HFC-125); these HFCs were also decomposed using oxygen and catalysts. It should be noted that both HFCs are interesting candidates that could be active fluorinating reagents under special conditions. Presumably, after temperature activation or under pyrolytic conditions, these HFCs can potentially be used as a source of fluorine for various fluorination purposes, taking into account that most published research and industry guidelines suggest that complete destruction of these HFCs requires high temperatures approaching 1000 °C.9
Among the general considerations for selecting the right fluorination or fluoroalkylation process, high-temperature pyrolytic decomposition of HFCs over heated carbon solids is a promising way that should be considered in the search for easily scalable fluorination methods that can be applied to various carbon solids.
Of all the carbon solids, activated carbon (AC) is an interesting model object that has been accepted to be tested for halogenation by reaction with double C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds. In principle, the carbon surface of AC, taken as is, can undergo addition reactions to double CC bonds, such as bromination,11 as is, or alternatively, these reactive CC bonds can be formed by thermal heating of the oxidized AC.12 In this approach, the surface oxygen-containing groups serve as sites for modification and can be replaced by other halogen groups.
C bonds. In principle, the carbon surface of AC, taken as is, can undergo addition reactions to double CC bonds, such as bromination,11 as is, or alternatively, these reactive CC bonds can be formed by thermal heating of the oxidized AC.12 In this approach, the surface oxygen-containing groups serve as sites for modification and can be replaced by other halogen groups.
The current synthetic strategy of functionalizing AC with HFCs in the gas phase, followed by heating and pyrolysis, presents a unique set of challenges and opportunities that are briefly outlined below. The challenges are related to controlling the degree of functionalization, as it is difficult to achieve the exact degree of functionalization because the gas phase reaction conditions must be carefully controlled to avoid over- or under-functionalization, which is the first problem.13
The second problem is uniformity, as it is difficult to ensure a uniform distribution of functional groups over the AC surface, resulting in heterogeneous properties. Reaction conditions are highly dependent on temperature management: maintaining optimal temperatures during heating of the AC and pyrolysis of the HFC is critical.14
Too high or too low temperatures can adversely affect the functionalization process and the structural integrity of the carbon solids. In addition, the reactivity of HFCs at high temperatures has not been extensively studied; the product of HFC thermolysis can be highly reactive and corrosive, requiring specialized equipment to handle it safely and effectively; in addition, there are environmental and health concerns with possible toxic by-products formed during pyrolysis that pose environmental and health risks; emissions from these processes require advanced scrubbing and filtration systems.15
The special case is the resulting material stability against degradation: the functional groups introduced via the HFC route may not be stable under all operating conditions, resulting in potential degradation of material performance over time, as has been observed with some fluorocarbons.16
Potentially, HFCs and their degradation products can affect structural integrity, as prolonged exposure to high temperatures and reactive gases can erode the porous architecture of carbon solids; similar processes can cause etching and cutting of carbon nanomaterials.17
In addition, there are issues of process cost and scalability, as the cost of HFCs and the specialized equipment required for gas-phase functionalization can be high, while scaling up the process from laboratory to industrial scale without losing efficiency or control over product quality is challenging, as previously demonstrated for related fluorine-free processes and certain carbon materials.18
Against this background, notable advances of the proposed synthesis are potentially enhanced functional properties of AC, since this strategy in principle allows targeted functionalization for the introduction of specific functional groups, as has already been done in similar routes for oxygen- and nitrogen-containing groups, improving adsorption properties, catalytic activity, and other desired properties of AC.19
In addition, the gas-phase approach can be adapted to introduce a wide range of functional groups, making it a versatile strategy for tailoring AC for different applications, as has been demonstrated for other processes such as gas-phase oxidation.20
If the gas-phase approach can be enhanced by process control with precision equipment, advances in reaction chamber design and temperature control systems will consequently improve the precision and reliability of the functionalization process, while the incorporation of real-time monitoring techniques, such as in situ/operando spectroscopy techniques,21 will allow better control and optimization of the functionalization process.
The other concerns mentioned above can be addressed with abatement technologies that reduce emissions, as the development of advanced scrubbers, thermal oxidizers and new photocatalytic approaches22 have mitigated some of the environmental and health concerns associated with halogenated compounds.
There are also potentially safer alternatives, and our research conducted with less toxic halogenated compounds and alternative functionalizing reagents is ongoing. It should be noted that the use of the right candidates could potentially reduce the environmental footprint of the process. Improved preparation methods can be used to stabilize introduced functional groups, resulting in longer lasting performance of functionalized AC with application specific tailoring. However, advances in the understanding of the interaction between functional groups and target HFC molecules or their residues will allow more precise tailoring of the AC surface for specific applications, such as water separation and purification and selective adsorption of gases.23
Economic feasibility shows the potential for cost reduction, as ongoing research is providing more cost-effective synthesis routes with bulk production techniques, gradually reducing the overall cost of the process. In terms of industrial adoption, as the technology matures, more industries can adopt gas-phase functionalization methods, leading to economies of scale and further cost reductions.
In summary, the synthetic strategy of functionalizing activated carbon with HFCs in the gas phase, followed by heating and pyrolysis, is a promising approach that offers significant potential for improving the properties of AC. However, it also presents several challenges related to process control, environmental impact, and material stability. Continued advances in reaction technology, environmental mitigation and materials science are addressing these challenges and making the process more viable and effective for industrial applications.
Since the chemical functionalization and transformation of the surface layer of AC and other carbon materials, under the influence of temperature-activated HFCs and the pyrolysis products of HFCs is, to the best of our knowledge, still terra incognita, we focused on the chemical processes of carbon surface modification. The following knowledge gaps were identified: (i) lack of information on the high temperature or thermally stimulated reactions between HFCs, which are used in many applications, and AC; (ii) the rules of fluorination of carbon surfaces are not studied, so that the possibility of producing surfaces with different F-containing coverage at medium to high temperatures, above 400 °C and below 900 °C, is questionable and not explored; (iii) the reactivity of carbon surfaces treated with HFC pyrolysis products at high temperatures to protect them from the oxidative action of hydrogen peroxide (H2O2) in water and alcohol media for continuous use in renewable energy applications is worth investigating.
In this study, we applied high-temperature pyrolysis of HFCs over Norit® granular AC. The pyrolysis treatment was performed in argon-diluted gaseous asymmetric 1,1,1,2-tetrafluoroethane (F4) or pentafluoroethane (F5). In contrast to our previous study,24 in which Norit 830 W was selected for the lower temperature treatment, the temperature range for the treatment was extended to 800 °C, and the products of the treatment with F4 and F5 were investigated by various physicochemical and spectrochemical methods. We hypothesize that the proposed high-temperature regime will result in a higher fluorine content in the resulting AC, at least an order of magnitude higher than that previously found.24,25 In contrast to the reported fluorine doping,24 we propose that novel fluorine-containing ACs obtained by a pyrolytic technique will combine the AC texture preservation with a fluorinated surface.
The specific catalytic behavior of the fluorinated carbon surface in the decomposition of H2O2 in protic and aprotic solvents is investigated. The pyrolysis treatment as a universal approach has the potential to prevent the degradation mechanisms that can occur with carbon electrodes in fuel cells during, e.g., during the oxygen reduction reaction.26
The sole decomposition of H2O2, as it is, has been partially studied only on some granular ACs; many works reported the oxidation of various organic pollutants with H2O2 over AC catalysts,27 suggesting that the process intensity can be improved as it depends on the particle size, specific surface area, pore size distribution, surface groups, and hydrophilicity/hydrophobicity of ACs.28
Typically, pH tests show that H2O2 has improved stability at low pH levels, which is of interest. An acidic medium is advantageous when using H2O2 as an oxidant because its decomposition is suppressed, and it has the highest oxidation potential. The latter decreases up to twofold with increasing pH, from E° = 1.63 V at pH = 0 to E° = 0.878 V at pH = 14.29 The decomposition of H2O2 can yield hydroxyl radicals HO˙, whose oxidation potential varies with the pH of the water solution and gives best results in the acidic pH range of 2.5 to 4 (E° = 2.8 V).30 For optimal stability, the pH range of pure H2O2 should be below 4.5. Above pH = 5, the H2O2 decomposition increases sharply, and it is known that a basic pH is favorable for the H2O2 decomposition to produce HO˙ and OH−.31
One hypothesis for the oxidation mechanism of organic pollutants in the presence of H2O2 is based on the porous structure of AC, which is thought to promote the formation of OH˙.27,28 However, the reaction mechanism of H2O2 decomposition, when AC is used as a catalyst, is not yet fully understood. Reactions (I) and (II) illustrate the catalytic decomposition of H2O2 in the presence of AC.
| AC + H2O2 → AC+ OH− + HO˙ | (I) |
 | (II) |
It is generally accepted that this process depends on both the porosity of the carbon material and the chemical properties of the surface.32
Surprisingly, no information is available on the effect of the proper fluorination or fluoroalkylation process on the catalytic behavior of the carbon surface with respect to the H2O2 decomposition reaction. Moreover, the catalytic potential of fluorinated carbons in the H2O2 decomposition reaction has not been studied at all; these materials are mainly used as hydrophobic supports and carriers for catalytically active metals or metal-non-metal compositions due to the possibility of controlling the strength of the catalyst–support interactions.33
2. Experimental
2.1 Materials
All reagent grade chemicals used and further mentioned were purchased from Merck and Sigma-Aldrich. Norit® granular AC 830 W was provided for use by EnergoChemService (Kyiv, Ukraine, http://www.exs.com.ua/). In the pretreatment step, the AC samples with an effective size D10 of 0.6–0.7 mm were deashed sequentially with 10% (w/v) hydrochloric acid (HCl), then intensively washed with 5% (w/w) sodium carbonate (Na2CO3), followed by double-distilled water until the wash solution had a neutral pH. The deashed powders were then filtered and air-dried at 120 °C for 12 hours. Each of the Arkema™ Forane® HFC, F4 (C2H2F4), or F5 (C2HF5) was mixed with pure argon gas (Air Gas, Kyiv, Ukraine, http://www.dpairgas.com.ua) to be used in the treatment as an equivolume of F4(F5)-Ar gas mixtures.2.2 Preparation
In a typical treatment procedure, a gas mixture of 50 vol% HFC in argon was introduced into the heated alundum reactor containing the AC sample. The total gas flow was set at 50 mL min−1§ under the control of Platon NGX flowmeters. The reactor temperature was set at 400, 500, 600, 700, and 800 °C using a programmable thermal controller. At the set temperature, the AC was maintained in the HFC gas (F4 or F5) for one hour.¶ The furnace was then naturally cooled to 350 °C. At this temperature, the gas supply was stopped, and the reactor was vented with argon until the furnace reached a temperature of 30 °C. The purpose of this procedure was to remove physisorbed HFC and HF. The fluoroalkylated AC powder was then collected and transferred from the reactor vessel to a Pyrex weighing bottle, where it was stored in a vacuum desiccator under CaCl2. The resulting fluoroalkylated AC was designated as AC-F#-T, where F# is F4 or F5, and T is the treatment temperature. Prior to characterization, the samples were intensively washed with rinse water, and then filtered and dried in an air dryer at 120 °C.2.3 Characterization
CHNS analysis was performed on a PerkinElmer Series II 2400 instrument according to the manufacturer's specifications. The fluorine concentration (CF) in the prepared samples was determined after their pyrohydrolytic decomposition by potentiometric ion-selective measurement of fluoride ions.34The fluorinated AC samples were examined by scanning and transmission electron microscopy (SEM and TEM). Surface morphology and carbon structure imaging were performed on Zeiss EVO-50 SEM and Jeol 1400F TEM instruments, respectively. Electron diffraction patterns (EDPs) were collected during the TEM experiments. The samples were analyzed by energy-dispersive X-ray analysis (EDX) using an Oxford INCA Energy 350 EDX System.
Fourier transform infrared attenuated total reflection (FTIR-ATR) spectra were recorded on a Shimadzu Prestige 21 IR spectrometer using the Pike MIRacle™ ATR accessory with a ZnSe crystal.
Fluorine chemistry (content) was analyzed by solid-state 19F NMR under magic angle spinning (19F-ssNMR). AC samples were ground to a fine powder and filled into Bruker ZrO2 rotors with an outer diameter of 2.5 mm. The 19F-ssNMR spectra were acquired with a Bruker Avance III spectrometer operating at a 564.7 MHz 19F resonance frequency (600 MHz for 1H). The spectra were acquired using a Bruker triple-tuned 1H/19F/X MAS probe and a 1H/19F frequency splitter, using single pulse excitation (2.5 μs 90° pulse length) under 22 kHz magic angle spinning (MAS) and 50 kHz 1H decoupling. All spectra were processed using TopSpin software (Bruker) without line broadening. The rotor 19F background signal was measured and subtracted from all samples. Spectra were referenced relative to CFCl3 using externally acquired signals from crystalline flufenamic acid.
Nitrogen adsorption–desorption isotherms were measured on a Quantachrome Autosorb-6 analyzer at −196 °C. Textural parameters were determined using a routine provided by the ASWin 2.0 porosimeter software. The pore size distribution (PSD) was simulated using the quenched solid density functional theory (QSDFT) method. Packing density (ϕ) was measured using the weighing technique.
X-ray photoelectron spectroscopy (XPS) analysis was performed on a SPECS spectrometer equipped with Al Kα monochromatized radiation at 1486.6 eV. The fitting procedure was performed within the SpecsLab2 Casa XPS software for C 1s, O 1s, and F 1s envelopes.35 Additional XPS spectra were recorded on a JSPM 4610 spectrometer.
Thermogravimetric analysis (TGA) in a dynamic argon gas was performed on dry samples. Thermogravimetric and differential thermal gravimetric (TG/DTG) thermograms were recorded at a heating rate of 10 °C min−1 under an argon flow of 100 mL min−1.
Powder X-ray diffraction (PXRD) patterns were collected at 25 °C on a DRON-3 X-ray diffractometer; diffraction intensities were measured using a continuous coupled 2θ/θ scan with Ni-filtered CuKα radiation (λ = 1.5406 Å).
2.4 Catalytic decomposition of H2O2
The catalytic decomposition of H2O2 was carried out at atmospheric pressure and under isothermal conditions at 25 °C. The kinetic patterns of H2O2 decomposition were measured using a volumetric method by recording the volume of O2 evolving from the reaction at different time intervals for at least 1 h. For these measurements, a thermostatic measuring cell was filled with a selected solvent (15 mL), deionized water, or methanol. Then, 100 mg of catalyst was dispersed in 15 mL of deionized water or methanol to prepare a suspension. The reaction system, kept in motion by magnetic stirrers with Teflon blades and continuously weighed by an automatic balance with PC data recording, was maintained at the desired temperature by a water bath. Each prepared reaction medium was buffered with phosphate buffer (pH 6.86). H2O2 decomposition was then initiated by injecting 0.5 mL of 7.69 M H2O2 solution into the reaction system. To monitor the progress of the catalytic reaction, the volume of oxygen evolved during the decomposition of H2O2 in the presence of the catalyst was measured with a precision gas volumeter, and the molar concentration of H2O2 at each time point was calculated using the formula
 | (1) |
3. Results and discussion
3.1 Microanalysis
Table 1 summarizes the quantitative analysis data. They show that the prepared samples contain fluorine from 0.4 to ∼4.4 at%. After the pyrolytic treatment with HFCs, the resulting carbon solids were deashed up to a residual ash content of 0.5 wt%, sulfur and nitrogen contents were reduced to 0–0.1 at% and 0.2 at%, respectively.| Sample | Quantitative analysis (at%) | |||||
|---|---|---|---|---|---|---|
| CHNS | EDX | |||||
| C | H | N | S | aO | F | |
| a Calculated as total sample weight corrected for ash content, minus total sample weight determined by CHNS analysis. | ||||||
| 830W24 | 94.6 | 0.7 | 0.3 | 0.2 | 4.2 | 0 |
| AC-F4-400 (ref. 24) | 95.0 | 0.6 | 0.3 | 0.1 | 4.0 | 0.4 |
| AC-F4-500 (ref. 24) | 95.5 | 0.4 | 0.2 | 0.1 | 3.8 | 0.5 |
| AC-F4-600 | 95.7 | 0.5 | 0.2 | ∼0 | 3.6 | 3.7 |
| AC-F4-700 | 95.7 | 0.4 | 0.2 | ∼0 | 3.7 | 3.5 |
| AC-F4-800 | 95.5 | 0.4 | 0.2 | ∼0 | 3.9 | 3.3 |
| AC-F5-400 (ref. 24) | 96.1 | 0.7 | 0.3 | ∼0 | 2.9 | 0.2 |
| AC-F5-500 (ref. 24) | 96.0 | 0.6 | 0.2 | ∼0 | 3.2 | 0.9 |
| AC-F5-600 | 95.3 | 0.5 | 0.3 | 0.1 | 3.8 | 4.4 |
| AC-F5-700 | 95.6 | 0.3 | 0.2 | ∼0 | 3.9 | 3.5 |
| AC-F5-800 | 95.3 | 0.5 | 0.2 | ∼0 | 4.0 | 3.2 |
For AC-F4-T series samples, the fluorine concentration increases with the treatment temperature in F4, reaching a maximum of 3.7 at% at 600 °C. However, it decreases significantly when the treatment temperature is increased to 700–800 °C. Similarly, for AC-F5-T series samples, the fluorine content shows a consistent increasing trend with the treatment temperature up to 600 °C. However, above this temperature, a gradual decrease in the fluorine content from 4.4 to 3.2 at% is observed.
Samples AC-F5-600 and AC-F4-600 showed the highest fluorine content. This result can be attributed to a synergistic action of two primary factors: the efficient pyrolysis of HFCs and the dominance of the strong adsorption (chemisorption) of fluorinated species on the carbon surface, which outweighs the thermal desorption processes at a temperature of 600 °C.
Conversely, elevated temperatures may be a reason for the pyrohydrolysis and thermal decomposition of certain fluorine-containing surface groups that occur when AC is treated with HFCs at temperatures above 600 °C. Pyrohydrolysis and thermal decomposition result in thermal desorption that removes surface hydrogen fluoride and other complex fluorine-containing compounds, respectively.
The limited reactivity of the carbon matrix at temperatures below 600 °C, along with the inability of the pyrolysis process to facilitate high levels of HFC conversion at such temperatures, may be a reason for incomplete engagement of the available active centers of the AC surface in fluorination and fluoroalkylation reactions at 400 and 500 °C.
After the high-temperature treatment in both HFCs, SEM micrographs showed surface relief without any visible morphological changes at the macroscale level (Fig. 1). The macroporous structure of the pristine AC is maintained after the pyrolytic treatment, suggesting the non-erosive effect of the high-temperature treatment in HFCs and the chemical grafting of fluorine onto the carbon surface.
 | ||
| Fig. 1 SEM micrographs of (a) pristine AC, (b) AC-F4-600, and (c) AC-F5-600. | ||
3.2 Porous structure analysis
Table 2 shows an increase in the packing density ϕ with increasing treatment temperature, which is found for both series of samples, indicating that the microporosity decreases at higher treatment temperatures. However, for the AC-F5 series samples, the ϕ value shows a certain decrease above 700 °C.| Sample | CF (mmol g−1) | ϕ (g cm−3) | Texture parameters | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Specific surface area (m2 g−1) | Specific volumes (cm3 g−1) | ||||||||||
| SBET | Smic | Smes | Smic/Smes | Vtot | Vmic | Vmes | Vmic/Vmes | VDR | |||
| AC24 | 0.0 | 0.433 | 1254 | 1156 | 98 | 11.80 | 0.630 | 0.508 | 0.122 | 4.16 | 0.447 |
| AC-F4-400 (ref. 24) | 0.11 | 0.424 | 1062 | 937 | 125 | 7.50 | 0.592 | 0.435 | 0.157 | 2.77 | 0.370 |
| AC-F4-500 (ref. 24) | 0.39 | 0.429 | 1110 | 998 | 112 | 8.91 | 0.565 | 0.441 | 0.124 | 3.56 | 0.384 |
| AC-F4-600 | 1.93 | 0.489 | 902 | 815 | 87 | 9.37 | 0.524 | 0.410 | 0.114 | 3.60 | 0.388 |
| AC-F4-700 | 1.87 | 0.525 | 807 | 727 | 80 | 9.09 | 0.449 | 0.348 | 0.101 | 3.45 | 0.325 |
| AC-F4-800 | 1.75 | 0.548 | 706 | 637 | 69 | 9.23 | 0.370 | 0.283 | 0.087 | 3.25 | 0.256 |
| AC-F5-400 (ref. 24) | 0.09 | 0.368 | 1269 | 995 | 274 | 3.63 | 0.724 | 0.460 | 0.264 | 1.74 | 0.422 |
| AC-F5-500 (ref. 24) | 0.72 | 0.385 | 1192 | 1031 | 161 | 6.40 | 0.634 | 0.465 | 0.169 | 2.75 | 0.417 |
| AC-F5-600 | 2.30 | 0.475 | 941 | 815 | 126 | 6.47 | 0.508 | 0.362 | 0.146 | 2.48 | 0.328 |
| AC-F5-700 | 2.94 | 0.500 | 1051 | 911 | 140 | 6.51 | 0.554 | 0.407 | 0.147 | 2.77 | 0.367 |
| AC-F5-800 | 1.69 | 0.459 | 1156 | 1005 | 151 | 6.66 | 0.596 | 0.447 | 0.149 | 3.00 | 0.404 |
As can be seen from Fig. 2a and b, all isotherms for the studied sorbents, AC treated with F4 and F5 at 400–800 °C, are of Type I and IV according to the IUPAC classification.36
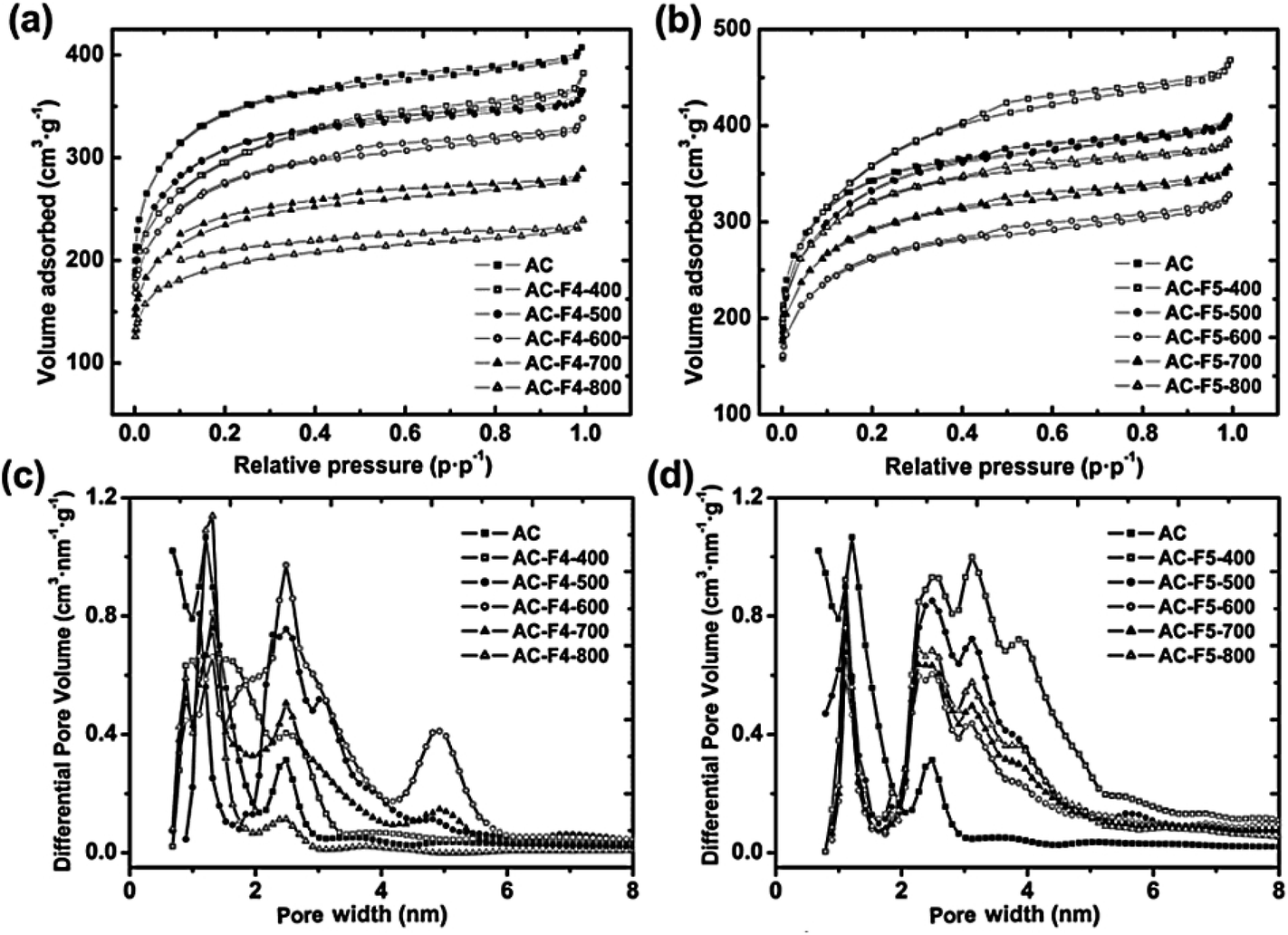 | ||
| Fig. 2 (a and b) N2 adsorption–desorption isotherms and (c and d) the differential pore volume vs. pore width for AC treated with F4 (a and c) or F5 (b and d). | ||
Changes in the microporous structure, caused by the fluorine grafting on the carbon surface, are responsible for the observed differences in the shape of the isotherms.
The recorded isotherms showed a hysteresis of H4 type from narrow slit-like pores of the irregular shape with a wide PSD. Table 2 compares the textural parameters of the samples in both series: the total pore volume (Vtot), the micropore volume (VDR) determined by the Dubinin–Radushkevich method, the Brunauer–Emmett–Teller specific surface area (SBET), the microporous and mesoporous parameters: the specific surface areas (Smic and Smes) and the corresponding specific volumes (Vmic and Vmes), and their ratios (Smic/Smes and Vmic/Vmes). Treatment with F4 at and above 600 °C caused a decrease in the AC specific surface areas and specific volumes. From the tabulated data, the values of the Smic/Smes and Vmic/Vmes ratios fluctuated around 9.2 and 3.5, respectively. On the other hand, the same pyrolytic treatment with F5 at 600 °C decreases all the textural parameters (except Smes) with respect to those of the pristine AC. A certain increase in mesoporosity and microporosity is observed after treatment with F5 at temperatures above 600 °C. The Smic/Smes and Vmic/Vmes values show an increase to 6.7 and 3.0, respectively. In fact, the AC taken as it is has the simplest PSD (Fig. 2c).
The function describing the micropore region has a peak at 1.5 nm, and only a narrow range can be assigned to the mesopores with a peak at 2.5 nm that are present in the pristine AC. Within the AC-F4 series, the PSD of AC-F4-800 is similar to that of pristine AC. However, the curve that fits the PSD is smoother and less intense. The mesopore region widens from 2 to 3 nm and shows a tail of low intensity in the range of 3 to 5 nm in the PSD (Fig. 2c). For AC-F4-600, the PSD shows a complex character. The microporous region in the PSD has a peak at 1.4 nm with a shoulder at about 0.9 nm. The mesoporous region in the PSD has a peak at 2.8 nm and two shoulders centered at 2.0 and 3.2 nm. Separate groups of mesopores can also be seen in this PSD, with a peak at 5 nm. For AC-F4-700, the increase in differential pore volume is less intense, and the corresponding fit function is smooth.
QSDFT simulations showed that all sorbents of the AC-F5-T series samples had four PSD regions (Fig. 2d). They included one PSD region with a peak at 1.4 nm, which was attributed to micropores, and three PSD regions with peaks at 2.5, 3.2, and 4 nm, which were attributed to mesopores. Once stopped after the treatment temperature of 500 °C, the contribution of microporosity to the differential pore volume continues to increase with the treatment temperature from 600 to 800 °C. Under these conditions, the mesopore range increases from 2 nm to 6 nm.
Compared to AC, the main results of the texture studies show a moderate decrease in the porosity for most of the AC samples treated with F4 or F5 with increasing the treatment temperature. In general, the reduction of the texture parameters of the studied sorbents correlates with the amount of fluorine chemisorbed on the carbon surface. Typically, the inner surface of small micropores is more involved in chemical reactions with HFC residues. For some samples, the reconstruction of the porous structure of the AC could be suggested; however, the changes in the shape of the PSD curves (Fig. 2c and d) are usually much more complex and indicate the formation of mesoporous substructures within the macroporous architecture of the AC, as a result of the treatment used. These changes are mostly related to the pore size in the range of 2–6 nm, i.e., they are related to the mesoporosity of the AC. At the same time, the contribution of the mesopore area and the mesopore specific volume to the total pore area and the total pore volume does not exceed 15% and 27%, respectively, for most of the modified AC samples. That is, all the modified AC samples remain microporous with a small number of mesopores. The changes in mesoporosity that occur as a result of modification compared to the unmodified AC are complex. The Smes for the F4 series samples increases insignificantly compared to the unmodified AC, only by 14–27 m2 g−1 for the AC-F4-400 and AC-F4-500 samples, and decreases for all the others. For the F5 series, the increase in Smes is more pronounced compared to the unmodified AC, but does not exceed 180 m2 g−1 (14% of SBET) for the AC-F5-400 sample and is smaller for the other samples of the series. Thus, the maximum changes in the contribution of mesopores (up to 15–30%) were recorded for the AC samples treated with F4 and F5 at the lowest temperatures (400 °C and 500 °C). Under these conditions, the modification probably leads to the chemisorption of HFC molecules without their noticeable thermal destruction. Apparently, the HFC molecules first enter the macropores, the most accessible part of the AC surface, and then can decompose inside them, and this decomposition can lead to the formation of new mesopores. The 19F ss-NMR spectra obtained (see Section 3.7) confirm the chemisorption of the HFC molecules, which is a reason for future changes in the porous structure of the AC.
3.3 TEM microstructure
TEM revealed the internal structure and the typical morphology of AC-F4-T and AC-F5-T particles (Fig. 3 and 4). Illustrative TEM images (Fig. 3c and g) show individual graphene-like planar segments (dark lines) oriented nearly parallel to the electron beam, indicating carbon lamellae that govern the bulk reactivity during oxidation.37,38 | ||
| Fig. 3 TEM images of (a–c) AC-F4-500 and (e–g) AC-F4-600 and their respective EDPs (d) and (h). | ||
 | ||
| Fig. 4 TEM images of (a–c) AC-F5-500 and (e–g) AC-F5-600 and respective EDPs (d) and (h). | ||
The length of the graphene-like structural units ranges from 0.5 nm up to 5 nm. These planar units, derived from the flat carbon molecular structures, are grouped in stacks with free space between them (TEM images a, b, e, and f in Fig. 3 and 4). This space is the source of the nanoporous structure. From the TEM experiments, the graphene-like units are the cause of the sp2 AC structure. The primary geometric analysis of the lamellae showed a wide variety of lamellar lengths, curvature, and different intra-lamellar spaces (TEM images b, c, f, and g in Fig. 3 and 4). Depending on the treatment conditions, different nanostructures that are intermediate between the perfect planar graphene-like orientation and those with a random orientation can be found in the TEM images (Fig. 3g and 4g). For AC treated with F4 and F5 at 500 °C, TEM studies have shown a radial variation in the carbon lamella length that increases outward (Fig. 3c and g).
Typically, the reactivity of the basal plane sites is lower than that of the edge sites. In terms of molecular oxygen addition, the reactivity of edge sites is from 10 to 100 times higher, depending on the oxidation temperature.38
Although nanoscale carbon structuring is strictly an internal property, it is related to the surface reactivity of carbon. Typically, we consider the surface reactivity to be related to the unbound, exposed lamellae and amorphous carbon.39 However, under gas-phase oxidation and fluoroalkylation chemical attack, the edge sites of graphene-like planes should determine the emerging reactivity, as is typical for edge structures.40
All of these issues are relevant to future applications of functionalized ACs. For example, in the fabrication of electrode materials, the carbon matrix compatibility, surface energy, and interaction (bonding) strength depend on the number of edge sites. In fact, the ratio of edge to basal sites is a parameter that controls the degree of surface functionalization. In AC-F4-T and AC-F5-T samples, the planar orientation of the layers can improve with increasing pyrolysis temperature. This increase is the reason for signs of partial graphitization (cf. EDPs, Fig. 3d, h and 4d, h). The TEM images suggest that the size of the 002 lattice fringes increases. A well-ordered stacking of the hexagonal carbon layers and graphite basal planes in different orientations can be seen in Fig. 3g and 4g. As the treatment temperature increases, their parallel stacking actually improves (Fig. 5). The formation of a strong cross-linking system between adjacent crystallites causes their dispersion in highly turbostratic arrangements, and the microtexture in samples AC-F4-800 and AC-F5-800 includes multiple aromatic layers with short in-plane dimensions and wavy orientation. The relative amount of the amorphous, turbostratic and graphitic carbon can be different presumably depending on the total porosity. EDPs showed a value of d002 in the range of about 0.37 to 0.35 nm. In contrast, for a single graphite crystal with perfect planar orientation of the lamellae and large hexagonal carbon layers, the d002 value is in the range of 0.3354–0.3370 nm.41 We have suggested that the influence of such partial structuring on the electrochemical behavior is relatively small, but it will correlate with changes in the electrical capacity of supercapacitors as observed in the case of partial graphitization.38
 | ||
| Fig. 5 TEM images of (a–c) AC-F4-800 and (e–g) AC-F5-800 and respective EDPs (d) and (h). | ||
3.4 PXRD analysis
Representative PXRD patterns of AC-F4-400, AC-F4-600, AC-F4-700, and AC-F4-800 sorbents are shown in Fig. S1.† All investigated samples show similar PXRD profiles, with only two broad diffraction features centered at 2θ ∼23.9–25.1° and 43.6–44.5°, Table S1.† They can be indexed by the Miller indices [002] and [100]/[101], which define the orientation of the plane in the general typical graphitic structure,42 and are associated with the crystallite height and width,43 respectively. Such broad patterns are typical of non-graphitized AC44 heat treated to 900 °C. Increasing the HFC pyrolysis temperature from 400 °C to 800 °C is accompanied by a general and gradual change, shifting the center position and sharpening the first diffraction feature, suggesting some increase in the pseudocrystallite height43 and a slight decrease in the lateral crystallite dimension (Fig. S1†). This suggests some ordering and the formation of stacking aromatic layers with more layers for the highest HFC treatment temperatures. The estimated interlayer spacings (d002 = 0.36–0.37 nm) are very similar to several reported data42,45 and are far from the typical graphitic dimensions of 0.335 nm.43 This observation indicates the presence of turbostratic and fully disordered carbon structures in the obtained samples. The reasons for this disorder are the presence of local stacking faults, random displacements between adjacent carbon layers, interlayer spacings that accept different values, unorganized (amorphous) carbon constituents that are not part of the layer structure, and strain in the carbon layers.45 HFC pyrolysis can be considered as a kind of activation accompanied by dehydration/gradual dehydrogenation, with the breakdown and thermal destruction of the 3-dimensional bonds between the different chemical components to form the skeleton of the completely disorganized graphene-like sheets. This process preserves the structural features of the parent AC produced by steam activation of coal, which can be considered as a kind of precursor of the final product. Increasing the temperature increases the degree of “pseudo” crystallinity, although it is very far from the crystalline orientation of graphite. The graphitization process actually requires much higher temperatures, typically 1000 °C and higher. This explains the very broad and diffuse profiles observed in most PXRD patterns in this case. At the high HFC pyrolysis temperature of 700 to 800 °C, the diffraction in the range of 2θ = 22–30° shows a possible diffraction contribution of components with maxima shifted to the graphitic 2θ value of 26.73°. This observation can be proposed to indicate fragmented, developed crystallites (as partially organized structures) as the half-maximum intensity of the first diffraction feature decreases. The second diffraction profiles around 2θ of 44° ± 0.5° are diffuse and broad, with weaker intensity. This indicates that the intragraphitic layers are less developed and that the lateral size of the graphene-like layers did not change significantly under the HFC pyrolysis conditions used with varying temperature parameters.Notably, XRD analysis over a relatively large volume of material compared to the TEM technique provides very average values that are representative of this complex microstructure.
3.5 ATR-FTIR spectroscopy
For AC treated with F4 at all temperatures, we observed an increase in intensity for typical IR bands of oxygen-containing groups (see Fig. 6a and Table 3 for band assignment). | ||
| Fig. 6 ATR-FTIR spectra of AC treated with F4 (a) and F5 (b). | ||
| Peak position (cm−1) | aAssignment46 | Group/structure |
|---|---|---|
| a Abbreviations: ν, stretching; δ, bending; as, asymmetric; s, symmetric; o and i, out-of-plane and in-plane. | ||
| 965 | δ(CR–H), δ(CH2), δ(CC), νo(COO) |
–CCH for 1,4 trans configuration, vinyl and carboxylic groups |
| 1027(1031) | ν(C–N), ν(=C–O–C), νo(CCO), ν(CO–O–CO) | Chromene, pyrone, ether, and anhydride groups |
| 1212(1217) | νs(C–F), νs(C–OH) | CFx and phenolic groups |
| 1368(1378) | δi(O–H), νas(C–F), δi(CH3) and δi(CH) | Phenolic and carboxylic groups, CF2 and CF3, methylene groups |
| 1517(1519) | ν(CC), νs(C–O), ν(CN) |
Aromatic rings |
| 1553 | ν(CC) |
Benzene rings |
| 1656(1678) | ν(CC), νas(CO) |
Alkenyl structure, carbonyl, pyrone, chromene groups |
| 1696 | ν(CN), ν(>CO) |
Amide and carbonyl groups |
| 1742(1746) | ν(CO) |
Carboxylic and carbonyl groups |
The characteristic stretching vibrations of the phenolic and CFx groups are indistinguishable due to absorption in the same wavenumber range. Here, at low levels of surface oxidation, AC has a more smoothed peak. However, at the treatment temperature of 400 °C, the number of oxygen-containing groups belonging to the anhydride type groups increases. At this temperature, phenolic groups participate in the fluorination reaction while carboxylic groups are converted to anhydride type groups. These anhydride type groups are not reactive in the fluoroalkylation reaction at and below 400 °C. The increase in intensity for the IR bands assigned to phenolic and carboxylic groups can be seen by comparing the ATR-FTIR spectra of AC and AC-F5-400 (Fig. 6b). For the AC-F5 series samples prepared at pyrolysis temperatures of 600–800 °C, the intense vibration is seen with a peak at 1217 cm−1. For these samples, the bending and stretching vibrations of the phenolic and carboxylic groups show an increase in intensity and a slight decrease is observed for those of the anhydride type groups.
3.6 TGA
Fig. 7 and Table 4 show the TGA results. Excluding AC-F4-500 and AC-F5-800, the total weight loss Δm of AC treated with F4 and F5 showed a slight increase of about 10% (Table 4 and Fig. 7a, c). On the TG/DTG thermograms (Fig. 7), three ranges of weight loss can be distinguished (Table 4). The first weight loss, Δm1, corresponds to the thermal desorption of physisorbed water and gases, and possibly fluoroalkylating reagents and their pyrolysis products. The second weight loss, Δm2, is due to weakly bound forms of grafted fluoroalkylating reagents that decompose below 500 °C. Fig. 7 and Table 4 show that the most significant high-temperature effect of Δm3 occurs between 500 °C and 1250 °C. In this temperature range, strongly bound fluoroalkylation products can decompose. | ||
| Fig. 7 (a and b) TG and (c and d) DTG thermograms. | ||
| Sample | Temperature range (°C) | |||
|---|---|---|---|---|
| 30–190 | 190–500 | 500–1250 | 30–1250 | |
| Weight loss (%) | ||||
| Δm1 | Δm2 | Δm3 | Δm | |
| 830W | 0.57 | 1.14 | 4.58 | 6.29 |
| AC-F4-400 | 0.53 | ∼0 | 7.42 | 7.95 |
| AC-F4-500 | 0.33 | ∼0 | 5.16 | 5.49 |
| AC-F4-600 | 0.94 | 0.84 | 7.33 | 9.11 |
| AC-F4-700 | 0.61 | 1.17 | 8.34 | 10.12 |
| AC-F4-800 | 0.62 | 0.43 | 7.05 | 8.10 |
| AC-F5-400 | 0.49 | ∼0 | 7.01 | 7.50 |
| AC-F5-500 | 0.49 | 0.07 | 8.65 | 9.21 |
| AC-F5-600 | 0.43 | 0.23 | 6.02 | 6.68 |
| AC-F5-700 | 0.41 | 0.40 | 8.19 | 9.00 |
| AC-F5-800 | 0.33 | ∼0 | 4.15 | 4.48 |
For each sample, several components corresponding to different types of functional groups can be isolated. However, it is difficult to correctly attribute the effects obtained to specific types of surface groups. There is no clear dependence of Δm3 on the pyrolysis temperature. This observation can be related to several types of fluorine-containing groups formed on the surface of AC. In fact, they have different thermal resistances and decompose at different temperatures. We also proposed the formation of new oxygen-containing groups by the mechanism of aging. This scenario can occur during the interaction of air oxygen with the active centers of the carbon surface when the fluoroalkylation treatment is stopped. Despite this possible oxidation, pyrolytic fluoroalkylation can introduce thermostable functional groups to modify the carbon surface.
As shown in Table 4, their decomposition temperature exceeds 500 °C. Fluoroalkylation with F4, unlike that with F5, may produce an insignificant amount, if any, of less thermostable surface groups. Their thermal decomposition takes place at temperatures between 30 and 480 °C. From the TGA and ATR-FTIR data obtained for the AC-F5 series, the pyrolytic treatment in HFC always removes all carboxyl, anhydride, and lactone groups. The same is true for undescribed but clearly detectable oxygen-containing groups whose thermal decomposition range peaks at 200 °C. The expected surface reaction is
|
–COOH + –OH → –C(O)–O– + H2O,
| (III) |
Overall, the TGA results are in agreement with the elemental analysis results. The larger amounts of volatiles evolved from the fluoroalkylated ACs in TGA represent the larger amount of heteroatoms, mainly oxygen and fluorine, in them.
3.7 19F ss-NMR spectroscopy
For both series, the main contribution to the overall NMR spectrum is provided by the CF3 and CF2 groups for AC-F#-600 samples and the CF groups for AC-F#-800 samples (Fig. 8a and b). We have uncovered the main isotropic signals and assigned each resonance to a specific surface functionality as shown in Fig. 8c. As reported by Weigert48 for CF3–CH2–F*, the signal (a) at −241 ppm can be assigned to –CH2F* groups. These groups can be found at any place and position on the edges of graphene-like sheets. The signal (b) at −227 ppm is from >CH–F* groups, or “aromatic” fluorine.49 Forming heterocyclic structures, –CF2– and –CF2–CF2– groups are responsible for the appearance of the signal (c) at −103 ppm.49 In Fig. 8a and b, the range from −64 to −99 ppm shows signals (d) from –CF3 andCF2 groups.48,49 Bonded to the carbon matrix at one end and connecting free rotated –CF3 or –CHF2 groups at the other end,  and –CF2– groups, labeled (e), are signaled at −137 to −150 ppm.49
and –CF2– groups, labeled (e), are signaled at −137 to −150 ppm.49
 | ||
| Fig. 8 Background-subtracted 19F ss-NMR spectra of the AC-F# series with their treatment temperatures (a) F4 and (b) F5. (c) Potential assignment of the fluorine-containing surface groups that could functionalize the edges of graphene-like sheets as revealed by 19F ss-NMR. | ||
Alternatively, it can be attributed to “semi-ionic” fluorine present in CF groups, the high amount of which was determined by XPS (see Section 3.9). The signals (f), between −50 and −100 ppm, could be attributed to –CF3 groups in highly fluorinated sites of the carbon edge.48,49 According to the 19F ss-NMR spectra (Fig. 8, signals (a, b, d, and e)), the tetra- and penta-fluoroalkyl groups can be formed on the carbon surface, e.g., by reaction with the surface –OH groups. Thermal treatment under pyrolytic or oxidative pyrolysis conditions resulted in rearrangement and dehydrofluorination reactions. Fragments of –CF2–CF2 and –CFCF2 can conjugate with the carbon matrix and introduce a carbon bridge (Fig. 8, signals (c, b) and signal (d)). For the samples with the highest fluorine content, we assumed the formation of highly fluorinated sites by the addition of HFC residues (Fig. 8 signal (f)).
We still cannot explain the drastic difference in the linewidth of certain resonances (note the sharpness of the signals (a, d, and e)), which can be attributed to highly mobile AC-trapped HFC molecules.24 However, since measures were taken to remove residual physisorbed HFC and the samples were intensively washed, we are more confident in the current assignment.
3.8 Thermal transformation of fluorine-containing groups: dehydrofluorination
Functionalization of the edges of graphene-like planes and associated temperature-induced structural rearrangements, such as dehydrofluorination, can be corroborated using 19F ss-NMR spectroscopy results. The thermodynamically favored dehydrofluorination accompanies any grafting of fluorine-containing groups onto the surface of the carbon matrix (Fig. 9). | ||
| Fig. 9 Schematic representation of thermally induced HFC addition, C–F bonds homolysis, further rearrangements and cascade reactions occurring during high temperature F4 (a) and F5 (b) treatments. | ||
For AC treated with HFC at 600 and 800 °C, a difference in chemical shifts can be registered ex situ according to the 19F ss-NMR data (Fig. 8). At elevated temperatures, organic fluorine reacts with the surface hydrogen to form HF. At the highest temperatures, under pyrolysis conditions, the F4 and F5 eliminate the HF to form the unsaturated fragments: trifluoroethylene and tetrafluoroethylene or tetrafluoroethane and pentafluoroethane, respectively. These molecules can couple the polyene fragments at the edges of the surface graphene-like sheets with the formation of functional groups similar to those schematized in Fig. 9a and b and govern their future transformations. It is easy to assume that the same process sutures the closest graphene-like layers, increasing the number of bonds within and between graphitic microcrystallites. This leads to an increase in the bulk density ϕ and an increase in mechanical strength, or one could expect the modulation of other material properties.
3.9 XPS
For the AC-F4 and AC-F5 series samples, XPS provides information on the charge state of the elements in the first few atomic layers of the functionalized carbon surface, which is most important for understanding the surface chemistry changes. As expected, the XPS spectra revealed the presence of intense carbon and oxygen signals (Fig. S2 and S3†) and smaller peaks of F 1s (Fig. 10), indicating that the fluorine content in the surface layer is low or comparable to that of oxygen. Fig. S2 and S3† show the C 1s and O 1s core level XP spectra of selected AC-F4 and AC-F5 samples. The experimental data for the C 1s core level XP spectra were fitted with 4–6 curves with a full width at half maximum of 1.5–2.5 eV (Table S2†). The binding energy (B.E.) values obtained for the maxima of each spectral component by the curve-fitting procedure were then assigned to specific functional groups according to the B.E. values found in the literature.50 | ||
| Fig. 10 Representative F 1s core level XP spectra. | ||
For the untreated AC, the resulting fit gives 4 components (β1C, β2C, β3C, β4C) corresponding to CC conjugated bonds (B.E. = 284.2–284.4 eV), C–O bonds that are part of phenolic groups (B.E. = 285.7–285.8 eV), C–O bonds corresponding to quinone surface groups (B.E. = 287.3–287.4 eV), and O–CO bonds corresponding to carboxyl, anhydride, and lactone groups of AC (B.E. = 289.5–289.9 eV), respectively. About one-third of the surface carbon atoms are involved in the oxygen-containing surface groups (β2C, β3C, and β4C components). Among the functional groups, phenolic groups (β2C) have the highest concentration on the surface, 15.1% (Table S2†). The experimental data for the O 1 s core-level XP spectra (Fig. S3†) and the ratio between β3O (B.E. = 532.9–533.1 eV), β2O (B.E. = 531.2–531.4 eV), and β1O (B.E. = 529.6–529.7 eV) for the AC sample are consistent with the ratios of the β4C, β3C, and β2C components, respectively (Table S3†).
Fluorination of AC at high temperatures leads to a redistribution between the components at the C 1s and O 1s core levels and to the formation of new components (Fig. S2 and S3†). For all fluorinated samples (Table S2†), the β5C (B.E. = 291.9–292.1 eV) and β6C (B.E. = 293.4–294.6 eV) components were detected, corresponding to C–F bonds in the CF2 and CF3 groups, respectively. A new form of O 1 s core level (β4O component with a peak at B.E. = 535.9–536.0 eV) also appears, and it can be assigned to oxygen forms chemisorbed near the fluorinated surface centers of the carbon matrix (Table S3†). For the samples of the AC-F4 and AC-F5 series, the contribution of the β4C and β1O components naturally decreases and disappears due to the decomposition of carboxyl groups. Based on the relative intensity of β5C and β6C, it can be concluded that fluorination with F4 and F5 has approximately the same efficiency in the formation of CF2 and CF3 groups.
Further analysis of the XP core level spectra reveals an increase in the relative intensity of the O 1s peak after pyrolysis treatment. On the one hand, this can confirm the formation of carbonyl and phenolic groups (Table S2 and Fig. S2†). As mentioned above, the unmodified AC contains some oxygen-containing constituents, and the surface oxygen groups are mainly in the form of hydroxyl/phenolic hydroxyl groups.24 However, carbonyl and carboxyl structures were also found, but in trace amounts. The latter may form as a result of the spontaneous oxidation of the sample with oxygen from the air during storage. There are significant changes in the oxygen chemistry of the studied AC before and after modification with F4 or F5. Many new carbonyl groups were formed on the carbon surface after functionalization. This is indicated by a significant increase in the total area of β2O peaks compared to those present in the O 1s core level XP spectrum of the unmodified AC. However, the content of β1O, β3O, and β4O components, which are considered as contributions from other types of oxygen-containing groups, decreases with the treatment temperature.
The obtained data for F 1s core level XPS spectra indicate the formation of four different forms of chemisorbed fluorine on the surface of fluorinated ACs (Fig. 10 and Table S4†). The β2F component with a binding energy of 685.4–685.5 eV, corresponding to different C–F groups, has the highest content. The content of this form is more than half of the total surface fluorine amount and does not strongly depend on the treatment temperature and fluorinating reagent. Obviously, the formation of C–F groups is possible due to the partial pyrolysis of F4 and F5 molecules in the gas phase and the interaction of the formed reactive fluorine-containing radical particles with the carbon surface. An alternative pathway for the formation of C–F groups involves the initial chemisorption of F4 and F5 molecules and their further chemical transformations in the carbon surface layer. In addition to C–F groups, many CF2 and CF3 groups (β3F and β4F components with binding energies of 688.4–688.6 and 690.7–690.9 eV, respectively) are formed on the surface of fluorinated ACs, which is a feature of this fluorination method. Although the use of traditional fluorination reagents (molecular fluorine and reactive fluorides) provides much larger amounts of chemisorbed fluorine on the surface of carbon materials, it results in the formation of only C–F groups. The use of F5 molecules and the treatment temperature of 600 °C ensure the formation of up to 35% of CF2 and CF3 groups on the carbon surface. The content of these groups naturally decreases with the increase of the treatment temperature to 800 °C. The β1F component corresponds to semi-ionic C–FSI bonds.
This type of bonding means the direct interaction of chemisorbed fluorine with the conjugated (aromatic) system of CC bonds of the carbon matrix and is typical for fluorinated graphite-like and nanomaterials.3,51–55 The content of semi-ionic forms of fluorine (C–FSI) increases significantly with increasing temperature and reaches 27–30% for AC-F4-800 and AC-F5-800 samples. Thus, for AC-F4-600 and AC-F5-600, the vast majority of the grafted fluorine (85–90%) forms C–F covalent bonds with the carbon matrix and is present on the surface in the form of C–F (β2F), CF2 (β3F), and CF3 (β4F) groups. With increasing treatment temperatures, there is a general tendency to increase the proportion of semi-ionic C–FSI groups due to a decrease in the content of CF2 and CF3 groups. It should be noted that the higher F/C ratio and % CF3 content observed in the present study for the samples of the AC-F5 series as compared to the samples of the AC-F4 series seem to be due to the higher F/H and F/C ratios in F5 compared to F4.
According to Tressaud et al.,51 the binding energy (B.E.) of 685.4 to 685.5 eV of the β1F component can be attributed to certain fluorine forms that are in an intermediate state between semi-ionic (SI) and covalent fluorine and are closer to semi-ionic forms. Despite the presence of CF3 and CF2 groups confirmed by 19F ssNMR, we registered unitary fluorine whose B.E. shifts outwards from the known B.E. for the C–F covalent bonds. This shift could be due to specific energy transfer and electron density fluctuations supported by graphene-like structures. The attribution of this peak to molecular HF is problematic because we used rigorous washing procedures for purification and intensive vacuuming prior to XPS studies.
Major β2F peaks at 687.1–687.2 eV correspond to the covalently bonded fluoroorganic species (namely CF and CFx). In the literature,52,53 such a shift has been attributed to the formation of weakened covalent bonds. These bonds correspond to a mixture of F–C sp2 and F–C sp3 bonds, implying that the C–F bond has a true covalent character. The formation of weakened C–F covalent bonds has been attributed to a strong electron density pull from carbon to fluorine, making the latter atom nearly ionic. Panich52 and Ewels et al.54 attributed this downshift of the F 1 s level to a low density of fluorine coverage of the graphene-like layer. Sato et al.55 and Struzzi et al.56 also attributed it to fluorine covalently bonded to the carbon surface at different surface coverage levels.
The so-called “covalent fluorine” peaks shown in the XP F 1 s spectra correspond to sharp NMR peaks (see Fig. 9). The broad NMR peaks correspond to “semi-ionic” fluorine. The narrow NMR peaks correspond to the “covalent” fluorine contained in the isolated perfluoroalkyl groups. Much of this “covalent” fluorine is present in AC-F4-400, AC-F5-400, and AC-F5-800 samples. Due to the low fluorine content and the functionalization of the developed porous structure, it is quite complex to acquire the XPS spectra of AC-F4-400 and AC-F5-400 samples with acceptable intensity to resolve the spectral components from them well. For the 19F ssNMR spectrum of the AC-F5-800 sample, we registered a combination of strong, narrow and broad NMR peaks. For the AC-F5-800 sample, we also found the prominent XPS peak of presumably “covalent” fluorine in the XP spectrum. The presence of “covalent” or “semi-ionic” fluorine is, in our opinion, determined by the rigidity of its bond to a graphene-like matrix. In the case of rigid C–F bonds, the fluorine-containing groups should be considered “semi-ionic” fluorine. Controversially, the fluorine groups are opposed to the graphene matrix plane by several saturated groups; which should be considered them as “covalent fluorine” forms. As can be seen, according to the XPS method, the predominant amount of fluorine is in the form of C–F groups, while according to the 19F ss-NMR data, the content of CF3 and CF2 groups is significant. The probable reason for this difference is the different functional composition on the outer surface (XPS) and in the sample volume (19F ss-NMR).
3.10 Catalytic decomposition of hydrogen peroxide
Cheap and effective electrochemical energy conversion devices are expected to play an increasing role in contemporary reversible energy storage applications. Various carbon materials are promising catalysts for the oxygen reduction reaction in fuel cells.57,58 The results of studies59 suggest that the main degradation mechanism in fuel cells is due to a chemical reaction with H2O2 generated during operation. Treatment with H2O2 reduces the oxygen reduction activity at high potential and/or reduces the transport properties at high current density. The effect is most pronounced in water as opposed to aprotic solutions. Therefore, the degradation of carbon materials induced by H2O2 has been studied by contacting them with water and methanol solutions. Depending on the chemical nature of the active carbon surface and the solvent used, the decomposition of H2O2 may be incomplete. The kinetic curves show a characteristic behavior where a slow process is followed by an initial fast phase (see Fig. 11 and S4†). The catalysts used showed a low degree of H2O2 conversion, up to 10–15% for 2–2.5 h of the reaction run. We believe that the incomplete decomposition of H2O2 may be related to some kind of chemical transformation of the active surface sites during the reaction process, which is accompanied by a decrease in catalytic activity. The most probable mechanism of deactivation of active centers and reduction of activity of the AC surface is the process of its oxidation under the action of H2O2, which leads to the formation of various functional groups, mainly phenolic and carboxylic. Under intense oxidation in solution, anhydride and lactone groups can also be formed. All these groups are acidic in nature, which has a certain inhibitory effect on the decomposition of H2O2 molecules. Since the HO2− anion is much more active than the H2O2 molecule, increasing the number of basic sites promotes the catalytic activity of the AC surface. Accordingly, H-type carbons, surface basicity, and high pH media were found to be the most favorable for performing H2O2 decomposition.59 Such behavior can be attributed to many factors, including: (1) on H-type carbons (with a basic nature), the available chromene groups initiate a chain mechanism of H2O2 decomposition; the dissociation of H2O2 as a weak acid is enhanced in alkaline media, while it is suppressed in acidic media. It has been suggested that an acidic medium stabilizes H2O2 and an alkaline medium promotes its decomposition. | ||
| Fig. 11 Representative conversion against time plots for H2O2 decomposition in (a) water and (b) methanol. | ||
Hydrogen peroxide decomposes to an extent that is independent of the porosity characteristics of the carbon. However, due to the intermediate formation of hydroxyl radicals, superoxide ions, and/or peroxide anions, the reaction is a function of the pH of the carbon surface rather than the porosity developed. The results confirm the driving role of the chemical nature of the surface in controlling the catalytic effect of AC on H2O2 decomposition. The kinetic data recorded during H2O2 decomposition cannot be described by a formal kinetic approach.
In order to imagine the H2O2 decomposition kinetics over the studied catalysts, we used mathematical models that take into account the irreversible partial decontamination of the active centers of the carbon surface with a scheme shown in Fig. 12.
 | ||
| Fig. 12 Scheme of catalytic decomposition of H2O2. | ||
In this scheme, [ ] is the active center on the carbon surface and [H2O2] is the surface complex of [ ] and H2O2. Assuming zero-order and first-order catalytic reaction models (depending on whether the second or first stage of the scheme in Fig. 12 is limiting), and considering the formation of intermediates between catalyst and reagent, we validated the following equations (see Text S1 and Fig. S5†)
 | (2) |
 | (3) |
 and CH2O2 are the initial and current concentrations of H2O2, kd is the decay constant, and k0i, k0f, k1i, and k1f are effective constants used to fit experimental data within two models. The indices (i and f) and (0 and 1) characterize the initial and final activity of the catalyst, respectively, and correspond to the model used.
and CH2O2 are the initial and current concentrations of H2O2, kd is the decay constant, and k0i, k0f, k1i, and k1f are effective constants used to fit experimental data within two models. The indices (i and f) and (0 and 1) characterize the initial and final activity of the catalyst, respectively, and correspond to the model used.
The chi-squared test parameter χ2/n was used to compare the observed data with the data we would expect to obtain according to eqn (2) and (3). Table 5 summarizes the results of the mathematical treatments; see also Fig. S5.† From Table 5, it is clear that the H2O2 conversions measured after 70 min from the start of the reaction (α70) show close values in the range of 1.25–10.9% in aqueous solutions and in the range of 3.37–6.12% in methanol solutions. The unmodified AC has the highest activity in the aqueous solution. Fluorination/fluoroalkylation of the AC causes a significant decrease in k0i and k1i, which are negatively correlated with the treatment temperature. Treatment with F4 has a greater negative effect on the catalytic activity of AC than treatment with F5. The decay constant kd has a value comparable to k1i only for selected catalysts. As a result of the decay, the catalytic activity is reduced by a factor of 3–4 for most of the samples studied. This can explain the low conversion of H2O2, about 10–15%.
| Sample | α70 (%) | k0i × 103 (mol min−1) | kd × 102 (min−1) | k0f × 104 (mol min−1) | a(χ2/n) × 10−6 | k1i × 103 (min−1) | kd × 102 (min−1) | k1f × 104 (min−1) | a(χ2/n) × 10−7 |
|---|---|---|---|---|---|---|---|---|---|
| a n is the total number of points on the curve that equals 100. | |||||||||
| H2O | |||||||||
| AC | 10.93 | 5.78 | 3.70 | 13.5 | 3.47 | 3.09 | 3.67 | 8.3 | 9.60 |
| AC-F4-500 | 4.42 | 2.38 | 4.10 | 6.1 | 0.24 | 1.26 | 4.10 | 3.4 | 0.65 |
| AC-F4-600 | 2.14 | 1.06 | 4.46 | 3.6 | 0.13 | 0.56 | 4.50 | 2.0 | 0.36 |
| AC-F4-700 | 1.80 | 1.03 | 10.20 | 3.9 | 0.03 | 0.55 | 10.44 | 2.1 | 0.09 |
| AC-F4-800 | 0.87 | 1.32 | 72.30 | 2.1 | 0.02 | 0.71 | 74.72 | 1.1 | 0.04 |
| AC-F5-600 | 1.76 | 4.44 | 39.89 | 3.3 | 0.03 | 2.37 | 40.28 | 1.7 | 0.08 |
| AC-F5-700 | 3.40 | 1.68 | 7.84 | 7.4 | 0.08 | 0.90 | 8.34 | 4.0 | 0.21 |
| AC-F5-800 | 12.66 | 5.99 | 4.43 | 22.5 | 0.30 | 3.19 | 4.56 | 13.8 | 0.74 |
| Carbofluor | 1.25 | 0.98 | 11.39 | 2.4 | 0.03 | 0.52 | 11.48 | 1.3 | 0.07 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||||||
| MeOH | |||||||||
| AC | 3.71 | 0.95 | 0.22 | 9.1 | 0.59 | 0.53 | 0.22 | 4.8 | 2.58 |
| AC-F4-500 | 6.12 | 7.24 | 13.65 | 9.4 | 0.25 | 3.99 | 13.70 | 5.4 | 0.75 |
| AC-F4-600 | 4.29 | 4.15 | 11.53 | 6.9 | 0.16 | 2.29 | 11.61 | 3.9 | 0.53 |
| AC-F4-700 | 2.81 | 0.73 | 66.92 | 6.9 | 0.10 | 0.41 | 66.10 | 3.3 | 0.25 |
| AC-F4-800 | 4.37 | 6.06 | 16.61 | 6.7 | 0.11 | 3.34 | 16.70 | 3.8 | 0.33 |
| AC-F5-600 | 4.29 | 3.68 | 11.61 | 7.7 | 0.25 | 2.02 | 11.71 | 4.3 | 0.81 |
| AC-F5-700 | 3.37 | 1.21 | 7.59 | 7.9 | 0.11 | 0.66 | 8.15 | 4.4 | 0.34 |
| AC-F5-800 | 9.87 | 7.33 | 7.22 | 14.2 | 0.62 | 4.04 | 7.14 | 8.5 | 0.18 |
| Carbofluor | 2.81 | 0.75 | 0.28 | 5.2 | 0.06 | 0.41 | 0.28 | 3.4 | 0.18 |
Compared to water solutions, the catalysts studied in methanol solutions showed opposite catalytic activity towards H2O2 decomposition. For example, the AC sample taken as is has low activity in the methanol solution. On the other hand, the catalysts of the AC-F4 series are much more efficient. Their activity increases by a factor of 3 to 4. However, their decay constant kd increases significantly, up to 2.5–3.3 times. The catalysts of the AC-F5 series show a decrease in the catalytic activity, as can be seen from the comparison of k0i and k1i values and kd. For all AC-F5 and AC-F4 catalysts studied, the constant k1f, which characterizes the catalytic activity of the final form of the active site in methanol solutions, has similar values of 3.8–5.4 min−1. Elemental analysis showed that 66% and 80% of the fluorine-containing groups present in the prepared catalysts survived the extensive H2O2 treatment.
The Carbofluor reference sample has a low catalytic activity in the H2O2 decomposition, which corresponds to the high fluorine content in the reference sample, which is commercially available powder of carbon monofluoride (CF1.12), and is consistent with the assumptions about the negative effect of many fluorine-containing C–F groups on the catalytic properties of the carbon surface in the target reaction.
Some representative data were obtained in order to show the pH dependence in water (Fig. S6 and Table S5†). The obtained conversion vs. time plots for H2O2 decomposition in water at different pH for AC-F4-500 and AC-F5-600 samples are shown in Fig. S6.† First of all, it should be noted that changing the pH of the medium affects the heterogeneous catalytic process in the same way as the homogeneous catalytic decomposition of peroxide: a decrease in pH inhibits the reaction, while an increase accelerates it. This is due to the much higher reactivity of HO2− ions compared to H2O2 molecules in the decomposition process. Reducing the pH to 4 significantly inhibits the process of peroxide decomposition. The effective constants k0i and k1i, which characterize the initial activity of the catalyst surface, decrease by a factor of 2.2–2.3 (Table S5†). The deactivation constant kd also decreases, but only by a factor of 1.5. Thus, the change from a neutral to an acid pH level inhibits the process of peroxide decomposition to a greater extent than the process of carbon surface decontamination under the action of H2O2. The creation of an alkaline medium does not accelerate the process of H2O2 so much—the increase of k0i and k1i constants is 1.8–2.0 times (Table S5†). The main reason is probably partial hydrolysis of active (chemisorbed) fluorine and surface rearrangement with loss of active centers. The deactivation constant also increases by a factor of 2.0–2.5 with increasing pH level. This confirms to some extent the assumption of a hydrolytic effect of an alkaline solution on the surface of the modified AC samples. Comparing the values of the ratios of the effective constants (k0i, k1i) to the deactivation constant kd (k0i/kd and k1i/kd), it is found that the values are the highest (optimal) at the neutral pH level.
Some insight was gained into the temperature dependence of H2O2 decomposition over selected catalysts in water. The obtained conversion vs. time plots for H2O2 decomposition at different temperatures for AC-F4-500 and AC-F5-600 samples are shown in Fig. S7.† From Table S6†, the α70 value and all the effective rate constants naturally increase with increasing temperature. The kinetic analysis confirmed the slightly better fit of the zero-order model compared to the first-order model (Table S6†), which allowed us to accurately determine the values of all the effective constants and to estimate the activation energies E.
The effective rate constants characterizing the catalytic activity of the surface carbon centers (k0i and k0f) have an activation energy E of 53–56 kJ mol−1. These values are lower than the activation energy of homogeneous decomposition of H2O2 (E = 75 kJ mol−1), which corresponds to the moderate catalytic activity of the catalysts used. The catalyst deactivation rate constant kd is characterized by a slightly higher activation energy E of about 60–61 kJ mol−1. Of course, only a minimal number of studies were carried out at three temperatures and the activation energies E that determined have a significant error. However, if the conclusion about the higher activation energy E of the catalyst deactivation process compared to the main process is correct, then an increase in temperature will have negative consequences. With an increase in temperature, the deactivation process becomes more intense than the main process of H2O2 decomposition, which is undesirable in the application.
Thus, both the increase in temperature and the creation of an alkaline environment, despite the noticeable acceleration of the H2O2 decomposition process, will be accompanied by even more intense decontamination of the active centers of AC-F4-500 and AC-F5-600 samples.
3.11 General analysis of the results
In summary, when AC is treated with F4 and F5 at low temperatures, the fluorine attached to the AC matrix is mainly in the form of perfluoroalkyl groups, which are electrochemically inert. In contrast, the fluorine attached at medium temperatures is mostly in the form of groups (C–F) directly bonded to the carbon matrix. This fluorine is sufficiently active for electrochemical purposes. At 600–700 °C, the fluorine-containing surface of AC is additionally covered with perfluoroalkyl groups. At the same time, at medium temperatures, the modification with F4 and F5 accompanies the thermal decomposition of the phenolic groups that takes place in this temperature range. As a result, water molecules are formed, which support the subsequent partial pyrohydrolysis of the fluorine-containing groups. The pyrohydrolytic action results in the appearance of a large amount of fluorine, which is believed to be electrochemically active. Thus, the composition and properties of the pyrolytically fluoralkylated carbon surface can be adjusted by changing the modification temperature. The increase in the catalytic activity of the pyrolytically fluoralkylated AC in the reaction of H2O2 decomposition in methyl alcohol can be related to three key factors. The first is the change in the electronic environment of the active center, the conjugated double CC bond, which becomes more electron-deficient under the influence of acceptor fluorine-containing groups. The second factor is the hydrophobization of the carbon surface due to the presence of the same groups, which leads to an increased wettability in low polar or non-polar solvents, such as methyl alcohol or its solutions, and a tendency not to adsorb or be wetted by water. The latter is of interest for the design of supercapacitors operating in non-aqueous solutions.60 The third factor is a partial graphitization of the surface under the influence of a fluorinating reagent at high temperatures.
In order to analyze the properties of AC-F4-T and AC-F5-T samples using the results obtained from the application of various methods, including ATR-FTIR, NMR, EDS, chemical analysis (CA), TG/DTG, and XPS, it is essential to have a clear understanding of the capabilities, sensitivities, and limitations of the methods. ATR-FTIR and XPS provide information about the outer surface; EDS analyzes the near-surface layer of the carbon matrix to a depth of about 5 microns; and NMR spectroscopy, CA, and TG/DTG allow the study of the entire sample. From the above characteristics, the results obtained may show discrepancies depending on the analytical approach used, e.g., the elemental composition and the presence of certain functional groups may differ. To illustrate this, we will examine how the fluoride concentration CF in the samples varies according to the method used. For convenience, all data are presented in mmol g−1 (Table S7†). As can be seen from the table, the outer surface of the modified samples shows enrichment in fluorine and oxygen. The EDS analysis showed that the near-surface layer (several microns) of the samples had fluorine concentrations 1.5 to 3 times higher than the average fluorine concentration observed in the bulk volume of the granules. The XPS analysis of the surface layer (outer surface) showed much higher fluorine concentration than the average fluorine concentration observed in the granule volume. A similar trend was observed for oxygen, with the outer surface containing 2.2 to 5.8 times more oxygen than the inner surface. The difference between the values of CF (XPS) and CF (CA) is moderate and amounts to 2–3.2 times for the AC-F#-700 and AC-F#-800 samples.
For the low temperature fluorinated AC-F#-400 and AC-F#-500 samples, the difference between CF (XPS) and CF (CA) is 9–36 times higher. This feature is explained by a different fluorination mechanism. At low temperatures of 400–500°C, the total concentration of chemisorbed fluorine CF (CA) is low and does not exceed 0.8 mmol g−1, regardless of the fluorinating reagent, F4 or F5. Under these conditions, only some of the active centers on the carbon surface can react with HFC molecules. Under such heating, only some of the oxygen-containing groups, namely, carboxyl groups and some anhydride and lactone groups, are decomposed. At the same time, the HFC molecules do not undergo significant thermolysis when heated to 400–500 °C; they are not sufficiently activated at these temperatures, so that their residues can form on the surface to be chemisorbed to the carbon surface in small amounts. This mechanism of fluorination is confirmed by the 19F ss-NMR spectra of AC-F4-400 and AC-F5-400 samples, which contain narrow, sharp peaks indicative of the energy homogeneity of the grafted fluorine groups.
At temperatures of 400–600 °C, fluoroalkyl, difluoromethylene CF2 and trifluoromethylene CF3 groups, which are very specific fluorine-containing groups, are grafted onto the carbon surface. They appear with a reasonable selectivity after fluorine chemisorption onto the carbon surface, confirmed by 19F ss-NMR and XPS results, which cannot be achieved by “traditional” harsh fluorination methods.61
Thus, F4 and F5 gases extend the arsenal of difluoromethylation and trifluoromethylation reagents, providing a versatile approach for the construction of (Csurf.–CF3/Csurf.–CF2–) bonds without the use of pre-functionalized substrates.
“Double activation” occurs at high modification temperatures of 600–800 °C: HFC molecules undergo significant pyrolysis, leading to the formation of more reactive fluorine-containing particles, and the decomposition of all types of oxygen-containing groups present in the surface layer of the carbon matrix becomes possible. According to TG/DTG and ATR-FTIR methods, phenolic and carbonyl (quinone) groups have the highest content on the surface of AC (about 1.6 mmol g−1), and their thermal decomposition adds a significant number of new active centers. Modification at high temperatures becomes much more effective, as evidenced by the most significant changes in structural and sorption parameters and the formation of various fluorine-containing surface groups (Table 2 and Section 3.7). According to the ATR-FTIR and XPS data, the outer surface of the samples contains various fluorine-containing groups, ranging from CF3 to C–F groups with a semi-ionic bonding type. In the 19F ss-NMR spectra of the samples, mainly broadened peaks were recorded, confirming the idea that different types of surface active centers are involved in the modification. Thus, the fluorination process becomes less selective at high temperatures.
The fluorination process preserves the developed nanoporosity, but the nanopore distribution differs from the original porous structure of the unmodified AC sample. The treatment temperature has a significant effect on both the efficiency of the fluorination process and the nature of the fluorine-containing groups formed on the carbon surface. The fluorination process is undoubtedly more localized on the outer surface of AC. The significant difference between the fluorine concentration on the outer surface and in the bulk solid is clear evidence of this. In terms of catalysis, this clearly indicates much better access to surface active centers on the outer surface and a higher concentration of fluorine-containing functional groups that can influence the decomposition of H2O2. The 19F ss-NMR and XPS results irrefutably show that the fluorine-containing surface layer undergoes a gradual transition from “covalent” to “semi-ionic” C–F bonds with increasing process temperature, which has not been reported previously. AC samples fluorinated with F4 and F5 and exposed to a temperature of 800 °C show fluorine in a semi-ionic state in amounts up to 30%. This fluorine is more susceptible to hydrolysis, which is detrimental to the catalytic properties of AC fluorinated at high temperatures.
Correlating the properties of the studied AC-F4-T and AC-F5-T catalysts with their activity in H2O2 decomposition, it was found that the best correlation between the α70 conversion degrees is observed with the effects of weight loss in the temperature range 500–1250°C (Δm3), see Fig. S8.† This effect is attributed to the thermal decomposition of the vast majority of functional groups located in the surface layer of the prepared samples of AC-F4-T and AC-F5-T series. Thus, the presence of a high concentration of surface groups leads to a decrease in the catalytic activity of the surface of the studied catalysts in the H2O2 decomposition reaction. Presumably, the functional groups block some of the surface centers active in the H2O2 decomposition reaction, and/or such active centers are deactivated by fluorination (oxidation). Other correlations between the properties of the surface layer of the modified samples and their catalytic activity are rather complex, which is due to several main reasons. First, the concentration of both fluorine-containing and oxygen-containing groups decreases significantly from the outer part of the granule to its center, which causes a difference in the activity and accessibility of the active surface areas. Second, the catalysts differ to some extent in structural and sorption parameters, which affect the transport of the reagent and the product of the H2O2 decomposition reaction. Third, some fluorine-containing groups may undergo hydrolysis and some active surface areas may undergo oxidation during the H2O2 decomposition reaction.
4. Conclusions
We have shown that the treatment of AC with HFC causes the temperature-dependent addition of fluorine-containing functionalities. In fact, the first low temperature process causes the carbon material to become enriched with fluorine. This includes the fluoroalkyl functionality, which is added in small amounts at 400 °C. At 600 °C, the fluorine content of almost all fluorinated AC is maximal. The vast majority of the chemisorbed fluorine forms covalent bonds with the carbon surface, and only 15–20% of the fluorine is in semi-ionic form. Fluorination at 600–700 °C, regardless of the fluorinating agent used, results in the formation of approximately 50% of the C–F groups and 25–35% of the fluoroalkyl CFx groups of the total amount of chemisorbed fluorine present. The formation of CFx groups distinguishes the proposed fluorination method from the treatment with elemental fluorine. By increasing the treatment temperature up to 800 °C, the fluorine-enriched matrix is prone to the formation of “simple” forms—covalent or semi-ionic C–F bonds.Based on these interesting results, further studies are underway to tune the treatment temperature in order to regulate the physicochemical parameters by changing the chemical nature of the functionalized groups. With regard to the reduction of microporosity, we propose the following reasoning to explain the cause of this observation. Both HFCs and the products of fluorine addition to the carbon matrix can form CF2 and tetrafluoroethylene diradicals, which tend to be long-lived at high temperatures when interacting with a conjugated π system. By migrating (laterally) across the damaged surface of the carbon matrix, these diradicals presumably interact with the surface radicals, forming cross-links of individual graphene-like layers and thus reducing the BET surface area.
The study reveals the negative correlation between the conversion degrees over AC-F4-T and AC-F5-T series samples used as H2O2 decomposition catalysts and the amount of functional groups decomposed in the temperature range of 500–1250 °C. The catalytic activity depends on the concentration of fluorine- and oxygen-containing groups, the structural and sorption parameters of the studied catalysts, and is affected by the potential hydrolysis of fluorine-containing groups or carbon surface oxidation. However, the hydrophobized surface showed the ability to effectively decompose H2O2 in methanol solutions.
In contrast to existing fluorinated carbon materials, we report a selective fluoroalkylation process and fluorinated/fluoroalkylated carbon materials. Since the fluorinated/fluoroalkylated carbon materials obtained at the treatment temperature of 800 °C contain the maximum amount of fluorine, of which up to 30% is semi-ionic, such materials are available (promising) precursors for obtaining sulfur-containing functional materials on their basis, e.g. in the process of replacing semi-ionic fluorine by sulfur described by Urbanová et al.62
These findings will broaden the scope of AC-based catalysts through surface engineering and consolidate the fundamental theory of catalytic surfaces for a wide range of environmental and energy-related applications.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and have been included as part of the ESI.†Author contributions
Gauhar Mussabek: Writing – review & editing, investigation, Saule Baktygerey: investigation, Yerzhan Taurbayev: investigation, Dana Yermukhamed: investigation, Nazym Zhylkybayeva: investigation, Alexander N. Zaderko: methodology, investigation, writing – original draft, Vitaliy E. Diyuk: writing – original draft, visualization, investigation, conceptualization, Sergii Afonin: methodology, investigation, writing – review & editing, resources, Gulmira Yar-Mukhamedova: resources, investigation, Ruslan T. Mariychuk: methodology, investigation. Liudmyla M. Grishchenko: investigation, Mária Kaňuchová: resources, investigation, Vladyslav V. Lisnyak: writing – review & editing, supervision, investigation, conceptualization.Conflicts of interest
There are no conflicts to declare. A. Zaderko is listed as the Fluocar® trademark owner of the US 5,479,088 and Ukraine 213,198 registration.Acknowledgements
This research was funded by the Committee of Science of the Ministry of Science and Higher Education of the Republic of Kazakhstan, Grant No. BR21882187. V. V. Lisnyak acknowledges the partial support of the Ministry of Education and Science of Ukraine under grant 0122U002023 and the support of the National Research Foundation of Ukraine (NRFU), the NRFU grant 2023-3-193. This work was partially supported by the Grant of the Ministry of Education and Science of Ukraine for perspective development of a scientific direction: “Mathematical Sciences and Natural Sciences” at the Taras Shevchenko National University of Kyiv. A. N. Zaderko acknowledges partial financial support of this work by the EU Horizon 2020 Research and Innovation Staff Exchange Program (RISE) under the Marie Skłodowska-Curie Action (project “UNAT”, No. 101008159) and the French Ministry of Education and the EU, The Programme for the Emergency Reception of Scientists in Exile (PAUSE). S. Afonin acknowledges financial support for NMR hardware from the German Research Foundation (DFG) infrastructure project “DFG: INST 121384/58-1 FUGG”. V. V. Lisnyak thanks Dr Miroslav Klinger from Institute of Physics of the Czech Academy of Science for the granting access to CrysTBox – Crystallographic Toolbox software.References
- L. F. Velasco, K. H. Kim, Y.-S. Lee and P. Lodewyckx, Front. Chem., 2021, 8, 593756 CrossRef CAS PubMed
; T. Wang, X. Zang, X. Wang, X. Gu, Q. Shao and N. Cao, Energy Storage Mater., 2020, 30, 367 CrossRef
- K. Guerin, M. Dubois, A. Houdayer and A. Hamwi, J. Fluorine Chem., 2012, 134, 11 CrossRef CAS
- New Fluorinated Carbons: Fundamentals and Applications, ed. O. V. Boltalina and T. Nakajima, Elsevier, Amsterdam, 2016 CrossRef PubMed
- M.-J. Jung, E. J. S. Kim, S. I. Lee, J.-S. Yood and Y.-S. Lee, J. Fluorine Chem., 2011, 132, 1127 CrossRef CAS
- Y. Liu, L. Jiang, H. Wang, H. Wang, W. Jiao and G. Chen, et al., Nanotechnol. Rev., 2019, 8, 573 Search PubMed
- C. Huang, C.-H. Pan, C.-Y. Tsai and I.-Y. Tseng, Surf. Coat. Tech., 2013, 231, 47 CrossRef CAS
- V. E. Diyuk, O. M. Zaderko and L. M. Grishchenko, Process for producing fluorine-containing carbon material, UA Patent, 110585, 2016 CrossRef CAS
- H. Groult, F. Leroux and A. Tressaud, Modern Synthesis Processes and Reactivity of Fluorinated Compounds, Progress in Fluorine Science, Elsevier, Amsterdam, 2016 Search PubMed
- M. Sheraz, A. Anus, V. C. T. Le, C. M. A. Swamidoss, E.-k. Kim and S. Kim, Greenhouse Gases: Sci. Technol., 2021, 11, 1118 CrossRef CAS
- M. Shin, D. Jang and J. Ha, J. Mater. Cycles Waste Manag., 2016, 18, 399 CrossRef CAS
- V. E. Diyuk, A. N. Zaderko, K. I. Veselovska, V. V. Lisnyak and J. T. Anal, Calorimetry, 2015, 120, 1665 CAS
- K. I. Veselovs’ka, V. L. Veselovs’kyi, O. M. Zaderko, V. E. Diyuk and O. V. Ishchenko, J. Superhard Mater., 2015, 37, 39 CrossRef
- A. J. Mathew, V. L. John and T. P. Vinod, Gas-Phase Modifications of Carbon Nanostructures, In Handbook of Functionalized Carbon Nanostructures, A. Barhoum and K. Deshmukh, Springer, Cham, 2024, pp. 1–32 Search PubMed
- Y. Xu, K. Zhang and X. Dai, et al., J. Therm. Sci., 2024, 33, 1990 CrossRef CAS
- E. Yli-Rantala, D. Behringer, D. Herzke, S. M. Mudge, M. Beekman, A. de Blaeij, J. Devilee, S. Gabbert, M. van Kuppevelt, M. Zare Jeddi, P. Gabrielsen and X. Trier, Fluorinated Polymers in a Low Carbon, Circular and Toxic-free Economy, Eionet Report - ETC/WMGE 2021/9, ed. M. Wahlström and E. Pohjalainen, European Environment Agency: European Topic Centre Waste and Materials in a Green Economy, Boeretang, Flanders, Belgium, 2021 CrossRef CAS
- E. Pameté, L. Köps, F. A. Kreth, S. Pohlmann, A. Varzi, T. Brousse, A. Balducci and V. Presser, Adv. Energy Mater., 2023, 13, 2301008 CrossRef CAS
- I. Pande, L. F. Pascual, A. Kousar, J. Sainio, H. Jiang and T. Laurila, Diam. Relat. Mater., 2024, 144, 111004 CrossRef CAS
- R. S. M. Chrystie, Chem. Rec., 2023, 23, e202300087 CrossRef CAS PubMed
- B. Petrovic, M. Gorbounov and S. M. Soltani, Carbon Capture Science & Technol., 2022, vol. 3, p. 100045 CrossRef CAS
- M. Jerigová, M. Odziomek and N. López-Salas, ACS Omega, 2022, 7, 11544 CrossRef CAS PubMed
- J. J. Bravo-Suarez and P. D. Srinivasan, Catal. Rev., 2017, 59, 295 CrossRef CAS
- C. A. Martínez-Huitle, M. A. Rodrigo, I. Sirés and O. Scialdone, Appl. Catal. B: Environ., 2023, 328, 122430 CrossRef CAS
- E. M. Can, A. Mufundirwa, P. Wang, S. Iwasaki, T. Kitahara, H. Nakajima, M. Nishihara, K. Sasaki and S. M. Lyth, J. Power Sour., 2022, 548, 232098 CrossRef CAS
- A. N. Zaderko, R. Y. Shvets, I. I. Grygorchak, S. Afonin, V. E. Diyuk, R. T. Mariychuk, O. Yu. Boldyrieva, M. Kaňuchová and V. V. Lisnyak, Appl. Surf. Sci., 2019, 470, 882 CrossRef CAS
- A. Zaderko, V. Prusov and V. Diyuk, Method for carbon material surface modification by the fluorocarbons and derivatives, US Pat., 10000382, 2015 Search PubMed
- H. Singh, S. Q. Zhuang, B. Ingis, B. B. Nunna and E. S. Lee, Carbon, 2019, 151, 160 CrossRef CAS
- L. B. Khalil, B. S. Girgis and T. A. Tawfik, J. Chem. Technol. Biotechnol., 2001, 76, 1132 CrossRef CAS
- A. Aguinaco, J. P. Pocostales, J. F. García-Araya and F. J. Beltrán, J. Chem. Technol. Biotechnol., 2011, 86, 595 CrossRef CAS
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem. Int. Edit., 2006, 45, 6962 CrossRef CAS PubMed
- J. L. Wang and L. J. Xu, Crit. Rev. Environ. Sci. Technol., 2012, 42, 251 CrossRef CAS
- G. Boczkaj and A. Fernandes, Chem. Eng. J., 2017, 320, 608 CrossRef CAS
- F. Lücking, H. Köser, M. Jank and A. Ritter, Water Res., 1998, 32, 260 CrossRef CAS
- Z. Zheng, X. Hong and D. Wu, et al., Adv. Compos. Hybrid Mater., 2023, 6, 90 CrossRef CAS
- V. E. Diyuk, A. N. Zaderko and L. M. Grishchenko, et al., Appl. Nanosci., 2022, 12, 2103 CrossRef CAS
- V. E. Diyuk, A. N. Zaderko and L. M. Grishchenko, et al., Appl. Nanosci., 2022, 12, 637 CrossRef CAS
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051 CrossRef CAS
- C. K. Gaddam, R. L. V. Wal, X. Chen, A. Yezerets and K. Kamasamudram, Carbon, 2016, 98, 545 CrossRef CAS
- A. Gutiérrez-Pardo, J. Ramírez-Rico, R. Cabezas-Rodríguez and J. Martínez-Fernández, J. Power Sources, 2015, 278, 18 CrossRef
- O. V. Khavryuchenko, V. D. Khavryuchenko, V. V. Lisnyak and G. H. Peslherbe, Chem. Phys. Lett., 2011, 513, 261 CrossRef CAS
- V. D. Khavryuchenko, O. V. Khavryuchenko and V. V. Lisnyak, Catal. Comm., 2010, 11, 340 CrossRef CAS
- B. Kwiecinska and H. I. Petersen, Int. J. Coal Geol., 2004, 57, 99 CrossRef CAS
- B. Sakintuna, Y. Yürüm and S. Çetinkaya, Energ Fuels, 2004, 18, 883 CrossRef CAS
- P. Puech, M. Jeanningros, D. Neumeyer and M. Monthioux, Carbon Trends, 2023, 13, 100311 CrossRef CAS
- B. S. Girgis, Y. M. Temerk, M. M. Gadelrab and I. D. Abdullah, Carbon Lett., 2007, 8, 95 CrossRef
- B. Sakintuna and Y. Yurum, Energy Fuels, 2004, 18, 883 CrossRef CAS
- A. Barroso-Bogeat, M. Alexandre-Franco, C. Fernández-González and V. Gómez-Serrano, Energy Fuels, 2014, 28, 4096 CrossRef CAS
- A. Barroso-Bogeat, M. Alexandre-Franco, C. Fernández-González and V. Gómez-Serrano, J. Anal. Appl. Pyrolysis, 2015, 116, 243 CrossRef CAS
- F. J. Weigert, J. Fluorine Chem., 1990, 46, 375 CrossRef CAS
- W. R. Dolbier Jr, Guide to Fluorine NMR for Organic Chemists, Wiley, Hoboken NJ, 2nd edn, 2016, pp. 239–240 Search PubMed
- T. R. Gengenbach, G. H. Major, M. R. Linford and C. D. Easton, J. Vac. Sci. Technol. A, 2021, 39, 013204 CrossRef CAS
- A. Tressaud, F. Moguet, S. Flandroiss, M. Chambon, C. Guimon, G. Nanset, E. Papireri, V. Gupta and P. Bahl, J. Phys. Chem. Solids, 1996, 57, 745 CrossRef CAS
- A. M. Panich, Synth. Met., 1999, 100, 169 CrossRef CAS
- I. P. Asanov, V. M. Paasonen, L. N. Mazalov and A. S. Nazarov, J. Struct. Chem., 1998, 39, 928 CrossRef CAS
- C. P. Ewels, G. Van Lier, J.-C. Charlier, M. I. Heggie and P. R. Briddon, Phys. Rev. Lett., 2006, 96, 216103 CrossRef PubMed
- Y. Sato, K. Itoh, R. Hagiwara, T. Fukunaga and Y. Ito, Carbon, 2004, 42, 3243 CrossRef CAS
- C. Struzzi, M. Scardamaglia, N. Reckinger, H. Sezen, M. Amati, L. Gregoratti, J.-F. Colomer, C. Ewels, R. Snyders and C. Bittencourt, Phys. Chem. Chem. Phys., 2017, 19, 31418 RSC
- M. Klingele, C. Van Pham, A. Fischer and S. Thiele, Fuel Cells, 2016, 16, 522 CrossRef CAS
- V. Goellner, V. Armel, A. Zitolo, E. Fond and F. Jaouena, J. Electrochem. Soc., 2015, 162, H403 CrossRef CAS
- G. I. Razdyakonova, V. A. Likholobov and O. A. Kokhanovskaya, Rcnsit, 2015, 7, 180 Search PubMed
- A. N. Zaderko, L. M. Grishchenko and D. Pontiroli, et al., Appl. Nanosci., 2022, 12, 361 CrossRef CAS
- B. J. Walder, N. B. Schorr, L. B. Brunke, M. P. Siegal, T. M. Alam, K. J. Fritzsching and T. N. Lambert, Solids, 2022, 3, 237 CrossRef CAS
- V. Urbanová, K. Holá, A. B. Bourlinos, K. Čépe, A. Ambrosi, A. H. Loo, M. Pumera, F. Karlický, M. Otyepka and R. Zbořil, Adv. Mater., 2015, 27, 2305 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04883k |
| ‡ Contributed equally. |
| § A gas flow rate of 50 mL min−1 is optimal in terms of HFC reagent consumption and maintenance of HFC reagent excess. |
| ¶ Increasing the treatment time to 3 hours, especially at 600–800 °C, reduces the total fluorine content in the resulting AC-F4 and AC-F5 samples by approx. up to 20%. Norit® GAC 830 W is produced by steam activation of coal; its superior hardness makes it particularly suitable for thermal reactivation (NORIT GAC 830, Norit Americas Inc., Marshall, Texas, U.S.A., 2023). |
| This journal is © The Royal Society of Chemistry 2024 |