
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigating the anti-inflammatory potential of N-amidic acid organoselenium candidates: biological assessments, molecular docking, and molecular dynamics simulations†
Hanan A. Althikrallah‡
 *a,
Saad Shaaban‡*ab,
Ayman Abo Elmaatyc,
Hussein Ba-Ghazala,
Mohammed N. Almarria,
Marwa Sharakyd,
Radwan Alnajjare and
Ahmed A. Al-Karmalawy‡*fg
*a,
Saad Shaaban‡*ab,
Ayman Abo Elmaatyc,
Hussein Ba-Ghazala,
Mohammed N. Almarria,
Marwa Sharakyd,
Radwan Alnajjare and
Ahmed A. Al-Karmalawy‡*fg
aDepartment of Chemistry, College of Science, King Faisal University, Al-Ahsa 31982, Saudi Arabia. E-mail: sibrahim@kfu.edu.sa; halhekrallh@kfu.edu.sa
bDepartment of Chemistry, Faculty of Science, Mansoura University, 35516 Mansoura, Egypt
cMedicinal Chemistry Department, Faculty of Pharmacy, Port Said University, Port Said, 42511, Egypt
dCancer Biology Department, Pharmacology Unit, National Cancer Institute (NCI), Cairo University, Cairo, Egypt
eCADD Unit, Faculty of Pharmacy, Libyan International Medical University, Benghazi, Libya
fDepartment of Pharmaceutical Chemistry, College of Pharmacy, The University of Mashreq, Baghdad 10023, Iraq
gPharmaceutical Chemistry Department, Faculty of Pharmacy, Horus University–Egypt, New Damietta, 34518, Egypt. E-mail: akarmalawy@horus.edu.eg
First published on 9th October 2024
Abstract
Inflammation is a complex process with many contributing factors, and it often causes pain. The pathophysiology of pain involves the release of inflammatory mediators that initiate pain sensation, as well as edema and other inflammation hallmarks. Selenium-containing compounds (OSe) are very promising for developing new medicines because they can treat many different diseases. In this study, we estimated the anti-inflammatory properties of maleanilic and succinanilic acids containing selenium (OSe). These molecules were designed by combining different strategies to enhance their anti-inflammatory properties. Hence, the anti-inflammatory impacts of compounds 8, 9, 10, and 11 were pursued using inflammatory markers COX-2, IL-1β, and IL-6. Notably, it was revealed that compounds 8, 9, 10, and 11 downregulated COX-2, IL-1β, and IL-6 by (2.01, 1.63, 2.26, and 2.05), (1.42, 1.64, 1.93, and 2.59), and (1.67, 2.54, 2.22, and 4.06)-fold changes, respectively. Moreover, molecular docking studies were conducted on compounds 8, 9, 10, and 11 to pursue their binding affinities for the COX-2 enzyme. Notably, very promising binding scores of compounds 8, 9, 10, and 11 towards the binding site of the COX-2 receptor were attained. Additionally, more accurate molecular dynamics simulations were performed for 200 ns for the docked complexes of compounds 8, 9, 10, and 11 to confirm the molecular docking findings, which ignore the protein's flexibility. Therefore, the exact stability of the N-amidic acids OSe compounds 8, 9, 10, and 11 towards the binding pocket of the COX-2 enzyme was examined and explained as well. Also, the MM-GBSA binding energy was calculated for equilibrated MD trajectory, and 200 snapshots were selected with a 50 ps interval for further analysis. Accordingly, the investigated compounds can be treated as prominent lead anti-inflammatory candidates for further optimization.
1. Introduction
Inflammation is a complex process with many contributing factors, and it often causes pain. It is the body's way of fighting back against invaders like germs, harmful chemicals, or physical injuries to tissues. Inflammation involves increased leakage from blood vessels, changes in cell membranes, and damage to proteins.1,2 The pathophysiology of pain involves unleashing inflammatory mediators that start pain sensation, as well as edema and other inflammation hallmarks.3Steroids are powerful tools to fight inflammation and the pain it causes. However, their use can be tricky. Steroids can have a range of side effects, and we cannot simply stop taking them abruptly once treatment is over; they need to be tapered off gradually.4 Meanwhile, NSAIDs (nonsteroidal anti-inflammatory drugs) like ibuprofen, diclofenac, and indomethacin are generally safe for short-term use. However, taking them for a long time can lead to serious gastrointestinal and renal problems.5,6
The way the body processes arachidonic acid is essential for understanding inflammation.7 Arachidonic acid is metabolized to thromboxane A2 and prostaglandins by the cyclooxygenase (COX) cascade or by the 5-lipoxygenase (5-LOX) pathway upon suitable stimulation of neutrophils. Arachidonic acid is released from phospholipid membranes and transformed into prostaglandins and leukotrienes by the COX or LOX cascades.8
The studies focused on discovering cyclooxygenase isozymes (COX-1 and COX-2) which have contributed significantly to our understanding of inflammatory mechanisms.9 When inflammation occurs, macrophages move in and can release a variety of signaling molecules. Some molecules, such as interleukin (IL)-1β, IL-6, tumor necrosis factor (TNF)-α, chemokines, and interferons, act like messengers that prompt the inflammatory response. These are called pro-inflammatory mediators.10
The inhibition of COX-1 is responsible mainly for the unfavorable gastrointestinal and renal side effects associated with NSAIDs. To address this issue, the “coxibs” classes were developed as selective inhibitors for COX-2. However, it's worth noting that “coxibs” themselves are associated with cardiovascular severe effects.11–13 However, it is increasingly suggested that these adverse effects are likely to be dependent on the drug itself rather than being inherent to the entire class.13,14
Besides, COX-2 is overexpressed in many cancers, including colon, stomach, liver, breast, ovary, lung, and prostate cancer. Hence, drugs that block COX-2 might help prevent the incidence of these cancers.15,16 Accordingly, COX-2 represents a promising antitumor target, particularly in cancer cells where it is overexpressed. Therefore, there is an ongoing demand for the synthesis of novel selective COX-2 inhibitors with enhanced gastric and renal profiles, aiming to minimize consequential side effects.3
Research on organoselenium (OSe) compounds is gaining attention due to their potential to shield cells from damage, thanks to their antioxidant properties.17,18 Besides, the literature unveiled that some OSe compounds were employed as anti-inflammatory candidates.19–23 For instance, phenyl diselenide (PhSe)2 I and its derivatives II and III, have shown interesting antinociceptive activities in vivo as well as good anti-inflammatory activities owing to their abilities to decrease proinflammatory cytokines (Fig. 1).24–26 Furthermore, 2-hydroxy-5-5,5′-diselanediylbis(2-hydroxybenzoic acid) IV and selenocyanatobenzoic acid V manifested good antinociceptive and anti-inflammatory activities in the croton-oil models (Fig. 1).20,27 Interestingly, their mode of action includes altering glutamatergic, nitrergic, and serotonergic pathways.20,28
 | ||
| Fig. 1 Biologically relevant amidic acid, dicarboxamide, and cyclic imides. | ||
On the other hand, the dicarboxamide-containing scaffolds and their cyclic imide analogues manifested potential anticoagulant and anti-inflammatory activities.29,30 For example, 1,3-dicarboxamide VI manifested promising antioxidant features by elevating the expression of the cytochrome P-450 enzymes in the liver (Fig. 1).30–33 Furthermore, the OSe-based isomaleimide VII developed in our house, has shown promising antiapoptotic activity as well as cytoprotective and antioxidant potential against oligodendrocytes (Fig. 1).34–36 Moreover, the OSe-containing N-succinimide VIII displayed good antioxidant and anticancer activity against HEPG2 cells (Fig. 1).35–37
We recently developed different OSe agents as potential SARS-CoV-2 MPro inhibitors.38 Interestingly, these compounds contain maleic and succinic acid fragments, and preliminary computational calculations data pointed out that they could bind within the active site of the 6LU7 protein pocket.38,39 These studies need further validation by pharmacological assessments. Furthermore, there is no available biological data for these compounds.
As this is an enormously unknown group of compounds, nothing is known about their anti-inflammatory potential and their biological targets. Furthermore, there is ample evidence in the literature indicating that OSe compounds (diorganyl diselenides, for instance, and particularly, diphenyl diselenide) have a wide range of applications as anti-inflammatory agents and some of these compounds have already entered clinical trials, e.g., ebselen and ethaselen.24–26,40
In continuation of our previous work, our objectives were expanded further to investigate the underlying anti-inflammatory activities of these compounds to determine whether these candidates may be useful for further medicinal studies in the future. Therefore, we aim to investigate their anti-inflammatory activities using inflammation-related markers such as COX-2, IL-6, and IL-1β. Finally, molecular docking studies were performed against the target apoptotic markers. They greatly recommended the potential activity of the examined candidates to induce apoptosis as a mechanism for their antitumor activity.
1.1. Rational of design
The design aimed to enhance anti-inflammatory properties by combining various lead optimization techniques. The starting compound (PhSe)2 presented several challenges. It is highly fat-soluble, making it difficult to be absorbed orally. Additionally, it caused unwanted effects (e.g., toxicity issues) due to its interactions with unintended targets, and its overall physicochemical properties limited its effectiveness.41,42 As a result, (PhSe)2 was simplified to 4-aminobenzeneselenol. Subsequently, a lead optimization tool was utilized employing various strategies, including substituent variation, chain elongation, and rigidification. In the substituent variation approach, selenium was substituted with different alkyl groups to explore changes in the anti-inflammatory activity. The 4-amino group was also replaced with an amido-butanoic acid motif as part of the chain elongation approach. This change aimed to improve how the molecule binds to its target receptor. The amido-butanoic acid motif likely achieves this by introducing additional interactions: hydrophobic interactions (attracted to other non-polar molecules) and hydrogen bonding. Furthermore, to understand how the molecule's flexibility affects its anti-inflammatory activity, rigid building blocks (olefinic bonds) were incorporated within the elongated chain of the butanoic acid group (between α and β carbons), as depicted in Fig. 2. This “rigidification approach” essentially can limit the molecule's flexibility in a specific area, changing its activity. | ||
| Fig. 2 The proposed design rationale for the studied N-amidic acids OSe compounds as anti-inflammatory candidates. | ||
2. Results and discussion
2.1. Chemistry
The increasing interest in developing new OSe agents is driven by their privileged anti-inflammatory and antioxidant properties.28,43,44 Thereby, there is an urgent demand for innovative procedures that efficiently give access to highly functionalized OSe compounds and estimate their potential bioactivities. Despite the considerable advancements noticed over the last decade in the chemistry of OSe compounds, their synthesis is usually associated with several challenges.18,43 These include the restriction to certain reaction conditions (e.g., under inert gas, dry conditions, or high temperature) as well as the incorporation of hazardous and toxic reagents.45–47 Hence, there is a high requirement for developing mild and simple protocols using stable OSe reagents compatible with a wide functional group. Diaryl diselenides are versatile precursors for synthesizing a wide range of multifunctional OSe intermediates, such as aryl selenide halide (ArSeX). The latter is used for the synthesis of structurally diverse selenaheterocycles.48 Indeed, diaryl diselenides are generally stable and safe to use and handle.40,48,49 Likewise, carboxamides are key scaffolds in several bioactive molecules such as peptides, pseudopeptides, enzymes, and pharmacologically active agents.37,50 Their potential bioactivities stem from their exceptional electronic properties as well as their ability to form hydrogen bonding. Therefore, incorporating carboxamides into the OSe scaffolds will enhance the overall anti-inflammatory properties. From a chemistry perspective, dimethyl 5,5′-diselanediylbis(2-aminobenzoate) (3) has three functional groups, i.e., diselenide (Se–Se), ester (COOMe), and amino (NH2) groups. It can be obtained in excellent yield (92%) and high purity and is also soluble in most organic solvents.37,39,51 Compound 3 is synthesized in two steps, starting from methyl 2-aminobenzoate (1) by selenocyanation and subsequent hydrolysis (Scheme 1). The reduction of OSe 3 by NaBH4 affords reactive nucleophile sodium arylselenolate intermediate. The reaction of sodium arylselenolate with alkyl halides such as iodomethane, α-chlorotoluene, and 2-chloroacetanilide furnished the respective para-substituted primary aromatic OSe amines 4, 5, and 6 in very good yields (up to 96%) (Scheme 1). The nucleophilic attack of the amine functionality of the OSe compounds 4, 5, and 6 on the maleic and succinic anhydride carbonyl carbon resulted in ring opening and the subsequent formation of the N-amidic acids 7–12 in good yields (up to 95%) as shown in Scheme 1. | ||
| Scheme 1 Synthesis of the target amidic acids (7–12). | ||
2.2. Biological assessments
First, the cytotoxic inhibitory concentration 50 (IC50) of the investigated compounds (7–12) was pursued against the human melanoma cancer (A375) cell line using the SRB assay.52 Their IC50 values were recorded as 7, 5, 4.8, 7, 5, and 17 μg mL−1, respectively.53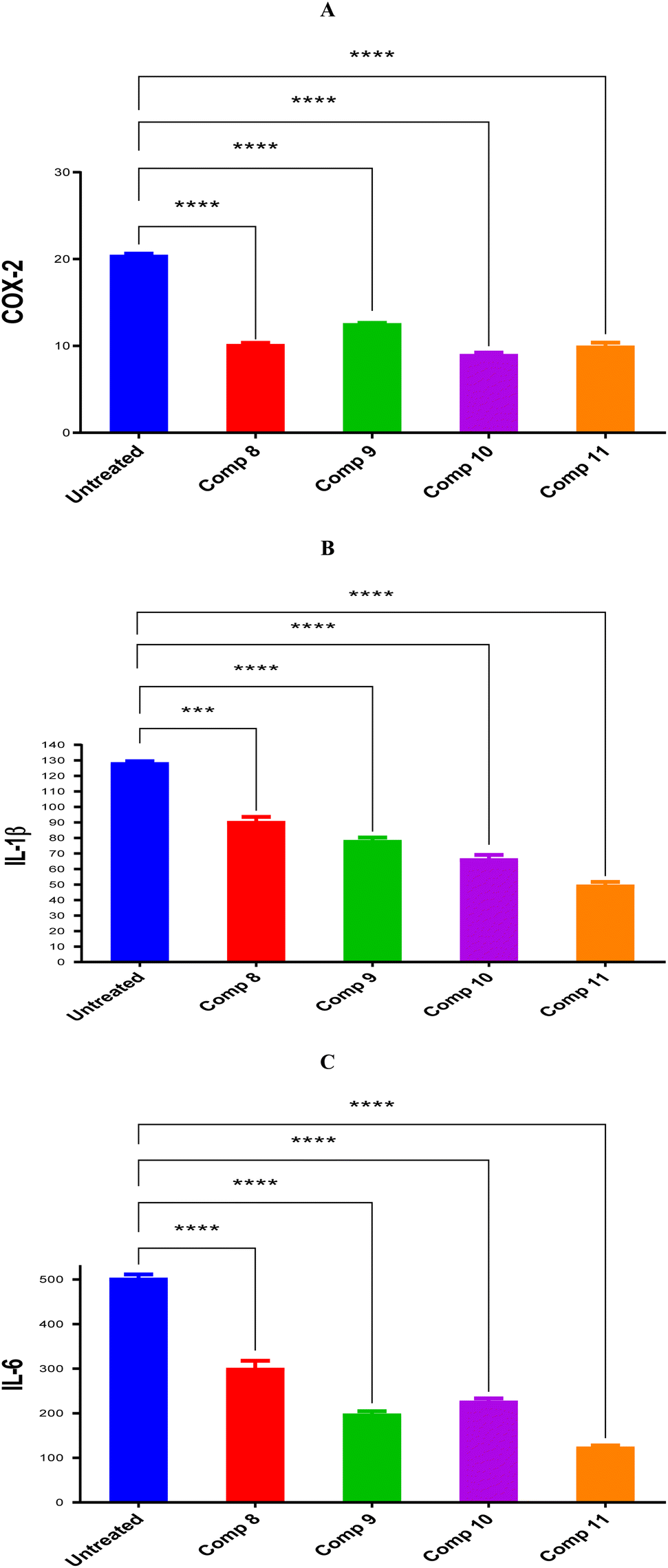 | ||
| Fig. 3 Compounds 8, 9, 10, and 11 protein expression levels for (A) COX-2, (B) IL-1β, and (C) IL-6 in comparison to untreated control cells with statistical analysis using one-way ANOVA (p < 0.5). | ||
2.3. In silico studies
The binding scores of compounds 8, 9, 10, and 11 were recorded as −8.21, −8.51, −6.51, and −7.29 kcal mol−1 at RMSD values of 1.05, 1.25, 1.42, and 1.60 Å, respectively. Compound 8 showed a hydrogen bond with Leu338; however, compound 9 described two hydrogen bonds with Leu338 and Phe504. On the other side, compound 10 represented two hydrogen bonds with Leu338 and Ser339, and compound 11 formed a hydrogen bond with Leu338 (Fig. 4).
 | ||
| Fig. 4 3D binding modes of compounds 8, 9, 10, and 11 within the binding pocket of COX-2 (PDB ID: 3LN1) compared to COX-2 co-crystallized inhibitor. | ||
In conclusion, the very promising binding scores of compounds 8, 9, 10, and 11 towards the binding site of COX-2 receptor. Besides, the nearly similar binding interactions towards the crucial amino acids responsible for the antagonistic activity significantly confirm the anti-inflammatory potential of compounds 8, 9, 10, and 11.
The complex's stabilities were monitored with respect to their Cα initial positions as a function of time. Fig. 5 represents the RMSD of the Cα of these complexes, and as can be seen, the stabilities could be observed with low levels of fluctuations (<3.00 Å), which indicates low conformational changes within the protein structure.
 | ||
| Fig. 5 The RMSD of the ligand-COX-2 (PDB ID: 3LN1) complexes as a function of time. | ||
Further, the behavior of ligands 8, 9, 10, and 11 inside the binding pocket of the COX-2 enzyme was monitored with respect to their original position inside the active site and was plotted as a function of time as well, Fig. 6. Briefly, the ligands' RMSD showed great stability with respect to the simulation time. Both compounds 8 and 9 showed RMSD of less than 3.00 Å, especially after 25 ns of the simulation time, indicating high stability of the ligands. However, the RMSD of compounds 10 and 11 reached 4.5 and 5.5 Å after 60 and 70 ns, respectively, indicating less but still acceptable stability behaviors. Besides, the RMSD of the co-crystallized inhibitor was recorded up to 4.00 Å, which was decreased to lower than 3.00 Å after 125 ns of the simulation time.
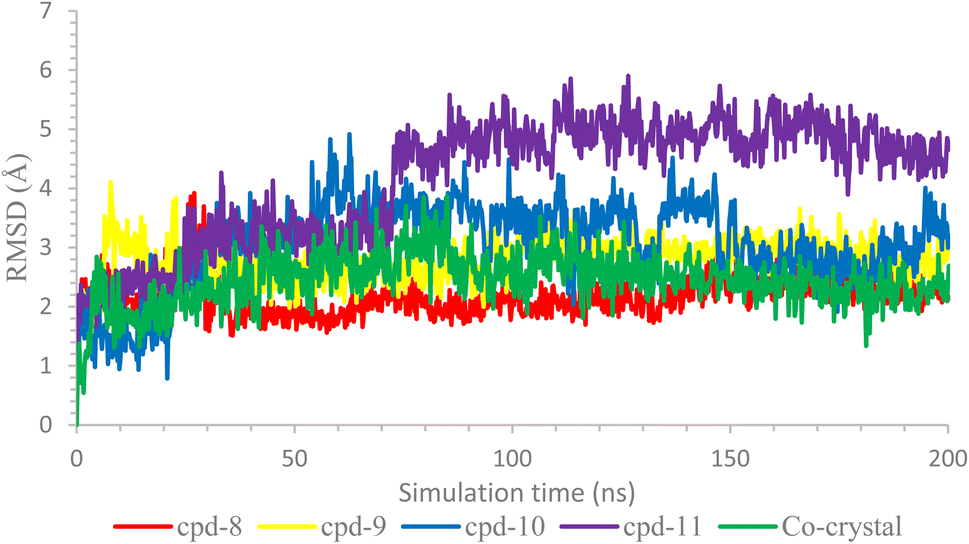 | ||
| Fig. 6 The RMSD of the ligands inside the active site of COX-2 (PDB ID: 3LN1). | ||
On the other side, the protein–ligand interactions were analyzed using the histogram tool to describe the most crucial amino acids of the COX-2 binding site responsible for the binding interactions with the examined ligands (Fig. 7).
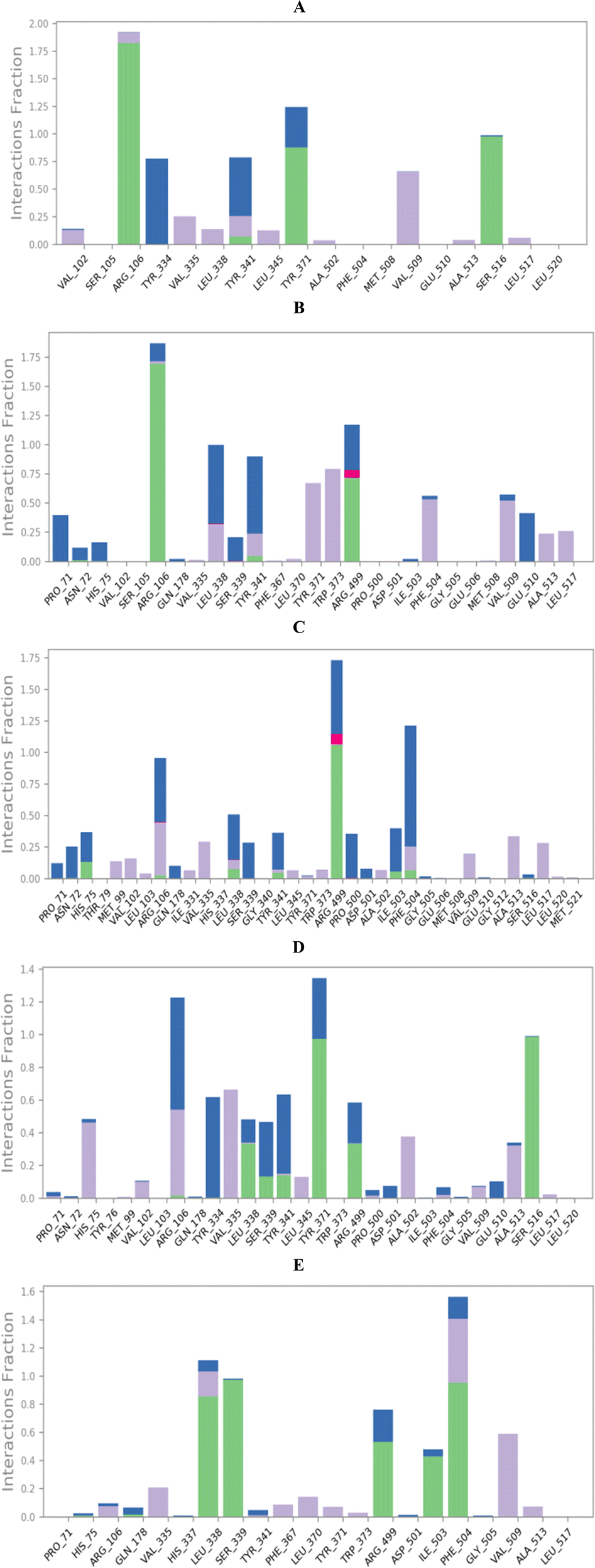 | ||
| Fig. 7 Histograms of compounds (A) 8, (B) 9, (C) 10, (D) 11, and (E) co-crystal with the active site of COX-2 (PDB ID: 3LN1). | ||
The histogram of compound 8-COX-2 complex showed that Arg106, Tyr371, and Ser516 were the most important residues to interact with at 190, 125, and 100% binding interaction, respectively. Also, the types of interactions between compound 8 and COX-2 residue were hydrogen and hydrophobic bonds (Arg106), hydrogen and water bridged hydrogen bonds (Tyr371), and hydrogen and water bridged hydrogen bonds (Ser516), Fig. 7A. Moreover, the histogram of compound 9-COX-2 complex described that Arg106, Arg499, and Leu338 were superior with 185, 125, and 100% binding interactions, respectively. The interactions between compound 9 and COX-2 were (hydrogen, hydrophobic, and water-bridged hydrogen bonds), (hydrogen, ionic, and water-bridged hydrogen bonds), and (hydrophobic, ionic, and water-bridged hydrogen bonds) for Arg106, Arg499, and Leu338, respectively (Fig. 7B). Furthermore, the compound 10-COX-2 histogram represented Arg449, Phe504, and Arg106 as the most important, with 175, 125, and 100% binding interactions, respectively. The types of interactions were (hydrogen, ionic, and water-bridged hydrogen bonds), (hydrogen, hydrophobic, and water-bridged hydrogen bonds), and (hydrogen, hydrophobic, ionic, and water-bridged hydrogen bonds) for Arg449, Phe504, and Arg106, respectively (Fig. 7C). In addition, compound 11-COX-2 histogram recorded Tyr371, Arg106, and Ser516 to be the best with 130, 120, and 100% interactions, respectively. The interactions were found to be (hydrogen and water-bridged hydrogen bonds), (hydrophobic and water-bridged hydrogen bonds), and (hydrogen and water-bridged hydrogen bonds) for Tyr371, Arg106, and Ser516, respectively (Fig. 7D). The co-crystallized inhibitor-COX-2 histogram clarified Phe504, Leu338, and Ser339 as the crucial amino acid residues for interactions with 155, 110, and 100% interactions, respectively. The interactions were as (hydrogen, hydrophobic, and water bridged hydrogen bonds), (hydrogen, hydrophobic, and water bridged hydrogen bonds), and (hydrogen and water bridged hydrogen bonds) for Phe504, Leu338, and Ser339, respectively (Fig. 7E).
At the same time, the heat maps for all complexes (8, 9, 10, and 11)-COX-2, which present the interactions of the compounds with each residue as a function of time, were discussed to understand the exact time of interactions further. Herein, the heat maps of both compounds 8 and 9-COX-2 complexes showed that (Arg106, Tyr371, and Ser516) and (Arg106, Arg499, and Leu338) contributed to the binding interaction for each ligand from the start until the end of the simulation time (Fig. 8A and B), respectively. Moreover, the heat map of compound 10-COX-2 complex clarified that only Arg449 interactions were all over the 200 ns of the simulation time; however, the interactions of Arg106 and Phe504 were clearer after 25 and 30 ns of the simulation time, respectively (Fig. 8C). Further, the heat map of compound 11-COX-2 complex represented that both Tyr371 and Ser516 interactions were clear from the start until the end of the simulation time. Besides, the interactions of Arg106 appeared from 25 ns until the end of the simulation time (Fig. 8D). Finally, the heat map of the co-crystallized-COX-2 complex showed that both Leu338 and Ser339 interactions were apparent from the start until the end of the simulation time, and the interactions of Phe504 started after 5 ns until the end of the simulation time (Fig. 8E).
 | ||
| Fig. 8 Heat maps of compounds (A) 8, (B) 9, (C) 10, (D) 11, and (E) co-crystal with the active site of COX-2 (PDB ID: 3LN1). | ||
| Comp. | ΔGBind | Coulomb | Covalent | H-bond | Lipo | Packing | Solv_GB | VdW |
|---|---|---|---|---|---|---|---|---|
| a Covalent: covalent binding energy; Coulomb: coulomb energy; Lipo: lipophilic energy; H-bond: hydrogen-bonding energy; Solv_GB: generalized Born electrostatic solvation energy; and VdW: van der Waals energy. | ||||||||
| 8 | −35.92 | 1.51 | 1.00 | −2.20 | −10.68 | −0.27 | 7.80 | −33.08 |
| 9 | −51.93 | −25.77 | 1.18 | −2.42 | −19.96 | −1.67 | 40.76 | −44.05 |
| 10 | −50.99 | 3.13 | 2.14 | −1.26 | −18.80 | 0.53 | 13.02 | −49.76 |
| 11 | −49.95 | −18.05 | 1.52 | −1.41 | −14.39 | −2.13 | 32.25 | −47.75 |
| Co-crystal | −60.61 | −19.00 | 1.69 | −2.03 | −19.57 | −1.10 | 21.66 | −42.27 |
The binding energy was calculated as follows:
| ΔGbind = ΔEMM + ΔGsolv + ΔGSA |
| ΔEMM = E(complex) − E(ligand) + E(receptor) |
As can be clarified from Table 1, compounds 9, 10, and 11 showed excellent binding energies of −51.93, −50.99, and −49.95 kcal mol−1, respectively, in comparison to the co-crystallized ligand with ΔG binding of −60.61 kcal mol−1. Moreover, compound 9 recorded better Coulomb, H-bond, lipophilic, packing, and generalized Born electrostatic solvation energies, compared to the co-crystallized inhibitor. Furthermore, compound 10 described frontier covalent and van der waals energies compared to the co-crystallized inhibitor.
3. Conclusion
By combining chain elongation, substituent variation, and rigidification approaches, the designed OSe compounds can be treated as outstanding anti-inflammatory candidates. The investigated OSe compounds 8, 9, 10, and 11 exhibited remarkable downregulation of anti-inflammatory markers i.e. COX-2, IL-1β, and IL-6, assuring their anti-inflammatory potential. Furthermore, the very promising binding scores of compounds 8, 9, 10, and 11 towards the binding site of the COX-2 receptor after conducting molecular docking, as well as, the nearly similar binding interactions towards the crucial amino acids responsible for the antagonistic activity confirm greatly the anti-inflammatory potential of compounds 8, 9, 10, and 11. Moreover, the molecular dynamics simulations recommended the stability of the examined complexes based on the low values of the RMSDs (<3.00 Å) indicating low conformational changes and the absence of protein denaturation. Besides, the ligands' RMSD showed great stability concerning the simulation time as well. Finally, the MM-GBSA calculations clarified that compounds 9, 10, and 11 showed superior binding energies of −51.93, −50.99, and −49.95 kcal mol−1, respectively, in comparison to the co-crystallized ligand with ΔG binding of −60.61 kcal mol−1. Moreover, compound 9 recorded better Coulomb, H-bond, lipophilic, packing, and generalized Born electrostatic solvation energies, and compound 10 described frontier covalent and van der Waals energies, compared to the co-crystallized inhibitor. There are more opportunities for multi-disciplinary experiments and investigations involving organic and medicinal chemistry as well as pharmacology. Furthermore, the chemical structure of active N-amidic acid compounds 8, 9, 10, and 11 offers significant scope for diversification, and the development of more bioactive agents is, therefore, now straightforward. The latter is a promising research point for future studies of their activity via the design, preparation, and assessment of structural variants of N-amidic acid compounds 8, 9, 10, and 11. Moreover, cell-based experiments are highly needed to evaluate their exact effect on neurons and to identify their possible biological targets. The potential of developing N-amidic acid small OSe libraries in a simple synthetic route will significantly improve and accelerate pharmacological studies. However, it is not possible to create other derivatives and test them now. Furthermore, more extensive research and additional studies are required to evaluate their exact mode(s) of action and to discover possible intracellular biological targets, which we are already in progress. Furthermore, to set up the entire picture of OSe candidates as possible anti-inflammatory agents, this direction of research should be shifted to animal studies. We are entirely aware that a comprehensive QSAR will require a more extensive and diverse set of candidates, not only OSe compounds. A clear QSAR, in our opinion, should also include organosulfur and organotellurium analogues to perform wider screening and analysis of their selectivity. Also, to generate consistent QSAR and to get a better insight into their possible mechanism, these compounds should be expanded to include broader organoselenocyanates as well. Moreover, N-amidic acid organoselenium compounds 8, 9, 10, and 11 that downregulate anti-inflammatory markers propose a startup for more structural variants of organoselenium compounds to develop more efficient anti-inflammatory candidates.4. Materials and methods
4.1. Synthesis of the N-amidic acids OSe compounds
The N-amidic acids OSe agents were synthesized following our published literature procedure (ESI, SI1†).38,39,51,55,564.2. Protein expression of the inflammation-related genes
The inhibitory potentials of compounds 8, 9, 10, and 11 were evaluated by measuring the protein expression of the inflammation-related genes (COX-2, IL-6, and IL-1β) in both the treated and untreated cells. These treatments were applied using the IC50 values of compounds 8, 9, 10, and 11 (5, 4.8, 7, and 5), respectively, toward the A375 cancer cell line.53 This protocol tends to measure and compare COX-2, IL-6, or IL-1β protein expression levels between the cells treated with the examined candidates and the negative control untreated cells (ESI, SI2†).4.3. In silico studies
Funding
This research work was funded by the Deputyship for Research & Innovation, Ministry of Education in Saudi Arabia for funding this research work through the project number INST222.Data availability
The data supporting this article have been included in the main manuscript and the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors extend their appreciation to the Deputyship for Research & Innovation, Ministry of Education in Saudi Arabia for funding this research work through the project number INST222.References
- H. Bartsch and J. Nair, Langenbeck's Arch. Surg., 2006, 391, 499–510 CrossRef PubMed.
- C. N. Serhan, Faseb. J., 2017, 31, 1273 CrossRef CAS PubMed.
- A. Sakr, S. Rezq, S. M. Ibrahim, E. Soliman, M. M. Baraka, D. G. Romero and H. Kothayer, J. Enzyme Inhib. Med. Chem., 2021, 36, 1810–1828 CrossRef CAS PubMed.
- G. S. Hassan, D. E. A. Rahman, E. A. Abdelmajeed, R. H. Refaey, M. A. Salem and Y. M. Nissan, Eur. J. Med. Chem., 2019, 171, 332–342 CrossRef CAS PubMed.
- T. K. Motawi, H. M. Abd Elgawad and N. N. Shahin, J. Biochem. Mol. Toxicol., 2007, 21, 280–288 CrossRef CAS PubMed.
- S. Bindu, S. Mazumder and U. Bandyopadhyay, Biochem. Pharmacol., 2020, 180, 114147 CrossRef CAS PubMed.
- U. S. Akula and B. Odhav, J. Med. Plants Res., 2008, 2, 207–212 Search PubMed.
- M. S. Jan, S. Ahmad, F. Hussain, A. Ahmad, F. Mahmood, U. Rashid, F. Ullah, M. Ayaz and A. Sadiq, Eur. J. Med. Chem., 2020, 186, 111863 CrossRef CAS PubMed.
- C. A. Rouzer and L. J. Marnett, J. Lipid Res., 2009, 50, S29–S34 CrossRef PubMed.
- J. Zhang, Z. Wang, J. Wang, X. Zhuo, L. Yu, T. Han, Y. Song, C. Gai, Y. Zou and Q. Meng, Eur. J. Med. Chem., 2024, 116272 CrossRef CAS PubMed.
- G. Bozimowski, J. Am. Assoc. Nurse Anesth., 2015, 83(6), 425 Search PubMed.
- C. Pereira-Leite, C. Nunes, S. K. Jamal, I. M. Cuccovia and S. Reis, Med. Res. Rev., 2017, 37, 802–859 CrossRef PubMed.
- J. S. Alotaibi, Y. S. Al-Faiyz and S. Shaaban, Pharmaceuticals, 2023, 16, 367 CrossRef CAS PubMed.
- Y. Zhang, Y. Wang, C. He, X. Liu, Y. Lu, T. Chen, Q. Pan, J. Xiong, M. She and Z. Tu, J. Med. Chem., 2017, 60, 4135–4146 CrossRef CAS PubMed.
- A.-M. Alaa, A. Angeli, A. S. El-Azab, M. E. Hammouda, M. A. El-Sherbeny and C. T. Supuran, Bioorg. Chem., 2019, 84, 260–268 CrossRef PubMed.
- M. M. El-Kerdawy, M. A. Ghaly, S. A. Darwish, H. A. Abdel-Aziz, A. R. Elsheakh, R. S. Abdelrahman and G. S. Hassan, Bioorg. Chem., 2019, 83, 250–261 CrossRef CAS PubMed.
- M. Sak, Y. S. Al-Faiyz, H. Elsawy and S. Shaaban, Antioxidants, 2022, 11, 1231 CrossRef CAS PubMed.
- S. Shaaban, H. E. Gaffer, M. Alshahd and S. S. Elmorsy, Int. J. Res. Dev. Pharm. Life Sci., 2015, 4, 1654–1668 CAS.
- D. S. Shelar, P. P. Dhavan, P. R. Singh, B. L. Jadhav, S. P. Vaidya and S. T. Manjare, J. Mol. Struct., 2021, 1244, 130914 CrossRef CAS.
- P. M. Chagas, S. G. Rosa, M. H. M. Sari, C. E. S. Oliveira, R. F. S. Canto, S. C. A. da Luz, A. L. Braga and C. W. Nogueira, Pharmacol., Biochem. Behav., 2014, 118, 87–95 CrossRef CAS PubMed.
- K.-M. Shin, B.-G. Park, J.-A. Jang, R.-G. Kim, L.-L. Shen, K.-T. Lee and J.-H. Jeong, 2002.
- A. A. Sarhan, N. A. Ashour and A. A. Al-Karmalawy, Inform. Med. Unlocked, 2021, 24, 100604 CrossRef PubMed.
- C. Nogueira, E. Quinhones, E. Jung, G. Zeni and J. Rocha, Inflammation Res., 2003, 52, 56–63 CrossRef CAS PubMed.
- C. Gallo-Rodriguez and J. B. Rodríguez, Synthesis, 2024, 56(15), 2295–2315 CrossRef CAS.
- C. Gallo-Rodriguez and J. B. Rodriguez, ChemMedChem, 2024, e202400063 CrossRef CAS PubMed.
- P. Zhang, S. Zhang, H. Hu, T. Hu, K. Shi, Y. Xu, G. Xu, H. Hu and S. Pan, Food Biosci., 2024, 58, 103651 CrossRef CAS.
- S. Shaaban, S. M. Shabana, Y. S. Al-Faiyz, G. Manolikakes and F. F. El-Senduny, Bioorg. Chem., 2021, 109, 104713 CrossRef CAS PubMed.
- S. Shaaban, M. A. Arafat, H. E. Gaffer and W. S. Hamama, Der. Pharma Chem., 2014, 6, 186–193 Search PubMed.
- E. Carosati, P. Ioan, M. Micucci, F. Broccatelli, G. Cruciani, B. Zhorov, A. Chiarini and R. Budriesi, Curr. Med. Chem., 2012, 19, 4306–4323 CrossRef CAS PubMed.
- F. F. El-Senduny, S. M. Shabana, D. Rösel, J. Brabek, I. Althagafi, G. Angeloni, G. Manolikakes and S. Shaaban, Future Med. Chem., 2021, 13, 1655–1677 CrossRef CAS PubMed.
- M. D. Mashkovskij, Lekarstvennye sredstva [Drugs], Novaya volna. [in Russian], Moscow, 2005 Search PubMed.
- M. Stasevych, V. Zvarych, O. Yaremkevych, M. Vovk, A. Vaskevych, T. Halenova and O. Savchuk, Acta Chim. Slov., 2022, 69, 584–595 CrossRef CAS PubMed.
- L. Kas’yan, A. Serbin, A. Kas’yan, D. Karpenko and E. Golodaeva, Russ. J. Org. Chem., 2008, 44, 340–347 CrossRef.
- S. Shaaban, H. Ferjani, H. M. Abd El-Lateef, M. M. Khalaf, M. Gouda, M. Alaasar and T. A. Yousef, Front. Chem., 2022, 10, 961787 CrossRef CAS PubMed.
- S. Shaaban, A. Zarrouk, D. Vervandier-Fasseur, Y. S. Al-Faiyz, H. El-Sawy, I. Althagafi, P. Andreoletti and M. Cherkaoui-Malki, Arabian J. Chem., 2021, 14 CAS.
- S. Shaaban, D. Vervandier-Fasseur, P. Andreoletti, A. Zarrouk, P. Richard, A. Negm, G. Manolikakes, C. Jacob and M. Cherkaoui-Malki, Bioorg. Chem., 2018, 80, 43–56 CrossRef CAS PubMed.
- M. Sak, Y. S. Al-Faiyz, H. Elsawy and S. Shaaban, Antioxidants, 2022, 11, 1231 CrossRef CAS PubMed.
- S. Shaaban, Y. S. Al-Faiyz, G. M. Alsulaim, M. Alaasar, N. Amri, H. Ba-Ghazal, A. A. Al-Karmalawy and A. Abdou, Inorganics, 2023, 11, 321 CrossRef CAS.
- B. Al-Abdallah, Y. S. Al-Faiyz and S. Shaaban, Inorganics, 2022, 10, 246 CrossRef CAS.
- J. M. Sonego, S. I. de Diego, S. H. Szajnman, C. Gallo-Rodriguez and J. B. Rodriguez, Chem.–A Euro. J., 2023, 29, e202300030 CrossRef CAS PubMed.
- O. d. R. A. Junior, E. Antônio, R. M. Mainardes and N. M. Khalil, J. Trace Elem. Med. Biol., 2017, 39, 176–185 CrossRef PubMed.
- C. W. Nogueira and J. B. Rocha, J. Braz. Chem. Soc., 2010, 21, 2055–2071 CrossRef CAS.
- S. Shaaban, H. Ba-Ghazal, Y. S. Al-Faiyz, A. A. Al-Karmalawy, N. Amri and I. Youssef, Tetrahedron, 2024, 133957 CrossRef CAS.
- S. Shaaban, H. M. A. El-Lateef, M. M. Khalaf, M. Gouda and I. Youssef, Polymers, 2022, 14, 2208 CrossRef CAS PubMed.
- G. I. Giles, F. H. Fry, K. M. Tasker, A. L. Holme, C. Peers, K. N. Green, L. O. Klotz, H. Sies and C. Jacob, Org. Biomol. Chem., 2003, 1, 4317–4322 RSC.
- F. H. Fry and C. Jacob, Curr. Pharm. Des., 2006, 12, 4479–4499 CrossRef CAS PubMed.
- C. A. Collins, F. H. Fry, A. L. Holme, A. Yiakouvaki, A. Al-Qenaei, C. Pourzand and C. Jacob, Org. Biomol. Chem., 2005, 3, 1541–1546 RSC.
- A. D. Sonawane, R. A. Sonawane, M. Ninomiya and M. Koketsu, Dalton Trans., 2021, 50, 12764–12790 RSC.
- P. N. Makhal, A. Nandi and V. R. Kaki, ChemistrySelect, 2021, 6, 663–679 CrossRef CAS.
- S. Shaaban, A. Negm, M. A. Sobh and L. A. Wessjohann, Anticancer Agents Med. Chem., 2016, 16, 621–632 CrossRef CAS PubMed.
- B. Al-Abdallah, Y. S. Al-Faiyz and S. Shaaban, Biomolecules, 2022, 12, 1765 CrossRef CAS PubMed.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, JNCI, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS PubMed.
- S. Shaaban, H. A. Althikrallah, A. Negm, A. Abo Elmaaty and A. A. Al-Karmalawy, RSC Adv., 2024, 14, 18576–18587 RSC.
- F. Farouk, A. A. Elmaaty, A. Elkamhawy, H. O. Tawfik, R. Alnajjar, M. A. S. Abourehab, M. A. Saleh, W. M. Eldehna and A. A. Al-Karmalawy, J. Enzyme Inhib. Med. Chem., 2023, 38, 2171029 CrossRef PubMed.
- S. Shaaban, M. S. S. Adam and N. M. El-Metwaly, J. Mol. Liq., 2022, 363, 119907 CrossRef CAS.
- S. Shaaban, A. Abdou, A. G. Alhamzani, M. M. Abou-Krisha, M. A. Al-Qudah, M. Alaasar, I. Youssef and T. A. Yousef, Life, 2023, 13, 912 CrossRef CAS PubMed.
- R. Huey, G. M. Morris and S. Forli, Using AutoDock 4 and AutoDock vina with AutoDockTools: a tutorial, The Scripps Research Institute Molecular Graphics Laboratory, 2012 Search PubMed.
- S. Yuan, H. S. Chan and Z. Hu, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 7, e1298 Search PubMed.
- A. A. Al-Karmalawy, M. Rashed, M. Sharaky, H. S. Abulkhair, M. M. Hammouda, H. O. Tawfik and M. A. Shaldam, Eur. J. Med. Chem., 2023, 115661, DOI:10.1016/j.ejmech.2023.115661.
- A. A. Elmaaty, K. M. Darwish, A. Chrouda, A. A. Boseila, M. A. Tantawy, S. S. Elhady, A. B. Shaik, M. Mustafa and A. A. Al-karmalawy, ACS Omega, 2022, 7, 875–899 CrossRef CAS PubMed.
- R. R. Ezz Eldin, M. A. Saleh, M. H. Alotaibi, R. K. Alsuair, Y. A. Alzahrani, F. A. Alshehri, A. F. Mohamed, S. M. Hafez, A. A. Althoqapy, S. K. Khirala, M. M. Amin, Y. A. F, A. H. AbdElwahab, M. S. Alesawy, A. A. Elmaaty and A. A. Al-Karmalawy, J. Enzyme Inhib. Med. Chem., 2022, 37, 1098–1119 CrossRef CAS PubMed.
- A. M. El-Naggar, A. M. A. Hassan, E. B. Elkaeed, M. S. Alesawy and A. A. Al-Karmalawy, Bioorg. Chem., 2022, 123, 105770 CrossRef CAS PubMed.
- S. Release, Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, 2017 Search PubMed.
- M. M. Hammoud, M. Khattab, M. Abdel-Motaal, J. Van der Eycken, R. Alnajjar, H. Abulkhair and A. A. Al-Karmalawy, J. Biomol. Struct. Dyn., 2022, 1–18 CrossRef PubMed.
- R. M. El-Masry, A. A. Al-Karmalawy, R. Alnajjar, S. H. Mahmoud, A. Mostafa, H. H. Kadry, S. M. Abou-Seri and A. T. Taher, New J. Chem., 2022, 46, 5078–5090 RSC.
- A. A. Al-Karmalawy, R. Alnajjar, A. A. Elmaaty, F. A. Binjubair, S. T. Al-Rashood, B. S. Mansour, A. Elkamhawy, W. M. Eldehna and K. A. Mansour, J. Biomol. Struct. Dyn., 2023, 1–13, DOI:10.1080/07391102.2023.2240419.
- A. Abo Elmaaty, A. A. Al-Karmalawy, M. S. Nafie, M. M. Shamaa, I. Zaki, R. Alnajjar and M. Y. Zakaria, Int. J. Pharm., 2023, 640, 122980 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and Methods; SI1. Chemistry; SI2. Anti-inflammatory markers assay (Enzyme-linked Immunosorbent Assay); SI3. Molecular dynamics simulations; SI4. MD trajectory analysis and prime MM-GBSA calculations. See DOI: https://doi.org/10.1039/d4ra04762a |
| ‡ These two authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |