
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the antitumor potential of novel quinoline derivatives via tubulin polymerization inhibition in breast cancer; design, synthesis and molecular docking†
Heba Abdelmegeeda,
Lina M. A. Abdel Ghany *b,
Amira Youssefc,
Abd-Allah S. El-Etrawycd and
Noha Ryadc
*b,
Amira Youssefc,
Abd-Allah S. El-Etrawycd and
Noha Ryadc
aChemistry of Natural Compounds Department, Pharmaceutical and Drug Industries Research Institute, National Research Centre, Giza 12622, Egypt
bPharmaceutical Chemistry Department, College of Pharmaceutical Sciences and Drug Manufacturing, Misr University for Science and Technology (MUST), 6th of October City, P.O. Box 77, Giza, Egypt. E-mail: Lina.ameen@must.edu.eg
cPharmaceutical Organic Chemistry Department, College of Pharmaceutical Sciences and Drug Manufacturing, Misr University for Science and Technology (MUST), 6th of October City, P.O. Box 77, Giza, Egypt
dDepartment of Chemistry, Basic Science, Misr University for Science and Technology (MUST), 6th of October City, P.O. Box 77, Giza, Egypt
First published on 12th July 2024
Abstract
A series of quinoline derivatives was designed and synthesized as novel tubulin inhibitors targeting the colchicine binding site. All the rationalized compounds 3a–e, 4a–e, 5a–e, and 6a–e have been chosen for screening their cytotoxic activity against 60 cell lines by NCI. Compounds 3b, 3c, 4c, 5c and 6c demonstrated the most notable antitumor activity against almost all cell lines. Compound 4c emerged as the most potent compound as an antiproliferative agent. This compound was subsequently chosen for five-dose testing and it exhibited remarkable broad-spectrum efficacy with strong antitumor activity against several cell lines. Compound 4c significantly induced cell cycle arrest in MDA-MB-231 cells at G2 and M phases where the cell population increased dramatically to 22.84% compared to the untreated cells at 10.42%. It also increased the population in MDA-MB-231 cells at both early and late stages of apoptosis. Compound 4c can successfully inhibit tubulin polymerization with an IC50 value of 17 ± 0.3 μM. The β-tubulin mRNA levels were notably reduced in MDA-MB-231 cells treated with compound 4c which is similar to the effect observed with colchicine treatment. Docking studies revealed that compound 4c interacted well with crucial amino acids in the active site.
1. Introduction
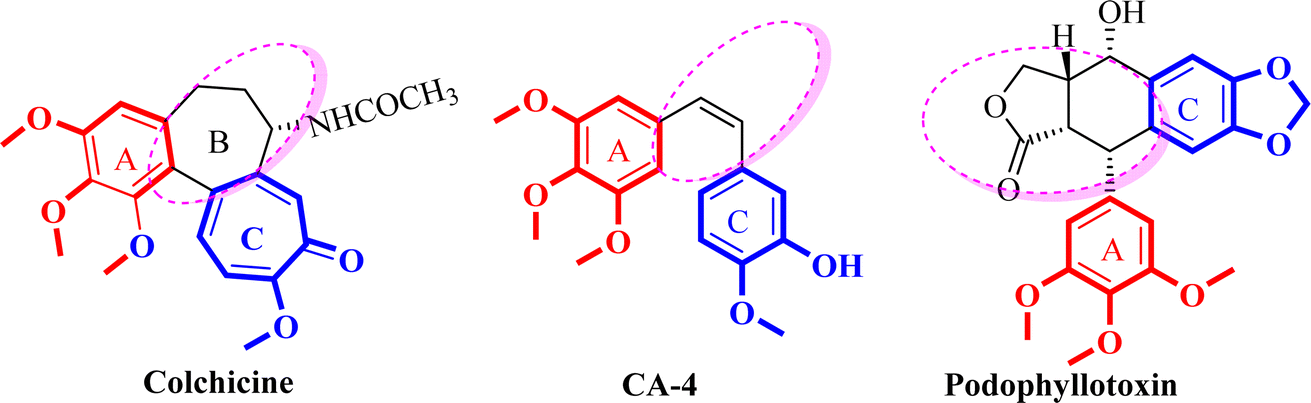
Cancer is the development of abnormal cells that proliferate uncontrollably and affect human health and it is one of the leading causes of death globally.1–3 The latest data indicate that breast cancer is one of the most prevalently diagnosed cancers among women with an estimated 2.3 million new cases (11.7%) and the 5th cause of cancer-related deaths with an estimated 6.9%.1–3 Currently, chemotherapy is the main approach for cancer treatment; since microtubules are essential for cell viability, especially for the fast division of cancer cells, drugs that interfere with the dynamics of microtubule/tubulin have become essential therapeutics.4,5 Most of these agents act by binding to the tubulin, an α/β heterodimer protein that forms the microtubule which is a major component of the eukaryotic cytoskeleton.6Microtubule targeting agents (MTA) are also named antimitotic agents which bind to the tubulin in the microtubules and prohibit the proliferation of the cells. There are two distinct categories of microtubule targeting agents; those that bind to the binding site of paclitaxel, such as paclitaxel which are identified as microtubule stabilizing agents or tubulin promotors.7 Conversely, agents that bind to the binding site of colchicine, such as colchicine, combretastatin A-4 (CA-4), and podophyllotoxin or to the binding site of vinca alkaloid, such as vincristine, are identified as tubulin inhibitors or destabilizing agents (Fig. 1).8
 | ||
| Fig. 1 Structure of tubulin destabilizing agents. | ||
The colchicine binding site (CBS), which sits at the interface between α and β-tubulin heterodimers, has been the subject of much research to identify potential anticancer drugs. Colchicine was the first tubulin destabilizing agent. It was extracted from the poisonous meadow saffron Colchicum autumnale and it binds with high affinity to β-tubulin subunit, interfering with microtubule polymerization and causing mitosis.5,9,10
Pharmacophoric patterns identified in studies of colchicine binding site inhibitors (CBSIs) consistently displayed specific structural characteristics and recurring interactions between tubulin and ligands at pharmacophoric points. These studies proposed that the different structural classes of CBSIs can be linked by a six to seven-point pharmacophore consisting of three hydrogen bond acceptors (HBA), one hydrogen bond donor (HBD), one or two hydrophobic center (HY), and/or one planar group.11 Ten top-scored hypothetical pharmacophores were generated in another study using the HypoGen algorithm, the training set consisted of 26 drugs with tubulin inhibitory activity ranging from 0.52 to 13.800 nM. One hydrogen-bond acceptor (HBA), one hydrogen-bond donor (HBD), one ring aromatic feature (RA), one hydrophobic feature (HY), and three excluded volumes (EV) made up the greatest hypothetical pharmacophore.12 Another training set for the 3D QSAR pharmacophore model used the structures of 21 different drugs defined four different feature types: the hydrophobic feature (HY), hydrophobic aromatic group (HY-AR), hydrogen bond acceptor (HBA), and hydrogen bond donor (HBD) (Fig. 2).13–15 The previously mentioned features are necessary to ensure stable and effective binding to the CBS.16 The presence of many hydrophobic and aromatic groups ensures a fit to the hydrophobic pocket of the β-tubulin subunit, while the HBA and HBD groups extend the inhibitors towards the hydrophilic α-tubulin subunit.
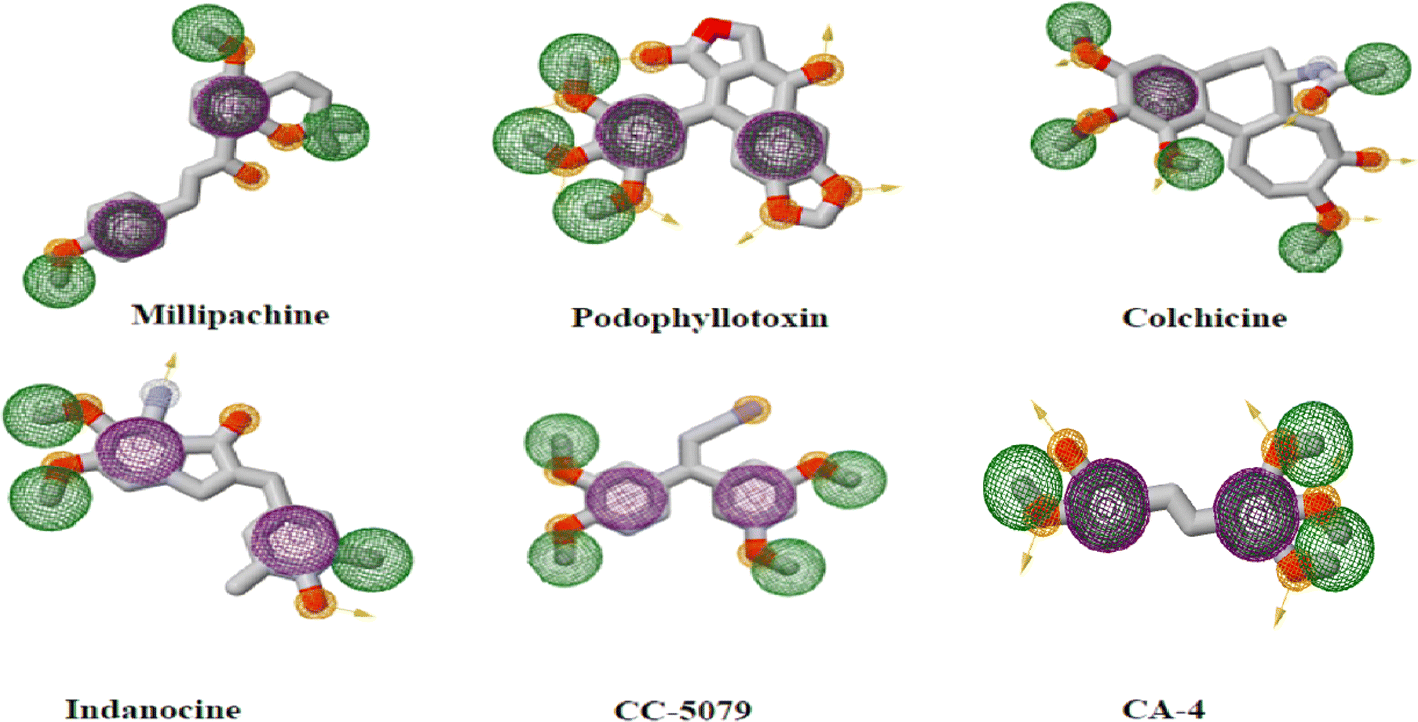 | ||
| Fig. 2 Two-dimensional structures of reported CBSI's with pharmacophoric features necessary to impart depolymerization upon tubulin where purple spheres represent aromatic pharmacophoric points (AR), the orange spheres with and without arrow denote the functional groups that act as hydrogen bond donor (HBD) and acceptor (HBA), respectively. Finally green spheres represent hydrophobic centers (HY). | ||
SAR analysis of colchicine revealed that the important trimethoxyphenyl group is oriented within β-tubulin. The interaction between ring A and CBS provides the strength of colchicine binding to tubulin. On the other hand, interactions between the CBS and the oxygen atoms on ring C control the inhibition. It is suggested that within the binding locus, ring A anchors and keeps the B and C rings oriented correctly.5,9,10
Moreover, colchicine showed its anticancer effect on apoptotic genes of human breast cancer cell lines.10,17,18 CA-4 binds at the CBS in a manner akin to that of colchicine.10 Nevertheless, the instability of the cis-double bond, which might change into an inactive trans-conformation, has hindered the development of CA-4.19 In recent years, various conformationally restricted analogs of CA-4 and colchicine-bearing triazolopyrimidine I, benzimidazole II, azetidine III, pyridine IV and VI, pyrazole V, pyrimidine VII, imidazopyridine VIII have been reported as potent tubulin inhibitors (Fig. 3).20–26
 | ||
| Fig. 3 Chemical structure of reported CA-4 analogs with cytotoxic activity. | ||
Quinoline is a privileged scaffold in the expansion of anticancer drugs as they have exhibited potent antiproliferative activity via different mechanisms of action including cell cycle arrest, induction of apoptosis, and inhibition of angiogenesis, and cell migration disruption. Many tubulin inhibitors IX–XIV also, possess quinoline motifs (Fig. 4), these compounds exhibited their antiproliferative activity because of tubulin polymerization inhibition and the disruption of microtubule assembly.27–32
 | ||
| Fig. 4 Some quinoline-containing antimitotic agents and tubulin polymerization inhibitors. | ||
In light of the aforementioned findings, we aimed to synthesize novel molecules with promising tubulin polymerization inhibition activity via structure optimization of lead compounds CA-4 and colchicine. As shown in our design strategy (Fig. 5), bioisosteric replacement of ring C in colchicine and CA-4 was performed with a quinoline ring. 3,4,5-Trimethoxyphenyl moiety (ring A) in colchicine and CA-4 was retained in certain synthesized derivatives, while others were embellished with 3,4-dimethoxyphenyl, 4-methoxyphenyl, 4-fluorophenyl or 4-bromophenyl moieties. Chain elongation in compounds 3a–e by the introduction of 2-propen-1-one instead of an olefinic bond aimed to increase the rigidity of the structure to overcome the drawbacks of CA-4. For more rigidification, cyclization with rigid core ring B was performed by 2-pyridinone 4a–e, pyridinethione 5a–e and pyrimidinethione ring 6a–e. Our designed plan was achieved according to Scheme 1 (Fig. 6). The novel compounds were also fully characterized for their cytotoxic activity on NCI 60-cell lines and additional mechanistic biochemical tests were conducted for the molecule with the highest potency. Moreover, docking studies were conducted in an attempt to interpret the outcomes of biological tests.
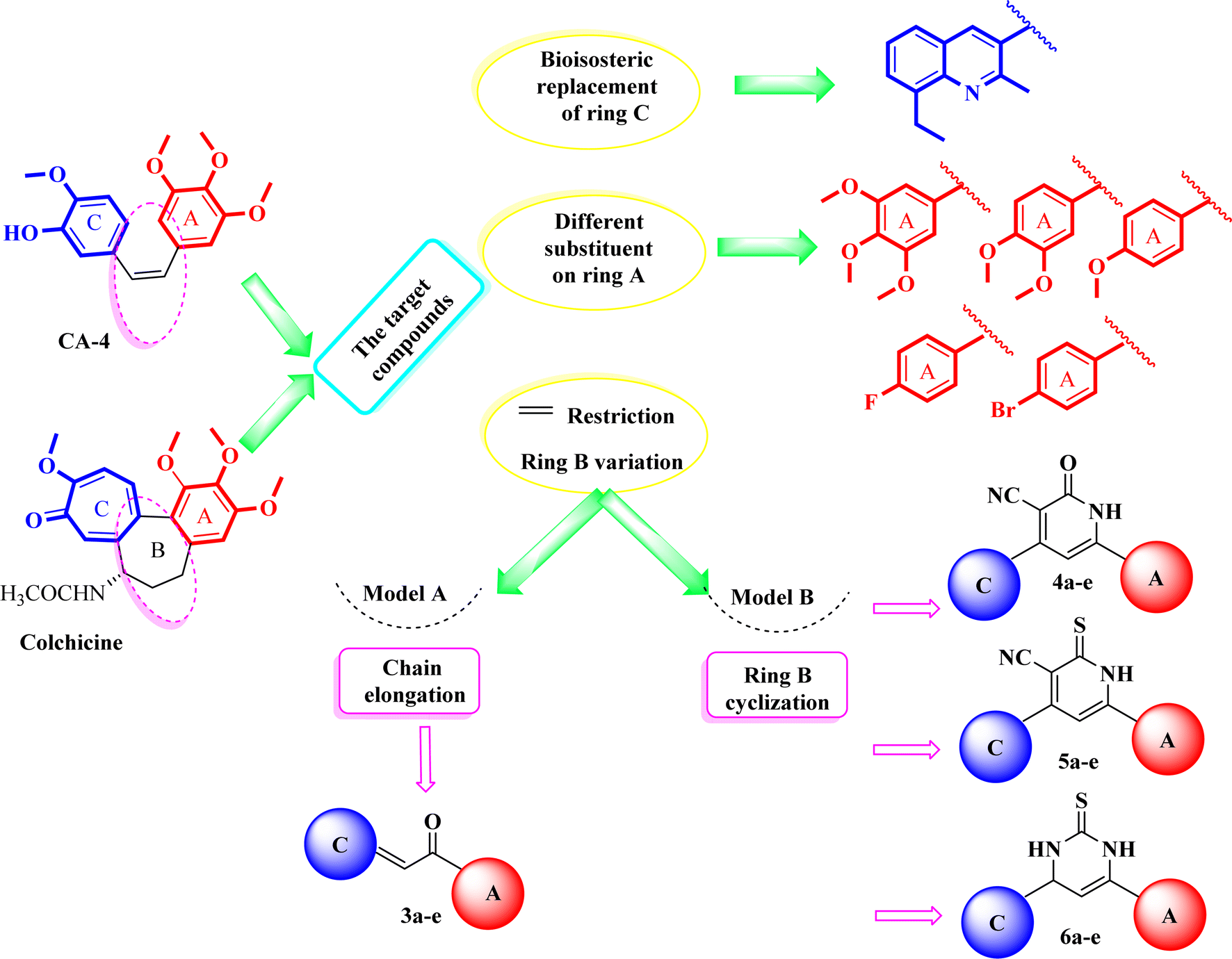 | ||
| Fig. 5 Design strategy of the rationalized compounds 3a–e, 4a–e, 5a–e, and 6a–e. | ||
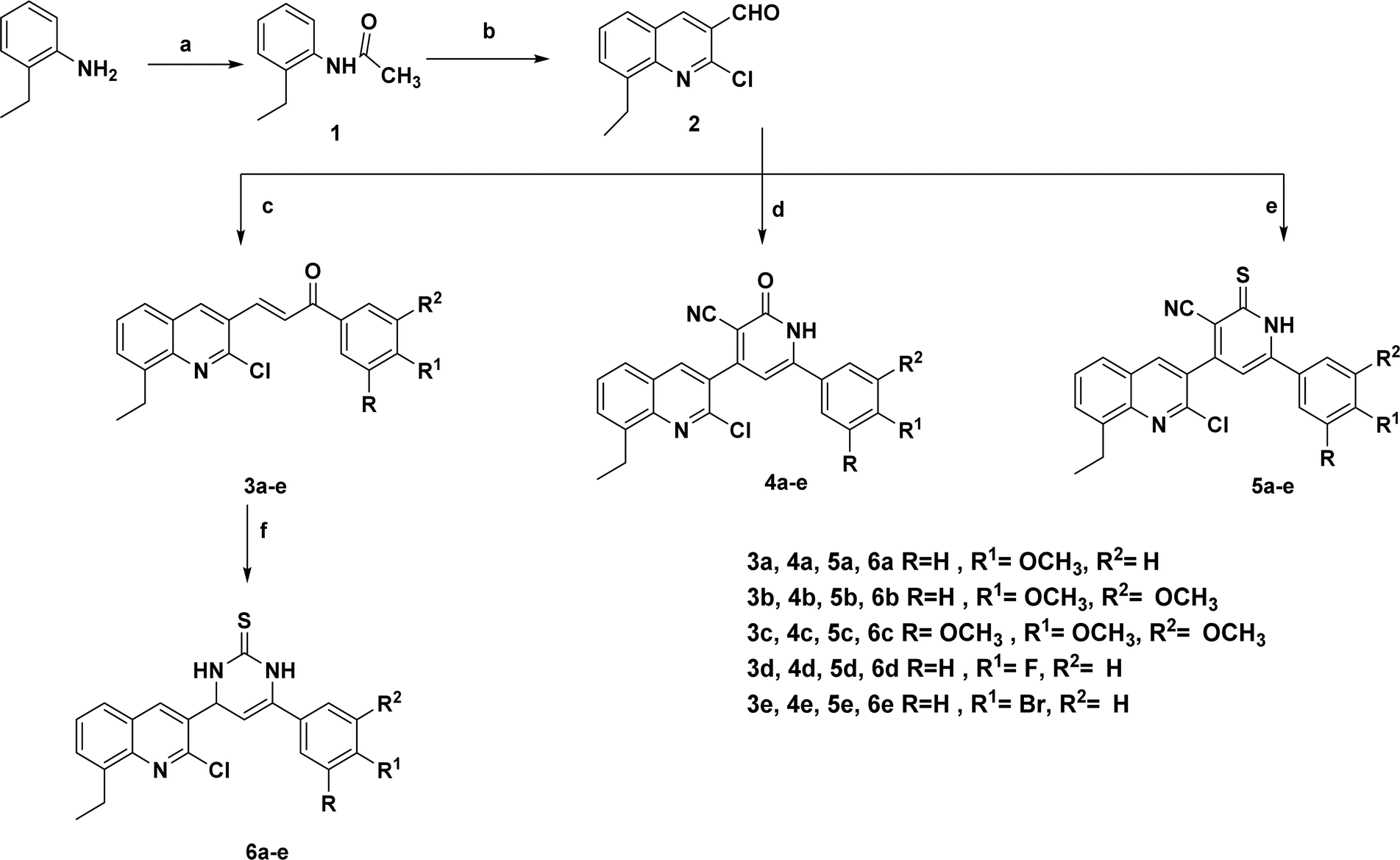 | ||
| Scheme 1 Synthesis of the target compounds 3a–e, 4a–e, 5a–e, and 6a–e. | ||
 | ||
| Fig. 6 Some of the designed titled compounds with common structural features to reported tubulin depolymerization agents. | ||
2. Results and discussion
2.1. Chemistry
The present study described the synthetic plan of novel compounds 3a–e, 4a–e, 5a–e, and 6a–e as illustrated in Scheme 1. Compound 1 and the key intermediate 2 were prepared in excellent yields as described in the reported literature.33–36 Chalcone derivatives 3a–e were prepared following the Claisen–Schmidt condensation reaction by reacting a mixture of the carbaldehyde derivative 2 with the appropriate substituted acetophenone in the presence of 50% aqueous potassium hydroxide. The IR spectrum of each synthesized compound exhibited a strong absorption band corresponding to the carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) functional group, which was observed in the range of 1600–1656 cm−1. 1H NMR charts showed signals corresponding to α, β alkene protons, as a doublet signal at δ 8.12–8.20 ppm referred to the α-CH alkene proton while another doublet appeared at δ 7.64–8.11 ppm corresponding to β-CH alkene proton. On the other hand, their 13C NMR spectra showed a peak referring to the carbonyl (CO) functional group at δ 186.8–188.0 ppm. Refluxing the 8-ethylquinoline-3-carbaldehyde derivative 2 with different acetophenones and either ethyl cyanoacetate or 2-cyanothioacetamide in the presence of ammonium acetate afforded the target pyridine derivatives 4a–e and 5a–e, respectively. The IR spectra of the carbonitrile derivatives 4a–e and 5a–e showed a peak at 2218–2239 cm−1 of the added CN group and a band of NH appeared at 3398–3433 cm−1. 1H NMR spectra of 2-oxo-1,2-dihydropyridine derivatives 4a–e showed a singlet at δ 6.85–7.16 ppm corresponding to the proton of 1,2-dihydropyridine ring, while 13C NMR spectra revealed signals at δ 116.4–118.2 and 156.0–159.6 ppm for nitrile and carbonyl groups, respectively. 1H NMR spectra of the isosteric analog; 2-thioxo-1,2-dihydropyridine derivatives 5a–e showed similar singlet at δ 6.78–7.17 ppm corresponding to the proton of 1,2-dihydropyridine ring, although, their 13C NMR spectra revealed signals at δ 116.0–117.0 and 156.5–162.1 ppm for nitrile and thiocarbonyl groups, respectively. Additionally, mass spectrometric analysis was conducted to further confirm the structure of the synthesized 1,2-dihydropyridine derivative compounds, designated as 4a–e and 5a–e. The mass spectrometric data showed the presence of molecular ion peaks that corresponded to the expected molecular weights of the respective synthesized compounds. 3,4-Dihydropyrimidinethione derivatives 6a–e were synthesized by refluxing the corresponding chalcone 3a–e with thiourea in an alkaline medium. The afforded compounds were prepared with an overall good yield of 70–74%. The IR spectra confirmed that the carbonyl functional group present in the starting materials 3a–e was no longer detected in the final synthesized compounds but an absorption band appeared at 3162–3169 cm−1 corresponding to NH. 1H NMR of 6a–e showed two doublets at δ 5.39–5.61 ppm which is characteristic to the dihydropyrimidine ring protons along with the exchangeable protons of 2NH that appeared at δ 9.10–9.10 and 10.04–10.20 ppm. Additionally, 13C NMR showed a peak at δ 96.5–99.7 ppm pointing to C5 of the pyrimidine ring and a peak at δ 174.0–177.0 ppm referring to CS. Finally, it is worth mentioning that all our novel compounds were subjected to elemental analyses for further authentication of the synthesized compounds.
O) functional group, which was observed in the range of 1600–1656 cm−1. 1H NMR charts showed signals corresponding to α, β alkene protons, as a doublet signal at δ 8.12–8.20 ppm referred to the α-CH alkene proton while another doublet appeared at δ 7.64–8.11 ppm corresponding to β-CH alkene proton. On the other hand, their 13C NMR spectra showed a peak referring to the carbonyl (CO) functional group at δ 186.8–188.0 ppm. Refluxing the 8-ethylquinoline-3-carbaldehyde derivative 2 with different acetophenones and either ethyl cyanoacetate or 2-cyanothioacetamide in the presence of ammonium acetate afforded the target pyridine derivatives 4a–e and 5a–e, respectively. The IR spectra of the carbonitrile derivatives 4a–e and 5a–e showed a peak at 2218–2239 cm−1 of the added CN group and a band of NH appeared at 3398–3433 cm−1. 1H NMR spectra of 2-oxo-1,2-dihydropyridine derivatives 4a–e showed a singlet at δ 6.85–7.16 ppm corresponding to the proton of 1,2-dihydropyridine ring, while 13C NMR spectra revealed signals at δ 116.4–118.2 and 156.0–159.6 ppm for nitrile and carbonyl groups, respectively. 1H NMR spectra of the isosteric analog; 2-thioxo-1,2-dihydropyridine derivatives 5a–e showed similar singlet at δ 6.78–7.17 ppm corresponding to the proton of 1,2-dihydropyridine ring, although, their 13C NMR spectra revealed signals at δ 116.0–117.0 and 156.5–162.1 ppm for nitrile and thiocarbonyl groups, respectively. Additionally, mass spectrometric analysis was conducted to further confirm the structure of the synthesized 1,2-dihydropyridine derivative compounds, designated as 4a–e and 5a–e. The mass spectrometric data showed the presence of molecular ion peaks that corresponded to the expected molecular weights of the respective synthesized compounds. 3,4-Dihydropyrimidinethione derivatives 6a–e were synthesized by refluxing the corresponding chalcone 3a–e with thiourea in an alkaline medium. The afforded compounds were prepared with an overall good yield of 70–74%. The IR spectra confirmed that the carbonyl functional group present in the starting materials 3a–e was no longer detected in the final synthesized compounds but an absorption band appeared at 3162–3169 cm−1 corresponding to NH. 1H NMR of 6a–e showed two doublets at δ 5.39–5.61 ppm which is characteristic to the dihydropyrimidine ring protons along with the exchangeable protons of 2NH that appeared at δ 9.10–9.10 and 10.04–10.20 ppm. Additionally, 13C NMR showed a peak at δ 96.5–99.7 ppm pointing to C5 of the pyrimidine ring and a peak at δ 174.0–177.0 ppm referring to CS. Finally, it is worth mentioning that all our novel compounds were subjected to elemental analyses for further authentication of the synthesized compounds.
2.2. Biology
 | ||
| Fig. 7 Heatmap of all the synthesized quinoline derivatives 3a–e, 4a–e, 5a–e and 6a–e demonstrating their effect on tumor cells viability of 60 different cancer cell lines. Red color indicates higher tumor cells viability, while blue color indicates less tumor cells viability. | ||
Pyridin-2-one 4c (trimethoxy-substituted) demonstrated the highest level of cytotoxic activity among the synthesized compounds. It possessed potent cytotoxic activity against wide range of tumor cells as leukemia (CCRF-CEM, MOLT-4, RPMI-8226, and SR) with GI values of 80.39, 76.43, 74.51 and 77.32%, respectively, non-small cell lung cancer (A549/ATCC and HOP-62) with GI values of 88.16 and 87.86%, respectively, CNS cancer (SF-295 and SNB-19) with GI values of 96.38 and 89.58%, respectively, melanoma (LOX IMVI, MALME-3M and SK-MEL-5) with GI values of 91.58, 89.41, 74.51 and 78.58%, respectively, ovarian cancer (OVCAR-8) with GI value of 92.11%, renal cancer (ACHN and CAKI-1) with GI values of 75.69 and 95.06%, respectively, prostate cancer (DU-145) with GI value of 80.55% and breast cancer (T-47D) with GI value of 91.56%. Its lethal effect was evident across a wide range of tumor cells including non-small cell lung cancer (HOP-92, NCI-H226 and NCI-H522), CNS cancer (SF-539, SNB-75 and U251), melanoma (UACC-62), ovarian cancer (OVCAR-3 and OVCAR-4), renal cancer (786-0, A498, RXF 393, SN12C, TK-10 and UO-31) and breast cancer (MDA-MB-231/ATCC and HS 578T). Other derivatives in the series including pyridin-2-one derivative 4a (methoxy-substituted) showed moderate activity over CNS cancer (SNB-75) with GI value of 54.69%, where, compound 4b (dimethoxy-substituted), 4d (fluoro-substituted) and 4e (bromo-substituted) possessed mild cytotoxic activity over multiple cell lines.
Pyridinethione derivative 5c displayed significant cytotoxic activity on non-small cell lung cancer (HOP-92) with GI value of 77.38%, while, its moderate cytotoxic activity appeared over numerous cell lines as leukemia (CCRF-CEM, HL-60(TB), MOLT-4 and RPMI-8226) with GI values of 57.63, 51.76, 61.16 and 63.06%, respectively, non-small cell lung cancer (NCI-H522) with GI value of 65.68%, renal cancer (CAKI-1) with GI value of 69.81%, and breast cancer (MDA-MB-231/ATCC, T-47D and MDA-MB-468) with GI values of 52.51, 60.59 and 50.55%, respectively. Additional derivatives bearing pyridinethione namely, 5a (methoxy-substituted), 5b (dimethoxy-substituted), 5d (fluoro-substituted) and 5e (bromo-substituted) showed a slight cytotoxic activity over multiple cell lines.
Finally, pyrimidine derivative 6c (trimethoxy-substituted) possessed moderate cytotoxic effect over leukemia (MOLT-4 and RPMI-8226) with GI values of 67.65 and 57.59%, respectively, non-small cell lung cancer (NCI-H226 and NCI-H522) with GI values of 57.97 and 50.3%, respectively and renal cancer (CAKI-1 and UO-31) with GI values of 64.11 and 67.70%, respectively. Pyrimidine derivative 6b (dimethoxy-substituted) possessed moderate cytotoxic effect over leukemia (MOLT-4) with GI value of 53.3%. Other derivatives bearing pyrimidine moiety specially, 6a (methoxy-substituted), 6d (fluoro-substituted) and 6e (bromo-substituted) showed limited cytotoxic activity over multiple cell lines.
In summary, upon comparing all the newly synthesized compounds (Fig. 7), clearly shows that 4c > 3c > 3b > 5c > 6c derivatives demonstrated the highest antitumor activity against almost all cell lines. Notably, compound 4c being the most potent compound as anti-proliferative agent. Interestingly, 4c derivative along with the other compounds with the highest activity (i.e., 3c, 5c, 6c) belonged to the c derivatives series which is characterized by trimethoxy group substitution in the main ring. Hence, the trimethoxy group substitution is shown to be superior to all other modifications in the main ring (methoxy, dimethoxy, fluorine, and bromine). On the other hand, cyclization with pyridine-2-one cyclization is more effective than (open chain > pyridinethione > pyrimidine). This suggests that trimethoxy group and pyridin-2-one are responsible for the compounds' high antitumor activity.
| Subpanel tumor cell lines | Activity | Subpanel tumor cell lines | Activity | ||||
|---|---|---|---|---|---|---|---|
| GI50 | TGI | LC50 | GI50 | TGI | LC50 | ||
| Leukemia | Melanoma | ||||||
| CCRF-CEM | 12.00 | >100 | >100 | M14 | 29.70 | >100 | >100 |
| HL-60(TB) | 13.40 | >100 | >100 | MDA-MB-435 | 32.60 | >100 | >100 |
| K-562 | 7.72 | 43.80 | >100 | SK-MEL-2 | 39.80 | >100 | >100 |
| MOLT-4 | 8.17 | 79.90 | >100 | SK-MEL-28 | 21.10 | >100 | >100 |
| RPMI-8226 | 5.16 | >100 | >100 | SK-MEL-5 | 17.50 | 90.20 | >100 |
| SR | 5.70 | >100 | >100 | UACC-257 | 42.60 | >100 | >100 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
UACC-62 | 17.50 | 60.10 | >100 | |||
| Non-small cell lung cancer | |
||||||
| A549/ATCC | 12.20 | 48.70 | >100 | Ovarian cancer | |||
| EKVX | 28.60 | >100 | >100 | IGROV1 | 12.20 | 46.30 | >100 |
| HOP-62 | 20.70 | 78.90 | >100 | OVCAR-3 | 14.20 | 29.80 | 62.30 |
| HOP-92 | 2.37 | 9.68 | >100 | OVCAR-4 | 17.20 | >100 | >100 |
| NCI-H226 | 10.90 | 32.60 | 97.70 | OVCAR-5 | 48.40 | >100 | >100 |
| NCI-H23 | 3.20 | >100 | >100 | OVCAR-8 | 18.50 | 93.60 | >100 |
| NCI-H322M | 10.50 | >100 | >100 | NCI/ADR-RES | 44.90 | >100 | >100 |
| NCI-H460 | 16.10 | 41.80 | >100 | SK-OV-3 | 26.00 | >100 | >100 |
| NCI-H522 | 19.10 | >100 | >100 | |
|||
|
Renal cancer | ||||||
| Colon cancer | 786-0 | 11.70 | 26.70 | 61.10 | |||
| COLO 205 | 45.60 | >100 | >100 | A498 | 15.80 | 45.20 | >100 |
| HCC-2998 | 35.70 | >100 | >100 | ACHN | 15.60 | 31.00 | 61.80 |
| HCT-116 | 14.10 | 33.30 | 78.40 | CAKI-1 | 10.70 | 22.90 | 49.20 |
| HCT-15 | 27.60 | >100 | >100 | RXF 393 | 2.21 | 6.69 | 35.40 |
| HT29 | 32.10 | >100 | >100 | SN12C | 10.30 | 25.20 | 61.80 |
| KM12 | 33.60 | >100 | >100 | TK-10 | 17.50 | 34.50 | >100 |
| SW-620 | 27.50 | >100 | >100 | UO-31 | 12.60 | >100 | >100 |
|
|
||||||
| CNS cancer | Prostate cancer | ||||||
| SF-268 | 13.80 | >100 | >100 | PC-3 | 40.30 | >100 | >100 |
| SF-295 | 19.40 | >100 | >100 | DU-145 | 15.50 | 38.00 | 93.50 |
| SF-539 | 12.00 | 24.80 | 51.30 | |
|||
| SNB-19 | 9.25 | 50.90 | >100 | Breast cancer | |||
| SNB-75 | 2.38 | 16.90 | 52.60 | MCF7 | 24.00 | >100 | >100 |
| U251 | 10.80 | 28.40 | 74.90 | MDA-MB-231/ATCC | 14.50 | 31.80 | 69.80 |
|
HS 578T | 2.38 | 7.26 | >100 | |||
| Melanoma | BT-549 | 4.11 | 28.10 | >100 | |||
| LOX IMVI | 13.50 | 33.20 | 81.30 | T-47D | 12.20 | 64.10 | >100 |
| MALME-3M | 10.10 | 40.00 | >100 | MDA-MB-468 | 12.70 | 61.60 | >100 |
 | ||
| Fig. 8 Dose response curves of 4c derivative against all 60 cell lines. | ||
Hence, we investigated the distribution of cells at the different phases of cell cycle after MDA-MB-231 cells treatment with the newly synthesized 4c derivative and the positive control colchicine which is a well-established tubulin polymerization inhibitor. Fig. 9A shows the histograms of the control non-treated cells in addition to 4c, and colchicine-treated cells demonstrating propidium iodide PI signals at each phase. Both 4c and colchicine histograms showed an increase in signal intensity at G2/M phase. Moreover, the distribution of cells in each phase of the cell cycle was measured. As illustrated in Fig. 9B, 4c and colchicine significantly induced cell cycle arrest at G2 and M phases where the cells population increased dramatically to 22.84% and 26.51%, respectively compared with control non-treated cells which were 10.42%. There was no significant difference between 4c and colchicine in inducing cell cycle arrest at G2 and M phases. Hence, 4c derivative exerts its cytotoxic action against breast cancer via inhibiting tubulin polymerization causing cell cycle arrest at G2 and M phases.
 | ||
| Fig. 9 (A) Histograms of cell cycle phase distribution of control, colchicine, and 4c treated cells using PI staining for FACS analysis. (B) Percentages of cells accumulation at G0–G1, S, and G2/M cell cycle phases induced by control, colchicine, and 4c. Data are represented as the mean ± SD of three independent experiments. Statistical analysis was conducted using two-way ANOVA followed by Tukey's multiple comparison test; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared to the control. | ||
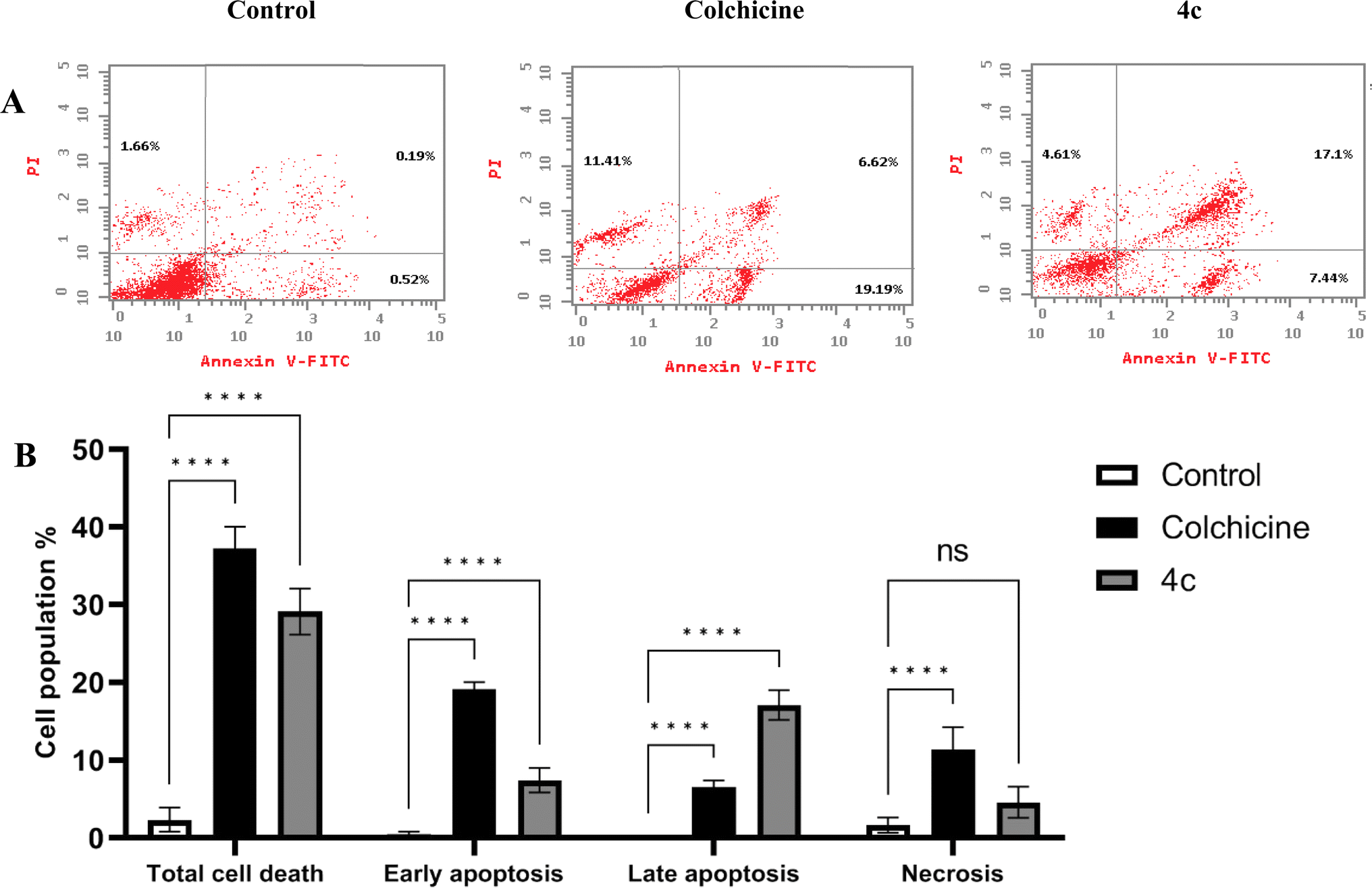 | ||
| Fig. 10 (A) Dot blots of apoptotic cells populations using PI/annexin V-FITC staining for FACS analysis. (B) Percentages of early apoptosis, late apoptosis and necrosis induced by 4c and colchicine in MDA-MB-231 cells compared with non-treated control cells. Data are represented as mean ± SD of three individual experiments. Statistical analysis was done by applying either two-way ANOVA followed by with Dunnett's multiple comparison test; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared to the control. | ||
| Compound | IC50 (μM) for tubulin polymerization inhibition |
|---|---|
| 4c | 17 ± 0.3 |
| Colchicine | 7.48 ± 0.11 |
| CA-4 | 4.647 ± 0.06 |
 | ||
| Fig. 11 Relative gene expression of β-tubulin induced by (A) 4c and (B) colchicine compared with the control. Data are represented as mean ± SD of three individual experiments. Statistical analysis was done by applying Mann–Whitney test; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared to the control. | ||
2.3. In silico studies
In the current simulation, redocking of colchicine was done which revealed the reported binding mode of the compound.51,52 After validation of our docking procedure, the titled compounds 3b, 3c, 4c, and 5c displayed correct binding modes into the CBS. Chalcone derivatives 3b and 3c which were lethal against non-small cell lung cancer cells; NCI-H226, CNS cancer cells; SF-539 and SNB-75 and ovarian cancer cells; OVCAR-3, showed a high binding interaction within the hydrophobic pocket of the β-tubulin subunit with the correct pose equal to −9.0 and −8.0 kcal mol−1, respectively. As can be deduced from (Fig. 12) both compounds revealed identical binding modes where they were deeply buried in the colchicine site at the α, β intradimer interface and formed mainly hydrophobic contacts with several residues of β-tubulin where the 2-chloro-8-ethylquinoline ring bonded through several π-alkyl and π-sigma interactions to βCys241, βLeu248, βAla250, βLeu255, βAla316, βIle318 and βLys352 (Table S4†). The dimethoxy and trimethoxyphenyl rings of 3b and 3c, respectively made H-bond interactions with αSer178 (3.33 and 3.29 Å, respectively) acting as hydrogen acceptors, both rings made π-alkyl interactions with αAla180. The dimethoxy group in 3b formed an extra H-bond with αGlu183 (3.48 Å) acting as a hydrogen donor while the trimethoxy group in 3c extended towards βLys254 forming π-alkyl interaction with the residue. The 2-oxo-1,2-dihydropyridine derivative 4c was the most cytotoxic synthesized compound against 60 cell lines. It inhibited the growth of most of the tested cells and was lethal towards many non-small cell lung cancer cell lines, CNS cancer cells, renal and breast cancer cells, while its isosteric derivative 2-thioxo-1,2-dihydropyridine containing compound 5c revealed less cytotoxic effect than 4c and exhibited reasonable cytotoxicity against most of the tested cell lines. Compound 4c revealed the highest predicted binding interaction with CBS of tubulin compared with 5c and colchicine equal to −11.5, −10.9 and −9.8 kcal mol−1, respectively, which support the results of the biological assay. Compounds 4c and 5c fitted comfortably into the active space of the CBS of tubulin, their mode of binding was almost identical and was like that of colchicine as revealed by superimposing both compounds (Fig. 13). The 2-chloro-8-ethylquinoline ring oriented itself towards the hydrophobic pocket of the CBS of tubulin forming multiple π-alkyl and π-sigma interactions with βCys241, βLeu248, βAla250, βLeu255 and βIle318. The trimethoxy phenyl ring in both compounds faced the hydrophilic region of CBS of tubulin. Both compounds are linked to αTyr224 and βGln247. Compound 4c formed hydrogen bond interactions with both residues (3.28 and 3.26 Å, respectively) acting as hydrogen acceptor and donor, respectively. Compound 5c bonded to the same two residues with π-alkyl and π-sigma interactions, respectively. Moreover, 4c made an additional hydrogen bond through its trimethoxy group with αSer178 (3.34 Å) acting as a hydrogen acceptor. π-Alkyl interaction occurred between βLys352 and the 2-oxo-1,2-dihydropyridine ring in compound 4c, as well as with the 2-thioxo-1,2-dihydropyridine ring in compound 5c. 2-Oxo-1,2-dihydropyridine moiety in 4c linked by three hydrogen bonds to αThr179, αAla180 and βAsn258 (3.37, 3.29, 4.12 Å, respectively) acting as hydrogen donor, acceptor and donor, respectively. Conversely, in compound 5c the 2-thioxo-1,2-dihydropyridine ring in 5c formed a hydrogen bond with αVal181 (3.65 Å) acting as hydrogen acceptor. Close analysis of the binding mode of 4c revealed that it formed many hydrogen bond interactions with the hydrophilic part of CBS of tubulin in addition to the hydrophobic interactions with the β-tubulin subunit. These interactions support the promising activity of compound 4c towards tubulin, as well as its cytotoxicity against numerous cancer cell lines (Table 3).
 | ||
| Fig. 12 2D and 3D interaction diagrams of (A) colchicine, (B) compound 3b and (C) compound 3c into the colchicine binding site of tubulin enzyme. | ||
 | ||
| Fig. 13 2D and 3D interaction diagrams of (A) compound 4c, (B) compound 5c into the colchicine binding site of tubulin enzyme and (C) aligned docking pose of colchicine (pink) and 4c into the CBS. | ||
| Compound | CDocker energy (kcal mol−1), tubulin enzyme (PDB ID: 4O2B) |
|---|---|
| 3b | −9.0 |
| 3c | −8.0 |
| 4c | −11.5 |
| 5c | −10.9 |
| Colchicine | −9.8 |
 | ||
| Fig. 14 Radar chart showing six predicted physicochemical properties of the tested compound 3b, 3c, 4c, 5c, colchicine and CA-4. | ||
| Comp. no. | TPSAa | PAINSb | WLOGPc | NRBd | HBDe | HBAf | GI absorptiong | BBB permeabilityh | Lipinskii |
|---|---|---|---|---|---|---|---|---|---|
| a Topological polar surface area.b Pan assay interference structures.c Lipophilicity petameter WLOGP.d Number of rotatable bonds.e Number of hydrogen bond acceptors.f Number of hydrogen bond donor.g Gastrointestinal absorption.h Blood–brain barrier permeability.i Lipiniki (drug likeness). | |||||||||
| 3b | 48.42 | 0 | 5.26 | 6 | 0 | 4 | High | Yes | 0 violation |
| 3c | 57.65 | 0 | 5.26 | 7 | 0 | 5 | High | Yes | 0 violation |
| 4c | 97.23 | 0 | 5.37 | 6 | 1 | 6 | High | No | 0 violation |
| 5c | 112.25 | 0 | 6.74 | 6 | 1 | 5 | Low | No | 0 violation |
| Colchicine | 83.09 | 0 | 2.55 | 6 | 1 | 6 | High | No | 0 violation |
| CA-4 | 77.38 | 0 | 2.38 | 7 | 2 | 6 | High | Yes | 0 violation |
 | ||
| Fig. 15 Boiled egg model of the tested compounds 3b, 3c, 4c, 5c, colchicine and CA-4. | ||
3. Conclusion
To sum up, a group of novel quinoline derivatives 3a–e, 4a–e, 5a–e, and 6a–e were designed and synthesized as tubulin polymerization inhibitors targeting the colchicine binding site. Their cytotoxic activity against 60 cell lines by NCI were evaluated. Compounds 3b, 3c, 4c, 5c and 6c possessed the most remarkable antitumor activity against almost all cell lines, especially compound 4c (pyridin-2-one ring substituted with 3,4,5-trimethoxy groups) which was the most potent compound as an antiproliferative agent. Compound 4c significantly induced cell cycle arrest at G2 and M phases and encouraged apoptosis, as evidenced by a rise in the number of cells in both early and late phases of apoptosis. Compound 4c successfully inhibited tubulin polymerization with IC50 value 17 ± 0.3 μM. The β-tubulin mRNA levels were markedly reduced in MDA-MB-231 cells treated with compound 4c, resembling the effect observed with colchicine treatment. Molecular docking exhibited the interaction mode of compound 4c with tubulin including formation of hydrogen bonds and hydrophobic interactions, which demonstrated energy score −11.5 kcal mol−1 in comparison to colchicine with energy score −9.8 kcal mol−1, and it interacted with essential amino acids in the active sites. Compound 4c could has high GI absorption, drug-like properties and the ability to penetrate the blood–brain barrier. Taken together, compound 4c is a highly promising candidate for further preclinical studies against breast cancer.4. Experimental
4.1. Chemistry
3-(2-Chloro-8-ethylquinolin-3-yl)-1-(4-methoxyphenyl)prop-2-en-1-one (3a). Yellow powder, yield 85%, mp 138–140 °C. IR (KBr, cm−1): 3022 (CH aromatic), 2960 (CH aliphatic), 1656 (C
O), 1568 (CN). 1H NMR (400 MHz, DMSO-d6), δ ppm: 1.30 (t, 3H, J = 8.0 Hz, CH2C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3), 3.11–3.16 (q, 2H, J = 8.0 Hz, C2CH3), 3.89 (s, 3H, OCH3), 7.12 (d, 2H, J = 8.0 Hz, Ar-H), 7.64 (t, 1H, J = 8.0 Hz, Ar-H), 7.71 (d, 1H, J = 8.0 Hz, Ar-H), 7.91 (d, 1H, J = 8.0 Hz, Ar-H), 8.02 (d, 1H, J = 16 Hz, CH alkene β proton), 8.16 (d, 1H, J = 16 Hz, CH alkene α proton), 8.21 (d, 2H, J = 8.0 Hz, Ar-H), 9.21 (s, 1H, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 15.2, 24.0, 56.0, 114.5 (2C), 126.2, 126.9, 127.0, 127.4, 128.1, 130.4, 130.6, 131.5 (2C), 137.3, 138.0, 141.6, 145.9, 149.1, 163.9, 187.2. Anal. calcd for C21H18ClNO2 (351.83): C, 71.69; H, 5.16; N 3.98; found: C, 71.85; H, 5.33; N, 4.21.
3), 3.11–3.16 (q, 2H, J = 8.0 Hz, C2CH3), 3.89 (s, 3H, OCH3), 7.12 (d, 2H, J = 8.0 Hz, Ar-H), 7.64 (t, 1H, J = 8.0 Hz, Ar-H), 7.71 (d, 1H, J = 8.0 Hz, Ar-H), 7.91 (d, 1H, J = 8.0 Hz, Ar-H), 8.02 (d, 1H, J = 16 Hz, CH alkene β proton), 8.16 (d, 1H, J = 16 Hz, CH alkene α proton), 8.21 (d, 2H, J = 8.0 Hz, Ar-H), 9.21 (s, 1H, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 15.2, 24.0, 56.0, 114.5 (2C), 126.2, 126.9, 127.0, 127.4, 128.1, 130.4, 130.6, 131.5 (2C), 137.3, 138.0, 141.6, 145.9, 149.1, 163.9, 187.2. Anal. calcd for C21H18ClNO2 (351.83): C, 71.69; H, 5.16; N 3.98; found: C, 71.85; H, 5.33; N, 4.21.
3-(2-Chloro-8-ethylquinolin-3-yl)-1-(3,4-dimethoxyphenyl)prop-2-en-1-one (3b). Yellow powder, yield 84%, mp 135–137 °C. IR (KBr, cm−1): 3060 (CH aromatic), 2964 (CH aliphatic), 1653 (C
O), 1577 (CN). 1H NMR (400 MHz, DMSO-d6), δ ppm: 1.29 (t, 3H, J = 8.0 Hz, CH2C3), 3.09–3.15 (q, 2H, J = 8.0 Hz, C2CH3), 3.87 (s, 3H, OCH3), 3.89 (s, 3H, OCH3), 7.13 (d, 1H, J = 8.0 Hz, Ar-H), 7.62 (t, 2H, J = 8.0 Hz, Ar-H), 7.70 (d, 1H, J = 8.0 Hz, Ar-H), 7.89–7.95 (m, 2H, Ar-H), 8.00 (d, 1H, J = 16 Hz, CH alkene β proton), 8.13 (d, 1H, J = 16 Hz, CH alkene α proton), 9.15 (s, 1H, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 15.3, 24.0, 56.1, 56.3, 111.2, 111.3, 124.3, 126.5, 127.0, 127.3, 127.5, 128.3, 130.5, 130.8, 137.4, 138.2, 141.7, 146.0, 149.2, 149.3, 154.0, 187.3. Anal. calcd for C22H20ClNO3 (381.86): C, 69.20; H, 5.28; N 3.67; found: C, 69.43; H, 5.40; N, 3.85.
3-(2-Chloro-8-ethylquinolin-3-yl)-1-(3,4,5-trimethoxyphenyl)prop-2-en-1-one (3c). White powder, yield 86%, mp 140–142 °C. IR (KBr, cm−1): 3064 (CH aromatic), 2958 (CH aliphatic), 1639 (C
O), 1583 (CN). 1H NMR (400 MHz, DMSO-d6), δ ppm: 1.28 (t, 3H, J = 8.0 Hz, CH2C3), 3.06–3.15 (q, 2H, J = 8.0 Hz, C2CH3), 3.79 (s, 3H, OCH3), 3.92 (s, 6H, 2× OCH3), 7.46 (s, 2H, Ar-H), 7.64 (t, 1H, J = 8.0 Hz, Ar-H), 7.72 (d, 1H, J = 8.0 Hz, Ar-H), 7.93 (d, 1H, J = 8.0 Hz, Ar-H), 8.04 (d, 1H, J = 16 Hz, CH alkene β proton), 8.12 (d, 1H, J = 12 Hz, CH alkene α proton), 9.14 (s, 1H, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 15.3, 24.0, 56.7, 60.7 (2C), 106.8 (2C), 126.1, 126.2, 127.0, 127.4, 130.8, 132.8, 138.2, 138.3, 141.7, 142.7, 145.9, 149.1, 150.7, 153.3 (2C), 188.0 Anal. calcd for C23H22ClNO4 (411.88): C, 67.07; H, 5.38; N 3.40; found: C, 67.41; H, 5.45; N, 3.62.
3-(2-Chloro-8-ethylquinolin-3-yl)-1-(4-fluorophenyl)prop-2-en-1-one (3d). White powder, yield 83%, mp 215–217 °C. IR (KBr, cm−1): 3078 (CH aromatic), 2964 (CH aliphatic), 1643 (C
O), 1598 (CN). 1H NMR (400 MHz, DMSO-d6), δ ppm: 1.31 (t, 3H, J = 8.0 Hz, CH2C3), 3.11–3.17 (q, 2H, J = 8.0 Hz, C2CH3), 7.46 (t, 2H, J = 8.0 Hz, Ar-H), 7.60 (d, 1H, J = 7.2 Hz, Ar-H), 7.64 (d, 1H, J = 16 Hz, CH alkene β proton), 7.74 (t, 1H, J = 8.0 Hz, Ar-H), 8.06–8.09 (m, 3H. Ar-H), 8.20 (d, 1H, J = 16 Hz, CH alkene α proton), 9.29 (s, 1H, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 15.5, 23.6, 115.3 (2C), 126.1, 126.9, 127.0, 127.5, 128.1, 130.4, 130.6, 131.6 (2C), 137.4, 138.1, 141.6, 146.0, 149.2, 167.3, 187.5. Anal. calcd for C20H15ClFNO (339.79): C, 70.70; H, 4.45; N 4.12; found: C, 70.59; H, 4.62; N, 4.37.
1-(4-Bromophenyl)-3-(2-chloro-8-ethylquinolin-3-yl)prop-2-en-1-one (3e). White powder, yield 83%, mp 210–212 °C. IR (KBr, cm−1): 3101 (CH aromatic), 2958 (CH aliphatic), 1600 (C
O), 1583 (CN). 1H NMR (400 MHz, DMSO-d6), δ ppm: 1.31 (t, 3H, J = 8.0 Hz, CH2C3), 3.06–3.15 (q, 2H, J = 8.0 Hz, C2CH3), 7.06 (d, 1H, J = 8 Hz, Ar-H), 7.36 (t, 1H, J = 8.0 Hz, Ar-H), 7.61 (m, 1H, Ar-H), 7.73 (d, 2H, J = 8.4 Hz, Ar-H), 7.91 (d, 2H, J = 8.4 Hz, Ar-H), 8.11 (d, 1H, J = 15.2 Hz, CH alkene β proton), 8.17 (d, 1H, J = 16 Hz, CH alkene α proton), 8.50 (s, 1H, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 15.9, 23.6, 126.2, 126.8, 127.1, 127.5, 128.2, 128.5 (2C), 129.1, 130.3, 130.6, 131.7 (2C), 137.9, 139.9, 142.3, 146.0, 150.3, 186.8. Anal. calcd for C20H15ClBrNO (400.70): C, 59.95; H, 3.77; N 3.50; found: C, 60.23; H, 3.91; N, 3.64.
4-(2-Chloro-8-ethylquinolin-3-yl)-6-(4-methoxyphenyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (4a). Yellow powder, yield 79%, mp 270–272 °C. IR (KBr, cm−1): 3421 (NH), 3095 (CH aromatic), 2962 (CH aliphatic), 2218 (CN), 1647 (C
O), 1514 (CN). 1H NMR (400 MHz, DMSO-d6), δ ppm: 1.34 (t, 3H, J = 8.0 Hz, CH2C3), 3.14–3.20 (m, 2H, C2CH3 + s, 1H, NH, D2O exchangeable), 3.84 (s, 3H, OCH3), 7.00 (s, 1H, CH pyridine), 7.08 (d, 2H, J = 8.8 Hz, Ar-H), 7.69 (t, 1H, J = 8.0 Hz, Ar-H), 7.80 (d, 1H, J = 6.8 Hz, Ar-H), 7.92 (d, 2H, J = 8.8 Hz, Ar-H), 7.96 (d, 1H, J = 8.0 Hz, Ar-H), 8.66 (s, 1H, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 15.3, 23.9, 55.7, 106.4, 115.0 (2C), 116.4, 124.8, 126.1, 126.8, 126.9, 127.8, 127.9, 128.6, 129.8, 129.9 (2C), 131.0, 140.3, 141.9, 145.9, 152.9, 156.7, 162.4. MS m/z (%): 417.40 (M + 2, 5.89), 415.49 (M+, 13.79), 125.14 (100.00). Anal. calcd for C24H18ClN3O2 (415.88): C, 69.31; H, 4.36; N 10.10; found: C, 69.08; H, 4.50; N, 10.29.
4-(2-Chloro-8-ethylquinolin-3-yl)-6-(3,4-dimethoxyphenyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (4b). Yellow powder, yield 71%, mp 280–282 °C. IR (KBr, cm−1): 3431 (NH), 3040 (CH aromatic), 2962 (CH aliphatic), 2239 (CN), 1641 (C
O), 1517 (CN). 1H NMR (400 MHz, DMSO-d6), δ ppm: 1.34 (t, 3H, J = 8.0 Hz, CH2C3), 2.52 (s, 1H, NH, D2O exchangeable), 3.17–3.26 (m, 2H, C2CH3), 3.80 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 6.85 (s, 1H, CH pyridine), 7.00 (d, 1H, J = 8.4 Hz, Ar-H), 7.60–7.67 (m, 3H, Ar-H), 7.75 (d, 1H, J = 6.8 Hz, Ar-H), 7.95 (d, 1H, J = 8.0 Hz, Ar-H), 8.52 (s, 1H, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 15.5, 22.6, 56.0, 56.1, 105.8, 110.5, 110.8, 111.0, 111.8, 181.2, 121.0, 123.7, 126.7, 127.1, 128.1, 130.4, 130.9, 140.0, 141.9, 145.8, 146.8, 149.3, 151.3, 156.1, 173.9. MS m/z (%): 447.88 (M + 2, 3.22), 445.74 (M+, 9.87), 313.27 (100.00). Anal. calcd for C25H20ClN3O3 (445.90): C, 67.34; H, 4.52; N 9.42; found: C, 67.53; H, 4.61; N, 9.68.
4-(2-Chloro-8-ethylquinolin-3-yl)-2-oxo-6-(3,4,5-trimethoxyphenyl)-1,2-dihydropyridine-3-carbonitrile (4c). Yellow powder, yield 72%, mp 297–299 °C. IR (KBr, cm−1): 3446 (NH), 3140 (CH aromatic), 2953 (CH aliphatic), 2220 (CN), 1645 (C
O), 1581 (CN). 1H NMR (400 MHz, DMSO-d6), δ ppm: 1.33 (t, 3H, J = 8.0 Hz, CH2C3), 3.14–3.23 (m, 3H (2H, C2CH3 + 1H, NH, D2O exchangeable)), 3.73 (s, 3H, OCH3), 3.86 (s, 6H, 2OCH3), 7.16 (s, 1H, CH pyridine), 7.26 (s, 2H, Ar-H), 7.69 (t 1H, J = 8.0 Hz, Ar-H), 7.80 (d, 1H, J = 8 Hz, Ar-H), 7.98 (d, 1H, J = 8 Hz, Ar-H), 8.66 (s, 1H, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 16.2, 23.9, 56.7, 60.8 (2C), 105.6 (2C), 107.1, 116.3, 116.7, 126.9, 127.6, 128.0, 128.6, 129.8, 130.7, 140.3, 140.5, 141.8, 145.9, 146.1, 152.6, 153.6 (2C), 156.7, 162.6. MS m/z (%): 477.68 (M + 2, 11.23), 475.05 (M+, 34.00), 42.89 (100.00). Anal. calcd for C26H22ClN3O4 (475.93): C, 65.62; H, 4.66; N 8.83; found: C, 65.49; H, 4.85; N, 9.07.
4-(2-Chloro-8-ethylquinolin-3-yl)-6-(4-fluorophenyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (4d). White powder, yield 77%, mp 260–262 °C. IR (KBr, cm−1): 3431 (NH), 3047 (CH aromatic), 2966 (CH aliphatic), 2222 (CN), 1656 (C
O), 1535 (CN). 1H NMR (400 MHz, DMSO-d6), δ ppm: 1.34 (t, 3H, J = 8.0 Hz, CH2C3), 3.11–3.21 (m, 2H, C2CH3 + s, 1H, NH, D2O exchangeable), 7.11 (s, 1H, CH pyridine),7.39 (t, 2H, J = 8.0 Hz, Ar-H), 7.63–7.71 (m, 1H, Ar-H), 7.80 (d, 1H, J = 6.8 Hz, Ar-H), 7.96–7.803 (m, 3H, Ar-H), 8.68 (s, 1H, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 16.6, 23.3, 106.2, 114.6 (2C), 117.6, 119.5, 120.6, 122.5, 124.2, 125.9, 127.2 (2C), 127.9, 129.2, 129.9, 131.3, 144.6, 147.3, 153.6, 159.7, 161.7, 162.4. MS m/z (%): 405.62 (M + 2, 4.18), 403.80 (M+, 11.20), 376.34 (100.00). Anal. calcd for C23H15ClFN3O (403.84): C, 68.41; H, 3.74; N, 10.41; found: C, 68.70; H, 3.86; N 10.64.
6-(4-Bromophenyl)-4-(2-chloro-8-ethylquinolin-3-yl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (4e). White powder, yield 78%, mp 290–262 °C. IR (KBr, cm−1): 3419 (NH), 3034 (CH aromatic), 2964 (CH aliphatic), 2222 (CN), 1647 (C
O), 1589 (CN). 1H NMR (400 MHz, DMSO-d6), δ ppm: 1.33 (t, 3H, J = 8.0 Hz, CH2C3), 1.87 (s, 1H, NH, D2O exchangeable), 3.06–3.15 (m, 2H, C2CH3), 7.00 (s, 1H, CH pyridine),7.69–7.70 (m, 3H, Ar-H), 7.79 (t, 1H, J = 8.0 Hz, Ar-H), 7.91–7.96 (m, 3H, Ar-H), 8.61 (s, 1H, Ar-H). MS m/z (%): 466.63 (M + 2, 29.98), 465.61 (M + 1, 54.96), 464.52 (M+, 48.74), 353.35 (100.00). Anal. calcd for C23H15BrClN3O (464.75): C, 59.44; H, 3.25; N 9.04; found: C, 59.32; H, 3.41; N, 9.29.
4-(2-Chloro-8-ethylquinolin-3-yl)-6-(4-methoxyphenyl)-2-thioxo-1,2-dihydropyridine-3-carbonitrile (5a). Yellow powder, yield 71%, mp 290–292 °C. IR (KBr, cm−1): 3433 (NH), 3070 (CH aromatic), 2962 (CH aliphatic), 2218 (CN), 1577 (C
N), 1147 (CS). 1H NMR (400 MHz, DMSO-d6), δ ppm: 1.33 (t, 3H, J = 8.0 Hz, CH2C3), 3.15–3.21 (q, 2H, J = 8.0 Hz, C2CH3 + s, 1H, NH, D2O exchangeable), 3.83 (s, 3H, OCH3), 6.99 (s, 1H, CH pyridine), 7.08 (d, 2H, J = 9.3 Hz, Ar-H), 7.68 (t, 1H, J = 8.0 Hz, Ar-H), 7.79 (d, 1H, J = 7.2 Hz, Ar-H), 7.92 (d, 2H, J = 8.8 Hz, Ar-H), 7.96 (d, 1H, J = 7.6 Hz, Ar-H), 8.65 (s, 1H, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 15.3, 24.3, 56.3, 106.7, 114.9 (2C), 117.0, 120.7, 121.4, 124.8, 125.4, 126.7, 126.9, 128.6, 130.0 (2C), 131.0, 140.3, 142.0, 145.8, 152.8, 156.5, 160.1, 162.4. MS m/z (%): 433.93 (M + 2, 16.24), 432.21 (M + 1, 40.09), 431.34 (M+, 50.68), 423.98 (100.00). Anal. calcd for C24H18ClN3OS (431.94): C, 66.74; H, 4.20; N 9.73; found: C, 66.83; H, 4.34; N, 9.91.
4-(2-Chloro-8-ethylquinolin-3-yl)-6-(3,4-dimethoxyphenyl)-2-thioxo-1,2-dihydropyridine-3-carbonitrile (5b). Yellow powder, yield 71%, mp 280–282 °C. IR (KBr, cm−1): 3446 (NH), 3151 (CH aromatic), 2964 (CH aliphatic), 2218 (CN), 1577 (C
N), 1145 (CS). 1H NMR (400 MHz, DMSO-d6), δ ppm: 1.33 (t, 3H, J = 8.0 Hz, CH2C3), 1.88 (s, 1H, NH, D2O exchangeable), 3.20 (s, 2H, C2CH3), 3.83 (s, 3H, OCH3), 3.84 (s, 3H, OCH3), 7.02 (s, 1H, CH pyridine), 7.08 (d, 1H, J = 8.4 Hz, Ar-H), 7.33–7.61 (m, 2H, Ar-H), 7.68 (t, 1H, J = 8.0 Hz, Ar-H), 7.79 (d, 1H, J = 6.8 Hz, Ar-H), 7.96 (d, 1H, J = 7.6 Hz, Ar-H), 8.63 (s, 1H, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 15.3, 24.3, 56.1, 56.2, 100.0, 106.5, 110.9, 112.3, 116.4, 121.6, 124.6, 124.7, 126.8, 126.9, 128.3, 129.9, 130.8, 140.3, 141.8, 145.9, 146.1, 149.3, 152.1, 156.7, 162.5 MS m/z (%): 463.93 (M + 2, 2.78), 462.63 (M + 1, 9.64), 461.68 (M+, 6.26), 43.21 (100.00). Anal. calcd for C25H20ClN3O2S (461.96): C, 65.00; H, 4.36; N 9.10; found: C, 64.88; H, 4.51; N, 9.32.
4-(2-Chloro-8-ethylquinolin-3-yl)-2-thioxo-6-(3,4,5-trimethoxyphenyl)-1,2-dihydropyridine-3-carbonitrile (5c). Yellow powder, yield 69%, mp 279–281 °C. IR (KBr, cm−1): 3446 (NH), 3051 (CH aromatic), 2966 (CH aliphatic), 2220 (CN), 1585 (C
N), 1128 (CS). 1H NMR (400 MHz, DMSO-d6), δ ppm: 1.31 (t, 3H, J = 8.0 Hz, CH2C3), 3.18–3.22 (m, 2H, C2CH3 + s, 1H, NH, D2O exchangeable), 3.73 (s, 3H, OCH3), 3.86 (s, 6H, 2× OCH3), 7.17 (s, 1H, CH pyridine), 7.25 (s, 2H, Ar-H), 7.67 (t, 1H, J = 8.0 Hz, Ar-H), 7.80 (d, 1H, J = 8 Hz, Ar-H), 7.98 (d, 1H, J = 8 Hz, Ar-H), 8.69 (s, 1H, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 14.9, 23.7, 56.8 (2C), 60.5, 101.8, 106.4, 109.2, 116.0 (2C), 126.1, 126.9, 127.4, 128.1, 129.6, 130.8, 132.7, 138.0, 138.4, 146.1, 149.3, 153.2 (2C), 159.5, 173.7, 175.8. MS m/z (%): 493.53 (M + 2, 26.83), 491.95 (M+, 72.17), 441.90 (100.00). Anal. calcd for C26H22ClN3O3S (491.99): C, 63.47; H, 4.51; N 8.54; found: C, 63.60; H, 4.63; N, 8.70.
4-(2-Chloro-8-ethylquinolin-3-yl)-6-(4-fluorophenyl)-2-thioxo-1,2-dihydropyridine-3-carbonitrile (5d). Yellow powder, yield 76%, mp 235–237 °C. IR (KBr, cm−1): 3398 (NH), 3049 (CH aromatic), 2966 (CH aliphatic), 2222 (CN), 1508 (C
N), 1165 (CS). 1H NMR (400 MHz, DMSO-d6), δ ppm: 1.34 (t, 3H, J = 8.0 Hz, CH2C3), 3.11–3.22 (m, 2H, C2CH3 + s, 1H, NH, D2O exchangeable), 6.78 (s, 1H, CH pyridine), 7.23 (t, 2H, Ar-H), 7.65 (d, 1H, J = 8.0 Hz, Ar-H), 7.73–7.77 (m, 1H, Ar-H) 7.94 (d, 1H, J = 8 Hz, Ar-H), 8.08 (d, 2H, J = 7.6 Hz, Ar-H), 8.49 (s, 1H, Ar-H). 13C NMR (100 MHz, DMSO-d6), δ ppm: 15.0, 22.0, 105.5, 115.9, 116.1 (2C), 118.3, 126.1, 126.7, 128.1, 128.3 (2C), 128.5, 129.7, 130.0, 133.9, 141.5, 145.2, 145.7, 149.7, 162.2, 166.4, 173.6. MS m/z (%): 421.12 (M + 2, 9.84), 419.78 (M+, 27.88), 64.08 (100.00). Anal. calcd for C23H15ClFN3S (419.90): C, 65.79; H, 3.60; N 10.01; found: C, 66.05; H, 3.74; N, 10.29.
6-(4-Bromophenyl)-4-(2-chloro-8-ethylquinolin-3-yl)-2-thioxo-1,2-dihydropyridine-3-carbonitrile (5e). Buff powder, yield 79%, mp charring 250 °C. IR (KBr, cm−1): 3446 (NH), 3126 (CH aromatic), 2910 (CH aliphatic), 2218 (CN), 1564 (C
N), 1195 (CS). 1H NMR (400 MHz, DMSO-d6), δ ppm: 1.33 (t, 3H, J = 8.0 Hz, CH2C3), 3.17–3.26 (m, 2H, C2CH3 + s, 1H, NH, D2O exchangeable), 6.99 (s, 1H, CH pyridine),7.05 (d, 2H, J = 9.2 Hz, Ar-H), 7.66 (t, 1H, J = 8.0 Hz, Ar-H), 7.77 (d, 1H, J = 7.2 Hz, Ar-H), 7.91–7.97 (m, 3H, Ar-H), 8.67 (s, 1H, Ar-H). MS m/z (%): 482.73 (M + 2, 19.45), 481.83 (M + 1, 17.34), 480.23 (M+, 41.55), 474.31 (100.00). Anal. calcd for C23H15BrClN3S (480.81): C, 57.46; H, 3.14; N 8.74; found: C, 57.70; H, 3.23; N, 8.97.
4-(2-Chloro-8-ethylquinolin-3-yl)-6-(4-methoxyphenyl)-3,4-dihydropyrimidine-2(1H)-thione (6a). White powder, yield 71%, mp above 300 °C. IR (KBr, cm−1): 3165 (NH), 3016 (CH aromatic), 2964 (CH aliphatic), 1130 (C
S). 1H NMR (400 MHz, DMSO-d6), δ ppm: 1.28 (t, 3H, J = 8.0 Hz, CH2C3), 3.12–3.19 (q, 2H, J = 8.0 Hz, C2CH3), 3.76 (s, 3H, OCH3), 5.39 (d, 1H, CH pyrimidine, J = 4.4 Hz), 5.57 (d, 1H, CH pyrimidine, J = 4.4 Hz), 6.91 (d, 2H, J = 8.8 Hz, Ar-H), 7.45 (d, 2H, J = 8.8 Hz, Ar-H), 7.60 (t, 1H, J = 8.0 Hz, Ar-H), 7.69 (d, 1H, J = 7.6 Hz, Ar-H), 7.89 (d, 1H, J = 7.6 Hz, Ar-H), 8.21 (s, 1H, Ar-H), 9.16 (s, 1H, NH, D2O exchangeable), 10.04 (s, 1H, NH, D2O exchangeable). 13C NMR (100 MHz, DMSO-d6), δ ppm: 16.3, 24.1, 55.3, 58.3, 99.7, 120.7 (2C), 124.1, 125.8, 126.5, 126.8, 128.8, 130.2, 131.9 (2C), 136.3, 139.3, 141.7, 145.8, 148.5, 159.4, 175.6. Anal. calcd for C22H20ClN3OS: C, 64.46; H, 4.92; N 10.25; found: C, 64.32; H, 5.11; N, 10.49.
4-(2-Chloro-8-ethylquinolin-3-yl)-6-(3,4-dimethoxyphenyl)-3,4-dihydropyrimidine-2(1H)-thione (6b). Orange powder, yield 74%, mp 237–239 °C. IR (KBr, cm−1): IR (KBr, cm−1): 3196 (NH), 3080 (CH aromatic), 2960 (CH aliphatic), 1172 (C
S). 1H NMR (400 MHz, DMSO-d6), δ ppm: 1.29 (t, 3H, J = 8.0 Hz, CH2C3), 3.12–3.16 (m, 2H, C2CH3), 3.75 (s, 3H, OCH3), 3.78 (s, 3H, OCH3), 5.42 (d, 1H, CH pyrimidine, J = 4 Hz), 5.58 (d, 1H, CH pyrimidine, J = 4 Hz), 6.92 (d, 1H, J = 8 Hz, Ar-H), 7.05–7.08 (m, 2H, Ar-H), 7.61 (d, 1H, J = 7.6 Hz, Ar-H), 7.69 (d, 1H, J = 7.2 Hz, Ar-H), 7.90 (d, 1H, J = 8 Hz, Ar-H), 8.22 (s, 1H, Ar-H), 9.10 (s, 1H, NH, D2O exchangeable), 10.16 (s, 1H, NH, D2O exchangeable). 13C NMR (100 MHz, DMSO-d6), δ ppm: 15.0, 24.1, 56.1, 56.2, 56.2, 97.5, 111.3, 124.0, 124.5, 126.3, 126.7, 127.1, 127.5, 128.2, 130.4, 130.8, 137.5, 146.0, 149.0, 149.4, 151.5, 154.0, 174.3. Anal. calcd for C23H22ClN3O2S: C, 62.79; H, 5.04; N 9.55; found: C, 62.51; H, 5.25; N, 9.76.
4-(2-Chloro-8-ethylquinolin-3-yl)-6-(3,4,5-trimethoxyphenyl)-3,4-dihydropyrimidine-2(1H)-thione (6c). Yellow powder, yield 72%, mp 235–237 °C. IR (KBr, cm−1): 3165 (NH), 3099 (CH aromatic), 2964 (CH aliphatic), 1130 (C
S). 1H NMR (400 MHz, DMSO-d6), δ ppm: 1.29 (t, 3H, J = 8.0 Hz, CH2C3), 3.12–3.18 (q, 2H, J = 8.0 Hz, C2CH3), 3.65 (s, 3H, OCH3), 3.80 (s, 6H, 2OCH3), 5.51 (d, 1H, CH pyrimidine, J = 4.4 Hz), 5.61 (d, 1H, CH pyrimidine, J = 4.4 Hz), 6.81 (s, 2H, Ar-H), 7.60 (t, 1H, J = 8.0 Hz, Ar-H), 7.69 (d, 1H, J = 8 Hz, Ar-H), 7.92 (d, 1H, J = 8 Hz, Ar-H), 8.31 (s, 1H, Ar-H), 9.10 (s, 1H, NH, D2O exchangeable), 10.16 (s, 1H, NH, D2O exchangeable). 13C NMR (100 MHz, DMSO-d6), δ ppm: 15.2, 24.1, 53.3, 56.0 (2C), 60.7, 99.4, 103.4 (2C), 126.3, 127.6, 128.2, 128.9, 129.8, 134.6, 135.7, 137.9, 138.6, 141.6, 145.2, 147.3, 153.0 (2C), 177.0. Anal. calcd for C24H24ClN3O3S: C, 61.33; H, 5.15; N 8.94; found: C, 61.08; H, 5.32; N, 9.09.
4-(2-Chloro-8-ethylquinolin-3-yl)-6-(4-fluorophenyl)-3,4-dihydropyrimidine-2(1H)-thione (6d). Orange powder, yield 70%, mp 220–222 °C. IR (KBr, cm−1): 3167 (NH), 3016 (CH aromatic), 2960 (CH aliphatic), 1197 (C
S). 1H NMR (400 MHz, DMSO-d6), δ ppm: 1.29 (t, 3H, J = 8.0 Hz CH2C3), 3.12–3.17 (q, 2H, J = 8.0 Hz, C2CH3), 5.44 (d, 1H, CH pyrimidine, J = 4.4 Hz), 5.59 (d, 1H, CH pyrimidine, J = 4.4 Hz), 7.19 (t, 2H, J = 8.0 Hz, Ar-H), 7.53–7.62 (m, 3H, Ar-H), 7.69 (d, 1H, J = 6.8 Hz, Ar-H), 7.90 (d, 1H, J = 8 Hz, Ar-H), 8.22 (s, 1H, Ar-H), 9.19 (s, 1H, NH, D2O exchangeable), 10.17 (s, 1H, NH, D2O exchangeable). 13C NMR (100 MHz, DMSO-d6), δ ppm: 16.0, 24.1, 59.2, 97.3, 115.8 (2C), 125.0, 126.6, 127.0, 128.2, 128.7, 129.9, 130.4 (2C), 131.1, 136.5, 145.5, 146.8, 149.7, 162.2, 174.0. Anal. calcd for C21H17ClFN3S: C, 63.39; H, 4.31; N 10.56; found: C, 63.54; H, 4.45; N, 10.78.
6-(4-Bromophenyl)-4-(2-chloro-8-ethylquinolin-3-yl)-3,4-dihydropyrimidine-2(1H)-thione (6e). Orange powder, yield 74%, mp 248–240 °C. IR (KBr, cm−1): 3162 (NH), 3018 (CH aromatic), 2964 (CH aliphatic), 1197 (C
S). 1H NMR (400 MHz, DMSO-d6), δ ppm: 1.29 (t, 3H, J = 8.0 Hz, CH2C3), 3.12–3.19 (m, 2H, C2CH3), 5.50 (d, 1H, CH pyrimidine, J = 4.4 Hz), 5.60 (d, 1H, CH pyrimidine, J = 4.4 Hz), 7.46 (d, 2H, J = 8.4 Hz, Ar-H), 7.54–7.62 (m, 3H, Ar-H), 7.69 (d, 1H, J = 7.2 Hz, Ar-H), 7.90 (d, 1H, J = 7.6 Hz, Ar-H), 8.25 (s, 1H, Ar-H), 9.18 (s, 1H, NH, D2O exchangeable), 10.20 (s, 1H, NH, D2O exchangeable). 13C NMR (100 MHz, DMSO-d6), δ ppm: 15.0, 24.5, 57.9, 96.5, 123.1, 123.8, 124.5, 129.1 (2C), 129.8, 130.5, 132.6 (2C), 133.3, 137.2, 139.7, 140.8, 148.9, 152.8, 153.5, 174.4. Anal. calcd for C21H17BrClN3S: C, 54.98; H, 3.73; N 9.16; found: C, 54.87; H, 3.90; N, 9.35.
4.2. Biology
| % Cytotoxicity = [1 − (AVx/AVNC)] × 100 |
4.2.1.1. Five doses testing of compound 4c. The methodology of the NCI anticancer screening has been described in details at (http://www.dtp.nci.nih.gov). The anticancer assay was performed in full NCI 60 cell lines derived from nine tumor subpanels, including leukemia, lung, colon, CNS, melanoma, ovarian, renal, prostate, and breast cancer cell lines.
The cell lines were first cultured for 48 h, after being exposed to a single dosage of the compound. Using the protein-binding dye sulforhodamine B dye, cell proliferation and viability were evaluated (SRB assay). The results were presented for each chemical as GI%. Compound that met NCI criteria for substantial activity were further evaluated using a five-dose experiment. The NCI60 cell lines were treated with the chemicals in this assay at five different concentration levels (0.01, 0.1, 1, 10, and 100 μM). Following cell line incubation, the results were depicted using three parameters: GI50, which signifies the molar concentration resulting in a 50% reduction in all cell count after incubation; TGI, indicating the molar concentration leading to total growth inhibition; and LC50, representing the molar concentration causing a 50% loss of the initial cells after incubation.
4.2.5.1. RNA isolation. The cells were seeded in a six-well plate at a concentration of 3 × 105 cells per well. After 24 h. incubation, the IC50 dose of compound 4c and the positive control colchicine was applied to each well for 48 h. The cells were washed, collected and centrifuged at 2500 rpm for 15 min at 4 °C. The total RNA isolation kit (RNeasy extraction kit – QIAGEN) was used to isolate RNA according to the manufacturer's instructions.
4.2.5.2. Gene expression analysis by quantitative real-time PCR. Gene expression of β-tubulin was determined using iScript One-Step RT-PCR kit with SYBR Green (BIORAD) according to the manufacturer protocol. Primers sequences used for qPCR are illustrated in (Table 5). The genes expression was normalized to β-actin as the housekeeping gene. The protocol used was cDNA synthesis for 10 min at 50 °C, iScript Reverse transcriptase inactivation for 5 min at 95 °C and PCR cycling and detection (30 to 45 cycles): 10 s at 95 °C and 30 s at 55 °C to 60 °C.
| Gene | Primer sequence |
|---|---|
| β-Tubulin | F: 5′-TCAGCGTCTACTACAACGAGGC-3′ |
| R: 5′-GCCTGAAGAGATGTCCAAAGGC-3′ | |
| β-Actin | F: 5′-CATTGCTGACAGGATGCAGAAGG-3′ |
| R: 5′-TGCTGGAAGGTGGACAGTGAGG-3′ |
4.3. In silico studies
 | ||
| Fig. 16 Alignment between the conformation of the co-crystallized ligand (colchicine) colored in grey and the best-docked pose (colored in blue) into α–β tubulin enzyme with RMSD value equal to 0.17 Å. | ||
Data availability
The authors of this article declare that and data sharing should be upon request.Conflicts of interest
There are no conflicts to declare.References
- R. B. Malabadi, M. Sadiya, K. P. Kolkar, S. S. Mammadova, R. K. Chalannavar and H. Baijnath, Int. J. Sci. Res. Arch., 2024, 11, 2502–2539, DOI:10.30574/ijsra.2024.11.1.0315.
- S. Łukasiewicz, M. Czeczelewski, A. Forma, J. Baj, R. Sitarz and A. Stanisławek, Cancers, 2021, 13, 1–30, DOI:10.3390/cancers13174287.
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71(3), 209–249, DOI:10.3322/caac.21660.
- E. Mukhtar, V. M. Adhami and H. Mukhtar, Mol. Cancer Ther., 2014, 13, 275–284, DOI:10.1158/1535-7163.MCT-13-0791.
- Y. Lu, J. Chen, M. Xiao, W. Li and D. D. Miller, Pharm. Res., 2012, 29, 2943–2971, DOI:10.1007/s11095-012-0828-z.An.
- M. Knossow, V. Campanacci, L. A. Khodja and B. Gigant, iScience, 2020, 23, 1–14, DOI:10.1016/j.isci.2020.101511.
- S. Khwaja, K. Kumar, R. Das and A. S. Negi, Bioorg. Chem., 2021, 116, 105320, DOI:10.1016/j.bioorg.2021.105320.
- S. Mirzaei, F. Hadizadeh, F. Eisvand, F. Mosaffa and R. Ghodsi, J. Mol. Struct., 2020, 1202, 127310, DOI:10.1016/j.molstruc.2019.127310.
- E. C. McLoughlin and N. M. O'Boyle, Pharmaceuticals, 2020, 13, 1–43, DOI:10.3390/ph13040072.
- E. A. Foumani, S. Irani, Y. Shokoohinia and A. Mostafaie, Cell J., 2022, 24, 647–656, DOI:10.22074/cellj.2022.8290.
- T. L. Nguyen, C. McGrath, A. R. Hermone, J. C. Burnett, D. W. Zaharevitz, B. W. Day, P. Wipf, E. Hamel and R. Gussio, J. Med. Chem., 2005, 48(19), 6107–6116, DOI:10.1021/jm050502t.
- M.-m. Niu, J.-y. Qin, C.-p. Tian, X.-f. Yan, F.-g. Dong, Z.-q. Cheng, G. Fida, M. Yang, H. Chen and Y.-q. Gu, Acta Pharmacol. Sin., 2014, 35(7), 967–979, DOI:10.1038/aps.2014.34.
- L.-H. Zhang, L. Wu, H. K. Raymon, R. S. Chen, L. Corral, M. A. Shirley, R. K. Narla, J. Gamez, G. W. Muller and D. I. Stirling, Cancer Res., 2006, 66(2), 951–959, DOI:10.1158/0008-5472.CAN-05-2083.
- J. Yang, W. Yan, Y. Yu, Y. Wang, T. Yang, L. Xue, X. Yuan, C. Long, Z. Liu and X. Chen, J. Biol. Chem., 2018, 293(24), 9461–9472, DOI:10.1074/jbc.RA117.001658.
- L. M. Leoni, E. Hamel, D. Genini, H. Shih, C. J. Carrera, H. B. Cottam and D. A. Carson, J. Natl. Cancer Inst., 2000, 92(3), 217–224, DOI:10.1093/jnci/92.3.217.
- Y.-K. Chiang, C.-C. Kuo, Y.-S. Wu, C.-T. Chen, M. S. Coumar, J.-S. Wu, H.-P. Hsieh, C.-Y. Chang, H.-Y. Jseng and M.-H. Wu, J. Med. Chem., 2009, 52(14), 4221–4233, DOI:10.1021/jm801649y.
- X. Lv, C. He, C. Huang, G. Hua, Z. Wang, S. W. Remmenga, K. J. Rodabough, A. R. Karpf, J. Dong, J. S. Davis and C. Wang, Mol. Cancer Ther., 2017, 16, 1080–1091, DOI:10.1158/1535-7163.MCT-16-0626.
- P. Dhyani, C. Quispe, E. Sharma, A. Bahukhandi, P. Sati, D. C. Attri, A. Szopa, J. Sharifi-Rad, A. O. Docea, I. Mardare, D. Calina and W. C. Cho, Cancer Cell Int., 2022, 22, 1–20, DOI:10.1186/s12935-022-02624-9.
- Y. Ren, Y. Ruan, B. Cheng, L. Li, J. Liu, Y. Fang and J. Chen, Bioorg. Med. Chem., 2021, 46, 116376, DOI:10.1016/j.bmc.2021.116376.
- X.-S. Huo, X.-E. Jian, J. Ou-Yang, L. Chen, F. Yang, D.-X. Lv, W.-W. You, J.-J. Rao and P.-L. Zhao, Eur. J. Med. Chem., 2021, 220, 113449, DOI:10.1016/j.ejmech.2021.113449.
- R. M. Noha, M. K. Abdelhameid, M. M. Ismail, M. R. Mohammed and E. Salwa, Eur. J. Med. Chem., 2021, 209, 112870, DOI:10.1016/j.ejmech.2020.112870.
- N. Maciejewska, M. Olszewski, J. Jurasz, M. Serocki, M. Dzierzynska, K. Cekala, E. Wieczerzak and M. Baginski, Sci. Rep., 2022, 12(1), 3703, DOI:10.1038/s41598-022-07691-6.
- S. Elmeligie, A. T. Taher, N. A. Khalil and A. H. El-Said, Arch. Pharmacal Res., 2017, 40, 13–24, DOI:10.1007/s12272-016-0849-y.
- M. Perużyńska, A. Borzyszkowska-Ledwig, J. G. Sośnicki, Ł. Struk, T. J. Idzik, G. Maciejewska, Ł. Skalski, K. Piotrowska, P. Łukasik and M. Droździk, Int. J. Mol. Sci., 2021, 22(5), 2462, DOI:10.3390/ijms22052462.
- X. Zhai, X. Wang, J. Wang, J. Liu, D. Zuo, N. Jiang, T. Zeng, X. Yang, T. Jing and P. Gong, Sci. Rep., 2017, 7(1), 43398, DOI:10.1038/srep43398.
- M. K. Abd elhameid, N. Ryad, A.-S. MY, M. R. Mohammed, M. M. Ismail and S. El Meligie, Chem. Pharm. Bull., 2018, 66(10), 939–952, DOI:10.1248/cpb.c18-00269.
- T. S. Ibrahim, M. M. Hawwas, A. M. Malebari, E. S. Taher, A. M. Omar, N. M. O'Boyle, E. McLoughlin, Z. K. Abdel-Samii and Y. A. Elshaier, Pharmaceuticals, 2020, 13(11), 393, DOI:10.3390/ph13110393.
- M. M. Amin, G. E.-D. A. Abuo-Rahma, M. S. A. Shaykoon, A. A. Marzouk, M. A. Abourehab, R. E. Saraya, M. Badr, A. M. Sayed and E. A. Beshr, Bioorg. Chem., 2023, 134, 106444, DOI:10.1016/j.bioorg.2023.106444.
- T. S. Ibrahim, M. M. Hawwas, A. M. Malebari, E. S. Taher, A. M. Omar, T. Neamatallah, Z. K. Abdel-Samii, M. K. Safo and Y. A. Elshaier, J. Enzyme Inhib. Med. Chem., 2021, 36(1), 802–818, DOI:10.1080/14756366.2021.1899168.
- W. Li, F. Xu, W. Shuai, H. Sun, H. Yao, C. Ma, S. Xu, H. Yao, Z. Zhu and D.-H. Yang, J. Med. Chem., 2018, 62(2), 993–1013, DOI:10.1021/acs.jmedchem.8b01755.
- W. Li, W. Shuai, H. Sun, F. Xu, Y. Bi, J. Xu, C. Ma, H. Yao, Z. Zhu and S. Xu, Eur. J. Med. Chem., 2019, 163, 428–442, DOI:10.1016/j.ejmech.2018.11.070.
- M. Hagras, M. A. El Deeb, H. S. Elzahabi, E. B. Elkaeed, A. B. Mehany and I. H. Eissa, J. Enzyme Inhib. Med. Chem., 2021, 36(1), 640–658, DOI:10.1080/14756366.2021.1883598.
- V. Ramesh, B. A. Rao, P. Sharma, B. Swarna, D. Thummuri, K. Srinivas, V. Naidu and V. J. Rao, Eur. J. Med. Chem., 2014, 83, 569–580, DOI:10.1016/j.ejmech.2014.06.013.
- D.-W. Wang, H.-Y. Lin, R.-J. Cao, T. Chen, F.-X. Wu, G.-F. Hao, Q. Chen, W.-C. Yang and G.-F. Yang, J. Agric. Food Chem., 2015, 63(23), 5587–5596, DOI:10.1021/acs.jafc.5b01530.
- A. Srivastava and R. Singh, Indian J. Chem., 2005, 44B(9), 1868–1875 CAS.
- T. S. Ibrahim, M. M. Hawwas, E. S. Taher, N. A. Alhakamy, M. A. Alfaleh, M. Elagawany, B. Elgendy, G. M. Zayed, M. F. Mohamed and Z. K. Abdel-Samii, Bioorg. Chem., 2020, 105, 104352, DOI:10.1016/j.bioorg.2020.104352.
- H. K. Matthews, C. Bertoli and R. A. de Bruin, Nat. Rev. Mol. Cell Biol., 2022, 23(1), 74–88, DOI:10.1038/s41580-021-00404-3.
- P. Lara-Gonzalez, F. G. Westhorpe and S. S. Taylor, Curr. Biol., 2012, 22(22), R966–R980, DOI:10.1016/j.cub.2012.10.006.
- M. N. Peerzada, M. S. Dar and S. Verma, Expert Opin. Ther. Pat., 2023, 33(11), 797–820, DOI:10.1080/13543776.2023.2291390.
- S. Łukasiewicz, M. Czeczelewski, A. Forma, J. Baj, R. Sitarz and A. Stanisławek, Cancers, 2021, 13(17), 4287, DOI:10.3390/cancers13174287.
- S. Masood, Women’s Health, 2016, 12(1), 103–119, DOI:10.2217/whe.15.99.
- K. J. Chavez, S. V. Garimella and S. Lipkowitz, Breast Dis., 2010, 32(1–2), 35, DOI:10.3233/BD-2010-0307.
- B. L. Witt and T. O. Tollefsbol, Life, 2023, 13(12), 2311, DOI:10.3390/life13122311.
- K. N. Bhalla, Oncogene, 2003, 22(56), 9075–9086, DOI:10.1038/sj.onc.1207233.
- S. Boichuk, A. Galembikova, K. Syuzov, P. Dunaev, F. Bikinieva, A. Aukhadieva, S. Zykova, N. Igidov, K. Gankova and M. Novikova, Molecules, 2021, 26(19), 5780, DOI:10.3390/molecules26195780.
- H. Aryapour, G. H. Riazi, S. Ahmadian, A. Foroumadi, M. Mahdavi and S. Emami, Pharm. Biol., 2012, 50(12), 1551–1560, DOI:10.3109/13880209.2012.695799.
- K. Liu, M. Mo, G. Yu, J. Yu, S.-m. Song, S. Cheng, H.-m. Li, X.-l. Meng, X.-p. Zeng and G.-c. Xu, Bioorg. Chem., 2023, 139, 106727, DOI:10.1016/j.bioorg.2023.106727.
- H. Zhu, W. Zhu, Y. Liu, T. Gao, J. Zhu, Y. Tan, H. Hu, W. Liang, L. Zhao and J. Chen, Eur. J. Med. Chem., 2023, 257, 115529, DOI:10.1016/j.ejmech.2023.115529.
- Y. Wang, H. Zhang, B. Gigant, Y. Yu, Y. Wu, X. Chen, Q. Lai, Z. Yang, Q. Chen and J. Yang, FEBS J., 2016, 283(1), 102–111, DOI:10.1111/febs.13555.
- J. Wang, D. D. Miller and W. Li, Drug Discovery Today, 2022, 27(3), 759–776, DOI:10.1016/j.drudis.2021.12.001.
- H. Sahakyan, N. Abelyan, V. Arakelov, G. Arakelov and K. Nazaryan, PLoS One, 2019, 14(8), e0221532, DOI:10.1371/journal.pone.0221532.
- A. E. Prota, F. Danel, F. Bachmann, K. Bargsten, R. M. Buey, J. Pohlmann, S. Reinelt, H. Lane and M. O. Steinmetz, J. Mol. Biol., 2014, 426(8), 1848–1860, DOI:10.1016/j.jmb.2014.02.005.
- M. Elagawany, L. M. Abdel Ghany, T. S. Ibrahim, A. S. Alharbi, M. S. Abdel-Aziz, E. M. El-Labbad and N. Ryad, J. Enzyme Inhib. Med. Chem., 2024, 39(1), 2311157, DOI:10.1080/14756366.2024.2311157.
- N. Ryad, A. A. Elmaaty, I. M. Ibrahim, A. H. A. Maghrabi, M. A. Y. Alahdal, R. M. Saleem, I. Zaki and L. M. A. A. Ghany, Future Med. Chem., 2024, 1–21 Search PubMed.
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61(8), 3891–3898, DOI:10.1021/acs.jcim.1c00203.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31(2), 455–461, DOI:10.1002/jcc.21334.
- E. W. Bell and Y. Zhang, J. Cheminf., 2019, 11, 1–9, DOI:10.1186/s13321-019-0362-7.
- BIOVIA Discovery Studio Visualizer – Dassault Systèmes (https://www.3ds.com).
- H. M. Abd El-Lateef, A. A. Elmaaty, L. M. Abdel Ghany, M. S. Abdel-Aziz, I. Zaki and N. Ryad, ACS Omega, 2023, 8(20), 17948–17965, DOI:10.1021/acsomega.3c01156.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04371e |
| This journal is © The Royal Society of Chemistry 2024 |