
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCl alloying improves thermal stability and increases luminescence in iodine-rich inorganic perovskites†
Deniz N. Cakan a,
Connor J. Dolana,
Eric Oberholtza,
Moses Kodura,
Jack R. Palmerb,
Hendrik M. Vosslerb,
Yanqi Luoc,
Rishi E. Kumarb,
Tao Zhoud,
Zhonghou Caic,
Barry Laic,
Martin V. Holtd,
Sean P. Dunfielda and
David P. Fenning*ab
a,
Connor J. Dolana,
Eric Oberholtza,
Moses Kodura,
Jack R. Palmerb,
Hendrik M. Vosslerb,
Yanqi Luoc,
Rishi E. Kumarb,
Tao Zhoud,
Zhonghou Caic,
Barry Laic,
Martin V. Holtd,
Sean P. Dunfielda and
David P. Fenning*ab
aAiiso Yufeng Li Family Department of Chemical and Nano Engineering, University of California, La Jolla, San Diego, California 92093, USA. E-mail: dfenning@ucsd.edu
bMaterials Science and Engineering Program, University of California, La Jolla, San Diego, California 92093, USA
cAdvanced Photon Source, Argonne National Laboratory, Lemont, IL 60439, USA
dCenter for Nanoscale Materials, Argonne National Laboratory, Lemont, IL 60439, USA
First published on 10th July 2024
Abstract
The inorganic perovskite CsPbI3 shows promising photophysical properties for a range of potential optoelectronic applications but is metastable at room temperature. To address this, Br can be alloyed into the X-site to create compositions such as CsPbI2Br that are stable at room temperature but have bandgaps >1.9 eV – severely limiting solar applications. Herein, in an effort to achieve phase stable films with bandgaps <1.85 eV, we investigate alloying chlorine into iodine-rich triple-halide CsPb(I0.8Br0.2−xClx)3 with 0 < x < 0.1. We show that partial substitution of iodine with bromine and chlorine provides a path to maintain broadband terrestrial absorption while improving upon the perovskite phase stability due to chlorine's smaller size and larger ionization potential than bromine. At moderate Cl loading up to ≈5%, X-ray diffraction reveals an increasingly smaller orthorhombic unit cell, suggesting chlorine incorporation into the lattice. Most notably, this Cl incorporation is accompanied by a significant enhancement over Cl-free controls in the duration of black-phase stability of up to 7× at elevated temperatures. Additionally, we observe up to 5× increased steady state photoluminescence intensity (PL), along with a small blue-shift. In contrast, at high loading (≈10%), Cl accumulates in a second phase that is visible at grain boundaries via synchrotron fluorescence microscopy and negatively impacts the perovskite phase stability. Thus, replacing small fractions of bromine for chlorine in the iodine-rich inorganic perovskite lattice results in distinct improvement thermal stability and optoelectronic quality while minimally impacting the bandgap.
1 Introduction
Metal halide perovskites of the form ABX3 exhibit excellent light absorption,1 long carrier-diffusion lengths,2 and – critically – a tunable electronic bandgap.3,4 These properties have made them a promising class of materials for light emitting diodes,5 photodetectors,6 and photovoltaics.7–12 However, their predominately I-rich organic-inorganic compositions suffer from phase instability, thermal instability, water-mediated decomposition, deleterious condensation reactions, and light-induced degradation that drastically limits their stability and thus application.13–15To improve upon phase stability, which can be understood heuristically through the Goldschmidt tolerance factor, the community has often alloyed smaller halides, such as Br, at the X-site.16,17 While alloying with Br is a viable approach to increase phase stability,18 too large a fraction of Br in organic–inorganic APb(I1−xBrx)3 has been shown to cause halide segregation under non-equilibrium conditions of light or potential bias.14,19,20 In contrast, alloying with Cl has been shown to improve phase stability without inducing light- or bias-driven segregation.21 Moreover, addition of Cl into organic–inorganic perovskites has been shown to reduce device voltage losses.10
However, the amount of Cl remaining in the final film depends greatly on the loading level, annealing condition, and cation composition – which can provide a reaction pathway for Cl to leave during crystallization. For example, when Cl is introduced in the presence of methylammonium (MA) or formamidinium (FA), MACl or FACl has been shown to volatilize to some degree during perovskite crystallization.22 Specifically, FACl has been shown to undergo weight loss at 161 °C via differential scanning calorimetry (DSC) and a phase change at 191 °C via thermogravimetric analysis (TGA).23 Even when the fraction of Cl that volatilizes is significant, Cl one-step addition24,25 and two-step addition26 to precursor inks has still been found to enhance grain growth,27,28 and increase luminescence,29 without substantially modifying the final material composition or bandgap.22,30–32 In contrast, when Cl lacks sufficient avenues for volatilization, it has been shown to both incorporate into the lattice and increase elemental heterogeneity, as indicated by diffraction10 and nano X-ray fluorescence (nXRF)33 respectively.
A-site alloys, now often FA-based system with small amounts of Cs and MA to modulate crystallization and improve phase stability, still suffer from many of the same chemical degradation mechanisms, albeit at slower rates. For example, Tan et al., showed that triple cation systems employing Cs, MA, and FA thermally degrade at two distinct rates, one associated with the volatilization of MA and one with the volatilization of FA.34 Consequentially, going fully inorganic at the A-site offers a distinct approach to addressing the issue of perovskite durability; by removing the A-site organic components, inorganic perovskites are no longer susceptible to the various condensation reactions35 or thermal degradation34 that has been shown occur in the mixed organic systems. All-inorganic perovskite systems have the strongest reported operational stability (equivalent to ≥5 years at 35 °C)36 and improved elevated stability under light and heat.37,38 Nevertheless, halide perovskites need to improve their operational durability on the order of a magnitude before they can become commercially viable.39
While all-inorganic perovskite systems avoid some of the chemical stability challenges intrinsic to their hybrid counterpart, all-inorganic systems, like CsPbI3, suffer from thermodynamic phase instability near room temperature. CsPbI3 can take on three photoactive perovskite phases (α-cubic, β-tetragonal, γ-orthorhombic) that are metastable to transformation to the non-photoactive non-perovskite phase (δ-orthorhombic) at room temperature.40,41 This instability is further exacerbated in ambient conditions as humidity catalyzes the γ-CsPbI3 to δ-CsPbI3 transition.42 To improve phase stability, the community has taken an analogous approach to the organic–inorganic systems by alloying smaller halides into the lattice. For example, Nasstrom et al. have shown that substituting I with Br to form CsPbI2Br delivers photo-active phase stability above ≈300 K.37
In these high Br fraction CsPbI2Br alloys, Cl alloying has been studied as a strategy toward further improvement of grain growth and black-phase stability in ambient conditions.43–47 However, the large Br site fraction in CsPbI2Br increases the bandgap to >1.9 eV, limiting terrestrial solar light absorption.45,48 Cl alloying has been found to enhance the steady state photoluminescence over Cl-free controls by up to ≈1.4× in all-inorganic12,40,49,50 and ≈4.2× in organic containing24,28,51–62 systems.
The need to enhance performance and maintain silicon-tandem relevant bandgaps without largely affecting bandgap motivates alloying Cl into higher I fraction inorganic compositions. Triple halide alloying at ≤1.85 eV bandgap in all-inorganic compositions has been only studied in very limited fashion to date but have shown some promise to enhance photovoltages.63 These compositions show promising enhancement to photovoltaic device voltages. However, the thermal stability of such triple halide inorganic phases is unknown.
Here, we investigate previously unexplored regimes of Cl loading in triple-halide inorganic alloys and identify triple-halide compositions of improved thermal stability and optoelectronic properties, focusing on full-spectrum visible-light absorbing ≈1.83 eV CsPb(I0.80Br0.20−xClx)3 films. Using a combination of diffraction, optical spectroscopy, and micro-spectroscopy, we show that Cl incorporation up until ≈5% Cl. With 5% Cl incorporation, we measure 5× enhanced steady state photoluminescence (PL) and 7× increased duration of black-phase stability at elevated temperatures (≥60 °C) over Cl-free controls. At ≈10% Cl, we observe evidence of secondary phases that negatively affect the stability of the film. Our observations show that modest Cl substitution in triple-halide inorganics can increase PL intensity and thermal stability without significant modification to bandgap, enabling material enhancement for broadband solar absorption.
2 Experimental
2.1 Materials
All materials were purchased from Sigma-Aldrich with specifications detailed in Table S1† and used without further purification.2.2 Fabrication: substrate preparation
TEC 8 ITO substrates (Biotain), float-glass (Thermo Fisher Scientific), and quartz (Chemglass) substrates were sonicated sequentially in a 2% detergent (Hellmanex-III) deionized water mix, deionized water, acetone, and isopropanol alcohol for 15 min each. At the end of the isopropanol sonication, substrates were removed one at a time and dried immediately with a filtered N2 stream to prevent organic residue on the surface of the films. Prior to deposition, substrates were cleaned by UV-ozone for 15 min with an O2 flow rate of 500 cc per min.2.3 Fabrication: perovskite
All-inorganic perovskite thin films with nominal stoichiometries of CsPb(I0.80Br0.20−xClx)3 (x = 0.10, 0.05, 0.01, 0) were targeted using a formamidine acetate (FAAc) templating strategy64 and excess lead iodide. Specifically, FAAc, CsI, PbI2, PbBr2, and PbCl2 precursor powders were mixed in appropriate ratios to obtain the compositions listed above for each Cl loading. Next, 25% mole fraction FAAc relative to A-site was added to each vial, which has been reported to template the high symmetry cubic phase at low temperature, and then volatilize off and exchange with Cs at high temperature to create a fully inorganic film.64 Finally, 12% excess of PbI2 was added to each vial in an attempt to ensure that film surfaces and domain boundaries were passivated.65 The precursor powders were then dissolved in DMSO at 1.33 M with respect to Cs using a vortex mixer. Prior to deposition, the solution was filtered into another vial using a 200 nm pore size PTFE membrane, and then dispensed onto substrates which were then spun at 500 rpm for 10 s and then at 3250 rpm for 50 s. At 5 s remaining in the second cycle, 200 μL of methyl-acetate (MeOAc) was dispensed in a continuous and uniform stream via a 1000 μL pipette; MeOAc was chosen due to its superior ability to decomplex Cs from DMSO.66 Wet films were left to dry in inert 25 °C conditions for 1 h and then annealed at 230 °C for 10 min, resulting in approximately 550 nm thick films (Fig. S1†). Hot plates used had temperature precision of <5%. All film preparation was performed in an inert nitrogen glovebox at <1 ppm water and oxygen.642.4 Characterization: benchtop X-ray diffraction (XRD)
XRD measurements were taken with a Rigaku SmartLab diffractometer using an 8.04 keV Cu Kα parallel beam light source with a Kβ filter. Float-glass/perovskite samples were coated with several μm of polyisobutylene rubber (PIB) (70 mg mL−1 in chlorobenzene) with no additives as a barrier layer to prevent moisture-induced degradation (Fig. S1†). DICVOL14 (ref. 67) software was used to index diffraction peaks according to an orthorhombic Pnma space group; lattice constants and satisfactory de Wolff figures of merit (≥10) of were obtained, as shown in Table S2.†,68 Tabulated lattice volumes can be found in Table S2.†2.5 Characterization: scanning electron microscopy (SEM)
SEM measurements were performed using a Thermo Fisher Apreo microscope, equipped with an in-lens backscattered electron detector. Images were scaled using real space pixel area value to match field of view area across all samples. Apparent grain domain size analysis was then conducted by summing pixels within a manually drawn convex hull in ImageJ, with apparent area calculated via the set pixel to real space scale.2.6 Characterization: synchrotron X-ray fluorescence spectroscopy
Synchrotron-based nanoprobe X-ray fluorescence (nXRF) measurements were taken using both a 270 nm full-width half-maximum (FWHM) focused beam at 7 keV and a 380 nm FWHM focused beam at 16.4 keV at beamline 2-IDD of the Advanced Photon Source (APS) at Argonne National Laboratory (ANL) in a helium environment. The lower 7 keV beam energy was employed to enable sensitivity to Cl. Mapping regions were aligned between the two measurement energies via gold fiducials, and maps generated by the smaller probe were blurred to match the larger probe size using a Gaussian kernel and then averaged.69 Similar to previous investigations,33 the nXRF measurement was conducted through the backside of a gold contact, with a full fluorescence spectrum collected point-by-point for each location on the map. Cl in soda lime glass substrate, which has a common range of 0.5–3 wt% Cl,70 was subtracted as background. For domain boundary analysis, nXRF measurements were taken utilizing a 30 nm FHWM focused beam at 8.5 keV at beamline 26-IDC of the APS at ANL in a vacuum environment. For all nXRF analysis, MAPS software was used to fit the background and perform peak deconvolution.33,71,72 A list of similar synchrotron light sources and their relevant capabilities toward characterization of perovskite can be found in a review by Kodur et al.732.7 Characterization: ultraviolet-visible (UV-vis) absorption spectroscopy
UV-vis absorption spectroscopy measurements of thin films were taken using an integrating sphere setup (PerkinElmer Lambda 1050). Following transmission and reflection measurements, absorption was calculated and direct transition Tauc plots were fit to estimate the optical bandgap of the absorber. For all UV-vis absorption spectroscopy measurements, float-glass/perovskite samples were encapsulated in glass as previously described. A total of 5 samples were measured for each condition (Fig. S2†), with the median sample shown in Fig. 3.2.8 Characterization: photoluminescence spectroscopy (PL)
PL measurements were taken with a Raman microscope (Renishaw inVia). For large-area sampling and single point spectra, a 633 nm laser was operated at 1.75 mW with a 6 μm spot size and a 600 mm−1 grating. Sampling was done on a 300 × 300 μm area with a point to point spacing of 10 μm. For μPL mapping measurements, a 532 nm laser was operated at 200 mW with a 865 nm spot size and a 1800 mm−1 grating. For all PL measurements, float-glass/perovskite samples were encapsulated between two sheets of glass using PIB (Quanex) edge seal pressed at 70 °C and 20 psi to prevent moisture-induced degradation.74 Raw spectra, shown in Fig. S3,† were smoothed via a 0.015 eV moving average Savitzky–Golay filter75 to remove interference effects of the glass encapsulating layer. Time series of peak PL emission energy and PL FWHM data was smoothed using a 100 s rolling average filter.2.9 Characterization: fourier transform infrared spectroscopy attenuated total reflectance (FTIR-ATR)
FTIR-ATR (PerkinElmer UATR Two) was carried out on thin films before their anneal and shortly after their anneal to record any loss in organic material. Background scans were taken between diamond ATR crystal and the probe head subsequent to each sample scan.2.10 Characterization: photoluminescence quantum yield (PLQY)
Photoluminescence quantum yield (PLQY) measurements were carried out on a homebuilt setup consisting of a 532 nm DPSS laser module controlled by a laser diode controller (SRS, Model LDC502) coupled into an integrating sphere (Newport, Model 819C-SF-4). The sphere was fitted with a switchable-gain Si photodetector (Thorlabs PDA100A2) with an automated filter slider (Thorlabs, ELLK6) loaded with a 600 nm longpass filter (Thorlabs FELH600, O.D. 6). To increase signal-to-noise ratio, the laser was modulated at 993 Hz about a 1-sun equivalent injection and photodetector signal was read out using a lock-in amplifier (SRS, Model SR830). PLQY was calculated using the method developed by de Mello et al.,76 with the responsivity of the Si photodetector accounted for using a photoluminescence spectrum of each sample and the responsivity of the detector. To calculate the quasi-Fermi level splitting, the following relationship was used, adapted from de Mello et al.:76
QFLS = Vlim + kT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(PLQY) ln(PLQY)
| (1) |
2.11 Characterization: thermal stability
Within 24 hours of formation at 230 °C, films of each composition were brought from room temperature to 60 °C, 85 °C, and 100 °C to probe the kinetics of phase transformation using the visual yellowing of the film as an indicator for the phase transformation from one of the photoactive black phases to the non-perovskite yellow δ-phase. Measurements were conducted in a glovebox with ≤1 ppm oxygen and moisture. Videos of the degrading films were taken utilizing a camera (GoPro) recording at 6 frames per minute. To process the data, the center 2 cm of each 2.5 cm substrate were averaged and converted to grayscale. Then, initial intensities within each Cl loading were normalized to account for nonuniform lighting in the glovebox. Variations in light intensity caused by variable ambient light in the laboratory were filtered out and the temporally-adjacent points were used to linearly interpolate across the filtered time points. Finally, the onset of discoloration was determined by fitting the linear portion of the discoloration curve and using the x-axis intercept as the onset value. For samples where the onset of discoloration was not observed, the total duration of the test was used as a conservative estimate for the onset value. Raw time lapse videos can be found in the ESI.†3 Results and discussion
Inorganic CsPb(I0.80Br0.20−xClx)3 films with x = 0 to 0.10 were investigated for their composition uniformity, optoelectronic quality, and phase stability especially at elevated temperatures and are hereafter referred to as s 0% Cl, 1% Cl, 5% Cl, and 10% Cl. To better understand the effect of adding Cl into the solution and its resultant effect on the final film, a series of analysis techniques were performed, including X-ray diffraction (XRD), scanning electron microscopy (SEM), synchrotron-based nano-X-ray fluorescence (nXRF), photoluminescence (PL), and thermal degradation tests.Analysis of XRD patterns reveals incorporation of Cl into the lattice until 5% Cl, followed by phase segregation and the emergence of a secondary Br- or/and Cl-rich phase (Fig. 1a). Extracted orthorhomobic (Pnma) lattice volumes as a function of Cl loading are shown in Fig. 1b. As can be seen in Fig. 1b, the lattice volume shrinks up to 5% Cl, followed by a lattice expansion at 10% Cl. To estimate the lattice volume, the full XRD patterns (Fig. 1a) were indexed using DICVOL14. Due to the texture of the thin films, discriminating the space group of the black phase is a challenge. The minor peaks appearing between 23° 2θ and 25° 2θ in the patterns (Fig. 1a) are consistent with the literature suggesting the Pnma γ-CsPbI3 phase is favored at room temperature.77 Williamson–Hall analysis of the diffraction patterns revealed relaxation of strain with increasing Cl loading (Fig. S4†). No measurable strain was present at 5 or 10% Cl compared to a strain of 5 × 10−3 in Cl free controls. No evidence of a non-perovskite δ-phase was found in any samples, e.g., indicated by the lack of a peak at ≈10° 2θ in Fig. 1a. However, a secondary peak emerges at 10% Cl at slightly larger 2θ relative to the main {002} peak. The emergence of a secondary peak at smaller d-spacing and the main phase's larger lattice volume at 10% Cl suggests that the main phase becomes more I-rich while a Cl- and Br-richer secondary phase emerges. This trend in the inorganic triple halide perovskite is analogous to the lattice volume behavior upon Cl incorporation reported by Xu et al. in recent work on triple halide organic–inorganic perovskites, who ascribed the observed trends to a solubility limit for Cl within the lattice.10 In short, it appears that up until 5% Cl, Cl successfully incorporates into the lattice, causing a decrease in lattice volume with no additional peaks/phases. Beyond 10% Cl, we expect continued evidence of secondary phases.
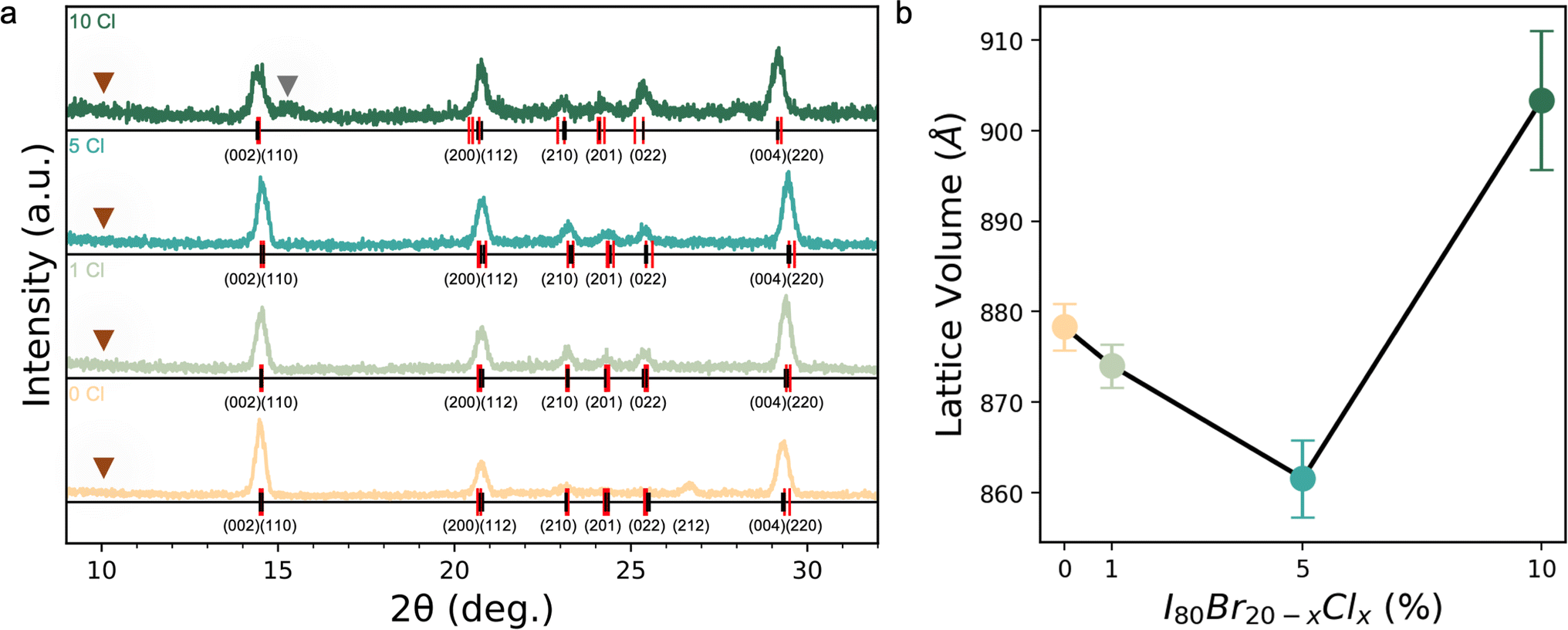 | ||
| Fig. 1 (a) Parallel beam thin film diffraction XRD of CsPb(I0.80Br0.20−xClx)3 films with Cl varied from 0% to 10%. The orthorhombic (Pnma) calculated peaks are marked as red vertical lines, with observed two theta peak location as black vertical lines. Emerging secondary phase in 10% Cl marked with gray arrow and the location of the non-perovskite δ-phase peak were it to be present marked by the brown triangle. 8.04 keV X-ray source. (b) Lattice volume calculated from DICVOL14 based on orthorhombic indexing. | ||
SEM results show that at low loading Cl changes film morphology relatively little, while at 10% Cl loading it increases the nucleation density as seen in Fig. 2a–d (full area SEM images in S5†). At 10% Cl, pinholes are eliminated and there is a reduction of median apparent grain domain size (Fig. S6†). Given that this change in morphology at 10% Cl appears to correlate well to the emergence of a secondary peak in XRD, we believed that many of these additional nucleation points may be from Cl-rich phases.
 | ||
| Fig. 2 (a–d) SEM images of CsPb(I0.80Br0.20−xClx)3 with Cl varied from 0% to 10% showing increasing apparent domain size until 1% followed by a decrease in size. Scale bar 2 μm. | ||
nXRF identifies three distinct Cl loading regimes. As can be seen in Table 1 the average Cl fraction measured in each film correlates well to its loading concentration, but is consistently lower. This reduction in Cl fraction of the final film indicates the likely volatilization of at least a fraction of FACl during the 230 °C anneal. FACl is known to undergo mass loss at 161 °C.23 The XRF maps used to tabulate the average values in Table 1 are shown in Fig. S7.† Additionally, we conducted FTIR-ATR (Fig. S8†) and observed the disappearance of characteristic vibrations at ≈3400 cm−1 and ≈1400–1500 cm−1 after the 230 °C anneal.78 The relatively similar Cl concentrations in the 0% and 1% Cl-loaded films may suggest that much of the Cl volatilizes away at low loading. This is consistent with reports in organic systems.79,80 It thus appears Cl loading has three distinct regimes: (1) at low Cl loading, most Cl volatilizes off, (2) at moderate Cl loading less than the solubility limit of Cl within the lattice, some Cl volatilizes off and some incorporates into the lattice as indicated by a smaller unit cell in XRD, and (3) at high Cl loading that exceeds the solubility limit of Cl within the lattice, some Cl volatilizes off, a fraction incorporates in to the main phase, but most nucleates out into a Cl-rich phase as evidenced by XRD.
| Cl% | Nominal I% | Measured I% | Nominal Br% | Measured Br% | Nominal Cl% | Measured Cl% above background |
|---|---|---|---|---|---|---|
| 0 | 81 | 84.2 | 19 | 15.9 | 0 | 0 |
| 1 | 80.8 | 82.9 | 18.3 | 17.2 | 0.9 | 0.02 |
| 5 | 81.3 | 84.1 | 14.1 | 14.0 | 4.5 | 1.98 |
| 10 | 80.8 | 81.0 | 9.6 | 13.3 | 9.6 | 5.72 |
To better understand how the three distinct regimes of Cl incorporation affect the optoelectronic properties of the films, UV-vis absorption, μPL, and PL quantum yield (PLQY) measurements were performed. The optical Tauc bandgap shows only a small ≈3 meV blue-shift when adding 5% Cl and a ≈17 meV red-shift at 10% Cl relative to the control, as seen in Fig. 3a. 1% Cl addition appears to lead to minimal changes in absorption onset. These trends are consistent in measurements of 5 samples at each loading, though the variation in the absorption onset increases with Cl loading (Tauc analysis in Fig. S2† and UV-vis spectra in Fig. S9†), which suggests increasing heterogeneity. Cl has been previously shown to incorporate heterogeneously in hybrid halide perovskite alloy thin films,33 and, similarly, heterogeneity appears to increase with increasing Cl content in the all-inorganic halide alloy.
 | ||
| Fig. 3 (a) Direct transition Tauc plots of CsPb(I0.80Br0.20−xClx)3 with Cl varied from 0% to 10% generated with best fit line drawn to the intercept. (b) PL spectra with vertical line denoting peak PL energy. (c) PLQY bar plots of films. (d) Calculated implied voltage of films using median bandgap from Tauc plot. | ||
Fig. 3b shows that a monotonic increase in PL intensity is seen with Cl addition but the energy of the peak emission first blue shifts a small amount up to 5% Cl, then at 10% Cl reverts to near the 0% Cl maximum. At 1% Cl, the PL emission energy is nearly identical but slightly blue-shifted from the control, consistent with limited Cl incorporation to the crystal structure. At 5% Cl, Cl incorporation into the lattice results in a 12.5 meV PL blue-shift in emission energy as well as increased intensity. Notably, at 5% Cl there is a larger increase in the PL emission energy (≈10 meV) than the shift in the optical bandgap (≈3 meV), corroborating an improvement in optoelectronic quality. At 10% Cl, the PL emission red-shifts, broadens, increases intensity, and becomes asymmetric, which is consistent with Cl leading to the precipitation of phases with varied halide content and therefore varied emission profiles. The red-shift with respect to the control seen in 10% Cl films is expected as the minor phase at smaller d-spacing likely contains Br, leading to the main phase becoming relatively richer in I.
To further quantify luminescence, we conducted PLQY (Fig. 3c) and observed supporting evidence of increased luminescence with Cl loading. PLQY values on the order of 0.01% indicate substantial non-radiative losses in the films, and are similar to the more commonly investigated all inorganic ≈1.7 eV CsPbI3 (ref. 81) (PLQY of ≈0.04%) yet less luminescent than hybrid ≈1.55 eV bandgap Cs0.05(FAxMAy)0.95Pb(IxBry)3 (ref. 82) (PLQY of ≈3%). But, across all Cl concentrations, PL intensity and PLQY increases with Cl loading. The corresponding implied voltage under 1-sun illumination is shown in (Fig. 3d), where due to the bandgap narrowing at 10% Cl, the implied voltage (eqn (1)) is greatest at 5% Cl amongst all conditions. Without evidence of secondary phases nor an increased in apparent domain size in the ≤5% Cl films, the increasing PLQY may be due to the increased passivation of Shockley–Read–Hall recombination centers similar to its effects in MAPbI3.25 Collectively, these results suggest that increasing the Cl loading increases quasi-Fermi level splitting, at least in the intensely light-emitting regions of the film, which become heterogeneously distributed in the 10% Cl films as described below.
Microscopic μPL and nXRF mapping on the 10% Cl sample reveals a distribution of Cl at apparent domain boundaries that appears to correlate well with PL intensity and emission energy. μPL maps as a function of intensity and emission energy are shown in Fig. 4a and b, while nXRF maps of the total fluorescence yield and Cl to I ratio are shown in Fig. 4c and d, respectively. As can be seen in the PL maps, there are substantial spatial variations in the PL intensity and peak emission energy, explaining the shoulders in the area-averaged PL emission spectra above. Moreover, the PL intensity appears to be maximal in regions where the emission energy changes most drastically (likely due to varied halide content) and at the edge of the apparent domains (e.g. Area 1 in Fig. 4a). nXRF shows reduced total fluorescence yield and a large concentration of Cl with respect to I at such domain boundaries. DeQuilettes et al. previously demonstrated a correlation between local Cl content in the film and PL intensity in MAPbI3.29 Moreover, the increase in PL intensity of the 10% Cl samples is analogous to the results in organic-inorganic perovskites of Frohna et al., who found evidence for a similar carrier funneling mechanism from Br-rich to I-rich areas in FA0.79MA0.16Cs0.05Pb(I0.83Br0.17)3.83 Together, the trends in the inorganic triple halide films are consistent with carrier funneling from a Cl-rich wider bandgap material at the domain boundaries to I-rich narrower material within the domains.
 | ||
| Fig. 4 (a) μPL maps of CsPb(I0.80Br0.10Cl0.10)3 showing contrast (1) of PL intensity with (b) spatially heterogeneous peak PL emission energy (scale bar 4 μm). (c) Corresponding nXRF total fluorescence yield (TFY) with 10 μm FoV showing clear apparent boundary regions (scale bar 2 μm) with (d) zoom (3 μm FoV) showing a high ratio of Cl to I at the boundary (scale bar 600 nm). | ||
To investigate the stability of these phases over time, prolonged PL measurements were conducted, which revealed that the peak emission energy and FWHM was relatively robust to illumination across 0% Cl through 5% Cl conditions (Fig. 5a and b). Conversely in 10% Cl, we observed a minor peak PL energy red-shift and FWHM broadening over the duration of the test. Full PL spectra over time are shown on edge-sealed glass packages in Fig. S10.† As can be seen, all films photodarken with no clear correlation to Cl loading. While surface reactions affecting passivation cannot be ruled out as a plausible explanation, the diminishing PL appears consistent with previous reports that illumination introduces defects into a CsPbI3 lattice.84 In contrast with the PL intensity, the peak emission energy for single phase conditions remain stable (Fig. 5a), suggesting that the halide distribution does not change with time. The lack of change in emission energy for single phase film compositions further suggests that the laser induced defects are not strongly affected by halide heterogeneity, while the multiple phase 10% Cl shows indication of halide instability. Thus, it appears that increasing Cl content up to 5% halide fraction increases PL intensity in the inorganic perovskites with a modest blue-shift in emission that is stable against light-induced halide redistribution/segregation. The stability of the PL emission energy of the inorganic triple halide perovskites shown here indicates the ability to maintain the absorber's applicability to a targeted spectral design.
 | ||
| Fig. 5 (a) Peak PL emission energy and PL FWHM (b) of CsPb(I0.80Br0.20−xClx)3 films collected from a single point (6 μm) over 545 s. Raw data is in lighter color (yellow is 0% Cl, light green is 1% Cl, teal is 5% Cl, and 10% Cl is dark green) with 100 s rolling average in darker color. Bar plots of fitted onset of discoloration of CsPb(I0.80Br0.20−xClx)3 of films held at (c) 100 °C, (d) 85 °C, and (e) 60 °C, black bar indicates respective standard deviation. For samples where no discoloration onset was observed, the onset of discoloration was conservatively estimated as the final test time. Bars encompassing such samples have the count of occurrences denoted by a number, accompanied by an asterisk, placed above them. A total of 2 samples per condition were observed for 100 °C and 85 °C temperatures, with 4 samples observed for 60 °C. | ||
Lastly, it appears that the perovskite phase stability at elevated temperatures increases with uniform Cl incorporation, but is adversely affected by secondary phases. To understand how Cl incorporation affects thermal stability, three sets of films were imaged over a 70 h period at 60 °C, 85 °C, and 100 °C (see ESI† for videos) to visualize color changes over time indicative of the time point at which the photoactive black phase (α, β or γ) begins to degrade (example onset fitting in Fig. S11†). As can be seen in Fig. 5c–e, at 60 °C, 85 °C, and 100 °C, Cl incorporation reproducibly enhances the phase stability of the films at low Cl loadings with the best thermal stability at 5% Cl, followed by a reduction in stability at higher loading. This reduction is likely explained by the initial presence of multiple phases in films with 10% Cl. At a lower temperature of 60 °C, Cl has a smaller effect on the photoactive phase thermal stability but many films did not degrade within the duration of the test. By the tolerance factor heuristic, Cl addition into the inorganic triple halide alloy should favor perovskite phase formation. Thus, it is expected that Cl incorporation should lower the temperature at which the black perovskite phase is thermodynamically stable, as appears to be the case from the 60 °C, 85 °C, and 100 °C data.
These photo- and thermal stability tests on bare films permit a direct determination of improvements in underlying phase stability in thin films as a function of composition of the perovskite. Unlike full devices, which include transport, contact and often passivation layers that can inhibit phase transformation or provide encapsulation,85,86 the tests shown here are on standalone perovskite films. Overall, the improvement of bare inorganic perovskite thin films thermal stability at elevated temperatures relevant to optoelectronic device operating conditions including terrestrial photovoltaics indicates Cl incorporation as an additional opportunity to overcome the metastability of perovskites.
4 Conclusion
Herein, we have explored Cl alloys of up to 10% Cl in ≈1.83 eV bandgap CsPb(I0.80Br0.20−xClx)3 films. Specifically, we have shown three distinct regimes of behavior dependent on Cl loading: (i) at low loadings of ≈1% Cl most all Cl volatilizes during crystallization annealing, (ii) at moderate Cl loading of ≈5% Cl, the Cl remaining after annealing incorporates uniformly into the lattice, and (iii) at higher Cl loading, most Cl precipitates out into a Cl-rich phase.We find that alloying Cl up through moderate loading shrinks the perovskite lattice, blue-shifts its PL modestly, increases PL intensity by a factor of 5×, and significantly enhances black-phase thermal stability at 60–100 °C by up to 7× while maintaining relatively stable PL emission energy under laser illumination. In contrast at higher loadings, we find evidence of a second phase leading to a narrowed bandgap of the main I-rich perovskite and low thermal and photostability. These data indicate that incorporating Cl into a single-phase alloy presents an opportunity to enhance the thermal stability and optoelectronic properties of interest in triple-halide inorganic perovskites relative to Br/I inorganic perovskites. These findings ultimately suggest that replacing ≈5% Br with Cl can result in distinct improvement in both thermal stability and optoelectronic quality of the CsPbI3 host crystal without significantly altering the bandgap.
Data availability
Data supporting the findings of this study are available from the corresponding author upon reasonable request.Author contributions
D. N. C. led all experiments, data curation, formal analysis, and visualizations. C. J. D., Y. L., R. E. K., T. Z., Z. C., B. L., M. H. assisted with synchrotron measurements. E. O. assisted with sample preparations. D. P. F. came up with the experimental framework and led funding acquisition and supervised the research. D. N. C., S. P. D. wrote original draft, while the final review and editing of the manuscript saw contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported in part by the California Energy Commission EPC-19-004 and the National Science Foundation CAREER award 1848371. Use of the Center for Nanoscale Materials and the Advanced Photon Source, both Office of Science user facilities, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract No. DE-AC02-06CH11357.References
- M. Kato, T. Fujiseki, T. Miyadera, T. Sugita, S. Fujimoto, M. Tamakoshi, M. Chikamatsu and H. Fujiwara, J. Appl. Phys., 2017, 121, 115501 CrossRef.
- G. W. P. Adhyaksa, L. W. Veldhuizen, Y. Kuang, S. Brittman, R. E. I. Schropp and E. C. Garnett, Chem. Mater., 2016, 28, 5259–5263 CrossRef CAS.
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. I. Seok, Nano Lett., 2013, 13, 1764–1769 CrossRef CAS.
- E. L. Unger, L. Kegelmann, K. Suchan, D. Sörell, L. Korte and S. Albrecht, J. Mater. Chem. A, 2017, 5, 11401–11409 RSC.
- D. Yang, B. Zhao, T. Yang, R. Lai, D. Lan, R. H. Friend and D. Di, Adv. Funct. Mater., 2022, 32, 2109495 CrossRef CAS.
- F. Ye, H. Lin, H. Wu, L. Zhu, Z. Huang, D. Ouyang, G. Niu and W. C. H. Choy, Adv. Funct. Mater., 2019, 29, 1806984 CrossRef.
- D. Yang, B. Zhao, T. Yang, R. Lai, D. Lan, R. H. Friend and D. Di, Adv. Funct. Mater., 2022, 32, 2109495 Search PubMed.
- Best Research-Cell Efficiency Chart, 2024, https://www.nrel.gov/pv/cell-efficiency.html Search PubMed.
- J. Jeong, M. Kim, J. Seo, H. Lu, P. Ahlawat, A. Mishra, Y. Yang, M. A. Hope, F. T. Eickemeyer, M. Kim, Y. J. Yoon, I. W. Choi, B. P. Darwich, S. J. Choi, Y. Jo, J. H. Lee, B. Walker, S. M. Zakeeruddin, L. Emsley, U. Rothlisberger, A. Hagfeldt, D. S. Kim, M. Grätzel and J. Y. Kim, Nature, 2021, 592, 381–385 Search PubMed.
- J. Xu, C. C. Boyd, Z. J. Yu, A. F. Palmstrom, D. J. Witter, B. W. Larson, R. M. France, J. Werner, S. P. Harvey, E. J. Wolf, W. Weigand, S. Manzoor, M. F. A. M. van Hest, J. J. Berry, J. M. Luther, Z. C. Holman and M. D. McGehee, Science, 2020, 367, 1097–1104 CrossRef CAS.
- K. Xiao and H. Tan, Nat. Energy, 2020, 5, 870–880 CrossRef CAS.
- K. Wang, Z. Jin, L. Liang, H. Bian, H. Wang, J. Feng, Q. Wang and S. F. Liu, Nano Energy, 2019, 58, 175–182 CrossRef CAS.
- D. Zhang, D. Li, Y. Hu, A. Mei and H. Han, Commun. Mater., 2022, 3, 58 CrossRef.
- R. E. Beal, N. Z. Hagström, J. Barrier, A. Gold-Parker, R. Prasanna, K. A. Bush, D. Passarello, L. T. Schelhas, K. Brüning, C. J. Tassone, H.-G. Steinrück, M. D. McGehee, M. F. Toney and A. F. Nogueira, Matter, 2020, 2, 207–219 CrossRef.
- S. P. Dunfield, L. Bliss, F. Zhang, J. M. Luther, K. Zhu, M. F. A. M. Hest, M. O. Reese and J. J. Berry, Adv. Energy Mater., 2020, 10, 1904054 CrossRef CAS.
- V. M. Goldschmidt, Naturwissenschaften, 1926, 14, 477–485 CrossRef CAS.
- J. Hieulle, X. Wang, C. Stecker, D.-Y. Son, L. Qiu, R. Ohmann, L. K. Ono, A. Mugarza, Y. Yan and Y. Qi, J. Am. Chem. Soc., 2019, 141, 3515–3523 CrossRef CAS PubMed.
- N. J. Jeon, J. H. Noh, Y. C. Kim, W. S. Yang, S. Ryu and S. I. Seok, Nat. Mater., 2014, 13, 897–903 CrossRef CAS PubMed.
- E. T. Hoke and M. D. McGehee, Chem. Sci., 2015, 6, 613–617 RSC.
- Z. Xu, R. A. Kerner, S. P. Harvey, K. Zhu, J. J. Berry and B. P. Rand, ACS Energy Lett., 2023, 8(1), 513–520 CrossRef CAS.
- F. Peña-Camargo, P. Caprioglio, F. Zu, E. Gutierrez-Partida, C. M. Wolff, K. Brinkmann, S. Albrecht, T. Riedl, N. Koch, D. Neher and M. Stolterfoht, ACS Energy Lett., 2020, 5, 2728–2736 CrossRef.
- H. Yu, F. Wang, F. Xie, W. Li, J. Chen and N. Zhao, Adv. Funct. Mater., 2014, 24, 7102–7108 CrossRef CAS.
- H. Li, J. Zhou, L. Tan, M. Li, C. Jiang, S. Wang, X. Zhao, Y. Liu, Y. Zhang, Y. Ye, W. Tress and C. Yi, Sci. Adv., 2022, 8, 7422 CrossRef PubMed.
- X. Liu, Y. Guo, Y. Cheng, S. Lu, R. Li and J. Chen, Chem. Commun., 2023, 59, 13394–13405 RSC.
- L. Fan, Y. Ding, J. Luo, B. Shi, X. Yao, C. Wei, D. Zhang, G. Wang, Y. Sheng, Y. Chen, A. Hagfeldt, Y. Zhao and X. Zhang, J. Mater. Chem. A, 2017, 5, 7423–7432 RSC.
- J. Bing, D. Lee, Y. Cho, J. Zheng, Y. Li, S. Tang, M. Zhang, S. Huang and A. Ho-Baillie, Mater. Today Energy, 2020, 18, 100551 CrossRef CAS.
- Q. Dong, Y. Yuan, Y. Shao, Y. Fang, Q. Wang and J. Huang, Energy Environ. Sci., 2015, 8, 2464–2470 RSC.
- M. Kim, G. H. Kim, T. K. Lee, I. W. Choi, H. W. Choi, Y. Jo, Y. J. Yoon, J. W. Kim, J. Lee, D. Huh, H. Lee, S. K. Kwak, J. Y. Kim and D. S. Kim, Joule, 2019, 3, 2179–2192 CrossRef CAS.
- D. W. DeQuilettes, S. M. Vorpahl, S. D. Stranks, H. Nagaoka, G. E. Eperon, M. E. Ziffer, H. J. Snaith and D. S. Ginger, Science, 2015, 348, 683–686 CrossRef CAS PubMed.
- W. Song, X. Zhang, S. Lammar, W. Qiu, Y. Kuang, B. Ruttens, J. D'Haen, I. Vaesen, T. Conard, Y. Abdulraheem, T. Aernouts, Y. Zhan and J. Poortmans, ACS Appl. Mater. Interfaces, 2022, 14, 27922–27931 Search PubMed.
- K. Xu, A. Al-Ashouri, Z.-W. Peng, E. Köhnen, H. Hempel, F. Akhundova, J. A. Marquez, P. Tockhorn, O. Shargaieva, F. Ruske, J. Zhang, J. Dagar, B. Stannowski, T. Unold, D. Abou-Ras, E. Unger, L. Korte and S. Albrecht, ACS Energy Lett., 2022, 7, 3600–3611 Search PubMed.
- S. Dastidar, D. A. Egger, L. Z. Tan, S. B. Cromer, A. D. Dillon, S. Liu, L. Kronik, A. M. Rappe and A. T. Fafarman, Nano Lett., 2016, 16, 3563–3570 CrossRef CAS PubMed.
- Y. Luo, S. Gamliel, S. Nijem, S. Aharon, M. Holt, B. Stripe, V. Rose, M. I. Bertoni, L. Etgar and D. P. Fenning, Chem. Mater., 2016, 28, 6536–6543 CrossRef CAS.
- W. Tan, A. R. Bowring, A. C. Meng, M. D. McGehee and P. C. McIntyre, ACS Appl. Mater. Interfaces, 2018, 10, 5485–5491 CrossRef CAS.
- X. Wang, Y. Fan, L. Wang, C. Chen, Z. Li, R. Liu, H. Meng, Z. Shao, X. Du, H. Zhang, G. Cui and S. Pang, Chem, 2020, 6, 1369–1378 CAS.
- X. Zhao, T. Liu, Q. C. Burlingame, T. Liu, R. Holley, G. Cheng, N. Yao, F. Gao and Y.-L. Loo, Science, 2022, 377, 307–310 CrossRef CAS PubMed.
- H. Näsström, P. Becker, J. A. Márquez, O. Shargaieva, R. Mainz, E. Unger and T. Unold, J. Mater. Chem. A, 2020, 8, 22626–22631 RSC.
- Y. Zhou and Y. Zhao, Energy Environ. Sci., 2019, 12, 1495–1511 RSC.
- H. Zhu, S. Teale, M. N. Lintangpradipto, S. Mahesh, B. Chen, M. D. McGehee, E. H. Sargent and O. M. Bakr, Nat. Rev. Mater., 2023, 8, 569–586 CrossRef.
- J. Ma, M. Qin, Y. Li, X. Wu, Z. Qin, Y. Wu, G. Fang and X. Lu, Matter, 2021, 4, 313–327 CrossRef CAS.
- W. Xiang, S. F. Liu and W. Tress, Energy Environ. Sci., 2021, 14, 2090–2113 RSC.
- M. Kumar, V. Pawar, P. K. Jha, P. A. Jha and P. Singh, J. Solid State Chem., 2022, 308, 122893 CrossRef CAS.
- U. Khan, Y. Zhinong, A. A. Khan, A. Zulfiqar and N. Ullah, Nanoscale Res. Lett., 2019, 14, 116 CrossRef PubMed.
- Z. Guo, S. Zhao, A. Liu, Y. Kamata, S. Teo, S. Yang, Z. Xu, S. Hayase and T. Ma, ACS Appl. Mater. Interfaces, 2019, 11, 19994–20003 CrossRef CAS.
- S. Chen, T. Zhang, X. Liu, J. Qiao, L. Peng, J. Wang, Y. Liu, T. Yang and J. Lin, J. Mater. Chem. C, 2020, 8, 3351–3358 RSC.
- L. Atourki, M. Bernabé, M. Makha, K. Bouabid, M. Regragui, A. Ihlal, M. Abd-lefdil and M. Mollar, RSC Adv., 2021, 11, 1440–1449 RSC.
- X. Wu, S. Wang, J. Zhang, H.-W. Shiu, Y.-J. Hsu, H. Yan, J. Zhu and X. Lu, Nano Energy, 2023, 117, 108907 CrossRef CAS.
- Y. Gu, H. Wang, J. Sun, Y. Lu, P. Luo and J. Hu, J. Alloys Compd., 2023, 963, 171291 CrossRef CAS.
- B. Li, Y. Zhang, L. Zhang and L. Yin, J. Power Sources, 2017, 360, 11–20 CrossRef CAS.
- Y. Gu, H. Wang, J. Sun, Y. Lu, P. Luo and J. Hu, J. Alloys Compd., 2023, 963, 171291 CrossRef CAS.
- J. Park, J. Kim, H. S. Yun, M. J. Paik, E. Noh, H. J. Mun, M. G. Kim, T. J. Shin and S. I. Seok, Nature, 2023, 616, 724–730 CrossRef CAS PubMed.
- P. Gostishchev, D. Saranin, L. Luchnikov, D. Muratov, A. Ishteev, M. Voronova, D. Gets, E. Argunov, T. S. Le, S. Didenko and A. Di Carlo, Sol. RRL, 2023, 7, 1–9 Search PubMed.
- H. Li, C. Zhang, C. Gong, D. Zhang, H. Zhang, Q. Zhuang, X. Yu, S. Gong, X. Chen, J. Yang, X. Li, R. Li, J. Li, J. Zhou, H. Yang, Q. Lin, J. Chu, M. Grätzel, J. Chen and Z. Zang, Nat. Energy, 2023, 8, 946–955 CrossRef CAS.
- M. K. Mohammed, M. S. Jabir, H. G. Abdulzahraa, S. H. Mohammed, W. K. Al-Azzawi, D. S. Ahmed, S. Singh, A. Kumar, S. Asaithambi and M. Shekargoftar, RSC Adv., 2022, 12, 20461–20470 RSC.
- J. Jin, H. Li, C. Chen, B. Zhang, L. Xu, B. Dong, H. Song and Q. Dai, ACS Appl. Mater. Interfaces, 2017, 9, 42875–42882 CrossRef CAS PubMed.
- Y. Zhao, F. Ma, Z. Qu, S. Yu, T. Shen, H. X. Deng, X. Chu, X. Peng, Y. Yuan, X. Zhang and J. You, Science, 2022, 377, 531–534 CrossRef CAS.
- Y. Sheng, Y. Hu, A. Mei, P. Jiang, X. Hou, M. Duan, L. Hong, Y. Guan, Y. Rong, Y. Xiong and H. Han, J. Mater. Chem. A, 2016, 4, 16731–16736 RSC.
- S. You, X. Xi, X. Zhang, H. Wang, P. Gao, X. Ma, S. Bi, J. Zhang, H. Zhou and Z. Wei, J. Mater. Chem. A, 2020, 8, 17756–17764 RSC.
- H. Min, M. Kim, S. U. Lee, H. Kim, G. Kim, K. Choi, J. H. Lee and S. I. Seok, Science, 2019, 366, 749–753 CrossRef CAS PubMed.
- M. Mateen, Z. Arain, X. Liu, A. Iqbal, Y. Ren, X. Zhang, C. Liu, Q. Chen, S. Ma, Y. Ding, M. Cai and S. Dai, Sci. China Mater., 2020, 63, 2477–2486 CrossRef CAS.
- R. Chen, M. Li, Y. Zhang, H. Gao, Y. Peng, S. Tang, L. Yu and W. Huang, J. Phys. Chem. C, 2021, 125, 2866–2874 Search PubMed.
- S. H. Chan, Y. H. Chang, M. H. Jao, K. C. Hsiao, K. M. Lee, C. S. Lai and M. C. Wu, Sol. RRL, 2022, 6, 2–11 Search PubMed.
- Q. Ye, Y. Zhao, S. Mu, F. Ma, F. Gao, Z. Chu, Z. Yin, P. Gao, X. Zhang and J. You, Adv. Mater., 2019, 31, 1905143 CrossRef CAS PubMed.
- J. Zhang and A. Hagfeldt, Joule, 2020, 4, 222–234 CrossRef CAS.
- Y. Chen, Q. Meng, Y. Xiao, X. Zhang, J. Sun, C. B. Han, H. Gao, Y. Zhang, Y. Lu and H. Yan, ACS Appl. Mater. Interfaces, 2019, 11, 44101–44108 CrossRef CAS PubMed.
- T. Moot and J. M. Luther, Adv. Energy Mater., 2020, 10, 1–9 Search PubMed.
- A. Boultif and D. Louër, J. Appl. Crystallogr., 2004, 37, 724–731 CrossRef CAS.
- P. M. de Wolff, J. Appl. Crystallogr., 1968, 1, 108–113 CrossRef.
- P. Virtanen, R. Gommers, T. E. Oliphant, M. Haberland, T. Reddy, D. Cournapeau, E. Burovski, P. Peterson, W. Weckesser, J. Bright, S. J. van der Walt, M. Brett, J. Wilson, K. J. Millman, N. Mayorov, A. R. J. Nelson, E. Jones, R. Kern, E. Larson, C. J. Carey, İ. Polat, Y. Feng, E. W. Moore, J. VanderPlas, D. Laxalde, J. Perktold, R. Cimrman, I. Henriksen, E. A. Quintero, C. R. Harris, A. M. Archibald, A. H. Ribeiro, F. Pedregosa, P. van Mulbregt and SciPy 1.0 Contributors, Nat. Methods, 2020, 17, 261–272 CrossRef CAS.
- D. Obranovic, D. Barham and R. G. V. H. On, Proceedings of the 33rd International Symposium on Archaeometry, Amsterdam, The Netherlands, 2017, pp. 4–7 Search PubMed.
- S. Vogt, J. Phys. IV, 2003, 104, 635–638 CAS.
- J.-P. Correa-Baena and D. P. Fenning, Science, 2019, 363, 627–631 CrossRef CAS PubMed.
- M. Kodur, R. E. Kumar, Y. Luo, D. N. Cakan, X. Li, M. Stuckelberger and D. P. Fenning, Adv. Energy Mater., 2020, 10, 1903170 CrossRef CAS.
- R. Runser, M. Kodur, J. H. Skaggs, D. N. Cakan, J. B. Foley, M. Finn, D. P. Fenning and D. J. Lipomi, ACS Appl. Energy Mater., 2021, 4, 10314–10322 CrossRef CAS.
- A. Savitzky and M. J. E. Golay, Anal. Chem., 1964, 36, 1627–1639 CrossRef CAS.
- J. C. de Mello, H. F. Wittmann and R. H. Friend, Adv. Mater., 1997, 9, 230–232 CrossRef CAS.
- R. J. Sutton, M. R. Filip, A. A. Haghighirad, N. Sakai, B. Wenger, F. Giustino and H. J. Snaith, ACS Energy Lett., 2018, 3, 1787–1794 CrossRef CAS.
- National Institute of Advanced Industrial Science and Technology (AIST), SDBS-3590: Formamidine Acetate – Collection of Spectral Data, Spectral Database for Organic Compounds, 1999, https://sdbs.db.aist.go.jp/sdbs/cgi-bin/landingpage?sdbsno=3590, Accessed on January 3, 2024 Search PubMed.
- S. T. Williams, F. Zuo, C.-C. Chueh, C.-Y. Liao, P.-W. Liang and A. K.-Y. Jen, ACS Nano, 2014, 8, 10640–10654 CrossRef CAS PubMed.
- B. Lee, T. Hwang, S. Lee, B. Shin and B. Park, Sci. Rep., 2019, 9, 4803 CrossRef PubMed.
- Z. Iqbal, F. Zu, A. Musiienko, E. Gutierrez-Partida, H. Köbler, T. W. Gries, G. V. Sannino, L. Canil, N. Koch, M. Stolterfoht, D. Neher, M. Pavone, A. B. Muñoz-García, A. Abate and Q. Wang, ACS Energy Lett., 2023, 8, 4304–4314 CrossRef CAS.
- F. Peña-Camargo, P. Caprioglio, F. Zu, E. Gutierrez-Partida, C. M. Wolff, K. Brinkmann, S. Albrecht, T. Riedl, N. Koch, D. Neher and M. Stolterfoht, ACS Energy Lett., 2020, 5, 2728–2736 CrossRef.
- K. Frohna, M. Anaya, S. Macpherson, J. Sung, T. A. S. Doherty, Y.-h. Chiang, A. J. Winchester, K. W. P. Orr, J. E. Parker, P. D. Quinn, K. M. Dani, A. Rao and S. D. Stranks, Nat. Nanotechnol., 2022, 17, 190–196 CrossRef CAS PubMed.
- Z. Lin, M. C. Folgueras, H. K. Le, M. Gao and P. Yang, Matter, 2022, 5, 1455–1465 CrossRef CAS.
- S. Wang, A. Wang and F. Hao, iScience, 2022, 25, 103599 CrossRef CAS PubMed.
- C. C. Boyd, R. Cheacharoen, T. Leijtens and M. D. McGehee, Chem. Rev., 2019, 119, 3418–3451 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04348k |
| This journal is © The Royal Society of Chemistry 2024 |