
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Syntheses, characterizations and reactions of acene-2,3-dicarbaldehydes†
Qian Liu
 and
Glen P. Miller
*
and
Glen P. Miller
*
Department of Chemistry, University of New Hampshire, 23 Academic Way, Durham, New Hampshire 03824, USA. E-mail: glen.miller@unh.edu
First published on 9th August 2024
Abstract
Here, we report improved syntheses, detailed characterizations and reactions of a series of acene-2,3-dicarbaldehydes including tetracene-2,3-dicarbaldehyde. DFT calculations corroborate and complement the experimental results. Tetracene-2,3-dicarbaldehyde and the benchmark organic semiconductor pentacene have isoelectronic π-systems and similar HOMO–LUMO gaps. Tetracene-2,3-dicarbaldehyde is soluble in a host of organic solvents (e.g., DMF, toluene, THF, chloroform, dichloromethane) and shows excellent photooxidative resistance in solution phases exposed to light and air. Further, it is readily sublimed from the solid-state without decomposition, and can be functionalized using different chemistries. We have demonstrated the utility of acene-2,3-dicarbaldehydes as reactants in the syntheses of novel α,α′-diaryl-2,3-acenedimethanols and acenotropones via Grignard reactions and double-aldol condensation reactions, respectively. The acenotropones were further reacted with concentrated H2SO4 to generate hydroxyacenotropylium ions that exhibit long wavelength absorption in the visible and near-IR regions. The optical gap measured for hydroxyanthracenotropylium ion is 1.3 eV. The results gained here implicate acene-2,3-dicarbaldehydes as potential high-value organic semiconductors and as precursors to a host of interesting molecules and materials.
 Qian Liu | Qian Liu received his B.E. in Pharmaceutical Engineering with honors from Changzhou University, China in July 2020. During his B.E. journey, he won 9 national awards including a China College Students' Entrepreneurship Competition Gold Award (2018), a National Competition of College Pharmaceutical Engineering Design Third Prize (2018, 2019), 3 provincial/municipal awards and 20+ school-level awards. He began his PhD studies in chemistry at the University of New Hampshire in August 2020. Unable to travel to the US during COVID-19 (July 2020 to August 2021), he worked as a senior chemistry teacher where he tutored 100+ students in grades 9 to 12. He began his in-person studies at the University of New Hampshire in August of 2021, and has since won 8 awards and scholarships recognizing his research and academic excellence. These include the Craig A. West Memorial Award for excellence in research and teaching (2024), the Charles Lathrop Parsons Vaughn Excellence in Chemistry Award (2023), the Mary Zoukis Papastavros '60 Chemistry Fellowship for research and academic excellence (2023), the UNH Bertram Husch Scholarship for international students (2022), and the Alvin R. Ingram Graduate Fellowship for excellence in academic performance and research (2022). Currently, he is a PhD candidate working on the design, synthesis and characterization of small molecule organic semiconductors in Prof. Glen P. Miller's research group. |
 Glen P. Miller | Glen P. Miller received his B.S. in chemistry in 1987 and his PhD in organic chemistry in 1990 at Clarkson University in Potsdam, New York. He received postdoctoral training at Exxon Research & Engineering Company in Annandale, New Jersey (1990–1992) before transitioning to Research Chemist (1992–1993) and Senior Chemist (1993–1995) at Exxon. He began his academic career at the University of New Hampshire in 1995 where he has held positions as Assistant Professor (1995–2001), Associate Professor (2001–2004), Professor (2004-present), Director of the Materials Research Program (2009–2015), Associate Director of the Nanoscale Science & Engineering Center for High-rate Nanomanufacturing (2004–2014) and Chair, Department of Chemistry (2015–2021). He is an authority in the area of nanostructured carbons including fullerenes, carbon nanotubes, graphene and large acenes. His group has successfully synthesized and characterized many nanostructures and organic compounds including C3v C60H18, bis and tris-[60]fullerene adducts of large acenes, hydrogenated SWNTs, fullerene nanowhiskers, photooxidatively resistant pentacene derivatives, exceptionally persistent heptacene and nonacene derivatives, and most recently several large starphenes. Miller's current research interests include the identification of design elements that enable the formation of organic semiconductors/semimetals with unusually small HOMO–LUMO gaps < 1 eV. |
1 Introduction
Select polycyclic aromatic hydrocarbons (PAHs) and their derivatives have been utilized as organic semiconductors in various electronic devices including organic field-effect transistors (OFETs),1–3 organic light-emitting diodes (OLEDs),4,5 organic photovoltaics (OPVs),6–8 organic photodetectors (OPDs)9–11 and organic sensors.12–15 Acenes such as pentacene have been considered for all of these applications owing to their small HOMO–LUMO gaps and their high charge carrier mobilities.Large acenes like pentacene are of interest because they exhibit relatively small HOMO–LUMO gaps and relatively high charge carrier mobilities. However, they suffer from several problems including (i) poor solubility in most solvents, and (ii) a propensity to photooxidize, especially when dissolved in solution phases exposed to light and air. With regards to (i), adequate solubility is important as it enables solution processing like spin coating, blade coating, spray coating, ink-jet printing, etc., for the construction of thin-film electronic devices.16,17 With regards to (ii), pentacene undergoes rapid photooxidation with a half-life of only 7.5 minutes18 in solution phases exposed to light and air. Solution processing is complicated by an organic semiconductor like pentacene. It is most desirable to utilize acenes that are both soluble and resistant to photooxidation.
The solubilities and photooxidative resistances of acenes can be improved through the judicious choice of substituents.18 One strategy to slow the photooxidation of acenes is to add electron-withdrawing substituents to their backbones thereby lowering the energy of their HOMOs and making them altogether less reactive with singlet oxygen. Halogenated acenes,16,17 organothio substituted acenes18–20 and TIPS substituted acenes17,21,22 are examples of derivatives with electron-withdrawing substituents that demonstrate enhanced photooxidative resistance. Depending upon the number and type of electron-withdrawing substituents included, the acene may switch from p-type to n-type.16,17
Aldehydes are well-known electron-withdrawing substituents and as such, they too should increase the photooxidative resistance of acenes. Aldehydes also react with a myriad of nucleophiles. Thus, it seems reasonable that aldehyde substituted acenes may show enhanced photooxidative resistance and also act as synthetic precursors for the synthesis of functional molecules and materials that include acene moieties. Indeed, o-phthalaldehyde maps directly onto larger acene-2,3-dicarbaldehydes like anthracene-2,3-dicarbaldehyde, tetracene-2,3-dicarbaldehyde and pentacene-2,3-dicarbaldehyde, and has proven useful for the synthesis of acene quinones,18,22 heteroarenes22,23 and isobenzoheteroles.24,25 The literature remains sparse, however, with regards to the synthesis, characterization and reactivity of naphthalene-2,3-dicarbaldehyde, anthracene-2,3-dicarbaldehyde, tetracene-2,3-dicarbaldehyde and pentacene-2,3-dicarbaldehyde (Fig. 1). Only one synthesis of tetracene-2,3-dicarbaldehyde with limited (i.e., UV-vis) characterization has been reported26 and there are no reported syntheses of pentacene-2,3-dicarbaldehyde.
 | ||
| Fig. 1 The number of references and reactions of acene-2,3-dicarbaldehydes reported on SciFinder up to and including May, 2024. | ||
A conventional synthetic plan for the formation of anthracene-2,3-dicarbaldehyde, 8, is illustrated in Scheme 1. Thus, Friedman demonstrated27 a sodium dichromate oxidation of 2,3-dimethylnapthalene to produce 2,3-naphthalenedicarboxylic acid in 87–93% yield. An analogous reaction starting with 2,3-dimethylanthracene, 1, should produce anthracene-2,3-dicarboxylic acid, 2. A borane-THF reduction of diacid 2 to anthracene-2,3-dimethanol 3 was demonstrated in near quantitative yield by Seo and co-workers.28 Alternatively, diethyl anthracene-2,3-dicarboxylate, 4, could be utilized. From naphthalene-2,3-dicarbaldehyde, 7, compound 4 was synthesized in two steps by Lin and co-workers29 and subsequently reduced to diol 3 in 77% yield using DIBAL. Wang and co-workers demonstrated separate mechanochemical IBX oxidations of naphthalene-2,3-dimethanol and 3 yielding naphthalene-2,3-dicarbaldehyde, 7, and anthracene-2,3-dicarbaldehyde, 8, respectively, in 81% yields.30
 | ||
| Scheme 1 Conventional methods to synthesize anthracene-2,3-dicarbaldehyde, 8, and by analogy tetracene-2,3-dicarbaldehyde, 9. | ||
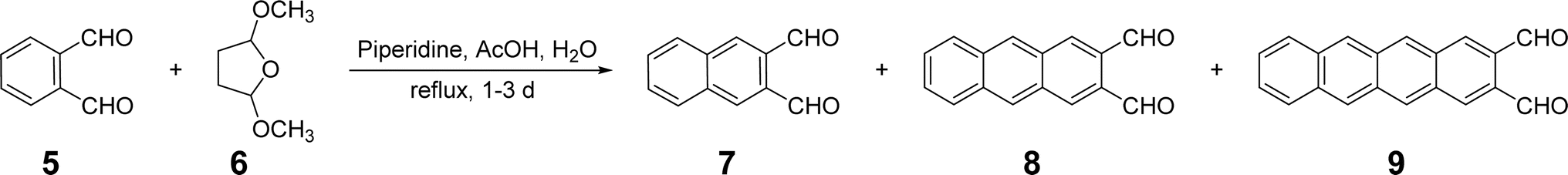
While a synthetic plan like that outlined in Scheme 1 seemingly could be modified for the synthesis of tetracene-2,3-dicarbaldehyde, 9, or larger acene-2,3-dicarbaldehydes, no such syntheses have been reported. Scheme 1 includes high-yielding reactions but it is nonetheless a multi-step synthesis utilizing starting materials that must either be synthesized in multiple steps or purchased at high cost. Alternatively, a method reported by Mallouli and Lepage26 for the synthesis of multiple acene-2,3-dicarbaldehydes utilizes a one-pot procedure involving only low cost, readily available starting materials: 5, 2,5-dimethoxytetrahydrofuran, 6, and piperidine (Scheme 2).
 | ||
| Scheme 2 A one-pot synthesis of acene-2,3-dicarbaldehydes demonstrated by Mallouli and Lepage26 leading to mixtures of naphthalene-2,3-dicarbaldehyde, 7, anthracene-2,3-dicarbaldehyde, 8, and tetracene-2,3-dicarbaldehyde, 9. | ||
Lin and co-workers properly noted a problem29 with the one-pot method of Mallouli and Lepage,26 namely a lack of selectivity. Thus, acene-2,3-dicarbaldehydes 7, 8 and 9 are all formed in the same pot with little selectivity. Of these, 9 is the most interesting as a potential organic semiconductor and likewise, a similar method to prepare it in good yield with good selectivity is desirable.
In this study, we modified the procedure of Mallouli and Lepage26 such that acene-2,3-dicarbaldehydes including anthracene-2,3-dicarbaldehyde, 8, and tetracene-2,3-dicarbaldehyde, 9, were prepared in higher combined yield. We also report conditions where 9 can be prepared with vastly improved selectivity. Our work shines a spotlight on tetracene-2,3-dicarbaldehyde, 9, which has avoided attention and a detailed characterization, until now. It has a π-system that is isoelectronic with pentacene and a similar HOMO–LUMO gap, but with far more agreeable properties. Dicarbaldehyde 9 is soluble in a host of organic solvents (e.g., DMF, toluene, THF, chloroform, dichloromethane) and shows excellent photooxidative resistance in solution phases exposed to light and air. Further, the dicarbaldehydes described here can be sublimed from the solid-state without decomposition, and can be functionalized using different chemistries to produce novel structures with interesting properties.
2 Results and discussion
2.1 A plausible reaction sequence for the one-pot synthesis of acene-2,3-dicarbaldehydes
The one-pot synthesis of acene-2,3-dicarbaldehydes demonstrated by Mallouli and Lepage26 is illustrated in Scheme 2. Thus, 5 and 6 are heated in aqueous acetic acid in the presence of a few drops of piperidine. All three acene-2,3-dicarbaldehydes (7, 8 and 9) were formed but with little selectivity and without thorough characterization.A plausible reaction sequence for this multi-stage, one-pot synthesis is proposed in Scheme 3. First, 6 can be converted reversibly to succinaldehyde under the acidic reaction conditions employed (step 1). Succinaldehyde can react reversibly with piperidine to form a conjugated 1,3-diene-1,4-diamine with nucleophilic α-carbons (step 2). Simultaneously, 5 can react reversibly with piperidine to produce an iminium ion intermediate with highly electrophilic carbons (step 3). The products of steps 2 and 3 can undergo reversible polar additions to form an intermediate which, upon irreversible elimination of multiple piperidine equivalents, yields 7 (step 4). In iterative fashion, 7, and eventually 8, can react with 6 and piperidine to form 8 and 9, respectively (step 5). The reaction essentially stops at the tetracene-2,3-dicarbaldehyde stage due to the poor solubility of 9 in the reaction medium (vide infra). Scheme 3 represents a plausible reaction sequence, not a detailed mechanism. For example, it is not necessary or even likely that both iminium ions generated from 5 form at the same time as in step 3. Likewise, it is not necessary that both enamines form in step 2 before reaction with an iminium ion, and so on.
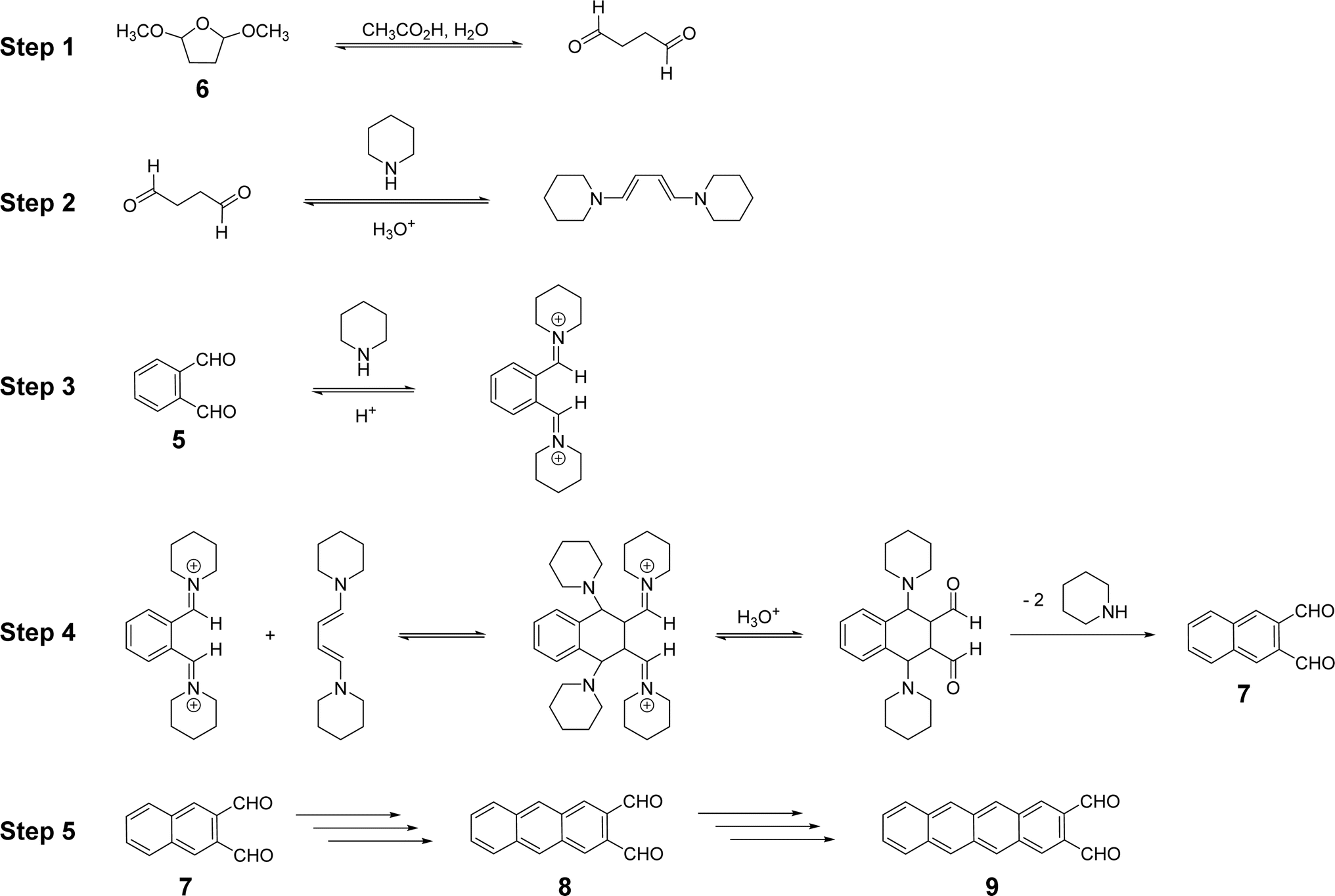 | ||
| Scheme 3 A plausible, iterative reaction sequence leading to naphthalene-2,3-dicarbaldehyde, 7, and by analogy, anthracene-2,3-dicarbaldehyde, 8, and tetracene-2,3-dicarbaldehyde, 9. | ||
2.2 An improved one-pot synthesis of acene-2,3-dicarbaldehydes
In experiments 1–3 of Table 1, we utilized essentially the same reaction conditions as employed by Mallouli and Lepage.26 Upon considering Scheme 3, we modified these conditions to increase the relative concentrations of 6 and piperidine, and by allowing for longer reaction times (experiments 4 and 5 in Table 1). In doing so, we observed the formation of 8 and 9 in greater combined yield (experiments 4 and 5) and found conditions leading to the highly selective formation of 9 (experiment 5). The conditions of experiment 5 in Table 1 lead to 9 in 48% yield, with only traces of other aceno-2,3-dicarbaldehydes present. Thus, according to the proposed reaction pathway of Scheme 3, increasing the concentrations of 6 and piperidine should lead to larger equilibrium concentrations of enamine and iminium ion reactants (steps 3 and 4 of Scheme 3). Longer reaction times (experiments 4 and 5 of Table 1) allow for iterative growth of acene-2,3-dicarbaldehydes up to but not beyond 9. Pentacene-2,3-dicarbaldehyde and larger acene-2,3-dicarbaldehydes were not observed to form, likely due to the poor solubility of 9 in the reaction medium. The irreversible elimination of piperidine (step 4 of Scheme 3) combined with the poor solubility of 9 in the reaction medium enables the highly selective formation and accumulation of 9, nearly free of other aceno-2,3-dicarbaldehydes.
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Exp. | 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6a :6a |
Piperidine | AcOH | H2O | Time/h | 7 | 8 | 9 |
| a Two grams (15 mmol) of 5 were utilized in each experiment. The ratios of 5:6 are molar ratios. All product percent yields refer to isolated product, after purification. |
||||||||
| 1 | 1:1 |
2 drops | 1 mL | 1.5 mL | 18 | 30% | 9% | — |
| 2 | 1:2 |
3 drops | 1.5 mL | 1.5 mL | 24 | 17% | 25% | — |
| 3 | 1:4 |
4 drops | 3 mL | 1 mL | 24 | 10% | 15% | 21% |
| 4 | 1:6 |
5 drops | 4 mL | 1 mL | 24 | 7% | 20% | 25% |
| 5 | 1:8 |
5 drops | 4 mL | 1 mL | 72 | — | Trace | 48% |
In addition to modifying the reactions conditions to produce 8 and 9 in higher combined yield, we found overall improved yields of acene-2,3-dicarbaldehydes upon modifying the work-up procedure reported by Mallouli and Lepage.26 Specifically, we noted product losses during vacuum filtration when using methanol or ether as wash solvents. To address this, we collected the filtrate suspension and subjected it to a second vacuum filtration to obtain a second-batch of filtered solid product. First-batch and second-batch filtered solids were subsequently sublimed at reduced pressures. Additional details are provided in the ESI.†
Acene-2,3-dicarbaldehydes 7, 8, and 9 are all robust molecules, even when subjected to elevated temperatures for prolonged times. As such, they may be separated from one another via high-temperature vacuum sublimations. We observe 132 °C/0.1 Torr/4–6 hours to be optimal conditions for the sublimation of 7, 180 °C/0.1 Torr/4–6 hours to be optimal conditions for the sublimation of 8, and 220 °C/0.1 Torr/4–12 hours to be optimal conditions for the sublimation of 9. It should also be noted that acene-2,3-dicarbaldehydes 5, 7, 8 and 9 are soluble in a variety of organic solvents (e.g., DMF, toluene, THF, chloroform and dichloromethane) and all show indefinite stability in solution phases exposed to light and air.
2.3 Optical properties of acene-2,3-dicarbaldehydes
The optical properties of acene-2,3-dicarbaldehydes 5, 7, 8 and 9 are illustrated in Fig. 2. In the solid state, the color of 5, 7, 8 and 9 graduate from pale yellow to deep orange/red (Fig. 2a). Solutions of acene-2,3-dicarbaldehydes 5 and 7 are essentially colorless under room light while those of 8 and 9 are golden yellow and orange/red, respectively (Fig. 2b). Upon irradiation with 365 nm UV light, 5, 7, 8 and 9 fluoresce leaving solutions that are purple, sky blue, golden yellow and orange/red in color (Fig. 2c). UV-vis spectra indicate that 5, 7, 8 and 9 have broad absorptions in the range of 250–550 nm with λmax values at 300, 340, 410 and 503 nm, respectively (Fig. 2d). The red-shifting λmax values for these molecules provide strong evidence for progressively smaller HOMO–LUMO gaps upon moving from 5 to 7 to 8 to 9. Fluorescence spectra show emissions in the range of 250–700 nm with maximum λem values of 315, 363, 455 and 572 nm for 5, 7, 8 and 9, respectively (Fig. 2e). Notably, 7 exhibits the most intensive fluorescence emissions. | ||
| Fig. 2 Acene-2,3-dicarbaldehydes: (a) in the solid state; (b) 1 × 10−3 M solutions in CH2Cl2 under room light; (c) 1 × 10−3 M solutions in CH2Cl2 irradiated with 365 nm UV light; (d) UV-vis spectra of 1 × 10−4 M solutions in CH2Cl2; (e) fluorescence spectra of 2 × 10−6 M solutions in CH2Cl2. | ||
2.4 DFT calculations of acene-2,3-dicarbaldehydes
To gain further insight into the structure and electronic properties of acene-2,3-dicarbaldehydes, time-dependence DFT (TD-DFT) calculations were performed at the B3LYP-D3(BJ)/6-311+G(d,p)/SMD(DCM)//B3LYP-D3(BJ)/6-31G(d)/SMD(DCM) level of theory using the Gaussian 09 program.31 Frontier orbital energy levels, HOMO–LUMO gaps, orbital distributions and UV-Vis spectra are shown in Fig. 3. The HOMO and LUMO orbital densities for acene-2,3-dicarbaldehydes 5, 7–9 and pentacene-2,3-dicarbaldehyde 10 are spread throughout each molecule including their terminal aldehyde groups (Fig. 3, top), consistent with highly conjugated, highly delocalized π-systems. Similar to unsubstituted acenes, as the number of atoms-in-conjugation grows, LUMO energies decrease slightly while HOMO energies show a modest rise. The result is a significant lowering of the HOMO–LUMO gaps from 4.27 eV to 1.99 eV (Fig. 3, top) and progressively longer wavelengths of absorptions (Fig. 3, bottom) as one progresses from 5 to 7 to 8 to 9 to 10. This trend is in good agreement with experimental results as shown in Table 2 where a detailed collection of optical and DFT calculated properties for acene-2,3-dicarbaldehydes is assembled. | ||
| Fig. 3 Calculated HOMO and LUMO orbitals (top) and UV-vis spectra (bottom) for acene-2,3-dicarbaldehydes 5, 7, 8, 9 and 10. Solvent model (dichloromethane) TD-DFT calculations were performed at the B3LYP-D3(BJ)/6-311+G(d,p)/SMD(DCM)//B3LYP-D3(BJ)/6-31G(d)/SMD(DCM) level of theory using the Gaussian 09 program. | ||
| Compd | λAbs(DFT)max/nm | λAbsmax/nm | λEmmax/nm | λonset/nm | Eoptg/eV | EDFTLUMO/eV | EDFTHOMO/eV | EDFTg/eV |
|---|---|---|---|---|---|---|---|---|
| 5 | 284 | 300 | 315 | 324 | 3.8 | −2.75 | −7.02 | 4.27 |
| 7 | 355 | 340 | 363 | 349 | 3.6 | −2.54 | −6.58 | 4.04 |
| 8 | 407 | 410 | 455 | 437 | 2.8 | −2.63 | −5.93 | 3.30 |
| 9 | 455 | 503 | 572 | 533 | 2.3 | −2.90 | −5.48 | 2.59 |
| 10 | 494 | — | — | — | — | −3.13 | −5.12 | 1.99 |
UV-vis spectra were recorded at 1 × 10−4 M in CH2Cl2 solution; fluorescence spectra were recorded at 2 × 10−6 M in CH2Cl2 solution; optical energy gaps were determined from the onset wavelengths (λonset) associated with the lowest-energy absorption bands; the onset is defined as the intersection between the baseline and a tangent line that touches the point of inflection; solvent model (dichloromethane) TD-DFT calculations were performed at the B3LYP-D3(BJ)/6-311+G(d,p)/SMD(DCM)//B3LYP-D3(BJ)/6-31G(d)/SMD(DCM) level of theory using the Gaussian 09 program.
As mentioned above, 5, 7, 8 and 9, each with a pair of electron-withdrawing aldehyde substituents, show excellent stability in the solid-state and in solution phases exposed to light and air. Large acenes (i.e., tetracene and larger) are known to sensitize singlet oxygen, 1O2, formation. This highly reactive oxygen species can then undergo a [4 + 2] cycloaddition across an embedded diene of the large acene.18 Electron-withdrawing substituents that lower HOMO energies can create a larger HOMOacene–LUMOoxygen energy difference, thereby slowing the rate of concerted cycloaddition. A comparison of calculated HOMO energies for acene-2,3-dicarbaldehydes and their corresponding unsubstituted acenes (Fig. 4) is informative. Thus, the HOMO energies associated with acene-2,3-dicarbaldehydes with two or more rings are lower by 0.2–0.4 eV than the corresponding unsubstituted acenes (Fig. 4) and this likely accounts for their reduced reactivity with singlet oxygen.
 | ||
| Fig. 4 HOMO energies for both acene-2,3-dicarbaldehydes (blue data points, blue line) and the corresponding unsubstituted acenes (red data points, red line) plotted against the number of 6-membered rings in each molecule. Solvent model (dichloromethane) TD-DFT calculations were performed at the B3LYP-D3(BJ)/6-311+G(d,p)/SMD(DCM)//B3LYP-D3(BJ)/6-31G(d)/SMD(DCM) level of theory using the Gaussian 09 program. | ||
It is worth noting that even with a modest lowering of their HOMO energies and improved photooxidative resistances, 9 and 10 both possess HOMO energies (Table 2, Fig. 2 and 3) that are consistent with some of the best known p-type organic semiconductors.32 Therefore, while it benefits from enhanced solubility and photooxidative resistance, we do not expect a switch from p-type to n-type organic semiconductor behavior for 9.
2.5 Further reactions of acene-2,3-dicarbaldehydes
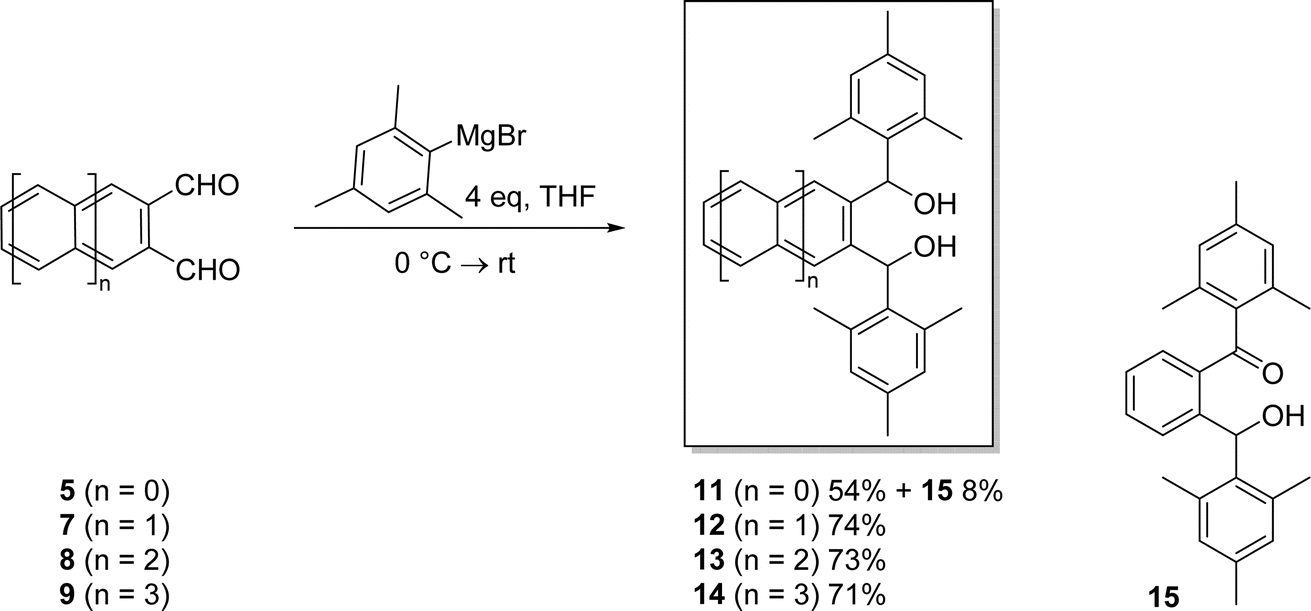 | ||
| Scheme 4 Synthesis of novel α,α′-dimesityl-2,3-acenedimethanols 11–14. | ||
Thus, diol 11 was formed in 54% yield by reacting 5 with mesityl magnesium bromide at 0 °C followed by stirring at room temperature for 5 hours. The ketone-alcohol by-product 15 was also formed in 8% yield, presumably via air oxidation of initially formed 11. Acene-2,3-dicarbaldehydes 7–9 were also reacted with mesityl magnesium bromide at 0 °C followed by stirring at 50 °C for 2 hours and then stirring at room temperature for 24 hours. In this way, novel α,α′-dimesityl-2,3-acenedimethanols 12–14 were obtained in good yields ranging from 71 to 74%. Ketone-alcohols akin to 15 were not observed in these reactions. Although acene-2,3-dicarbaldehydes are all soluble in a host of organic solvents, α,α′-dimesityl-2,3-acenedimethanols 11–14 are highly soluble in many of the same solvents. Their excellent solubilities are likely due to the conformational requirement that the terminal mesityl groups rotate out of the plane, thus reducing intermolecular π–π stacking that can otherwise promote agglomeration and precipitation.
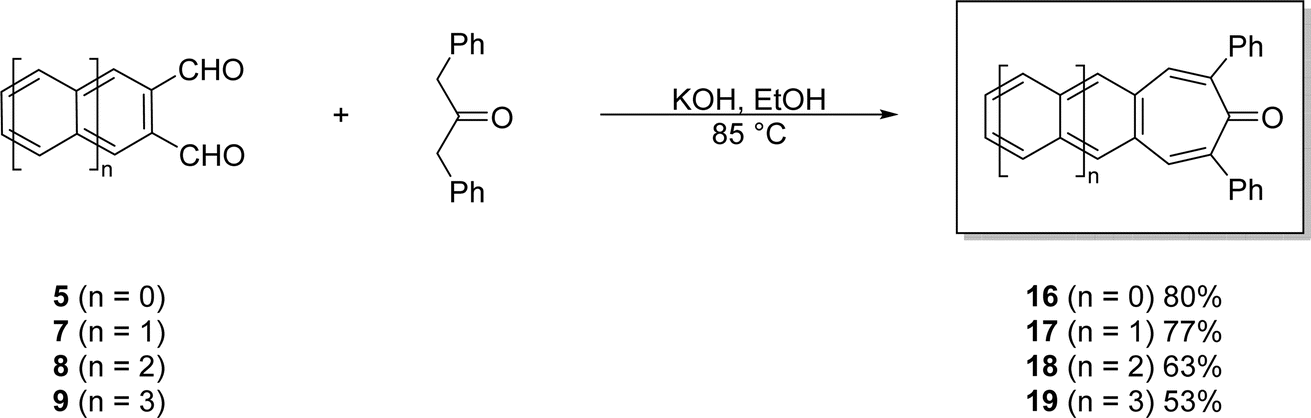 | ||
| Scheme 5 Efficient, one-pot syntheses of acenotropones 16–19 via double-aldol condensations of acene-2,3-dicarbaldehydes 5 and 7–9. | ||
These base-mediated reactions afford the corresponding 2,7-diphenyl-4,5-acenotropones 16–19 in yields ranging from 53 to 80%. Combined with a simple work up (vacuum filtration followed by solvent washes), the reactions are scalable. The color of the acenotropones deepen from yellow to red with increasing acene length. Each of the 2,7-diphenyl-4,5-acenotropones, 16–19, are stable in the solid-state. However, unlike its precursor 9, tetracenotropone 19 does photooxidize if left in solution phases exposed to light and air.
We were compelled to react acenotropones 16–18 with concentrated H2SO4 in order to generate the corresponding hydroxyacenotropylium ions 20–22 (Fig. 5). Acenotropylium ions are highly interesting, unexplored molecules with potential applications as n-type organic semiconductors in organic electronics. While 1 × 10−4 M solutions of acenotropones 16–18 in CH2Cl2 are either colorless (16 and 17) or yellow (18), the corresponding hydroxyacenotropylium ions generated in concentrated H2SO4, 20–22, are yellow (20), red (21) and green (22), respectively (Fig. 5a). The UV-vis spectra for acenotropones 16–18 in CH2Cl2 solution (Fig. 5b) show absorptions in the range of 250–500 nm (λmax values at 284, 306 and 402 nm, respectively) while those for the corresponding hydroxyacenotropylium ions 20–22 (Fig. 5c) are significantly red-shifted (λmax values of 506, 524 and 978 nm, respectively). It is noteworthy that hydroxyanthracenotropylium ion 22 shows weak but dramatically red-shifted absorptions in the near-IR region with λmax values of 860 and 978 nm (Fig. 5d). The optical gap measured for 22 is a mere 1.3 eV, placing it in the rare category of organic semiconductors with optical Eg values below 1.5 eV. For comparison, we observe that unsubstituted tropone has a λmax value of 298 nm in CH2Cl2 (Fig. 5b, purple line) while the hydroxytropylium ion prepared in concentrated H2SO4 (Fig. 5c, purple line) shows a slight red shift (λmax = 304 nm), consistent with a literature report.46
 | ||
| Fig. 5 (a) 1 × 10−4 M solutions of acenotropones 16–18 in CH2Cl2 solution and 1 × 10−4 M solutions of hydroxyacenotropylium ions 20–22 in conc. H2SO4; (b) UV-vis spectra of tropone and acenotropones 16–18 in CH2Cl2 solution; (c) UV-vis spectra of hydroxytropylium ion and hydroxyacenotropylium ions 20–22 in conc. H2SO4; (d) UV-vis spectra of hydroxyanthracenotropylium ion 22 at several different concentrations in conc. H2SO4. | ||
To further investigate the structure and electronic properties of acenotropones 16–19 and hydroxyacenotropylium ions 20–23, gas phase time-dependence DFT (TD-DFT) calculations were performed at the B3LYP/6-311+G(d,p)//B3LYP/6-31G(d) level using the Gaussian 09 program.31 Frontier orbital energy levels, HOMO–LUMO gaps, orbital distributions and UV-Vis spectra were all calculated (Fig. 6). HOMO and LUMO orbital densities for acenotropones 16–19 and hydroxyacenotropylium ions 20–23 are spread throughout each molecule (Fig. 6a and c), consistent with highly conjugated, highly delocalized π-systems. Interestingly, the phenyl substituents show progressively less HOMO and LUMO orbital densities with increasing acene size (Fig. 6a and c) despite similar degrees of rotation out of plane. Similar to the acene-2,3-dicarbaldehydes of Fig. 3, a significant lowering of HOMO–LUMO gaps is observed for both acenotropones 16–19 (3.98 eV for 16; 2.34 eV for 19) and hydroxyacenotropylium ions 20–23 (3.09 eV for 20; 1.45 eV for 23) as the number of rings-in-conjugation grows (Fig. 6a and c). Likewise, the calculated wavelengths of absorption increase as the number of rings-in-conjugation grows (Fig. 6b and d). The shortest calculated HOMO–LUMO gaps and the longest calculated wavelengths of absorption are observed for the hydroxyacenotropylium ions (Fig. 6b and d).
 | ||
| Fig. 6 Calculated HOMO and LUMO orbitals and UV-Vis spectra for acenotropones 16–19 (a and b) and hydroxyacenotropylium ions 20–23 (c and d). Gas phase TD-DFT calculations were performed at the B3LYP/6-311+G(d,p)//B3LYP/6-31G(d) level of theory using the Gaussian 09 program. | ||
A detailed collection of optical and DFT calculated properties for acenotropones 16–19 and hydroxyacenotropylium ions 20–23 is assembled in Table 3. DFT calculated HOMO–LUMO gaps for the neutral acenotropones 16–18 are in good agreement with experimental results. However, the DFT calculated HOMO–LUMO gaps for the charged hydroxyacenotropylium ions 20–22 are roughly 0.7 eV higher than those measured experimentally. The discrepancy may be due, in part, to the gas phase calculations employed for the charged hydroxyacenotropylium ions 20–22 (Fig. 6 and Table 3). In the future, we plan to compare and contrast calculated structures and electronics associated with (i) gas phase, (ii) polar solvent phase and (iii) solid-state structures containing multiple hydroxyacenotropylium salts.
| Compd | λAbs(DFT)max/nm | λAbsmax/nm | λonset/nm | Eoptg/eV | EDFTLUMO/eV | EDFTHOMO/eV | EDFTg/eV |
|---|---|---|---|---|---|---|---|
| a UV-Vis spectra of acenotropones were recorded at 1 × 10−5 M in CH2Cl2 while hydroxyacenotropylium ions 20 and 21 were recorded at 1 × 10−5 M in conc. H2SO4.b The UV-vis spectrum of 22 was recorded at 1 × 10−3 M in conc. H2SO4. Optical energy gaps were determined from the onset of the lowest-energy absorption band (λonset), the onset is defined as the intersection between the baseline and a tangent line that touches the point of inflection. Gas phase TD-DFT calculations were performed at the B3LYP/6-311+G(d,p)//B3LYP/6-31G(d) level of theory using the Gaussian 09 program. | |||||||
| 16 | 320, 248 | 284 | 322 | 3.9 | −2.27 | −6.25 | 3.98 |
| 17 | 319, 275 | 306 | 344 | 3.6 | −2.41 | −6.02 | 3.61 |
| 18 | 360, 310 | 402, 332 | 458 | 2.7 | −2.81 | −5.70 | 2.89 |
| 19 | 385, 334 | — | — | — | −3.05 | −5.39 | 2.34 |
| 20 | 386, 266 | 506, 288 | 546 | 2.3 | −6.60 | −9.69 | 3.09 |
| 21 | 311, 243 | 524, 336 | 650 | 1.9 | −6.57 | −9.24 | 2.67 |
| 22b | 358, 276 | 978, 860, 584, 338 | 982 | 1.3 | −6.51 | −8.52 | 2.01 |
| 23 | 403, 300 | — | — | — | −6.46 | −7.91 | 1.45 |
3 Conclusions
A series of acene-2,3-dicarbaldehydes including anthracene-2,3-dicarbaldehyde, 8, and tetracene-2,3-dicarbaldehyde, 9, have been synthesized, purified and characterized using an improved one-pot synthetic method. The dicarbaldehydes are all fluorophores that show good solubility and excellent photooxidative resistance in solution phases exposed to light and air. Experimental and computational (DFT) results show that acene-2,3-dicarbaldehydes possess red-shifted UV-vis absorptions and progressively smaller HOMO–LUMO gaps as the number of rings-in-conjugation grows. Dicarbaldehydes 8 and 9 possess optical HOMO–LUMO gaps of 2.8 and 2.3 eV, respectively. Dicarbaldehyde 9 and the benchmark organic semiconductor pentacene have isoelectronic π-systems and similar HOMO–LUMO gaps. Dicarbaldehyde 9 is soluble in a host of organic solvents (e.g., DMF, toluene, THF, chloroform, dichloromethane) and is readily sublimed from the solid-state at 220 °C without decomposition. We demonstrated the synthetic utility of acene-2,3-dicarbaldehydes by reacting them with mesityl magnesium bromide to form novel α,α′-dimesityl-2,3-acenedimethanols in 54–74% yield, and by reacting them with 1,3-diphenylacetone in one-pot, double-aldol condensation reactions to form acenotropones in 53–80% yield. The acenotropones were further reacted with concentrated H2SO4 to generate hydroxyacenotropylium ions that show long wavelength absorptions in the visible and near-IR regions. The optical gap measured for hydroxyanthracenotropylium ion, 22, is a mere 1.3 eV.Data availability
ESI† is available and includes experimental data, 1H NMR spectra, 13C NMR spectra and high-resolution mass spectra (HRMS) for key compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the University of New Hampshire.References
- J. T. E. Quinn, J. Zhu, X. Li, J. Wang and Y. Li, J. Mater. Chem. C, 2017, 5, 8654–8681 RSC.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- C. Zheng, T. Tong, Y. Hu, Y. Gu, H. Wu, D. Wu, H. Meng, M. Yi, J. Ma, D. Gao and W. Huang, Small, 2018, 14, 1800756 CrossRef PubMed.
- T. Yu, L. Liu, Z. Xie and Y. Ma, Sci. China: Chem., 2015, 58, 907–915 CrossRef CAS.
- X.-H. Zhu, J. Peng, Y. Cao and J. Roncali, Chem. Soc. Rev., 2011, 40, 3509–3524 RSC.
- B. Kan, Y. Kan, L. Zuo, X. Shi and K. Gao, InfoMat, 2021, 3, 175–200 CrossRef CAS.
- A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS PubMed.
- O. Inganäs, Adv. Mater., 2018, 30, 1800388 CrossRef PubMed.
- J. Miao and F. Zhang, Laser Photonics Rev., 2019, 13, 1800204 CrossRef.
- D. Yang and D. Ma, Adv. Opt. Mater., 2019, 7, 1800522 CrossRef.
- H. Ren, J.-D. Chen, Y.-Q. Li and J.-X. Tang, Adv. Sci., 2021, 8, 2002418 CrossRef CAS PubMed.
- L. Torsi, M. Magliulo, K. Manoli and G. Palazzo, Chem. Soc. Rev., 2013, 42, 8612–8628 RSC.
- M. Berggren and A. Richter-Dahlfors, Adv. Mater., 2007, 19, 3201–3213 CrossRef CAS.
- T. Someya, Z. Bao and G. G. Malliaras, Nature, 2016, 540, 379–385 CrossRef CAS PubMed.
- D. T. Simon, E. O. Gabrielsson, K. Tybrandt and M. Berggren, Chem. Rev., 2016, 116, 13009–13041 CrossRef CAS PubMed.
- M. L. Tang and Z. Bao, Chem. Mater., 2011, 23, 446–455 CrossRef CAS.
- A. N. Lakshminarayana, A. Ong and C. Chi, J. Mater. Chem. C, 2018, 6, 3551–3563 RSC.
- I. Kaur, W. Jia, R. P. Kopreski, S. Selvarasah, M. R. Dokmeci, C. Pramanik, N. E. McGruer and G. P. Miller, J. Am. Chem. Soc., 2008, 130, 16274–16286 CrossRef CAS PubMed.
- I. Kaur, N. N. Stein, R. P. Kopreski and G. P. Miller, J. Am. Chem. Soc., 2009, 131, 3424–3425 CrossRef CAS PubMed.
- I. Kaur, M. Jazdzyk, N. N. Stein, P. Prusevich and G. P. Miller, J. Am. Chem. Soc., 2010, 132, 1261–1263 CrossRef CAS PubMed.
- O. Lobanova Griffith, N. E. Gruhn, J. E. Anthony, B. Purushothaman and D. L. Lichtenberger, J. Phys. Chem. C, 2008, 112, 20518–20524 CrossRef CAS.
- M. Müller, L. Ahrens, V. Brosius, J. Freudenberg and U. H. F. Bunz, J. Mater. Chem. C, 2019, 7, 14011–14034 RSC.
- J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS PubMed.
- H. Tozawa, K. Kitamura and T. Hamura, Chem. Lett., 2017, 46, 703–706 CrossRef CAS.
- T. Hamura, in Middle Molecular Strategy: Flow Synthesis to Functional Molecules, ed. K. Fukase and T. Doi, Springer, Singapore, 2021, pp. 203–223 Search PubMed.
- A. Mallouli and Y. Lepage, Synthesis, 1980, 1980, 689 CrossRef.
- L. Friedman, Org. Synth., 1963, 43, 80 CrossRef CAS.
- U. R. Seo, Y. K. Chung and C. Lee, ChemCatChem, 2016, 8, 1051–1054 CrossRef CAS.
- C.-H. Lin, K.-H. Lin, B. Pal and L.-D. Tsou, Chem. Commun., 2009, 803–805 RSC.
- C. Wang, C. Yue, A. Smith and J. Mack, J. Organomet. Chem., 2022, 976, 122430 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09 (Revision E.01), Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- H. Dong, C. Wang and W. Hu, Chem. Commun., 2010, 46, 5211–5222 RSC.
- S. Matsuoka, S. Jung, K. Miyakawa, Y. Chuda, R. Sugimoto and T. Hamura, Chem.–Eur. J., 2018, 24, 18886–18889 CrossRef CAS PubMed.
- Q. Liu and G. P. Miller, Beilstein J. Org. Chem., 2024, 20, 1099–1110 CrossRef CAS PubMed.
- K. Kitamura, R. Kudo, H. Sugiyama, H. Uekusa and T. Hamura, Chem. Commun., 2020, 56, 14988–14991 RSC.
- R. Sivasakthikumaran, S. M. Rafiq, E. Sankar, J. A. Clement and A. K. Mohanakrishnan, Eur. J. Org. Chem., 2015, 2015, 7816–7835 CrossRef CAS.
- B. J. Herold, A. F. N. Correia and J. D. S. Veiga, J. Am. Chem. Soc., 1965, 87, 2661–2665 CrossRef CAS.
- E. Van Der Drift, A. J. Dammers, J. Smidt, M. Plato and K. Möbius, J. Magn. Reson., 1980, 40, 551–579 CAS.
- J. Yang, MS thesis, University of New Hampshire, 2015.
- T. Kodama, Y. Kawashima, K. Uchida, Z. Deng and M. Tobisu, J. Org. Chem., 2021, 86, 13800–13807 CrossRef CAS PubMed.
- D. Meuche, H. Strauss and E. Heibronner, Helv. Chim. Acta, 1958, 41, 2220–2229 CrossRef CAS.
- J. Thiele and J. Schneider, Adv. Cycloaddit., 1909, 369, 287–299 CAS.
- M. Kudoh, T. Satoh, H. Ikeda, T. Nakazawa, T. Miyashi, S. Katagiri and S. Sudoh, Bull. Chem. Soc. Jpn., 2009, 82, 70–75 CrossRef CAS.
- W. Ried and H. J. Schwenecke, Chem. Ber., 1958, 91, 566–572 CrossRef CAS.
- D. L. Crossley, C. D. Gabbutt, B. Mark Heron, P. Kay and M. Mogstad, Dyes Pigm., 2012, 95, 62–68 CrossRef CAS.
- S. H. Gill and K. M. Harmon, J. Mol. Struct., 2000, 525, 113–122 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04273e |
| This journal is © The Royal Society of Chemistry 2024 |