
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and evaluation of sulfadiazine derivatives as potent dual inhibitors of EGFRWT and EGFRT790M: integrating biological, molecular docking, and ADMET analysis†
Hany M. Abd El-Lateef‡
*ab,
Hend A. A. Ezelarab‡ *c,
Ali M. Alib,
Azhaar T. Alsaggafd,
Wael A. Mahdie,
Sultan Alshehrie,
Mohamed A. El Hamd*fg and
Moustafa O. Aboelez*h
*c,
Ali M. Alib,
Azhaar T. Alsaggafd,
Wael A. Mahdie,
Sultan Alshehrie,
Mohamed A. El Hamd*fg and
Moustafa O. Aboelez*h
aDepartment of Chemistry, College of Science, King Faisal University, Al-Ahsa 31982, Saudi Arabia. E-mail: hmahmed@kfu.edu.sa
bDepartment of Chemistry, Faculty of Science, Sohag 82524, Egypt
cDepartment of Medicinal Chemistry, Faculty of Pharmacy, Minia University, 61519-Minia, Egypt. E-mail: hend_aly@minia.edu.eg
dDepartment of Chemistry, Taibah University, Madinah, Saudi Arabia
eDepartment of Pharmaceutics, College of Pharmacy, King Saud University, Riyadh 11451, Saudi Arabia
fDepartment of Pharmaceutical Chemistry, College of Pharmacy, Shaqra University, Shaqra 11961, Saudi Arabia. E-mail: aboelhamdmohamed@su.edu.sa
gDepartment of Pharmaceutical Analytical Chemistry, Faculty of Pharmacy, South Valley University, Qena 83523, Egypt
hDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Sohag University, Sohag, Egypt. E-mail: drmoustafaaboelez@yahoo.com
First published on 6th September 2024
Abstract
A series of derivatives (5–14) were synthesized through the diazotization of sulfadiazine with active methylene compounds. The chemical structures of these newly designed compounds were validated through spectral and elemental analysis techniques. The antiproliferative potential of derivatives 5–14 was assessed against three distinct cancer cell lines (A431, A549, and H1975) using the MTT assay. The results revealed that compounds 8, 12, and 14 exhibited the most potent antiproliferative activity, with IC50 values ranging from 2.31 to 7.56 μM. Notably, these values were significantly lower than those of known EGFR inhibitors, including erlotinib, gefitinib, and osimertinib, suggesting the potential of these derivatives as novel antiproliferative agents. Furthermore, compound 12 was identified as the most potent inhibitor of both EGFRWT and EGFRT790M protein kinases, with IC50 values of 14.5 and 35.4 nM, respectively. These results outperformed those of gefitinib and osimertinib, which exhibited IC50 values of 18.2 and 368.2 nM, and 57.8 and 8.5 nM, respectively. Molecular docking studies of compounds 8, 12, and 14 within the ATP-binding sites of both EGFRWT and EGFRT790M corroborated the in vitro results when compared to erlotinib, gefitinib, and osimertinib. The docking results indicated that compound 8 exhibited a favorable binding affinity for both EGFRWT and EGFRT790M, with binding scores of −6.40 kcal mol−1 and −7.53 kcal mol−1, respectively, which were comparable to those of gefitinib and osimertinib, with binding scores of −8.01 and −8.72 kcal mol−1, respectively. Furthermore, an assessment of the most promising EGFR inhibitors (8, 12, and 14) using the egg-boiled method for their in silico ADME properties revealed significant lipophilicity, blood–brain barrier (BBB) penetration, and gastrointestinal (GIT) absorption. Collectively, our designed analogs, particularly compounds 8, 12, and 14, exhibit promising dual antiproliferative and EGFRWT and EGFRT790M kinase inhibitory properties, positioning them as efficient candidates for further therapeutic development.
1. Introduction
Molecular hybridization is a pivotal strategy in drug design,1–5 involving the deliberate fusion of two or more biologically relevant pharmacophores, often bridged by a spacer or linker, to yield a novel hybrid entity with significantly enhanced activity and efficacy compared to the parent molecule.6–9 This strategy can also decrease undesirable side effects, thereby improving the overall therapeutic index.10–13 Moreover, recent investigations have revealed that sulfonamides possess potent growth inhibitory activity against a diverse range of tumor lines and tyrosine kinase (TK) enzymes, underscoring their potential as promising anticancer agents.The utilization of pyrimidines as a scaffold for producing new EGFR inhibitors that target multiple pathways, particularly oncogenesis, has been correlated with the significant anticancer properties of sulfonamides. These compounds have been shown to exhibit effective activity across a multitude of pathways, including the inhibition of carbonic anhydrase, matrix metalloproteinase, NADPH reductase, histone deacetylase, and PI3K. Notably, compounds I–III demonstrated more pronounced activities against both HER2 and EGFR compared to erlotinib, with concomitant increases in Bcl-2, Bax, and caspase-3 expression levels and the induction of cell cycle arrest in the G2/M phase12–16 (Fig. 1).
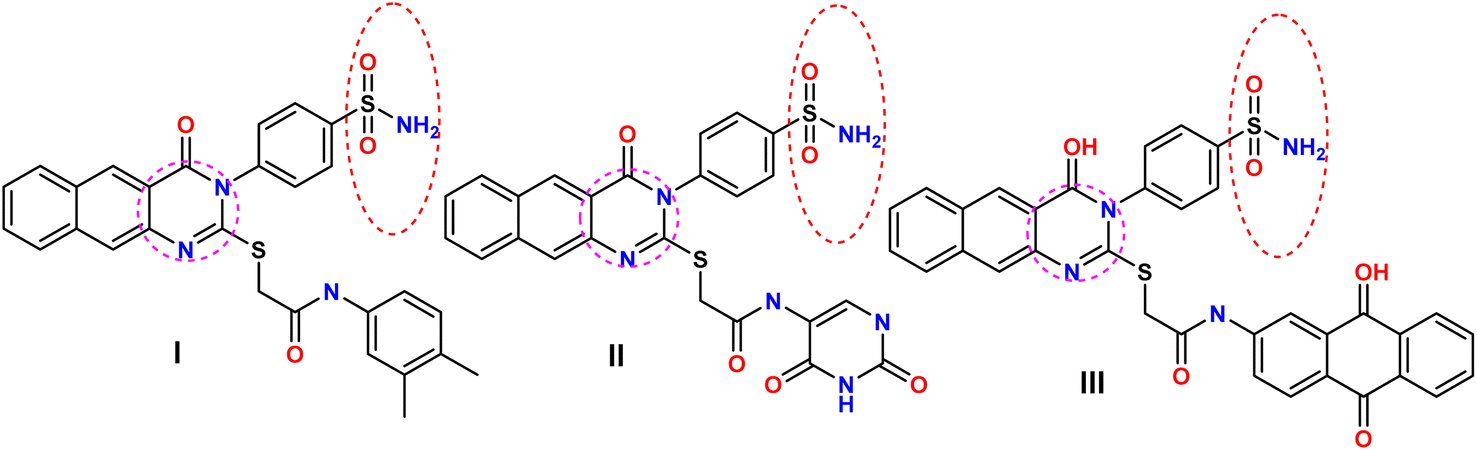 | ||
| Fig. 1 Examples of sulfonamide-based derivatives as EGFR inhibitors. | ||
EGFR, a receptor TK, plays a significant role in maintaining epithelial tissue homeostasis and is essential for generating and regulating cellular processes. However, the overexpression of the EGFR family members can lead to aberrant cell signaling, contributing to the progression and development of certain solid tumors, including non-small cell lung cancer (NSCLC) and breast cancers.17,18 The EGFR-TK domain comprises five distinct sites, including the ATP-binding pocket, which is comprised of (1) the adenine binding pocket, where H-bonds are formed with the adenine ring through residue amino acids; (2) the hydrophilic pocket (ribose region); (3) the hydrophobic region I, which is critical for receptor inhibition activity, although it is not targeted by ATP; (4) the hydrophobic region II, which is also essential and frequently targeted by EGFR-TKIs (tyrosine kinase inhibitors) with a Y-shaped structure; and (5) the phosphate-binding region, which influences the pharmacokinetic features of inhibitors. Importantly, most EGFR-TKIs exhibit a Y-shaped structure and possess four critical pharmacophoric properties, which are essential for their inhibitory activity.19–22
The first generation of EGFR-TKIs, comprising gefitinib and erlotinib, significantly enhanced the therapy landscape for NSCLC, however they were also associated with adverse effects, such as corneal damage, trichomegaly, and occasionally fatal interstitial lung disease (ILD). Furthermore, the development of acquired resistance due to EGFR-TK mutations compromised the antitumor efficacy of these treatments.22–25 This type of mutation diminishes the selectivity of ATP-competitive agents for the kinase while increasing EGFR's sensitivity to ATP, thereby facilitating the rapid emergence of drug-resistant mechanisms.26 Particularly, the T790M “gatekeeper” mutation in EGFR is a prevalent mechanism of resistance, frequently observed in patients who have developed resistance to 1st generation EGFR-TKIs.27–29
The development of second-generation EGFR-TKIs, including pelitinib and neratinib, was driven by the need to address resistance in NSCLC treatment regimens resulting from EGFR-TK mutations.30–33 These inhibitors interact with Cys797 near the EGFR's ATP-binding groove via electrophilic Michael-acceptor moieties, thereby inactivating the protein. However, their non-selective inhibition of both EGFRWT and EGFRT790M led to a relatively lower maximum tolerated dose (MTD) and adverse clinical outcomes.34–36
Third generation EGFR-TKIs, including olmutinib37,38 and osimertinib (AZD9291),39 are characterized as irreversible covalent inhibitors with enhanced binding affinity for EGFRT790M and lower affinity for EGFRWT. These inhibitors exhibit sustained sub-nanomolar IC50 values for prevalent activating mutations (del19 and L858R). Due to its favorable biological properties, the FDA approved AZD9291 in 2015 for patients with NSCLC therapeutic regimens with mutant EGFR. However, a 2016 safety study reported two cases of Stevens-Johnson syndrome and severe epidermal necrolysis.40–42 AZD9291 is currently the primary 3rd generation authorized as a first-line therapy for patients with EGFR-positive activating mutations (del19 and L858R), NSCLC, and metastatic EGFRT790M-positive NSCLC, as identified by an approved FDA test.
Investigations into the mechanisms underlying resistance to 3rd generation have revealed the presence of both primary and acquired resistant mechanisms.40 In order to inform subsequent treatment strategies, patients experiencing NSCLC progression after 3rd generation therapies should undergo comprehensive next-generation sequencing (NGS) analysis of plasma or tissue samples to elucidate the specific resistance mechanisms employed.35 Further research into the mechanisms of resistance to 3rd generation is imperative for the development of effective therapeutic strategies. This study examines the findings on 3rd generation resistance in aggressive NSCLC, providing a detailed examination of the underlying molecular mechanisms and discussing novel treatment approaches for addressing this complex issue. Mobocertinib (Fig. 2), the latest addition to the 3rd generation family, has been granted accelerated approval by the FDA in 2021 for the treatment of locally advanced or metastatic NSCLC harboring exon 20 insertions, thereby offering a promising therapeutic option for patients with this specific molecular subtype.
 | ||
| Fig. 2 The chemical structures of reported EGFR-TKIs. | ||
Our study aimed to design novel EGFR-TKI derivatives, incorporating the four primary pharmacophoric properties of EGFR-TKIs, with the goal of enhancing their efficacy and selectivity. To achieve this, we synthesized a series of derivatives by structurally modifying erlotinib at four distinct positions (Fig. 3). Building upon our previous research focused on the scope of azole and pyrimidine derivative-based scaffolds, which have demonstrated promising antiproliferative activity against both EGFRWT and EGFRT790M, we intended to further investigate the potential of these scaffolds in the development of next-generation EGFR-TKIs.
 | ||
| Fig. 3 Rationale design of the target derivatives utilizing gefitinib, osimertinib, and erlotinib. | ||
2. Results and dissection
2.1. Chemistry
In the present study, we initiated the synthesis by reacting 4-acetamidobenzene sulfonyl chloride (1) with 2-aminopyrimidine (2), resulting in the formation of N-(4-(N-(pyrimidin-2-yl)sulfamoyl)phenyl)acetamid (3). Subsequently, compound (3) was subjected to hydrolysis, yielding sulfadiazine (4). The next step involved the diazotization of sulfadiazine, which was then coupled with a diverse array of compounds, including ethyl acetoacetate, acetylacetone, ethyl benzoylacetate, benzoyl acetone, ethyl cyanoacetate, cyanothioacetamide, a dimer of malononitrile, diethyl malonate, N-[4-amino-5-cyano-6-(methylthio)pyridin-2-yl]-3-oxobutanamide, and 4,6-diamino-2-(methylthio)nicotinonitrile, in the presence of pyridine as a solvent. This reaction sequence led to the successful synthesis of hydrazone derivatives 5–14, as illustrated in Scheme 1. | ||
| Scheme 1 Synthesis of sulfadiazine-based derivatives 5–14. | ||
The chemical identities of the synthesized derivatives 5–14 were unequivocally confirmed through a combination of various spectral analyses. The IR spectra revealed the presence of NH2 and NH groups, which were identified by absorption bands in the range of 3457–3103 cm−1. Additionally, the IR spectra showed characteristic absorption bands for CN groups between 2215–2211 cm−1 and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups between 1725–1655 cm−1. The 1H NMR spectra of 5–14 displayed distinct signals for NH groups, which resonated between δ 13.65–11.34 ppm. Furthermore, the 1H NMR spectra of derivatives 5, 7, 9, and 12 exhibited quartet and triplet signals for ester groups, which appeared between δ 4.37–4.23 and δ 1.29–1.24 ppm, respectively. Especially, the SCH3 group in compound 14 was identified as a singlet at δ 2.52 ppm. The 13C NMR spectra provided additional confirmation of the structures of all compounds. For example, the 13C NMR spectrum of 13 showed a signal at δ 12.87 ppm, which was attributed to the SCH3 group, and a signal at δ 79.92 ppm, which was assigned to the C–CN group. Similarly, the 13C NMR spectrum of 12 revealed two distinct signals for CO groups at δ 195.22 and 196.65 ppm, as well as a signal for a CH3 group at δ 25.39 ppm. The elemental analysis data further verified the proposed structures of the sulfadiazine derivatives, providing a comprehensive confirmation of their chemical identities.
O groups between 1725–1655 cm−1. The 1H NMR spectra of 5–14 displayed distinct signals for NH groups, which resonated between δ 13.65–11.34 ppm. Furthermore, the 1H NMR spectra of derivatives 5, 7, 9, and 12 exhibited quartet and triplet signals for ester groups, which appeared between δ 4.37–4.23 and δ 1.29–1.24 ppm, respectively. Especially, the SCH3 group in compound 14 was identified as a singlet at δ 2.52 ppm. The 13C NMR spectra provided additional confirmation of the structures of all compounds. For example, the 13C NMR spectrum of 13 showed a signal at δ 12.87 ppm, which was attributed to the SCH3 group, and a signal at δ 79.92 ppm, which was assigned to the C–CN group. Similarly, the 13C NMR spectrum of 12 revealed two distinct signals for CO groups at δ 195.22 and 196.65 ppm, as well as a signal for a CH3 group at δ 25.39 ppm. The elemental analysis data further verified the proposed structures of the sulfadiazine derivatives, providing a comprehensive confirmation of their chemical identities.
2.2. Biological evaluation
| Comp. | IC50 (μM)a | ||
|---|---|---|---|
| A549 | A431 | H1975 | |
| a IC50 = Mean ± SD, n = 3. | |||
| 5 | 19.24 ± 3.09 | 18.21 ± 0.46 | 20.12 ± 2.21 |
| 6 | 16.60 ± 0.92 | 17.21 ± 0.52 | 15.70 ± 0.21 |
| 7 | 11.25 ± 1.09 | 13.89 ± 0.80 | 10.32 ± 1.80 |
| 8 | 4.78 ± 0.12 | 4.12 ± 0.54 | 2.31 ± 0.23 |
| 9 | 14.98 ± 1.27 | 14.70 ± 0.18 | 13.14 ± 0.67 |
| 10 | 19.67 ± 0.34 | 18.23 ± 2.09 | 15.80 ± 2.48 |
| 11 | 13.56 ± 1.32 | 15.09 ± 0.58 | 14.13 ± 1.23 |
| 12 | 5.15 ± 0.30 | 6.38 ± 0.12 | 6.12 ± 0.24 |
| 13 | 15.45 ± 1.34 | 20.31 ± 2.12 | 17.09 ± 1.62 |
| 14 | 7.56 ± 0.27 | 6.15 ± 0.23 | 4.59 ± 0.43 |
| Erlotinib | 17.50 ± 2.34 | 11.09 ± 0.67 | 12.86 ± 1.23 |
| Gefitinib | 15.75 ± 1.09 | 9.60 ± 0.43 | 10.15 ± 0.39 |
| Osimertinib | — | 5.96 ± 0.63 | 0.93 ± 0.09 |
| Comp. | IC50 (nM) | |
|---|---|---|
| EGFRWT | EGFRT790M | |
| 5 | 49.2 ± 6.7 | 93.6 ± 8.5 |
| 6 | 52.6 ± 9.3 | 66.8 ± 10.4 |
| 7 | 38.9 ± 8.3 | 114.6 ± 9.6 |
| 8 | 23.2 ± 0.1 | 9.6 ± 3.2 |
| 9 | 43.5 ± 0.2 | 17.1 ± 0.1 |
| 10 | 31.5 ± 5.8 | 60.8 ± 6.2 |
| 11 | 47.7 ± 11.4 | 132.8 ± 16.2 |
| 12 | 14.5 ± 7.5 | 35.4 ± 8.6 |
| 13 | 41.7 ± 8.2 | 214.9 ± 15.2 |
| 14 | 15.3 ± 3.1 | 24.3 ± 3.4 |
| Gefitinib | 18.2 ± 2.2 | 368.2 ± 25.6 |
| Osimertinib | 57.8 ± 10.2 | 8.5 ± 2.7 |
3. In silico studies
3.1. Molecular docking study
In silico simulation tests were conducted on derivatives 8, 12, and 14 at the active ATP binding sites of both EGFRWT and EGFRT790M protein kinases (PDB: 4HJO and PDB: 3W2O, respectively). The protein models were obtained from the PDB website using the MOE 2019.01 program. The results showed that the newly designed compounds docked against the EGFRWT kinase with good binding affinity scores, ranging from −6.40 to −7.10 kcal mol−1 (Table 3). The outcomes of this simulation approach were elucidated secondarily by docking erlotinib and TAK-285 as the co-localized ligands at the active sites of both EGFRWT and EGFRT790M. The root mean square deviation (RMSD) values of erlotinib and TAK-285 secondly docked analogs and co-localized conformers, respectively, were 1.4 and 1.85 Å, respectively, illustrating the rationality of this docking technique (Fig. 1S and 5S, respectively, in ESI†).| Comp. | ΔG score kcal mol−1 | RMSD_refine (Å) | Amino acid/bond | Distance (Å) | E (kcal mol−1) |
|---|---|---|---|---|---|
| 8 | −6.40 | 1.99 | Met769/pi–H | 3.98 | −0.5 |
| Lys721/H acceptor | 3.39 | −2.5 | |||
| 12 | −7.10 | 1.05 | Met769/H acceptor | 3.34 | −1.5 |
| Lys692/H acceptor | 3.21 | −1.4 | |||
| Lys704/H acceptor | 2.95 | −8.3 | |||
| Val702/pi–H | 4.11 | −0.5 | |||
| Val702/pi–H | 4.24 | −0.7 | |||
| HOH Thr766/H acceptor | 3.17 | −0.7 | |||
| HOH Gln767/H acceptor | 3.17 | −0.7 | |||
| 14 | −6.53 | 2.14 | Met769/H donor | 2.98 | −0.6 |
| Lys721/H acceptor | 2.91 | −8.1 | |||
| Leu694/pi–H | 3.91 | −0.7 | |||
| Val702/pi–H | 3.84 | −0.8 | |||
| Erlotinib | −6.83 | 1.4 | Met769/H acceptor | 2.95 | −4.4 |
| HOH Thr766/H donor | 3.03 | −1.6 | |||
| HOH Gln767/H donor | −1.1 | ||||
| Gefitinib | −8.01 | 1.33 | Met769/H acceptor | 3.03 | −3.3 |
| HOH Thr766/H donor | 3.10 | −1.5 | |||
| HOH Gln767/H donor | 3.10 | −1.5 | |||
| Leu694/pi–H | 4.3 | −0.8 | |||
| Leu820/pi–H | 4.56 | −0.7 | |||
| Osimertinib | −7.01 | 2.38 | Lys692/pi–cation | 4.38 | −0.7 |
| Val702/pi–H | 3.77 | −0.7 |
To investigate the potential dual EGFR inhibitory activities of gefitinib and osimertinib, these reversible first- and irreversible third-generation EGFR inhibitors were docked against wild-type and mutant EGFR, respectively. Additionally, erlotinib, the co-localized ligand, was re-docked into its corresponding co-crystalized protein model (PDB code: 4HJO) to evaluate the ability of MOE to replicate the native ligand superimposition to the wild-type EGFR protein active site (Fig. 1S and 2S,† and Table 3).
The re-docked erlotinib structure exhibited a high degree of similarity with the native ligand, with an energy score (ΔG) of −6.83 kcal mol−1 and a good RMSD value of 1.4 Å. Particularly, the re-docked erlotinib structure formed one H-bond (hydrogen bond) with Met769 and two H2O-dependent H-bonds with Gln767 and Thr766, highlighting the accuracy of the docking protocol.
Osimertinib and gefitinib, their binding interactions with the active EGFRWT binding site were investigated through molecular docking studies. Osimertinib was found to form one π–H bond with Val702, and one π–cation contact with Lys692 AAs (amino acids), resulting in an energy score (ΔG) of −7.01 kcal mol−1 and 2.38 Å as its RMSD value (Fig. 3S† and Table 3). However, these interactions were not sufficient to inhibit the EGFRWT protein kinase, consistent with previous literature data indicating that osimertinib has a higher affinity for the mutant EGFRT790M protein kinase and no activity against the EGFRWT.
In contrast, gefitinib was found to form hydrogen bonds with Met769 and H2O-assisted H-bonds with Thr766 and Gln767 amino acids, similar to erlotinib. Additionally, gefitinib formed two hydrophobic bonds with Leu694 and Leu820 AAs within the EGFR binding site, resulting in an energy score (ΔG) of −8.01 kcal mol−1 and an RMSD value of 1.33 Å (Fig. 4S† and Table 3). These interactions suggest that gefitinib can afford good inhibitory activity within the EGFRWT protein active site, consistent with its use as reference drug in the in vitro experiments.
Derivatives 5, 12, and 14, their binding interactions with the active EGFRWT binding site (PDB code: 4HJO) were investigated. Derivative 5 was able to bind to the EGFRWT active site with a satisfactory interaction affinity, exhibiting an energy score (ΔG) of −6.40 kcal mol−1 and 1.99 Å as its RMSD value. The binding mode of derivative 5 involved one H bond with Lys721 AA and one π–H bond with the major amino acid Met769 (Fig. 4 and Table 3). Derivative 12 demonstrated a good interaction affinity relative to the reference erlotinib, with an energy score (ΔG) of −7.10 kcal mol−1 and 1.05 Å as its RMSD value (Fig. 5 and Table 3). The binding mode of derivative 12 involved hydrogen bonds with Met769, Lys692, Lys704, and H2O-assisted H-bonds with Thr766 and Gln767 amino acids, similar to erlotinib. Additionally, derivative 12 formed two hydrophobic bonds with Val702 amino acid within the EGFR binding site. Derivative 14 exhibited a different binding mode than erlotinib, with an acceptable interaction affinity relative to erlotinib, characterized by an energy score (ΔG) of −6.53 kcal mol−1 and 2.14 Å as its RMSD value (Fig. 6 and Table 3). The binding interaction mode of derivative 14 involved a H-bond with the major amino acid Met769 and another hydrogen bond with Lys721 AA inside the EGFRWT active site. Furthermore, derivative 14 formed two hydrophobic interactions with Leu694 and Val702 AAs.
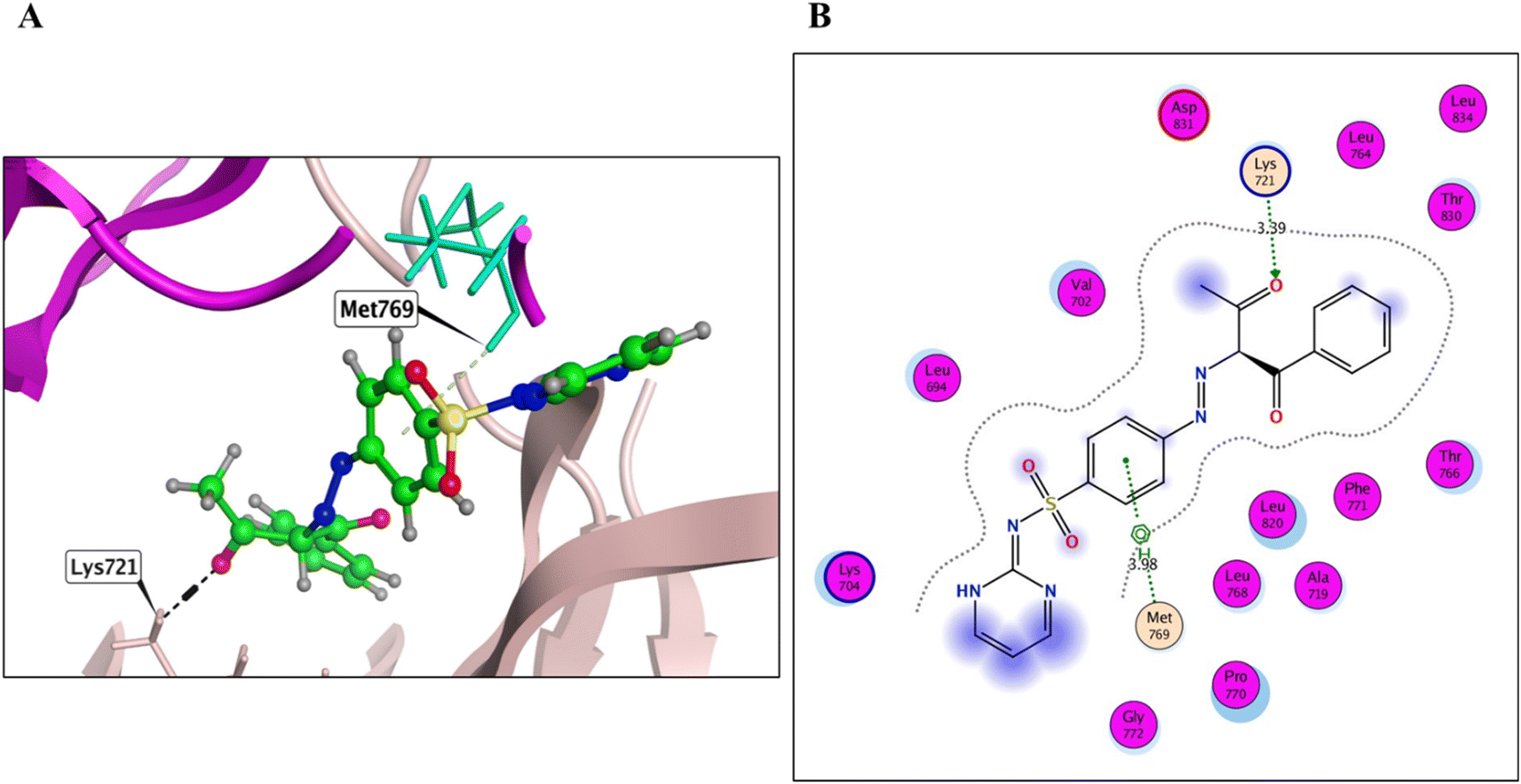 | ||
| Fig. 4 3D (A) and 2D images (B) of 8 (green sticks) within the wild EGFR kinase. | ||
 | ||
| Fig. 5 3D (A) and 2D images (B) of 12 (green sticks) within the wild EGFR kinase. | ||
 | ||
| Fig. 6 3D (A) and 2D images (B) of 14 (green sticks) within the wild EGFR kinase. | ||
The binding interactions of derivatives 8, 12, and 14 with the EGFRT790M mutant model (PDB code: 3W2O) were investigated and the outcomes illustrated that these derivatives displayed good binding affinities versus EGFRT790M, with energy scores ranging from −6.33 to −7.53 kcal mol−1 (Table 4). Relying on previous literature data about the EGFRT790M mutant model, Met790 amino acid in the EGFRT790M mutant model can adopt specific conformations to fit with various inhibitors, while the R858 residue can afford conformational activation loop variants. These outcomes provide a deeper understanding of the SARs that can facilitate the development of more efficient inhibitory agents against sensitive and resistant drug-dependent EGFR mutations.51 Regarding the validation of the docking protocol of the co-crystallized ligand TAK-285, we re-docked it into its corresponding EGFRT790M adenine binding pocket of the co-crystal protein model (PDB code: 3W2O). The results showed that the re-docked TAK-285 binding score (ΔG) was −7.35 kcal mol−1, with a good RMSD value of 3.01 Å (Fig. 5S† and Table 4). The binding mode of TAK-285 involved one H-bond with each Met793, Asp800, Leu788, and Lys745 amino acids. Additionally, the pyrimidine ring of TAK-285 formed one H2O-assisted H-bond with Met790, with 3.01 Å as the bond length value.
| Comp. | ΔG score kcal mol−1 | RMSD-refine (Å) | Amino acid/bond | Distance (Å) | E (kcal mol−1) |
|---|---|---|---|---|---|
| 8 | −7.53 | 2.03 | Met790/H donor | 3.56 | −0.9 |
| Gly796/H acceptor | 3.38 | −0.5 | |||
| Gly796/H acceptor | 3.46 | −0.5 | |||
| 12 | −6.62 | 1.73 | Met790/H donor | 4.15 | −0.5 |
| Ile752/H donor | 4.30 | −0.7 | |||
| Val726/pi–H | 4.37 | −0.7 | |||
| HOH Met790/H acceptor | 2.93 | −0.8 | |||
| 14 | −6.33 | 1.06 | HOH Met790/H acceptor | 2.79 | −0.8 |
| Asp855/H donor | 2.99 | −1.3 | |||
| Gefitinib | −5.98 | 1.25 | Met790/H donor | 3.62 | −0.5 |
| TAK-285 | −7.35 | 1.85 | HOH Met790/H acceptor | 3.01 | −0.8 |
| Met793/H acceptor | 3.25 | −3.1 | |||
| Asp800/H donor | 3.10 | −2.3 | |||
| Leu788/H donor | 3.39 | −1.3 | |||
| Lys745/H acceptor | 3.17 | −0.8 | |||
| Osimertinib | −8.72 | 1.53 | Met790/H donor | 3.81 | −1.1 |
| HOH Met790/H acceptor | 3.31 | −0.9 | |||
| Glu762/H donor | 3.24 | −0.7 | |||
| Asp855/ionic | 3.90 | −0.7 | |||
| Asp855/ionic | 3.81 | −0.9 | |||
| Leu718/pi–H | 4.39 | −0.5 | |||
| Leu718/pi–H | 4.34 | −0.5 | |||
| Leu718/pi–H | 3.96 | −1.0 | |||
| Erlotinib | −6.54 | 2.06 | Met769/H donor | 3.84 | −0.5 |
| Lys745/H acceptor | 3.04 | −4.9 |
The binding interactions of gefitinib, osimertinib, and erlotinib with the EGFRT790M mutant model (PDB code: 3W2O) were investigated as well. Gefitinib, utilized as a reference in the in vitro experiments, was found to form only one significant hydrogen bond with Met790 AA, with 3.62 Å a distance value and an energy score of −5.98 kcal mol−1 (Fig. 6S† and Table 4). In contrast, osimertinib was found to bind to the EGFRT790M mutant model in a more potent manner, with a binding score (ΔG) of −8.72 kcal mol−1 and an RMSD value of 1.53 Å (Fig. 7S† and Table 4). The binding mode of osimertinib involved two H-bonds with Met790, one H-bond with Glu762, two ionic bonds with Asp855, and three hydrophobic bonds with Leu718 AA within the mutant EGFRT790M binding site. These results suggest that osimertinib can afford potent inhibitory activity within the mutant EGFRT790M protein active site, consistent with its auspicious IC50 value in the in vitro EGFRT790M biological investigation. Erlotinib was also re-docked against the mutant EGFRT790M binding site to evaluate its affinity. The results elucidated that erlotinib made H-bonds with only two non-essential amino acids, Met769 and Lys745, with distances of 3.84 and 3.04 Å, respectively (energy score = −6.54 kcal mol−1) (Fig. 8S† and Table 4). These findings are consistent with literature data, which suggest that erlotinib has affinity only for the wild-type EGFR protein tyrosine kinase, not the mutant EGFRT790M 50.
Derivative 8 was found to bind to the active EGFRT790M site via three main H-bonds: one with Met790 and two with Gly796 AAs, with distance values of 3.56, 3.38, and 3.46 Å, respectively (energy score = −7.53 kcal mol−1) (Fig. 7 and Table 4). This binding mode suggests that derivative 8 has a strong affinity for the EGFRT790M mutant model. Derivative 12 was found to bind to the EGFRT790M mutant model in a similar manner to TAK-285, with a binding mechanism that involved hydrogen bonding with only two essential amino acids, Met790 and Ile752, with distance values of 4.15 and 4.30 Å (energy score = −6.62 kcal mol−1) (Fig. 8 and Table 4). Additionally, derivative 12 formed one hydrophobic interaction with Val726. Derivative 14 was found to bind to the EGFRT790M mutant model via two fundamental H-bonds with Met790 and Asp855, with distance values of 2.79 and 2.99 Å, respectively (energy score = −5.51 kcal mol−1) (Fig. 9 and Table 4). These binding modes highlight the significance of the hydrogen bond donating and accepting moieties in the designed ligands to act as efficient dual EGFRWT and EGFRT790M inhibitors. The results suggest that the designed ligands have the potential to dually inhibit EGFRWT and EGFRT790M protein tyrosine kinases, which is a critical aspect of cancer therapy.
 | ||
| Fig. 7 3D (A) and 2D images (B) of 8 (green sticks) within the mutant EGFRT790M kinase. | ||
 | ||
| Fig. 8 3D (A) and 2D images (B) of 12 (green sticks) within the mutant EGFRT790M kinase. | ||
 | ||
| Fig. 9 3D (A) and 2D images (B) of 14 (green sticks) within the mutant EGFRT790M kinase. | ||
3.2. Structure–activity relationship (SAR) study
Our rationalized drug design approach involved a comprehensive study of SAR of the target derivatives as potential antiproliferative agents. Specifically, we investigated the impact of R1 and R2 substituents on the biological activity of the scaffolds (Fig. 10). The results of our study revealed that compounds 8, 12, and 14 exhibited highly potent IC50 values, indicating significant in vitro antiproliferative activities and dual EGFRWT and EGFRT790M inhibitory activity. Furthermore, molecular docking studies demonstrated that these compounds formed strong interactions with the target protein, which correlated with their high biological activity. A key finding of our SAR analysis was the importance of hydrogen bond acceptor (HBA) functional groups in achieving effective dual EGFR inhibition. The presence of HBA groups in compounds 8, 12, and 14 was found to be crucial for their outstanding potency and selectivity towards both EGFRWT and EGFRT790M. These results afford valuable information that greatly helps in the design of novel EGFRIs and illustrate the importance of careful consideration of the SAR of target compounds in the production of promising cancer therapies. | ||
| Fig. 10 SAR study of sulfadiazine-based hydrazones 8, 12, and 14. | ||
Lapatinib has been shown to interact with the inactive tyrosine kinase domain of EGFR due to its large, sterically hindered [(3-fluorobenzyl)oxy] core scaffold on the peripheral aniline residue. This prevents it from fitting into the small-sized ATP-binding site, instead allowing it to reach and fit into the allosteric-binding site that is re-opened by the dislocation of the αC helix in the inactive conformation model.52 Similarly, the large-sized R1 and R2 side chains in compounds 8, 12, and 14, particularly in compound 12, prevent them from fitting into the small ATP-binding pocket. As a result, compound 12 selectively interacts with the inactive EGFR conformation, but not the active one. Compound 12 was detected to be the most active inhibitor of both EGFRWT and EGFRT790M kinases among all tested derivatives. This is attributed to its binding mode, dimensions, and stable docking scores, which are superior to those of the reference inhibitors erlotinib and gefitinib for wild-type EGFR, and TAK-285 and osimertinib for mutant EGFRT790M. These outcomes are coherent with the in vitro inhibition of compound 12 against both EGFRWT and EGFRT790M protein kinases, demonstrating its potential as a potent and selective EGFR inhibitor.
3.3. Pharmacokinetic characteristics and ADMET predict
To further investigate the potential of the synthesized scaffolds as orally bioavailable candidates,52 their absorption, distribution, metabolism, and excretion (ADME) properties were evaluated using the ADME Swiss prediction free online tool.51–56 The boiled egg diagram of the WLOGP versus TPSA (Topological Polar Surface Area) of the uploaded derivatives 8 and 12 revealed that both compounds have low GI absorption and are unable to pass through the BBB relative to the reference compounds erlotinib and gefitinib (Fig. 11). This suggests that these compounds are unlikely to cause central nervous system (CNS) negative impacts due to their low BBB permeability. In contrast, scaffold 14, a highly polar compound, was found to be out of range and vanished from the boiled egg diagram, highlighting the need for optimization to make it an effective orally bioavailable candidate. The ADME Swiss prediction tool also suggested that derivative 8 is unlikely to be a P-glycoprotein (PGP) substrate, which means that it may not be exposed to drug resistance mechanisms. However, scaffold 12 is predicted to be a PGP substrate, indicating a possibility of exposure to drug resistance mechanisms. The TPSA values of the designed scaffolds 8, 12, and 14 ranged from 138 to 216.04 Å2, while their log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values were between 1.85 and 3.93 (Table 5). These values provide further insight into the lipophilicity and bio-molecular polarity of the designed scaffolds.
P values were between 1.85 and 3.93 (Table 5). These values provide further insight into the lipophilicity and bio-molecular polarity of the designed scaffolds.
 | ||
| Fig. 11 The boiled-egg diagram for 8, 12, 14, erlotinib, and gefitinib. | ||
| Comp. | M. wt | Fraction Csp3 | RB | HBA | HBD | MR | TPSA | LogP |
GI absorption | BBB perm. | Lip. V. | Bio. Sc. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 8 | 423.45 | 0.05 | 8 | 7 | 2 | 111.67 | 138.86 | 3.22 | Low | No | Yes | 0.55 |
| 12 | 435.48 | 0.00 | 6 | 7 | 2 | 114.36 | 169.64 | 3.93 | Low | No | Yes | 0.55 |
| 14 | 441.49 | 0.06 | 6 | 8 | 4 | 119.04 | 216.04 | 1.85 | Low | No | Yes | 0.55 |
| Erlotinib | 393.44 | 0.27 | 10 | 6 | 1 | 111.40 | 74.73 | 3.07 | High | Yes | Yes | 0.55 |
| Gefitinib | 446.90 | 0.36 | 8 | 7 | 1 | 121.66 | 68.74 | 4.32 | High | Yes | Yes | 0.55 |
The bioavailability diagram for scaffolds 8, 12, and 14 is displayed in Fig. 12(A–E). This radar plot consists of six axes representing six critical oral bioavailability characteristics: solubility (INSOLU), flexibility (FLEX), size (SIZE), lipophilicity (LIPO), saturation (INSATU), and polarity (POLAR) (Table S1†). The pink area in the radar plot represents the optimum property parameters for oral bioavailability. The red colored lines represent scaffolds 8 (Fig. 12A), 12 (Fig. 12B), and 14 (Fig. 12C), which are nearly within the pink area relative to erlotinib (Fig. 12D) and gefitinib (Fig. 12E). This suggests that the designed scaffolds have acceptable assumed oral bioavailability. Scaffolds 8, 12, and 14 are predicted to afford a suitable bioavailability with a score value equal to 0.55, similar to the reference compounds erlotinib and gefitinib. Additionally, scaffolds 8 and 12 follow the Lipinski rule of 5 with no detected violations, similar to the reference drugs. However, derivative 14 obeys the Lipinski rule of 5 with only one detected violation (number of polar atoms; nitrogen or oxygen atoms higher than 10) compared to the reference compounds (Table S1†). These results demonstrate that the designed compounds have promising pharmacokinetic and drug-like properties. As shown in Fig. 12(A–E) and Table 5, analogs 8 and 12 have good lipophilic characters compared to the reference compounds, unlike compound 14, which has lower lipophilicity. The unsaturation property of scaffolds 8, 12, and 14 is nearly out of the tinted area, indicating that these compounds may require optimization to improve their saturation characteristics. The fraction Csp3 values of scaffolds 8, 12, and 14 were calculated at 0.06, indicating that these compounds have a low ratio of sp3 hybridized carbons over all the carbon numbers. For saturation character, the ratio of sp3 hybridized carbons over all the carbon numbers of the analog (fraction Csp3) should not be lower than 0.25.
 | ||
| Fig. 12 Pink zoned oral bioavailability radars for 8 (A), 12 (B), 14 (C), erlotinib (D), and gefitinib (E). | ||
Hydrazones 8, 12, and 14 exhibited low saturation character, with fraction Csp3 values of 0.05, 0.00, and 0.06, respectively. Conversely, their hydrophilicity ranged from moderately soluble to very soluble, relative to the reference compounds erlotinib and gefitinib, which had solubility ranging from moderately soluble to poorly soluble. Unlike erlotinib and gefitinib, which showed inhibitory activity against CYP2C9, CYP2C19, CYP2D6, and CYP3A4, hydrazones 8, 12, and 14 did not exhibit inhibitory activity against CYP1A2 and CYP2D6. Additionally, scaffold 14 did not show inhibitory activity against CYP2C9 and CYP2C19, unlike derivatives 8 and 12. According to PAINS and Brenk filters, hydrazones 8, 12, and 14 are suitable for biological experiments, exhibiting potent chemical reactivity, low hazardous doses, and adequate metabolic stability. Scaffolds 8, 12, and 14 demonstrated lead-likeness, unlike erlotinib and gefitinib, and their synthetic procedures were categorized as accessible. Compounds 8, 12, and 14 had better safety profiles than the reference compounds erlotinib and gefitinib (Table S1†). These compounds are classified as non-AMES toxic, hERG I inhibitors, and skin sensitization agents. Hydrazones 8, 12, and 14 exhibited hepatotoxicity and parameters indicating carcinogenic agents, which may cause severe hepatic side effects, similar to the reference compounds erlotinib and gefitinib. Compounds 8 and 12 demonstrated hERG II K+ channel inhibitory activity, similar to the reference compounds erlotinib and gefitinib. In contrast, scaffold 14 did not exhibit hERG II K+ channel inhibitory activity (Table S1†).
4. Experimental part
4.1. General chemistry
All general information were provided in the ESI.†4.2. Synthesis of sulfadiazine 4
A mixture of an equimolar amount of 4-acetamidobenzene sulfonyl chloride (1) with 2-aminopyrimidine (2) was refluxed in pyridine for 1 hour, then the mixture was added to 5 mL of H2O/HCl in ratio 2:1, then filtered the mixture, washed with H2O several times, dried, and recrystallized from EtOH, m.p. 251–253 °C (reported m.p. 252–256 °C).37
4.3. Synthesis of sulfadiazine 5–14
A solution of sodium nitrite (0.1 g, 1.3 mmol) in water (2 mL) was gradually added to a solution of sulfadiazine (0.25 g, 1 mmol) in conc. HCl (3 mL) at 0–5 °C. The formed diazonium was mixed under stirring with an ice-cooled solution of appropriate coupling compounds (1 mmol), namely, ethyl acetoacetate, acetylacetone, ethyl benzoylacetate, benzoyl acetone, ethyl cyanoacetate, cyanothioacetamide, dimer of malononitrile, diethyl malonate, N-[4-amino-5-cyano-6-(methylthio)pyridin-2-yl]-3-oxobutanamide and 4,6-diamino-2-(methylthio)nicotinonitrile in pyridine (20 mL). The mixture was kept for 30 minutes in an ice bath and then mixed into 100 mL of cold water. The product was filtered off, washed with distilled water, dried, and recrystallized from EtOH.Brownish yellow solid, yield (91%), m.p. 160–162 °C. IR (KBr) ν: 3404, 3256 (2NH), 3089, 3039 (C–H Aromatic), 2974, 2938, 2874, 2811, 2734 (C–H aliphatic), 1690 (br, C
 O), 1584 (CN); 1H NMR δ: 1.25–1.29 (t, 3H, J = 8 Hz, C
O), 1584 (CN); 1H NMR δ: 1.25–1.29 (t, 3H, J = 8 Hz, C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3 ester), 2.41 (s, 3H, C3), 4.29–4.34 (q, 2H, J = 8 Hz, CH2), 7.03–7.06 (t, 1H, J = 6 Hz, Carom), 7.54–7.56 (d, 2H, J = 8 Hz, Carom), 7.97–8.00 (d, 2H, J = 12 Hz, Carom), 8.50–8.51 (d, 2H, J = 4 Hz, Carom), 11.59 (s, 1H, N, D2O exchangeable), 11.76 (br, 1H, N, D2O exchangeable); 13C NMR δ: 14.28 (CH3 ester), 25.76 (CH3), 61.84 (CH2), 115.06, 116.21, 129.97, 134.22, 134.62, 146.55, 157.44, 158.75, 162.60 (CO), 194.31 (CO); anal. calcd for C16H17N5O5S (391.40): C, 49.10; H, 4.38; N, 17.89. Found: C, 49.22; H, 4.26; N, 17.75.
3 ester), 2.41 (s, 3H, C3), 4.29–4.34 (q, 2H, J = 8 Hz, CH2), 7.03–7.06 (t, 1H, J = 6 Hz, Carom), 7.54–7.56 (d, 2H, J = 8 Hz, Carom), 7.97–8.00 (d, 2H, J = 12 Hz, Carom), 8.50–8.51 (d, 2H, J = 4 Hz, Carom), 11.59 (s, 1H, N, D2O exchangeable), 11.76 (br, 1H, N, D2O exchangeable); 13C NMR δ: 14.28 (CH3 ester), 25.76 (CH3), 61.84 (CH2), 115.06, 116.21, 129.97, 134.22, 134.62, 146.55, 157.44, 158.75, 162.60 (CO), 194.31 (CO); anal. calcd for C16H17N5O5S (391.40): C, 49.10; H, 4.38; N, 17.89. Found: C, 49.22; H, 4.26; N, 17.75.Yellow solid, yield (80%), m.p. 228–230 °C. IR ν: 3422, 3312 (2NH), 3082, 3033 (C–H aromatic), 2936, 2865, 2808, 2729 (C–H aliphatic), 1677 (C
 O), 1585 (CN); 1H NMR δ 2.42 (s, 3H, C3), 2.47 (s, 3H, C3), 7.04–7.05 (m, 1H, Carom), 7.68–7.70 (d, 2H, J = 8 Hz, Carom), 8.00–8.02 (d, 2H, J = 8 Hz, Carom), 8.50 (s, 2H, Carom), 11.70 (br, 1H, N, D2O exchangeable), 13.51 (s, 1H, N, D2O exchangeable); 13C NMR δ: 26.75 (CH3), 31.70 (CH3), 116.25, 129.88, 135.83, 135.95, 145.95, 157.34, 158.83, 159.04, 196.92 (CO), 197.92 (CO); anal. calcd for C15H15N5O4S (361.37): C, 49.85; H, 4.18; N, 19.38. Found: C, 49.73; H, 4.27; N, 19.51.
O), 1585 (CN); 1H NMR δ 2.42 (s, 3H, C3), 2.47 (s, 3H, C3), 7.04–7.05 (m, 1H, Carom), 7.68–7.70 (d, 2H, J = 8 Hz, Carom), 8.00–8.02 (d, 2H, J = 8 Hz, Carom), 8.50 (s, 2H, Carom), 11.70 (br, 1H, N, D2O exchangeable), 13.51 (s, 1H, N, D2O exchangeable); 13C NMR δ: 26.75 (CH3), 31.70 (CH3), 116.25, 129.88, 135.83, 135.95, 145.95, 157.34, 158.83, 159.04, 196.92 (CO), 197.92 (CO); anal. calcd for C15H15N5O4S (361.37): C, 49.85; H, 4.18; N, 19.38. Found: C, 49.73; H, 4.27; N, 19.51.Yellow solid, yield (76%), m.p. 205–207 °C; IR ν: 3224, 3172 (2NH), 3083, 3039 (C–H aromatic), 2984, 2942, 2874, 2813, 2738 (C–H aliphatic), 1684, 1662 (C
 O), 1582 (CN); 1H NMR δ: 1.24–1.27 (t, 3H, J = 6 Hz, C3), 4.32–4.37 (q, 2H, J = 6 Hz, C2), 7.01–7.04 (m, 1H, Carom), 7.39–7.41 (d, 2H, J = 8 Hz, Carom), 7.53–7.60 (m, 3H, Carom), 7.65–7.72 (m, 1H, Carom), 7.91–7.98 (m, 3H, Carom), 7.49–8.51 (m, 2H, Carom), 11.66 (br, 1H, N), 11.99 (br, 1H, N); 13C NMR δ: 14.27 (CH3 ester), 61.91 (CH2), 115.12, 116.20, 128.28, 129.56, 129.97, 130.37, 133.43, 134.73, 137.04, 146.44, 157.41, 158.76, 162.43 (CO), 192.49 (CO); anal. calcd for C21H19N5O5S (453.47): C, 55.62; H, 4.22; N, 15.44. Found: C, 55.47; H, 4.38; N, 15.32.
O), 1582 (CN); 1H NMR δ: 1.24–1.27 (t, 3H, J = 6 Hz, C3), 4.32–4.37 (q, 2H, J = 6 Hz, C2), 7.01–7.04 (m, 1H, Carom), 7.39–7.41 (d, 2H, J = 8 Hz, Carom), 7.53–7.60 (m, 3H, Carom), 7.65–7.72 (m, 1H, Carom), 7.91–7.98 (m, 3H, Carom), 7.49–8.51 (m, 2H, Carom), 11.66 (br, 1H, N), 11.99 (br, 1H, N); 13C NMR δ: 14.27 (CH3 ester), 61.91 (CH2), 115.12, 116.20, 128.28, 129.56, 129.97, 130.37, 133.43, 134.73, 137.04, 146.44, 157.41, 158.76, 162.43 (CO), 192.49 (CO); anal. calcd for C21H19N5O5S (453.47): C, 55.62; H, 4.22; N, 15.44. Found: C, 55.47; H, 4.38; N, 15.32.Orange solid, yield (75%), m.p. 235–237 °C. IR ν: 3268, 3113 (2NH), 3084, 3038 (C–H aromatic), 2943, 2873, 2813, 2739 (C–H aliphatic), 1655 (C
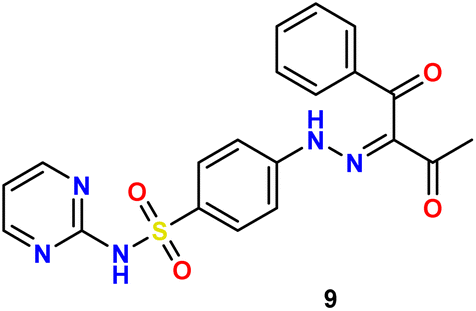 O), 1582 (CN); 1H NMR δ 2.51 (s, 3H, CH3), 7.03–7.05 (t, 1H, J = 4 Hz, Carom), 7.50–7.58 (m, 4H, Carom), 7.69–7.72 (t, 1H, J = 6 Hz, Carom), 7.80–7.82 (d, 2H, J = 8 Hz, Carom), 7.95–7.97 (d, 2H, J = 8 Hz, CHarom), 8.50–8.51 (d, 2H, J = 4 Hz, Carom), 11.34 (s, 1H, N), 13.65 (s, 1H, N); 13C NMR δ: 25.39 (CH3), 114.58, 116.22, 129.21, 129.54, 129.94, 133.84, 134.94, 135.85, 141.45, 147.22, 157.22, 158.77, 195.22 (CO), 196.65 (CO); anal. calcd for C20H17N5O4S (423.44): C, 56.73; H, 4.05; N, 16.54. Found: C, 56.57; H, 4.21; N, 16.47.
O), 1582 (CN); 1H NMR δ 2.51 (s, 3H, CH3), 7.03–7.05 (t, 1H, J = 4 Hz, Carom), 7.50–7.58 (m, 4H, Carom), 7.69–7.72 (t, 1H, J = 6 Hz, Carom), 7.80–7.82 (d, 2H, J = 8 Hz, Carom), 7.95–7.97 (d, 2H, J = 8 Hz, CHarom), 8.50–8.51 (d, 2H, J = 4 Hz, Carom), 11.34 (s, 1H, N), 13.65 (s, 1H, N); 13C NMR δ: 25.39 (CH3), 114.58, 116.22, 129.21, 129.54, 129.94, 133.84, 134.94, 135.85, 141.45, 147.22, 157.22, 158.77, 195.22 (CO), 196.65 (CO); anal. calcd for C20H17N5O4S (423.44): C, 56.73; H, 4.05; N, 16.54. Found: C, 56.57; H, 4.21; N, 16.47.Yellow solid, yield (78%), m.p. 225–227 °C; IR ν: 3387, 3218 (2NH), 3036, 3032 (C–H aromatic), 2972, 2935, 2866, 2806 (C–H aliphatic), 2214 (C

![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1725 (CO), 1582 (CN); 1H NMR δ: 1.27–1.30 (t, 3H, J = 6 Hz, C3), 4.27–4.32 (q, 2H, J = 6 Hz, C2), 7.03–7.06 (t, 1H, J = 6 Hz, Carom), 7.59–7.61 (d, 2H, J = 8 Hz, CHarom), 8.01–8.03 (d, 2H, J = 8 Hz, Carom), 8.50–8.51 (d, 2H, J = 4 Hz, Carom), 11.84 (br, 1H, N, D2O exchangeable) 12.45 (br, 1H, NH, D2O exchangeable); 13C NMR δ: 14.35 (CH3), 62.29 (CH2), 106.78 (CN), 111.49, 116.20, 116.64, 129.95, 135.97, 145.81, 157.31, 158.81, 160.89 (CO); anal. calcd for C15H14N6O4S (374.37): C, 48.12; H, 3.77; N, 22.45. Found: C, 48.28; H, 3.63; N, 22.57.
N), 1725 (CO), 1582 (CN); 1H NMR δ: 1.27–1.30 (t, 3H, J = 6 Hz, C3), 4.27–4.32 (q, 2H, J = 6 Hz, C2), 7.03–7.06 (t, 1H, J = 6 Hz, Carom), 7.59–7.61 (d, 2H, J = 8 Hz, CHarom), 8.01–8.03 (d, 2H, J = 8 Hz, Carom), 8.50–8.51 (d, 2H, J = 4 Hz, Carom), 11.84 (br, 1H, N, D2O exchangeable) 12.45 (br, 1H, NH, D2O exchangeable); 13C NMR δ: 14.35 (CH3), 62.29 (CH2), 106.78 (CN), 111.49, 116.20, 116.64, 129.95, 135.97, 145.81, 157.31, 158.81, 160.89 (CO); anal. calcd for C15H14N6O4S (374.37): C, 48.12; H, 3.77; N, 22.45. Found: C, 48.28; H, 3.63; N, 22.57.Reddish brown solid, yield (69%), m.p. 275–277 °C. IR ν: 3360, 3228, 3188 (NH + NH2), 3041 (C–H aromatic), 2211 (C
 N), 1581 (CN); 1H NMR δ: 7.06–7.08 (t, 1H, J = 4 Hz, Carom), 7.86–7.88 (d, 2H, J = 8 Hz, Carom), 7.93–7.95 (d, 2H, J = 8 Hz, CHarom), 8.51–8.52 (d, 2H, J = 4 Hz, Carom), 9.50 (s, 1H, N2, D2O exchangeable), 9.87 (s, 1H, N2, D2O exchangeable), 11.82 (br, 1H, N, D2O exchangeable); 13C NMR δ: 111.30 (CN), 115.28, 116.31, 116.52, 129.49, 135.25, 146.12, 157.36, 158.86, 188.03 (CS); anal. calcd for C13H11N7O2S2 (361.40): C, 43.20; H, 3.07; N, 27.13. Found: C, 43.32; H, 3.19; N, 27.02.
N), 1581 (CN); 1H NMR δ: 7.06–7.08 (t, 1H, J = 4 Hz, Carom), 7.86–7.88 (d, 2H, J = 8 Hz, Carom), 7.93–7.95 (d, 2H, J = 8 Hz, CHarom), 8.51–8.52 (d, 2H, J = 4 Hz, Carom), 9.50 (s, 1H, N2, D2O exchangeable), 9.87 (s, 1H, N2, D2O exchangeable), 11.82 (br, 1H, N, D2O exchangeable); 13C NMR δ: 111.30 (CN), 115.28, 116.31, 116.52, 129.49, 135.25, 146.12, 157.36, 158.86, 188.03 (CS); anal. calcd for C13H11N7O2S2 (361.40): C, 43.20; H, 3.07; N, 27.13. Found: C, 43.32; H, 3.19; N, 27.02.Yellow solid, yield (77%), m.p. 295–297 °C; IR ν: 3370, 3303, 3214 (NH + NH2), 3088, 3050 (C–H aromatic), 2215 (br, C
 N), 1634 (CN); 1H NMR δ: 7.06–7.09 (t, 1H, J = 4 Hz, Carom), 7.53 (br, 1H, N), 7.76–7.78 (d, 2H, J = 8 Hz, Carom), 8.22–8.24 (d, 2H, J = 8 Hz, Carom), 8.54–8.55 (d, 2H, J = 4 Hz, Carom), 10.04 (br, 3H, NH + NH2); 13C NMR δ: 79.43 (C–(CN)2), 112.91 (CN), 114.48 (CN), 116.02, 117.29 (CN), 127.46. 129.05, 141.51, 144.12, 148.59, 152.58, 157.35, 158.78; anal. calcd for C16H11N9O2S (393.38): C, 48.85; H, 2.82; N, 32.05. Found: C, 48.72; H, 2.67; N, 32.22.
N), 1634 (CN); 1H NMR δ: 7.06–7.09 (t, 1H, J = 4 Hz, Carom), 7.53 (br, 1H, N), 7.76–7.78 (d, 2H, J = 8 Hz, Carom), 8.22–8.24 (d, 2H, J = 8 Hz, Carom), 8.54–8.55 (d, 2H, J = 4 Hz, Carom), 10.04 (br, 3H, NH + NH2); 13C NMR δ: 79.43 (C–(CN)2), 112.91 (CN), 114.48 (CN), 116.02, 117.29 (CN), 127.46. 129.05, 141.51, 144.12, 148.59, 152.58, 157.35, 158.78; anal. calcd for C16H11N9O2S (393.38): C, 48.85; H, 2.82; N, 32.05. Found: C, 48.72; H, 2.67; N, 32.22.Brownish red solid, yield (72%), m.p. 148–150 °C. IR ν: 3225, 3103 (2NH), 3077, 3034 (C–H aromatic), 2981, 2934, 2870, 2806, 2732 (C–H aliphatic), 1722, 1681 (C
 O), 1578 (CN); 1H NMR δ: 1.25–1.30 (m, 6H, 2C3), 4.23–4.35 (qq, 4H, J = 6.6 Hz, 2CH2), 7.02–7.04 (t, 1H, J = 4 Hz, Carom), 7.50–7.52 (d, 2H, J = 8 Hz, CHarom), 7.98–8.00 (d, 2H, J = 8 Hz, CHarom), 8.50–8.51 (d, 2H, J = 4 Hz, CHarom), 11.71 (s, 1H, N), 11.89 (s, 1H, N); 13C NMR δ: 14.33 (CH3), 14.46 (CH3), 61.54 (CH2), 62.01 (CH2), 115.06, 116.20, 125.15, 129.98, 134.75, 146.34, 157.40, 158.75, 161.69 (CO), 162.52 (CO); anal. calcd for C17H19N5O6S (421.42): C, 48.45; H, 4.54; N, 16.62. Found: C, 48.59; H, 4.41; N, 16.53.
O), 1578 (CN); 1H NMR δ: 1.25–1.30 (m, 6H, 2C3), 4.23–4.35 (qq, 4H, J = 6.6 Hz, 2CH2), 7.02–7.04 (t, 1H, J = 4 Hz, Carom), 7.50–7.52 (d, 2H, J = 8 Hz, CHarom), 7.98–8.00 (d, 2H, J = 8 Hz, CHarom), 8.50–8.51 (d, 2H, J = 4 Hz, CHarom), 11.71 (s, 1H, N), 11.89 (s, 1H, N); 13C NMR δ: 14.33 (CH3), 14.46 (CH3), 61.54 (CH2), 62.01 (CH2), 115.06, 116.20, 125.15, 129.98, 134.75, 146.34, 157.40, 158.75, 161.69 (CO), 162.52 (CO); anal. calcd for C17H19N5O6S (421.42): C, 48.45; H, 4.54; N, 16.62. Found: C, 48.59; H, 4.41; N, 16.53.Orange solid, yield (80%), m.p. 298–300 °C. IR ν: 3457, 3302, 3205 (NH + NH2), 3075, 3027 (C–H aromatic), 2935, 2867, 2812, 2734 (C–H aliphatic), 2215 (C
 N), 1574 (CN); 1H δ 2.57 (s, 3H, C3), 7.08 (s, 1H, Carom), 7.03–7.19 (m, 6H, 4Carom + N2, D2O exchangeable), 8.52–8.53 (m, 3H, 2CHarom+ N, D2O exchangeable), 9.19 (br, 1H, NH), 11.91 (br, 1H, N, D2O exchangeable), 13.37 (s, 1H, N, D2O exchangeable); 13C NMR δ: 12.87 (CH3), 79.92 (C–CN), 114.59, 116.16 (2C), 122.51, 129.08, 139.92, 153.79, 154.48, 155.11, 157.32, 158.88, 168.04; anal. calcd for C17H15N9O2S2 (441.49): C, 46.25; H, 3.42; N, 28.55. Found: C, 46.38; H, 3.54; N, 28.41.
N), 1574 (CN); 1H δ 2.57 (s, 3H, C3), 7.08 (s, 1H, Carom), 7.03–7.19 (m, 6H, 4Carom + N2, D2O exchangeable), 8.52–8.53 (m, 3H, 2CHarom+ N, D2O exchangeable), 9.19 (br, 1H, NH), 11.91 (br, 1H, N, D2O exchangeable), 13.37 (s, 1H, N, D2O exchangeable); 13C NMR δ: 12.87 (CH3), 79.92 (C–CN), 114.59, 116.16 (2C), 122.51, 129.08, 139.92, 153.79, 154.48, 155.11, 157.32, 158.88, 168.04; anal. calcd for C17H15N9O2S2 (441.49): C, 46.25; H, 3.42; N, 28.55. Found: C, 46.38; H, 3.54; N, 28.41.Yellow solid, yield (83%), m.p. > 300 °C, IR ν: 3415, 3386, 3341 (NH + NH2), 3039 (C–H aromatic), 2930, 2871, 2816, 2734 (C–H aliphatic), 2208 (C
 N), 1668 (br, CO), 1582 (CN); 1H NMR δ: 2.52 (s, 3H, C3), 7.06–7.09 (t, 1H, J = 4 Hz, Carom), 7.19 (br, 2H, N2), 7.38 (s, 1H, Cpyridyl), 7.65–7.67 (d, 2H, J = 8 Hz, Carom), 8.01–8.03 (d, 2H, J = 8 Hz, Carom), 8.52–8.53 (d, 2H, J = 4 Hz, Carom), 11.38 (s, 1H, N), 11.82 (br, 1H, N), 13.37 (s, 1H, N); anal. calcd for C21H19N9O4S2 (525.56): C, 47.99; H, 3.64; N, 23.99. Found: C, 48.11; H, 3.51; N, 23.83.
N), 1668 (br, CO), 1582 (CN); 1H NMR δ: 2.52 (s, 3H, C3), 7.06–7.09 (t, 1H, J = 4 Hz, Carom), 7.19 (br, 2H, N2), 7.38 (s, 1H, Cpyridyl), 7.65–7.67 (d, 2H, J = 8 Hz, Carom), 8.01–8.03 (d, 2H, J = 8 Hz, Carom), 8.52–8.53 (d, 2H, J = 4 Hz, Carom), 11.38 (s, 1H, N), 11.82 (br, 1H, N), 13.37 (s, 1H, N); anal. calcd for C21H19N9O4S2 (525.56): C, 47.99; H, 3.64; N, 23.99. Found: C, 48.11; H, 3.51; N, 23.83.4.4. Biological evaluation
4.5. Docking studies and ADMET estimation
All procedures were provided in the ESI.†57–625. Conclusion
A new series of sulfadiazine (5–14) was effectively synthesized via diazotization of sulfadiazine with some active methylene compounds. The cytotoxic activity of all products was investigated against A431, A549, and H1975 cancer cell panels via the MTT assay technique, utilizing erlotinib, gefitinib, and osimertinib as positive controls. Specifically, compounds 8, 12, and 14 were revealed to be the most potent analogs on A431, A549, and H1975 cancer cell panels with the IC50 values of (4.12, 4.78, and 2.31 μM), (6.38, 5.15, and 6.12 μM), and (6.15, 7.56, and 4.59 μM) compared to erlotinib (IC50 = 11.09, 17.50, and 12.86 μM), gefitinib (IC50 = 9.60, 15.75, and 10.15 μM), and osimertinib (IC50 = 5.96, NT, and 0.93 μM), respectively. Moreover, compound 12 was the most potent tested one versus both EGFRWT and EGFRT790M protein kinases with IC50 values of 14.5 and 35.4 nM, respectively, compared to gefitinib and osimertinib as reference drugs with IC50 values of 18.2 and 368.2 nM and 57.8 and 8.5 nM, respectively. The molecular-docking results of scaffolds 8, 12, and 14 inside both the EGFRWT and EGFRT790M ATP active sites elucidated their ability to afford the critical fitting interactions made within the kinase domain by the references erlotinib, gefitinib, and osimertinib, respectively. Furthermore, investigating the physicochemical characteristics of the most outstanding EGFR inhibitors, compounds 8, 12, and 14, through the Swiss ADME web server illustrated good GIT absorption, lipophilicity, and low BBB penetration characteristics. The valuable outcomes from this simulation study are that scaffolds 8 and 12 are assumed to be the most promising analogs regarding their dual EGFRWT and EGFRT790M inhibitory activity, as well as promising antiproliferative lead compounds with good pharmacokinetic properties. These findings suggest that these scaffolds should be further investigated and optimized for more potent and selective antiproliferative activity in the future.Data availability
All data generated or analyzed during this study are included in this published article (and its ESI†).Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Deanship of Scientific Research, Vice Presidency for Graduate Studies and Scientific Research, King Faisal University, Saudi Arabia [Grant KFU241525]. The authors are thankful to the Researchers Supporting Project number (RSP2024R516) at King Saud University, Riyadh, Saudi Arabia. The authors would like to thank the Deanship of Scientific Research at Shaqra University for supporting this work.References
- L. Kujtan and J. Subramanian, Epidermal growth factor receptor tyrosine kinase inhibitors for the treatment of non-small cell lung cancer, Expert Rev. Anticancer Ther., 2019, 19(7), 547–559 CrossRef CAS PubMed.
- H. Hayashi, E. Nadal, J. E Gray, A. Ardizzoni, N. Caria, T. Puri and C. Grohe, Overall treatment strategy for patients with metastatic NSCLC with activating EGFR mutations, Clin. Lung Cancer, 2022, 23(1), e69–e82 CrossRef CAS PubMed.
- L. Hu, M. Fan, S. Shi, X. Song, F. Wang, H. He and B. Qi, Dual target inhibitors based on EGFR: Promising anticancer agents for the treatment of cancers, Eur. J. Med. Chem., 2022, 227, 113963 CrossRef CAS PubMed.
- A. Y. Helena and M. E Arcila, Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers, Clin. Cancer Res., 2013, 19(8), 2240–2247 CrossRef PubMed.
- P. C. C. Lv, F. Zhou and J. Chen, Design, synthesis and biological evaluation of thiazolidinone derivatives as potential EGFR and HER-2 kinase inhibitors, Bioorg. Med. Chem., 2010, 18(1), 314–329 CrossRef CAS PubMed.
- H. Gourdeau, L. Leblond, B. Hamelin, C. Desputeau, K. Dong, I. Kianicka, D. Custeau, C. Boudreau, L. Geerts and S.-X. Cai, Antivascular and antitumor evaluation of 2-amino-4-(3-bromo-4, 5-dimethoxy-phenyl)-3-cyano-4 H-chromenes, a novel series of anticancer agents, Mol. Cancer Ther., 2004, 3, 1375–1384 CrossRef CAS PubMed.
- S. Sogabe, Y. Kawakita, S. Igaki, H. Iwata, H. Miki, D. R. Cary, T. Takagi, S. Takagi, Y. Ohta and T. Ishikawa, Structure-Based Approach for the Discovery of Pyrrolo[3,2- d ]pyrimidine-Based EGFR T790M/L858R Mutant Inhibitors, ACS Med. Chem. Lett., 2013, 4, 201–205 CrossRef CAS PubMed.
- J. Xu, J. Wang and S. Zhang, et al., Mechanisms of resistance to irreversible epidermal growth factor receptor tyrosine kinase inhibitors and therapeutic strategies in non-small cell lung cancer, Oncotarget, 2017, 8(52), 90557 CrossRef PubMed.
- M. G. Kumar, S. Arora and G. Joshi, et al., Design and Synthesis of Non-Covalent Imidazo [1, 2-a] quinoxaline-Based Inhibitors of EGFR and Their Anti-Cancer Assessment, Molecules, 2021, 26(5), 1490–1503 CrossRef CAS PubMed.
- A. N. Hata, M. J. Niederst and H. L Archibald, Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition, Nat. Med., 2016, 22(3), 262–269 CrossRef CAS PubMed.
- M. O. Aboelez, A. Belal, G. Xiang and X. Ma, Design, synthesis, and molecular docking studies of novel pomalidomide-based PROTACs as potential anti-cancer agents targeting EGFRWT and EGFRT790M, J. Enzyme Inhib. Med. Chem., 2022, 37, 1196–1211 CrossRef PubMed.
- X. Zhang, F. Xu and L. Tong, et al., Design and synthesis of selective degraders of EGFRL858R/T790M mutant, Eur. J. Med. Chem., 2020, 192, 112199 CrossRef CAS PubMed.
- M. S. Kamel, A. Belal, M. O. Aboelez, E. K. Shokr, H. Abdel-Ghany, H. S. Mansour, A. M. Shawky and M. A. El-Remaily, Microwave-Assisted Synthesis, Biological Activity Evaluation, Molecular Docking, and ADMET Studies of Some Novel Pyrrolo [2, 3-b] Pyrrole Derivatives, Molecules, 2022, 27(7), 2061 CrossRef CAS PubMed.
- M. E. Shoman, M. O. Aboelez, M. S. A. Shaykhon, S. A. Ahmed, G. E.-D. A. Abuo-Rahma and O. M. Elhady, New nicotinic acid-based 3, 5-diphenylpyrazoles: Design, synthesis and antihyperlipidemic activity with potential NPC1L1 inhibitory activity, Mol. Diversity, 2021, 25, 673–686 CrossRef CAS PubMed.
- T. Yang, W. Zhang and S. Cao, Discovery of highly potent and selective EGFR(T790M/L858R) TKIs against NSCLC based on molecular dynamic simulation, Eur. J. Med. Chem., 2022, 228, 113984 CrossRef CAS PubMed.
- F. Sherbiny, A. Bayoumi and A. El-Morsy, Design, Synthesis, biological Evaluation, and molecular docking studies of novel Pyrazolo[3,4-d]Pyrimidine derivative scaffolds as potent EGFR inhibitors and cell apoptosis inducers, Bioorg. Chem., 2021, 5(116), 105325 CrossRef PubMed.
- S. A. Ahmed, M. S. Kamel, M. O. Aboelez, X. Ma, A. A. Al-Karmalawy, S. A. Mousa, E. K. Shokr, H. Abdel-Ghany, A. Belal and M. A. El Hamd, Thieno [2, 3-b] thiophene derivatives as potential EGFRWT and EGFRT790M inhibitors with antioxidant activities: Microwave-assisted synthesis and quantitative in vitro and in silico studies, ACS Omega, 2022, 7, 45535–45544 CrossRef CAS PubMed.
- T. Yadav, S. G. Moin and M. A. Kumar, Review on Fused Pyrimidine Systems as EGFR Inhibitors and Their Structure-Activity Relationship, Front. Chem., 2022, 2(10), 861288 CrossRef PubMed.
- M. M. Elsayed, M. O. Aboelez, B. E. Elsadek, H. A. Sarhan, K. A. Khaled, A. Belal, A. Khames, Y. A. Hassan, A. A. Abdel-Rheem and E. B. Elkaeed, Tolmetin sodium fast dissolving tablets for rheumatoid arthritis treatment: preparation and optimization using Box-Behnken design and response surface methodology, Pharmaceutics, 2022, 14, 880 CrossRef CAS PubMed.
- N. Jain, J. Xu, R. M. Kanojia, F. Du, G. Jian-Zhong, E. Pacia, M.-T. Lai, A. Musto, G. Allan, G. Reuman and M. Cousineau, Identification and Structure− Activity Relationships of Chromene-Derived Selective Estrogen Receptor Modulators for Treatment of Postmenopausal Symptoms, J. Med. Chem., 2009, 52, 7544–7569 CrossRef CAS PubMed.
- A.-E.-B. A Ghattas, A. Khodairy, H. M. Moustafa, B. R. Hussein, M. M. Farghaly and M. O. Aboelez, Synthesis, in vitro antibacterial and in vivo anti-inflammatory activity of some new pyridines, Pharm. Chem. J., 2017, 51, 652–660 CrossRef.
- A. H. Moustafa, W. W. Ahmed, M. F. Awad, M. O. Aboelez, A. Khodairy and A. A. Amer, Eco-friendly and regiospecific intramolecular cyclization reactions of cyano and carbonyl groups in N, N-disubstituted cyanamide, Mol. Diversity, 2022, 26(5), 2813–2823 CrossRef CAS PubMed.
- L. Kujtan and J. Subramanian, Epidermal growth factor receptor tyrosine kinase inhibitors for the treatment of non-small cell lung cancer, Expert Rev. Anticancer Ther., 2019, 19(7), 547–559 CrossRef CAS PubMed.
- H. Ezelarab, H. Hassan, S. Abbas, T. Ali and E. Beshr, The main biotargets of indole or 2-oxoindole-based hybrids acting as promising antiproliferative agents, J. Adv. Biomedical Pharm. Sci., 2023, 6, 174–183, DOI:10.21608/jabps.2023.217559.1190.
- H. A. A. Ezelarab, et al., New antiproliferative 3-substituted oxindoles inhibiting EGFR/VEGFR-2 and tubulin polymerization, Mol. Diversity, 2024, 28, 563–580 CrossRef CAS PubMed.
- M. O. Aboelez, M. S. Kamel, A. Belal, A.E. Abdel-Aziz, H. Abourehab, A. M. Abdel-Ghany, E. Hamd and M. A. E. A. El-Remaily, Microwave-assisted synthesis, spectroscopic characterization, and biological evaluation of fused thieno [2, 3-d] pyrimidines as potential anti-cancer agents targeting EGFRWT and EGFRT790M, Mol. Diversity, 2022, 27(2) DOI:10.1007/s11030-022-10477-7.
- R. Rosell and E. Carcereny, et al., Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial, Lancet Oncol., 2012, 13(3), 239–246 CrossRef CAS PubMed.
- V. Di Noia, A. D'Aveni, E. D'Argento, S. Rossi, P. Ghirardelli, P. L. Bortolotti, V. Vavassori, E. Bria and G. Ceresoli, Treating disease progression with osimertinib in EGFR-mutated non-small-cell lung cancer: novel targeted agents and combination strategies, ESMO Open, 2021, 6(6), 100280 CrossRef CAS PubMed.
- G. Xu and M. C. Abad, et al., 4-Amino-6-arylamino-pyrimidine-5-carbaldehyde hydrazones as potent ErbB-2/EGFR dual kinase inhibitors, Bioorg. Med. Chem. Lett., 2008, 18, 4615–4619 CrossRef CAS PubMed.
- M. S. Kamel, M. O. Aboelez, M. R. Elnagar, E. K. Shokr, H. M. R. M. Selim, H. E. Selim, H. E. Abdel-Ghany, A. M. A. Drar, M. A. BelalEl Hamd and A. A. Abd El Aleem, Green Synthesis Design, Spectroscopic Characterizations, and Biological Activities of Novel Pyrrole Derivatives: An Application to Evaluate Their Toxic Effect on Cotton Aphids, ChemistrySelect, 2022, 7(40), e202203191 CrossRef CAS.
- S. Sigismund, D. Avanzato and L. Lanzetti, Emerging functions of the EGFR in cancer, Mol. Oncol., 2018, 12(1), 3–20 CrossRef PubMed.
- H. A. A. Ezelarab, S. H. Abbas, M. A. S. Abourehab, M. Badr, S. Sureram, P. Hongmanee, P. Kittakoop, G. E.-D. A. Abuo-Rahma and H. A. Hassan, Novel antimicrobial ciprofloxacin-pyridinium quaternary ammonium salts with improved physicochemical properties and DNA gyrase inhibitory activity, Med. Chem. Res., 2021, 30, 2168–2183, DOI:10.1007/s00044-021-02798-3.
- F. Francesca, T. Annachiara, C. Valentina, P. Luca, M. Luca, P. Daniele and R. Giulio, Structure-activity exploration of a small-molecule allosteric inhibitor of T790M/L858R double mutant EGFR, J. Enzyme Inhib. Med. Chem., 2023, 38(1), 239–245 CrossRef PubMed.
- E. Abdelkreem, S. M. Mahmoud, M. O. Aboelez and M. Abd El Aal, Nebulized magnesium sulfate for treatment of persistent pulmonary hypertension of newborn: a pilot randomized controlled trial, Indian J. Pediatr., 2021, 88, 771–777 CrossRef PubMed.
- K. K. Ang, B. A. Berkey and X. Tu, et al., Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma, Cancer Res., 2002, 62(24), 7350–7356 CAS.
- P. Matada, N. Abbas and S. Dhiwar, et al., Design, Synthesis, In Silico and In Vitro Evaluation of Novel Pyrimidine Derivatives as EGFR Inhibitors, Anti-Cancer Agents Med. Chem., 2021, 21(4), 451–461 CrossRef PubMed.
- H. A. El-Sayeda, A. H Moustafaa, A. A. Faddab and K. E. Abd El-Rahmana, Pyrazole and Nicotinonitrile Derivatives Synthesized from Sulfa Drugs, and Their Antibacterial Activity, Russ. J. Gen. Chem., 2019, 89(2), 339–347 CrossRef.
- K. Terufumi, C. Ignacio and C. Manuel, et al., Targeted treatment for unresectable EGFR mutation-positive stage III non-small cell lung cancer: Emerging evidence and future perspectives, Lung Cancer, 2024, 187, 107414, DOI:10.1016/j.lungcan.2023.107414.
- W. Yahua, D. Bin., L. Chengliu, J. Xiaohui and L. Jinhuo, LAPS score for individualized treatment of advanced EGFR-mutated non-small cell lung cancer receiving EGFR-TKIs with or without bevacizumab, Ann. Med., 2023, 55(2), 2257227, DOI:10.1080/07853890.2023.2257227.
- C. H. Dai and Y. Shu, et al., YM155 sensitizes non-small cell lung cancer cells to EGFR-tyrosine kinase inhibitors through the mechanism of autophagy induction, Biochim. Biophys. Acta, Mol. Basis Dis., 2018, 1864(12), 3786–3798 CrossRef CAS PubMed.
- A. Kassab, Pyrazolo[3,4-d]pyrimidine scaffold: A review on synthetic approaches and EGFR and VEGFR inhibitory activities, Arch. Pharm., 2023, 356(1), 2200424 CrossRef CAS PubMed.
- A. Gazdar, Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors, Oncogene, 2009, 28(1), S24–S31 CrossRef CAS PubMed.
- Y. Guo, L. Yang and L. Liu, et al., Comparative study of clinicopathological characteristics and prognosis between combined and pure small cell lung cancer (SCLC) after surgical resection, Thorac. Cancer, 2020, 11(10), 2782–2792 CrossRef CAS PubMed.
- X. Wang, Y. Guo and L. Liu, et al., YAP1 protein expression has variant prognostic significance in small cell lung cancer (SCLC) stratified by histological subtypes, Lung Cancer, 2021, 2(160), 166–174 Search PubMed.
- A. Belal, M. A. Elanany, E. Y Santali, A. A. Al-Karmalawy, M. O. Aboelez, A. H. Amin, M. H. Abdellattif, A. B. Mehany and H. Elkady, Screening a Panel of Topical Ophthalmic Medications against MMP-2 and MMP-9 to Investigate Their Potential in Keratoconus Management, Molecules, 2022, 27, 3584 CrossRef CAS PubMed.
- W. Pao, V. A. Miller and K. A. Politi, et al., Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain, PLoS Med., 2005, 2(3), 73–92 CrossRef PubMed.
- H. A. A. Ezelarab, T. F. S. Ali, S. H. Abbas, H. A. Hassan and E. A. M. Beshr, Indole-based FLT3 inhibitors and related scaffolds as potential therapeutic agents for acute myeloid leukemia, BMC Chem., 2023, 17, 73 CrossRef CAS PubMed.
- M. M. A. Elsayed, M. O. Aboelez, M. S. Mohamed, R. A. Mahmoud and A. A. El-Shenawy, et al., Tailoring of Rosuvastatin Calcium and Atenolol Bilayer Tablets for the Management of Hyperlipidemia Associated with Hypertension: A Preclinical Study, Pharmaceutics, 2022, 14(8), 1629 CrossRef CAS PubMed.
- B. Kaghazchi, L. Um and M. Elshani, Spatial Analysis of NQO1 in Non-Small Cell Lung Cancer Shows Its Expression Is Independent of NRF1 and NRF2 in the Tumor Microenvironment, Biomolecules, 2022, 12(11), 122–135 CrossRef PubMed.
- D. Todsaporn, A. Zubenko, V. Kartsev, T. Aiebchun, P. Mahalapbutr, A. Petrou, A. Geronikaki, L. Divaeva, V. Chekrisheva, L. Yildiz, K. Choowongkomon and T. Rungrotmongkol, Discovery of Novel EGFR Inhibitor Targeting Wild-Type and Mutant Forms of EGFR: In Silico and In Vitro Study, Molecules, 2023, 28(7), 3014 CrossRef CAS PubMed.
- S. Sogabe, Y. Kawakita, S. Igaki, H. Iwata, H. Miki, D. R. Cary, T. Takagi, S. Takagi, Y. Ohta and T. Ishikawa, Structure-Based Approach for the Discovery of Pyrrolo[3,2- d ]Pyrimidine-Based EGFR T790M/L858R Mutant Inhibitors, ACS Med. Chem. Lett., 2013, 4(2), 201–205 CrossRef CAS PubMed.
- M. S. Abdelbaset, M. Abdel-Aziz and M. Ramadan, Discovery of novel thienoquinoline-2-carboxamide chalcone derivatives as antiproliferative EGFR tyrosine kinase inhibitors, Bioorg. Med. Chem., 2019, 27, 1076–1086 CrossRef CAS PubMed.
- F. Doganc, I. Celik, G. Eren, M. Kaiser, R. Brun and H. Goker, Synthesis, in vitro antiprotozoal activity, molecular docking and molecular dynamics studies of some new monocationic guanidinobenzimidazoles, Eur. J. Med. Chem., 2021, 221, 113545 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7, 1–13 CrossRef PubMed.
- A. Mavani, A. Ovung and A. Luikham, et al., Biophysical and molecular modeling evidences for the binding of sulfa molecules with hemoglobin, J. Biomol. Struct. Dyn., 2023, 41(9), 3779–3790 CrossRef CAS PubMed.
- K. AL Azzam, SwissADME and pkCSM Webservers Predictors: an integrated Online Platform for Accurate and Comprehensive Predictions for In Silico ADME/T Properties of Artemisinin and its Derivatives, Compl. Use of Min. Resour., 2022, 325(2), 14–21 Search PubMed.
- A. Daina and V. Zoete, A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules, ChemMedChem, 2016, 11, 1117–1121 CrossRef CAS PubMed.
- C. A. Lipinski, F. B. Lombardo, W. Dominy and P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Delivery Rev., 2012, 64, 4–17 CrossRef.
- L. Yuanyuan, K. Wang, M. Jiaxuan, W. Zhengcong, Z. Qiujin and J. Yongguo, Effect of yolk spheres as a key histological structure on the morphology, character, and oral sensation of boiled egg yolk gel, Food Chem., 2023, 424, 136380 CrossRef PubMed.
- H. A. A. Ezelarab, H. A. Hassan, G. E.-D. A. Abuo-Rahma and S. H. Abbas, Design, synthesis, and biological investigation of quinoline/ciprofloxacin hybrids as antimicrobial and anti-proliferative agents, J. Iran. Chem. Soc., 2023, 20, 683–700 CrossRef CAS.
- M. O. Aboelez, H. A. A. Ezelarab and G. Alotaibi, et al., Inflammatory setting, therapeutic strategies targeting some pro-inflammatory cytokines and pathways in mitigating ischemia/reperfusion-induced hepatic injury: a comprehensive review, Naunyn-Schmiedeberg's Arch. Pharmacol., 2024, 1, 17 Search PubMed.
- M. A. E. El-Remaily, M. O. Aboelez and H. A. A. Ezelarab, Guanidine dicycloamine-based analogs: green chemistry synthesis, biological investigation, and molecular docking studies as promising antibacterial and antiglycation leads, Mol. Diversity, 2024, 1, 23 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: IR spectra, 1H NMR spectra, and 13CNMR spectra. See DOI: https://doi.org/10.1039/d4ra04165h |
| ‡ Equal contributed. |
| This journal is © The Royal Society of Chemistry 2024 |