
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZinc acetate-catalyzed, green and efficient synthesis of xanthene derivatives under ultrasound irradiation: X-ray crystallographic analysis and in silico study†
Rayene Sayada,
Abdeslem Bouzina*a,
Yousra Ouafa Bouonea,
Dounia Beldjezziaa,
Abdelhak Djemelb,
Malika Ibrahim-Ouali c,
Nour-Eddine Aoufa and
Zineb Aouf*a
c,
Nour-Eddine Aoufa and
Zineb Aouf*a
aLaboratory of Applied Organic Chemistry, Bioorganic Chemistry Group, Sciences Faculty, Chemistry Department, Badji Mokhtar-Annaba University, Box 12, 23000 Annaba, Algeria. E-mail: zineb.aouf@univ-annaba.dz
bResearch Unit in Medicinal Plants, URPM, Attached to Research Center of Biotechnology, CRBt, 3000 Laghouat, 25000 Constantine, Algeria
cAix Marseille Univ., CNRS, Marseille F-13397, France
First published on 6th August 2024
Abstract
A simple, one-pot method using zinc acetate and ultrasound irradiation has been developed to synthesize xanthene derivatives from cyclic diketones and aromatic aldehydes, yielding good to excellent results. This method offers advantages like mild conditions, high atom economy, easy isolation, and a recyclable catalyst. All xanthene derivatives, including two new molecules, were confirmed using standard spectroscopic methods, with X-ray crystallographic data provided for compound 3r. The synthesized molecules were shown to inhibit the VEGFR-2 enzyme, confirmed by molecular docking studies. A 200 ns molecular dynamics simulation validated these findings, showing significant stability for the 3e-VEGFR-2 complex after 1 ns and the 3p-VEGFR-2 complex for 8 ns. DFT calculations were used to analyze electronic and geometric properties, including HOMO and LUMO bandgap energies and molecular electrostatic potential surfaces. Additionally, the absorption, distribution, metabolism, and excretion (ADME) properties of the synthesis compounds were assessed.
Introduction
Xanthenes represent a distinct class of tricyclic compounds that incorporate oxygen, and they are encountered in natural products, synthetic bioactive substances, and fluorescent dyes.1 Xanthene derivatives have garnered considerable attention in the fields of medicinal chemistry and organic synthesis transformations over recent decades.2 It is noted that many reviews focusing on the synthetic strategies and biological activities of xanthene derivatives have been published.3–6 These molecules exhibit a range of biological activities, including anti-tumor,7 anticancer,8 antibacterial,9 antiviral,10 antifungal,11 analgesic and anti-inflammatory properties.12 Moreover, owing to their advantageous spectroscopic characteristics, they find applications as dyes in laser technology13 and fluorescent materials for biomolecule visualization within organisms.14 Given the substantial interest in xanthene derivatives, a multitude of synthetic protocols has emerged due to their diverse array of applications. Over the past two decades, various methods and numerous catalysts have been documented for xanthene derivatives synthesis such as CsF,15 nanoparticles,16 L-proline,17 ionic liquid,18 tetra-n-butylammonium fluoride,19 CeCl3·7H2O,20 cellulose sulfuric acid,21 TMSCl,22 2-pyridonium hydrogen sulfate,23 β-CD-BSA,24 Co(HSO4)2,25 NaHSO4·SiO2,26 2,4,6-trichloro-1,3,5-triazine,27 as well as SmCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 28 have been employed in these processes. However, many of these methods exhibit various drawbacks, including prolonged reaction times, costly catalysts, harsh conditions, low product yields, and the use of toxic organic solvents.
28 have been employed in these processes. However, many of these methods exhibit various drawbacks, including prolonged reaction times, costly catalysts, harsh conditions, low product yields, and the use of toxic organic solvents.
Recently, Zn(OAc)2 has emerged as a remarkable Lewis acid catalyst for organic synthesis,29 demonstrating excellent results. In contrast to traditional Lewis acids, Zn(OAc)2 present several advantages, including widespread commercial availability, low price, recyclability, ease of use as an organo-catalyst, environmental friendliness, and a high degree of compatibility with oxygen- and nitrogen-containing substrates and various functional groups.30
As part of our ongoing research efforts aimed at pioneering environmentally sustainable methodologies, we report a green and efficient synthesis of xanthene derivatives catalyzed by zinc acetate using ultrasound irradiation, which is a green source of energy for activating chemical reactions.
Further, all synthesized compounds were subjected to in silico molecular docking study and molecular dynamics simulation against the vascular endothelial growth factor receptor-2 (VEGFR-2) enzyme to assess their potential for inhibiting it. VEGFR-2 is one of the human enzymes that are considered as interesting targets for drug development of cancer chemotherapy. It plays a crucial role in regulating of tumor angiogenesis with VEGFR-2-related kinases considering as the principal transducers of VEGF-dependent angiogenesis. Consequently, the inhibition of the VEGF/VEGFR signalling pathway is deemed a promising therapeutic strategy for impeding tumor angiogenesis and subsequent tumor growth.31
Furthermore, we conducted a comprehensive investigation of the chemical reactivity of the studied compounds using DFT calculations. Additionally, ADME analyses were carried out to evaluate the physicochemical properties and performance of the tested compounds.
Results and discussion
Chemistry
In recent years, the combination of sonochemistry with catalytic systems has become a powerful strategy that integrates the advantages of ultrasound and catalysis to enhance chemical reactions. This synergistic approach has demonstrated significant benefits in various fields, including organic synthesis, pharmaceuticals industries, environmental chemistry, and materials sciences.32 This combined system provides advantages that include reduced reaction duration, improved heat and mass transfer, resulting in increased chemical reaction rates, yields, and selectivity.33In this context, several methodologies have been documented for the synthesis of xanthene compounds from dimedone and aldehydes, using a combination of sonochemistry and catalytic system under various reaction conditions. Table 1 provides a summary of selected studies described in the literature.
| No. | Catalysts | Conditionsa | Times (min)/yields (%) |
|---|---|---|---|
| a All reactions was achieved with benzaldehyde and dimedone (mole rate 1:2). |
|||
| 1 | Ceric(IV) ammonium nitrate CAN (5 mol%) | US (40 kHz), 50 °C, isopropanol (2 mL) | 35/98 (ref. 34) |
| 2 | MCM-41-SO3H (5 mol%) | US (25 kHz), 60 °C, water (5 mL) | 60/95 (ref. 35) |
| 3 | TiO2 (5 mol%) | US (50 kHz), r.t., ACN (2 mL) | 6/90 (ref. 36) |
| 4 | Melamine trisulfonic acid MSTA (7.5 mol%) | US (34–37 kHz), 60 °C, ethyl acetate (3 mL) | 110/86 (ref. 37) |
| 5 | 2-Pyrodidonium hydrogen sulfate [H-NMP]+[HSO4]− (20 mol%) | US (20 kHz), 60 °C, water (20 mL) | 50/86 (ref. 38) |
| 6 | 1-Carboxymethyl-3-methylimidazolium tetrafluoroborate [cmmim][BF4] (200 mg) | US (50 kHz), 30 °C, methanol (1 mL) | 50/87 (ref. 39) |
| 7 | Zinc acetate Zn(OAc)2 (10 mol%) | US (40 kHz), 60 °C, EtOH (2 mL) | 15/95 (our work) |
Based on previous research, our objective in this study was to develop an innovative method for the synthesis of xanthene derivatives using Zn(OAc)2/ultrasonic irradiation as a combined system. To facilitate the optimization of the model reaction, we selected dimedone (1a) and benzaldehyde (2a) as model reactants while subjecting then to ultrasonic irradiation. We then investigated the influence of Zn(OAc)2 under various reaction conditions (Scheme 1 and Table 2).
 | ||
| Scheme 1 Zn(OAc)2/US as a combined system for the preparation of xanthene derivatives. | ||
| Entry | Catalyst (mol%) | Solvents | Conditionsa | Times (min)/yieldsb (%) |
|---|---|---|---|---|
| a Reaction conditions: dimedone (2 equiv.), benzaldehyde (1 equiv.).b Isolated yields: the yields were calculated after recrystallization in diethyl ether/n-hexane in (6:4). |
||||
| 1 | Catalyst-free | Neat | US (40 kHz), r.t. | 240/trace |
| 2 | Zn(OAc)2 (2 mol%) | Neat | US (40 kHz), r.t. | 120/25 |
| 3 | Zn(OAc)2 (5 mol%) | Neat | US (40 kHz), r.t. | 120/50 |
| 4 | Zn(OAc)2 (10 mol%) | Neat | US (40 kHz), r.t. | 120/69 |
| 5 | Zn(OAc)2 (15 mol%) | Neat | US (40 kHz), r.t. | 120/69 |
| 6 | Zn(OAc)2 (10 mol%) | DCM (2 mL) | US (40 kHz), r.t. | 30/78 |
| 7 | Zn(OAc)2 (10 mol%) | THF (2 mL) | US (40 kHz), r.t. | 30/71 |
| 8 | Zn(OAc)2 (10 mol%) | Toluene (2 mL) | US (40 kHz), r.t. | 30/74 |
| 9 | Zn(OAc)2 (10 mol%) | ACN (2 mL) | US (40 kHz), r.t. | 30/70 |
| 10 | Zn(OAc)2 (10 mol%) | Water (2 mL) | US (40 kHz), r.t. | 30/82 |
| 11 | Zn(OAc)2 (10 mol%) | EtOH (2 mL) | US (40 kHz), r.t. | 30/87 |
| 12 | Zn(OAc)2 (10 mol%) | EtOH (2 mL) | US (40 kHz), 40 °C | 15/90 |
| 13 | Zn(OAc)2 (10 mol%) | EtOH (2 mL) | US (40 kHz), 60 °C | 15/95 |
| 14 | Zn (OAc)2 (10 mol%) | EtOH (2 mL) | Without US (agitation, r.t.) | 120/trace |
| 15 | Zn(OAc)2 (10 mol%) | EtOH (2 mL) | Without US (agitation, reflux) | 120/35 |
| 16 | Zn(OTf)2 (10 mol%) | EtOH (2 mL) | Without US (agitation, r.t.) | 120/trace |
| 17 | Zn(OTf)2 (10 mol%) | EtOH (2 mL) | US (40 kHz), 60 °C | 120/15 |
Based on previous research, our objective in this study was to develop an innovative method for the synthesis of xanthene derivatives using Zn(OAc)2/ultrasonic irradiation as a combined system.
To facilitate the optimization of the model reaction, we selected dimedone (1a) and benzaldehyde (2a) as model reactants while subjecting then to ultrasonic irradiation. We then investigated the influence of Zn(OAc)2 under various reaction conditions (Scheme 1 and Table 2).
Initially, the reaction was performed in catalyst and solvent free conditions, with the application of ultrasound irradiation (40 kHz) at room temperature. A trace yield of the desired product (3a) was attained after long reaction time (entry 1, Table 2). These adverse outcomes encouraged us to employ a commercially accessible, inexpensive and environmentally friendly catalyst such as zinc acetate Zn(OAc)2. For this reason, various concentrations of zinc acetate, specifically at 2, 5, 10, and 15 mol% (entry 2–5, Table 2) were tested in our study. The results showed that the reaction was less efficient when the catalyst was used at a concentration of 2 or 5 mol%, even though the reaction was subjected for a longer time. Simultaneously, when the catalyst concentration was set at 10 or 15 mol%, a good product yield was achieved. Thus, the utilization of 10 mol% of Zn(OAc)2 proves adequate to promote the reaction. Then, to evaluate the influence of the solvent, several solvents such as dichloromethane, tetrahydrofuran, toluene, acetonitrile, water and ethanol were applied for the model reaction (entry 6–11, Table 2), the desired product (3a) was produced with yields ranging from (70–87%) after 30 min. As evidenced in Table 2, ethanol proved to be the most effective solvent for the reaction, with the highest yield of product (3a).
Additionally, we explored the impact of temperature variation by employing different temperature settings in this reaction (entry 12 and 13, Table 2). The highest yield, reaching 95%, was achieved at 60 °C in 15 minutes under ultrasound irradiation, representing an improvement in yield compared to the use of 40 °C and room temperature.
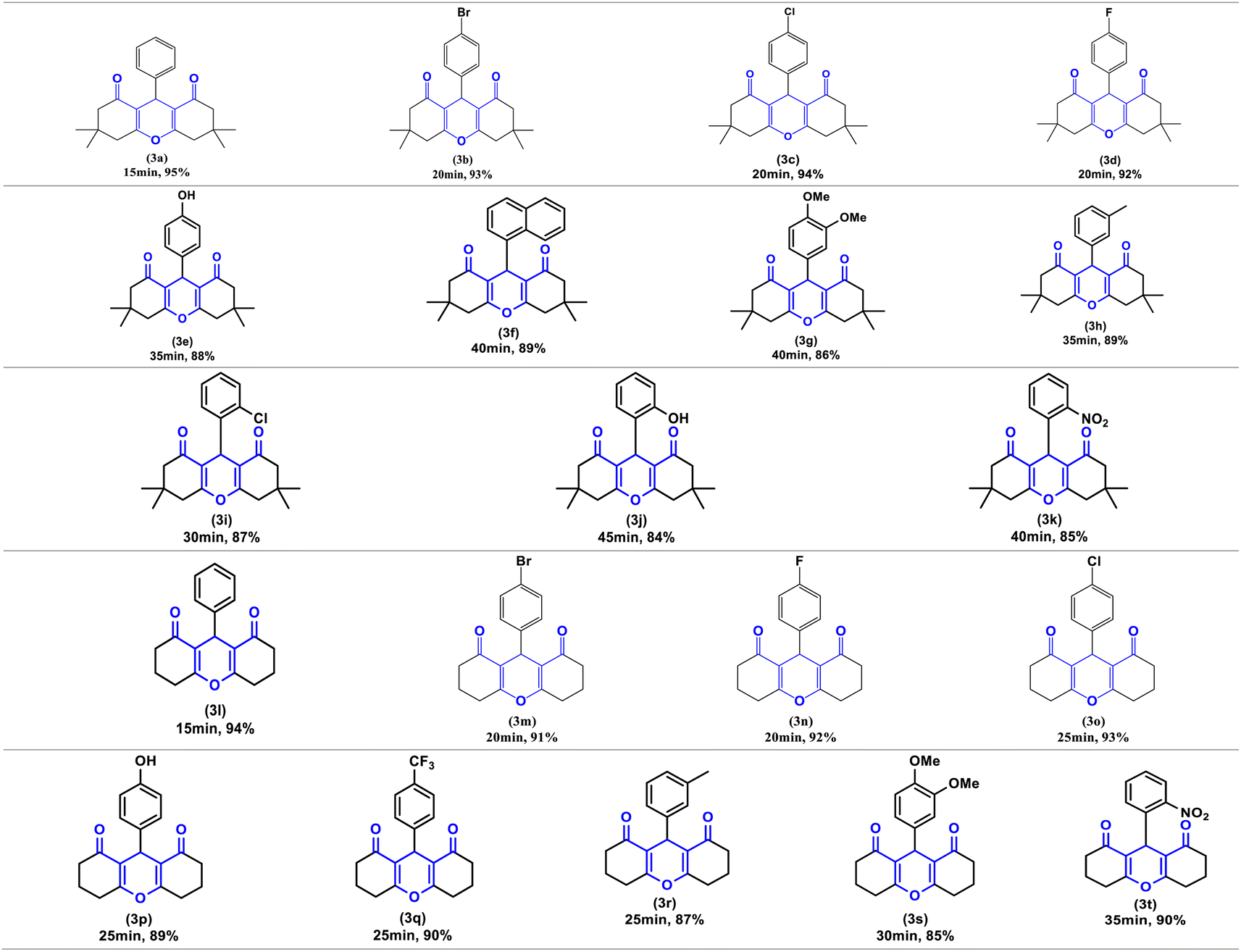
Finally, to demonstrate the influence of ultrasonic irradiation in this reaction, we examined the synthesis of (3a) in the presence of 10 mol% Zn(OAc)2 in ethanol without employing ultrasonic irradiation, both at room temperature and under reflux conditions (entry 14 and 15, Table 2). In the absence of sonication, the formation of product (3a) at 60 °C resulted in a low yield (35%), and at room temperature, the reaction did not progress. Based on these findings, ultrasound irradiation emerges as a promising approach to enhance the synthesis of the xanthene derivatives under conditions previously reported. To investigate the efficiency and versatility of Zn(OAc)2/ultrasound as a combined system for the synthesis of 1,8-dioxo-octahydroxanthenes derivatives. Numerous aromatic aldehydes with different substituents, were subjected with dimedone or 1,3-cyclohexanedione in the optimal reaction conditions to produce the desired products (3a–3t) in excellent yields (95–84%) between (15–45 min), the resulting products are outlined in Table 3.
| a Reaction conditions: dimedone or 1,3-cyclohexanedione (2 equiv.), aldehydes (1 equiv.); the yields were calculated after recrystallization in diethyl ether/n-hexane in (6:4). |
|---|
 |
To investigate the influence of the catalytic anion, the acetate ion, we synthesized xanthene using a zinc precursor catalyst under similar reaction conditions, both with and without ultrasound irradiation. Initially, we attempted the reaction without ultrasound irradiation using zinc triflate as the catalyst at room temperature (entry 16, Table 2), but no product was observed after 2 hours. We then repeated the procedure with ultrasound irradiation and zinc triflate in ethanol (entry 17, Table 2), which produced a small amount of the desired compound within 2 hours, likely due to residual starting material in the reaction mixture. The reaction progress was monitored using thin-layer chromatography (TLC) on silica gel plates with a dichloromethane/methanol (9:1) eluent.
Notably, when comparing zinc triflate with zinc acetate under similar reaction conditions, we identified two major drawbacks. Firstly, zinc triflate acts as a homogeneous catalyst dissolved in ethanol, while zinc acetate is a heterogeneous catalyst that can be easily recovered by filtration. Moreover, the use of zinc acetate is more cost-effective compared to zinc triflate (in terms of time, purification methods, and yields).
As illustrated in this table, electron-withdrawing and electron-donating substituents on the aromatic ring of aldehydes can partially influence reaction times and yields. Aldehydes with electron-withdrawing groups produce the highest yields (94–90%) in the shortest amount of time (20–25 min), compared to aldehydes with electron-donating substituents. Additionally, substituted aldehydes in para positions were found to be more reactive than the ortho and meta positions. The highest yield (95%) was obtained with benzaldehyde (3a) in 15 minutes, while the lowest yield (84%) was obtained with 2-hydroxybenzaldehyde (3j) in 45 minutes.
Furthermore, compounds 3e, 3j and 3p bearing hydroxyl groups did not react with zinc acetate due to the delocalization of the free electron pairs of the hydroxyl groups, which participate in the mesomerism of the aromatic nuclei, weakening their reactivity. Consequently, we obtain only one product, that of condensation, characterized by usual spectroscopic methods.
The usual spectroscopic methods, including IR and NMR (1H, 13C), were employed to confirm the structures of all the compounds. The IR spectra of compounds (3a–3t) showed absorption bands at 1665 ± 30 cm−1 and 1450 ± 30 cm−1, which are attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CC stretching vibrations, respectively. The CH proton signal (between two dimedone or 1,3-cyclohexanedione) of all compounds appeared as a singlet in the 1H NMR spectrum, in the range of 4.80–4.20 ppm. In the 13C NMR spectrum, the carbon situated at the bridge between two dimedone or cyclohexanedione rings exhibits a chemical shift at ∼50.6 ppm and the carbonyl carbon appears approximately at ∼196.3 ppm.
O and CC stretching vibrations, respectively. The CH proton signal (between two dimedone or 1,3-cyclohexanedione) of all compounds appeared as a singlet in the 1H NMR spectrum, in the range of 4.80–4.20 ppm. In the 13C NMR spectrum, the carbon situated at the bridge between two dimedone or cyclohexanedione rings exhibits a chemical shift at ∼50.6 ppm and the carbonyl carbon appears approximately at ∼196.3 ppm.
Zinc acetate can act as a mild Lewis acid, coordinating with the carbonyl oxygen of aldehydes. This increases the carbonyl carbon's electrophilicity to some extent, facilitating nucleophilic attack. While less common than stronger Lewis acids for direct activation, zinc acetate finds use in specific reactions due to its advantageous properties like chemoselectivity. The versatility of zinc acetate is illustrated by the numerous reviews dedicated to its applications in organic synthesis and catalysis.40
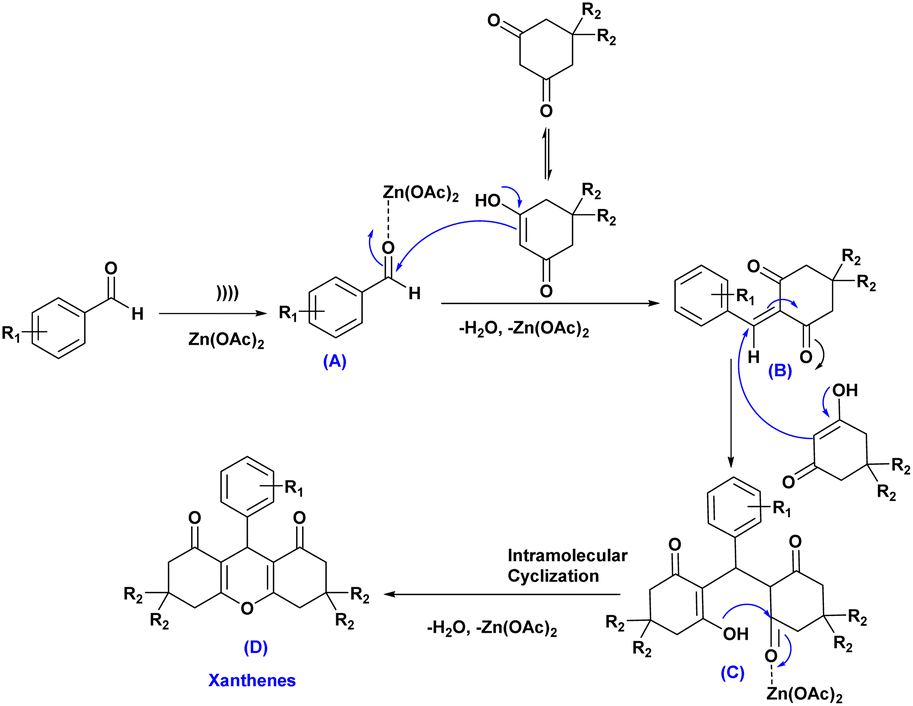
A plausible mechanism for the synthesis of xanthene derivatives using Zn(OAc)2 has been shown in Scheme 2. Firstly, ultrasound cavitation activates the coordination of Zn(OAc)2 with aldehydes, generating the zinc oxide-aldehydes intermediate (A), which then reacts with dimedone/1,3-cyclohexanedione to form intermediate (B) after dehydration and removal of Zn(OAc)2. Afterward, the active methylene group of the second molecule of dimedone/1,3-cyclohexanedione reacted with intermediate (B) through a conjugate Michael addition, forming intermediate (C). This intermediate then underwent intermolecular cyclization with (H2O, Zn(OAc)2 elimination, resulting in the formation of the final products (D).
 | ||
| Scheme 2 Proposed mechanism for the synthesis of xanthene derivatives. | ||
X-ray study
Suitable crystals of compound 3r were obtained after crystallization in diethyl ether/n-hexane and were exposed to X-ray diffraction analysis in order to completely reveal its structural composition.As displayed in the ORTEP diagram represented in Fig. 1, the asymmetric unit of the crystal structure comprises one molecule of compound 3,3,6,6-tetramethyl-9-(m-tolyl)-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione that crystallizes in the orthorhombic system with the Pca21 space group.
 | ||
| Fig. 1 ORTEP diagram of compound 3r. | ||
No conventional hydrogen bonds were observed since there is no hydrogen linked to an electronegative heteroatom. However, many short contacts were noticed (Table 4) in the structure that contribute to the linkage of the crystal components including intermolecular contacts of type C–H…O with lengths comprised between 2.609 and 2.654 Å, and C–H…C with a length of 2.847 Å. A crystal-packing diagram was built using Mercury software (Fig. 2), which shows a general view of the repartition of the molecules that constitutes the crystal structure.
| D–H…A | d(D–H) | d(H…A) | d(D–A) | D–H–A | Symmetry |
|---|---|---|---|---|---|
| C9–H9A…O2 | 0.970 | 2.613 | 3.416(5) | 140.2 | x, y, z; 1 − x, 1 − y, −1/2 + z |
| C11–H11B…O2 | 0.970 | 2.654 | 3.488(5) | 144.3 | x, y, z; 1 − x, 1 − y, −1/2 + z |
| C6–H6B…O3 | 0.970 | 2.609 | 3.432(4) | 142.8 | x, y, z; 1 − x, 1 − y, −1/2 + z |
| C15–H15…C13 | 0.930 | 2.847 | 3.702(4) | 153.2 | x, y, z; 1 − x, 1 − y, −1/2 + z |
 | ||
| Fig. 2 Crystal packing diagram viewed along (b) axis represented as ellipsoids drawn at 40% probability level. Contacts are represented as green dashed sticks. | ||
A general comparison between the structure of compound 3r and some previously reported xanthene derivatives differentiated by substituents in various positions indicates resemblances in geometry, bond lengths, and angle values.41 The bond lengths in the structure closely match those of analogous cyclohexanedione and dimedone-based xanthene compounds. For instance, carbonyl bonds lengths of cyclohexenone ring C3–O2 and C12–O3 are respectively equal to 1.225 and 1.211 Å, which is consistent with previous studies that state a length for carbonyl bond within the range of (1.221–1.229 Å). Similarly, C–O bonds lengths of the central pyranic heterocycle in compound 3r citing C7–O1 (1.380 Å) and C8–O1 (1.376 Å) align with literature values ranging from 1.376 to 1.380 Å. Cyclohexenone rings adopt half-chair conformations in which C5 and C10 deviated from the planes A and C with distances equal to 0.631 and 0.643 Å respectively. While the pyranic ring displays an almost planar geometry, with atom C1 distancing from the plane B by a length of 0.287 Å (Fig. 3). These findings are in perfect reliability with crystallographic identifications of similar xanthenedione rings.42 Furthermore, torsions angles compared to the aromatic ring are near to 180°, which suggests an antiperiplanar conformation.
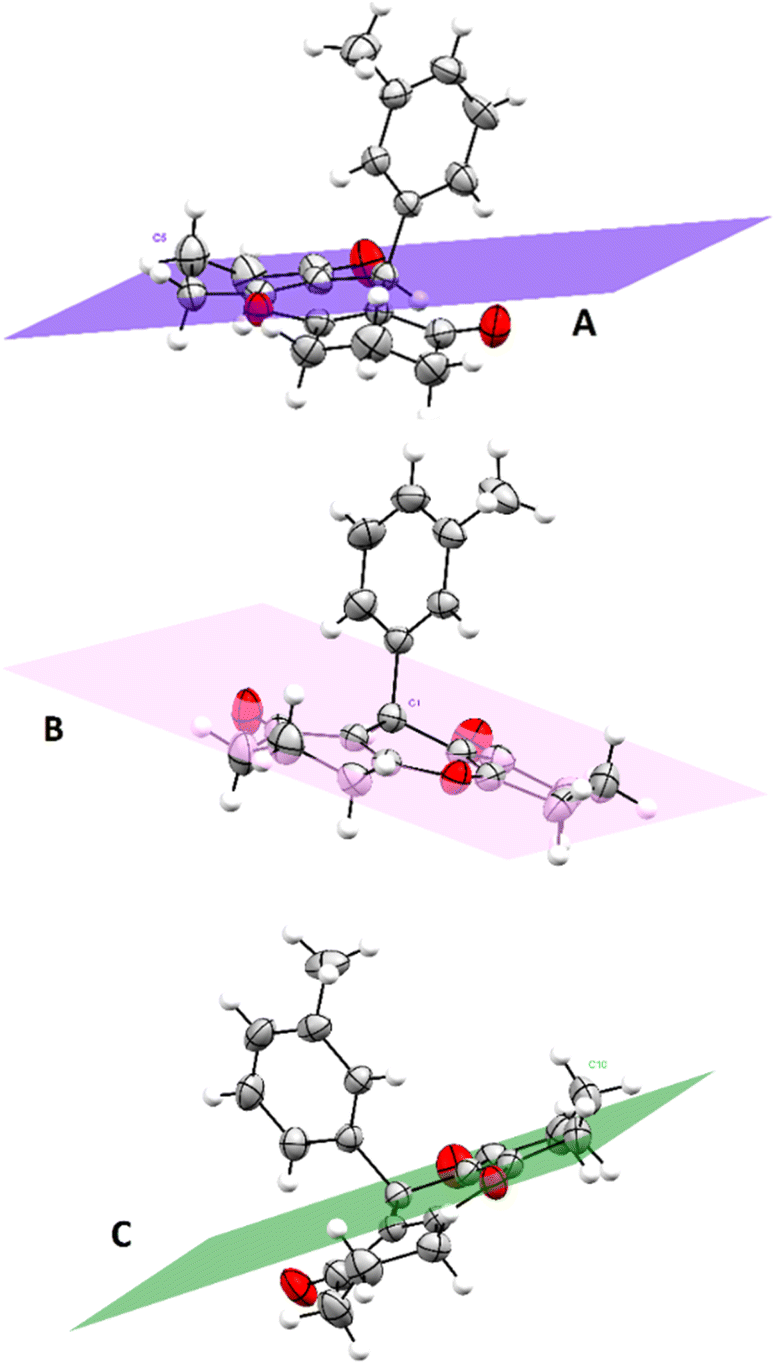 | ||
| Fig. 3 Graphical representation of planes A, C corresponding to cyclohexenones rings and plane B corresponding to the pyranic cycle. (Plane A: calculated through C2–C3–C4–C6–C7, plane B: calculated through C2–C7–O1–C8–C13, and plane C: calculated through C8–C9–C11–C12–C13). Obtain permission and include the acknowledgement required by the copyright holder if a figure is being reproduced from another source. | ||
In silico study
The field of molecular docking, when applied to VEGFR-2, focuses on how small molecules, often potential drug candidates, interact with this receptor at the atomic and molecular level. By employing advanced computational methods and docking software, researchers can predict and evaluate the binding affinity and structural interactions between VEGFR-2 and these molecules. This provides valuable insights into the potential efficacy of compounds in modulating the receptor's activity.
Molecular docking studies involving VEGFR-2 also can help researchers identify and prioritize compounds that can selectively target VEGFR-2, either by inhibiting or activating its signalling pathways. This selectivity is vital because it allows for the development of precise, targeted therapies with reduced off target effects and minimal harm to healthy tissues.
Furthermore, the insights gained from molecular docking studies can guide the design and optimization of potential drug candidates, ensuring their maximum efficacy and safety. VEGFR-2, as a key player in angiogenesis regulation, is a focal point for molecular docking investigations. These studies offer a gateway to the development of tailored therapies for cancer and angiogenesis-related disorders, potentially improving patient outcomes and the quality of medical treatments.
In order to gain a comprehensive understanding of the interactions between the active site of VEGFR-2 and ligands, we conducted molecular docking study to explore the binding modes of compounds under investigation. Our studies were performed using Schrodinger Suite (version 11.8) and Chimera X programs. We utilized 4-amino-furo[2,3-d] pyrimidine as the reference ligand to investigate the binding mode. The accuracy of the docking protocol was assessed through the re-docking of the reference ligand into the active site of VEGFR-2. Fig. 4 illustrates the docked reference ligand and the co-crystallized one occupying almost the same position within the receptor (RMSD = 0.38 Å). This confirms the validation of our docking protocol, which utilized the standard precision (SP) scoring function, in absence of water molecules.
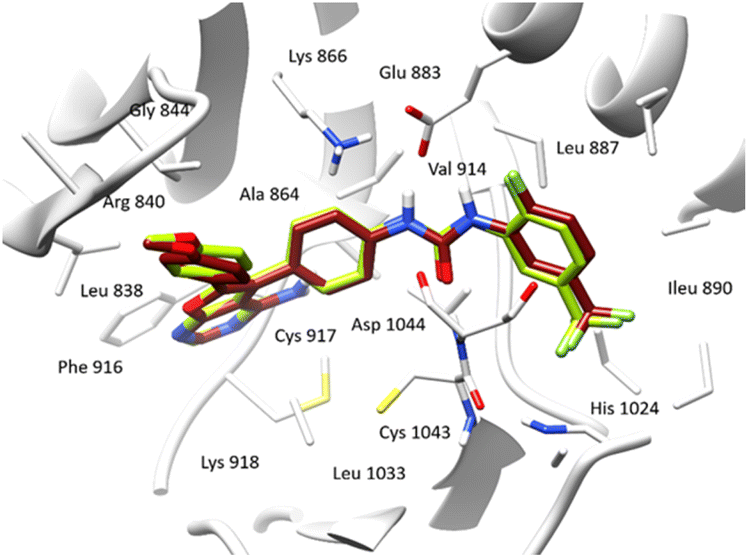 | ||
| Fig. 4 Docked and co-crystalized 4-amino-furo[2,3-d] pyrimidine in the active site of VEGFR-2 enzyme after self-docking calculation. (Co-crystallized ligand in green sticks and the docked ligand in brown sticks). | ||
The results of this study, including the estimated glide score of the docked positions, are provided in Table 5 and Fig. 5. Our molecular docking analysis of all studied compounds revealed that the majority of the compounds exhibit a good stability within the binding cavity. Specifically, compounds 3t, 3p, 3e, 3i, 3b, 3m and 3c displayed superior glide scores compared to the other when binding to the target protein.
| Compound code | Docking score |
|---|---|
| 3a | −5.08 |
| 3b | −6.24 |
| 3c | −6.12 |
| 3d | −6.02 |
| 3e | −6.52 |
| 3f | −4.80 |
| 3g | −6.01 |
| 3h | −5.28 |
| 3i | −6.34 |
| 3j | −5.80 |
| 3k | −5.12 |
| 3l | −6.10 |
| 3m | −6.20 |
| 3n | −5.94 |
| 3o | −5.20 |
| 3p | −6.80 |
| 3q | −5.80 |
| 3r | −6.05 |
| 3s | −5.40 |
| 3t | −6.99 |
| Reference ligand −13.11 | |
 | ||
| Fig. 5 Superimposition of the most stable xanthene derivatives in the active site of VEGFR-2 enzyme. | ||
These compounds exhibited an average binding score ranging from −6.99 to −6.12 kcal mol−1. On the other hand, compounds 3f, 3a and 3o demonstrated lower stability compared to the rest, with glide score of −4.80, −5.08 and −5.20 kcal mol−1, respectively. These findings provide valuable insights into the binding affinity of these compounds with the target protein.
A thorough analysis of the molecular docking results revealed that hydrogen bonds, hydrophobic forces, and electrostatic attractions primarily drove the interactions within the active site of the VEGFR-2 enzyme.
The obtained results play a crucial role in determining the binding affinity and specificity of molecules within the enzyme's binding pocket, shedding light on the potential effectiveness of various ligands in modulating VEGFR-2 activity. The docking outcomes for the xanthene compounds, as well as the reference ligand, have been presented in Table 6.
| Code | Hydrogen bond | Hydrophobic interaction | Pi-cation |
|---|---|---|---|
| 3a | — | Val897, Cys1043, Ala864, Val846, Val914, Phe916, Cys917, Leu838, Leu1033 | — |
| 3b | Lys866 | Val846, Phe843, Leu838, Leu1033, Cys917, Phe916, Val914, Val912, Leu887, Ala864, Val865 | Lys866 |
| 3c | Lys866 | Val846, Leu838, Leu1033, Cys917, Phe916, Cys1043, Val897, Val914, Val912, Leu887, Ala864, Val865 | Lys866 |
| 3d | Lys866 | Val846, Phe843, Leu838, Leu1033, Cys917, Phe916, Val914, Val912, Val897, Leu887, Ala864 | Lys866 |
| 3e | Lys866, Glu883 | Val846, Phe843, Leu838, Leu1033, Cys917, Phe916, Val914, Val912, Leu887, Cys1043, Ala864 | Lys866 |
| 3f | — | Val846, Leu838, Leu1033, Cys917, Phe916, Val914, Val897, Leu887, Cys1043, Ala864 | — |
| 3g | Lys866 | Ala864, Val897, Val914, Leu1033, Cys1043, Val846, Phe843, Ala842 | Lys866 |
| 3h | — | Val897, Cys1043, Ala864, Val914, Phe916, Cys917, Leu838, Leu1033, Val846 | — |
| 3i | Lys866 | Val846, Leu838, Leu1033, Cys917, Phe916, Val914, Cys1043, Val897, Ala864, Leu887 | — |
| 3j | Leu838 | Val846, Ala864, Val914, Phe916, Cys917, Leu838, Leu1033, Cys1043, Val897 | — |
| 3k | Leu887, Cys1043, Val914, Val912, Val265, Val897, Phe916, Cys917, Ala864, Leu1033, Leu818, Val846 | ||
| 3l | — | Leu887, Cys1043, Val914, Val912, Val265, Val897, Phe916, Cys917, Ala864, Leu1033, Leu818, Val846 | — |
| 3m | — | Leu887, Cys1043, Val914, Val912, Val265, Val897, Phe916, Cys917, Ala864, Leu1033, Leu818, Val846 | — |
| 3n | — | Val846, Leu838, Cys917, Phe916, Val914, Val912, Leu1033, Val897, Cys1043, Leu887, Val865, Ala864 | Lys866 |
| 3o | — | Val846, Leu838, Cys917, Phe916, Val914, Val912, Val897, Cys1043, Leu1033, Leu887, Ala864, Val865 | — |
| 3p | Glu883 | Val846, Leu838, Val914, Phe916, Cys917, Val897, Leu1033, Cys1043, Ala864, Leu887 | — |
| 3q | — | Val914, Phe916, Cys917, Ala864, Val846, Leu838, Leu1033, Cys1043, Val897, Leu887 | — |
| 3r | — | Val846, Leu838, Cys917, Phe916, Val914, Ile913, Val912, Leu1033, Val897, Cys1043, Val865, Ala864 | — |
| 3s | Cys917 | Val846, Leu838, Val914, Phe916, Cys917, Val897, Leu1033, Cys1043, Ala864, Leu887 | Lys866 |
| 3t | — | Val846, Leu838, Val914, Phe916, Cys917, Val897, Leu1033, Cys1043, Ala864, Leu887 | — |
| Ref. ligand | Glu883, Cys917, Glu915, Asp1044 | Ile896, Leu887, Ile890, Leu1017, Ile1042, Cys1043, Val896, Val897, Val914, Phe916, Cys917, Ala864, Leu1033, Leu838, Val846 | — |
The reference ligand exhibits outstanding stability within the active pocket, as indicated by a docking score of −13.11 kcal mol−1, owing to the presence of functional groups in its structure. These functional groups are capable of forming four crucial hydrogen bonds with Glu883, Cys917, Glu915 and Asp1044 residues, which play a significant role in inhibiting the VEGFR-2 enzyme. Additionally, the reference ligand engages in hydrophobic interactions with several residues, including Phe 916, Leu 383, and Cys 1043, creating strong attractions with the aromatic rings of the reference ligand (Fig. 6).
 | ||
| Fig. 6 3D binding interactions of the reference ligand after docking calculations in the active site of VEGFR-2 enzyme. The amino acid residues were shown as grey stick model and H-bonds were shown as black lines. | ||
Both compounds 3e and 3p exhibited notable stability within the active site of the VEGRF-2 enzyme owing to the presence of a hydroxyl group in the para position of their aromatic rings, forming a crucial hydrogen bond with the key residue Glu803.
This hydroxyl group significantly enhances the stability of these ligands within the active pocket. Additionally, compound 3e established an additional hydrogen bond and cation interaction with Lys866, a residue contributing to the enzyme's inhibition (Fig. 7).
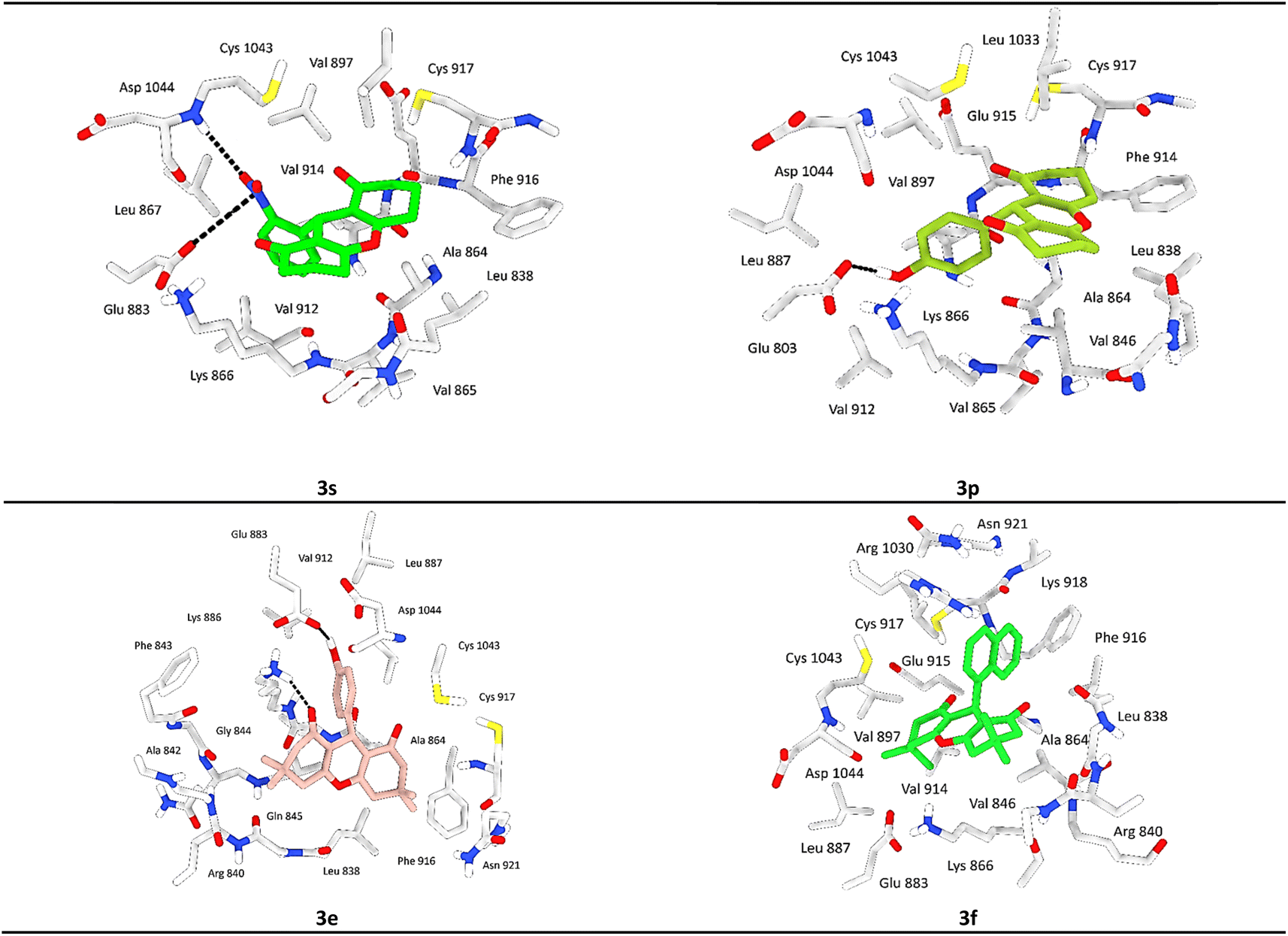 | ||
| Fig. 7 3D binding interactions of compounds 3s, 3p, 3e and 3f after docking calculations in the active site of VEGFR-2 enzyme. The amino acid residues were shown as grey stick model and H-bonds were shown as black lines. | ||
Moreover, both compounds displayed significant hydrophobic interactions with residues Phe916, Cys917, Val897, Leu1033, and Cys1043, which are known to play a role in inhibiting the VEGRF-2 enzyme. These interactions elucidate the molecular docking values and contribute to the understanding of the compounds inhibitory mechanisms.
Compound 3s showed equal significance to compounds 3e and 3p, exhibiting considerable stability within the active site with acceptable docking scores. 3s forms a crucial hydrogen bond with the residue Asp1044, known among the residues responsible for inhibiting the VEGRF-2 enzyme, and an ionic bond with the key residue Glu883. Additionally, it participates in notable hydrophobic interactions, particularly through a π–cation interaction with the residue Lys886.
After analyzing other compounds, we have observed that the nature of the substituent on the phenyl ring significantly influences stability within the active site. Compounds containing a halogen substituent on the aromatic ring or a secondary aromatic ring, such as in the case of a naphthalene group, exhibit average stability compared to other compounds within the pocket. Compound 3f exemplifies this characteristic.
In our investigation, we employed in silico studies to analyze the interaction of our compounds with the VEGFR-2 enzyme, a known cancer target, aiming to elucidate their inhibitory mechanisms. The xanthene pharmacophore, comprised of three rings, served as the basis for our modifications, primarily focused on substituents on the aromatic ring.
The base molecule 3a, which does not carry any electron-donating or electron-withdrawing substituents at the aromatic ring level, presented an inhibition of the VEGFR-2 enzyme with an average docking score of −5.08 kcal mol−1. This is due to the fact that the ligand 3a does not form hydrogen bonds with key active site residues. On the other hand, the introduction of substituents such as fluorine, chlorine, methoxy, hydroxy, and others confers several advantages, particularly enhancing inhibitory potency. These derivatives also share the common xanthene pharmacophore, which is intricately linked to a ring system. Notably, the positions and types of substituents on the aromatic ring exert a significant influence on the outcomes of molecular docking studies.
Our findings indicate that the incorporation of a hydroxyl substituent at the para position of the aromatic ring in compounds 3e (−6.52 kcal mol−1) and 3p (−6.80 kcal mol−1) demonstrates significant in silico inhibition of the VEGFR-2 enzyme due to the formation of hydrogen bonds with key active site residues. Additionally, electron-withdrawing groups, such as the methoxy group and halogens like fluorine, chlorine, and bromine at the para position, also exhibit notable inhibition.
Conversely, groups such as nitro, methyl, and methoxy show comparatively lesser inhibition. Furthermore, substituents at position ortho of the aromatic ring interfere with molecular stabilization within the active pocket, consequently diminishing enzyme inhibition.
Our investigation involved assessing system stability through variations in RMSD values, the count of hydrogen bonds between the selected ligands and VEGFR-2 enzyme and the protein–ligand contacts analysis. The other parameters such as the root mean square fluctuation (RMSF), the radius of gyration and DSSP, and monitoring the protein secondary structure elements (SSE) like alpha helices and beta-strands throughout the simulation will be presented in the ESI.†
Specifically in biology, the predictive assessment of the gap between the ligand and the protein relies on evaluating PL-RMSD (Protein and Ligand Root Mean Square Deviation) extracted from MD simulation trajectories. Deviations falling within the 1 to 3 Å range are supposed acceptable for small molecules. PL-RMSD serves as a metric to measure the distance fluctuations between the protein and its associated ligand across the simulation duration. Initially, all 1000 protein frames are aligned to the reference frame backbone (frame 0), following which the RMSD is computed based on C-Alpha atoms.
Continuous monitoring of protein RMSD offers insights into structural variations throughout the simulation, shedding light on whether the simulation maintains equilibrium and whether its fluctuations revolve around a certain thermal average. Notably, higher RMSD values suggest potential instability within the protein system.
The comparative RMSD analysis between the VEGFR-2 protein and ligand 3e bound indicates consistent values for both structures from the onset of the MD simulation. It is notable that equilibrium was achieved by all systems after 1 ns (Fig. 8). Subsequently, we divided our productive phase into time intervals ranging from 1 to 200 ns across all simulations. Beyond the initial 1 ns of MD simulation, the observed ligand-RMSD values closely mirrored those of the protein's RMSD, implying a strong and sustained stability between ligand 3e and the protein within its binding pocket.
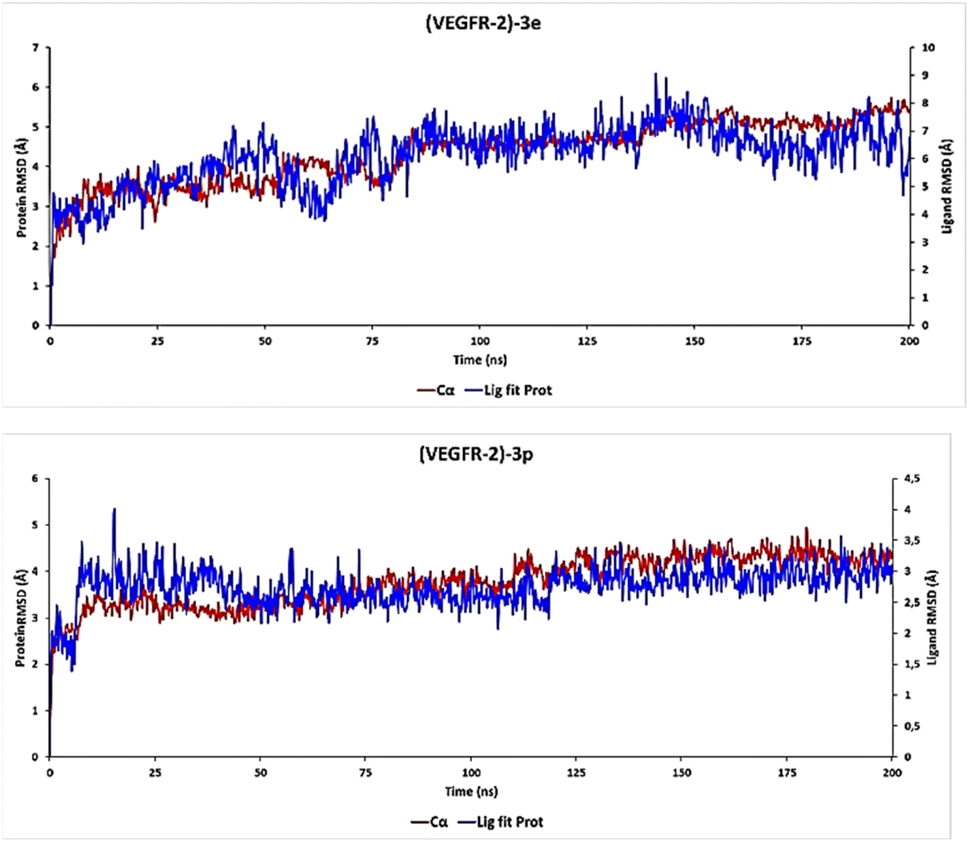 | ||
| Fig. 8 (PL-RMSD) obtained from the MD simulation trajectories (3e and 3p). | ||
Furthermore, an examination of the PL-RMSD values underscores continuous backbone stability throughout the MD simulation while ligand 3e remains bound to the protein. Typically, if the observed values significantly exceed the RMSD of the protein, it suggests that the ligand may have diffused away from its initial binding site. However, in the case of ligand 3e, minimal variance (ranging from 1.0 to 2.0 Å) is observed between the protein and ligand RMSD, indicating that the ligand remains proximate to its original position throughout the 200 ns of simulation time.
For the ligand 3p, substantial stability was observed after 8 ns of MD simulation, marking the start of the productive phase divided into time intervals spanning from 8 to 200 ns across all simulations (Fig. 8). This stability is clearly evidenced by the ligand-RMSD values closely aligning with the RMSD of the protein, indicative of a consistent and robust interaction between the 3p ligand and the protein within its binding pocket. Such alignment suggests a persistent and favorable binding mode.
During the productive phase, minimal variance (ranging from 0 to 0.6 Å) was observed between the protein and ligand RMSD, underscoring that the 3p ligand remains in close proximity to its original position throughout the 200 ns of simulation time.
Hydrogen bonds play a crucial role in the binding of ligands to protein residues. Thus, considering the properties of hydrogen bonding is essential in drug design due to their significant impact on drug specificity, metabolism, and adsorption. In our study, we present the hydrogen bonds formed between the studied ligands and the VEGFR-2 enzyme during the 200 ns of MD simulation, as depicted in Fig. 9.
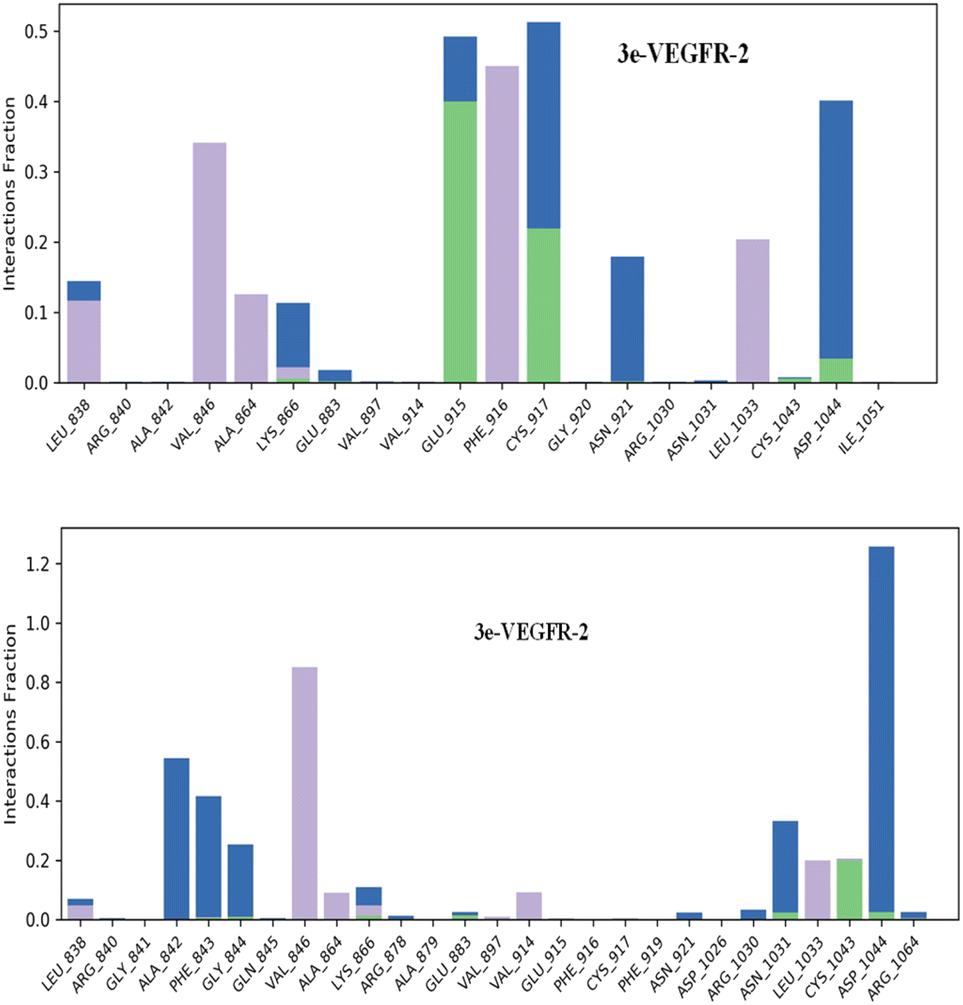 | ||
| Fig. 9 Protein–ligand contacts obtained from the MD simulation trajectories. | ||
The results showed that the ligand 3e can form up to 5 hydrogen bonds on average during the 200 ns of MD simulation with Lys 866, Glu915, Cys917, Cys1043 and Asp1044 residues; and the ligand 3p can form up to 5 hydrogen bonds with Lys866, Glu883, Asn1031, Cys1043, and Asp1044.
Regarding hydrophobic interactions, which play a key role in stabilizing the ligand–protein complex, they are typically classified into three subtypes: π–cation, π–π stacking, and other non-specific interactions. Typically, these interactions involve a hydrophobic amino acid and either an aromatic or aliphatic group on the compound. The analysis revealed that both compounds 3e and 3p exhibit strong bonding interactions within the active site of the VEGFR-2 enzyme. The identification of a π–cation interaction between the ligand 3e and the Lys 866 residue led us to an explanation of why the ligand 3e attains a superior docking score compared to the ligand 3p, which lacks π–cation or π–π stacking interactions.
In completion of our in silico study, and in order to associate the predicted binding mode of xanthene derivatives and their structural properties, we conducted a computational investigation through density functional theory (DFT) in which we used the B3LYP/6-31G(d,p) model contained within Gaussian 09 to obtain optimized molecular geometries, determine electron density of compounds, and define their different global chemical reactivity descriptors citing chemical hardness (η), molecular softness (s), electronic chemical potential (μ), electronegativity (χ), and electrophilicity index (ω), results are depicted in Table 7.
| Molecular descriptors (gas phase) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | logP |
μ (D) | EHOMO (eV) | ELUMO (eV) | ΔEgap | (η) | (S) | (μ) | (χ) | (ω) |
| a Dipole moment (μ), energy of the highest occupied molecular orbital (EHOMO), energy of the lowest unoccupied molecular orbital (ELUMO), the HOMO–LUMO gap (ΔEgap), chemical hardness (η) = (ELUMO − EHOMO)/2, molecular softness (S) = 1/η, electronic chemical potential (μ) = −χ = (EHOMO + ELUMO)/2, electronegativity (χ) = −(EHOMO + ELUMO)/2, electrophilicity index (ω) = μ2/2η. | ||||||||||
| 3a | 2.08 | 6.0432 | −6.1471 | −1.5445 | 4.6025 | 2.3013 | 0.4345 | −3.8458 | 3.8458 | 3.2135 |
| 3b | 2.91 | 7.3494 | −6.1416 | −1.6936 | 4.4480 | 2.2240 | 0.4496 | −3.9176 | 3.9176 | 3.4505 |
| 3c | 2.64 | 7.4033 | −6.2042 | −1.6920 | 4.5122 | 2.2561 | 0.4432 | −3.9481 | 3.9481 | 3.4545 |
| 3d | 2.24 | 6.8678 | −6.1149 | −1.6262 | 4.4888 | 2.2444 | 0.4456 | −3.8705 | 3.8705 | 3.3375 |
| 3e | 1.69 | 4.7750 | −5.6145 | −1.5124 | 4.1021 | 2.0511 | 0.4876 | −3.5635 | 3.5635 | 3.0955 |
| 3f | 3.08 | 5.7832 | −5.5130 | −1.5818 | 3.9312 | 1.9656 | 0.5087 | −3.5474 | 3.5474 | 3.2011 |
| 3g | 1.83 | 7.1153 | −5.4768 | −1.4887 | 3.9881 | 1.9941 | 0.5015 | −3.4828 | 3.4828 | 3.0415 |
| 3h | 2.57 | 5.6706 | −6.0861 | −1.5233 | 4.5628 | 2.2814 | 0.4383 | −3.8047 | 3.8047 | 3.1725 |
| 3i | 2.64 | 7.1479 | −6.1710 | −1.6098 | 4.5612 | 2.2806 | 0.4385 | −3.8904 | 3.8904 | 3.3183 |
| 3j | 1.69 | 8.1477 | −5.4975 | −1.8278 | 3.6697 | 1.8349 | 0.5450 | −3.6627 | 3.6627 | 3.6556 |
| 3k | 2.87 | 9.1839 | −6.3639 | −1.9323 | 4.4316 | 2.2158 | 0.4513 | −4.1481 | 4.1481 | 3.8827 |
| 3l | 0.48 | 6.0239 | −6.1612 | −1.5666 | 4.5946 | 2.2973 | 0.4353 | −3.8639 | 3.8639 | 3.2493 |
| 3m | 1.31 | 7.2722 | −6.1509 | −1.7173 | 4.4336 | 2.2168 | 0.4511 | −3.9341 | 3.9341 | 3.4909 |
| 3n | 0.64 | 6.8094 | −6.1272 | −1.6490 | 4.4782 | 2.2391 | 0.4466 | −3.8881 | 3.8881 | 3.3758 |
| 3o | 1.04 | 7.3246 | −6.2162 | −1.7162 | 4.4999 | 2.2500 | 0.4444 | −3.9662 | 3.9662 | 3.4958 |
| 3p | 0.09 | 7.1185 | −5.6295 | −1.5350 | 4.0945 | 2.0472 | 0.4885 | −3.5822 | 3.5822 | 3.1341 |
| 3q | 1.41 | 7.8681 | −6.4736 | −1.7832 | 4.6904 | 2.3452 | 0.4264 | −4.1284 | 4.1284 | 3.6337 |
| 3r | 0.97 | 6.1091 | −6.0251 | −1.5429 | 4.4823 | 2.2411 | 0.4462 | −3.7840 | 3.7840 | 3.1945 |
| 3s | 0.23 | 7.1539 | −5.4820 | −1.5075 | 3.9745 | 1.9872 | 0.5032 | −3.4948 | 3.4948 | 3.0729 |
| 3t | 0.86 | 9.2091 | −6.4031 | −1.9497 | 4.4534 | 2.2267 | 0.4491 | −4.1764 | 4.1764 | 3.9166 |
Dipole moment that defines charge separation in a molecule and its polarity are included in the range [4.7750–8.1477D] for the studied xanthene-based molecules. Compound 3j was noted as the most polar and compound 3e as the least polar among investigated compounds. Optimized molecular geometries as well as frontier molecular orbitals are represented in Fig. 10, HOMO–LUMO energy gaps ranges between 3.6697 and 4.6904 eV in which compound 3q showed the highest value, hence, it is the most stable compound among xanthene derivatives.
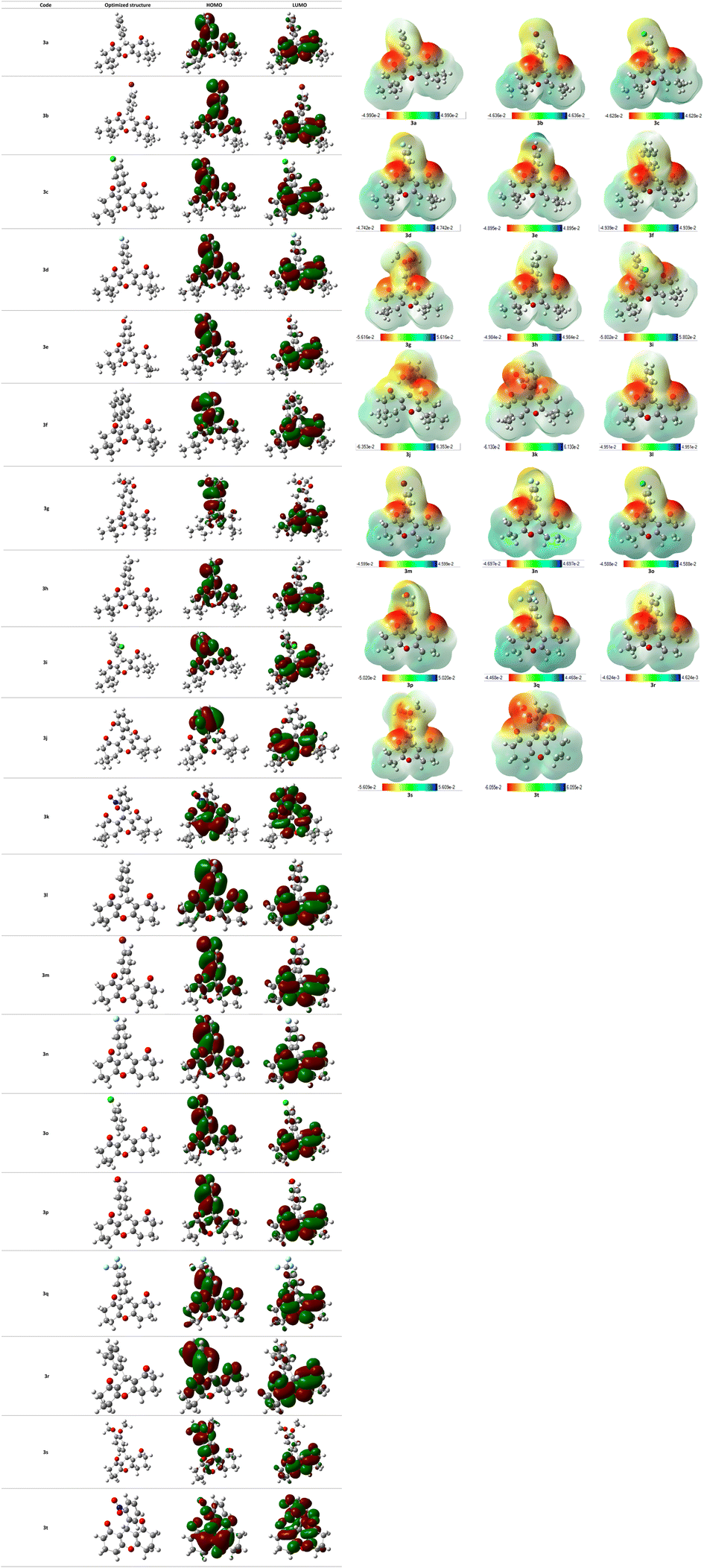 | ||
| Fig. 10 Representation of HOMO/LUMO orbitals and optimized structures at B3LYP/6-31G(d,p) level in gas phase. | ||
The molecular electrostatic potential (MEP) is an effective tool to predict electron-rich and electron-deficient positions in a structure, and learn more about the behavior of a molecule towards other molecules or biological systems including reactivity and the ability of hydrogen bonds formation. MEP surface of all compounds were determined from DFT structure optimization results using B3LYP method at 6-31G(d,p) level and were visualized as shown in Fig. 10. Every derivative has a color code lying in a specific range between the deepest red and the deepest blue. Red color in the MEP indicates the most nucleophilic (negative) parts of the molecules while the blue color designates the most electrophilic (positive) fragments.
logP (octanol–water partition coefficient) ≤5, molecular weight ≤500, number of hydrogen bond acceptors ≤10, and number of hydrogen bond donors ≤5.In this study, the drug likeness and ADME properties of the investigated compounds were explored, and compared with rhodomyrtone drug. The predicted physico-chemical properties are summarized in Table 8. The molecular weight of rhodomyrtone and the studied compounds (3a–3t) ranged from 294.34 to 442.54 g mol−1 (<500). Low molecular weight drug molecules (<500) can be transported, diffused, and absorbed more easily compared to heavier molecules. Molecular weight plays a crucial role in therapeutic drug action; if it increases significantly, it can impact the drug's effectiveness. The number of hydrogen bond acceptors and donors in the tested compounds was found to be within Lipinski's limit. The hydrogen bond acceptor count ranged from 3 to 6 (<10), and the hydrogen bond donor count was less than 5 in all molecules (<5). Additionally, most compounds have one rotatable bond, except for compounds (3q, 3k, and 3t) which have 2, and compounds 3g and 3s, which have 3 rotatable bonds, respectively (<10). These compounds exhibited favorable lipophilicity, as indicated by their ilogP values ranging from 2.83 to 3.81, which are less than <5.
| Properties | Molecular weight (g mol−1) | Rotatable bonds | H-bond donor | H-bond acceptor | Violations | logPo/W ilogP |
logS ESOL |
GI | BBB | logKp |
Bioavailability score | TPSA (Å) | DLS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a logPo/W ilogP: the octanol/water partition coefficient, logS ESOL: the decimal logarithm of the molar solubility in water, BBB: blood–brain barrier, logKp: the skin permeability coefficient, TPSA (Å): the topological polar surface area, DLS: dynamic lipophilicity. |
|||||||||||||
| Rhodomyrtone | 442.54 | 5 | 2 | 6 | 0 | 3.76 | −6.02 | High | No | −4.91 | 0.56 | 100.90 | −0.33 |
| 3a | 350.45 | 1 | 0 | 3 | 0 | 3.40 | −4.62 | High | Yes | −5.62 | 0.85 | 43.37 | −1.31 |
| 3b | 429.35 | 1 | 0 | 3 | 0 | 3.81 | −5.54 | High | Yes | −5.54 | 0.85 | 43.37 | −1.17 |
| 3d | 368.44 | 1 | 0 | 4 | 0 | 3.54 | −4.79 | High | Yes | −5.66 | 0.85 | 43.37 | −0.98 |
| 3c | 384.90 | 1 | 0 | 3 | 0 | 3.71 | −5.22 | High | Yes | −5.38 | 0.85 | 43.37 | −0.77 |
| 3e | 366.45 | 1 | 1 | 4 | 0 | 3.16 | −4.48 | High | Yes | −5.97 | 0.85 | 63.60 | −0.72 |
| 3f | 400.51 | 1 | 0 | 3 | 0 | 3.61 | −5.79 | High | No | −5.04 | 0.85 | 43.37 | −1.10 |
| 3g | 410.50 | 3 | 0 | 5 | 0 | 3.85 | −4.80 | High | Yes | −6.93 | 0.85 | 61.83 | −0.41 |
| 3h | 364.48 | 1 | 0 | 3 | 0 | 3.44 | −4.93 | High | Yes | −5.45 | 0.85 | 43.37 | −0.99 |
| 3i | 384.90 | 1 | 0 | 3 | 0 | 3.62 | −5.22 | High | Yes | −4.13 | 0.85 | 43.37 | −0.87 |
| 3j | 366.45 | 1 | 1 | 4 | 0 | 3.02 | −4.48 | High | Yes | −5.97 | 0.85 | 63.60 | −0.78 |
| 3k | 395.45 | 2 | 0 | 5 | 0 | 3.07 | −4.71 | High | No | −6.01 | 0.56 | 89.19 | −1.25 |
| 3l | 310.34 | 1 | 1 | 4 | 0 | 2.40 | −3.12 | High | Yes | −6.81 | 0.85 | 63.60 | −0.54 |
| 3m | 373.24 | 1 | 0 | 3 | 0 | 3.09 | −4.17 | High | Yes | −6.45 | 0.85 | 43.37 | −0.99 |
| 3n | 312.33 | 1 | 0 | 4 | 0 | 2.83 | −3.42 | High | Yes | −6.49 | 0.85 | 43.37 | −0.80 |
| 3o | 328.79 | 1 | 0 | 3 | 0 | 3.00 | −3.85 | High | Yes | −6.23 | 0.85 | 43.37 | −0.62 |
| 3p | 294.34 | 1 | 0 | 3 | 0 | 2.70 | −3.26 | High | Yes | −6.46 | 0.85 | 43.37 | −1.16 |
| 3q | 362.34 | 2 | 0 | 6 | 0 | 3.06 | −5.38 | High | No | −6.25 | 0.85 | 43.37 | −1.04 |
| 3r | 308.37 | 1 | 0 | 3 | 0 | 2.88 | −3.56 | High | Yes | −6.29 | 0.85 | 43.37 | −0.63 |
| 3s | 354.40 | 3 | 0 | 5 | 0 | 3.15 | −3.43 | High | Yes | −6.86 | 0.85 | 61.83 | 0.07 |
| 3t | 339.34 | 2 | 0 | 5 | 0 | 2.70 | −3.33 | High | No | −6.86 | 0.56 | 89.19 | −0.98 |
Furthermore, these compounds demonstrated excellent water solubility, as evidenced by logS ESOL values ranging from −3.12 to −5.79. A value below −10 is considered insoluble, whereas a value above −4 is considered soluble according to SwissADME solubility measurements (logS). In fact, a soluble molecule simplifies various aspects of drug development, especially handling and formulation. Regarding oral administration, researchers have noted that solubility plays a crucial role in determining absorption.
On the other hand, the skin permeability coefficient (Kp) is linearly related to molecule size and lipophilicity, and logKp (cm s−1) measures a molecule's permeability through the skin. A higher value of logKp indicates a lower likelihood of the molecules permeating the skin. Among the aforementioned compounds, 3g exhibited the lowest skin permeability (−6.93 cm s−1), whereas 3l showed high skin permeability (−4.13 cm s−1). Compounds with high skin permeability are generally less suitable for oral medication. Meanwhile, the topological polar surface area ranged between 43.37 and 89.19 Å2 compared to the topological polar surface area of the rhodomyrtone drug, which is 100.90 Å2, all of these values, fell within the recommended ranges for oral bioavailability.
Dynamic Lipophilicity (DLS) is a parameter that assesses how well a compound aligns with the characteristics of drug molecules. Interestingly, compound 3s exhibited the highest DLS value of 0.07 even better than rhodomyrtone drug (DLS = −0.33), consistent with experimental bioactivity results. In contrast, compound 3a displayed the lowest DLS value (−1.31). Among these compounds, 3s demonstrated the most favorable pharmacokinetic properties (Fig. 11).
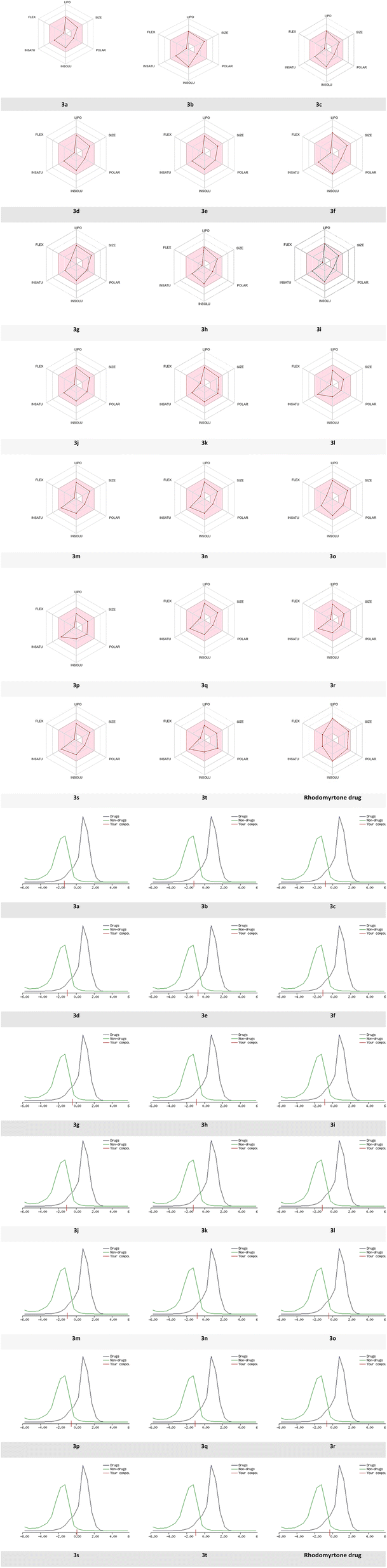 | ||
| Fig. 11 Radar related to physicochemical properties of the synthesized molecules, rhodomyrtone drug and DLS score. | ||
Based on the previous results, bioavailability radar visualization was used to predict the oral bioavailability of the investigated ligands.
The results showed that all compounds failed the oral bioavailability test. The pink area of the plot defines the drug-likeness of compounds limits for the six physicochemical properties: lipophilicity, size, polarity, solubility, flexibility, and saturation.
Experimental
General information
:acetone (9:1) eluent.Upon completion of the reaction, when the TLC indicated the desired outcome, Zn(OAc)2 was precipitated and then filtered out. Subsequently, the reaction mixture was concentrated by evaporation under vacuum. To isolate the pure product, a mixture of diethyl ether and n-hexane in a 6:4 ratio was added to the concentrated solution. The solution was then subjected to overnight cooling at 6 °C, leading to the crystallization of the desired product. This procedure was conducted to synthesize and isolate all compounds.
3,3,6,6-Tetramethyl-9-phenyl-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3a). The product (3a, C23H26O3) was obtained as a white powder; 95% yield; Rf = 0.71 (DCM/acetone) (9
:1); FT-IR (KBr, cm−1): 1667 (CO), 1458 (CC); 1H NMR (400 MHz, CDCl3) δ 0.99 (s, 6H, 2CH3), 1.10 (s, 6H, 2CH3), 2.12–2.20 (q, JH–H = 16.4 Hz, 4H, 2CH2-CC), 2.46 (s, 4H, 2CH-CO), 4.75 (s, 1H, *CH), 7.09 (t, JH–H = 7.6 Hz, 1H, CHAr), 7.20 (t, JH–H = 6.4 Hz, 2H, 2CHAr), 7.28 (t, JH–H = 6.4 Hz, 2H, 2CHAr) ppm; 13C NMR (100 MHz, CDCl3) δ 27.5 (2CH3), 29.4 (2CH3), 31.9 (2C-(CH3)2), 32.3 (2CH2-CC), 41.0 (*CH), 50.9 (2CH2-CO), 115.8 (2CCO), 126.4 (CHAr), 128.1 (CHAr), 128.5 (CHAr), 144.2 (CAr), 162.3 (2C-O), 196.4 (2CO) ppm.
9-(4-Bromophenyl)-3,3,6,6-tetramethyl-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3b). The product (3b, C23H23O3Br) was obtained as a white cristal; 93% yield; Rf = 0.78 (DCM/acetone) (9
:1); FT-IR (KBr, cm−1): 1662 (CO), 1469 (CC); 1H NMR (400 MHz, CDCl3) δ 0,98 (s, 6H, 2CH3), 1.03 (s, 6H, 2CH3), 2.08 (d, JH–H = 8.0 Hz, 2H, CH2-CC), 2.25 (d, JH–H = 8.0 Hz, 2H, CH2-CC), 2.50–2.56 (m, 4H, 2CH2-CO), 4.50 (s, 1H, *CH), 7.20 (d, JH–H = 7.8 Hz, 2H, 2CHAr), 7.28 (d, JH–H = 7.8, 2H, 2CHAr) ppm; 13C NMR (100 MHz, CDCl3) δ 26.9 (2CH3), 29.0 (2CH3), 31.4 (2C-(CH3)2), 32.3 (2CH2-CC), 40.6 (*CH), 50.4 (2CH2-CO), 114.4 (2C-CO), 128.3 (CHAr), 130.3 (CHAr), 134.7 (CAr-Br), 143.7 (CAr), 163.5 (2C-O), 196.5 (2CO) ppm.
9-(4-Chlorophenyl)-3,3,6,6-tetramethyl-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3c). The product (3c, C23H23O3Cl) was obtained as a white powder; 94% yield; Rf = 0.80 (DCM/acetone) (9
:1); FT-IR (KBr, cm−1): 1665 (CO), 1467 (CC); 1H NMR (400 MHz, CDCl3) δ 0,99 (s, 6H, 2CH3), 1.09 (s, 6H, 2CH3), 2.12–2.25 (q, 4H, JH–H = 16.4 Hz, 2CH2-CC), 2.45 (s, 4H, 2CH2-CO), 4.66 (s, 1H, *CH), 6.53 (d, JH–H = 8.4 Hz, 2H, 2CHAr), 7.05 (d, JH–H = 8.0 Hz, 2H, 2CHAr) ppm; 13C NMR (100 MHz, CDCl3) δ 27.5 (2CH3), 28.5 (2CH3), 30.6 (2C-(CH)2), 32.3 (2CH2-CC), 41.0 (*CH), 49.2 (2CH2-CO), 115.3 (2C-CO), 117.1 (2CHAr), 129.4 (2CHAr), 135.3 (CAr-Cl), 155.2 (CAr), 163.4 (C-O), 195.7 (2CO) ppm.
9-(4-Fluorophenyl)-3,3,6,6-tetramethyl-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3d). The product (3d, C23H23O3F) was obtained as a white powder; 92% yield; Rf = 0.81 (DCM/acetone) (9
:1); FT-IR (KBr, cm−1): 1659 (CO), 1507 (CC); 1H NMR (400 MHz, CDCl3) δ 0,94 (s, 6H, 2CH3), 1.08 (s, 6H, 2CH3), 2.00 (d, JH–H = 7.8 Hz, 2H, CH2-CC), 2.26 (d, JH–H = 8.0 Hz, 2H, CH2 CC), 2.52–2.57 (m, 4H, 2CH2-CO), 4.51 (s, 1H, *CH), 7.01–7.15 (m, 2H, 2CHAr), 7.20–7.25 (m, 2H, 2CHAr) ppm; 13C NMR (100 MHz, CDCl3) δ 26.9 (2CH3), 29.1 (2CH3), 31.1 (2C-(CH3)2), 32.2 (2CH2-CC), 40.4 (*CH), 50.5 (2CH2-CO), 114.6 (2C-CO), 114.9 (CHAr), 115.2 (CHAr), 130.3 (d, JC–F = 9.0 Hz, CAr-F), 140.9 (CAr), 163.4 (2C-O), 196.5 (2CO) ppm.
9-(4-Hydroxyphenyl)-3,3,6,6-tetramethyl-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3e). The product (3e, C23H26O4) was obtained as a white powder; 88% yield; Rf = 0.80 (DCM/acetone) (9
:1); FT-IR (KBr, cm−1): (OH) 3411, 1659 (CO), 1512 (CC); 1H NMR (400 MHz, CDCl3) δ 1,00 (s, 6H, 2CH3), 1.09 (s, 6H, 2CH3), 2.16–2.27 (q, 4H, JH–H = 16.4 Hz, 2CH2-CC), 2.45 (s, 4H, 2CH2-CO), 4.67 (s, 1H, *CH), 6.29 (s, 1H, OH), 6.55 (d, JH–H = 8.8 Hz, 2H, 2CH-Ar), 7.06 (d, JH–H = 8.8 Hz, 2H, 2CH-Ar) ppm; 13C NMR (100 MHz, CDCl3) δ 26.8 (2CH3), 29.3 (2CH3), 31.9 (2C-(CH3)2), 33.0 (2CH2-CC), 41.0 (*CH), 50.9 (2CH2-CO), 115.3 (2C-CO), 116.5 (CHAr), 130.3 (CHAr), 136.4 (CAr), 154.7 (CAr-OH), 162.9 (2C-O), 197.5 (2CO) ppm.
3,3,6,6-Tetramethyl-9-(naphthalen-1-yl)-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3f). The product (3f, C27H28O3) was obtained as a white powder; 89% yield; Rf = 0.70 (DCM/acetone) (9
:1); FT-IR (KBr, cm−1): 1668 (CO), 1508 (CC); 1H NMR (400 MHz, CDCl3) δ 0,99 (s, 6H, 2CH3), 1.11 (s, 6H, 2CH3), 2.12–2.26 (q, 4H, JH–H = 16.4 Hz, 2CH2-CC), 2.50 (s, 4H, 2CH2-CO), 4.92 (s, 1H, *CH), 7.34–7.46 (m, 3H, 3CHAr), 7.69–7.77 (m, 4H, 4CHAr) ppm; 13C NMR (100 MHz, CDCl3) δ 27.4 (2CH3), 29.4 (2CH3), 32.1 (2C-(CH3)2), 32.3 (CH-CC), 41.1 (*CH), 50.9 (2CH2-CO), 115.8 (2C-CO), 125.4 (CHAr), 125.7 (CHAr), 127.0 (CHAr), 127.2 (CHAr), 127.6 (CHAr), 127.8 (CHAr), 128.1 (CHAr), 132.5 (CAr), 133.5 (CAr), 141.7 (CAr), 162.4 (2C-O), 196.4 (2CO) ppm.
9-(3,4-Dimethoxyphenyl)-3,3,6,6-tetramethyl-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3g). The product (3g, C25H30O5) was obtained as a white powder; 86% yield; Rf = 0.80 (DCM/acetone) (9
:1); FT-IR (KBr, cm−1): 1644 (CO), 1514 (CC); 1H NMR (400 MHz, DMSO-d6) δ, 0.97 (s, 6H, 2CH3), 1.09 (s, 6H, 2CH3), 2.11 (d, JH–H = 16.0 Hz, 2H, CH2–CC), 2.26 (d, JH–H = 16.2 Hz, 2H, CH2-CC), 2.55 (q, JH–H = 17.6 Hz, 4H, 2CH2-CO), 3.72 (s, 6H, 2CH3-O), 4.52 (s, 1H, *CH), 6.94–6.56 (m, 3H, 3CHAr) ppm; 13C NMR (100 MHz, DMSO-d6) δ 26.4 (2CH3), 28.6 (2CH3), 30.4 (2C-(CH3)2), 31.6 (2CH2-CC), 39.8 (*CH), 49.5 (2CH2-CO), 55.3 (2CH3-O), 111.2 (CHAr), 111.8 (CHAr), 114.5 (2C-CO), 118.4 (CHAr), 136.7 (CAr), 147.1 (CAr-OCH3),148.0 (CAr-OCH3), 162.7 (2C-O), 196.7 (2CO) ppm.
3,3,6,6-Tetramethyl-9-(m-tolyl)-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3h). The product (3h, C24H28O3) was obtained as a white powder; 89% yield; Rf = 0.75 (DCM/acetone) (9
:1); FT-IR (KBr, cm−1): 1667 (CO), 1463 (CC); 1H NMR (400 MHz, CDCl3) δ 0.99 (s, 6H, 2CH3), 1.10 (s, 6H, 2CH3), 2.14–2.24 (q, 4H, JH–H = 16.2 Hz (CH2-CC), 2.28 (s, 3H, CH3), 2.46 (s, 4H, 2CH2-CO), 4.71 (s, 1H, *CH), 6.89 (d, JH–H = 7.2 Hz, 1H, CHAr), 7.01 (d, JH–H = 8.0 Hz, 1H, CHAr), 7.06–7.13 (m, 2H, 2CHAr) ppm; 13C NMR (100 MHz, CDCl3) δ 21.6 (CH3-Ar), 27.5 (2CH3), 29.4 (2CH3), 31.9 (2C-(CH3)2), 32.4 (2CH2-CC), 41.1 (*CH), 51.0 (2CH2-CO), 116.0 (2C-CO), 125.4 (CHAr), 127.5 (CHAr), 128.1 (CHAr), 129.6 (CHAr), 137.6 (CAr-CH3), 144.2 (CAr), 162.4 (2C-O), 197.0 (2CO) ppm.
9-(2-Chlorophenyl)-3,3,6,6-tetramethyl-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3i). The product (3i, C23H23O3Cl) was obtained as a white powder; 87% yield; Rf (DCM/acetone) (9
:1); FT-IR (KBr, cm−1): 1665 (CO), 1467 (CC) 1H NMR (400 MHz, CDCl3) δ 1,01 (s, 6H, 2CH3), 1.09 (s, 6H, 2CH3), 2.13–2.24 (q, 4H, JH–H = 16.4 Hz, 2CH2-CC), 2.42 (s, 4H, 2CH2-CO), 4.99 (s, 1H, *CH), 7.05 (t, 1H, JH–H = 7.6 Hz, CHAr), 7.16 (t, 1H, JH–H = 7.6 Hz, CHAr), 7.21 (d, 1H, JH–H = 7.6 Hz, CHAr), 7.42 (d, 1H, JH–H = 7.6 Hz, CHAr) ppm; 13C NMR (100 MHz, CDCl3) δ 27.5 (2CH3), 29.4 (2CH3), 31.8 (2C-(CH3)2), 32.8 (2CH2-CC), 40.9 (*CH), 50.8 (2CH2-CO), 113.8 (2C-CO), 126.4 (CHAr), 128.5 (CHAr), 129.2 (CHAr), 130.3 (CHAr), 132.5 (CAr-Cl), 140.1 (CAr), 163.0 (2C-O), 196.1 (2CO) ppm.
9-(2-Hydroxyphenyl)-3,3,6,6-tetramethyl-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3j). The product (3j, C23H26O4) was obtained as a white powder; 84% yield; Rf = 0.8 (DCM/acetone) (9
:1); FT-IR (KBr, cm−1): 3400 (OH), 1637 (CO), 1458 (CC); 1H NMR (400 MHz, CDCl3) δ 0,99 (s, 6H, 2CH3), 1.03 (s, 3H, CH3), 1.12 (s, 3H, CH3), 1.90–2.00 (q, 2H, JH–H = 16.4 Hz, CH2-CC), 2.33 (s, 2H, CH2-CC), 2.35–2.61 (q, 4H, JH–H = 17.6 Hz, 2CH2-CO), 4.76 (s, 1H, *CH), 6.95–7.07 (m, 3H, CHAr), 7.11–7.19 (m, 1H, HAr), 10.44 (s, 1H, OH) ppm; 13C NMR (100 MHz, CDCl3) δ 27.3 (CH3), 27.9 (CH3), 29.2 (CH3), 29.9 (CH3), 31.1 (2C-(CH3)2), 32.4 (2CH2-CC), 41.7 (2CH2 CC), 50.0 (*CH), 50.8 (2CH2-CO), 111.2 (2C-CO), 115.9 (CHAr), 124.7 (CHAr), 127.6 (CHAr), 128.1 (CHAr), 151.2 (CAr), 169.2 (2C-O), 170.7 (C-OH), 201.0 (CO) ppm.
9-(4-Fluorophenyl)-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3n). The product (3n, C19H17FO3) was obtained as a white powder; 92% yield; Rf = 0.75 (DCM/acetone) (9
:1); FT-IR (KBr, cm−1): 1656 (CO), 1501 (CC); 1H NMR (400 MHz, DMSO-d6) δ 1.82–1.89 (m, 2H, CH2-CH2), 1.91–2.00 (m, 2H, CH2-CH2), 2.25–2.33 (m, 4H, 2CH2-CC), 2.53–2.69 (m, 4H, 2CH2-CC), 4.57 (d, JH–F = 11.9 Hz, 1H, *CH), 6.94 (t, JH–H = 8.6 Hz, 1H, CHAr), 7.15 (d, JH–H = 8.0 Hz, 1H, CHAr), 7.24 (t, JH–H = 6.0 Hz, 1H, CHAr), 7.33 (d, JH–H = 8.0 Hz, 1H, CHAr) ppm; 13C NMR (100 MHz, DMSO-d6) δ 20.7 (2CH2-CH2), 26.9 (2CH2-CC), 34.2 (2CH2-CO), 50.7 (*CH), 112.9 (2C-CO), 114.1 (CHAr), 115.0 (CHAr), 116.0 (CHAr), 129.5 (CHAr), 130.5 (CAr),163.9 (2C-O), 164.5 (C-F), 195.8 (CO), 205.6 (CO) ppm.
9-(4-Chlorophenyl)-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3o). The product (3o, C19H17ClO3) was obtained as a yellow powder; 93% yield; Rf = 0.74 (DCM/acetone) (9
:1); FT-IR (KBr, cm−1): 1645 (CO),1510 (CC); 1H NMR (400 MHz, DMSO-d6) δ 1.83–1.96 (m, 4H, 2CH2-CH2), 2.23–2.30 (m, 4H, 2CH2-CC), 2.60–2.68 (m, 4H, 2CH2-CO), 4.50 (s, 1H, *CH), 6.58 (d, JH–H = 8.0 Hz, 2H, 2CHAr), 6.97 (d, JH–H = 8.0 Hz, 2H, 2CHAr) ppm; 13C NMR (100 MHz, DMSO-d6) δ 20.6 (2CH2-CH2), 26.3 (2CH2-CC), 30.9 (2CH2-CO), 35.3 (*CH), 113.9 (2C-CO), 117.1 (CHAr), 128.7 (CHAr), 135.7 (C-Cl), 157.1 (CAr), 163.9 (2C-O), 196.2 (2CO) ppm.
9-(4-Hydroxyphenyl)-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3p). The product (3p, C19H18O4) was obtained as a white powder; 89% yield; Rf = 0.85 (DCM/acetone) (9
:1); FT-IR (KBr, cm−1): 3353 (OH), 1644 (CO), 1512 (CC),1H NMR (400 MHz, DMSO-d6) δ 1.79–1.98 (m, 4H, 2CH2-CH2), 2.21–2.30 (m, 4H, 2CH2-CC), 2.54–2.69 (m, 4H, 2CH2-CO), 4.49 (s, 1H, *CH), 6.57 (d, JH–H = 8.0 Hz, 2H, 2CHAr), 6.95 (d, JH–H = 8.0 Hz, 2H, 2CHAr), 9.09 (s, 1H, OH) ppm; 13C NMR (101 MHz, DMSO-d6) δ 19.8 (2CH2-CH2), 26.3 (2CH2-CC), 29.7 (2CH2-CO), 36.0 (*CH), 114.6 (2C-CO), 116.9 (CHAr), 128.7 (CHAr), 135.6 (C-Cl), 155.9 (CAr), 163.6 (2C-O), 195.8 (2CO) ppm.
9-(4-(Trifluoromethyl)phenyl)-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3q). The product (3q, C24H25F3O3) was obtained as a white powder; 90% yield; Rf = 0.73 (DCM/acetone) (9
:1); FT-IR (KBr, cm−1): 1663 (CO), 1420 (CC); 1H NMR (400 MHz, CDCl3) δ 1.96–2.11 (m, 4H, 2CH2-CH2), 2.27–2.42 (m, 4H, 2CH2-CC), 2.54–2.74 (m, 4H, 2CH2-CO), 4.85 (s, 1H, *CH), 7.41 (d, JH–H = 8.2 Hz, 2H, 2CHAr), 7.47 (d, JH–H = 8.2 Hz, 2H, 2CHAr) ppm; 13C NMR (100 MHz, CDCl3) δ 20.4 (2CH2-CH2), 27.2 (2CH2-CC), 33.0 (2CH2-CO), 37.0 (*CH), 116.4 (2C-CO), 125.2 (CF3), 130.1 (C-CF3), 128.9 (CHAr), 149.3 (CAr), 164.4 (2C-O), 196.5 (2CO) ppm.
9-(m-Tolyl)-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3r). The product (3r, C20H20O3) was obtained as a white powder; 87% yield; Rf = 0.77 (DCM/acetone) (9
:1); FT-IR (KBr, cm−1): 1665 (CO), 1486 (CC), 1H NMR (400 MHz, CDCl3) δ 1.94–2.05 (m, 4H, 2CH2-CH2), 2.25–2.38 (m, 4H, 2CH2-CC), 2.28 (s, 3H, CH3), 2.50–2.66 (m, 4H, 2CH2-CO), 4.77 (s, 1H, *CH), 6.91 (d, JH–H = 7.6 Hz, 1H, CHAr), 7.02 (d, JH–H = 8.0 Hz, 1H, CHAr), 7.07 (d, JH–H = 7.6 Hz, 1H, CHAr), 7.12 (s, 1H, CHAr) ppm; 13C NMR (100 MHz, CDCl3) δ 20.4 (2CH2-CH2), 21.5 (CH3), 27.2 (2CH2-CC), 31.6 (2CH2-CO), 37.0 (*CH), 117.1 (2C-CO), 125.3 (CHAr), 127.9 (CHAr), 128.0 (CHAr), 129.4 (CHAr), 137.5 (C-CH3), 143.6 (CAr), 163.9 (2C-O), 196.5 (2CO) ppm.
9-(3,4-Dimethoxyphenyl)-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (3s). The product (3s, C21H22O5) was obtained as a white powder; 85% yield; Rf = 0.82 (DCM/acetone) (9
:1); FT-IR (KBr, cm−1): 1668 (CO), 1512 (CC); 1H NMR (400 MHz, DMSO-d6) δ 1.81–1.99 (m, 4H, 2CH2-CH2), 2.24–2.39 (m, 4H, 2CH2-CC), 2.54–2.69 (m, 4H, 2CH2-CO), 3.67 (s, 3H, O-CH3), 3.69 (s, 3H, O-CH3), 4.54 (s, 1H, *CH), 6.63 (d, JH–H = 6.0 Hz, 1H, CHAr), 6.76 (JH–H = 7.6 Hz, 2H, 2CHAr) ppm; 13C NMR (100 MHz, DMSO-d6) δ 19.8 (2CH2-CH2), 26.3 (2CH2-CC), 29.6 (2CH2-CO), 37.6 (*CH), 56.7 (2CH3-O), 111.6 (CHAr), 113.3 (CHAr), 115.9 (CHAr), 119.6 (2C-CO), 137.1 (CAr), 146.9 (C-OCH3), 148.1 (C-OCH3), 165.9 (2C-O), 197.1 (2CO) ppm.
X-ray crystallography
Crystallographic data for the studied compound 3r was collected on a SuperNova, Dual, Cu at home/near, AtlasS2 four-circle diffractometer equipped with an AtlasS2 CCD detector using Cu Kα (micro-focus sealed tube) radiation (λ = 1.54184 Å), the crystal was kept at a temperature of 295 K during data collection.The crystallographic data and experimental details for structural analysis are summarized in Table 9. The reported structure was solved with the SHELXT-2014/5 (ref. 47) solution program by Intrinsic Phasing with Olex2 (ref. 48) as the graphical interface. The model was refined with SHELXL-2018/3 (ref. 49) using full matrix least-squares minimization on F2. All absorption corrections were performed with the CrysAlisPro 1.171.42.51a50 using spherical harmonics implemented in SCALE3 ABSPACK scaling algorithm. Crystal structure visualization and construction of crystal packing diagrams were performed using Mercury 4.0 software.51
| Moiety formula | C20H20O3 |
| Sum formula | C20H20O3 |
| Formula weight (g mol−1) | 308.36 |
| Crystal habit, color | Needle, colourless |
| Crystal system | Orthorhombic |
| Space group | Pca21 |
| a (Å) | 18.2645(5) |
| b (Å) | 9.2078(2) |
| c (Å) | 9.6666(2) |
| α (°) | 90 |
| β (°) | 90 |
| γ (°) | 90 |
| Volume (Å3) | 1625.69(7) |
| Z, Z′ | 4, 0 |
| Density (calculated) (g cm−3) | 1.260 |
| Absorption coefficient (mm−1) | 0.670 |
| F(000) | 656.0 |
| Crystal size (mm) | 0.22 × 0.04 × 0.02 |
| 2θ range for data collection (°) | 9.606 to 149.024 |
| Reflections collected | 6225 |
| Independent reflections | 2390 |
| Rint | 0.0363 |
| Number of parameters | 209 |
| Goodness-of-fit on F2 | 1.045 |
| Final R indices [I ≥ 2σ(I)] | R1 = 0.042, wR2 = 0.1010 |
| R indexes [all data] | R1 = 0.0555, wR2 = 0.1106 |
| Largest difference peak and hole (Å3) | 0.13, −0.12 |
| CCDC deposition no. | CCDC 2308384 |
CCDC number: 2308384 contains the supplementary crystallographic data for compound 3r.52
Computational methods
The docking complexes for the 3e and 3p compounds were taken as the initial coordinates for MD simulations. A 10 Å orthorhombic box with a dimension of 10.0 × 10.0 × 10.0 nm3 employed TIP3P model contains 10567 water molecules/36350 atoms for 3e, and 10551 water molecules/36290 3p was used for the solvation of the system with OPLS-2005 force field. To neutralize the system, sodium ions were introduced as counter ions. Following solvation, the system underwent energy minimization, with a convergence threshold set at 1 kcal mol−1 Å−1. Subsequently, pre-equilibration was performed using Desmond's default six-step relaxation protocol. The initial two steps involved energy minimization, with the first step restraining the solute and the second step removing these restraints. The subsequent steps consisted of short MD simulations, each lasting 12 ps, 12 ps, and 24 ps, respectively, within the NPT ensemble at temperatures of 10 K, 10 K, 300 K, and 300 K.
A 200 ns MD simulation production run was then executed, applying periodic boundary conditions and simulating under isothermal and isobaric conditions with a relaxation time of 0.2 ps. The MD simulation was pivotal in identifying regions of the protein influenced by the presence of the ligand, making it valuable for validating docking poses. The simulations treated the entire protein–ligand system as flexible, providing insights into potential conformational changes and binding interactions.
Visualization of the protein–ligand complexes and analysis of the MD trajectory were carried out using Maestro. In-depth analyses were conducted using Desmond's Simulation Event Analysis tool, offering a comprehensive exploration of the simulation data.58
Regarding this part of the work, we used the SwissADME web tool via the link: https://www.swissadme.ch/,63 that offers free access to a pool of quick yet reliable predictive models for physicochemical properties, pharmacokinetics, drug-likeness and medicinal chemistry friendliness, including in-house effective techniques like the BOILED-Egg, iLOGP, and bioavailability radar. The Drug Likeness Score (DLS) results were determined using the Molsoft web tool via the link: https://www.molsoft.com.64
Conclusion
In summary, we have developed an environmentally friendly and efficient method for synthesizing xanthene derivatives using ultrasound irradiation as an energy source and zinc acetate as a mild catalyst. The synthesized compounds were prepared in high yields (84–95%) within a short reaction time. Structural confirmation of all compounds in this series was established through conventional spectroscopic methods. Additionally, we conducted in silico studies on these compounds to determine their significance. The results from the molecular docking study demonstrated satisfactory interactions of the synthesized compounds with the VEGRF-2 enzyme. Furthermore, a molecular dynamics simulation was carried out to validate the binding mode predicted by the molecular docking of compounds 3e and 3p within the active site of VEGRF-2. Notably, the two selected ligands exhibited consistent and favorable results, maintaining the same hydrogen bonds and preserving hydrophobic interactions with the residues even after a 200 ns simulation. The compounds were optimized using the DFT-B3LYP method at the 6-31G(d,p) level, revealing interesting energy gaps for most of them, suggesting their potential stability. Furthermore, all compounds were assessed based on the Lipinski Rule of Five for their bioactivity, molecular descriptors, and drug-likeness, demonstrating favorable oral bioavailability.Data availability
All data generated or analyzed during this study are included in this published article and its ESI files.†Author contributions
All authors contributed significantly to the design, execution, and interpretation of the reported experiments. Rayene Sayad: wrote the original draft, executed the experimental section, and conducted the ADME study. Abdeslem Bouzina: conducted docking and dynamic studies, played a pivotal role in validation, and contributed to the review and editing process. Yousra Ouafa Bouone: drafted the sections related to Density Functional Theory (DFT) and X-ray analysis. Dounia Beldjezzia: managed the curation of data and conducted formal analysis. Abdelhak Djemel and Malika Ibrahim-Ouali: provided essential resources for the study and conducted analyses of the obtained products (NMR, IR, and X-ray diffraction). Nour-Eddine Aouf and Zineb Aouf: contributed to the review and editing process, covering aspects such as the introduction, results, discussion, and other relevant sections. All authors have read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The General Directorate for Scientific Research and Technological Development (DG-RSDT), Algerian Ministry of Scientific Research, Applied Organic Chemistry Laboratory (FNR 2000), supported this work financially.References
- (a) M. R. Taghartapeh, N. N. Pesyan, H. Rashidnejad, H. R. Khavasi and A. Soltani, J. Mol. Struct., 2017, 1149, 873 CrossRef; (b) L. Jeyaseeli, A. D. Gupta, K. A. Kumar, K. Mazumdar, N. K. Dutta and S. G. Dastidar, Int. J. Antimicrob. Agents, 2006, 27, 62 CrossRef PubMed; (c) T. Yıldız and H. B. Küçük, RSC Adv., 2017, 7, 6649 RSC.
- A. Chaudhary and J. M. Khurana, Curr. Org. Synth., 2018, 15, 369 CrossRef.
- A. G. Ghahsare, Z. S. Nazifi and S. M. R. Nazifi, Curr. Org. Synth., 2019, 16, 7 Search PubMed.
- A. S. Burange, K. G. Gadam, P. S. Tugaonkar, S. D. Thakur, R. K. Soni, R. R. Khan, M. S. Tai and C. S. Gopinath, Environ. Chem. Lett., 2021, 19, 3314 CrossRef.
- G. Shabir, A. Saeed and P. A. Channar, Mini-Rev. Org. Chem., 2018, 15, 197 CrossRef.
- M. Maia, D. I. S. P. Resende, F. Duraes, M. M. M. Pinto and E. Sousa, Eur. J. Med. Chem., 2021, 210, 113085 CrossRef CAS PubMed.
- R. Sangwan, M. Saini, R. Verma, S. Kumar, M. Banerjee and S. Jain, J. Mol. Struct., 2020, 1208, 127786 CrossRef CAS.
- N. Mulakayala, P. V. N. S. Murthy, D. Rambabu, M. Aeluri, R. Adepu, G. R. Krishna, C. M. Reddy, K. R. S. Prasad, M. Chaitanya, C. S. Kumar, M. V. B. Rao and M. Pal, Bioorg. Med. Chem. Lett., 2012, 22, 2191 Search PubMed.
- J. J. Omolo, M. M. Johnson, S. F. Van Vuuren and C. B. de Koning, Bioorg. Med. Chem. Lett., 2011, 21, 7088 CrossRef.
- J. M. Jamison, K. Krabill, A. Hatwalkar, E. Jamison and C. C. Tsai, Cell Biol. Int. Rep., 1990, 14, 1084 Search PubMed.
- J. M. Khurana, D. Magoo, K. Aggarwal, N. Aggarwal, R. Kumar and C. Srivastava, Eur. J. Med. Chem., 2012, 58, 477 CrossRef PubMed.
- H. N. Hafez, M. I. Hegab, I. S. Ahmed-Farag and A. B. A. El Gazzar, Bioorg. Med. Chem. Lett., 2008, 18, 4543 Search PubMed.
- G. S. Shankarling and K. J. Jarag, Laser dyes, Resonance, 2010, 15, 818 CrossRef.
- (a) S. H. Guo, T. H. Leng, K. Wang, C. Y. Wang, Y. J. Shen and W. H. Zhu, Talanta, 2018, 185, 364 CrossRef; (b) J. Liu, X. Chen, Y. Zhang, G. Gao, X. Zhang and S. Hou, J. Lumin., 2018, 204, 484 CrossRef; (c) H. Qi, H. Takano, Y. Kato, Q. Wu, C. Ogata, B. Zhu, Y. Murata and Y. Nakamura, Biochim. Biophys. Acta, Gen. Subj., 2011, 1810, 712 CrossRef; (d) T. M. Ebaston, A. Rozovsky, A. Zaporozhets, A. Bazylevich, H. Tuchinsky, V. Marks, G. Gellerman and L. D. Patsenker, ChemMedChem, 2019, 14, 1734 CrossRef.
- K. M. Khan, I. Khan, S. Perveen and M. I. Malik, J. Fluorine Chem., 2014, 1581, 5 Search PubMed.
- (a) H. F. Niya, N. Hazeri, M. Fatahpour and M. T. Maghsoodlou, Res. Chem. Intermed., 2020, 46, 3666 Search PubMed; (b) D. Thirumalai and S. Gajalakshmi, Res. Chem. Intermed., 2020, 46, 2668 CrossRef; (c) N. Srinivasan, A. Yurek-George and A. Ganesan, Mol. Diversity, 2005, 9, 291 CrossRef CAS.
- D. Prasad, A. Preetam and M. Nath, C. R. Chim., 2013, 16, 1157 Search PubMed.
- (a) M. Salami and A. Ezabadi, Res. Chem. Intermed., 2020, 46, 4626 CrossRef; (b) S. E. S. Sorkhi, M. M. Hashemi and A. Ezabadi, Res. Chem. Intermed., 2020, 46, 2246 Search PubMed; (c) H. Naeimi and Z. S. Nazifi, J. Ind. Eng. Chem., 2014, 20, 1050 CrossRef.
- E. Yoshioka, S. Kohtani and H. Miyabe, Angew. Chem., Int. Ed., 2011, 50, 6642 CrossRef.
- G. Sabitha, K. Arundhathi, K. Sudhakar, B. S. Sastry and J. S. Yadav, Synth. Commun., 2008, 38, 3446 Search PubMed.
- B. S. Kuarm, J. V. Madhav, S. V. Laxmi, B. Rajitha, Y. T. Reddy, P. N. Reddy and P. A. Crooks, Synth. Commun., 2011, 41, 669 Search PubMed.
- S. Kantevari, R. Bantu and L. Nagarapu, Arkivoc, 2006, 16, 148 Search PubMed.
- H. Naeimi and Z. S. Nazifi, J. Ind. Eng. Chem., 2013, 20, 1050 Search PubMed.
- K. Gong, H. Wang, S. Wang, Y. Wang and J. Chen, Catal., 2015, 36, 1255 Search PubMed.
- H. Eshghi, M. Eftekhar, M. Rahimizadeh, M. Hassanpour and M. Bakavoli, Org. Chem. Res., 2016, 2, 27 Search PubMed.
- B. Das, K. Laxminarayana, M. Krishnaiah and Y. Srinivas, Synlett, 2007, 20, 3112 Search PubMed.
- P. Zhang, Y. D. Yu and Z. H. Zhang, Synth. Commun., 2008, 38, 4479 Search PubMed.
- A. Ilangovan, S. Malayappasamy, S. Muralidharan and S. Maruthamuthu, Chem. Cent. J., 2011, 5, 87 CrossRef PubMed.
- (a) R. K. Vohra, J. L. Renaud and C. Bruneau, Collect. Czech. Chem. Commun., 2005, 70, 1952 CrossRef; (b) I. Węglarz, M. Szewczyk and J. Mlynarskia, Adv. Synth. Catal., 2020, 8, 1536 Search PubMed.
- (a) J. Y. Liu, G. E Cao, W. Xu, J. Cao and W. L. Wang, Appl. Organomet. Chem., 2010, 24, 691 Search PubMed; (b) Q. Yuancheng, Z. Dan and M. Li, Lett. Org. Chem., 2012, 9, 272 Search PubMed.
- S. J. Modi and V. M. Kulkarni, Med. Drug Discovery, 2019, 2, 100009 CrossRef.
- I. V. Machado, J. R. N. dos Santos, M. A. P. Januario and A. G. Corrêa, Ultrason. Sonochem., 2021, 78, 105704 CrossRef CAS PubMed.
- W. Bonrath, Ultrason. Sonochem., 2005, 12, 106 CrossRef.
- N. Mulakayala, G. P. Kumar, D. Rambabu, M. Aeluri, M. V. B. Rao and M. Pal, Tetrahedron Lett., 2012, 53, 6926 Search PubMed.
- S. Rostamizadeh, A. M. Amani, G. H. Mahdavinia, G. Amiri and H. Sepehrian, Ultrason. Sonochem., 2010, 17, 309 CrossRef PubMed.
- S. Kumari, A. Shekhar and D. D. Pathak, Chem. Sci. Trans., 2014, 3, 663 Search PubMed.
- A. R. M. Zarea, M. Rezaeib, M. Merajoddinb, H. Hamidianb, A. Zareb and M. K. Rostamic, Sci. Iran., 2014, 21, 2058 Search PubMed.
- H. Naeimi and Z. S. Nazifi, J. Ind. Eng. Chem., 2014, 20, 1049 Search PubMed.
- (a) B. P. Reddy, V. Vijayakumar, T. Narasimhamurthy, J. Suresh and P. L. Lakshman, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, o916 Search PubMed; (b) M. A. Bigdeli, G. H. Mahdavinia and V. Amani, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, E63, o3493 Search PubMed.
- (a) M. M. Alam, R. Varala and V. Seema, Mini-Rev. Org. Chem., 2023, 21, 555 CrossRef; (b) M. Neetha, K. R. Rohit, S. Saranya and G. Anilkuma, ChemistrySelect, 2020, 5, 1054 CrossRef CAS.
- M. L. da Silva, R. R. Teixeira, L. de Azevedo Santos, F. T. Martins and T. C. Ramalho, Arab. J. Chem., 2020, 13, 974 CrossRef CAS.
- A. N. Dadhania, V. K. Patel and D. K. Raval, C. R. Chim., 2012, 15, 383 CrossRef.
- A. G. A. El-Helby, H. Sakr, I. H. Eissa, H. Abulkhair, A. A. Al-Karmalawy and K. El-Adl, Arch. Pharm., 2019, 352, 1900113 CrossRef CAS.
- (a) S. Limsuwan, E. N. Trip, K. Trhm, S. Piersma, A. Hiranrat, W. Mahabusarakam, S. P. Voravuthikunchai, J. M. V. Dijl and O. Kayser, Phytomedicine, 2009, 16, 651 CrossRef; (b) M. Kaya, E. Demir and H. Bekci, J. Enzyme Inhib. Med. Chem., 2012, 28, 893 Search PubMed.
- (a) R. Sangwan, M. Saini, R. Verma, S. Kumar, M. Banerjee and S. Jain, J. Mol. Struct., 2020, 1208, 127786 CrossRef CAS; (b) N. Mulakayala, P. V. N. S. Murthy, D. Rambabu, M. Aeluri, R. Adepu, G. R. Krishna, C. M. Reddy, K. R. S. Prasad, M. Chaitanya, C. S. Kumar and M. B. Rao, Bioorg. Med. Chem. Lett., 2012, 22, 2191 Search PubMed; (c) F. Al-Omran, R. M. Mohareb and A. Abou El-Khair, Med. Chem. Res., 2014, 23, 1633 Search PubMed.
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 341 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 8 Search PubMed.
- Rigaku Oxford Diffraction, CrysAlisPro Software System, 2022 Search PubMed.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, Mercury 4.0: from visualization to analysis, design and prediction, J. Appl. Crystallogr., 2020, 53, 235 CrossRef PubMed.
- N. Aouf, CCDC 2308384: Experimental Crystal Structure Determination, 2023 Search PubMed.
- Y. Miyazaki, S. Matsunaga, J. Tang, Y. Maeda, M. Nakano, R. J. Philippe, M. Shibahara, W. Liu, H. Sato, L. Wangb and R. T. Nolteb, Bioorganic Med. Chem. Lett., 2005, 15, 2207 CrossRef.
- S. Release, 2. LigPrep, Version 3.4. Schrödinger, LLC, New York, NY, 2015, p. 26400175 Search PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, D. E. Shaw, M. Shelley, J. K. Perry, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1749 CrossRef.
- (a) E. C. Meng, T. D. Goddard, E. F. Pettersen, G. S. Couch, Z. J. Pearson, J. H. Morris and T. E. Ferrin, Protein Sci., 2023, 32, 4792 CrossRef; (b) E. F. Pettersen, T. D. Goddard, C. C. Huang, E. C. Meng, G. S. Couch, T. I. Croll, J. H. Morris and T. E. Ferrin, Protein Sci., 2021, 30, 82 CrossRef.
- (a) E. Chow, C. A. Rendleman, J. K. Bowers, R. O. Dror, D. H. Hughes, J. Gullingsrud, F. D. Sacerdoti and D. E. Shaw, Desmond performance on a cluster of multicore processors, DE Shaw Research Technical Report, New York, 2008, DESRES/TR-2008-01, https://deshawresearch.com/ Search PubMed; (b) Schrödinger Release 2023-4: Desmond Molecular Dynamics System, D. E. Shaw Research, Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, 2023 Search PubMed.
- R. V. Krems, Phys. Chem. Chem. Phys., 2019, 21, 13410 RSC.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5652 Search PubMed; (b) M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3665 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, Gaussian-09 Revision A.02, Gaussian Inc, Wallingford CT, 2009 Search PubMed.
- G. Zhang and C. B. Musgrave, J. Phys. Chem. A, 2007, 111, 1561 Search PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 64, 17 Search PubMed.
- (a) A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed; (b) https://www.swissadme.ch/.
- https://www.molsoft.com.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2308384. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra04135f |
| This journal is © The Royal Society of Chemistry 2024 |