
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Levofloxacin reposition-based design: synthesis, biological evaluation of new levofloxacin derivatives targeting topoisomerase II beta polymerase as promising anticancer agents, molecular docking, and physicochemical characterization†
Zeinab Mahmoud a,
Mohamed M. Ismailb,
Mona Kamela and
Amira Youssef*b
a,
Mohamed M. Ismailb,
Mona Kamela and
Amira Youssef*b
aPharmaceutical Organic Chemistry Department, Faculty of Pharmacy, Cairo University, 11561 Cairo, Egypt
bPharmaceutical Organic Chemistry Department, College of Pharmaceutical Sciences and Drug Manufacturing, Misr University for Science and Technology, P. O. Box 77 Giza, Egypt. E-mail: amira.youssef@must.edu.eg; Tel: +201285266644
First published on 3rd September 2024
Abstract
Repositioning of already approved medications through repurposing or re-profiling for new medical uses after certain structural modifications is a novel approach in drug discovery. Fluoroquinolone antibiotics are one of the cardinal agents investigated for potential anticancer activities. In this work, levofloxacin was repositioned for anticancer activities. A series of levofloxacin-based compounds were designed and synthesized through the derivatization of levofloxacin's carboxylic acid functionality. The newly synthesized compounds were screened for cytotoxic activities against breast, liver, and leukemia cancer cell lines. Their effect on normal cells was also investigated. The target compounds were evaluated for their proliferative inhibitory activity toward topoisomerase II beta polymerization. Compound 5 showed higher inhibitory activity against a breast cancer cell line (MCF-7) with IC50 = 1.4 μM and lesser side effects on a normal breast cell line (MCF-10a) with IC50 = 30.40 μM than reference drugs. The best activity against a liver cancer cell line (Hep3B) was exhibited by compounds 3c, 4b, 5, 7, 8, 13a and 13c with IC50 values ranging from 0.43 to 8.79 μM. Regarding the effect of compounds 5 and 13a on a leukemia cancer cell line (L-SR), their IC50 values were 0.96 and 3.12 μM, respectively. Compounds 3c and 5 showed Topo2β inhibitory effects on Hep3B cells (81.33% and 83.73%, respectively), which was better than levofloxacin and etoposide as reference drugs. Cytometry cell cycle analysis revealed that compounds 3c and 5 arrested the cell cycle at the S phase (37.56% and 39.09%, respectively). Compounds 3c and 5 exhibited an elevation in active caspase-3 levels by 4.9 and 4.5 folds, respectively. Molecular modeling simulation of compounds 3c and 5 demonstrated energy scores (−29.77 and −20.46 kcal mol−1, respectively) more than those of the reference drugs as they interact with the most essential amino acids required for good affinity towards human topoisomerase II beta enzyme (PDB ID: 3QX3). Physicochemical characteristics of the most promising cytotoxic compounds 3c and 5 were investigated and compared to etoposide and levofloxacin as reference drugs. However, they showed high gastrointestinal absorption and could not penetrate the blood–brain barrier.
1. Introduction
The high costs coupled with the increased attrition rate in developing new and efficacious anti-cancer agents for clinical use necessitate alternative thinking.1–4 There is increasing interest towards the repositioning of existing drugs and reevaluating them for new biological activities.5–8 It makes the repositioning of fluoroquinolones as antitumor agents justifiable by their apoptotic potential, anti-proliferative activity and induced cell cycle arrest in addition to their use for decades as antimicrobial agents with different derivatives.9–14 Fluoroquinolones exhibit the required features in cancer treatment, such as lower incidence of resistance development, reduced toxicity, diminished tendency for the development of drug-induced secondary tumors, and higher potency than other topoisomerase II inhibitors15–18 (Fig. 1).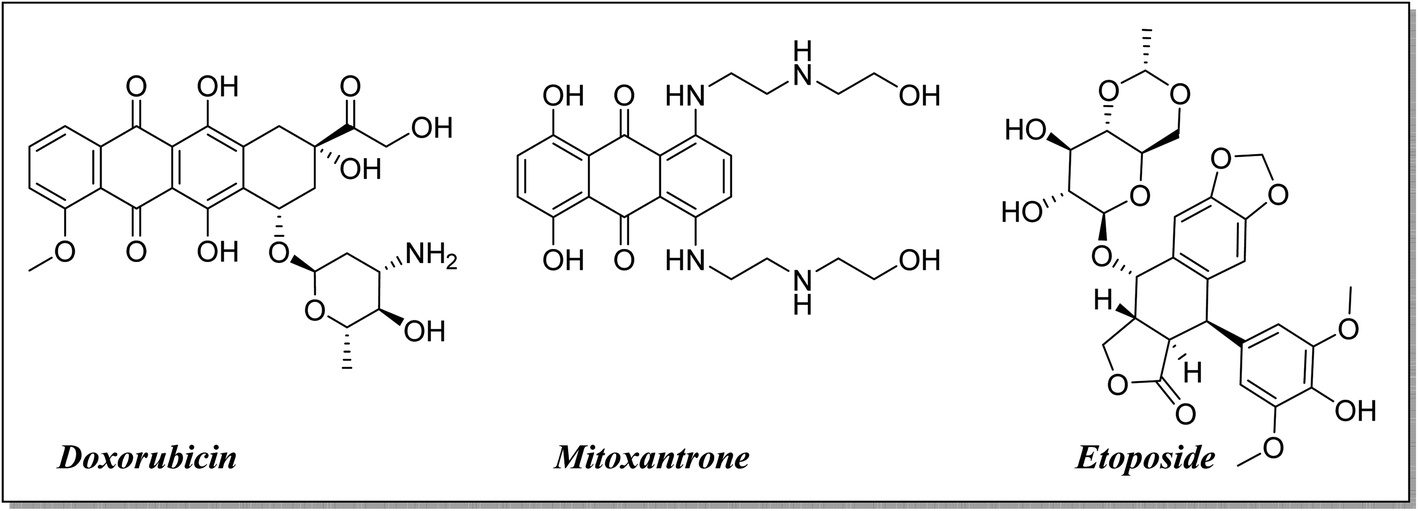 | ||
| Fig. 1 Significant topoisomerase II inhibitors. | ||
Moreover, fluoroquinolone derivatives are characterized by their ease and flexibility of synthesis using different procedures and building blocks to afford several useful chemical structures.15 It was reported that replacement of the C-3 carboxylic group of fluoroquinolone antibacterial analogs with a heterocyclic ring can yield novel antitumor analogs. More specifically, the replacement of the carboxylic group of fluoroquinolones with a heterocyclic ring, hydrazones, or Mannich bases furnished a promising approach for developing novel anticancer agents.10,17,19,20 The aim of this study was to design and synthesize new levofloxacin derivatives with improved biological activities.
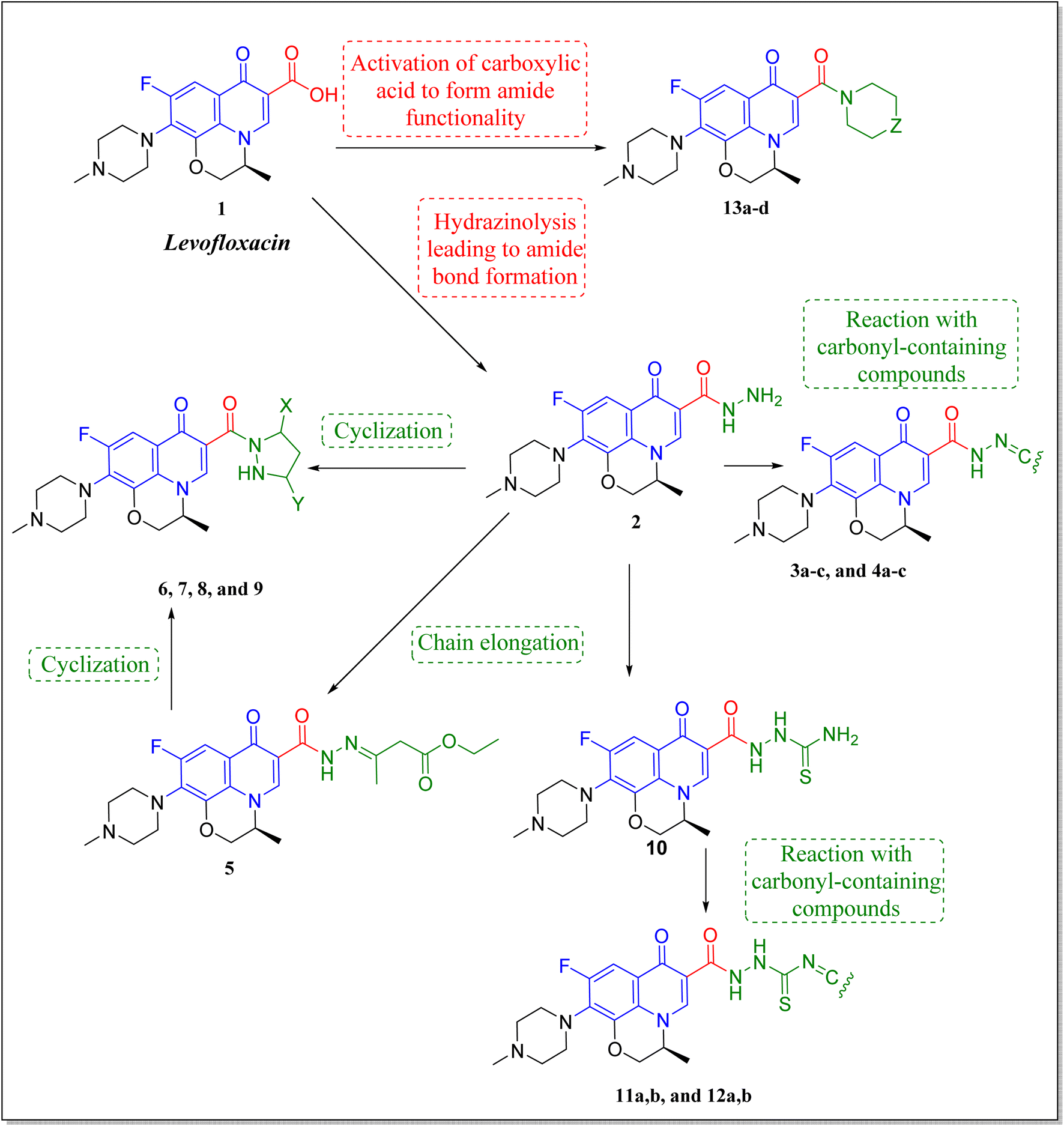
Levofloxacin is a well-known, widely used synthetic fluoroquinolone antibacterial agent. Moreover, many research studies highlighted its biological effect as an anticancer agent,21–24 through the inhibition of human topoisomerase II,25–28 involving topoisomerase II beta.28 It was concluded that the activity of levofloxacin on Topo2β is more potent than that of Topo2α, as it interacted with the TRP931 amino acid residue with a binding energy of −8.06 on Topo2α, while the binding with Topo2β showed interaction with 2 amino acid residues (SER480 and ASP479) with a better binding energy of −11.78. Moreover, levofloxacin was investigated for cell cycle analysis showing cell cycle arrest primarily in the S and G2/M phases of progression with a significant increase in caspase-3 levels.29–32 Structural features required for the anticancer activity of levofloxacin have been well defined.33 Most of the required chemical modifications to reposition levofloxacin from its antibacterial activity into its anticancer one were at its carboxylic group functionality.15,25,33–36 Based on the illustrated findings, this study aimed to design levofloxacin-based selective mammalian topoisomerase II beta (Topo2β) enzyme inhibitors as potent anticancer agents according to the denoted tactic in Fig. 2.
 | ||
| Fig. 2 Rational strategy for repositioning levofloxacin towards anticancer activity. | ||
2. Results and discussion
2.1. Chemistry
The deigned levofloxacin-based compounds were synthesized according to four schemes. In Scheme 1, levofloxacin 1 undergoes two main reactions. The hydrazinolysis of levofloxacin with hydrazine hydrate (80%) afforded the levofloxacin hydrazide 2 according to the reported procedure.35 Compound 2 is considered as the cornerstone for synthesizing all new members in Schemes 1–3. All the newly synthesized compounds were elucidated by 1H NMR, 13C NMR and IR spectroscopic techniques, and are in agreement with the rational correspondence of the proposed structures. Accordingly, refluxing compound 2 with different carbonyl-containing compounds in absolute ethanol in the presence of glacial acetic acid afforded the N′-benzylidinequinoline-6-carbohydrazide series 3 and 4. It was reported that fluoroquinolone Schiff's bases could be obtained by simple condensation of fluoroquinolone hydrazides with different carbonyl compounds in absolute ethanol in the presence of glacial acetic acid under reflux.37,38 Compounds 3a–c and 4a–c were prepared via the conventional pathway by reacting levofloxacin hydrazide 2 with the appropriate carbonyl-containing compounds namely, 4-chlorobenzaldehyde, 4-fluorobenzaldehyde and anisaldehyde to give 3a–c, and isatin, 5-bromoisatin and 5-chloroisatin to afford 4a–c, respectively. Interestingly, tracking the appearance and disappearance with the accurate integration of amino groups (NH) and (NH2) was so helpful in identifying the newly synthesized compounds. The D2O-influenced disappearance of the (NH) singlet peak in the range of δ 13.37–10.64 ppm corresponding to only one proton integration in addition to the drastic increase in the aromatic protons' integration confirmed the success of the reactions of this pathway. Compounds 4a–c showed two D2O exchangeable singlet signals around δ 10.77–13.85 ppm corresponding to two NH protons and exhibited aromatic protons in the range of δ 6.86–7.60 ppm in 1H NMR spectroscopy. The 13C NMR spectrum displayed the appearance of three peaks in the range of δ 174.6–159.1 ppm.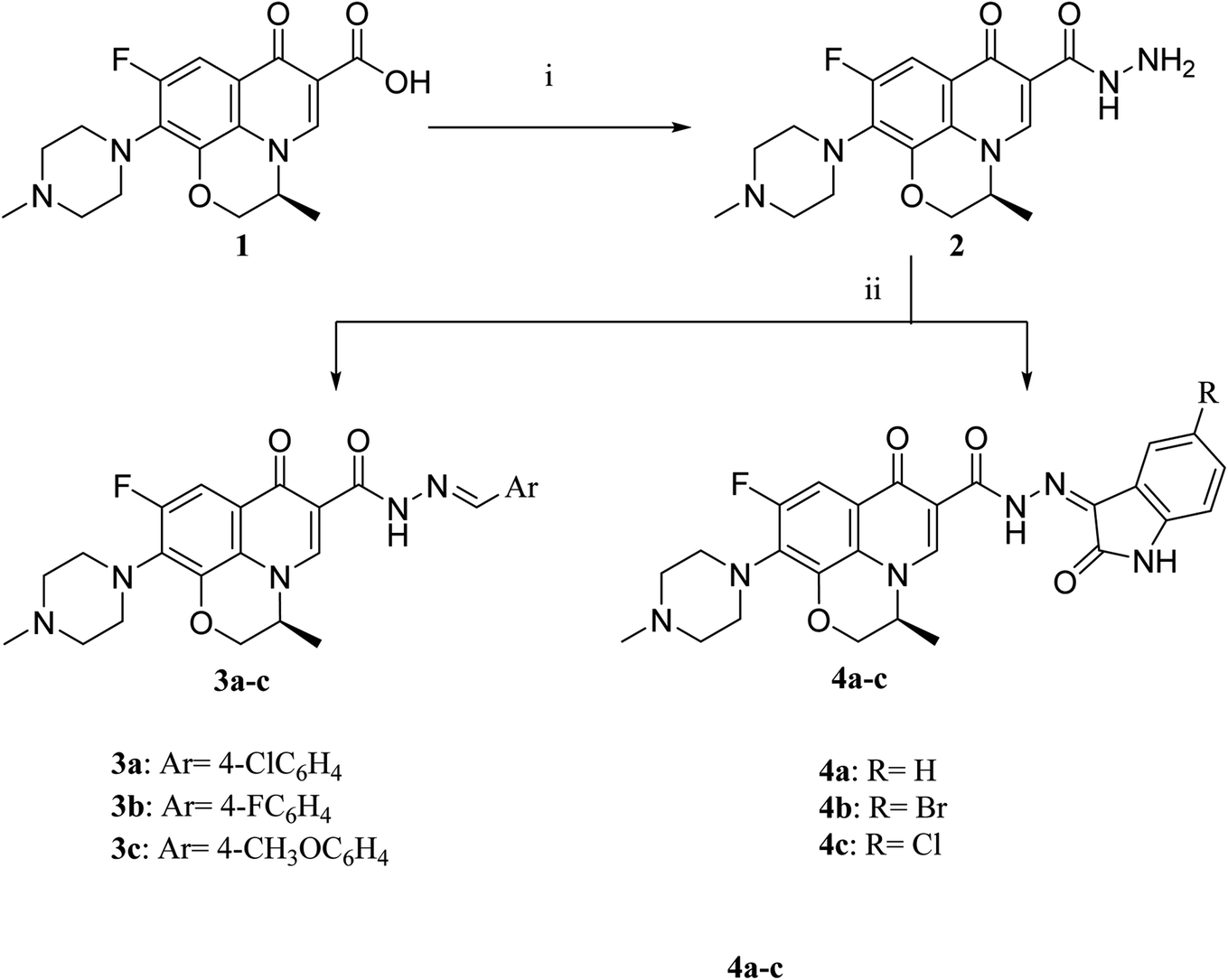 | ||
| Scheme 1 Preparation of levofloxacin derivatives 3a–c and 4a–c. Reagents and conditions: (i) 80% NH2NH2, reflux 24 h; (ii) appropriate carbonyl-containing compounds, absolute ethanol, glacial acetic acid, reflux 6 h. | ||
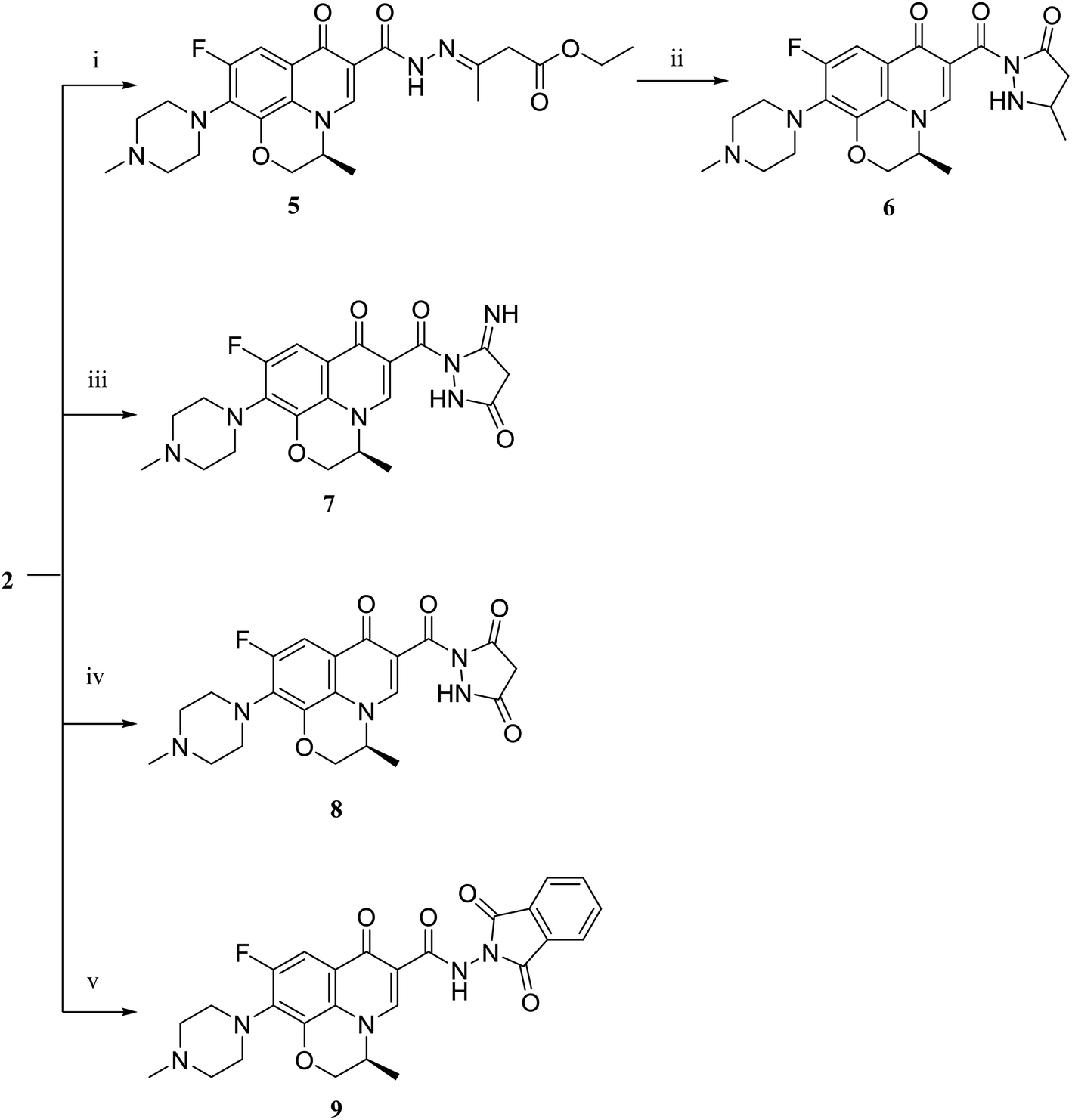 | ||
| Scheme 2 Preparation of levofloxacin derivatives 5, 6, 7, 8 and 9. Reagents and conditions: (i) ethyl acetoacetate, absolute ethanol, reflux 12 h; (ii) reflux 90 h; (iii) ethyl cyanoacetate, absolute ethanol, piperidine, reflux 90 h; (iv) diethyl malonate, absolute ethanol, sodium ethoxide, reflux 7 h; (v) phthalic anhydride, absolute ethanol, glacial acetic acid, reflux 4 h. | ||
 | ||
| Scheme 3 Preparation of levofloxacin derivatives 10, 11a,b and 12a,b. Reagents and conditions: (i) KSCN, absolute ethanol, conc. HCl, reflux 8 h; (ii) appropriate carbonyl-containing compounds, absolute ethanol, sodium acetate, reflux 12 h. | ||
Literature review showed that the related cyclization reactions of hydrazides with ethyl acetoacetate have been performed by heating under reflux in absolute ethanol,39–41 or in glacial acetic acid.42,43 In contrast, in Scheme 2, the reaction of compound 2 with ethyl acetoacetate ester afforded time-dependent. More clearly, levofloxacin hydrazide 2 reacted with ethyl acetoacetate in absolute ethanol under reflux for 12 hours and the reaction was monitored by TLC. The spectral data of the obtained compound 5 revealed that it is the open uncyclized product. However, upon extending the reaction time, a change in color was observed. This continuous change in color encouraged us to continue the reaction up to 90 hours under reflux to afford the cyclized pyrazolidone ring compound 6. The spectral data of both compounds 5 and 6 were in accordance with the predicted assumption. The 1H NMR spectrum of compound 5 revealed the presence of the triplet signal at δ 1.22 ppm corresponding to the methyl group of OCH2CH3 and the quartet signal of the two protons at δ 4.12 ppm related to the presence of OCH2CH3. Meanwhile, the signal of the two protons at δ 3.26 ppm corresponding to N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(CH3)CH2 and a singlet signal corresponding to three protons at δ 2.02 confirm the presence of NC(CH3)CH2. On the contrary, the 1H NMR spectrum of compound 6 showed the expected signals for the two CH2 groups of the pyrazolidone ring protons at δ 1.84 and 1.89 ppm, and the multiplet signal of three protons in the range of δ 2.02–2.09 ppm corresponding to the presence of CH3 of pyrazolidone ring. The 1H NMR spectra showed two D2O exchangeable singlet signals at δ 10.62 and 10.65 corresponding to the tautomeric OH, while the NH proton of pyrazolidone ring appeared at δ 3.26 ppm. Furthermore, refluxing the hydrazide derivative 2 with either ethyl cyanoacetate or diethyl malonate afforded the cyclic amides 7 and 8, sequentially. Compound 7 was prepared through the reaction of levofloxacin hydrazide 2 with ethyl cyanoacetate and a few drops of piperidine in absolute ethanol, following the reported methods.42 The 1H NMR spectrum of compound 7 revealed the presence of a multiplet signal of two protons in the range of δ 4.13–4.18 ppm corresponding to the CH2 protons of pyrazolidone ring, and two D2O exchangeable singlet signals in the range of δ 10.60–11.86 ppm due to two NH protons. The respective levofloxacin pyrazolidine-3,5-dione 8 was prepared via refluxing the hydrazide derivative 2 with diethyl malonate in the presence of sodium ethoxide, following the reported steps.42 Compound 8 1H NMR spectrum exhibited a multiplet signal corresponding to two protons of CH2 of pyrazolidone ring in the range of δ 4.54–4.57 ppm and the D2O exchangeable proton corresponding to the signal of NH proton at δ 10.62 ppm. The 13C NMR spectrum revealed the presence of the 4 carbonyl groups in the range of δ 154.4–174.2 ppm. Reviewing the literature revealed that the reaction of hydrazides with phthalic anhydride is achieved under different conditions, namely, fusion,44 or in the presence of anhydrous sodium acetate to give the cyclized ring structure.42 Adopting these reported procedures failed to give the target compound 9. Refluxing phthalic anhydride with compound 2 for 5 hours in the presence of glacial acetic acid yielded compound 9 as a straightforward product, according to the illustrated procedures.38,45 1H NMR spectrum revealed the presence of a D2O exchangeable singlet signal at δ 11.67 ppm due to two NH protons, and multiplet signals corresponding to the aromatic protons in the range of δ 7.48–7.60 ppm and δ 7.93–7.96 ppm. The appearance of four carbonyl carbons in the range of δ 165.6–174.2 ppm in the 13C NMR spectrum provided evidence. The 1H NMR spectrum revealed the presence of a D2O exchangeable singlet signal at δ 11.67 ppm due to two NH protons, and multiplet signals corresponding to the aromatic protons in the range of δ 7.48–7.60 ppm and δ 7.93–7.96 ppm, respectively. The appearance of four carbonyl carbons in the range of δ 165.6–174.2 ppm in the 13C NMR spectrum confirmed the formation of compound 9. The aim of the reactions of Scheme 3 was to bring about the chain elongation strategy for the design. In accordance, refluxing compound 2 with potassium thiocyanate afforded the hydrazinecarbothioamide derivative 10, according to the following procedure.42 The 1H NMR spectrum of compound 10 revealed the presence of a singlet signal equivalent to four D2O exchangeable protons at δ 7.89, 9.40, 11.20 and 11.39 ppm corresponding to two NH and NH2 protons. In the 13C NMR spectrum, the thiocarbonyl carbon appeared at δ 174.2 ppm. The condensation reaction of compound 10 with different carbonyl-containing compounds pulled off the production of the thioamide derivatives 11a,b and 12a,b according to the reported procedure.46 Series 11 and 12 IR spectrum showed the two NH absorption bands in the range of ν 3523–3419 cm−1. Additionally, the 1H NMR spectrum of compounds 11a and 11b revealed the presence of singlet signals of the two D2O exchangeable protons in the range of δ 9.38–13.20 ppm, and the presence of a singlet signal of the NCH proton at δ 8.42 and 8.35 ppm, respectively. Moreover, the 1H NMR spectrum of compounds 12a and 12b revealed the presence of a singlet signal of the NC–CH3 proton at δ 2.35 and 1.85 ppm, sequentially, and the doublet signals corresponding to the aromatic protons in the range of δ 7.00–7.81 ppm. However, the 13C NMR spectrum of series 11 and only compound 12b showed the presence of a carbon monosulphide atom in the range of δ 174.4–188.5 ppm. Unfortunately, the poor solubility of compound 12a hindered running any 13C NMR experiment.
C(CH3)CH2 and a singlet signal corresponding to three protons at δ 2.02 confirm the presence of NC(CH3)CH2. On the contrary, the 1H NMR spectrum of compound 6 showed the expected signals for the two CH2 groups of the pyrazolidone ring protons at δ 1.84 and 1.89 ppm, and the multiplet signal of three protons in the range of δ 2.02–2.09 ppm corresponding to the presence of CH3 of pyrazolidone ring. The 1H NMR spectra showed two D2O exchangeable singlet signals at δ 10.62 and 10.65 corresponding to the tautomeric OH, while the NH proton of pyrazolidone ring appeared at δ 3.26 ppm. Furthermore, refluxing the hydrazide derivative 2 with either ethyl cyanoacetate or diethyl malonate afforded the cyclic amides 7 and 8, sequentially. Compound 7 was prepared through the reaction of levofloxacin hydrazide 2 with ethyl cyanoacetate and a few drops of piperidine in absolute ethanol, following the reported methods.42 The 1H NMR spectrum of compound 7 revealed the presence of a multiplet signal of two protons in the range of δ 4.13–4.18 ppm corresponding to the CH2 protons of pyrazolidone ring, and two D2O exchangeable singlet signals in the range of δ 10.60–11.86 ppm due to two NH protons. The respective levofloxacin pyrazolidine-3,5-dione 8 was prepared via refluxing the hydrazide derivative 2 with diethyl malonate in the presence of sodium ethoxide, following the reported steps.42 Compound 8 1H NMR spectrum exhibited a multiplet signal corresponding to two protons of CH2 of pyrazolidone ring in the range of δ 4.54–4.57 ppm and the D2O exchangeable proton corresponding to the signal of NH proton at δ 10.62 ppm. The 13C NMR spectrum revealed the presence of the 4 carbonyl groups in the range of δ 154.4–174.2 ppm. Reviewing the literature revealed that the reaction of hydrazides with phthalic anhydride is achieved under different conditions, namely, fusion,44 or in the presence of anhydrous sodium acetate to give the cyclized ring structure.42 Adopting these reported procedures failed to give the target compound 9. Refluxing phthalic anhydride with compound 2 for 5 hours in the presence of glacial acetic acid yielded compound 9 as a straightforward product, according to the illustrated procedures.38,45 1H NMR spectrum revealed the presence of a D2O exchangeable singlet signal at δ 11.67 ppm due to two NH protons, and multiplet signals corresponding to the aromatic protons in the range of δ 7.48–7.60 ppm and δ 7.93–7.96 ppm. The appearance of four carbonyl carbons in the range of δ 165.6–174.2 ppm in the 13C NMR spectrum provided evidence. The 1H NMR spectrum revealed the presence of a D2O exchangeable singlet signal at δ 11.67 ppm due to two NH protons, and multiplet signals corresponding to the aromatic protons in the range of δ 7.48–7.60 ppm and δ 7.93–7.96 ppm, respectively. The appearance of four carbonyl carbons in the range of δ 165.6–174.2 ppm in the 13C NMR spectrum confirmed the formation of compound 9. The aim of the reactions of Scheme 3 was to bring about the chain elongation strategy for the design. In accordance, refluxing compound 2 with potassium thiocyanate afforded the hydrazinecarbothioamide derivative 10, according to the following procedure.42 The 1H NMR spectrum of compound 10 revealed the presence of a singlet signal equivalent to four D2O exchangeable protons at δ 7.89, 9.40, 11.20 and 11.39 ppm corresponding to two NH and NH2 protons. In the 13C NMR spectrum, the thiocarbonyl carbon appeared at δ 174.2 ppm. The condensation reaction of compound 10 with different carbonyl-containing compounds pulled off the production of the thioamide derivatives 11a,b and 12a,b according to the reported procedure.46 Series 11 and 12 IR spectrum showed the two NH absorption bands in the range of ν 3523–3419 cm−1. Additionally, the 1H NMR spectrum of compounds 11a and 11b revealed the presence of singlet signals of the two D2O exchangeable protons in the range of δ 9.38–13.20 ppm, and the presence of a singlet signal of the NCH proton at δ 8.42 and 8.35 ppm, respectively. Moreover, the 1H NMR spectrum of compounds 12a and 12b revealed the presence of a singlet signal of the NC–CH3 proton at δ 2.35 and 1.85 ppm, sequentially, and the doublet signals corresponding to the aromatic protons in the range of δ 7.00–7.81 ppm. However, the 13C NMR spectrum of series 11 and only compound 12b showed the presence of a carbon monosulphide atom in the range of δ 174.4–188.5 ppm. Unfortunately, the poor solubility of compound 12a hindered running any 13C NMR experiment.
The activation of levofloxacin carboxylic acid functionality by esterification was another tactic for amide bond synthesis. The synthesis of methyl levofloxacin ester was reported47 via heating under reflux levofloxacin in methanol in the presence of few drops of concentrated sulphuric acid for about 7–8 hours. To obtain the corresponding amide, aromatic amines were then added in the same round flask with continuous stirring and the reaction was continued to be refluxed for about 2–3 hours. Adopting this procedure in this study failed to afford the target compounds. In Scheme 4, levofloxacin 1 was reacted with ethyl chloroformate in the presence of triethylamine in methanol at 0 °C to give the unseparated intermediate ester, which was directly reacted with the appropriate secondary amines to afford series 13a–d, following the reported method.48 1H NMR spectra showed the aliphatic hydrogen signals of piperazine and azinanylcarbonyl protons in the aliphatic region in the range of δ 2.31–3.86 ppm. The 1H NMR spectra of compound 13c revealed the appearance of the specific signal corresponding to the methyl group at δ 2.31 ppm, while the spectrum of compound 13d showed the appearance of 5 aromatic protons of the N-phenyl group in the range of δ 6.92–6.99 ppm.
 | ||
| Scheme 4 Preparation of levofloxacin derivatives 13a–d. Reagents and conditions: (i) ClCOOEt, triethylamine (TEA), methanol, 0–5 °C, stirring 30 min; (ii) the appropriate secondary amine, 0 °C, srirring 12 h. | ||
2.2. Cytotoxic activity discussion
All the test compounds were evaluated for their cytotoxic activity against human breast cancer (MCF-7), human liver cancer (Hep3B) and leukemia (L-SR) cell lines. The new compounds showed promising cytotoxic activity compared with etoposide and levofloxacin as reference drugs. The in vitro antitumor activities of compounds 3a–c, 4a–c, 5, 6, 7, 8, 9, 10, 11a,b, 12a,b and 13a–d against the three cell lines were evaluated by an MTT assay. The drug concentration required to inhibit the cell growth by 50% (IC50) was evaluated, and the results are displayed and represented graphically (Tables 1, 2 and Fig. 3). For comparison, the IC50 values of reference drugs, etoposide and levofloxacin, are displayed. The addition of aldehydic derivatives to compound 2 results in compounds 3a–c exhibiting low cytotoxic activity against the breast cancer cell line MCF-7. Compounds 3a and 3b (4-chlorobenzaldehyde and 4-fluorobenzaldehyde), respectively, displayed high cytotoxic activity against leukemia (L-SR), as compound 3c (4-methoxybenzaldehyde derivative) showed high cytotoxic activity against the liver cancer cell line with an IC50 value of 0.43 μM. The addition of isatin derivatives to compound 2 displayed low to moderate cytotoxic activity against selected cancer cell lines. Compounds 4b (5-bromoisatin) exhibited high cytotoxic activity against the liver cancer cell line Hep3B with an IC50 value of 8.79 μM in comparison to reference drugs. The open uncyclized compound 5 showed excellent cytotoxic activity against MCF-7, Hep3B and L-SR cell lines (IC50 values of 1.4 μM, 3.77 μM, and 0.96 μM, correspondingly). Furthermore, the cyclized pyrazolidone ring compound 6 exhibited low cytotoxic activity against all cancer cell lines. The cyclized iminopyrazolidone and pyrazolidine-3,5-dione compounds 7 and 8, respectively, showed a moderate cytotoxic activity against MCF-7 (IC50 values of 4.37 and 3.51 μM) and excellent cytotoxic activity against Hep3B (IC50 values of 6.99 and 7.69 μM) compared with the reference drugs. The addition of ethyl cyanoacetate and diethyl malonate to compound 2 results in compounds 9 and 10, respectively, exhibiting low cytotoxic activity against all cancer cells. The addition of carbonyl-containing compounds to compound 10 results in compounds 11a,b and 12a, which exhibited low to moderate cytotoxic activity against cancer cell lines. Levofloxacin amide compounds 13a–d exhibited high to moderate cytotoxic activity against most cancer cell lines in comparison to etoposide and levofloxacin as the reference drugs.| Compound no. | Cytotoxicity IC50 (μM) | ||
|---|---|---|---|
| MCF-7 | Hep3B | L-SR | |
| 3a | 24.77 ± 1.61 | 24.67 ± 0.81 | 2.75 ± 0.07 |
| 3b | 62.99 ± 2.8 | 11.95 ± 0.39 | 5.64 ± 0.27 |
| 3c | 40.59 ± 3.17 | 0.43 ± 0.01 | 22.35 ± 1.34 |
| 4a | 39.1 ± 1.81 | 18.7 ± 0.87 | 49.6 ± 2.99 |
| 4b | 5.59 ± 0.26 | 8.79 ± 0.41 | 10.9 ± 0.66 |
| 4c | 15.8 ± 0.76 | 36.3 ± 1.68 | 4.32 ± 0.26 |
| 5 | 1.4 ± 0.06 | 3.77 ± 0.17 | 0.96 ± 0.06 |
| 6 | 40.6 ± 1.88 | 13.1 ± 0.61 | 28.9 ± 1.75 |
| 7 | 4.37 ± 0.2 | 6.99 ± 0.32 | 12.1 ± 0.73 |
| 8 | 3.51 ± 0.16 | 7.69 ± 0.36 | 5.49 ± 0.33 |
| 9 | 19.3 ± 0.9 | 42.2 ± 1.96 | 23.4 ± 1.41 |
| 10 | 95.4 ± 4.42 | 37.9 ± 1.76 | 66.0 ± 3.98 |
| 11a | 54.5 ± 2.52 | 80.2 ± 3.71 | 19.9 ± 1.2 |
| 11b | 15.5 ± 0.72 | 14.6 ± 0.67 | 40.3 ± 2.43 |
| 12a | 45.4 ± 2.1 | 23.6 ± 1.09 | 23.8 ± 1.43 |
| 12b | 5.21 ± 0.24 | 17.6 ± 0.81 | 34.2 ± 2.06 |
| 13a | 12.1 ± 0.56 | 3.46 ± 0.16 | 3.12 ± 0.19 |
| 13b | 26.8 ± 1.24 | 11.2 ± 0.52 | 18.3 ± 1.11 |
| 13c | 7.26 ± 0.34 | 1.04 ± 0.05 | 13.6 ± 0.82 |
| 13d | 9.59 ± 0.44 | 24.5 ± 1.14 | 15.1 ± 0.91 |
| Etoposide | 16.32 ± 0.72 | 21.93 ± 0.72 | 3.38 ± 0.13 |
| Levofloxacin | 2.79 ± 0.16 | 8.94 ± 0.29 | 7.38 ± 0.31 |
| Compound no. | MCF-7 (μM) | Topo2β% inhibition |
|---|---|---|
| 5 | 1.40 | 78.30 |
| Etoposide | 16.32 | 63.18 |
| Levofloxacin | 2.79 | 59.80 |
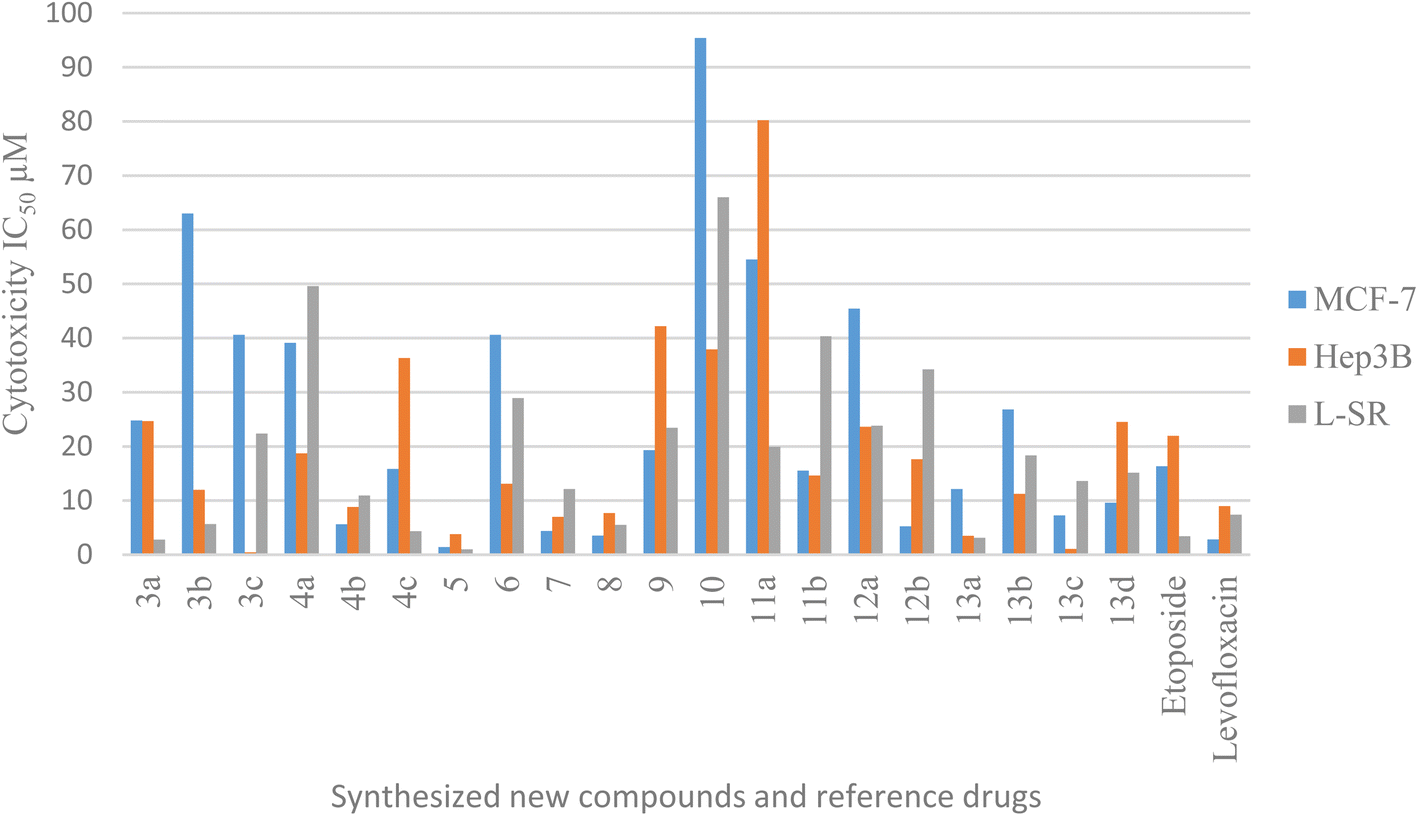 | ||
| Fig. 3 Graphical bar presentation of IC50 for the target compounds against MCF-7, Hep3B and L-SR cell lines using MTT assay. | ||
In conclusion, compound 5 showed high inhibitory activity against the MCF-7 cell line (IC50 = 1.4 μM; cf. etoposide IC50 value = 16.32 μM and levofloxacin IC50 value = 2.79 μM). Furthermore, compounds 3c, 4b, 5, 7, 8, 13a and 13c against the Hep3B cell line exhibited quite promising cytotoxic activity with IC50 values ranging from 0.43 to 8.79 μM in comparison to etoposide, and levofloxacin 21.93 and 8.94 μM, sequentially. Regarding the effect of compounds 5 and 13a against the L-SR cell lines, their IC50 values were 0.96 and 3.12 μM, respectively (cf. etoposide IC50 value = 3.38 μM and levofloxacin IC50 value = 7.38 μM).
2.3. Topoisomerase II beta (Topo2β) polymerase inhibition assay
This assay was employed to predict the inhibitory effects of novel compounds on Topo2β. Etoposide and levofloxacin were used as reference drugs. The percentage inhibition on topoisomerase II beta polymerization of the most active compounds against cancer cell lines was measured. Accordingly, compound 5 with good cytotoxic activity against human breast cancer cell line (MCF-7) was tested for inhibiting Topo2β polymerization using etoposide and levofloxacin as reference drugs. Compound 5 has proven to be a Topo2β polymerase inhibitor with 78.30% inhibition against MCF-7, in comparison with 63.18% and 59.80% for etoposide and levofloxacin, sequentially (Table 2). In the case of compounds that revealed excellent cytotoxic activity against the human liver cancer cell line Hep3B, a Topo2β polymerization inhibition assay was carried out for representative compounds 3c, 5, 7, 13a and 13c. Compounds 3c and 5 showed 81.33% and 83.73% inhibition, respectively against the human liver cancer cell line Hep3B, much more potent than etoposide and levofloxacin (73.22% and 73.59%, correspondingly) (Table 3). In addition, compounds 5 has the highest cytotoxic activity against leukemia cell line (L-SR). It showed Topo2β inhibition against leukemia cell line (L-SR) with 80.29% (cf. 69.85% and 70.40%) for etoposide and levofloxacin, respectively (Table 4). The selected compounds 3c and 5 with outstanding Topo2β polymerization inhibitory activity have the following features: 4-methoxyphenyl and opened uncyclized levofloxacin hydrazide. Furthermore, they exhibited remarkable inhibitory activity as well as great potencies against Hep3B. Therefore, the liver cancer cell line Hep3B was selected for further assays.| Compound no. | Hep3B (μM) | Topo2β % inhibition |
|---|---|---|
| 3c | 0.43 | 81.33 |
| 5 | 3.77 | 83.73 |
| 7 | 6.99 | 71.92 |
| 13a | 3.46 | 68.77 |
| 13c | 1.04 | 70.93 |
| Etoposide | 21.93 | 73.22 |
| Levofloxacin | 8.94 | 73.59 |
| Compound no. | L-SR (μM) | Topo2β% inhibition |
|---|---|---|
| 5 | 0.96 | 80.29 |
| 13a | 3.12 | 69.38 |
| Etoposide | 3.38 | 69.85 |
| Levofloxacin | 7.38 | 70.40 |
2.4. Cell cycle analysis
Different fluoroquinolone molecules, involving levofloxacin, were found to disturb the normal function of eukaryotic DNA topoisomerases either by stabilization of DNA cleavable complexes to induce the formation of DNA breaks (S phase) or through intercalation of DNA that results in the inhibition of the cell division (G2/M phase) and induction of apoptosis.49–53 Interestingly, it was informed that there was a significant increase in the percentage of apoptotic cells primarily at the S and G2/M phases for etoposide54,55 and levofloxacin derivatives on Hep3B cells after the modification of the carboxylic acid group of levofloxacin.25,33 Accordingly, the cell cycle analysis for the most promising compounds 3c and 5 against the human liver cancer cell line Hep3B was studied, as the carboxylic acid group of levofloxacin was modified, yielding n′-benzylidinequinoline-6-carbohydrazide and an open-chained compound respectively. The IC50 value of positive controls (etoposide and levofloxacin) used in this assay is 21.93 and 8.94 μM, respectively. Before treatment of Hep3b cells with the targeted compounds, it was noticed that the high percentages of cell cycle arrest of levofloxacin (39.87%) and etoposide (37.62%) were observed at the G2/M phases. Furthermore, the inhibition by etoposide and levofloxacin at the S phase was 26.24% and 30.47%, respectively. After treatment with compounds 3c and 5 with IC50 = 0.43 and 3.77 μM, sequentially, there was a significant increase in the percentage of apoptotic cells at the S phase to 37.56% and 39.09%, compared to etoposide (26.24%) and levofloxacin (30.47%). At the G2/M phase, it was showed that the percentages of apoptotic cells after treatment with compounds 3c and 5 decreased (18.91% and 22.42%, respectively), in comparison to etoposide and levofloxacin as reference drugs (37.62% and 39.87%). Only a significant increase in the percentage of apoptotic cells at the G0–G1 phase from 36.14% for etoposide and 29.66% for levofloxacin to 43.53% was detected upon treatment with compound 3c, but it could not inhibit the cell cycle through arresting at the G0–G1 phase as its percentage is less than the negative control Hep3B percentage (46.82%). Hence, it can be concluded that compounds 3c and 5 inhibit the cell cycle proliferation through arresting the cell cycle at the S phase and inducing the cell death (Table 5, Fig. 4 and 5).| Compound no. | IC50 | DNA content | ||
|---|---|---|---|---|
| %G0–G1 | %S | %G2/M | ||
| 3c/Hep3B | 0.43 | 43.53 | 37.56 | 18.91 |
| 5/Hep3B | 7.7 | 38.49 | 39.09 | 22.42 |
| Etoposide/Hep3B | 21.93 | 36.14 | 26.24 | 37.62 |
| Levofloxacin/Hep3B | 8.94 | 29.66 | 30.47 | 39.87 |
| Control Hep3B | — | 46.82 | 27.61 | 25.57 |
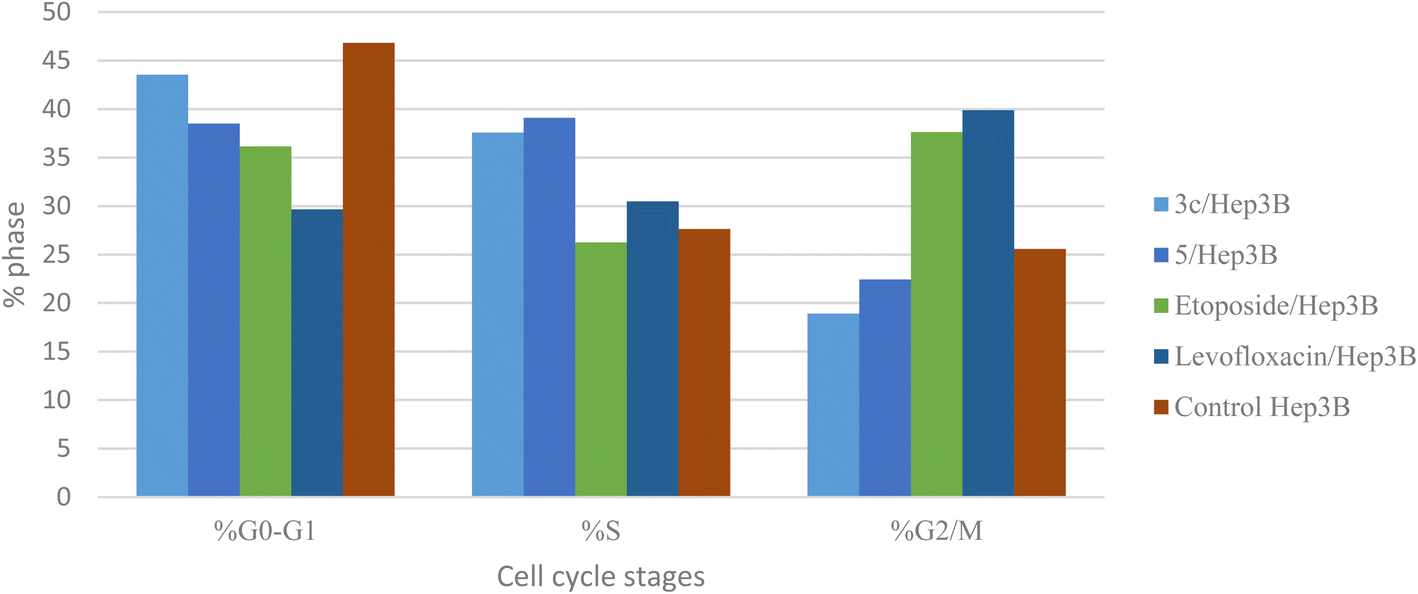 | ||
| Fig. 4 Graphical representation of the cell cycle analysis for compounds 3c, 5, control, etoposide, and levofloxacin against the human liver cancer cell line Hep3B. | ||
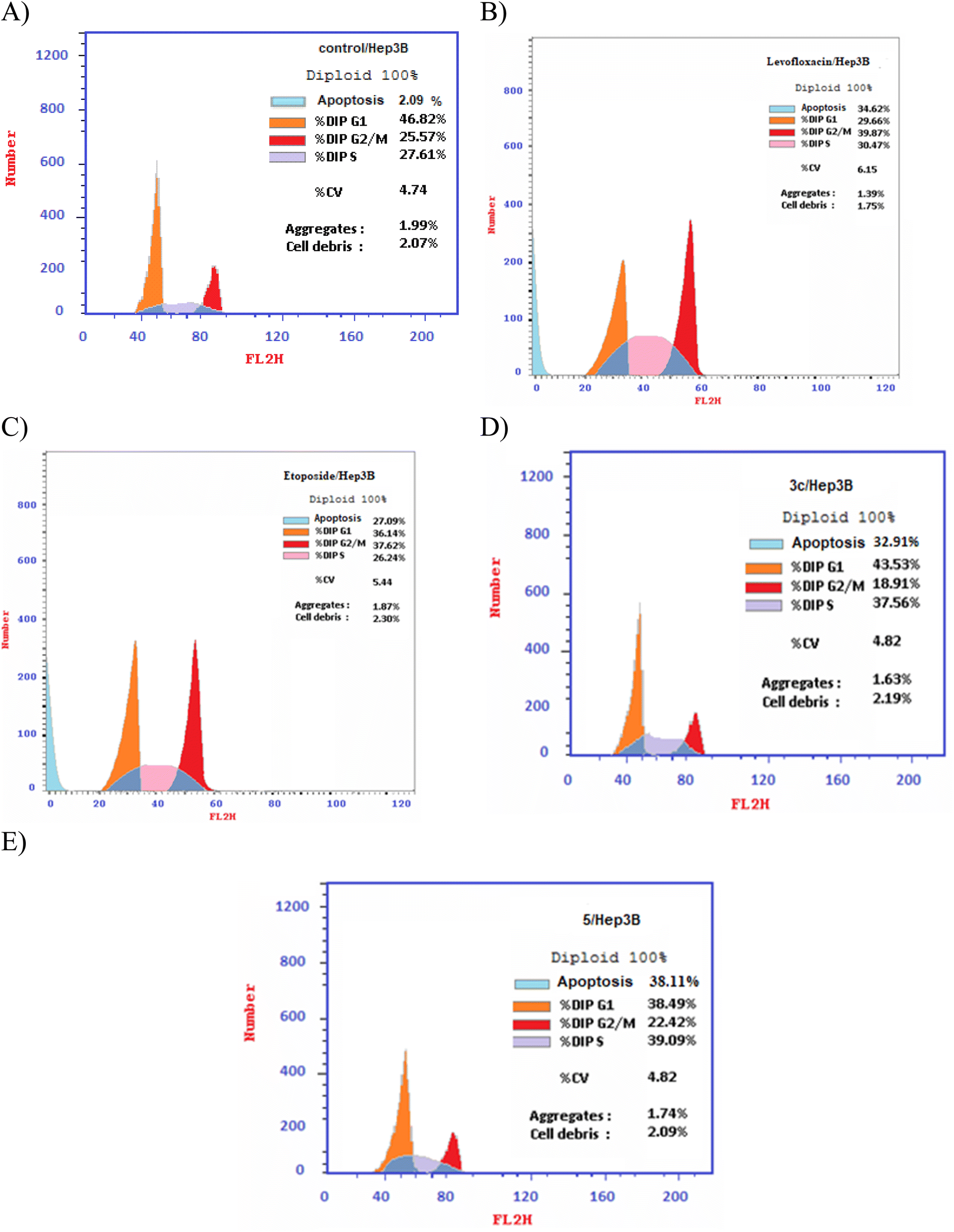 | ||
| Fig. 5 DNA-ploidy flow cytometric analysis for (A) control, (B) levofloxacin, (C) etoposide, (D) compound 3c, and (E) compound 5 against the human liver cancer cell line Hep3B. | ||
2.5. Annexin V-FITC/propidium iodide (PI) staining assay
Using Annexin V fluorescein isothiocyanate (FITC)-conjugate (Annexin V-FITC) is a standard procedure to investigate the apoptosis progression.56,57 A biometric flow cytometric analysis was performed for compounds 3c and 5 after treatment of the liver cancer cell line Hep3B with the tested compounds at their detected IC50 to ensure their ability to cause apoptosis. As shown in Fig. 6, quadrant plots were divided into four parts: Q1 lower left = total cells, Q2 lower right = early apoptotic cells, Q3 upper right = late apoptotic cells, and Q4 upper left = necrotic cells. The results from these plots showed the best significant rise in the percentage of apoptotic cells (summation of Q2 and Q3) by 25.87 and 32.21 for compounds 3c and 5 respectively compared to control, etoposide, and levofloxacin with apoptotic percentages of 0.85 and 23.60, respectively as illustrated in Table 6, Fig. 6 and 7.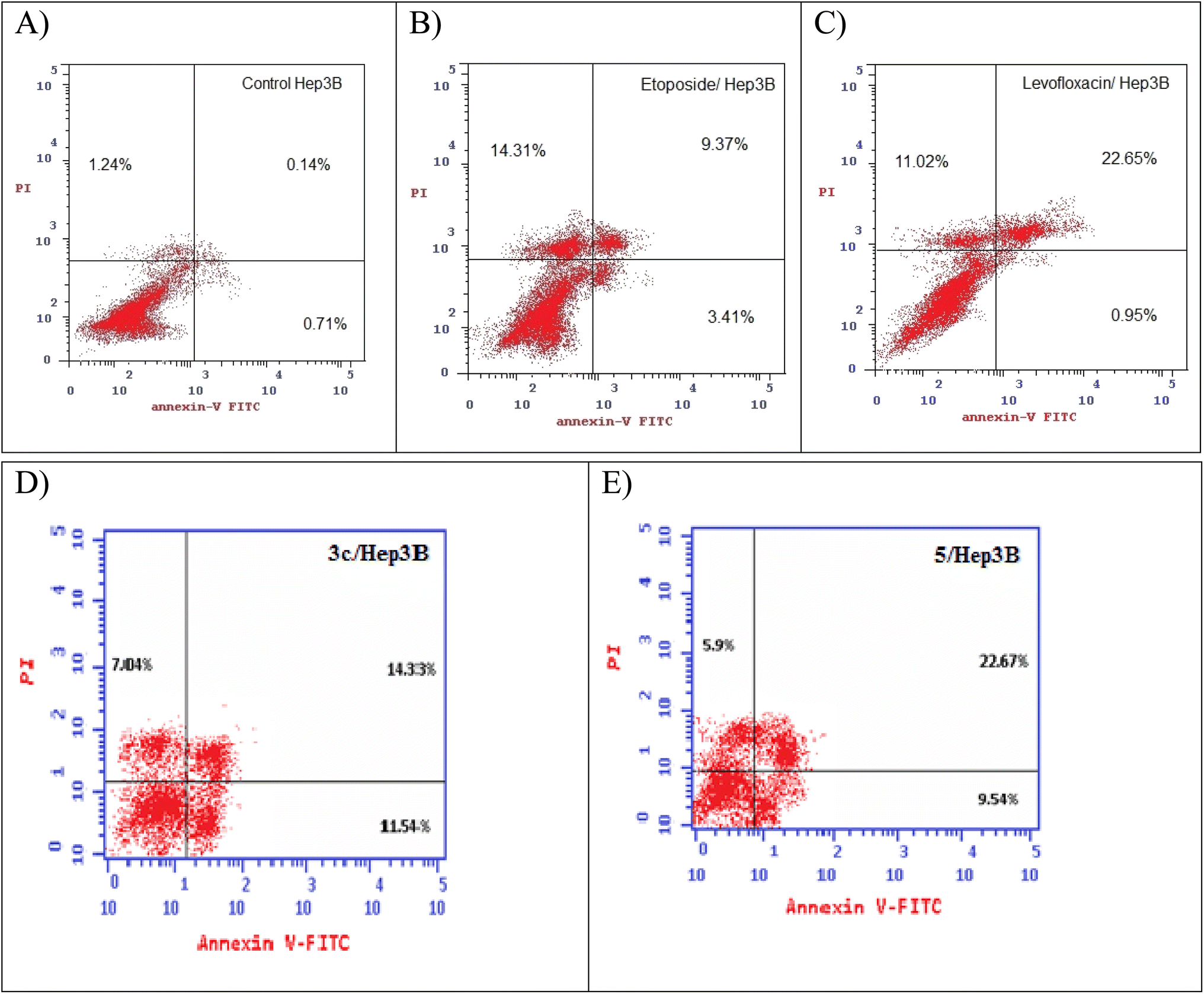 | ||
| Fig. 6 Representative dot plots of the liver cancer cell line Hep3B treated with (A) control, (B) etoposide, (C) levofloxacin, (D) compound 3c, and (E) compound 5 adopting flow cytometric analysis after Annexin V/propidium iodide (PI) staining. | ||
| Compound no. | Total % | Early apoptosis % | Late apoptosis % | Necrosis % |
|---|---|---|---|---|
| 3c/Hep3B | 32.91 | 11.54 | 14.33 | 7.04 |
| 5/Hep3B | 38.11 | 9.54 | 22.67 | 5.9 |
| Etoposide/Hep3B | 27.09 | 3.41 | 9.37 | 14.31 |
| Levofloxacin/Hep3B | 34.62 | 0.95 | 22.65 | 11.02 |
| Cont. Hep3B | 2.09 | 0.71 | 0.14 | 1.24 |
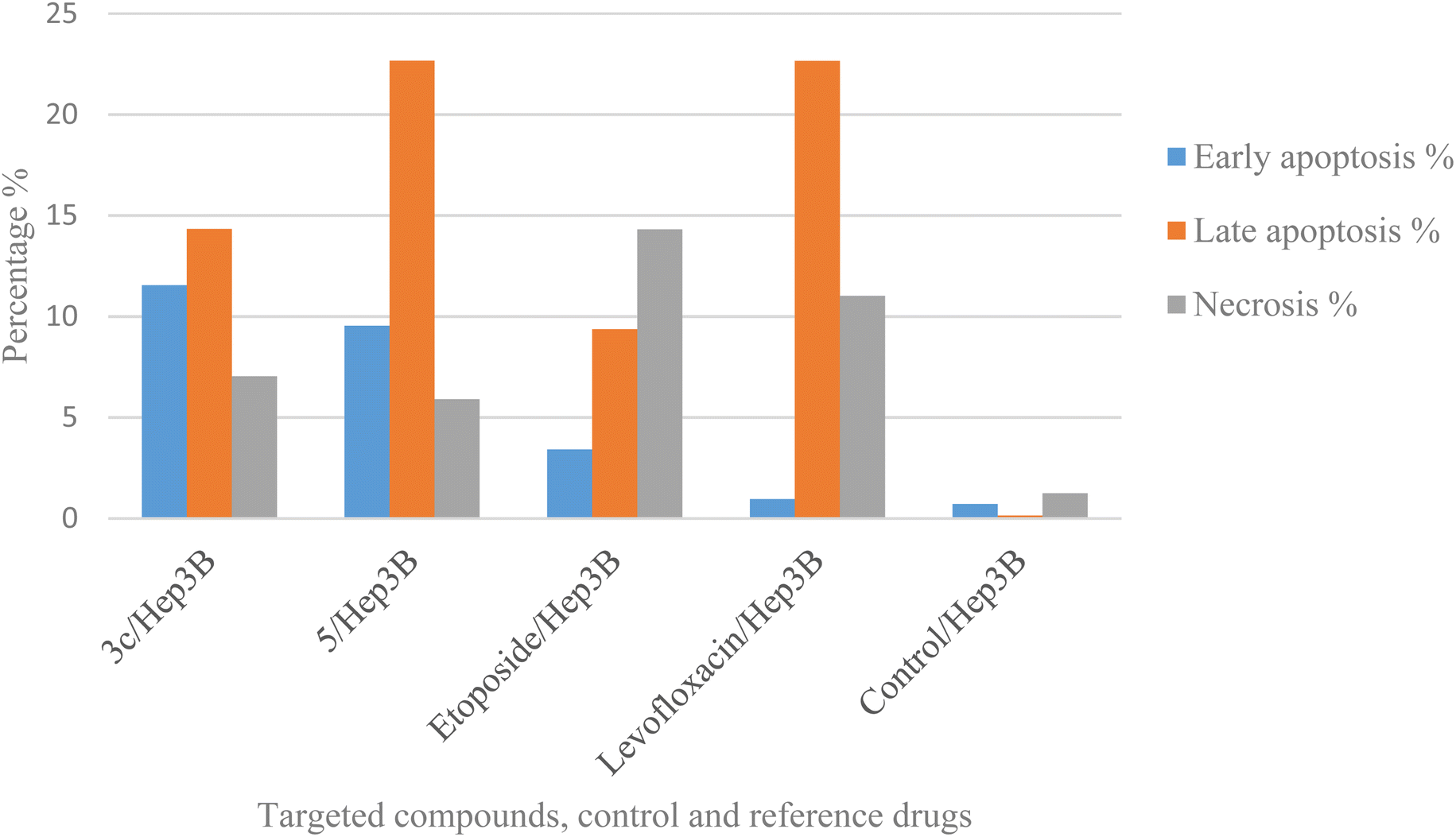 | ||
| Fig. 7 Graphical representation of the percentage of apoptosis and necrosis of compounds 3c, 5, control, etoposide, and levofloxacin in the liver cancer cell line Hep3B. | ||
2.6. Caspase-3 analysis
Caspases are crucial mediators of apoptosis. Among them, caspase-3 is considered as a key effector caspase in inducing cell apoptosis and serves as a marker of programmed cell death.58 Compounds 3c and 5 were tested for their effect on caspase-3 levels in the most sensitive liver cancer cell line Hep3B. The treatment of Hep3B cells with compounds 3c and 5 showed elevation in caspase-3 levels by 4.9 and 4.5 folds, respectively in comparison to values 5.5 and 4.7 and one-fold for etoposide, levofloxacin, and control, sequentially. These results supported cell cycle analysis and Annexin V studies and suggest that compounds 3c and 5 induced apoptosis through activation of caspase-3 (Table 7 and Fig. 8).| Compound no. | Caspase-3 | |
|---|---|---|
| Conc. Pg mL−1 | Folds | |
| 3c/Hep3B | 407.3 ± 7.55 | 4.9 |
| 5/Hep3B | 375.8 ± 16.6 | 4.5 |
| Etoposide/Hep3B | 460 ± 11.3 | 5.5 |
| Levofloxacin/Hep3B | 391.7 ± 5.86 | 4.7 |
| Control Hep3B | 55.28 ± 3.25 | 1 |
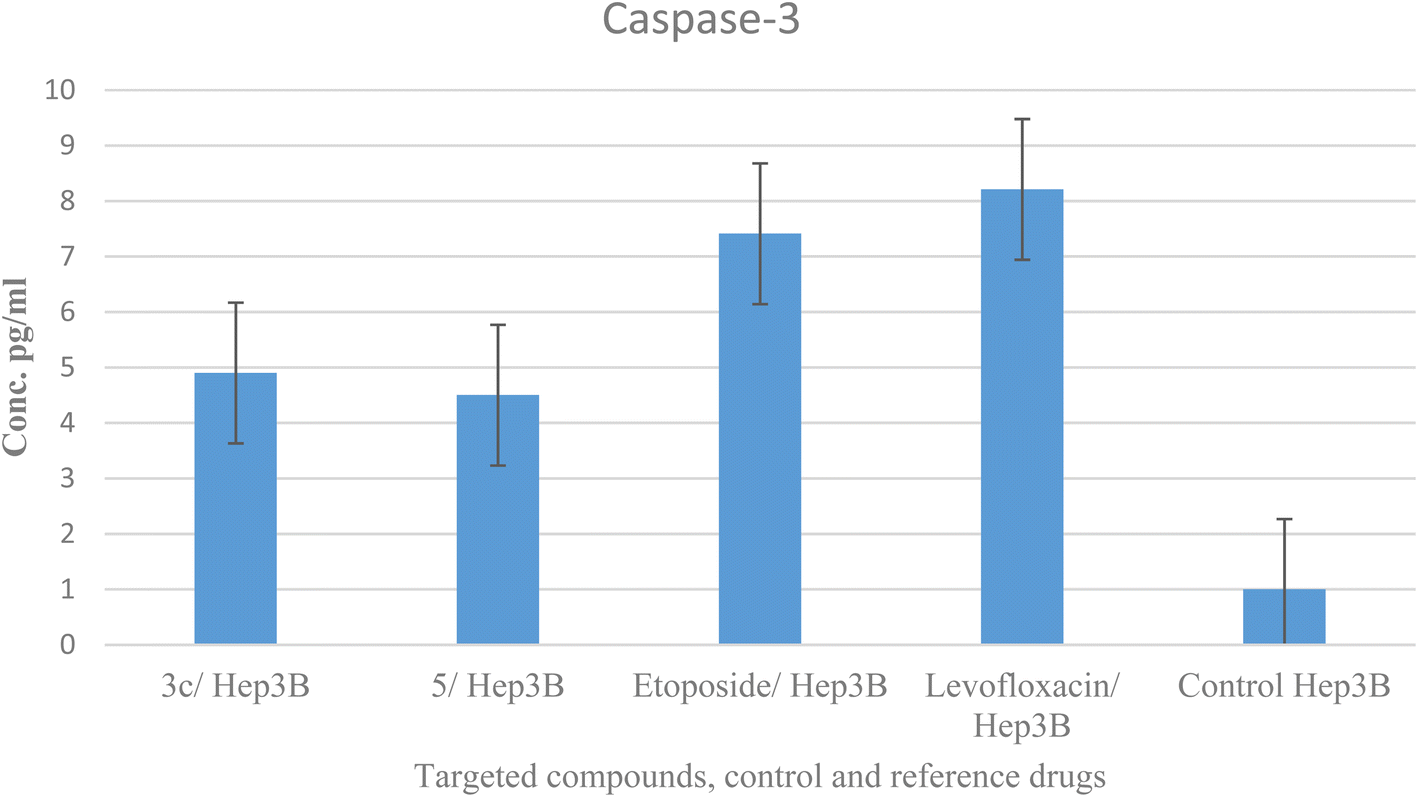 | ||
| Fig. 8 Graphical representation of compounds 3c, 5, control Hep3B and reference drugs on caspase-3. | ||
2.7. Effect on normal human cell lines
One of the serious problems of cancer chemotherapy is the undesired damage of normal cells. This is attributed to the undistinguishing cytotoxicity of anticancer drugs.59 Representative target compounds that were most active against the MCF-7, Hep3B and L-SR cell lines and causing the highest Topo2β percentage inhibition were selected. The following normal cell lines were selected: normal breast MCF-10a, normal liver THLE2 and normal lymphocyte PCS-800-011. Etoposide and levofloxacin were selected as reference drugs. Five representative compounds were evaluated for their toxicity towards the normal liver cell line THLE2, using etoposide and levofloxacin as reference drugs. In addition, only compound 5 was evaluated for its effect on the normal breast cell line MCF-10a, and compounds 5 and 7 were evaluated for their toxic effects on the normal lymphocyte cell line PCS-800-011 via an MTT assay, and the IC50 doses of all the representative target compounds against normal cell lines selected were calculated. In accordance, compound 5 (IC50 = 30.40 μM) has lesser side effects on the normal breast cell line MCF-10a than those of the reference drugs. Regarding the normal liver cell line THLE2, compounds 5 and 7 exhibited IC50 = 40.56 μM and 68.52 μM, respectively in comparison with 35.20 and 29.95 for etoposide and levofloxacin, sequentially. Finally, for the normal lymphocyte cell line PCS-800-011, compound 7 (IC50 = 32.06 μM) exhibited IC50 values exceeding those of levofloxacin, and the results are provided in Table 8.| Compound no. | Cytotoxicity IC50 (μM) | ||
|---|---|---|---|
| THLE2 | PCS-800-011 | MCF-10a | |
| 3c | 19.98 ± 0.76 | NA | NA |
| 5 | 40.56 ± 2.28 | 19.68 ± 1.03 | 30.40 ± 1.66 |
| 7 | 68.52 ± 3.86 | 32.06 ± 1.68 | NA |
| 13a | 29.93 ± 1.68 | NA | NA |
| 13c | 24.02 ± 1.35 | NA | NA |
| Etoposide | 35.20 ± 2.19 | 37.06 ± 1.79 | 23.88 ± 1.55 |
| Levofloxacin | 29.95 ± 1.52 | 20.23 ± 0.85 | 22.25 ± 1.41 |
2.8. Molecular modeling study
Computer-aided drug design is an important tool in designing selective and potent inhibitors.60 Docking study can be used to illustrate the molecular interaction of novel compounds at the protein–ligand interface.61 A molecular modeling simulation study was conducted to investigate the probable binding modes of the most active compound 5 on Topo2β enzyme of breast, 3c and 5 on Topo2β enzyme of liver cancer cell lines and compound 5 on Topo2β enzyme of leukemia cancer cell lines. In this study, we selected PDB ID: 3QX362 human topoisomerase II beta in complex with DNA and etoposide. DNA chains were retained from the active site. The molecular docking setup was validated by carrying out the docking of etoposide and fluoroquinolones in Topo2β, as reported in a previous study.28 It was reported that ASP479, SER480, ARG820, ARG503, LYS456 and GLN778 amino acid residues of Topo2β are involved in binding with fluoroquinolones. Etoposide demonstrated energy score (S) = −9.38 kcal mol−1 and showed interaction with DNA nucleotides: DG10, DG11, DA12 and DC11 in Topo2β complex, which is essential for binding affinities.28 Levofloxacin demonstrated energy score (S) = −11.78 kcal mol−1 and showed a hydrogen-bond interaction with ASP479 and SER480 in Topo2β complex.28 The results obtained from docking imitation are presented in Fig. 9.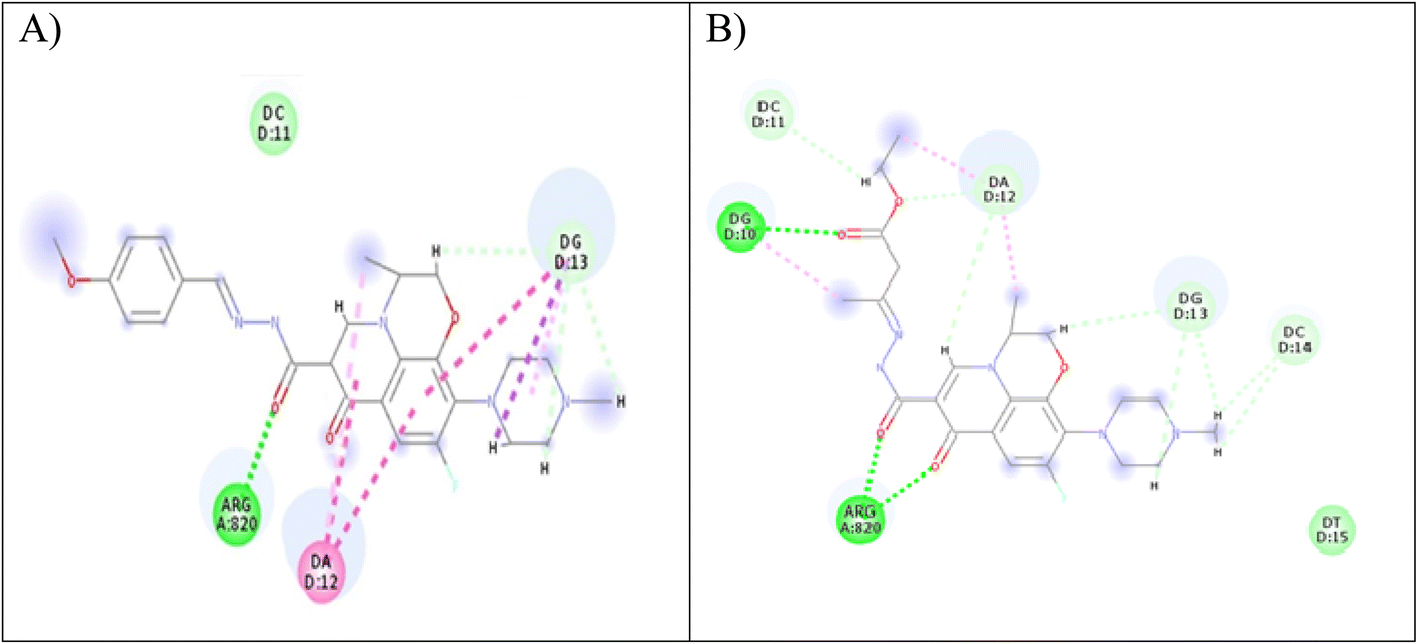 | ||
| Fig. 9 (A) Two-dimensional interaction of compound 3c at Topo2β binding sites (PDB code: 3QX3). (B) Two-dimensional interaction of compound 5 in Topo2β binding sites (PDB code: 3QX3). | ||
Compound 3c demonstrated energy score (S) = −29.77 kcal mol−1, as it showed one conventional hydrogen bond interaction with ARG820, three carbon–hydrogen bond interactions with DG13, and two pi-alkyl bonds with DG13 and DA12. Furthermore, compound 5 exhibited energy score (S) = −20.46 kcal mol−1, as it showed three conventional hydrogen bonds, one with DG10 and two with ARG820, three carbon–hydrogen bonds, one with DC11 and two with DA12, and two pi-alkyl bonds with DA12.
2.9. Physicochemical properties
The physicochemical characteristics of the most promising cytotoxic compounds 3c and 5 on various tested cell lines were investigated compared to etoposide and levofloxacin as reference drugs. The results of the ADME revealed six physicochemical properties: lipophilicity, size, polarity, solubility, saturation, and flexibility. Compounds 3c, 5 and levofloxacin showed no deviation in all physicochemical properties, while etoposide showed a deviation in size and polarity (Fig. 10). Other physicochemical parameters include topological polar surface area, pan assay interference structures, lipophilicity petameter WLOGP, number of rotatable bonds, number of hydrogen bond acceptors, number of hydrogen bond donors, gastrointestinal absorption blood–brain barrier permeability and drug likeness (Table 9). In addition, compounds 3c, 5, etoposide and levofloxacin were good substrates for P-glycoprotein. Furthermore, compounds 3c, 5 and levofloxacin showed high GI absorption as they were located in the boiled egg white, but etoposide showed low GI absorption. Finally, compounds 3c, 5, levofloxacin and etoposide are located outside the boiled egg yolk, so they cannot penetrate the blood–brain barrier (Fig. 11).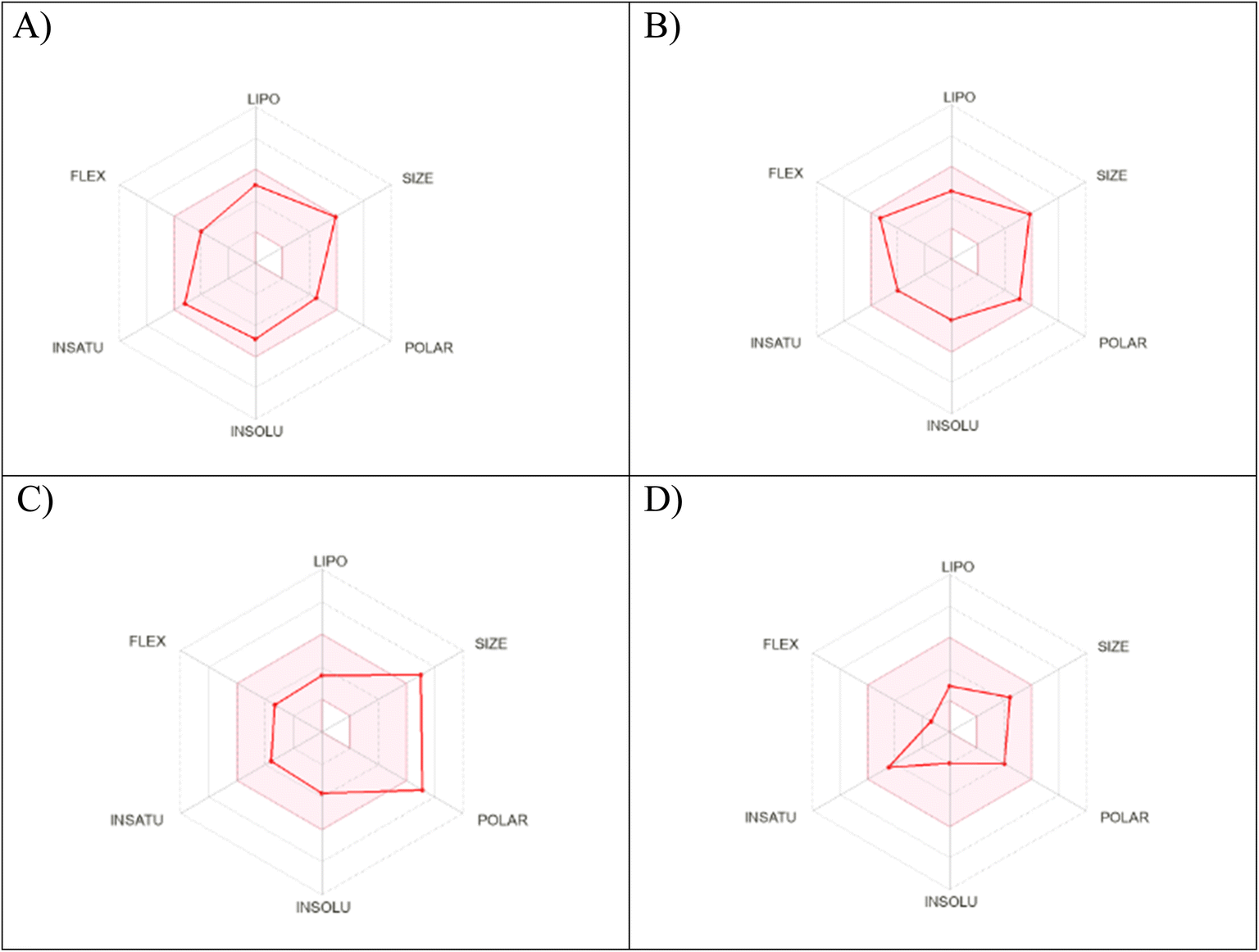 | ||
| Fig. 10 Radar chart showing six physicochemical properties of the tested (A) compound 3c, (B) compound 5, (C) etoposide and (D) levofloxacin. | ||
| Compound no. | TPSAa | PAINSb | WLOGPc | NRBd | HBDe | HBAf | GI abs.g | BBB perm.h | Lipinskii |
|---|---|---|---|---|---|---|---|---|---|
| a Topological polar surface rea.b Pan assay interference structures.c lipophilicity petameter WLOGP.d Number of rotatable bonds.e Number of hydrogen bond acceptors.f Number of hydrogen bond donor.g Gastrointestinal absorption.h Blood–brain barrier permeability.i Lipinski (drug likeness). | |||||||||
| 3c | 88.40 | 0 | 2.28 | 6 | 1 | 7 | High | No | 0 Violation |
| 5 | 105.47 | 0 | 1.56 | 8 | 1 | 8 | High | No | 0 Violation |
| Etoposide | 160.83 | 0 | 1.01 | 5 | 3 | 13 | Low | No | 0 Violation |
| Levofloxacin | 75.01 | 0 | 1.20 | 2 | 1 | 6 | High | No | 0 Violation |
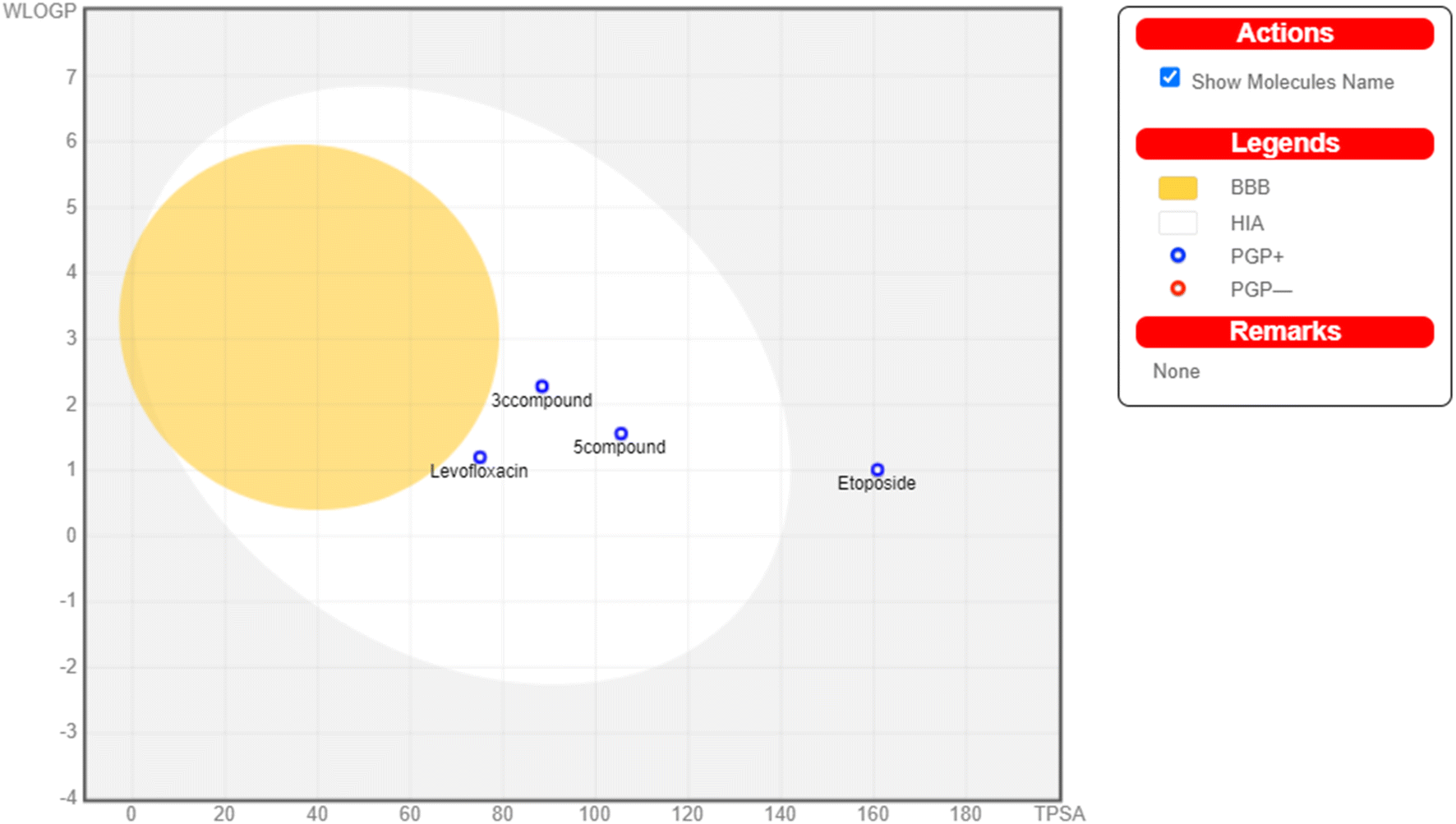 | ||
| Fig. 11 Boiled egg model of the tested compounds 3c, 5, etoposide and levofloxacin. | ||
3. Experimental
3.1. Chemistry
The melting points were determined using a Stuart apparatus, and the values given are uncorrected. IR spectra were recorded using a Shimadzu IR 4000s spectrophotometer (KBr, cm−1) at the College of Pharmaceutical Sciences and Drug Manufacturing, Misr University for Science and Technology, Egypt. 1H NMR spectra were recorded using a Bruker 400 MHz (Bruker Corp., Billerica, MA, USA) spectrophotometer, Faculty of Pharmacy, Cairo University, Cairo, Egypt and the Centre of Drug Discovery Research & Development, Faculty of Pharmacy, Ain shams University, and a Jeol 500 MHz spectrophotometer at the National Research Center. Chemical shifts were recorded in ppm on a δ scale, coupling constants (J) were given in Hz and peak multiplicities are designed as follows: s, singlet; d, doublet; t, triplet; m, multiplet. 13C NMR spectra were recorded using a Bruker 100 MHz spectrophotometer, Faculty of Pharmacy, Cairo University and the Centre of Drug Discovery Research & Development, Faculty of Pharmacy, Ain shams University, and a Jeol 500 MHz spectrophotometer at the National Research Center. Chemical shifts were recorded in ppm on a δ scale. Microanalyses for C, H, and N were carried out at the Regional Center for Mycology and Biotechnology, Faculty of Pharmacy, Al-Azhar University. C, H, and N analysis values were accepted within the range of ± 0.4 of the calculated percentages. The reactions' progress was monitored by TLC using aluminum sheets precoated with UV fluorescent silica gel (MERCK 60F 254), and spots were visualized using a UV Lamp. The used eluting system was methanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane with a ratio of 9.5/0.5 respectively. Scientific microwave MAS-II was used under 300 W microwave irradiation, operating at 80 °C. The evaluation of the cytotoxic activity was performed at VACSERA, Giza, Egypt, using MCF-7, Hep3B and leukemia SR cell lines. Enzyme assay, cell cycle analysis, apoptosis and caspase-3 assay were carried out at VACSERA, Giza, Egypt using an SEB870Hu 96 Tests Enzyme-linked Immunosorbent Assay Kit for Topoisomerase II Beta. The purity of compound 5 was detected by HPLC at the Lambda max of it. Levofloxacin was obtained from Amoun Pharmaceutical Industries Company. Compound 2 was prepared according to the reported procedure,35 while the levofloxacin ester intermediate was prepared according to similar reported method.48
:hexane with a ratio of 9.5/0.5 respectively. Scientific microwave MAS-II was used under 300 W microwave irradiation, operating at 80 °C. The evaluation of the cytotoxic activity was performed at VACSERA, Giza, Egypt, using MCF-7, Hep3B and leukemia SR cell lines. Enzyme assay, cell cycle analysis, apoptosis and caspase-3 assay were carried out at VACSERA, Giza, Egypt using an SEB870Hu 96 Tests Enzyme-linked Immunosorbent Assay Kit for Topoisomerase II Beta. The purity of compound 5 was detected by HPLC at the Lambda max of it. Levofloxacin was obtained from Amoun Pharmaceutical Industries Company. Compound 2 was prepared according to the reported procedure,35 while the levofloxacin ester intermediate was prepared according to similar reported method.48
3.1.1.1. N′-(4-Chlorobenzylidene)-9-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-2,3-dihydro-7H-[1,4]oxazino[2,3,4-ij]quinoline-6-carbohydrazide (3a). Yield, 50.2%; mp = 260–262 °C; IR (KBr) ν = 3446 (NH), 3005 (CH aromatic), 2935 (CH aliphatic), 1668, 1640 (2C
O), 1618 (CN) cm−1. 1H NMR (DMSO-d6) ppm δ: 1.45 (d, 3H, J = 6.4 Hz, CH3 of oxazine ring), 2.23 (s, 3H, CH3 of piperazine ring), 2.44 (t, 4H, J = 4 Hz, 2CH2 of piperazine ring), 3.27 (t, 4H, J = 4 Hz, 2CH2 of piperazine ring), 4.37 (d, 1H, J = 10.4 Hz, CH2 of oxazine ring), 4.55 (d, 1H, J = 10.8 Hz, CH2 of oxazine ring), 4.89–4.91 (m, 1H, CH of oxazine ring), 7.52 (d, 2H, J = 7.6 Hz, Ar–H), 7.54 (d, 1H, Ar–H), 7.76 (d, 2H, J = 8 Hz, 2Ar–H), 8.41 (s, 1H, NCH), 8.89 (s, 1H, Ar–H), 13.24 (s, 1H, NH, D2O exchangeable). 13C NMR (DMSO-d6) δ: 174.5, 161.6, 154.6, 150.1, 146.9, 145.9, 140.8, 134.9, 133.8, 131.8, 130.9, 129.3, 126.9, 109.6, 103.8, 68.6, 55.8, 54.8, 50.6, 46.5, 18.4. Calcd for C25H25ClFN5O3 (497.96): C, 60.30; H, 5.06; N, 14.06. Found: C, 60.47; H, 5.28; N, 14.32.
3.1.1.2. 9-Fluoro-N′-(4-fluorobenzylidene)-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-2,3-dihydro-7H-[1,4]oxazino[2,3,4-ij]quinoline-6-carbohydrazide (3b). Yield, 62.3%; mp = 266–268 °C; IR (KBr) ν = 3446 (NH), 3041 (CH aromatic), 2964 (CH aliphatic), 1675 (C
O), 1600 (CN) cm−1. 1H NMR (DMSO-d6) ppm δ: 1.45 (d, 3H, J = 6.7 Hz, CH3 of oxazine ring), 2.22 (s, 3H, CH3 of piperazine ring), 2.42 (t, 4H, J = 4 Hz, 2CH2 of piperazine ring), 3.26 (t, 4H, J = 5 Hz, 2CH2 of piperazine ring), 4.37 (d, 1H, J = 9.6 Hz, CH2 of oxazine ring), 4.56 (d, 1H, J = 11.4 Hz, CH2 of oxazine ring), 4.90–4.92 (m, 1H, CH of oxazine ring), 7.28–7.32 (m, 2H, 2Ar–H), 7.53 (d, 1H, J = 12.5 Hz, Ar–H), 7.79–7.82 (m, 2H, 2Ar–H), 8.41 (s, 1H, NCH), 8.88 (s, 1H, Ar–H), 13.21 (s, 1H, NH, D2O exchangeable).13C NMR (DMSO-d6) δ: 174.5, 164.8, 162.3, 161.4, 157.0, 154.5, 147.1, 145.9, 140.6, 131.5, 129.8, 124.7, 121.8, 116.2, 103.8, 68.5, 55.8, 54.7, 50.6, 46.5, 18.4. Calcd for C25H25F2N5O3 (481.50): C, 62.36; H, 5.23; N, 14.55. Found: C, 62.59; H, 5.35; N, 14.81.
3.1.1.3. 9-Fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-N′-(4-methoxybenzylidene)-7-oxo-2,3-dihydro-7H-[1,4]oxazino[2,3,4-ij]quinoline-6-carbohydrazide (3c). Yield, 44.6%; mp = 245–247 °C; IR (KBr) ν = 3446 (NH), 3050 (CH aromatic), 2933 (CH aliphatic), 1668, 1643 (2C
O), 1533 (CN) cm−1. 1H NMR (DMSO-d6) ppm δ: 1.45 (d, 3H, J = 6.7 Hz, CH3 of oxazine ring), 2.23 (s, 3H, CH3 of piperazine ring), 2.43 (t, 4H, J = 4 Hz, 2CH2 of piperazine ring), 3.26 (t, 4H, J = 5 Hz, 2CH2 of piperazine ring), 3.81 (s, 3H, OCH3), 4.36 (d, 1H, J = 9.5 Hz, CH2 of oxazine ring), 4.55 (d, 1H, J = 11.4 Hz, CH2 of oxazine ring), 4.90–4.93 (m, 1H, CH of oxazine ring), 7.02 (d, 2H, J = 8.8 Hz, 2Ar–H), 7.55 (d, 1H, J = 12.5 Hz, Ar–H), 7.69–7.80 (m, 2H, 2Ar–H), 8.34 (s, 1H, NCH), 8.88 (s, 1H, Ar–H), 13.13 (s, 1H, NH, D2O exchangeable). 13C NMR (DMSO-d6) δ: 174.5, 161.2, 157.0, 154.6, 148.0, 145.8, 140.6, 131.6, 129.3, 127.4, 124.8, 121.9, 114.8, 109.8, 103.8, 68.6, 55.8, 54.7, 50.61, 50.57, 46.5, 18.4. Calcd for C26H28FN5O4 (493.53): C, 63.27; H, 5.72; N, 14.19. Found: C, 63.04; H, 5.91; N, 14.47.
3.1.2.1. 9-Fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-N′-(2-oxoindolin-3-ylidene)-2,3-dihydro-7H-[1,4]oxazino[2,3,4-ij]quinoline-6-carbohydrazide (4a). Yield, 89.2%; mp <300 °C; IR (KBr) ν = 3429 (NH), 3080 (CH aromatic), 2930 (CH aliphatic), 1732, 1678, 1649 (3C
O), 1618 (CN) cm−1. 1H NMR (DMSO-d6) ppm δ: 1.47 (d, 3H, J = 9 Hz, CH3 of oxazine ring), 2.24 (s, 3H, CH3 of piperazine ring), 2.44 (t, 4H, J = 4 Hz, 2CH2 of piperazine ring), 3.27 (t, 4H, J = 4 Hz, 2CH2 of piperazine ring), 4.39 (d, 1H, J = 9.7 Hz, CH2 of oxazine ring), 4.56 (d, 1H, J = 10.6 Hz, CH2 of oxazine ring), 4.89–4.94 (m, 1H, CH of oxazine ring), 6.90–7.58 (m, 4H, 4Ar–H), 8.43 (d, 1H, J = 7.7 Hz, Ar–H), 9.00 (s, 1H, Ar–H), 10.77, 13.77 (s, 2H, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ: 165.4, 163.0, 161.3, 146.5, 144.1, 142.9, 140.5, 138.2, 133.1, 131.8, 126.3, 124.6, 122.6, 121.2, 111.2, 109.3, 104.4, 104.1, 68.5, 55.7, 55.1, 50.4, 46.4, 18.2. Calcd for C26H25FN6O4 (504.51): C, 61.90; H, 4.99; N, 16.66. Found: C, 61.73; H, 5.12; N, 16.80.
3.1.2.2. N′-(5-Bromo-2-oxoindolin-3-ylidene)-9-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-2,3-dihydro-7H-[1,4]oxazino[2,3,4-ij]quinoline-6-carbohydrazide (4b). Yield, 72.8%; mp <300 °C; IR (KBr) ν = 3446 (NH), 3080 (CH aromatic), 2933 (CH aliphatic), 1732, 1720, 1641 (3C
O), 1612 (CN) cm−1. 1H NMR (DMSO-d6) ppm δ: 1.47 (d, 3H, J = 6.5 Hz, CH3 of oxazine ring), 2.23 (s, 3H, CH3 of piperazine ring), 2.44 (t, 4H, J = 4 Hz, 2CH2 of piperazine ring), 3.27 (t, 4H, J = 4 Hz, 2CH2 of piperazine ring), 4.40 (d, 1H, J = 8.9 Hz, CH2 of oxazine ring), 4.56 (d, 1H, J = 10.6 Hz, CH2 of oxazine ring), 4.93 (m, 1H, CH of oxazine ring), 6.86–6.90 (m, 1H, Ar–H), 7.51 (d, 1H, J = 11 Hz, Ar–H), 7.60 (d, 1H, J = 8.9 Hz, Ar–H), 8.64 (s, 1H, Ar–H), 9.01 (s, 1H, Ar–H), 10.90, 13.83 (s, 2H, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ: 174.6, 165.2, 163.4, 148.8, 146.8, 145.0, 143.2, 140.6, 137.7, 137.3, 135.3, 127.1, 124.8, 117.5, 114.4, 113.2, 108.9, 104.2, 68.6, 55.7, 55.2, 50.4, 46.4, 18.3. Calcd for C26H24BrFN6O4 (583.41): C, 53.53; H, 4.15; N, 14.41. Found: C, 53.72; H, 4.39; N, 14.67.
3.1.2.3. N′-(5-Chloro-2-oxoindolin-3-ylidene)-9-fluoro-3-methyl-10-(4 methylpiperazin-1-yl)-7-oxo-2,3-dihydro-7H-[1,4]oxazino[2,3,4-ij]quinoline-6-carbohydrazide (4c). Yield, 69.6%; mp <300 °C; IR (KBr) ν = 3444 (NH), 3050 (CH aromatic), 2930 (CH aliphatic), 1732, 1722, 1680 (3C
O), 1612 (CN) cm−1. 1H NMR (DMSO-d6) ppm δ: 1.47 (d, 3H, J = 6.6 Hz, CH3 of oxazine ring), 2.25 (s, 3H, CH3 of piperazine ring), 2.46 (t, 4H, J = 4 Hz, 2CH2 of piperazine ring), 3.28 (t, 4H, J = 4 Hz, 2CH2 of piperazine ring), 4.39 (d, 1H, J = 9.2 Hz, CH2 of oxazine ring), 4.56 (d, 1H, J = 10.6 Hz, CH2 of oxazine ring), 4.90–4.95 (m, 1H, CH of oxazine ring), 6.91–6.96 (m, 1H, Ar–H), 7.38 (d, 1H, J = 8.3 Hz, Ar–H), 7.48–7.53 (m, 1H, Ar–H), 8.52 (s, 1H, Ar–H), 9.02 (s, 1H, Ar–H), 10.90, 13.85 (s, 2H, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ: 170.1, 163.1, 159.1, 149.5, 144.9, 143.0, 142.1, 140.7, 137.3, 132.4, 129.0, 126.6, 125.73, 124.6, 112.5, 108.9, 108.0, 104.0, 68.6, 55.7, 55.2, 50.4, 46.3, 18.3. Calcd for C26H24ClFN6O4 (538.96): C, 57.94; H, 4.49; N, 15.59. Found: C, 57.80; H, 4.63; N, 15.73.
3.1.3.1. Ethyl 3-(2-(9-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-2,3-dihydro-7H-[1,4]oxazino[2,3,4-ij]quinoline-6-carbonyl)hydrazineylidene)butanoate (5). Yield, 36.9%; mp = 100–102 °C; IR (KBr) ν = 3420 (NH), 3040 (CH aromatic), 2960 (CH aliphatic), 1740, 1670, 1660 (3C
O), 1606 (CN) cm−1. 1H NMR (DMSO-d6) ppm δ: 1.22 (t, 3H, J = 7.2 Hz, aliphatic OCH2CH3), 1.43 (d, 3H, J = 6.4 Hz, CH3 of oxazine ring), 2.02 (s, 3H, NC–CH3), 2.22 (s, 3H, CH3 of piperazine ring), 2.42 (t, 4H, J = 4 Hz, 2CH2 of piperazine ring), 3.25–3.26 (m, 5H, J = 4.4 Hz, NC–CH2 + 2CH2 of piperazine ring), 3.35 (s, 1H, CH2 of piperazine overlapped), 4.12 (q, 2H, J = 7.2 Hz, OCH2), 4.36 (d, 1H, J = 9.6 Hz, CH2 of oxazine ring), 4.55 (d, 1H, J = 12.8 Hz, CH2 of oxazine ring), 4.87–4.89 (m, 1H, CH of oxazine ring), 7.51 (d, 1H, J = 6 Hz, Ar–H), 8.87 (s, 1H, Ar–H), 12.91 (s, 1H, NH, D2O exchangeable). 13C NMR (DMSO-d6) δ: 174.7, 170.2, 161.2, 154.5, 150.3, 145.7, 140.5, 131.6, 124.7, 121.8, 109.8, 103.9, 68.5, 61.0, 55.7, 54.8, 50.5, 46.4, 44.1, 18.3, 16.9, 14.5. Calcd for C24H30FN5O5 (487.53): C, 59.13; H, 6.20; N, 14.37. Found: C, 59.41; H, 6.12; N, 14.50. HPLC purity = 96.87%.
3.1.3.2. 9-Fluoro-3-methyl-6-((3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)carbonyl)-10-(4-methylpiperazin-1-yl)-2,3-dihydro-7H-[1,4]oxazino[2,3,4-ij]quinolin-7-one (6). Yield, 38.3%; mp = 178–180 °C; IR (KBr) ν = 3419 (NH), 3055 (CH aromatic), 2978 (CH aliphatic), 1720, 1654 (2C
O) cm−1. 1H NMR (DMSO-d6) ppm δ: 1.42 (d, 3H, J = 8.8 Hz, CH3 of oxazine ring), 1.84, 1.89 (2s, 2H, CH2 tautomeric of pyrazolidone ring), 2.02–2.09 (m, 4H, 1H + 3H of CH3 of pyrazolidone ring), 2.23 (s, 3H, CH3 of piperazine ring), 2.44 (t, 4H, J = 4 Hz, 2CH2 of piperazine ring), 3.28 (t, 4H, J = 4 Hz, 2CH2 of piperazine ring), 4.34 (d, 1H, J = 6 Hz, CH2 of oxazine ring), 4.54 (d, 1H, J = 7.2 Hz, CH2 of oxazine ring), 4.84–4.87 (m, 1H, CH of oxazine ring), 7.55 (d, 1H, J = 9.6 Hz, Ar–H), 8.77 (s, 1H, Ar–H), 10.62, 10.65 (2s, 1H, OH tautomeric, D2O exchangeable). 13C NMR (DMSO-d6) δ: 178.3, 174.3, 164.2, 158.9, 154.5, 144.8, 140.6, 131.4, 124.8, 110.2, 68.5, 55.7, 54.5, 50.5, 46.5, 37.0, 23.1, 19.7, 18.4, 11.5. Calcd for C22H24FN5O4 (443.48): C, 59.58; H, 5.91; N, 15.79. Found: C, 59.80; H, 6.13; N, 16.03.
3.1.4.1 9-Fluoro-6-((5-imino-3-oxopyrazolidine-1-yl)carbonyl)-3-methyl-10-(4-methylpiperazin-1-yl)-2,3-dihydro-7H-[1,4]oxazino[2,3,4-ij]quinolin-7-one (7). Yield, 49.7%; mp = 101–103 °C; IR (KBr) ν = 3419, 3392 (2NH), 3018 (CH aromatic), 2980 (CH aliphatic), 1745, 1654, 1620 (3C
O) cm−1. 1H NMR (DMSO-d6) ppm δ: 1.43 (d, 3H, J = 5.6 Hz, CH3 of oxazine ring), 2.84 (s, 3H, CH3 of piperazine ring), 3.37–3.46 (m, 6H, 3CH2 of piperazine ring), 3.83 (s, 1H, CH2 of piperazine ring), 3.99 (s, 1H, CH2 of piperazine ring), 4.13–4.18 (m, 2H, CH2 of pyrazolidone ring), 4.36 (d, 1H, J = 9.2 Hz, CH2 of oxazine ring), 4.54 (d, 1H, J = 6.8 Hz, CH2 of oxazine ring), 4.85–4.86 (m, 1H, CH of oxazine ring), 7.49 (d, J = 12.8 Hz, 1H, Ar–H), 8.75–8.80 (m, 1H, Ar–H), 10.60, 11.86 (2 s, 1H, NH/OH, D2O exchangeable). 13C NMR (DMSO-d6) δ: 174.1, 166.4, 164.2, 162.3, 160.1, 144.8, 140.7, 124.6.4, 118.2, 116.1, 110.0, 103.8, 68.6, 62.4, 56.5, 54.8, 49.1, 44.5, 26.8, 18.3. Calcd for C21H23FN6O4 (442.45): C, 57.01; H, 5.24; N, 18.99. Found: C, 57.23; H, 5.40; N, 19.17.
3.1.5.1 9-Fluoro-6-((3,5-dioxopyrazolidine-1-yl)carbonyl)-3-methyl-10-(4-methylpiperazin-1-yl)-2,3-dihydro-7H-[1,4]oxazino[2,3,4-ij]quinolin-7-one (8). Yield, 37.6%; mp = 228–230 °C; IR (KBr) ν = 3419 (NH), 3005 (CH aromatic), 2960 (CH aliphatic), 1740, 1710, 1653, 1620 (4C
O) cm−1. 1H NMR (DMSO-d6) ppm δ: 1.41 (d, 3H, J = 6.7 Hz, CH3 of oxazine ring), 2.23 (s, 3H, CH3 of piperazine ring), 2.44 (t, 4H, J = 4.3 Hz, 2CH2 of piperazine ring), 3.25 (t, 4H, J = 4.4 Hz, 2CH2 of piperazine ring), 4.33–4.35 (m, 1H, CH of oxazine ring), 4.52–4.54 (m, 1H, CH2 of oxazine ring), 4.54–4.57 (m, 2H, CH2 of pyrazolidine ring), 4.82–4.87 (m, 1H, CH of oxazine ring), 7.50 (d, 1H, J = 12.7 Hz, Ar–H), 8.76 (s, 1H, Ar–H), 10.62 (s, 1H, NH, D2O exchangeable).13C NMR (DMSO-d6) δ: 174.2, 164.2, 156.9, 154.4, 144.8, 140.6, 131.4, 124.7, 122.1, 110.0, 103.9, 103.7, 68.5, 60.2, 55.8, 54.5, 50.6, 46.5, 18.4. Calcd for C21H22FN5O5 (443.44): C, 56.88; H, 5.00; N, 15.79. Found: C, 57.09; H, 5.14; N, 15.83. MS (m/z, %): 443.34 [M+, 47.50], 79.28 [100.00].
3.1.6.1 9-Fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-N-(1,3-dioxoisoindolin-2-yl)-2,3-dihydro-7H-[1,4]oxazino[2,3,4-ij]quinoline-6-carboxamide (9). Yield, 61.3%; mp = 138–140 °C; IR (KBr) ν = 3431 (NH), 3032 (CH aromatic), 2980 (CH aliphatic), 1735, 1654, 1620 (3C
O) cm−1. 1H NMR (DMSO-d6) ppm δ: 1.43 (d, 3H, J = 12.4 Hz, CH3 of oxazine ring), 2.64 (s, 3H, CH3 of piperazine ring), 3.00 (t, 4H, J = 4 Hz, 2CH2 of piperazine ring), 3.41 (s, 4H, J = 4 Hz, 2CH2 of piperazine ring), 4.38 (d, 1H, J = 11.6 Hz, CH2 of oxazine ring), 4.55 (d, 1H, J = 11.6 Hz, CH2 of oxazine ring), 4.85–4.88 (m, 1H, CH of oxazine ring), 7.48–7.60 (m, 3H, 3Ar–H), 7.93–7.99 (m, 1H, Ar–H), 8.14 (m, 1H, Ar–H), 8.77 (s, 1H, Ar–H), 10.61, 11.68 (2s, 1H, NH, D2O exchangeable). 13C NMR (DMSO-d6) δ: 174.2, 172.7, 168.9, 165.6, 144.8, 135.6, 135.1, 133.1, 132.7, 130.9, 130.4, 124.7, 124.1, 122.6, 108.5, 103.9, 68.7, 56.6, 54.9, 48.8, 44.3, 18.9. Calcd for C26H24FN5O5 (505.51): C, 61.78; H, 4.79; N, 13.85. Found: C, 61.52; H, 4.95; N, 14.09. MS (m/z, %): 505.75 [M+, 62.61], 189.33 [100.00].
3.1.7.1 2-[(9-Fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-2,3-dihydro-7H-[1,4]oxazino[2,3,4-ij]quinoline-6-carbonyl)]hydrazin-1-carbothioamide (10). Yield, 64.4%; mp >300 °C; IR (KBr) ν = 3444, 3419 (2NH), 3385 (NH2), 3050 (CH aromatic), 2960 (CH aliphatic), 1670 (C
O) cm−1. 1H NMR (DMSO-d6) ppm δ: 1.44 (d, 3H, J = 4 Hz, CH3 of oxazine ring), 2.80 (s, 3H, CH3 of piperazine ring), 3.15 (t, 2H, J = 4 Hz, CH2 of piperazine), 3.43–3.64 (m, 6H, 3CH2 of piperazine ring), 4.38 (d, 1H, J = 9.6 Hz, CH2 of oxazine ring), 4.58 (d, 1H, J = 10.6 Hz, CH2 of oxazine ring), 4.90–4.92 (m, 1H, CH of oxazine ring), 7.57 (d, 1H, J = 12.2 Hz, Ar–H), 8.83 (s, 1H, Ar–H), 7.89, 9.40, 11.20, 11.39 (4 broad s, 4H, NH/NH2, D2O exchangeable). 13C NMR (DMSO-d6) δ: 174.2, 156.7, 154.2, 145.7, 140.9, 130.0, 124.6, 122.8, 109.7, 104.0, 68.8, 56.5, 54.6, 53.5, 47.6, 42.8, 18.5. Calcd for C19H23FN6O3S (434.49): C, 52.52; H, 5.34; N, 19.34. Found: C, 52.79; H, 5.61; N, 19.12.
3.1.8.1. N-(4-Chlorobenzylidene)-2-[(9-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-2,3-dihydro-7H-[1,4]oxazino[2,3,4-ij]quinoline-6-carbonyl)]hydrazine-1-carbothioamide (11a). Yield, 76.3%; mp >300 °C; IR (KBr) ν = 3446, 3431 (2NH), 3082 (CH aromatic), 2991 (CH aliphatic), 1678, 1647 (2C
O), 1608 (CN) cm−1. 1H NMR (DMSO-d6) ppm δ: 1.45 (d, 3H, J = 6.4 Hz, CH3 of oxazine ring), 2.80 (s, 3H, CH3 of piperazine ring), 3.18 (broad s, 2H, CH2 of piperazine ring), 3.35 (s, 2H, CH2 of piperazine overlapped), 3.56 (t, 4H, J = 4 Hz, 2CH2 of piperazine ring), 4.40 (d, 1H, J = 9.2 Hz, CH2 of oxazine ring), 4.59 (d, 1H, J = 10.4 Hz, CH2 of oxazine ring), 4.91–4.95 (m, 1H, CH of oxazine ring), 7.52 (d, 2H, J = 8.8 Hz, 2Ar–H), 7.57 (d, 1H, J = 12 Hz, Ar–H), 7.77 (d, 2H, J = 8.8 Hz, 2Ar–H), 8.42 (s, 1H, NCH), 8.92 (s, 1H, Ar–H), 11.32, 13.20 (s, 2H, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ: 174.4, 161.5, 156.8, 154.3, 147.1, 146.0, 141.0, 134.9, 133.7, 129.3, 129.3, 124.7, 122.7, 109.75, 104.0, 103.8, 68.8, 54.8, 53.6, 47.6, 42.9, 18.4. Calcd for C26H26ClFN6O3S (557.04): C, 56.06; H, 4.70; N, 15.09. Found: C, 56.30; H, 4.87; N, 15.26.
3.1.8.2. 2-[(9-Fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-2,3-dihydro-7H-[1,4]oxazino[2,3,4-ij]quinoline-6-carbonyl)]N-(4-methoxybenzylidene)hydrazine-1-carbothioamide (11b). Yield, 36.2%; mp >300 °C; IR (KBr) ν = 3523, 3446 (2NH), 3030 (CH aromatic), 2989 (CH aliphatic), 1668, 1650 (2C
O), 1604 (CN) cm−1. 1H NMR (DMSO-d6) ppm δ: 1.45 (d, 3H, J = 8.8 Hz, CH3 of oxazine ring), 2.27 (s, 3H, CH3 of piperazine ring), 3.29 (t, 5H, CH2 of piperazine ring), 3.35 (s, 3H, CH2 of piperazine overlapped), 3.82 (s, 3H, OCH3), 4.37 (d, 1H, J = 10 Hz, CH2 of oxazine ring), 4.56 (d, 1H, J = 12.4 Hz, CH2 of oxazine ring), 4.91–4.93 (m, 1H, CH of oxazine ring), 7.03 (d, 2H, J = 9.6 Hz, 2Ar–H), 7.55 (d, 1H, J = 10 Hz, Ar–H), 7.71 (d, 2H, J = 11.6 Hz, Ar–H), 8.35 (s, 1H, NCH), 8.89 (s, 1H, Ar–H), 13.13 (s, 1H, NH, D2O exchangeable). 13C NMR (DMSO-d6) δ: 174.5, 161.3, 161.3, 154.6, 148.1, 145.8, 140.7, 140.7, 131.6, 129.3, 127.4, 124.8, 121.9, 121.86, 114.8, 109.8, 103.8, 68.6, 55.8, 54.7, 50.4, 46.4, 18.4. Calcd for C27H29FN6O4S (552.63): C, 58.68; H, 5.29; N, 15.21. Found: C, 58.92; H, 5.46; N, 15.39.
3.1.9.1. 2-[(9-Fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-2,3-dihydro-7H-[1,4]oxazino[2,3,4-ij]quinoline-6-carbonyl)]-N-(1-(4-methoxyphenyl)ethylidene)hydrazine-1-carbothioamide (12a). Yield, 75%; mp >300 °C; IR (KBr) ν = 3444, 3419 (2NH), 3043 (CH aromatic), 2987 (CH aliphatic), 1660, 1640 (2C
O), 1597 (CN) cm−1. 1H NMR (DMSO-d6) ppm δ: 1.45 (d, 3H, J = 6.8 Hz, CH3 of oxazine ring), 2.35 (s, 3H, NC_CH3), 2.67 (s, 3H, CH3 of piperazine ring), 3.11 (broad s, 3H, CH2 of piperazine ring), 3.35 (s, 2H, CH2 of piperazine overlapped), 3.52 (broad s, 3H, CH2 of piperazine ring), 3.81 (s, 3H, OCH3), 4.39 (d, 1H, J = 9.6 Hz, CH2 of oxazine ring), 4.58 (d, 1H, J = 10.8 Hz, CH2 of oxazine ring), 4.95–4.96 (m, 1H, CH of oxazine ring), 7.00 (d, 2H, J = 8.8 Hz, 2Ar–H), 7.62–7.65 (m, 1H, Ar–H), 7.81 (d, 2H, J = 8.8 Hz, 2Ar–H), 8.94 (s, 1H, CH Ar), 10.42, 13.10 (s, 2H, 2NH, D2O exchangeable). Calcd for C28H31FN6O4S (566.65): C, 59.35; H, 5.51; N, 14.83. Found: C, 59.47; H, 5.62; N, 15.04.
3.1.9.2. N-(1-(3,4-Dimethoxyphenyl)ethylidene)-2-[(9-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-2,3-dihydro-7H-[1,4]oxazino[2,3,4-ij]quinoline-6-carbonyl)]hydrazine-1-carbothioamide (12b). Yield, 71.2%; mp = 222–224 °C; IR (KBr) ν = 3523, 3446 (2NH), 3080 (CH aromatic), 2990 (CH aliphatic), 1668, 1650 (2C
O) cm−1. 1H NMR (DMSO-d6) ppm δ: 1.42 (d, 3H, J = 6.8 Hz, CH3 of oxazine ring), 1.85 (s, 3H, NC_CH3), 2.22 (s, 3H, CH3 of piperazine ring), 2.43 (t, 4H, J = 12.8 Hz, 2CH2 of piperazine ring), 3.26–3.27 (m, 6H, 2OCH3), 3.81 (t, 4H, J = 4 Hz, 2CH2 of piperazine ring), 4.35 (d, 1H, J = 10.4 Hz, CH2 of oxazine ring), 4.54 (d, 1H, J = 10.8 Hz, CH2 of oxazine ring), 4.87–4.88 (m, 1H, CH of oxazine ring), 7.00 (d, 1H, J = 8.4 Hz, Ar–H), 7.37–7.43 (m, 1H, Ar–H), 7.50 (s, 1H, Ar–H), 7.54 (d, 1H, J = 5.6 Hz, Ar–H), 8.78 (s, 1H, Ar–H), 9.38, 11.25 (s, 2H, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ: 188.5, 179.8, 175.9, 168.6, 165.8, 162.1, 156.7, 154.5, 152.9, 140.6, 135.4, 124.7, 121.6, 120.5, 114.8, 111.3, 104.3, 103.5, 68.6, 55.7, 54.6, 50.6, 50.5, 46.5, 23.6, 18.4. Calcd for C29H33FN6O5S (596.68): C, 58.38; H, 5.57; N, 14.08. Found: C, 58.50; H, 5.69; N, 14.31.
3.1.10.1. 9-Fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-6-((piperidin-1-yl)carbonyl)-2,3-dihydro-7H-[1,4]oxazino[2,3,4-ij]quinolin-7-one (13a). Yield, 39.7%; mp = 238–240 °C; IR (KBr) ν = 3080 (CH aromatic), 2935 (CH aliphatic), 1718, 1620 (2C
O) cm−1. 1H NMR (DMSO-d6) ppm δ: 1.57 (d, 3H, J = 4 Hz, CH3 of oxazine ring), 1.87 (s, 3H, CH3 of piperazine ring), 2.54 (s, 6H, 3CH2 of piperidine ring), 3.13 (broad s, 2H, CH2 of piperazine ring), 3.33–3.40 (m, 6H, 3CH2 of piperazine ring), 3.87 (s, 2H, CH2 of oxazine ring), 4.37–4.43 (m, 1H, CH of oxazine ring), 4.58 (broad s, 4H, 2CH2 of piperidine ring), 7.60 (d, 1H, J = 8 Hz, Ar–H), 8.60 (s, 1H, Ar–H). 13C NMR (DMSO-d6) δ: 177.1, 172.9, 167.4, 156.8, 155.1, 145.4, 144.8, 139.5, 133.2, 124.8, 120.4, 109.5, 107.9, 105.0, 68.3, 55.6, 54.9, 50.6, 46.4, 44.7, 22.7, 18.4. Calcd for C23H29FN4O3 (428.51): C, 64.47; H, 6.82; N, 13.08. Found: C, 64.70; H, 6.95; N, 13.31.
3.1.10.2. 9-Fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-6-((morpholin-4-yl)carbonyl)-2,3-dihydro-7H-[1,4]oxazino[2,3,4-ij]quinolin-7-one (13b). Yield, 46.5%; mp = 235–237 °C; IR (KBr) ν = 3041 (CH aromatic), 2935 (CH aliphatic), 1720, 1624 (2C
O) cm−1. 1H NMR (DMSO-d6) ppm δ: 1.58 (d, 3H, CH3 of oxazine ring), 2.36 (s, 3H, CH3 of piperazine ring), 2.56 (s, 6H, 3CH2 of piperazine ring), 3.34–3.40 (m, 8H, 4CH2 of morpholine ring), 3.86 (s, 2H, CH2 of piperazine ring), 4.37–4.56 (m, 3H, CH2 of oxazine ring + CH of oxazine ring), 7.59 (d, 1H, J = 12 Hz, Ar–H), 8.57 (s, 1H, Ar–H). 13C NMR (DMSO-d6) δ: 177.1, 172.8, 167.1, 166.2, 157.0, 155.1, 145.3, 144.7, 139.5, 133.2, 124.8, 120.4, 107.8, 105.2, 68.3, 55.7, 55.6, 50.58, 50.5, 46.4, 18.4. Calcd for C22H27FN4O4 (430.48): C, 61.38; H, 6.32; N, 13.02. Found: C, 61.60; H, 6.46; N, 13.29.
3.1.10.3. 9-Fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-6-((4-methylpiperazin-1-yl)carbonyl)-2,3-dihydro-7H-[1,4]oxazino[2,3,4-ij]quinolin-7-one (13c). Yield, 45.1%; mp = 239–241 °C; IR (KBr) ν = 3080 (CH aromatic), 2968 (CH aliphatic), 1718, 1620 (2C
O) cm−1. 1H NMR (DMSO-d6) ppm δ: 1.58 (d, 3H, J = 4 Hz, CH3 of oxazine ring), 2.31 (s, 3H, CH3 of piperazine ring), 2.36 (s, 3H, CH3 of piperazine ring), 2.49–2.60 (m, 8H, 4CH2 of piperidine ring), 3.33–3.41 (m, 8H, 4CH2 of piperazine ring), 3.87 (s, 2H, CH2 of oxazine ring), 4.32–4.43 (m, 1H, CH of oxazine ring), 7.61 (d, 1H, J = 8 Hz, Ar–H), 8.58 (s, 1H, Ar–H). 13C NMR (DMSO-d6) δ: 177.1, 167.2, 144.8, 139.7, 133.2, 124.8, 123.9, 120.4, 109.6, 107.8, 105.0, 68.3, 55.7, 54.8, 54.0, 53.3, 52.0, 50.6, 46.1, 18.4. Calcd for C23H30FN5O3 (443.52): C, 62.29; H, 6.82; N, 15.79. Found: C, 62.58; H, 7.10; N, 16.05.
3.1.10.4. 9-Fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-6-((4-phenylpiperazin-1-yl)carbonyl)-2,3-dihydro-7H-[1,4]oxazino[2,3,4-ij]quinolin-7-one (13d). Yield, 31.7%; mp = 246–248 °C; IR (KBr) ν = 3030 (CH aromatic), 2966 (CH aliphatic), 1718, 1620 (2C
O) cm−1. 1H NMR (DMSO-d6) ppm δ: 1.57 (d, 3H, J = 16 Hz, CH3 of oxazine ring), 2.37 (s, 3H, CH3 of piperazine ring), 3.19–3.28 (m, 8H, 4CH2 of piperidine ring), 3.37–3.42 (m, 8H, 4CH2 of piperazine ring), 3.89 (s, 2H, CH2 of oxazine ring), 4.02–4.55 (m, 1H, CH of oxazine ring), 6.92–6.99 (m, 5H, 5CH Ar), 7.65 (d, 1H, J = 8 Hz, Ar–H), 8.61 (s, 1H, Ar–H). 13C NMR (DMSO-d6) δ: 177.2, 167.3, 157.1, 151.2, 145.4, 144.7, 142.3, 139.5, 129.3, 124.8, 120.5, 117.2, 116.6, 108.0, 105.2, 68.2, 55.7, 55.6, 50.6, 49.2, 46.4, 45.2, 18.4. Calcd for C28H32FN5O3 (505.59): C, 66.52; H, 6.38; N, 13.85. Found: C, 66.41; H, 6.52; N, 14.11.
3.2. Cytotoxic activity
The newly synthesized compounds were evaluated for their in vitro growth inhibition of three human tumor cell lines representing different tumor types, namely, breast adenocarcinoma (MCF-7), liver hepatocellular carcinoma (Hep3B) and leukemia-SR. Etoposide and levofloxacin were taken as reference drugs, and an MTT assay was applied. Moreover, the toxic effect of representative target compounds was evaluated on normal breast (MCF10a), normal liver (THLE2) and normal lymphocyte (PCS-800-011) cell lines using an MTT assay. The in vitro IC50 antitumor activity for the newly synthesized compounds was established in VACSERA, Egypt. The breast tumor cell line (MCF-7), hepatic tumor cell line (Hep3B) and leukemia-SR cell line were obtained from American Type Culture Collection. The cells were cultured using Dulbecco's modified Eagle's medium (DMEM) (Invitrogen/Life Technologies) supplemented with 10% fetal bovine serum (FBS) (Hyclone), 10 μg mL−1 of insulin (Sigma), and 1% penicillin–streptomycin. All chemicals and reagents were purchased from Sigma-Aldrich or Invitrogen. For 24 hours before to the MTT experiment, plate cells (densities of 1.2–1.8 × 10000 cells per well) were layered in a 96-well plate containing 100 μL of the tested chemical per well and 100 μL of full growth media.
3.3. In vitro topoisomerase II beta polymerase inhibition assay
This assay employs a quantitative sandwich enzyme immunoassay technique. Target compounds were selected to be evaluated against topoisomerase II [MBS#942146], using a human DNA topoisomerase II beta (Topo2β) ELISA kit according to the manufacturer's instructions. Antibody specific for Topo2β was pre-coated onto a microplate. Standards and samples were pipetted into the wells and any Topo2β present was bound by the immobilized antibody. After removing any unbound substances, a biotin-conjugated antibody specific for Topo2β was added to the wells. After washing, avidin-conjugated Horseradish Peroxidase (HP) was added to the wells. Following washing to remove any unbound avidin–enzyme reagent, a substrate solution was added to the wells and color develops in proportion to the amount of Topo2β bound in the initial step. The color development was stopped, and the intensity of the color was measured. The minimum detectable dose of human Topo2β is typically less than 4.69 pg mL−1. The sensitivity of this assay or lower limit of detection (LLD) is defined as the lowest protein concentration that could be differentiated from zero. The mean OD value of 20 replicates of the zero standard was determined and added to their three standard deviations.643.4. Cell cycle analysis by propidium iodide (PI) staining
An apoptosis study was performed at the Cell Culture Unit, VACSERA, Cairo, Egypt. These studies were employed against compounds 3c and 5 to investigate their cytotoxic effect on the most sensitive liver hepatocellular carcinoma (Hep3B) cell line. The treated cells were washed twice with ice-cold phosphate-buffered saline (PBS), centrifuged and then fixed in ice-cold 70% (v/v) ethanol to prevent clustering of cells during fixation. Subsequently, cells were washed with PBS, resuspended with 0.1 mg mL−1 RNaseA, stained using 50 μg mL−1 propidium iodide (PI) and incubated for 40 min at 37 °C. Then 3 mL of PBS was added, and the cells were centrifuged to remove the supernatant. The cells were analyzed by flow cytometry using a FACSort Calibur (Becton Dickinson Biosciences, CA, USA). Finally, the Cell Quest software was used to determine the cell cycle distribution.65,663.5. Annexin-V-FITC apoptosis assay protocol
An Annexin-V-FITC apoptosis assay was used to determine apoptosis and necrosis. The cells were incubated with Annexin-V-FITC, centrifuged and then resuspended in 500 μL of 1× binding buffer. Then 5 μL of Annexin-V-FITC and 5 μL of propidium iodide (PI) were added, and the cells were then trypsinized and washed once with serum-containing media before incubation with Annexin-V-FITC at room temperature for 5 min in the dark. A FACSort Calibur flow cytometer (Becton Dickinson Biosciences) was used to analyze apoptosis.56,573.6. Caspase-3 enzyme assay
Caspase-3 is one of the crucial mediators of programmed cell death (apoptosis) as it is essential for the formation of apoptotic bodies.67–70 Active caspase-3 level was measured in pg ml−1 protein using an Invitrogen ELISA kit (catalog KHO1091). The following procedures were performed. First, 100 μL of the standard diluent buffer was added to the zero standard wells. Then, 100 μL of standards and controls or diluted samples were added to the appropriate microtitre wells. Cover wells were covered and incubated for 2 h at room temperature. The solution was decanted from wells. Then 100 μL of active caspase-3 detection antibody solution was pipetted into each well. The plate was covered and incubated for 1 h at room temperature. After that, 100 μL of Anti-Rabbit IgG HP working solution was added to each well. The wells were covered with a plate cover and incubated for 30 min at room temperature. Then, 100 μL of stabilized chromogen was added to each well. The liquid in the wells will begin to turn blue, and the wells were incubated for 30 min at room temperature. A stop solution was added to each well. The solution in the wells should change from blue to yellow. The plate was read within 2 h after adding the stop solution. Curve fitting software was used to generate the standard curve.3.7. Molecular modeling study
All docking studies were performed using Discovery Studio 2016. PDB (code: 3QX3) was chosen as a template for the modeling study of targeted compounds bound to DNA topoisomerase II beta binding sites.71 The crystal structure was obtained from the RCSB protein data bank. Generally, the protein structure was prepared and followed by adding hydrogen bonds. The prepared molecules were minimized and docked into the protein structure using the CDOCKER protocol. The following steps were used for the present docking study using the Discovery Studio software:(1) Loading of Topo2β complex: the 3D protein structure of the target Topo2β enzyme (3QX3) complexed with its ligand with a resolution of 2.3 Å was downloaded from the RCSB protein data bank as a PDB file.
(2) Preparation of the protein: the PDB file was opened in Discovery Studio. Water molecules were retained, and the native ligand was deleted. Protonation step is prepared from protocol/macromolecule/prepare protein. Identify the binding site from tools/define and edit binding site/define receptor then script/select ligand.
(3) Preparation of the test set: the test set molecules were drawn in 2D structures using the ChemBioDraw Ultra 11.0 software and saved as an MDL file. These molecules were opened on Discovery Studio. Energy minimization of targeted compounds was run from tools/full minimization. From tools/apply force field.
(4) Docking of the test set: from protocol/macromolecule/docking using CDOCKER protocol.
(5) Analysis of docking result: after running the protocol, a report about 10 different conformers of each molecule was obtained and then the best conformer was selected according to the – CDOCKER energy. The binding mode and interaction of the tested compounds were visualized at the binding sites.
3.8. Physicochemical properties
The SwissADME web tool was used to estimate the physicochemical and pharmacokinetic characteristics in silico (Swissadme. Available online: https://www.swissadme.ch/).4. Conclusion
To sum up, a group of structurally related levofloxacin compounds have been synthesized. Their cytotoxic activities against the three cell lines, namely, MCF-7, Hep3B and L-SR were investigated by an MTT method. Compounds 3c, 4b, 5, 7, 8, 13a and 13c possessed the highest cytotoxic activity against the Hep3b cancer cell line. Nevertheless, compounds 3c, 5 and 13a showed the highest cytotoxic activity against the L-SR cancer cell line, while compounds 3c and 7 exhibited the best cytotoxic activity against the MCF-7 cancer cell line. The inhibition of Topo2β polymerase enzyme was also investigated. Compounds 3c and 7 showed the best Topo2β inhibition activity, compared to etoposide and levofloxacin as reference drugs. An Annexin-V-FITC apoptosis assay protocol was used to investigate the cell cycle apoptosis profile. Compound 3c and compound 5 showed cell cycle arrest at the S phase. Compounds 3c and 5 showed elevation in caspase-3 levels. The effects on normal cell lines were evaluated in comparison to etoposide and levofloxacin. Compound 5 manifested lesser side effects on the normal breast cell line MCF-10a and on the normal liver cell line THLE2.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- M. Siddiqui and S. V. Rajkumar, The high cost of cancer drugs and what we can do about it, Mayo Clin. Proc., 2012, 87, 935–943, DOI:10.1016/j.mayocp.2012.07.007.
- H. Maeda and M. Khatami, Analyses of repeated failures in cancer therapy for solid tumors: poor tumor-selective drug delivery, low therapeutic efficacy and unsustainable costs, Clin. Transl. Med., 2018, 7, 1–20, DOI:10.1186/s40169-018-0185-6.
- G. Tran and S. Y. Zafar, Financial toxicity and implications for cancer care in the era of molecular and immune therapies, Ann. Transl. Med., 2018, 6, 166, DOI:10.21037/atm.2018.03.28.
- D. Sun, W. Gao, H. Hu and S. Zhou, Why 90% of clinical drug development fails and how to improve it?, Acta Pharm. Sin. B, 2022, 12, 3049–3062, DOI:10.1016/j.apsb.2022.02.002.
- S. Murteira, Z. Ghezaiel, S. Karray and M. Lamure, Drug reformulations and repositioning in pharmaceutical industry and its impact on market access: reassessment of nomenclature, J. Mark. Access Heal. Policy, 2013, 1, 21131, DOI:10.3402/jmahp.v1i0.21131.
- A. Talevi, Drug repositioning: Current approaches and their implications in the precision medicine era, Expert Rev. Precis. Med, Drug Dev., 2018, 3, 49–61, DOI:10.1080/23808993.2018.1424535.
- E. A. M. de Oliveira and K. L. Lang, Drug repositioning: Concept, classification, methodology, and importance in rare/orphans and neglected diseases, J. Appl. Pharm. Sci., 2018, 8, 157–165, DOI:10.7324/JAPS.2018.8822.
- K. Shameer, B. S. Glicksberg, R. Hodos, K. W. Johnson, M. A. Badgeley, B. Readhead, M. S. Tomlinson, T. O'Connor, R. Miotto, B. A. Kidd, R. Chen, A. Ma'Ayan and J. T. Dudley, Systematic analyses of drugs and disease indications in RepurposeDB reveal pharmacological, biological and epidemiological factors influencing drug repositioning, Briefings Bioinf., 2018, 19, 656–678, DOI:10.1093/bib/bbw136.
- A. Naeem, S. Badshah, M. Muska, N. Ahmad and K. Khan, The Current Case of Quinolones: Synthetic Approaches and Antibacterial Activity, Molecules, 2016, 21, 268, DOI:10.3390/molecules21040268.
- M. Samir, M. Ramadan, M. Hamed, M. Osman and G. AbouRahma, Recent Strategies in Design of Antitumor and Antibacterial Fluoroquinolones, J. Adv. Biomed. Pharm. Sci., 2021, 4, 134–151, DOI:10.21608/jabps.2021.68305.1124.
- N. K. Boyd, C. Teng and C. R. Frei, Brief Overview of Approaches and Challenges in New Antibiotic Development: A Focus On Drug Repurposing, Front. Cell. Infect. Microbiol., 2021, 11, 1–12, DOI:10.3389/fcimb.2021.684515.
- E. Qashou, Y. Al-Hiari, V. Kasabri, R. AlBashiti, S. AlAlawi, A. Telfah and A. AlHadid, Antiproliferative Activities of Lipophililic Fluoroquinolones-Based Scaffold Against a Panel of Solid and Liquid Cancer Cell Lines, Asian Pacific, J. Cancer Prev., 2022, 23, 1529–1537, DOI:10.31557/APJCP.2022.23.5.1529.
- M. A. F. Salih, Y. Al-Hiari, V. Kasabri, R. Darwish, H. Abumansour, L. Bourghli, S. Al Alawi and R. Albashiti, Newly Substituted Anilino-Fluoroquinolones with Proliferation Inhibition Potential against a Panel of Cancer Cell Lines, Asian Pacific, J. Cancer Prev., 2022, 23, 2507–2521, DOI:10.31557/APJCP.2022.23.7.2507.
- T. Kloskowski, Z. Fekner, K. Szeliski, M. Paradowska, D. Balcerczyk, M. Rasmus, P. Dąbrowski, Ł. Kaźmierski, T. Drewa and M. Pokrywczyńska, Effect of four fluoroquinolones on the viability of bladder cancer cells in 2D and 3D cultures, Front. Oncol., 2023, 13, 1–17, DOI:10.3389/fonc.2023.1222411.
- M. A. A. Abdel-Aal, S. A. Abdel-Aziz, M. S. A. Shaykoon and G. E. A. Abuo-Rahma, Towards anticancer fluoroquinolones: A review article, Arch. Pharm., 2019, 352, 1800376, DOI:10.1002/ardp.201800376.
- G. Bisacchi, and M. Hale, A “Double-Edged” Scaffold: Antitumor Power within the Antibacterial Quinolone, 2016, DOI:10.2174/0929867323666151223095839.
- M. E. Adly, E. M. Gedawy, A. A. El-Malah and O. M. Khalil, Design, synthesis and in vitro anticancer activity of some new lomefloxacin derivatives, Sci. Rep., 2024, 14, 1–19, DOI:10.1038/s41598-024-56313-w.
- N. Ferrario, E. Marras, V. Vivona, F. Randisi, A. M. Antonino Nicolò Fallica, G. Perletti and M. B. Gariboldi, Mechanisms of the Antineoplastic Effects of New Fluoroquinolones in 2D and 3D Human Breast and Bladder Cancer Cell Lines, Cancers, 2024, 16, 2227 CrossRef.
- C. Mugnaini, S. Pasquini and F. Corelli, The 4-Quinolone-3-Carboxylic Acid Motif as a Multivalent Scaffold in Medicinal Chemistry, Curr. Med. Chem., 2009, 16, 1746–1767, DOI:10.2174/092986709788186156.
- T. Li, Z. Gao Liu, Y. Xie, Y. Feng, Q. Yan, S. Wu, L. Ni, H. Zhao, W. Huang and G. Hu, Synthesis and anti-proliferative activity of fluoroquinolone C-3 fused heterocyclic α,β-unsaturated ketones derived from ciprofloxacin, Yaoxue Xuebao, 2015, 50, 569–573 CAS.
- V. Yadav and P. Talwar, Repositioning of fluoroquinolones from antibiotic to anti-cancer agents: An underestimated truth, Biomed. Pharmacother., 2019, 111, 934–946, DOI:10.1016/j.biopha.2018.12.119.
- M. Yu, R. Li and J. Zhang, Repositioning of antibiotic levofloxacin as a mitochondrial biogenesis inhibitor to target breast cancer, Biochem. Biophys. Res. Commun., 2016, 471, 639–645, DOI:10.1016/j.bbrc.2016.02.072.
- M. Song, H. Wu, S. Wu, T. Ge, G. Wang, Y. Zhou, S. Sheng and J. Jiang, Antibiotic drug levofloxacin inhibits proliferation and induces apoptosis of lung cancer cells through inducing mitochondrial dysfunction and oxidative damage, Biomed. Pharmacother., 2016, 84, 1137–1143, DOI:10.1016/j.biopha.2016.10.034.
- W. H. Ismail, H. Abumansour, O. H. Abusara, W. Khalil, H. Abul-Futouh, A. I. Ibrahim, M. K. Harb and D. H. Abulebdah, Biological evaluation of levofloxacin and its thionated derivatives: antioxidant activity, aldehyde dehydrogenase enzyme inhibition, and cytotoxicity on A549 cell line, N. Schmied. Arch. Pharmacol., 2024, 13, 1–11 Search PubMed.
- J. ping Sun, Z. yu Shi, S. meng Liu, Y. hua Kang, G. qiang Hu, C. shen Huangfu, J. bo Deng and B. Liu, Trimethoxy-benzaldehyde levofloxacin hydrazone inducing the growth arrest and apoptosis of human hepatocarcinoma cells, Cancer Cell Int., 2013, 13, 1–10, DOI:10.1186/1475-2867-13-67.
- V. L. Tunitskaya, A. R. Khomutov, S. N. Kochetkov, S. K. Kotovskaya and V. N. Charushin, Inhibition of DNA Gyrase by Levofloxacin and Related Fluorine-Containing Heterocyclic Compounds, Acta Naturae, 2011, 3, 94–99, DOI:10.32607/20758251-2011-3-4-94-99.
- T. Kloskowski, K. Szeliski, Z. Fekner, M. Rasmus, P. Dąbrowski, A. Wolska, N. Siedlecka, J. Adamowicz, T. Drewa and M. Pokrywczyńska, Ciprofloxacin and levofloxacin as potential drugs in genitourinary cancer treatment—the effect of dose–response on 2D and 3D cell cultures, Int. J. Mol. Sci., 2021, 22, 1–27, DOI:10.3390/ijms222111970.
- A. K. Jadhav and S. M. Karuppayil, Molecular docking studies on thirteen fluoroquinolines with human topoisomerase II a and b, In Silico Pharmacol., 2017, 5, 1–12, DOI:10.1007/s40203-017-0024-2.
- X. He, Q. Yao, D. Fan, Y. You, W. Lian, Z. Zhou and L. Duan, Combination of levofloxacin and cisplatin enhances anticancer efficacy via co-regulation of eight cancer-associated genes, Discover Oncol., 2022, 13, 76, DOI:10.1007/s12672-022-00541-x.
- L. Cantwell and J. Perkins, Infectious Disease Emergencies in Oncology Patients, Emerg. Med. Clin., 2018, 36, 795–810, DOI:10.1016/j.emc.2018.06.009.
- S. D. Yang, Z. L. Bai, F. Zhang, L. Ma, D. L. Yang and W. Y. Ding, Levofloxacin increases the effect of serum deprivation on anoikis of rat nucleus pulposus cells via Bax/Bcl-2/caspase-3 pathway, Toxicol. Mech. Methods, 2014, 24, 688–696, DOI:10.3109/15376516.2014.963772.
- S. Charshafian and S. Y. Liang, Infectious Disease Emergencies in the Cancer Patient- Rapid Fire, Emerg. Med. Clin., 2018, 36, 493–516, DOI:10.1016/j.emc.2018.04.001.Infectious.
- A. El-Malah, A. Youssef, M. Ismail, M. Kamel and Z. Mahmoud, New promising levofloxacin derivatives: Design, synthesis, cytotoxic activity screening, Topo2β polymerase inhibition assay, cell cycle apoptosis profile analysis, Bioorg. Chem., 2021, 113, 105029, DOI:10.1016/j.bioorg.2021.105029.
- J. Azéma, B. Guidetti, A. Korolyov, R. Kiss, C. Roques, P. Constant, M. Daffé and M. Malet-Martino, Synthesis of lipophilic dimeric C-7/C-7-linked ciprofloxacin and C-6/C-6-linked levofloxacin derivatives. Versatile in vitro biological evaluations of monomeric and dimeric fluoroquinolone derivatives as potential antitumor, antibacterial or antimycobacterial agents, Eur. J. Med. Chem., 2011, 46, 6025–6038, DOI:10.1016/j.ejmech.2011.10.014.
- G. Q. Hu, G. Q. Wang, N. N. Duan, X. Y. Wen, T. Y. Cao, S. Q. Xie and W. L. Huang, Design, synthesis and antitumor activity of fluoroquinolone C3 heterocyclic bis-oxadiazole methylsulfide derivatives derived from levofloxacin, Chem. Res. Chin. Univ., 2012, 28, 980–984 CAS.
- S. E. Hamideh Ahadi, M. Shokrzadeh, Z. Hosseini-khah, N. G. Barghi and M. Ghasemian, Conversion of antibacterial quinolone drug levofloxacin to potent cytotoxic agents, J. Biochem. Mol. Toxicol., 2023, 37, e23334 CrossRef.
- G. Q. Hu, X. K. Wu, G. Q. Wang, N. N. Duan, X. Y. Wen, T. Y. Cao, Y. Jun, W. Wei, S. Q. Xie and W. L. Huang, Synthesis and antitumor and antibacterial evaluation of fluoro-quinolone derivatives (III): Mono- and bis-Schiff-bases, Chin. Chem. Lett., 2012, 23, 515–517, DOI:10.1016/j.cclet.2012.01.029.
- O. M. Khalil, E. M. Gedawy, A. A. El-Malah and M. E. Adly, Novel nalidixic acid derivatives targeting topoisomerase II enzyme; Design, synthesis, anticancer activity and effect on cell cycle profile, Bioorg. Chem., 2019, 83, 262–276, DOI:10.1016/j.bioorg.2018.10.058.
- M. I. Marzouk, S. A. Shaker, A. A. A. Hafiz and K. Z. El-Baghdady, Design and synthesis of new phthalazinone derivatives containing benzyl moiety with anticipated antitumor activity, Biol. Pharm. Bull., 2016, 39, 239–251, DOI:10.1248/BPB.B15-00656.
- R. Antre, A. Cendilkumar, R. Nagarajan, D. Goli and R. J. Oswal, Synthesis , Antitumor and Antimicrobial Activities of Some Novel 1-(Substituted)-3-Methyl-1H-Pyrazol-5(4H)-One, J. Sci. Res., 2012, 5, 3–12, DOI:10.3329/jsr.v4i1.7027.
- A. I. Hashem, W. S. I. Abou-Elmagd, E. A. El-Bordany and A. Abdel Aziz, Synthesis and Reactions of a 2(5H)-Furanone Bearing Two Furyl Substituents, J. Heterocycl. Chem., 2019, 56, 218–225, DOI:10.1002/jhet.3398.
- A. A. Helwa, E. M. Gedawy, A. T. Taher, A. K. E. D. El-ansary and M. Sahar, Synthesis and biological evaluation of novel pyrimidine-5-carbonitriles featuring morpholine moiety as antitumor agents, Future Med. Chem., 2020, 12, 403–421 CrossRef CAS PubMed.
- S. A. Shaker and M. I. Marzouk, Utilization of cyanoacetohydrazide and oxadiazolyl acetonitrile in the synthesis of some new cytotoxic heterocyclic compounds, Molecules, 2016, 21, 1–25, DOI:10.3390/molecules21020155.
- A. Kajal, S. Bala, S. Kamboj and V. Saini, Synthesis, characterization, andcomputational studies on phthalic anhydride-based benzylidene-hydrazide derivatives as novel,potential anti-inflammatory agents, Med. Chem. Res., 2014, 23, 2676–2689, DOI:10.1007/s00044-013-0848-1.
- F. Hassanzadeh, E. Jafari, F. Shojaei and H. Sadeghi-Aliabadi, Synthesis and cytotoxic activity evaluation of some new 1, 3, 4-oxadiazole, 1, 3, 4-thiadiazole and 1, 2, 4- triazole derivatives attached to phthalimide, Res. Pharm. Sci., 2021, 16, 634–642, DOI:10.4103/1735-5362.327509.
- P. Majumdar, A. Pati, M. Patra, R. K. Behera and A. K. Behera, Acid Hydrazides , Potent Reagents for Synthesis of Oxygen - , Nitrogen - , and/or Sulfur-Containing Heterocyclic Rings, Chem. Rev., 2014, 114, 2942–2977, DOI:10.1021/cr300122t.
- N. Sultana, M. S. Arayne, S. Bushra, S. Rizvi and M. A. Mesaik, Synthesis , Characterization and Biological Evaluation of a Series of Levofloxacin Carboxamide Analogues, Bull. Korean Chem. Soc., 2009, 30, 2294–2298 CrossRef CAS.
- T. Noguch, Y. Yokoo and S. Jung, Convenient Preparation of Primary Amides via Activation of Carboxylic Acids with Ethyl Chloroformate and Triethylamine under Mild Conditions, Chem. Lett., 2013, 42, 580–582, DOI:10.1246/cl.130096.
- Ž. Skok, N. Zidar, D. Kikelj and J. Ilaš, Dual Inhibitors of Human DNA Topoisomerase II and Other Cancer-Related Targets, J. Med. Chem., 2020, 63, 884–904, DOI:10.1021/acs.jmedchem.9b00726.
- K. Buzun, A. Bielawska, K. Bielawski and A. Gornowicz, DNA topoisomerases as molecular targets for anticancer drugs, J. Enzyme Inhib. Med. Chem., 2020, 35, 1781–1799, DOI:10.1080/14756366.2020.1821676.
- Z. Topcu, DNA topoisomerases as targets for anticancer drugs, J. Clin. Pharm. Ther., 2001, 26, 405–416, DOI:10.1046/j.1365-2710.2001.00368.x.
- A. Ryckebusch, D. Garcin, A. Lansiaux, J.-F. Goossens, B. Baldeyrou, R. Houssin, C. Bailly and J.-P. Hénichart, Synthesis, Cytotoxicity, DNA Interaction, and Topoisomerase II Inhibition Properties of Novel Indeno[2,1-c]quinolin-7-one and Indeno[1,2-c]isoquinolin-5,11-dione Derivatives, J. Med. Chem., 2008, 51, 3617–3629, DOI:10.1021/jm800017u.
- M. Abdel-Aziz, S.-E. Park, G. E.-D. A. A. Abuo-Rahma, M. A. Sayed and Y. Kwon, Novel N-4-piperazinyl-ciprofloxacin-chalcone hybrids: Synthesis, physicochemical properties, anticancer and topoisomerase I and II inhibitory activity, Eur. J. Med. Chem., 2013, 69, 427–438, DOI:10.1016/j.ejmech.2013.08.040.
- S. H. Yoo, Y. G. Yoon, J. S. Lee, Y. S. Song, J. S. Oh, B. S. Park, T. K. Kwon, C. Park, Y. H. Choi and Y. H. Yoo, Etoposide induces a mixed type of programmed cell death and overcomes the resistance conferred by Bcl-2 in Hep3B hepatoma cells, Int. J. Oncol., 2012, 41, 1443–1454, DOI:10.3892/ijo.2012.1585.
- J. Y. Gao, C. S. Chang, J. C. Lien, T. W. Chen, J. L. Hu and J. R. Weng, Synthetic tryptanthrin derivatives induce cell cycle arrest and apoptosis via akt and mapks in human hepatocellular carcinoma cells, Biomedicines, 2021, 9, 1527, DOI:10.3390/biomedicines9111527.
- O. M. van, G. Koopman, C. P. Reutelingsperger, G. A. Kuijten, R. M. Keehnen and S. T. Pals, Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis, Blood, 1994, 84, 1415–1420 CrossRef.
- C. Haanen, H. Steffens-nakken and C. Reutelingsperger, A novel assay for apoptosis Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V, J. Immunol. Methods, 1995, 184, 39–51 CrossRef.
- A. G. Porter and R. U. Jänicke, Emerging roles of caspase-3 in apoptosis, Cell Death Differ., 1999, 6, 99–104, DOI:10.1038/sj.cdd.4400476.
- S. El-Meligie, A. T. Taher, A. M. Kamal and A. Youssef, Design, synthesis and cytotoxic activity of certain novel chalcone analogous compounds, Eur. J. Med. Chem., 2017, 126, 52–60, DOI:10.1016/j.ejmech.2016.09.099.
- L. G. Ferreira, R. N. Dos Santos, G. Oliva and A. D. Andricopulo, Molecular docking and structure-based drug design strategies, Molecules, 2015, 20, 13384–13421 CrossRef CAS PubMed.
- J. De Ruyck, G. Brysbaert, R. Blossey and M. F. Lensink, Molecular docking as a popular tool in drug design, an in silico travel, Adv. Appl. Bioinf. Chem., 2016, 9, 1–11, DOI:10.2147/AABC.S105289.
- Protein Data Bank, https://www.rcsb.org/pdb.
- T. Mosmann, Rapid Colorimetric Assay for Cellular Growth and Survival : Application to Proliferation and Cytotoxicity Assays, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- A. E. Kassab and E. M. Gedawy, Novel ciprofloxacin hybrids using biology oriented drug synthesis (BIODS) approach: Anticancer activity, effects on cell cycle profile, caspase-3 mediated apoptosis, topoisomerase II inhibition, and antibacterial activity, Eur. J. Med. Chem., 2018, 150, 403–418, DOI:10.1016/j.ejmech.2018.03.026.
- Z. Darzynkiewicz, E. Bedner and P. Smolewski, Flow Cytometry in Analysis of Cell Cycle and Apoptosis, Semin. Hematol., 2001, 38, 179–193 CrossRef CAS PubMed.
- M. F. Tolba, A. Esmat, A. M. Al-abd and S. S. Azab, Caffeic Acid Phenethyl Ester Synergistically Enhances Docetaxel and Paclitaxel Cytotoxicity in Prostate Cancer Cells, IUBMB Life, 2013, 65, 716–729, DOI:10.1002/iub.1188.
- K. C. Zimmermann, D. R. Green and S. Diego, How cells die : Apoptosis pathways, J. Allergy Clin. Immunol., 2001, 108, 99–103, DOI:10.1067/mai.2001.117819.
- S. Elmore, Apoptosis : A Review of Programmed Cell Death, J. Allergy Clin. Immunol., 2007, 108, 495–516, DOI:10.1080/01926230701320337.
- B. Khalilzadeh, N. Shadjou, H. Afsharan, M. Eskandani and H. N. Charoudeh, Reduced graphene oxide decorated with gold nanoparticle as signal amplification element on ultra-sensitive electrochemiluminescence determination of caspase-3 activity and apoptosis using peptide based biosensor, BioImpacts, 2016, 6, 135–147, DOI:10.15171/bi.2016.20.
- B. Khalilzadeh, H. Nozad and N. Shadjou, Sensors and Actuators B : Chemical Ultrasensitive caspase-3 activity detection using an electrochemical biosensor engineered by gold nanoparticle functionalized MCM-41 : Its application during stem cell differentiation, Sens. Actuators, B, 2016, 231, 561–575, DOI:10.1016/j.snb.2016.03.043.
- C. C. Wu, T. K. Li, L. Farh, L. Y. Lin, T. S. Lin, Y. J. Yu, T. J. Yen, C. W. Chiang and N. L. Chan, Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide, Science, 2011, 333, 459–462, DOI:10.1126/science.1204117.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra03975k |
| This journal is © The Royal Society of Chemistry 2024 |