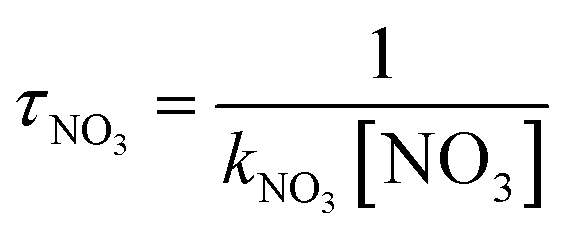DOI:
10.1039/D4RA03922J
(Paper)
RSC Adv., 2024,
14, 25472-25480
Theoretical investigation on degradation of CH![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e002.gif) CCH2OH by NO3 radicals in the atmosphere†
CCH2OH by NO3 radicals in the atmosphere†
Received
28th May 2024
, Accepted 29th June 2024
First published on 23rd August 2024
1. Introduction
In recent years, environmental pollution caused by organic pollutants has been of wide concern from all sectors of society. It has a complex origin and diverse formation mechanisms, and is widely present in the atmosphere, soil, and water.1 In the past few decades, air pollution caused by solid particulate matter (PM) and volatile organic compounds (VOCs) has become a serious environmental problem threatening human health.2 Volatile organic compounds have the characteristics of toxicity, persistence, or difficult degradation, which have a certain impact on human health and the growth of animals and plants, and have become important organic pollutants in the environment.3–5 Research has shown that volatile organic compounds are important substances that affect atmospheric environmental pollution and are also important precursors of secondary organic aerosols (SOA).6 VOCs not only damage the central nervous system, but also carry the risk of carcinogenesis and mutagenesis.7 Thus, the degradation of VOCs is urgent. The most common oxidants in the atmosphere are mainly reactive free radicals, such as OH and NO3 radicals and Cl atoms.8 Nitrate (NO3) radicals are the main oxidants at night and play an important role in the chemistry of the lower troposphere.9 The study of its reaction mechanism and reaction kinetics with pollutants in the atmospheric environment is helpful to better understand the atmospheric chemical process and improve the atmospheric environment. At present, the mechanism and kinetics for the reaction of olefins and saturated alcohols induced with OH and NO3 radicals and Cl atoms have been extensively studied.10 In 2011, Thanh Lam Nguyen et al.11 studied the reaction mechanism and kinetics of C2H4 + NO3, and calculated that the thermal rate constant and experimental data were the total reaction rate constant. In 2020, Inmaculada Colmenar et al.12 studied the reaction mechanism and kinetic properties of 2-ethoxyethanol with OH and NO3 radicals and Cl atoms, and determined the reaction rate constants of OH and NO3 radicals and Cl under specific conditions, and calculated the atmospheric life of 2-ethoxy ethanol and proposed the degradation mechanism as well. In 2021, Inmaculada Aranda et al.13 explored the reaction of 3-ethoxy-1-propanol with Cl, OH and NO3 in the atmosphere. However, this type of oxidation of alkynes has not been reported. CH![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCH2OH, also known as 2-propylene-1-alcohol, is a compound with both alkynyl and hydroxyl functional groups. CHCCH2OH has been widely used as corrosion inhibitors in industry.14–16 As an important unsaturated VOC, it has a certain impact on the atmospheric environment and endangers human health. Therefore, the degradation of such air pollutants has become an ongoing concern, although they are widely used as monomers and intermediates in industrial chemicals synthesis. To the best of our knowledge, there have been no theoretical studies concerning the reaction of CHCCH2OH with NO3 radicals up to now. According to the current research status of CHCCH2OH, the reaction mechanisms and the potential energy surface (PES) of the radical with NO3 are studied by quantum chemistry calculations at the CCSD(T)17/cc-pVTZ//B3LYP18,19/6-311++G(d,p)20,21 level of theory, the main contents are as follows: the intermediate substances, transition states and products are identified, and the potential barriers of each reaction step are obtained to evaluate the favorable reaction path. In addition, we have obtained the rate constant of CHCCH2OH + NO3 at 200–2000 K, including the most important product branches, the values of the calculated rate constants reproduce remarkably well the available experiment data. Meanwhile, the atmospheric fate of CHCCH2OH are also researched using the same level of theory.22,23
CCH2OH, also known as 2-propylene-1-alcohol, is a compound with both alkynyl and hydroxyl functional groups. CHCCH2OH has been widely used as corrosion inhibitors in industry.14–16 As an important unsaturated VOC, it has a certain impact on the atmospheric environment and endangers human health. Therefore, the degradation of such air pollutants has become an ongoing concern, although they are widely used as monomers and intermediates in industrial chemicals synthesis. To the best of our knowledge, there have been no theoretical studies concerning the reaction of CHCCH2OH with NO3 radicals up to now. According to the current research status of CHCCH2OH, the reaction mechanisms and the potential energy surface (PES) of the radical with NO3 are studied by quantum chemistry calculations at the CCSD(T)17/cc-pVTZ//B3LYP18,19/6-311++G(d,p)20,21 level of theory, the main contents are as follows: the intermediate substances, transition states and products are identified, and the potential barriers of each reaction step are obtained to evaluate the favorable reaction path. In addition, we have obtained the rate constant of CHCCH2OH + NO3 at 200–2000 K, including the most important product branches, the values of the calculated rate constants reproduce remarkably well the available experiment data. Meanwhile, the atmospheric fate of CHCCH2OH are also researched using the same level of theory.22,23
2. Quantum chemical methods
The B3LYP method with the 6-311++g(d,p) basis set was applied to optimize the geometries of reactants, products, intermediates and products. Moreover, vibration frequency computations were also implemented to make sure of the presence of intermediates with no imaginary frequency, and transition states with one and only one imaginary frequency. To make sure that the transition states linked with designated intermediate, the intrinsic reaction coordinate (IRC) computations24,25 were also executed at the same level. The polarizable continuum model (PCM) has been successfully applied to simulate aqueous-phase reactions.26–28 For the reaction of CHCCH2OH with the NO3 radical, the aqueous structures were optimized directly at the B3LYP/6-311++g(d,p) level using the solvent PCM. CCSD(T)/cc-pVTZ method to used to gain more trustworthy relative energies of all the species involved in the NO3 with CHCCH2OH reaction in gaseous and aqueous phase reactions. All computations were executed with Gaussian 09 package of program.29 Rice–Ramsperger–Kassel–Marcus theory and transition-state theory was used to compute the rate constants of the NO3 with CHCCH2OH reaction.30 The eco-toxicity of important intermediates and products are appraised employing the program of ECOSAR developed by USEPA.31
3. Results and discussion
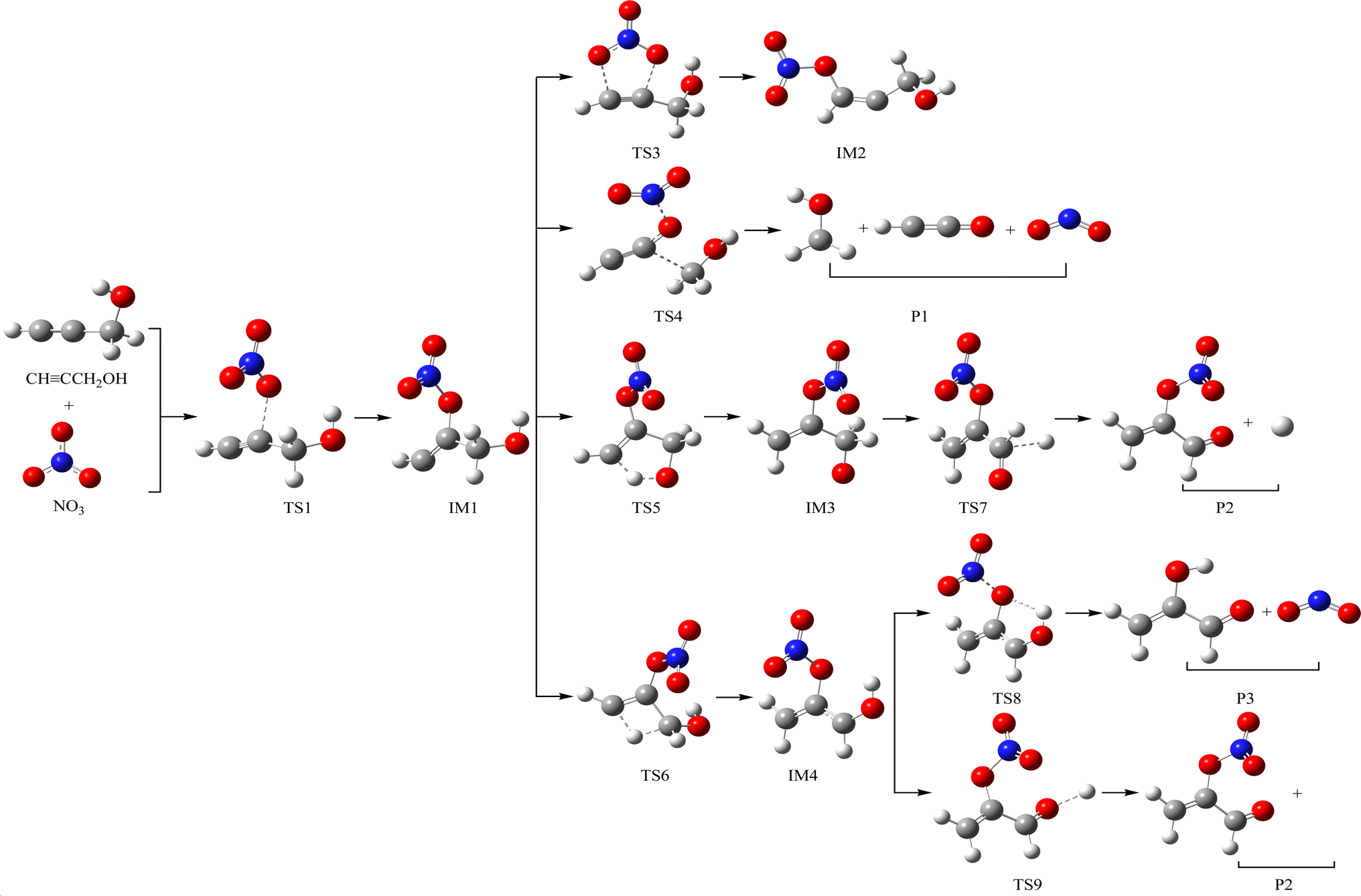
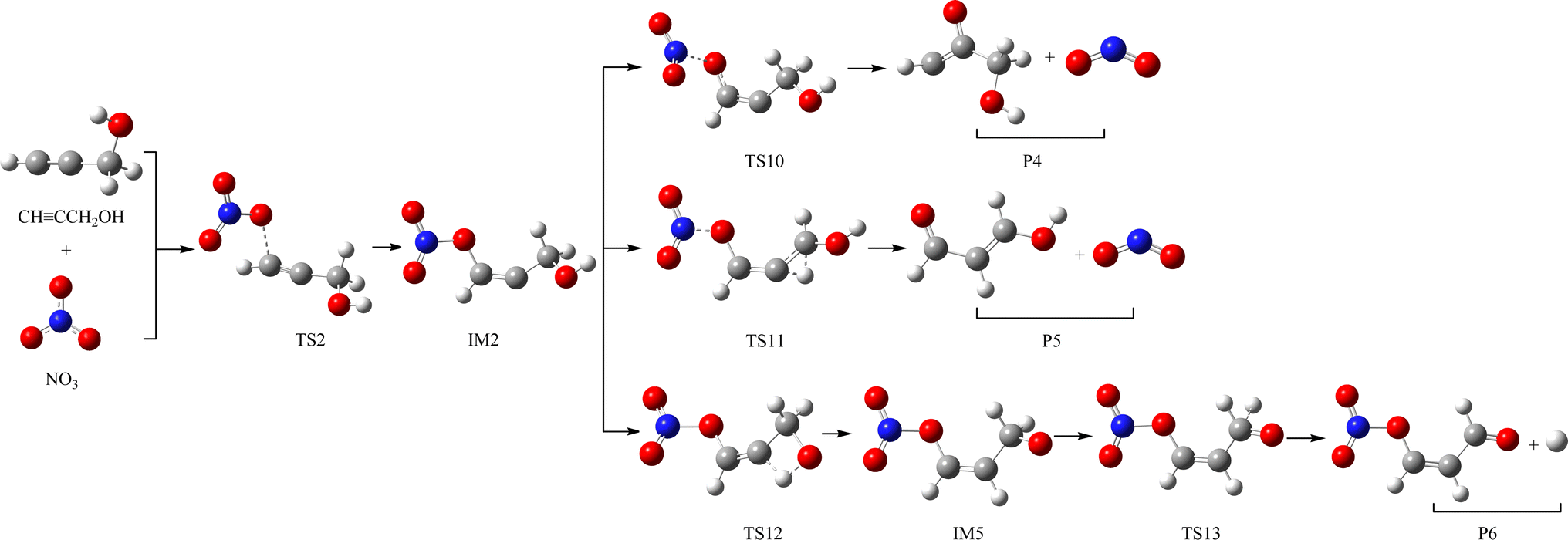
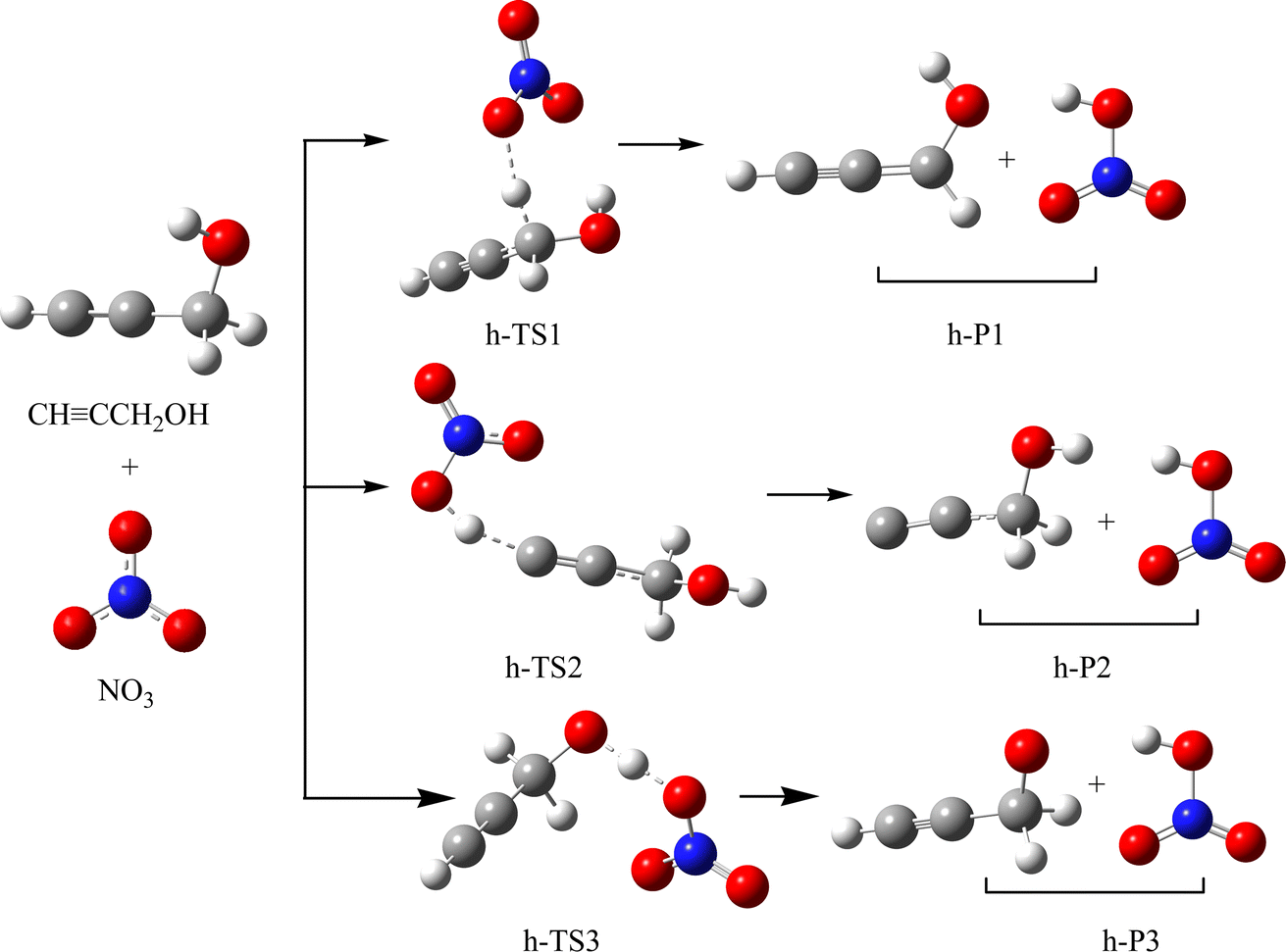
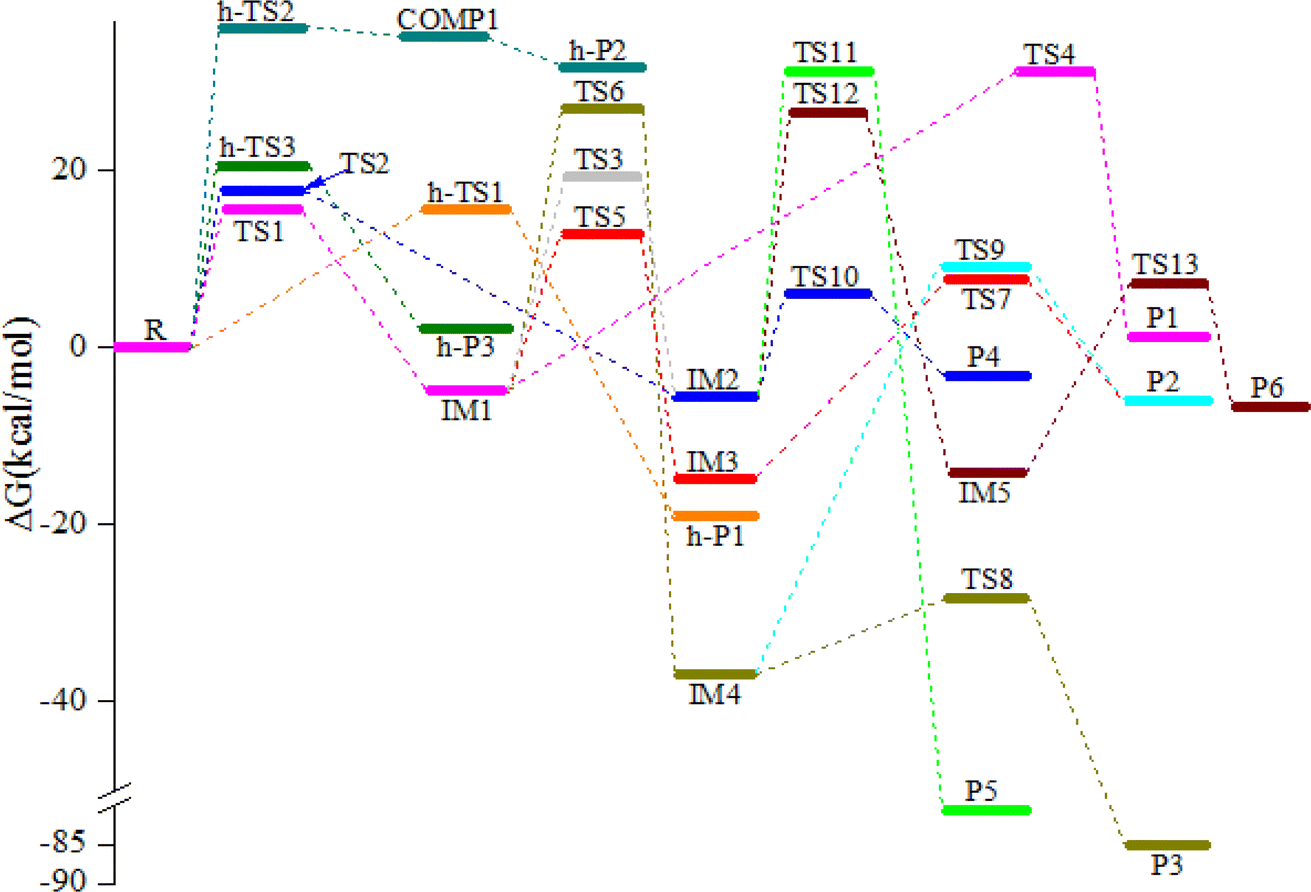
The schematic illustration of the reaction starting from IM1 and IM2 are displayed in Fig. 1 and 2. The schematic illustration of the hydrogen abstraction reaction is displayed in Fig. 3. The optimized geometric configuration of all the intermediates, transition states, reactants and all products involved in the CHCCH2OH with NO3 reaction are presented in Fig. S1 and S2,† respectively. The potential energy surface (PES) of the CHCCH2OH with NO3 reaction are presented in Fig. 4. The gaseous and aqueous structures are highly similar. The Zero Point Energies (ZPE), T1 diagnostics values, relative energies (ΔE), relative enthalpies (ΔH) and Gibbs free energy (ΔG) in gas and aqueous phase reactions are summarized in Tables S1 and S2.† The harmonic vibrational frequencies, moment of inertia, rotational constants, and numbers of the number of imaginary frequencies involved in this reaction are shown in Table S3.† The Z-matrix Cartesian coordinates of all species found on the PESs are shown in Table S4.† Unless otherwise stated, the geometric parameters and energies used in this work are respective obtained at the B3LYP/6-311++G(d,p) and CCSD(T)//B3LYP levels.
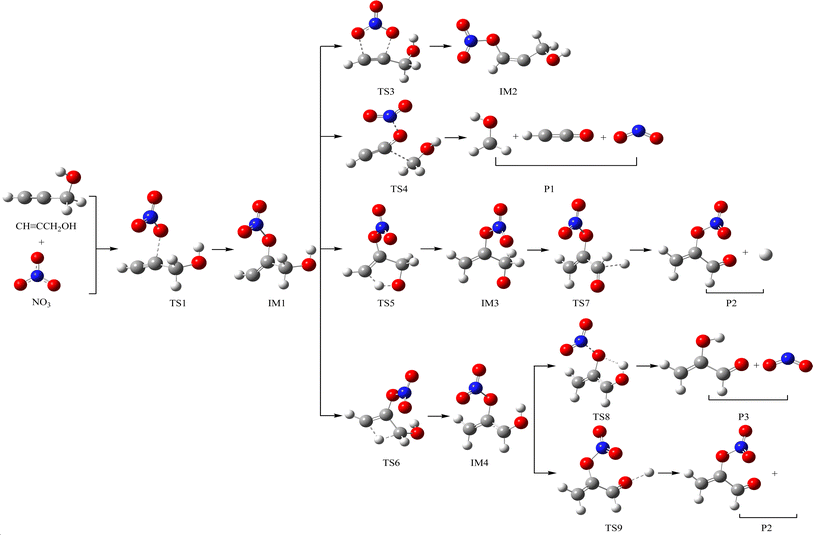 |
| | Fig. 1 The schematic illustration of the reaction starting from IM1 in the atmosphere, which obtained at the CCSD//B3LYP level. | |
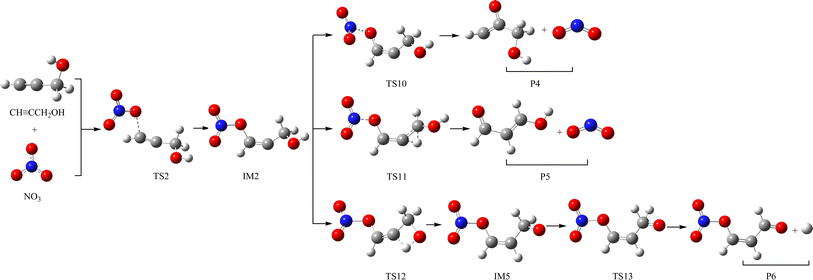 |
| | Fig. 2 The schematic illustration of the reaction starting from IM2 in the atmosphere, which obtained at the CCSD//B3LYP level. | |
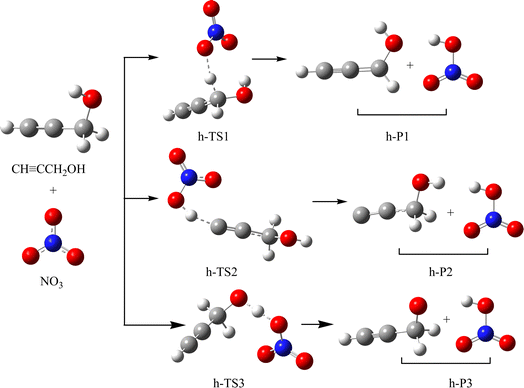 |
| | Fig. 3 The schematic illustration of the hydrogen abstraction reaction in the atmosphere, which obtained at the CCSD//B3LYP level. | |
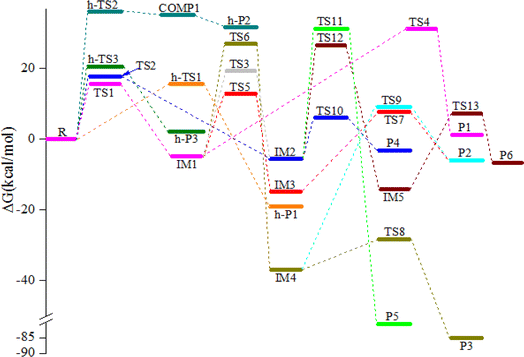 |
| | Fig. 4 The potential energy surface (PES) for the NO3 with CHCCH2OH reaction in the gas-phase at 298 K. | |
3.1. The addition/elimination mechanisms
The NO3-initial reaction of CHCCH2OH generate the rich energy intermediates IM1 (CHCONO2CH2OH) and IM2 (CHONO2CCH2OH) when the O atom in NO3 addition to the central-C and the terminal-C atom of CHCCH2OH, respectively. The involved corresponding transition states are labeled as TS1 and TS2, which are displayed in Fig. 1 and 2. The new generation C–O bonds in TS1 and TS2 are 2.002 Å and 2.047 Å. The energy of TS1 and TS2 are 4.76 and 8.22 kcal mol−1 higher than the reactants (CHCCH2OH + NO3). The exothermicity of IM1 (CHCONO2CH2OH) and IM2 (CHONO2CCH2OH) generation at the addition processes are estimated to be 16.77 and 16.45 kcal mol−1. IM1 and IM2 could transform into each other through annular transition state TS3. The free energy barrier of IM1 → TS3 → IM2 and IM2 → TS3 → IM1 are 24.26 and 24.90 kcal mol−1. This conversion process is unimportant owing to the high barrier heights. The internal energies of IM1 and IM2 are 16.05 and 15.85 kcal mol−1, which could generate corresponding products through subsequent unimolecular decomposition and isomerization processes.
IM1 could further directly dissociate to products P1 (CH2OH + HCCO + NO2) through TS4, which involves the simultaneous breakage of C–C and N–O bonds. The breaking C–C and N–O bonds in TS4 are 2.200 and 1.518 Å. The free energy barrier of IM1 → TS4 → P1 is 35.95 kcal mol−1, which prohibited the occurrence of the reaction. In addition, two internal rearrangement channels have been located for IM1. One is the H atom in –OH group shifting to the C atom of –CH group, generating IM3 (CH2CONO2CH2O) through a five-membered-ring TS5. The other isomerization channel of IM1 involves the H atom in –CH2OH group shifting to the C atom of –CH group, generating IM4 (CH2CONO2CHOH) through a four-membered-ring TS6. In TS5 and TS6, the breaking O–H and C–H bonds are 1.205 and 1.392 Å, and the generating C–H bonds are 1.336 and 1.475 Å, respectively. The free energy barriers of IM1 → TS5 → IM3 and IM1 → TS6 → IM4 are 17.60 and 31.80 kcal mol−1. Thus, the isomerization of IM1 to IM3 is better than the isomerization of IM1 to IM4. IM3, with the energy of 26.38 kcal mol−1, could dissociate to products P2 (CH2CONO2CHO + H) by cleavage the C–H bond through TS7 surmounting the free energy barrier of 22.55 kcal mol−1. Beginning with IM4, two probable reaction scenarios are located. IM4 could respective dissociate N–O bond and C–H bond through TS8 and TS9 to generate products P3 (CH2COHCHO + NO2) and P2. The free energy barrier of IM4 → TS8 → P3 is 8.50 kcal mol−1, which are 37.59 and 14.05 kcal mol−1 lower than that of IM4 → TS9 → P2 and IM3 → TS7 → P2, respectively. However, due to the higher free energy barrier of the isomerization of IM1 to IM4 (31.80 kcal mol−1), the most feasible channel from IM1 is IM1 → IM3 → P2.
As for IM2 (CHONO2CCH2OH), it could directly dissociate the N–O bond to generate P4 (CHOCCH2OH + NO2) through TS10 surmounting a free energy barrier of 11.64 kcal mol−1, and this dissociation process is exothermic by 2.84 kcal mol−1. Moreover, P5 (CHOCHCHOH + NO2) could be generated from IM2 through TS11 involving one of the H atom bonded the C atom of –CH2OH group shifting to the central-C atom accompanied by breaking the N–O bond. The breaking C–H bond and N–O bond are 1.330 and 1.475 Å, and the new generating C–H bond is 1.326 Å. However, owning to the higher dissociation free energy barrier height (36.72 kcal mol−1), the channel of generating P5 from IM2 is dynamically prohibited. In addition, IM2 could isomerizate to intermediate IM5 (CHONO2CHCH2O) through TS12 with the alcohol hydrogen shifting to the central-C atom overcoming the free energy barrier of 32.06 kcal mol−1. Subsequently, IM5 undergo simply C–H bond rupture generating the final products P6 (CHONO2CHCHO + H) through transition state TS13. In TS13, the rupturing C–H bond is 1.828 Å. The free energy barrier of IM5 → TS13 → P6 is 21.22 kcal mol−1. Thus, the pathway of generation of P6 from IM2 contributes less to the reaction.
3.2. The H-abstraction mechanisms
As revealed in Fig. 3 and 4, CHCCH2OH possesses three different types of H atoms: namely, the H atoms in –CH2–, –CH and –OH group, respectively. OH radical could abstract the H atom from the –CH and –CH2– group to generate h-P1 (CHCCHOH + HNO3) and h-P2 (CCCH2OH + HNO3). The corresponding transition states are presented as h-TS1 and h-TS2 in Fig. S1,† respectively. In h-TS1 and h-TS2, the breaking C–H bond are 1.174 and 1.518 Å; and the generating O–H bond are 1.580 and 1.094 Å, respectively. In addition, the OH radical could also abstract the H atom in –OH group through h-TS3. In h-TS3, the breaking and generating O–H bonds are 1.181 and 1.207 Å, respectively. The free energy barrier heights for the above three hydrogen abstraction channels are 15.66, 36.12 and 20.66 kcal mol−1, respectively. Therefore, the most feasible H-abstraction pathway is generation h-P1. Moreover, the free barrier of generation h-P1 is only 0.14 kcal mol−1 higher than that of the NO3 addition to the center-C atom of CHCCH2OH. Hence, the pathway of H-abstraction and addition/elimination could compete with each other.
3.3. The influence of the water environment
The reaction mechanism and advantageous channels of the CHCCH2OH with NO3 radical in aqueous solution are the same as those of gaseous reactions under the implicit water model (PCM). It is worth mentioning that, despite our tremendous efforts, transition states TS2 and TS4 were not found. Moreover, the free energy barriers in the gaseous phase are usually lower than those of aqueous reactions except for the processes of IM4 → TS9 → P2, IM2 → TS11 → P5 and IM5 → TS13 → P6. The free energy barriers of IM4 → TS9 → P2, IM2 → TS11 → P5 and IM5 → TS13 → P6 in the aqueous phase are 0.37, 2.39 and 1.18 kcal mol−1 lower than those in the gaseous phase. In addition, the free energy barriers in the gaseous phase for the advantageous channels (R → TS1 → IM1 → TS5 → IM3 → TS7 → P2) are 0.24, 1.12 and 0.12 kcal mol−1 lower than those in the aqueous phase, respectively. This result manifested that the water environment has a negative influence on the CHCCH2OH with NO3 reaction. Similar phenomena have also been found in some reactions.32,33
3.4. The dynamic calculations
To preferably comprehend the fate of CHCCH2OH in the atmosphere, we used the Fortran program and RRKM theory to compute the rate coefficient of key element reactions (pathways 1–4) at 10 Torr and 298 K. The specifics of the computation process are given in ESI.† The dominant reaction pathways using for the dynamic computations were displayed in Fig. 5.
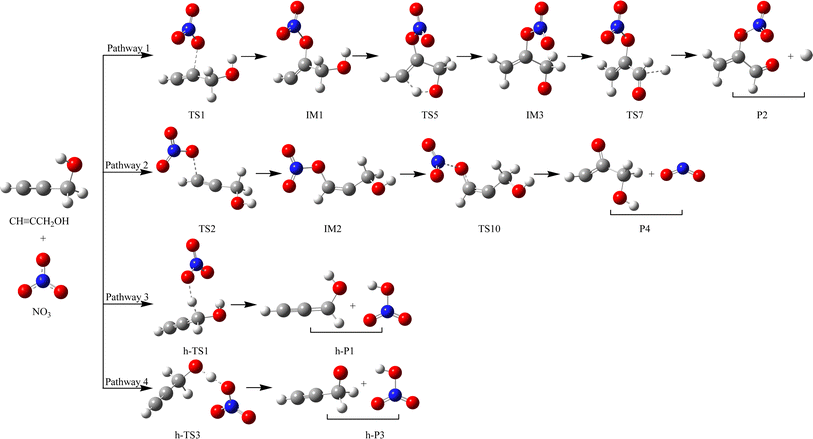 |
| | Fig. 5 The dominant reaction pathways using for the dynamic computations. | |
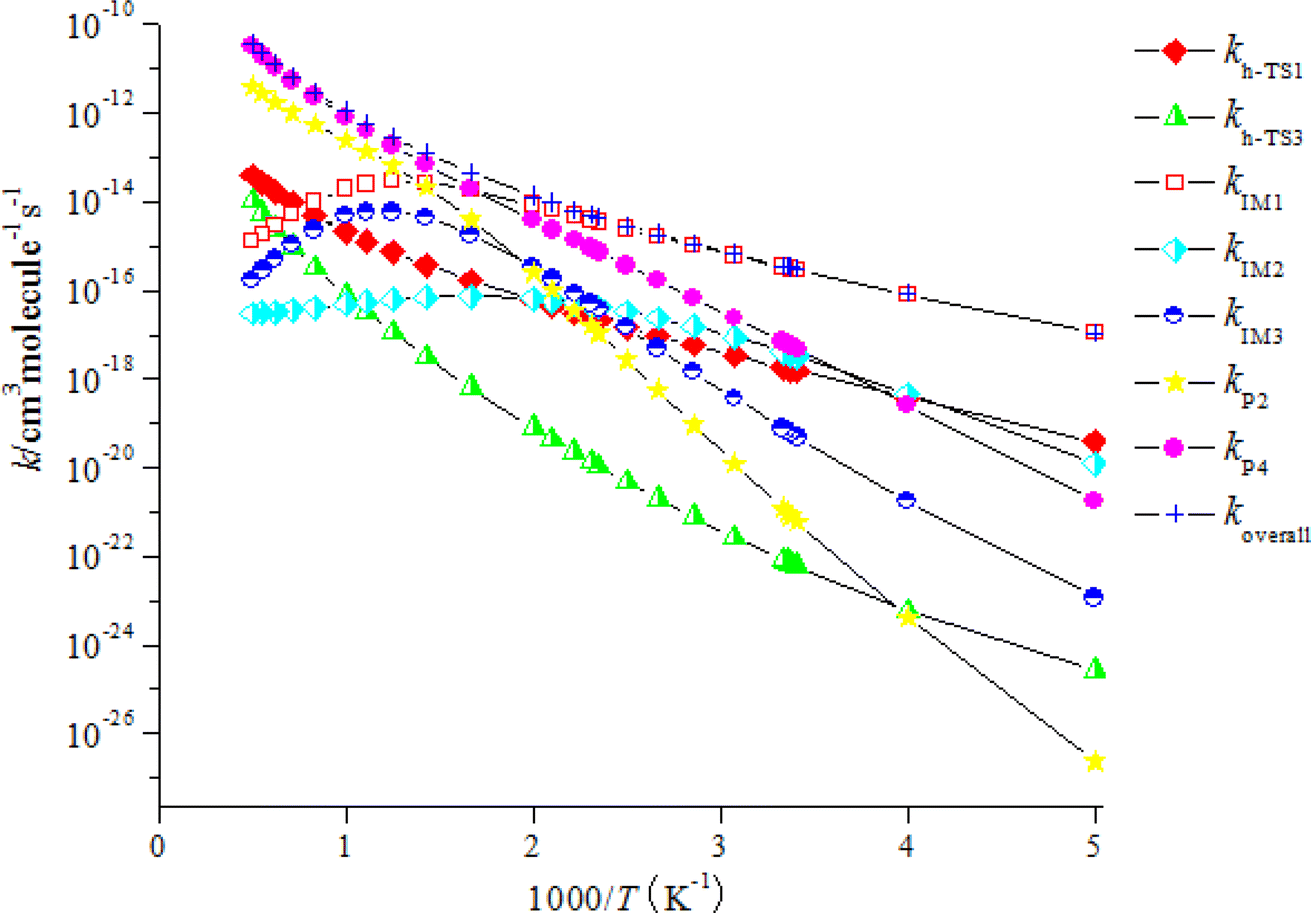
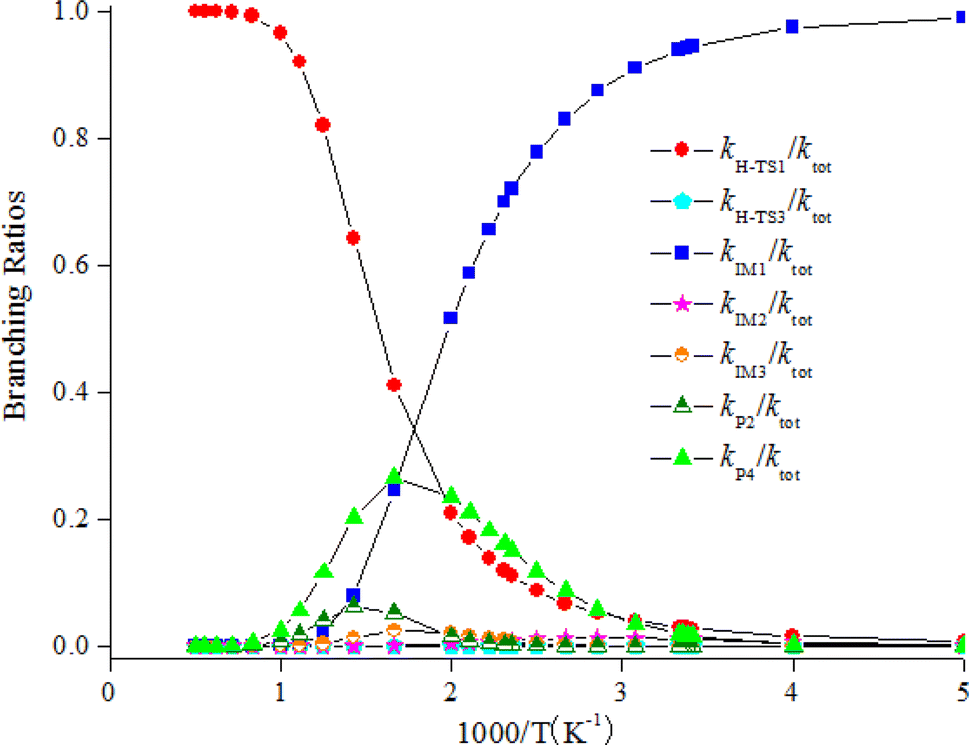
The computed rate coefficient of the NO3-initiated reaction of CHCCH2OH are listed in Table S5.† The temperature dependence of the branching rate coefficients and overall rate coefficients at 200–2000 K are displayed in Fig. 6. Fig. 6 reveals that the rate coefficients for the H-abstraction pathways (kh-TS1 and kh-TS3) and the pathways of generating P2 and P4 (kP2 and kP4) present positive temperature dependence. However, the rate coefficients of IM1 (CHCONO2CH2OH), IM2 (CHONO2CCH2OH) and IM3 (CH2CONO2CH2O) collisional stabilization channels increase firstly, and then decrease quickly with rising temperatures. The rate constants at 298 K is 3.48 × 10−16 cm3 per molecule per s. Fig. 7 displayed the branching ratios at the same temperature range and pressure. The low-temperature decomposition is occupied by the generation of IM1 (CHCONO2CH2OH) at 200–500 K. The channel of generating h-P1 (CHCCHOH + HNO3) through H-abstracting is contribute to the reaction at T > 500 K. The generation of the P4 (CHOCCH2OH + NO2) contribute to the reaction within a certain temperature range. The contribution of other channels to the reaction can be negligible.
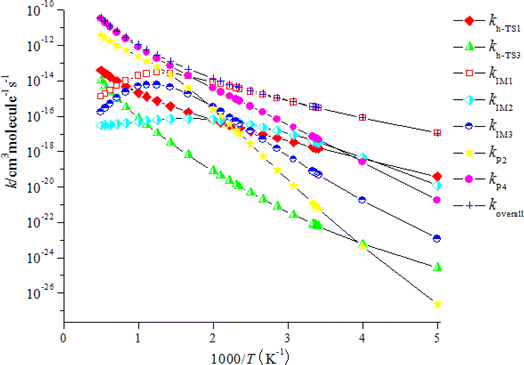 |
| | Fig. 6 Temperature dependence of the total and individual rate constants for the NO3 + CHCCH2OH reaction at 1 Torr of He, 200–2000 K. | |
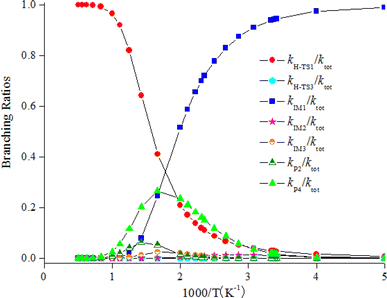 |
| | Fig. 7 Branching ratios of the significant product pathways for the NO3 + CHCCH2OH reaction at 200–2000 K. | |
3.5. Atmospheric implications of CH![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) CCH2OH
CCH2OH
The tropospheric lifetimes (τ) of CHCCH2OH could be assessed by reacting with NO3 radicals. The atmospheric lifetimes could be written as:
where kNO3 is the rate coefficient of the CHCCH2OH with NO3 reaction at 298 K as 3.48 × 10−16 cm3 per molecule per s. The lifetime of CHCCH2OH is estimated to be 58.35 days at 298 K using the average atmospheric concentration of NO3 (5.70 × 108 molecule per cm3).34
3.6. Ecological implications
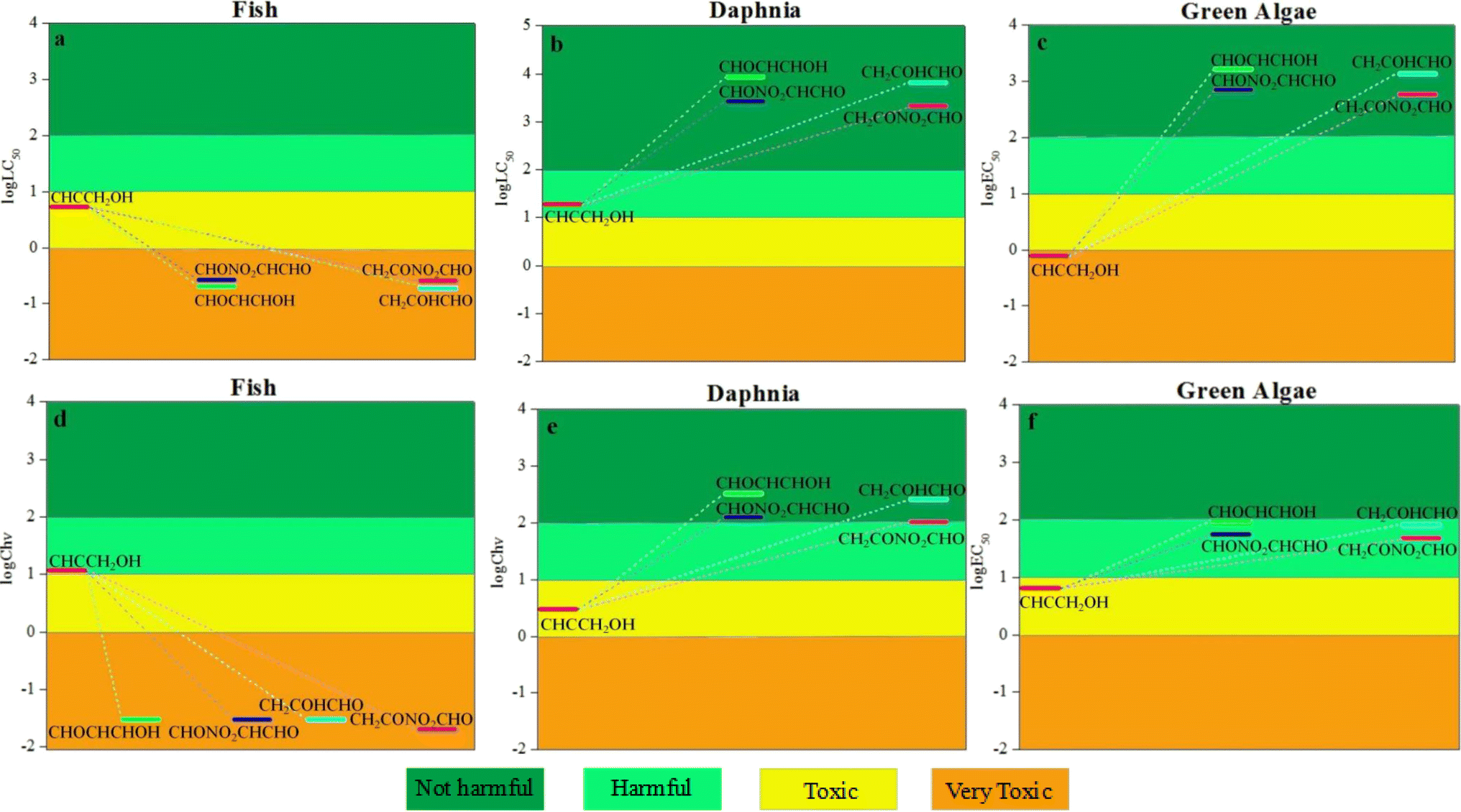
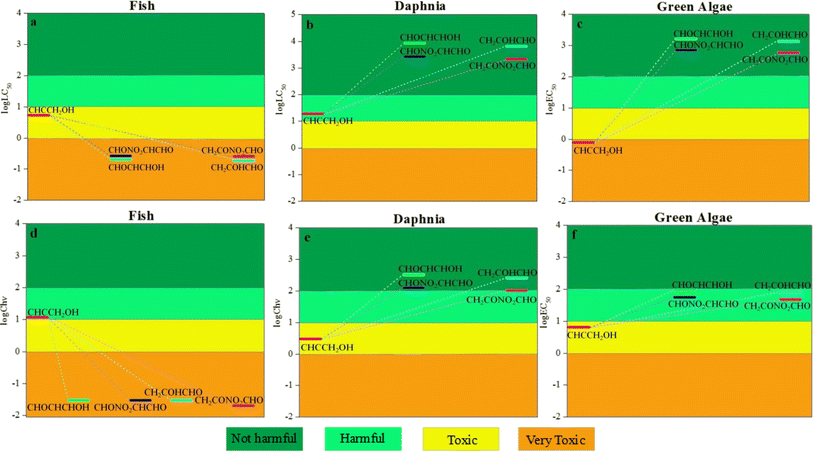
3.6.1. Acute toxicity and chronic toxicity to aquatic organisms. The computed acute toxicity log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) LC50 of propargyl alcohol to fish and daphnia and the logEC50 to green algae were 0.73, 1.27 and −0.11 mg L−1, respectively, manifesting that propargyl alcohol is toxic to the three aquatic organisms. As seen from Fig. 8, for fish, the values of logLC50 and logChv are negative, manifesting that the toxicity of propargyl alcohol is increased after degradation. For daphnia and green algae, the values of logLC50, logEC50 and logChv are larger than of propargyl alcohol, manifesting that the toxicity of propargyl alcohol is diminished after degradation. Furthermore, the order of acute toxicity for the products of the degradation in water (CHOCHCHOH, CHONO2CHCHO, CH2COHCHO and CH2CONO2CHO) were identical to chronic toxicity.
LC50 of propargyl alcohol to fish and daphnia and the logEC50 to green algae were 0.73, 1.27 and −0.11 mg L−1, respectively, manifesting that propargyl alcohol is toxic to the three aquatic organisms. As seen from Fig. 8, for fish, the values of logLC50 and logChv are negative, manifesting that the toxicity of propargyl alcohol is increased after degradation. For daphnia and green algae, the values of logLC50, logEC50 and logChv are larger than of propargyl alcohol, manifesting that the toxicity of propargyl alcohol is diminished after degradation. Furthermore, the order of acute toxicity for the products of the degradation in water (CHOCHCHOH, CHONO2CHCHO, CH2COHCHO and CH2CONO2CHO) were identical to chronic toxicity.
 |
| | Fig. 8 Acute and chronic toxicity of CHCCH2OH and its products of the degradation in water to aquatic organisms (mg L−1). | |
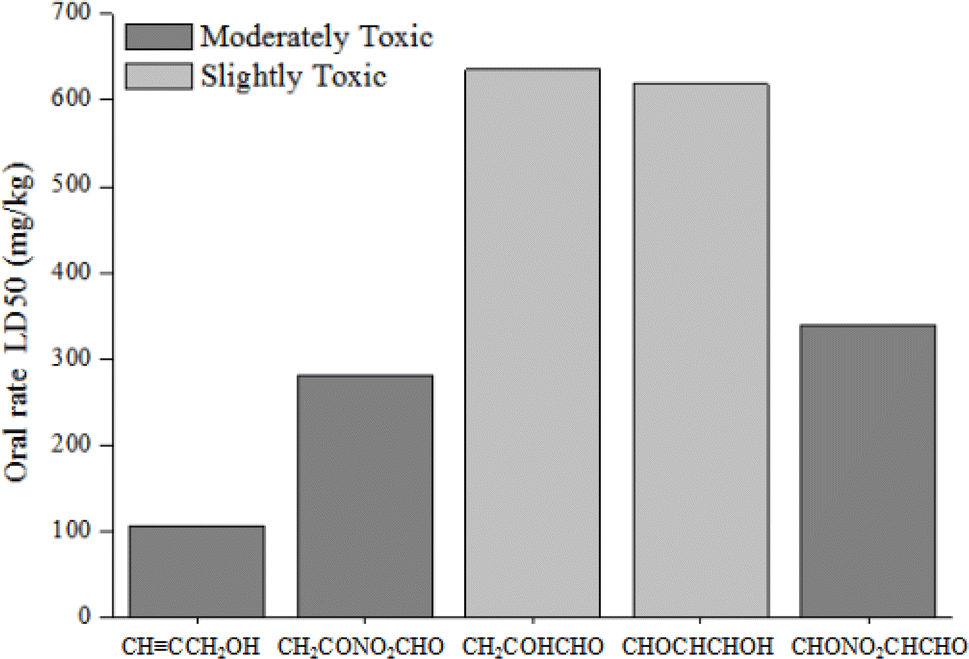
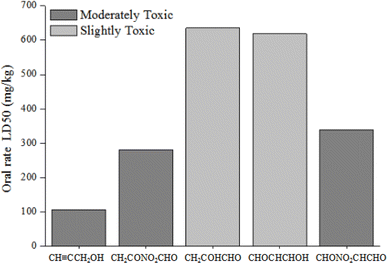
3.6.2. Acute toxicity to rats. The rat (oral) LD50 values for propargyl alcohol and its products of the degradation in water are presented in Fig. 9. Obviously, propargyl alcohol, CH2CONO2CHO and CHONO2CHCHO belong to the category of moderately toxic compounds (50 ≤ LD50 < 500 mg L−1). CH2COHCHO and CHOCHCHOH classify as slightly toxic compounds (500 ≤ LD50 < 5000 mg L−1). As seen from Fig. 9, the toxicity to rats of degradation products of propargyl alcohol are reduced.
 |
| | Fig. 9 Oral rate LD50 for CHCCH2OH and its products of the degradation in water. | |
3.7. Ecological implications
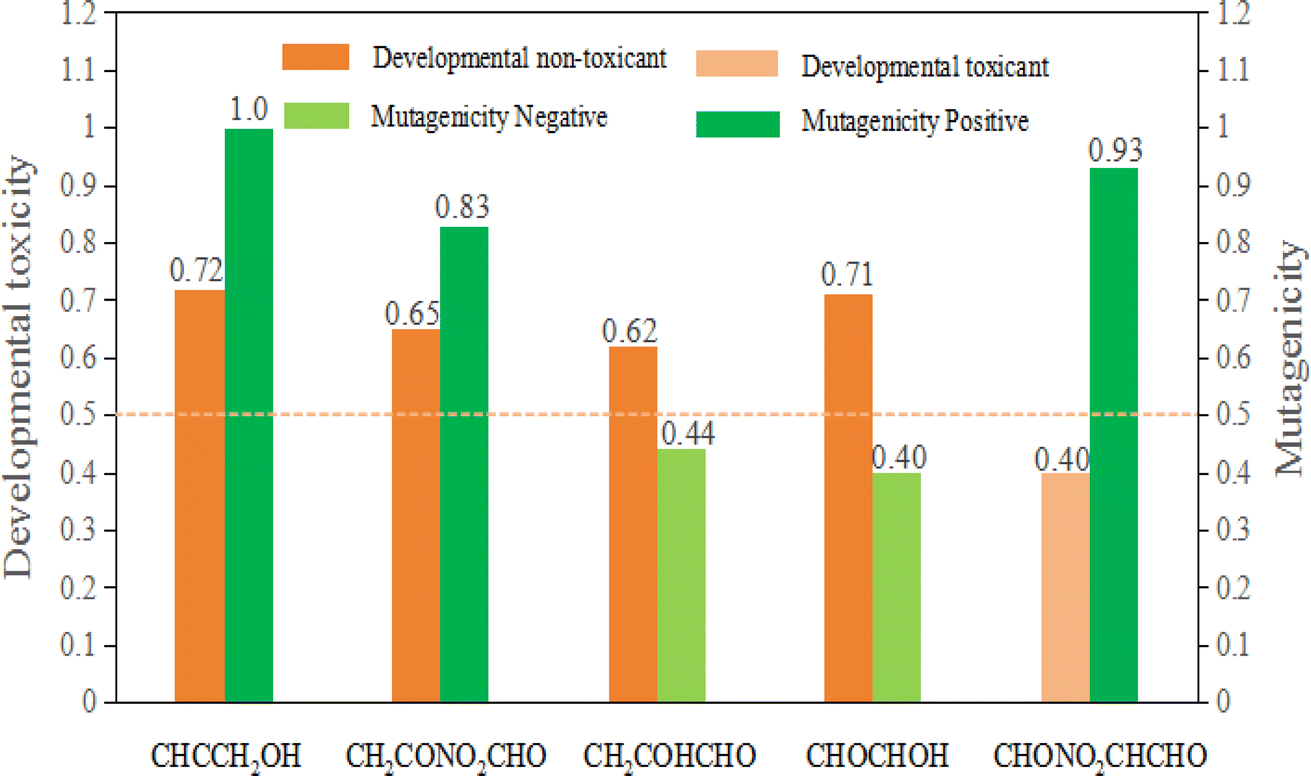
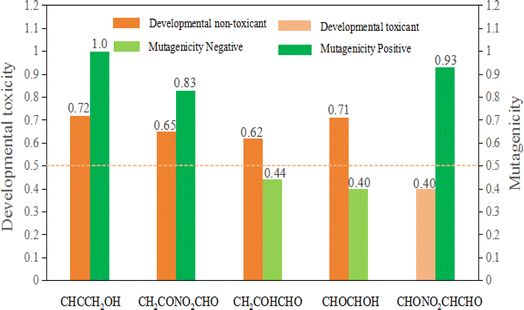
Apart from researching the toxicity of aquatic organisms and rats of propargyl alcohol and its products of the degradation in water, the evaluation results of developmental toxicity, mutagenicity, and bioaccumulation were forecasted taking advantaging of QSAR method (see Fig. 10). The value of developmental toxicity for propargyl alcohol (CHCCH2OH) is 0.72, which is higher than 0.50. Therefore, CHONO2CHCHO is developmental toxicant. Similarly, the value of its degradation products in water CH2CONO2CHO, CH2COHCHO and CHOCHCHOH are 0.65, 0.62 and 0.71, and also are developmental toxicants. While the value of degradation products CHONO2CHCHO is 0.40, which is less than 0.5. Thus, CHONO2CHCHO is developmental non-toxicant. Meanwhile, CHCCH2OH, CH2CONO2CHO, CHONO2CHCHO are Ames-positive compound (Ames mutagenicity > 0.50: mutagenicity positive). CH2COHCHO and CHOCHCHOH are Ames-negative compound (Ames mutagenicity < 0.50: mutagenicity negative). Thus, except CH2COHCHO and CHOCHCHOH, the degradation products of propargyl alcohol are harmful to the genes of organisms.
 |
| | Fig. 10 Oral rate LD50 for CHCCH2OH and its products of the degradation in water. | |
4. Conclusion
The potential energy surface (PES) and mechanism of the NO3 with CHCCH2OH reaction has been researched at the CCSD(T)/cc-pVTZ//B3LYP/6-311++G(d,p) level. The rate constants containing the most significant product branches starting from NO3 + CHCCH2OH have been gained at 200–2000 K using RRKM-TST theory. The useable experimental dynamic data could be quantitatively reproduced through computations. Addition/elimination and H-abstraction mechanisms have been discovered in the title reaction. IM1 (CHCONO2CH2OH) are the primary products below 500 K. A significant portion of the NO3 with CHCCH2OH reaction produces CHCCHOH and H2O at T > 500 K. The total rate constants reveal positive temperature dependence at 200–2000 K. Moreover, the calculated rate constants are consistent with the experimental value. The lifetime of CHCCH2OH is estimated to be 58.35 days at 298 K. Finally, the acute and chronic toxicity of CHCCH2OH during the degradation process was increased to fish, and decreased to daphnia and green algae based on the QSAR model.
Data availability
The data that support the findings of this study are available on request from the corresponding author.
Author contributions
The conception and design of calculation: Yunju Zhang, Jikang Gao and Meilian Zhao. The execution and analysis of calculation: Yaru Wang and Junchao Liao. Paper writing: Yunju Zhang, Meilian Zhao and Jikang Gao. All authors have reviewed the manuscript.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the Natural Science Foundations of China (No. 21707062), Scientific Research Starting Foundation of Mianyang Normal University (No. QD2016A007). Sichuan Province Science and Technology Project Support Program (No. 2023NSFSC1119).
References
- J. H. Zhu, D. S. Liu, C. S. Li, B. J. Zhang, J. L. Wang, W. J. Wu, J. W. Ji and Y. Q. Ma, RSC Adv., 2021, 11, 35408–35414 RSC.
- Y. W. Feng, L. L. Ling, J. H. Nie, K. Han, X. Y. Chen, Z. F. Bian, H. X. Li and Z. L. Wang, ACS Nano, 2017, 11, 12411–12418 CrossRef CAS PubMed.
- O. Faroon, J. Taylor, N. Roney, M. E. Fransen, S. Bogaczyk and G. Diamond, Toxicological Profile for Carbon Tetrachloride, Department of Health and Human Services, Public Health Service Agency for Toxic Substances and Disease Registry, Atlanta, GA, USA, 2005 Search PubMed.
- C. Walgraeve, Measuring volatile organic compounds and oxygenated polycyclic aromatic hydrocarbons in air: method development and application, Ghent University, 2012 Search PubMed.
- S. C. Lu, Q. L. Liu, R. Han, M. Guo, J. Q. Shi, C. F. Song, N. Ji, X. B. Lu and D. G. Ma, J. Environ. Sci., 2021, 105, 184–203 CrossRef CAS PubMed.
- Q. Liu, Y. Gao, W. W Huang, Z. H. Ling, Z. Wang and X. M. Wang, Atmos. Res., 2022, 274, 106184 CrossRef CAS.
- P. Nagaraju, S. H. Puttaiah and K. Wantala, et al., Appl. Water Sci., 2020, 10, 1–15 CrossRef.
- J. F. Sun, Q. Mu, H. Kimura, V. Murugadoss, M. X. He, W. Du and C. X. Hou, Adv. Compos. Hybrid Mater., 2022, 5, 627–640 CrossRef.
- S. Archer-Nicholls, R. Allen, N. L. Abraham, P. T. Griffiths and A. T. Archibald, Atmos. Chem. Phys., 2023, 23, 5801–5813 CrossRef CAS.
- C. H. Sun, L. Q. Lv and S. W. Zhang, Theor. Chem. Acc., 2016, 135, 1–9 Search PubMed.
- T. L. Nguyen, J. Park, K. Lee, K. Song and J. R. Barker, J. Phys. Chem. A, 2011, 115, 4894–4901 CrossRef CAS PubMed.
- I. Colmenar, S. Salgado, P. Martín, I. Aranda, A. Tapia and B. Cabañas, Atmos. Environ., 2020, 224, 117367 CrossRef CAS.
- I. Aranda, S. Salgado, P. Martin, F. Villanueva, E. Martinez and B. Cabanas, Chemosphere, 2021, 281, 130755–130764 CrossRef CAS PubMed.
- Y. Feng, K. S. Siow, W. K. Teo and A. K. Hsieh, Corros. Sci., 1999, 41, 829–852 CrossRef CAS.
- E. Duwell, J. Electrochem. Soc., 1962, 109, 1013–1017 CrossRef CAS.
- B. B. Pati, P. Chatterjee, T. B. Singh and D. D. N. Singh, Corrosion, 1990, 46, 354–359 CrossRef CAS.
- Y. S. Lee, S. A. Kucharski and R. J. Bartlett, J. Chem. Phys., 1984, 81, 5906–5912 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 14, 294–301 CrossRef.
- B. Feng, C. H. Sun, W. W. Zhao and S. W. Zhang, Environ. Sci.: Processes Impacts, 2020, 22, 1554–1565 RSC.
- J. T. Ye, F. Y. Bai and X. M. Pan, Environ. Sci. Pollut. Res., 2016, 23, 23467–23484 CrossRef CAS PubMed.
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027–2094 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, P. W. M. Gill, B. G. Johnson, M. A. Robb, J. R. Cheeseman, T. A. Keith, G. A. Petersson, J. A. Pople, et al., Gaussian 09, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- K. A. Holbrook, M. J. Pilling and S. H. Robertson, Unimolecular Reactions, J. Wiley, Chichester, UK, 1996 Search PubMed.
- ECOSAR v2.0, https://www.epa.gov/oppt/newchems/tools/21ecosar.htm Search PubMed.
- F. Y. Bai, S. Ni, Y. Z. Tang, X. M. Pan and Z. Zhao, Phys. Chem. Chem. Phys., 2019, 21, 17378–17392 RSC.
- F. Y. Bai, S. Ni, Y. Ren, Y. Z. Tang, Z. Zhao and X. M. Pan, J. Hazard. Mater., 2020, 386, 121636 CrossRef CAS PubMed.
- R. Atkinson, J. Phys. Chem. Ref. Data, 1991, 20, 459–507 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a