
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGreen synthesis, in silico modeling, and biological evaluation of N-substituted (Z)-5-arylidene imidazolidine/thiazolidine-2,4-dione/4-thione derivatives catalyzed by Bu SO3H core–shell nanostructures†
Malihe Akhavan a,
Zohreh Esamab,
Atefeh Mirshafac,
Maryam Lotfid,
Saeed Pourmande,
Froug Ashoria,
Motahare Rabania,
Golbahar Ekbatania,
Saeed Tourania,
Reza Beheshtia,
Zahra Keshavarziana,
Zahra Ghanbarimasirf and
Ahmadreza Bekhradnia*a
a,
Zohreh Esamab,
Atefeh Mirshafac,
Maryam Lotfid,
Saeed Pourmande,
Froug Ashoria,
Motahare Rabania,
Golbahar Ekbatania,
Saeed Tourania,
Reza Beheshtia,
Zahra Keshavarziana,
Zahra Ghanbarimasirf and
Ahmadreza Bekhradnia*a
aPharmaceutical Sciences Research Center, Department of Medicinal Chemistry, Mazandaran University of Medical Sciences, Sari, Iran. E-mail: abekhradnia@mazums.ac.ir; abekhradnia@gmail.com
bDepartment of Medicinal Chemistry, School of Pharmacy, Babol University of Medical Sciences, Babol, Iran
cRamsar Campus, Mazandaran University of Medical Sciences, Ramsar, Iran
dDepartment of Chemistry, NOVA, School of Science and Technology, Universidade NOVA de Lisboa, Portugal
eDepartment of Chemical Engineering, Tabriz University of Chemical Engineering, Tabriz, Iran
fDepartment of Organic Chemistry, Faculty of Chemistry, University of Mazandaran, Babolsar, Iran
First published on 19th July 2024
Abstract
In this effort, the immobilization of guanidine–sulfonate on the surface of Fe3O4 MNPs (magnetic nanoparticles) as a novel acid nanocatalyst has been successfully reported for the synthesis of N-substituted (Z)-5-arylidene thiazolidine-2,4-dione and related cyclic derivatives, including rhodanine (RHD) and hydantoin (HYD) via a one-pot multiple-component reaction under green conditions. The products were characterized by SEM, TEM, TGA, EDS, BET techniques, VSM, and FTIR. The novel compounds synthesized using this methodology, designated as series La (1-9), Lb (1-8), and Lc (1-8), were subjected to anticancer screening against A549 and MCF7cell lines via MTT assays. Notably, several compounds (L1a, L2a, L3a, L1b, L2b, L3b, and L4b) exhibited potent antiproliferative activities, with observed IC50 values as low as 1.23 μM and 1.02 μM against MCF-7 cells, thereby outperforming the established anticancer drugs doxorubicin and cisplatin. Molecular docking and dynamics simulations revealed that ligands L1a, L2a, and L3a strongly interact with the protein target 3CD8, with L1a displaying significant hydrophobic and hydrogen bonding interactions and L2a engaging in unique pi–pi stacking with key residues. For protein 2WGJ, ligand L4b exhibited exceptional binding affinity, characterized by robust hydrogen bonding, hydrophobic interactions, and additional stabilizing mechanisms such as water bridges and pi interactions. Hence, N-substituted (Z)-5-arylidene thiazolidine-2,4-dione and its cyclic derivatives may serve as promising candidates for further exploration in the development of new multi-target cancer chemotherapy agents. These findings introduce promising anticancer agents and establish Fe3O4 MNPs as a versatile and environmentally sustainable catalytic platform in drug discovery.
1 Introduction
Adenocarcinoma human alveolar basal epithelial cells and the human breast cancer cell line exemplify the complexity and aggressiveness of epithelial carcinoma. These malignancies highlight a critical challenge in oncotherapy: the necessity for effective and specific treatments that can overcome drug resistance and minimize toxicity associated with conventional chemotherapy. Consequently, the development of novel therapeutic strategies is crucial, with particular focus on targeting receptor tyrosine kinases (RTKs), which play a central role in cancer cell proliferation , angiogenesis, and metastasis.1–7 TZDs and their cyclic analogs, including RHD and HYD, have come to the fore due to their structural diversity and consequent biological activity against diverse RTKs. Recognizing the adaptability of TZDs as a pharmacophore, research has steered toward fine-tuning these molecules to dock effectively within the ATP binding sites of key RTKs implicated in A549 and MCF7 carcinomas.8–10 These compounds, through their refined interaction with the ATP pocket, obstruct the kinase's phosphorylation activity, crucial for RTK signaling.On the other hand, nitrogen-based heteroaromatic ring quinoxaline, as a bioisostere of privileged functional scaffolds such as quinoline, benzimidazole, and benzothiophene, has been found to render a wide variety of biological and therapeutic applications.1,2 Numerous synthetic quinoxaline derivatives, particularly 2-amino quinoxaline-containing compounds, have revealed remarkable medicinal roles such as antiproliferative agents, especially via kinase-inhibitor activities.3,4 In recent years, there have been advancements in creating new and more effective medications with quinoxaline structures. Additionally, there have been significant developments in building protocols for these pharmacologically active scaffolds.11,12 A new class of chemotherapeutic agents, called quinazoline-based derivatives, includes quinoxaline bioisostere, which has anticancer properties. These compounds block different protein kinases and other molecular targets. The advent of targeted cancer therapies revolutionized clinical paradigms, beginning with the approval of imatinib (1a), a milestone event (Fig. 1a).
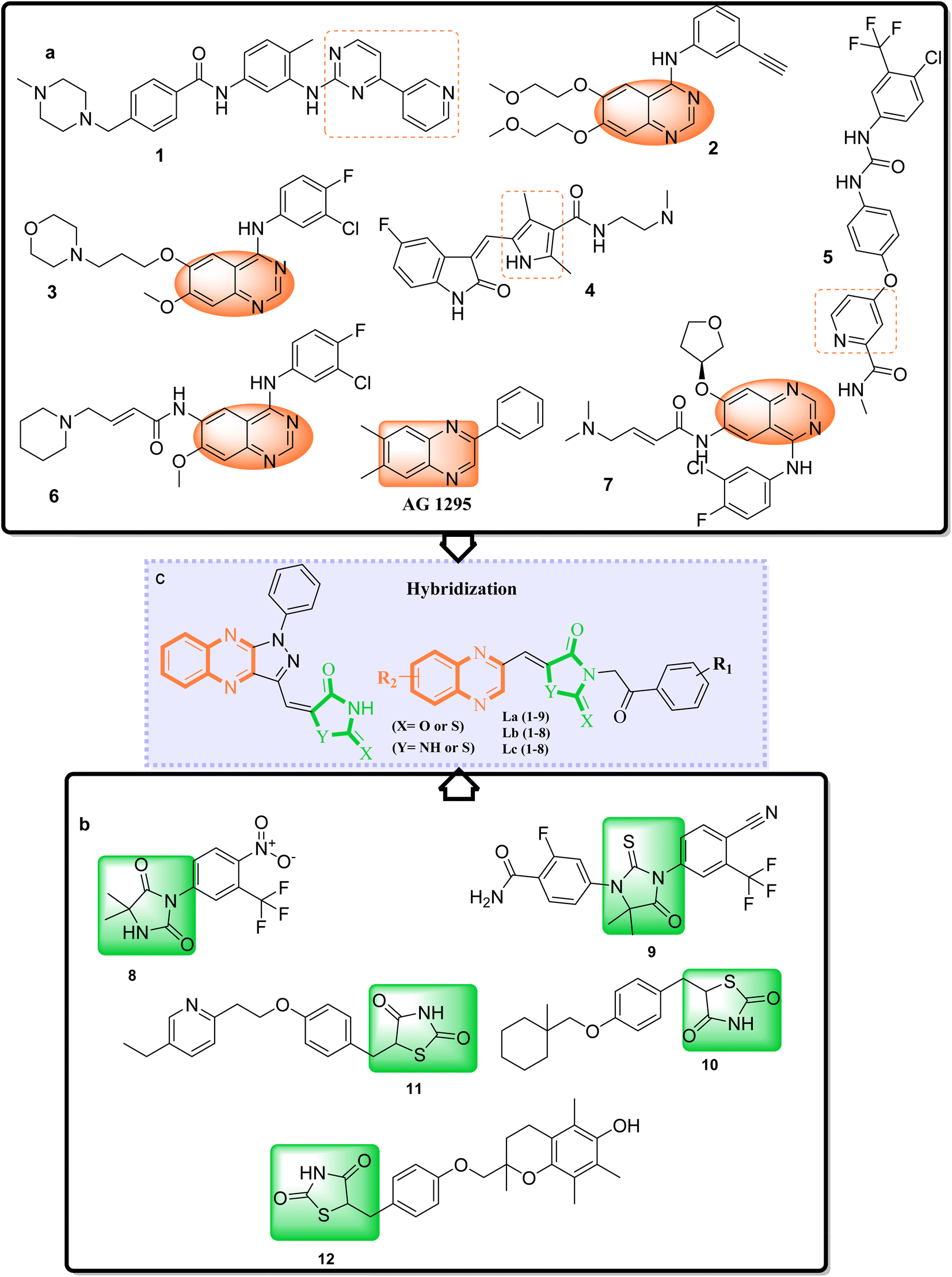 | ||
| Fig. 1 (a) Some prominent examples of small-molecule kinase inhibitors. (b) Nilutamide (8b) enzalutamide (9b), ciglitazone (10b), pioglitazone (11b) and troglitazone (12b) are drugs based on TZDs/RHD and HYD scaffolds. (c) Molecular hybridization strategy on the design, synthesis, of TZDs/RHD and HYD and quinoxaline derivatives as a c-Met RTK inhibitor. | ||
Erlotinib (2a) and gefitinib (3a), targeting the epidermal growth factor receptor (EGFR), significantly improved the management of non-small cell lung cancer (NSCLC) in patients with specific EGFR mutations. This success led to the development of multi-kinase inhibitors like sorafenib (4a) and sunitinib (5a), which are effective in treating renal cell carcinoma (RCC) and expanded kinase-centric therapeutic strategies. Afatinib (6a) and dacomitinib (7a) also target EGFR by occupying its ATP-binding site, similar to gefitinib. AG 1295, a quinoxaline derivative, selectively inhibits EGFR kinase and reverses transformed phenotypes in certain cancer models (Fig. 1a).8–14 TZD derivatives such as phenytoin are known anticonvulsants that modulate sodium channels to reduce neuronal hyperexcitability. In oncology, TZDs like nilutamide (8b) and enzalutamide (9b) serve as androgen receptor antagonists for prostate cancer, while ciglitazone (10b) attenuates VEGF production to impede angiogenesis. Additionally, TZDs like pioglitazone (11b) and troglitazone (12b) are established antidiabetic agents used in managing type 2 diabetes (Fig. 1b).15,16
A promising medicinal chemistry approach involves hybridizing TZDs and their cyclic analogs with quinoxaline derivatives. This combination leverages the anti-diabetic properties of TZDs, mediated through PPARγ activation, and the anti-tumor capabilities of quinoxalines. The resulting hybrid molecules offer dual modes of action, simultaneously modulating distinct cellular pathways for a comprehensive anticancer strategy. These TZD–quinoxaline hybrids target tyrosine kinases, crucial enzymes in cell division and survival pathways. Inhibiting these kinases can halt cancer cell proliferation, induce apoptosis, and impair angiogenesis, key processes in tumor development. By integrating the multifunctional effects of TZDs and the tyrosine kinase inhibitory properties of quinoxalines, these hybrids present a robust pharmacological profile capable of effectively undermining cancer cell viability.17–21
What adds to the importance of our research is describing a green and efficient synthesis route for preparing a leak-free and highly efficient multifunctional heterogeneous magnetic nanocatalyst via grafting sulfonic groups onto the surface of MNPs coated with a novel Schiff-base layer consisting of 3-chloropropyl trimethoxysilane (CPTMS) and guanidine–sulfonate as a productive bridge (Scheme 1). The core of these nanostructures is comprised of Fe3O4 MNPs, known for their ease of separation via an external magnetic field, thereby preventing the need for cumbersome filtration techniques and offering straightforward catalyst recovery. The Fe3O4 magnetic core is then modified with a layer of CPTMS, which serves as a linker to bridge the magnetic core with the functional outer shell. The terminal chloropropyl groups of CPTMS are adept at reacting with nucleophiles, facilitating the formation of a robust covalent bond with guanidine-based moieties. This functionalization endows the MNPs with a surface rich in guanidine groups, greatly enhancing their catalytic activity. The crowning layer of the shell is guanidine–BuSO3H, which bestows the nanostructures with strong acidity due to the sulfonic acid functional group. The synergy between the unique core–shell architecture and the guanidine–sulfonic acid functionalities maximizes the catalytic performance and extends the catalyst lifespan through the protection afforded by the shell. The stability of these core–shell nanostructures, coupled with their magnetic properties, allows for catalyst reuse with minimal activity loss over successive cycles. Then, we researched the usefulness of a newly created organic–inorganic hybrid (Fe3O4@CPTMS@guanidine–BuSO3H) as a core–shell-structured nanocatalyst for producing new quinoxaline-containing hybrids through a multi-step reaction. Based on the logic governing the structure design of these hybrid ligands, molecular docking and molecular dynamics simulation studies on c-Kit receptor tyrosine kinase were used to probe the mechanisms of their observed significant antiproliferative activities.
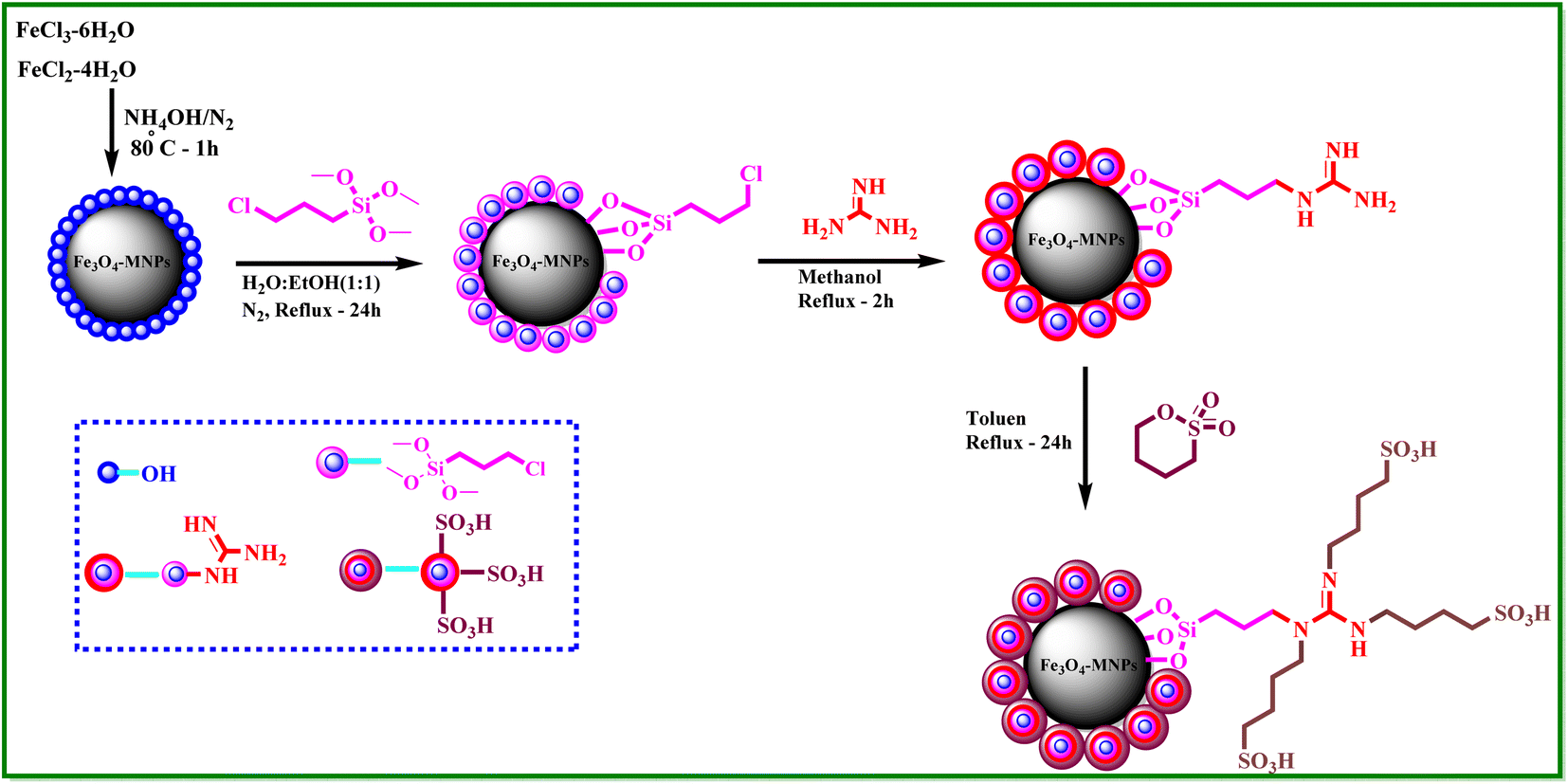 | ||
| Scheme 1 Synthesis steps of Fe3O4@CPTMS@guanidine–BuSO3H MNPs as a new heterogeneous catalyst. | ||
2 Experimental
2.1 Chemistry
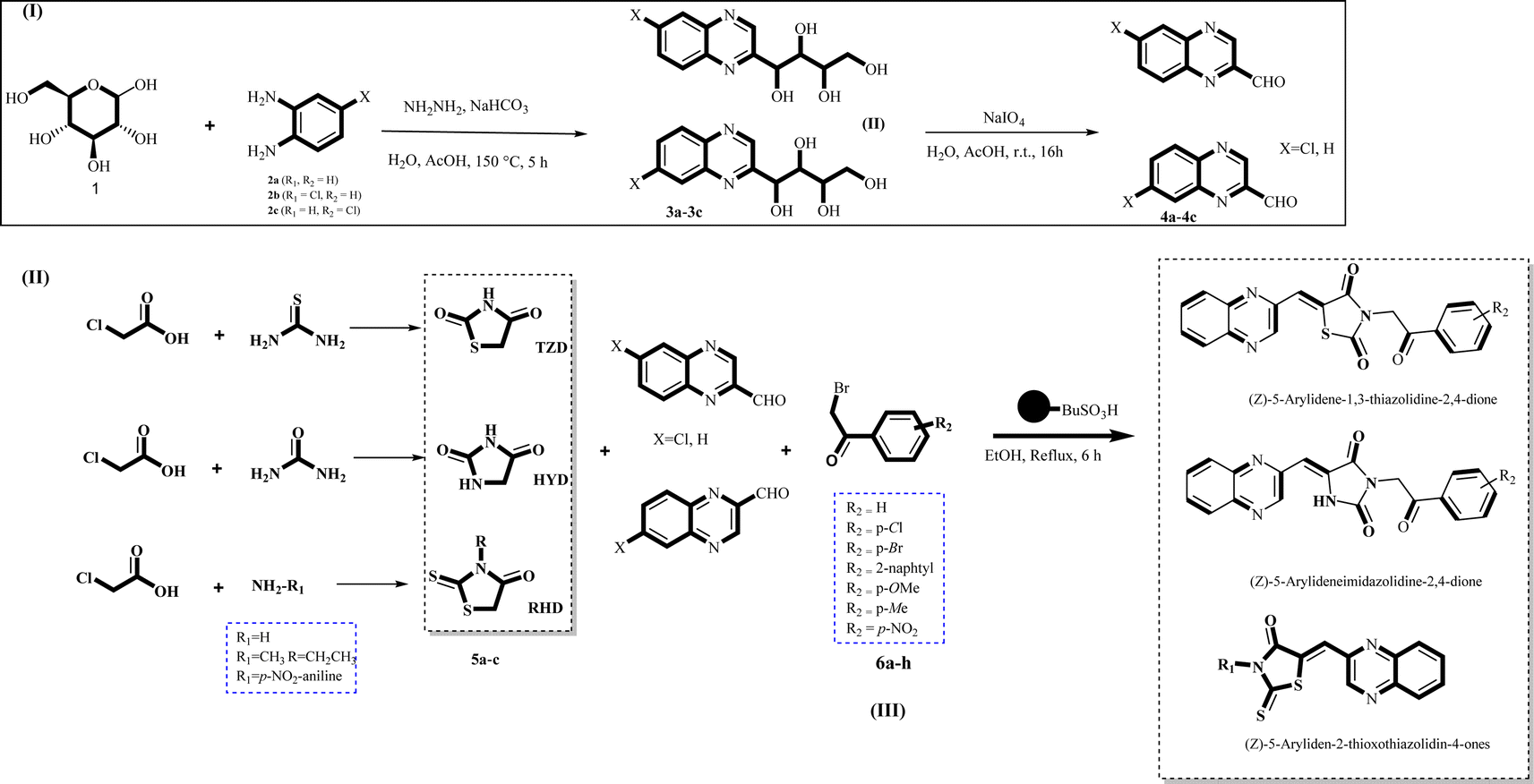 | ||
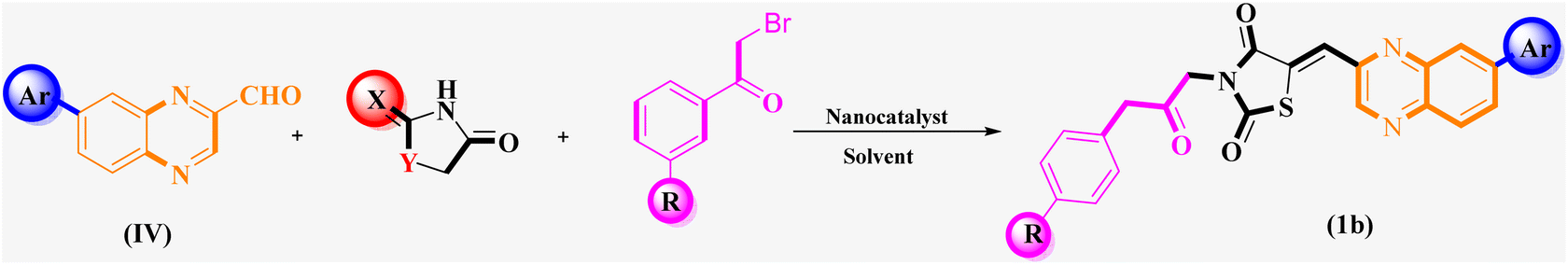
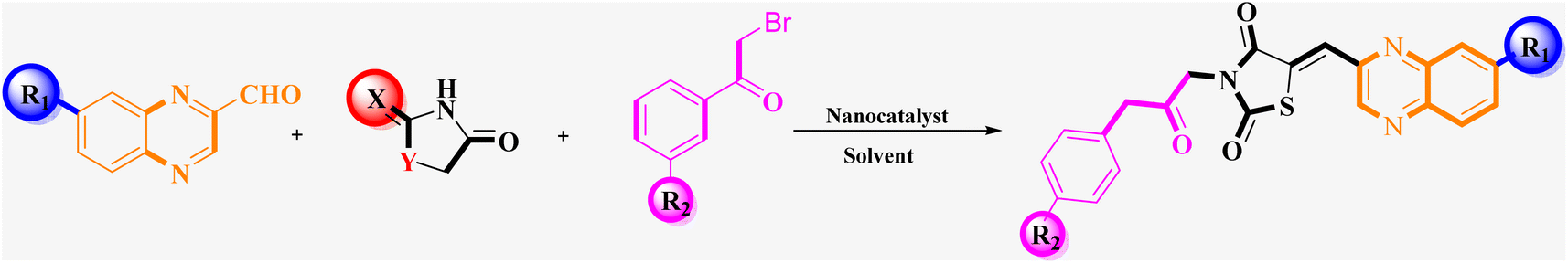
| Scheme 2 General synthesis pathway of the evaluated hybrid MCR product La (1-9), Lb (1-8), and Lc (1-8). The final products are derived from RHD/HYD and TZD as the core scaffold, attached to the quinoxaline-2-carboxaldehydes (4a–4c) and bromoacetophenone (6a–h). | ||
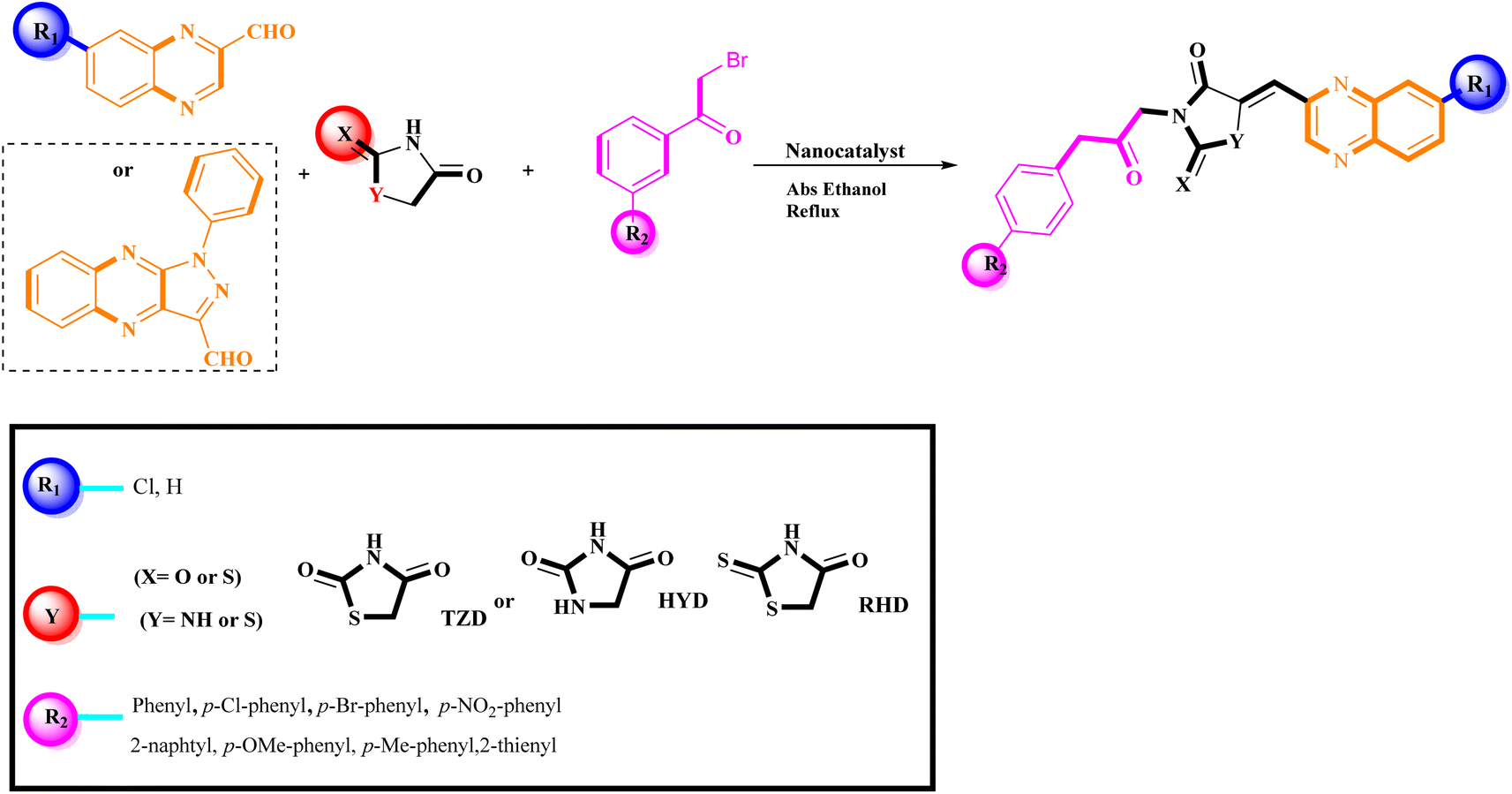 | ||
| Scheme 3 Synthesis of N-substituted (Z)-5-arylidene thiazolidine-2,4-dione and related cyclic derivatives by Fe3O4@CPTMS@guanidine–BuSO3H MNPs. | ||
2.2 Cytotoxicity MTT assay
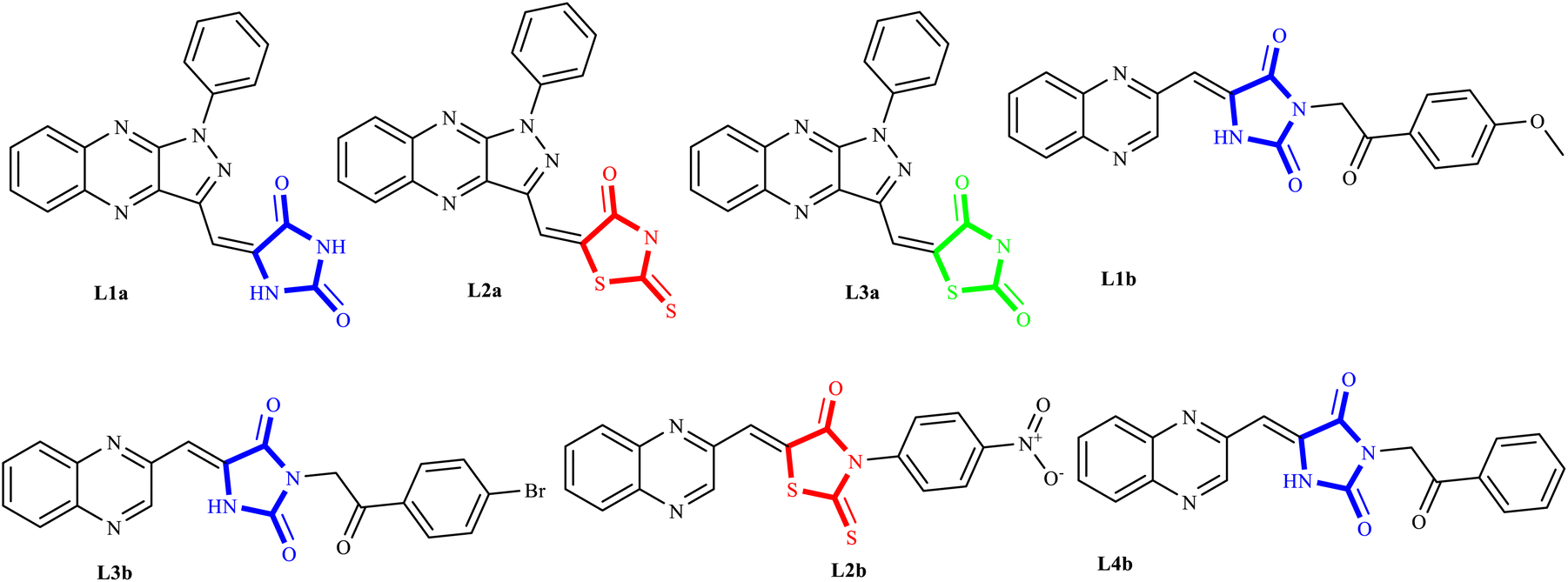
The investigation of the effects of multicomponent reaction (MCR) derivatives La (1-9), Lb (1-8), and Lc (1-8) on proliferation involved their application at concentrations ranging from 50 to 500 nM against two human cancer cell types, A549 and MCF7. This study employed a conventional MTT assay in a controlled in vitro setting.26,27 Doxorubicin and cisplatin served as the positive controls, whereas dimethyl sulfoxide (DMSO) was used as the negative control. The cancer cells were initially propagated in a growth medium supplemented with dialyzed fetal bovine serum (FBS), then suspended in Minimum Essential Medium (MEM) and subsequently seeded into a 96-well plate at a density of 10 × 104 viable cells per well. Following a 24 hour incubation period at 37 °C under a 5% CO2 atmosphere, which ensured proper cell adhesion, the compounds, now spanning concentrations of 50–500 nM, were introduced to the cell cultures and incubated for an additional 24/48 hours. Post incubation, the cells were rinsed with FBS. Thereafter, 20 μL of a 5 mg mL−1 solution of MTT was dispensed into each well, which was incubated for a further 4 hours under the aforementioned conditions. Subsequent to this step, the medium was evacuated, and around 70 μL of DMSO was added to dissolve the formazan product, a consequence of MTT reduction, thus yielding a colored solution. The plates were then subjected to thorough mixing to ensure the complete dissolution of the formed crystals. The absorbance for each well was recorded at 492 nm, indicative of the MTT formazan product, and 630 nm, serving as the reference wavelength, utilizing an ELISA reader. For experimental consistency, the stock solutions for all compounds were prepared in DMSO. Nonetheless, the final dilutions performed for the experiments were achieved using distilled water to confirm that the DMSO concentration did not exceed 0.5% in any well. The resultant data were analyzed, and the dose–response curves were generated using the Graph Pad Prism software as outlined in the "Result and Discussion" (3.6 section).272.3 Molecular docking
The X-ray crystallography structures of PF-02341066 (PDB ID: 2WGJ) and c-Met (PDB ID: 3CD8) was obtained from the Protein Data Bank (https://www.rcsb.org)28 as targets. Moreover, the .sdf format of 3D structure of ligands, Fig. 1, downloaded from the PubChem server29 and converted to .pdb format using Avogadro software and then optimized by the Gaussian 09W software using the B3LYP hybrid density functional theory at the 6-31G level.30 AutoDock 4.2.2 software was utilized for the molecular docking of studied ligands against targets. To create input files for proteins, water molecules, ligands, and ions were removed from .pdb files. Polar hydrogen atoms and Gasteiger charges were added to each protein. The binding sites were defined using a grid of size 22 Å × 22 Å × 22 Å and center [19 Å, 10 Å, 58 Å] for PDB: 3CD8. While for PDB: 2WGJ the grid box parameters were set to size 22 Å × 22 Å × 22 Å and centre [19 Å, 81 Å, 6 Å]. Molecular docking simulations were performed through virtual screening with AutoDock Vina, and the results, in terms of binding energy, were reported. Plip web server was applied in order to analyze and visualization docking results and intermolecular interactions between ligands and the target molecules.312.4 Molecular dynamic simulation
To assess the stability of both complexes, Molecular Dynamics (MD) simulations were conducted using GROMACS version 2020.32 The AMBER03 all-atom force field33 on the Ubuntu operating system (version 20) was employed for protonation and minimization steps. The SPC water model was selected to simulate docked complexes' molecular dynamics (MD) in explicit solvation. To neutralize the system, the proper amount of Na+ ions and Cl− atoms were introduced into the solution. Equilibration was performed under a pressure of 1 bar and a temperature of 300 K through two consecutive 100 ps simulations using canonical NVT ensembles and isobaric NPT ensembles, each lasting 1 ns. The particle mesh Ewald approximation was employed for the long-range electrostatic interaction, with a cut-off of 1 nm for computing Coulomb and van der Waals interactions.34 Subsequent to a 100 ns simulation run, coordinates were saved at every 2 fs time frame, and GROMACS tools were utilized for trajectory analysis.3 Results and discussion
3.1 Synthesis and characterization of the catalyst
A stoichiometric mixture of Fe(II) and Fe(III) salts was dissolved in deionized water under a nitrogen atmosphere. The dropwise addition of ammonium hydroxide then initiated Fe3O4 MNP precipitation.24 Upon synthesis, the Fe3O4 MNPs were functionalized with CPTMS to introduce reactive chloropropyl groups onto their surface.23 After this, a substitution reaction was engaged with N,N′-disubstituted guanidine for the chloropropyl-functionalized nanoparticles, and butane sulfonic acid was incorporated to yield guanidine sulfonate functionalities. Reaction conditions were meticulously controlled to ensure the complete replacement of chloropropyl groups and successful attachment of guanidine–BuSO3H moieties, culminating in the formation of Fe3O4@CPTMS@guanidine–BuSO3H MNPs. FT IR spectra, SEM, TEM, EDS, XRD, TGA, VSM, and BET confirmed the characterization data of the final product.The SEM images (Fig. 2) show particles with a homogeneous size and shape, averaging around 30 nm. This suggests a consistent synthesis process, where the particles exhibit uniform morphologies. The uniformity in size and shape is crucial for applications that require predictable and reproducible material properties. In contrast, the TEM analysis shown in Fig. 3 reveals uniformly sized, spherical particles with diameters ranging from 10 to 50 nm. The smaller average particle size observed in TEM compared to SEM suggests potential aggregation or slight morphological changes during SEM sample preparation. This could be due to the different preparation techniques and the interaction of the particles with the electron beam during SEM imaging. The aggregation or changes can lead to apparent size increases in SEM measurements. Nonetheless, TEM provides a more accurate estimation of particle size distribution and morphology due to its higher resolution and ability to visualize the internal structure of the particles. The uniform spherical morphology observed in TEM confirms the successful synthesis of nanoparticles with the desired characteristics. Furthermore, the size discrepancy between SEM and TEM highlights the importance of using complementary analytical techniques to obtain a holistic understanding of nanoparticle properties. By combining information from both SEM and TEM, we can better understand the synthesis process and the real morphology of particles.
 | ||
| Fig. 2 SEM image of Fe3O4@CPTMS@guanidine–BuSO3H MNPs. | ||
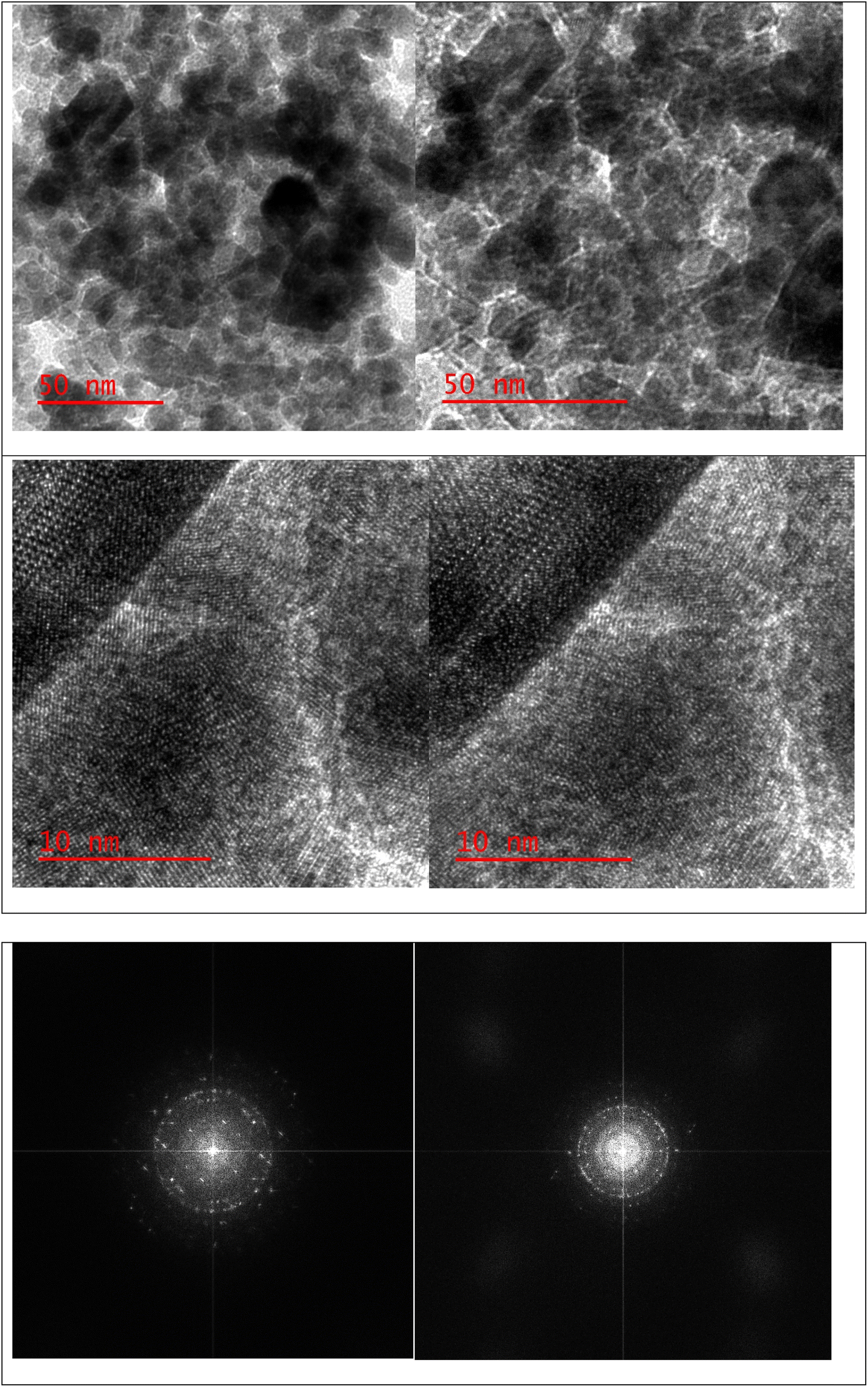 | ||
| Fig. 3 TEM image of Fe3O4@CPTMS@guanidine–BuSO3H MNPs. | ||
Fig. 4A represents the comparative FTIR spectra of (a) Fe3O4, (b) Fe3O4@CPTMS, (c) Fe3O4@CPTMS@guanidine, and (d) Fe3O4@CPTMS@guanidine–BuSO3H. The Fe3O4 and Fe3O4@CPTMS FT-IR results are consistent with the previous reports.22,23,35 Conjugation with guanidine is evidenced by the appearance of specific absorption bands at 1652 cm−1, 1427 cm−1, and 830 cm−1 in the FTIR spectrum. These bands correspond to the stretching vibrations of (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), (C–N), and bending vibrations of (–NH2) groups, thereby corroborating the successful modification of Fe3O4@CPTMS nanoparticles with guanidinium groups. Moreover, the FTIR spectrum exhibits a broad and prominent peak centered around 3462 cm−1, which can generally be attributed to O–H stretching vibrations at the presence of SO3H functional groups. These detailed observations in the FTIR spectra provide a clear insight into the chemical modifications and confirm the successful synthesis of Fe3O4@CPTMS@guanidine–BuSO3H nanoparticles.
N), (C–N), and bending vibrations of (–NH2) groups, thereby corroborating the successful modification of Fe3O4@CPTMS nanoparticles with guanidinium groups. Moreover, the FTIR spectrum exhibits a broad and prominent peak centered around 3462 cm−1, which can generally be attributed to O–H stretching vibrations at the presence of SO3H functional groups. These detailed observations in the FTIR spectra provide a clear insight into the chemical modifications and confirm the successful synthesis of Fe3O4@CPTMS@guanidine–BuSO3H nanoparticles.
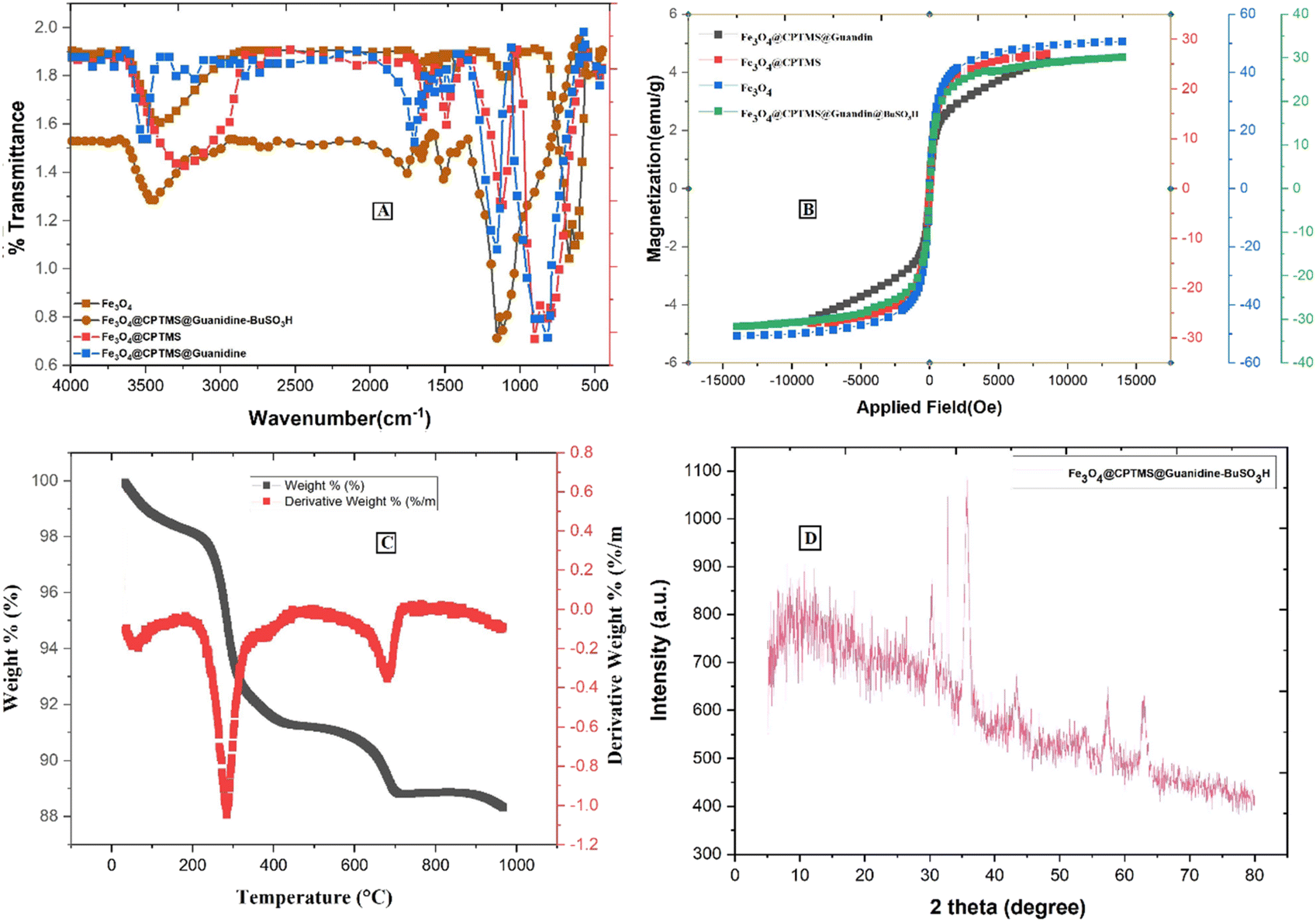 | ||
| Fig. 4 Spectra of Fe3O4@CPTMS@guanidine–BuSO3H MNPs: (A) IR spectrum, (B) VSM analysis, (C) TGA curve, and (D) XRD pattern. | ||
The magnetic properties of nanoparticles Fe3O4@CPTMS@guanidine–BuSO3H MNPs were investigated by vibrating a sample magnetometer (VSM) at room temperature. As shown in Fig. 4B, there is a discernible decrease in the saturation magnetization values among the synthesized nanoparticle variants, which are quantitatively evaluated to be 58 emu g−1 for uncoated Fe3O4 nanoparticles, 30 emu g−1 post-CPTMS functionalization, 28 emu g−1 following guanidinylation, and 25 emu g−1 after the final sulfonation step to yield Fe3O4@CPTMS@guanidine–BuSO3H MNPs. These measurements provide insights into the impact of sequential surface modifications on the magnetic properties, confirming that nanoparticles' magnetization decreases with the progressive addition of organic functional layers. This trend can be attributed to the non-magnetic nature of the added organic moieties and the resultant effective mass increase of the particles. It is also important to note that despite the decrease in saturation magnetization, the functionalized nanoparticles retain sufficient magnetic properties for potential applications. The properties are still adequate for applications requiring magnetic responsiveness, such as targeted drug delivery or as catalytic supports in magnetic separation processes.
Thermogravimetric analysis (TGA) was employed to assess the thermal stability and content integrity of Fe3O4@CPTMS@guanidine–BuSO3H up to 1000 °C (Fig. 4C). Initial weight loss observed from ambient to ∼200 °C is attributed to the desorption of water molecules indicative of the nanoparticles' hygroscopic nature and the guanidine and sulfonic acid groups' ability to hydrogen bond with water. Substantial weight reduction occurring between 200 °C and 550 °C confirms the decomposition of organic moieties grafted to the MNPs, with a recorded loss aligning with the expected 8.5% to 16.8% range, highlighting the decomposition of silane, guanidinium, and butane sulfonic acid structures. The grafting density of the coupled organic material is approximated around 0.70 mmol g−1.23
X-ray diffraction (XRD) analysis has been known as an analytical method for phase identification, phase purity, and unit cell dimensions of crystalline materials.23 XRD pattern reveals that Fe3O4 and Fe3O4@CPTMS@guanidine–BuSO3H MNPs were crystalline and consistent with our previous reports for Fe3O4 MNPs (Fig. 4D). This confirms that the synthesized Fe3O4 nanoparticles maintain their crystalline magnetite structure post-functionalization. For the Fe3O4@CPTMS@guanidine–BuSO3H MNPs, the XRD patterns exhibit similar diffraction peaks, indicating that the core crystalline structure of magnetite is preserved even after successive functionalization steps. Furthermore, utilizing the Scherrer equation with XRD pattern data, the size of the crystalline Fe3O4@CPTMS@guanidine–BuSO3H was estimated to be 30 nm.23,24 The nanoparticles' relatively uniform and small size distribution is beneficial for various applications, including drug delivery, catalysis, and magnetic resonance imaging. Furthermore, retaining this size post-functionalization indicates that the chemical modifications did not induce significant aggregation or alteration in particle size.
The EDX spectrum and elemental mapping analysis of the Fe3O4@CPTMS@guanidine–BuSO3H MNPs are shown in Fig. 5. The presence of different elements, including C, N, O, Si, S, and Fe was demonstrated in the Fe3O4@CPTMS@guanidine–BuSO3H MNPs. The maps show a homogeneous dispersion of C, N, O, Si, S, and Fe, indicating that the functional groups are uniformly distributed on the Fe3O4 nanoparticles. This uniform distribution is vital for the reproducibility and effectiveness of the functionalized MNPs in various applications such as catalysis, targeted drug delivery, and magnetic separation. These results corroborate the successful functionalization processes discussed in the previous sections.
 | ||
| Fig. 5 EDS and elemental mapping analysis of Fe3O4@CPTMS@guanidine–BuSO3H MNPs. | ||
In Fig. 6, the surface area and porosity characteristics of the compound Fe3O4@CPTMS@guanidine–BuSO3H are comprehensively analyzed using the Brunauer–Emmett–Teller (BET) method and the Barret–Joyner–Halenda (BJH) model. These analyses are pivotal to understanding the textural properties of the material, which are crucial for applications such as catalysis and adsorption. The BET analysis, performed with nitrogen gas adsorption–desorption at 77 K, indicates that the specific surface area of Fe3O4@CPTMS@guanidine–BuSO3H MNPs is approximately 82 m2 g−1. This substantial surface area suggests many accessible surface sites vital for catalytic activities. A high surface area typically increases active site availability, potentially enhancing the material's performance in various chemical processes. In addition to surface area, understanding the material's porosity is essential. The BJH analysis provided data on pore volume and pore size distribution. The analyses show a 0.2672 cm3 g−1 pore volume and an average pore radius of around 21.137 nm. The total pore volume indicates the material's capacity to hold or transport fluids and gases. A pore volume of 0.2672 cm3 g−1 shows that the material has significant void spaces, thereby effectively accommodating molecular diffusion and storage. An average pore radius of 21.137 nm classifies the material within the mesoporous range (2–50 nm). Mesoporous materials are particularly beneficial due to their balance between high surface areas and accessible pores, facilitating the diffusion of larger molecules more efficiently than microporous materials. This characteristic can greatly enhance catalytic efficacy and adsorption capacity by allowing more effortless movement of reactants and products. The combined results from the BET and BJH analyses signify that Fe3O4@CPTMS@guanidine–BuSO3H MNPs is a highly porous material with a substantial surface area, making it suitable for applications requiring high surface reactivity and efficient molecular transport. The mesoporous nature of the material indicates its potential to host various guest molecules, which is beneficial for heterogeneous catalysis, separation processes, and even drug delivery systems. The BET and BJH analyses provide critical insights into the surface and textural properties of Fe3O4@CPTMS@guanidine–BuSO3H MNPs. The material's high surface area and mesoporous structure underpin its potential in various scientific and industrial applications, making it an intriguing candidate for further research and development.
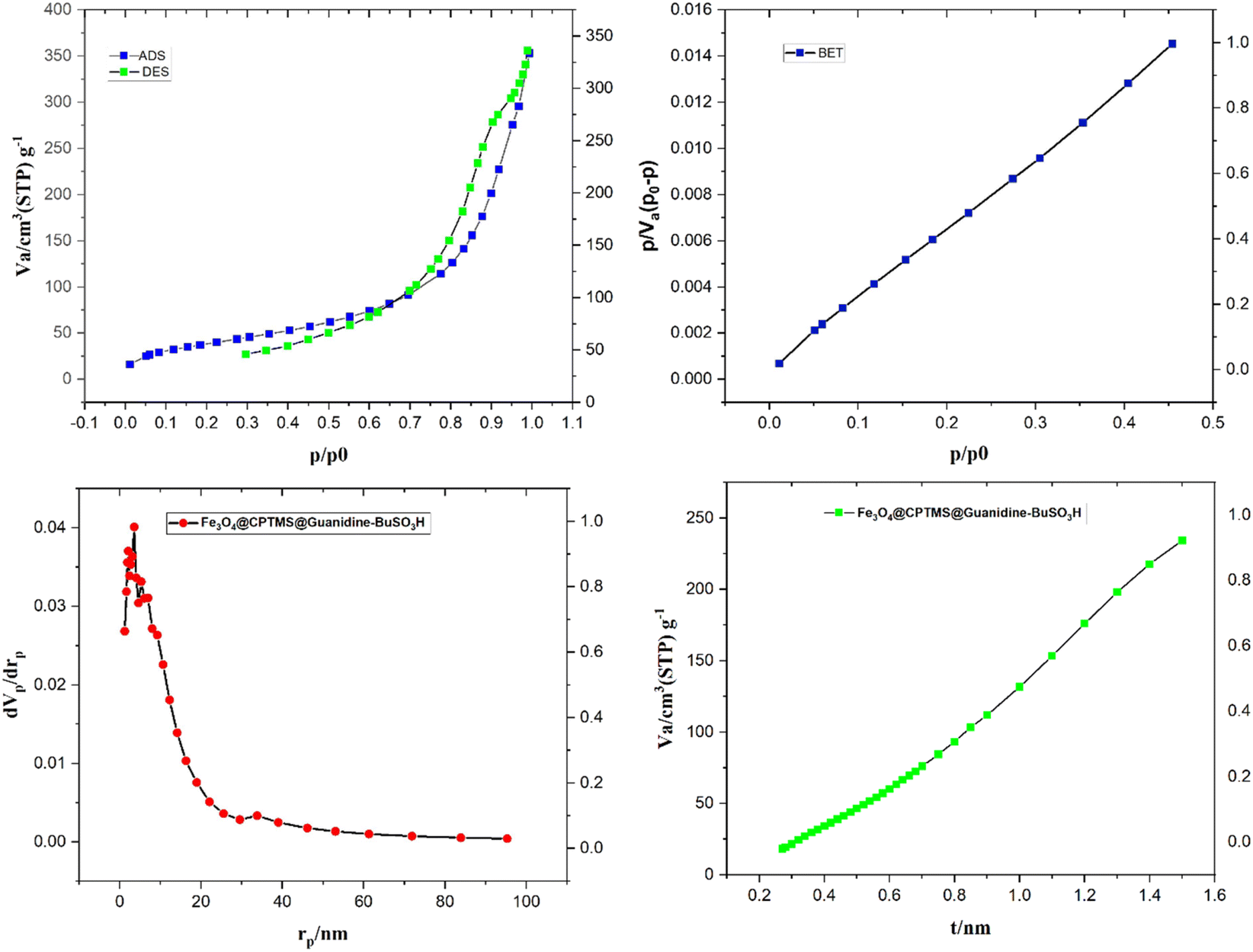 | ||
| Fig. 6 The nitrogen adsorption–desorption isotherm and pore size distribution of Fe3O4@CPTMS@guanidine–BuSO3H MNPs. | ||
In Fig. 7, the Raman spectra of the composite material Fe3O4@CPTMS@guanidine–BuSO3H MNPs are presented, highlighting the various contributions of its components. A detailed analysis of these spectra provides insights into the material's structural and chemical properties. Characteristic shifts attributable to the Fe3O4 core have been typically observed within the low wavenumber domain (<700 cm−1), indicative of metal–oxygen vibrational modes. Bands representing the symmetric stretching of tetrahedral (FeO4) and octahedral (FeO6) sites within the Fe3O4 spinel structure are expected to be discerned. Upon the incorporation of CPTMS, Raman active modes corresponding to organosilane entities, specifically those associated with Si–C and Si–O–Si stretching vibrations, have been identified. These distinct peaks are normally within the intermediate spectral range (∼800–1100 cm−1). When attention is shifted towards the guanidine functionalities grafted onto the structure, C–N single bond stretching and N–H bending modes have emerged within the Raman spectrum. The C–N stretching modes have typically appeared within the 1000–1400 cm−1 range, and the bending modes of NH groups within the 1500–1650 cm−1 window have been observed. Further complexity in the spectral features has been imparted by integrating BuSO3H groups onto the surface of the MNPs. A distinctive, strong SO symmetric stretching mode has been frequently identified at higher wavenumber regions (>1000 cm−1). Additionally, Raman bands ascribed to the C–H stretching and bending vibrations emanating from the butyl chain are usually located between 2800–3000 cm−1 and 1350–1450 cm−1, respectively. The Raman spectra in Fig. 7 provide a comprehensive understanding of the composite material's multicomponent structure. The distinct vibrational modes corresponding to the Fe3O4 core, CPTMS, guanidine functionalities, and BuSO3H groups indicate successful synthesis and functionalization.
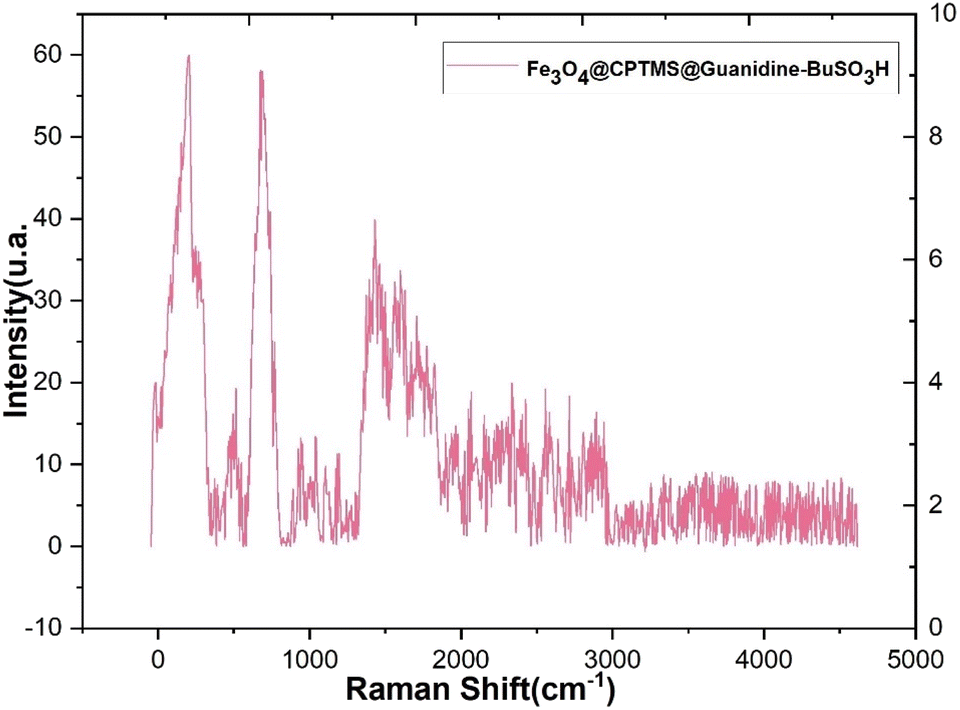 | ||
| Fig. 7 Raman spectra of Fe3O4@CPTMS@guanidine–BuSO3H MNPs. | ||
3.2 Evaluation of the catalytic activity of Fe3O4@CPTMS@guanidine–BuSO3H MNPs through the synthesis of the novel (Z)-5-arylidene imidazolidine/thiazolidine-2,4-dione/4-thione derivatives
The investigation into the efficiency of Fe3O4@CPTMS@guanidine–BuSO3H MNPs as a catalyst was carried out through a three-component assembly involving quinoxaline-2-carbaldehyde (IV), TZD, and 2-bromo-1-phenylethan-1-one, each at a concentration of 1 mmol, as outlined in model reaction one (Table 1). Optimization of the reaction conditions entailed exploring various catalyst amounts, temperatures, and solvents. Trials conducted without the nanocatalyst yielded suboptimal results. Experiments utilizing varying quantities of Fe3O4@CPTMS@guanidine–BuSO3H MNPs at 50 °C and 100 °C in absolute ethanol, a solvent recognized for its benign nature, demonstrated that yields could be enhanced from 32% to 96% by escalating catalyst load from 10 mg to 40 mg (Table 1, entries 3–6). Beyond a 40 mg threshold, an increase to 80 mg did not improve yields (Table 1, entry 7). Consistent with predictions, a reduction in temperature correlated with a decrease in reaction efficiency. Notably, the issue of generating undesired by-products was minimized when employing absolute ethanol compared to ethanol. These conditions were thus deemed optimal for the execution of such a three-component coupling reaction (Table 1, entry 6).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (g) | Solvent | Temperature (°C) | Time (h) | Yieldb (%) |
| a Reaction conditions: quinoxaline-2-carbaldehyde (IV) (1 mmol), TZD (1 mmol), and 2-bromo-1-phenylethan-1-one (1 mmol), and catalyst in the solvent.b Isolated yield after recrystallization.c Hard workup.d Unwanted by-product. | |||||
| 1 | — | Ethanol | 50, reflux | 48 h | — |
| 2 | Fe3O4 (50 mg) | Ethanol | 50, reflux | 24 h | — |
| 3 | MNPs (10 mg) | Ethanol | Reflux | 1 h | 32% |
| 4 | MNPs (20 mg) | Ethanol absolute | Reflux | 1 h | 64% |
| 5 | MNPs (30 mg) | Ethanol absolute | Reflux | 1 h | 78% |
| 6 | MNPs (40 mg) | Ethanol absolute | Reflux | 1 h | 96% |
| 7 | MNPs (80 mg) | Ethanol absolute | Reflux | 1 h | 95% |
| 8c,d | GAA (a few drops) | Ethanol | Reflux | Over night | Trace |
| 9 | Recovered MNPs | Ethanol absolute | Reflux | 1 h | 94 |
In the quest to enhance reaction parameters, the synthesis of compound 4b was achieved via the condensation of equimolar amounts of quinoxaline-2-carbaldehyde (IV), HYD, and 2-bromo-1-phenylethan-1-one. Optimization efforts are delineated in Table 2. Absolute ethanol emerged as the superior solvent choice due to its contributions toward high product yield, environmental compatibility, cost-effectiveness, and alignment with principles of green chemistry. It was observed that increments in the quantity of the catalyst did not proportionately influence the yield. The most favorable conditions for this single-step synthesis are detailed in Table 2, entry 6, which reports a 94% yield.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (g) | Solvent | Temperature (°C) | Time (h) | Yielda,b (%) |
| a Reaction conditions: quinoxaline-2-carbaldehyde (IV) (1 mmol), HYD (1 mmol), and 2-bromo-1-phenylethan-1-one (1 mmol).b Isolated yield after recrystallization.c Hard workup.d Unwanted by-product.e Fe3O4@CPTMS@guanidine–BuSO3H MNPs. | |||||
| 1d | — | Ethanol | Rt & reflux | 48 h | — |
| 2 | Nano-Fe3O4 (50 mg) | Ethanol | Rt & reflux | 48 h | — |
| 3 | MNPse (10 mg) | Ethanol | Reflux | 1 h | 28 |
| 4 | MNPs (10 mg) | Ethanol absolute | Reflux | 1 h | 42 |
| 5 | MNPs (30 mg) | Ethanol absolute | Reflux | 1 h | 55–67 |
| 6 | MNPs (40 mg) | Ethanol absolute | Reflux | 1 h | 94 |
| 7 | MNPs (40 mg) | Ethanol | Reflux | 1 h | 71 |
| 8 | MNPs (80 mg) | Ethanol absolute | Reflux | 1 h | 94 |
| 9c,d | GAA (a few drops) | Ethanol absolute | Reflux | Over night | Trace |
| 10 | Nanocatalyst recover | Ethanol absolute | Reflux | 1 h | 94 |
3.3 Comparison
The assessment of the acidic, heterogeneous, organic–inorganic nanocatalyst's performance, alongside the employed methodology, reveals that the Fe3O4@CPTMS@guanidine–BuSO3H MNPs enhance both the yield and the rate of reaction. This is achieved through straightforward reaction conditions, a simplified workup process, and a reduction in the formation of by-products, as detailed in Table 3. Data contained within the table illustrates that catalyzing the condensation of rhodanine with quinoxaline-2-carbaldehyde (the model reaction 1) with Fe3O4@CPTMS@guanidine–BuSO3H MNPs results in increased product yields, reduced reaction durations, and the imposition of less stringent reaction conditions.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Reaction condition | Time (h) | Yield and product | Ref. |
| 1 | Piperidine | HOAc | Microwave | 2 h | 50% compound | 18 |
| 2 | BiCl3 | Ethanol | Microwave | 8 h | 86% compound | 36 |
| 3 | Base | Ethanol | Reflux | 5 h | 13% compound | 37 |
| 4 | Alum | Ethanol | Microwave | 16 h | 55% compound | 38 and 39 |
| 5 | 2-HEAP | — | Heating | 8 h | 72% compound | 40 |
| 6 | Nanocatalyst (40 mg) | Ethanol | Reflux | 1 h | 90% compound | This work |
Utilizing the established optimum conditions, the corresponding MCRs were synthesized with the catalysis of Fe3O4@CPTMS@guanidine–BuSO3H MNPs. The reactions were conducted under reflux conditions for approximately one hour and achieved high yields ranging from 90% to 96%. Various substituted 2-bromoacetophenones, quinoxaline-2-carbaldehyde derivatives featuring a chlorine substituent, and (TZD), (RHD), and (HYD) were employed as substrates. Detailed outcomes of these syntheses are presented in Schemes 2, 3 and Table 4.
| Ligands | Chemical structure | Time, yield mp |
|---|---|---|
| L1a |  |
Time (min): 62, yield (93%) mp 118–120 °C |
| L2a | 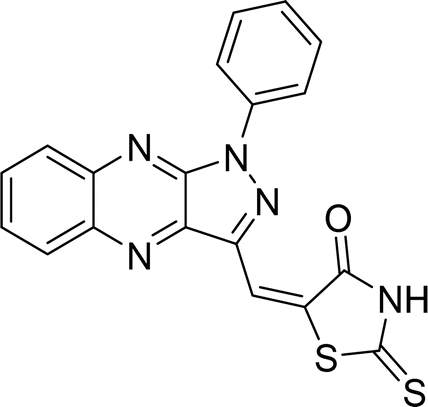 |
Time (min): 48, yield (96%) mp 127–129 °C |
| L3a | 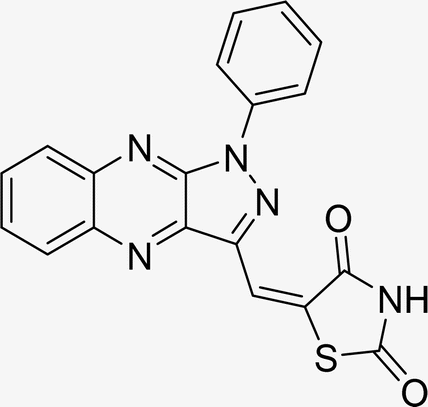 |
Time (min): 45, yield (90%) mp 128–130 °C |
| L4a | 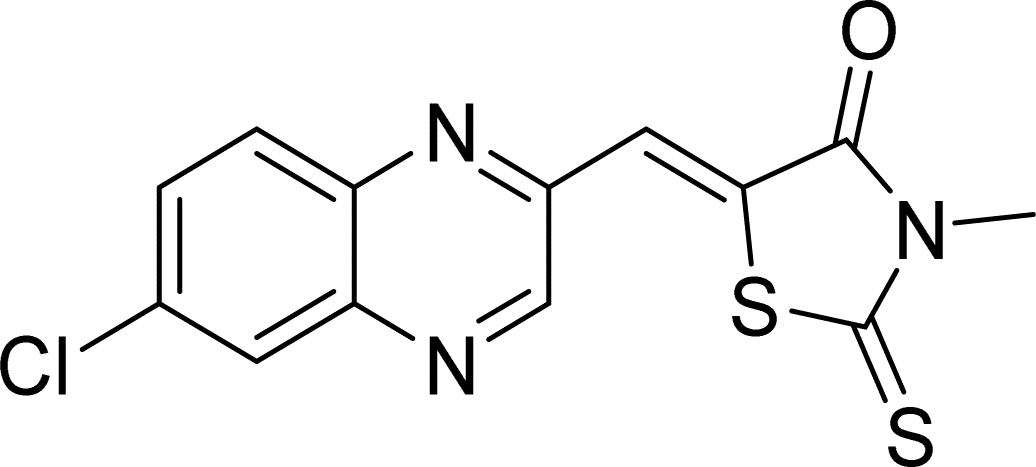 |
Time (min): 58, yield (90%) mp 171–173 °C |
| L5a | 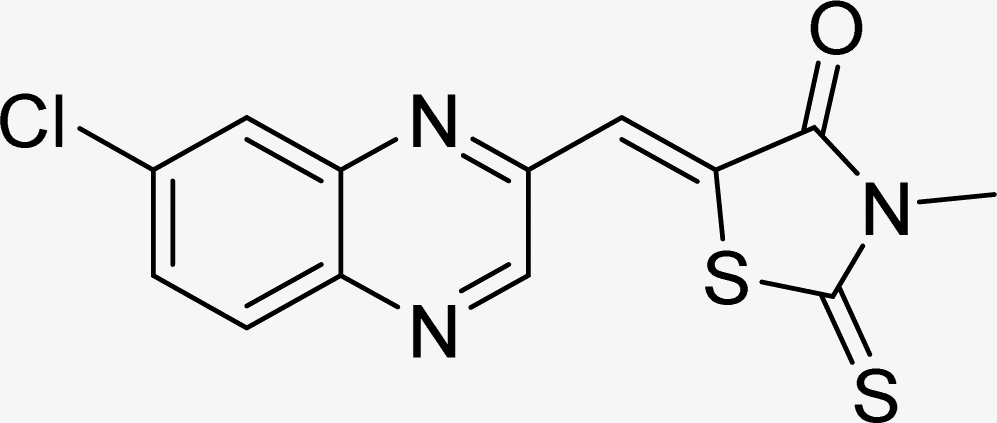 |
Time (min): 58, yield (94%) mp 286–289 °C |
| L6a | 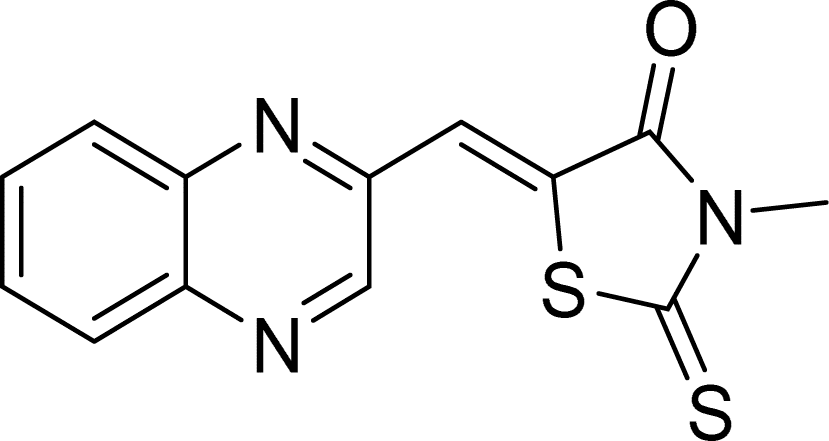 |
Time (min): 63, yield (95%) mp 175–177 °C |
| L7a | 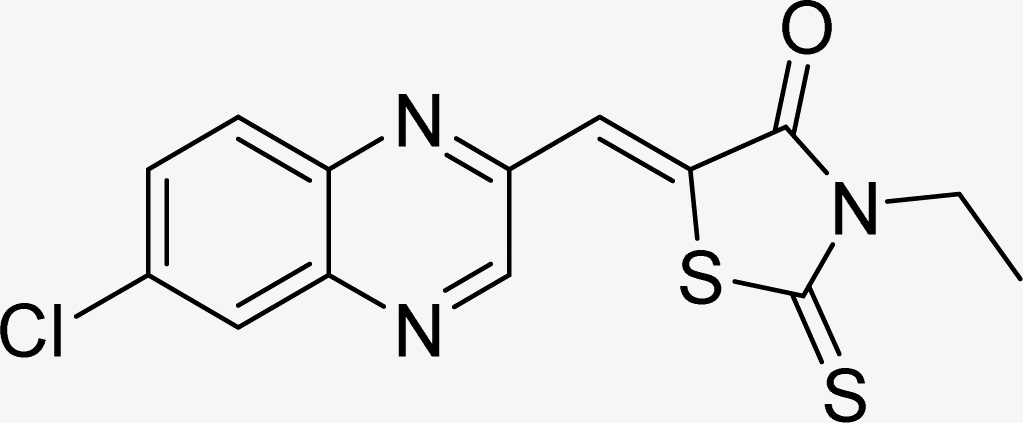 |
Time (min): 60, yield (90%) mp 165–168 °C |
| L8a | 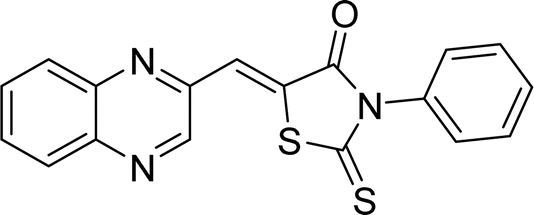 |
Time (min): 60, yield (94%) mp 132–137 °C |
| L9a |  |
Time (min): 53, yield (90%) mp 191–194 °C |
| L1b | 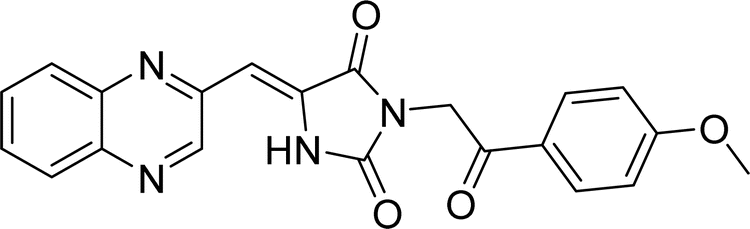 |
Time (min): 60, yield (90%) mp 212–215 °C |
| L2b | 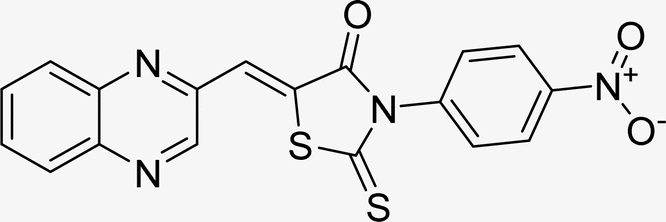 |
Time (min): 67, yield (96%) mp 176–179 °C |
| L3b | 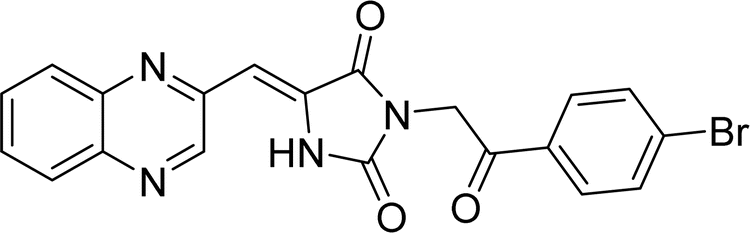 |
Time (min): 58, yield (90%) mp 208–110 °C |
| L4b | 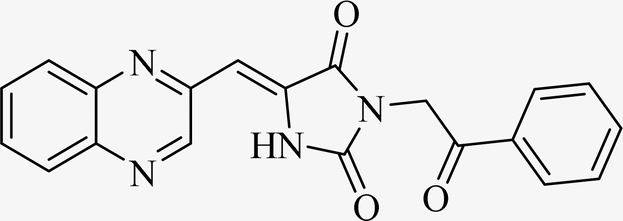 |
Time (min): 60, yield (94%) mp 149–152 °C |
| L5b | 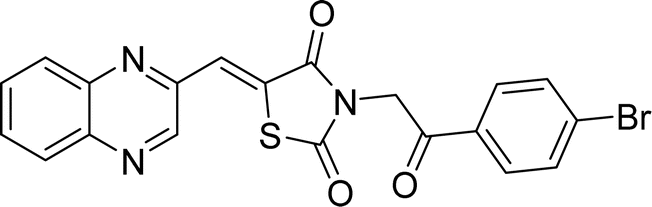 |
Time (min): 60, yield (90%) mp 148–150 °C |
| L6b | 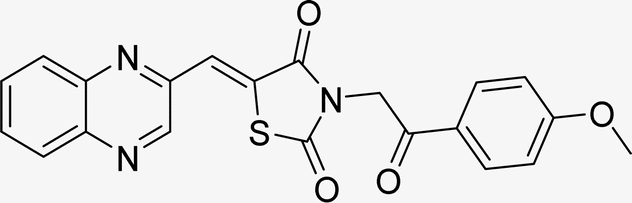 |
Time (min): 55, yield (90%) mp 160–162 °C |
| L7b |  |
Time (min): 69, yield (96%) mp 296–298 °C |
| L8b | 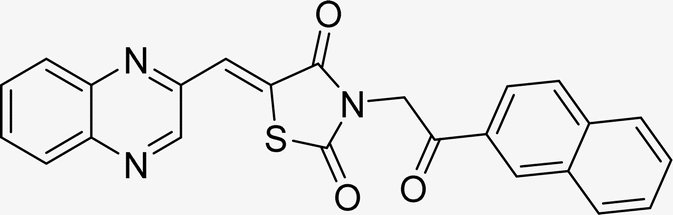 |
Time (min): 65, yield (90%) mp 168–170 °C |
| L1c | 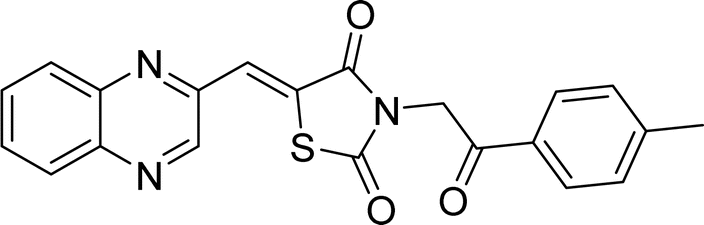 |
Time (min): 54, yield (89%) mp 267–269 °C |
| L2c | 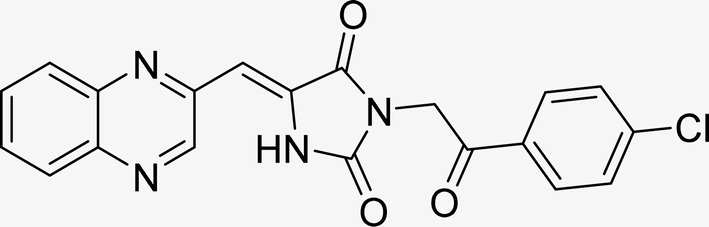 |
Time (min): 68, yield (90%) mp 138–140 °C |
| L3c | 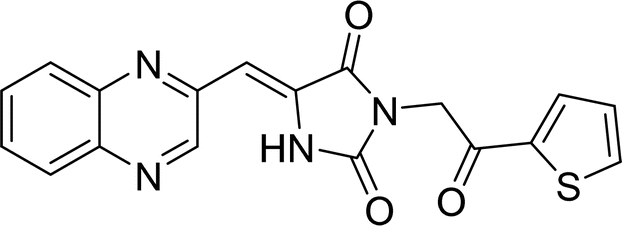 |
Time (min): 65, yield (93%) mp 138–140 °C |
| L4c | 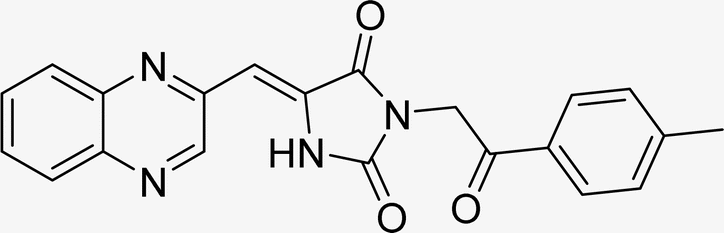 |
Time (min): 60, yield (90%) mp 218–220 °C |
| L5c | 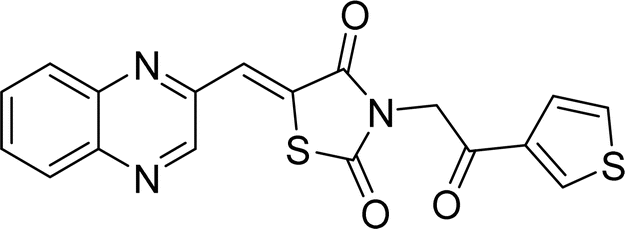 |
Time (min): 58, yield (90%) mp 163–166 °C |
| L6c | 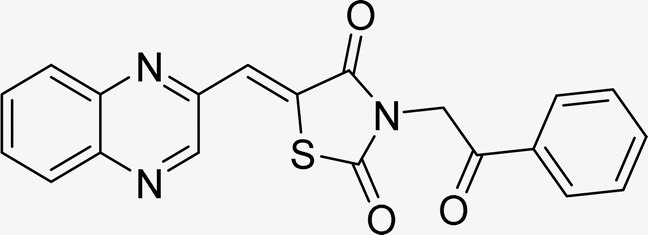 |
Time (min): 60, yield (96%) mp 178–182 °C |
| L7c |  |
Time (min): 68, yield (90%) mp 160–162 °C |
| L8c |  |
Time (min): 65, yield (90%) mp 198–202 °C |
3.4 Hot filtration test
To ascertain the heterogeneous character of the Fe3O4@CPTMS@guanidine–BuSO3H MNPs, a hot filtration test was performed during the synthesis of 6c using quinoxaline-2-carbaldehyde (IV), TZD, and 2-bromo-1-phenylethan-1-one as a representative reaction. The reaction proceeded under optimal conditions with the aforementioned MNPs (40 mg) up to the midpoint of the established time frame (30 minutes), achieving a product yield of 54%. Subsequently, the catalyst was removed from the reaction milieu via magnetic decantation, and the reaction was allowed to proceed for an additional 30 minutes without the catalyst. A marginal increase in product conversion (<2%) was observed, reinforcing the inference that the nanocatalyst operates in a truly heterogeneous manner. Furthermore, there was no detectable release of the Fe3O4@CPTMS@guanidine–BuSO3H MNPs into the reaction mixture throughout the process.233.5 Catalyst recyclability
In the realm of green chemistry, the ability to recycle and reuse catalysts is of paramount importance. The recycling potential of the Fe3O4@CPTMS@guanidine–BuSO3H MNPs was evaluated during the synthesis of compound 6c. After each reaction cycle, the magnetic catalyst was conveniently retrieved via an external magnet and subsequently conditioned for ensuing use. Demonstrating consistent and favorable catalytic performance, the magnetically recoverable nanoparticles maintained activity through five cycles, as depicted in Fig. 8. | ||
| Fig. 8 Reusability of the nanocatalyst for synthesis compound 1c. | ||
3.6 Colorimetric MTT cell viability assay: anti-proliferation effects on the novel (Z)-5-arylidene imidazolidine/thiazolidine-2,4-dione/4-thione derivatives vs. doxorubicin and cisplatin
The antiproliferative effects of the synthesized series of compounds, denoted La (1-9), Lb (1-8), and Lc (1-8), were systematically assessed in vitro against two human cancer cell lines: A549 (adenocarcinoma human alveolar basal epithelial cells) and MCF7 (human breast cancer cells). Each compound was treated using a gradient of concentrations (50, 100, 200, 300, 400, and 500 nM). As a part of the biological evaluation, doxorubicin and cisplatin acted as the established positive control standards. Cell viability was quantified following 24 and 48 hours of incubation utilizing the MTT colorimetric assay.25 After each incubation interval, the IC50 values, denoting the concentration at which cell viability is inhibited by 50%, were ascertained for each compound across the specified concentration range. These values and cell viability percentages were graphically represented, as depicted in Fig. 9. Such data are crucial in establishing the biological effectiveness and potential therapeutic value of the MCR derivatives. The presented findings are statistically expressed as the mean ± standard deviation (SD), based on a minimum of three independent experiments that yielded consistent results. Additionally, a control group using dimethyl sulfoxide (DMSO) at equivalent conditions (n = 2 ± standard error of the mean, SEM) accompanied each cell line assay to ensure validity. A standard curve was formulated on GraphPad Prism 8 software, with drug concentration plotted on the X-axis against the relative cell viability on the Y-axis, calculated as follows: cell viability (%) = (mean optical density (OD) of treated cells/control OD) × 100%.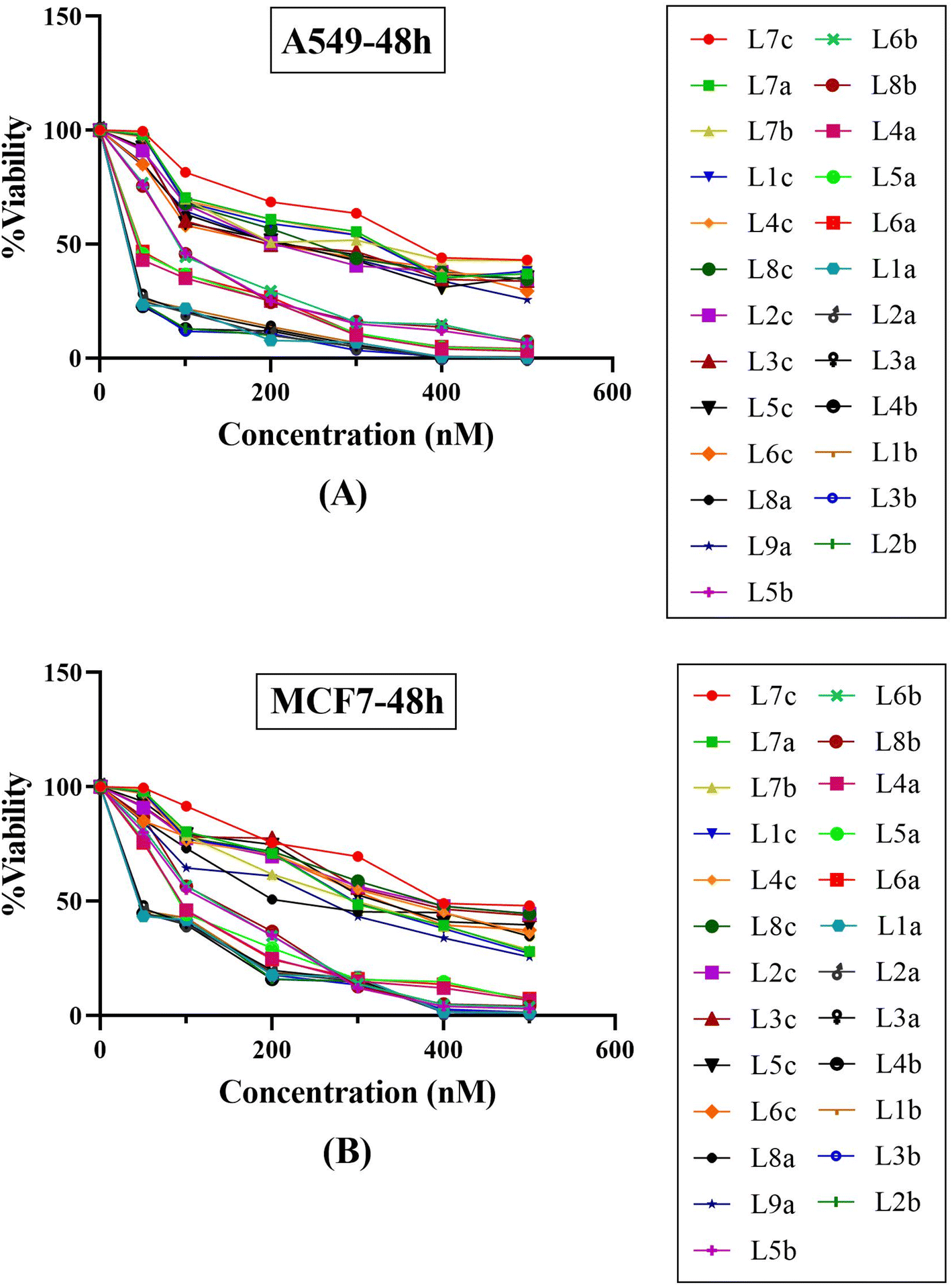 | ||
| Fig. 9 Compound concentration versus percentage cell viability plots for twenty-five synthetic final compounds against (A) adenocarcinoma human alveolar basal epithelial cells (A549) and (B) human breast cancer cell line (MCF7) after 48 hours incubation at various concentrations of 50, 100, 200, 300, 400 and 500 nM. | ||
The experimental findings presented in Fig. 9 and Table 5 demonstrate that the compounds under study exerted substantial cytotoxic effects in a manner dependent on both concentration and time against the tested cancer cell lines A549 and MCF7. These effects were significant when compared with the cytotoxic activity of established chemotherapeutic agents such as doxorubicin and cisplatin, which served as positive controls. Empirical observations revealed a spectrum of cytotoxic activities among the compounds, with IC50 values extending from 43.12 nM to 388.85 nM in A549 cells and from 67.07 nM to 448.28 nM in MCF7 cells, indicating variable potencies against these cell lines. Notably, compounds L1b, L2b, L3b, and L4b demonstrated potent activities in both cell lines, with IC50 values below 50 nM, highlighting their strong anticancer properties. A comparison between the two cell lines reveals that certain compounds demonstrate more pronounced cytotoxicity against one cell line over the other. For instance, compounds from the L5b–L8b series show lower IC50 values for the MCF7 cell line than the A549 cell line, suggesting a heightened sensitivity or potentially distinct biochemical interactions in the MCF7 cell line. It is particularly striking that all the tested compounds outperformed doxorubicin and cisplatin in lower IC50 values in the A549 cell line. In the case of MCF7, only three compounds have higher IC50 values than cisplatin, indicating differential cytotoxic profiles among cell lines and suggesting that these novel compounds could offer an advantage over traditional chemotherapy agents. Within the data set, a trend can be observed where the introduction of specific structural features may correlate with differences in potency. Indeed, the series L5b–L8b and L4a–L6a show more excellent inhibitory effects in both cell lines. This insight can be leveraged for further structure–activity relationship studies. The tested chemical entities exhibit substantial cytotoxic potential against A549 and MCF7 cell lines, indicating their ability to act as antineoplastic agents. These preliminary data lay the groundwork for successive phases of drug development, including further in vitro characterization, explorations of mechanistic pathways, in vivo efficacy studies, and studies on toxicological profiles. Specific focus should be given to the most potent compounds, namely those in the L1a–L4b range, which have showcased remarkable efficacy and may represent lead compounds for further optimization and development. In conclusion, these findings epitomize a significant stride in discovering viable chemotherapeutic alternatives and underscore the value of structurally diverse thiazolidinediones derivatives as potent inhibitors of cancer cell proliferation. Their promising anticancer activity warrants subsequent research to elucidate their molecular targets, modes of action, and potential applications in clinical oncology.
| Compound | IC50 value (nM) | |
|---|---|---|
| A549 cell line | MCF7 cell line | |
| L7c | 388.85 | 448.28 |
| L7a | 301.95 | 306.53 |
| L7b | 259.62 | 293.69 |
| L1c | 282.74 | 306.74 |
| L4c | 301.01 | 357.73 |
| L8c | 239.75 | 401.37 |
| L2c | 205.77 | 393.66 |
| L3c | 191.93 | 386.30 |
| L5c | 190.32 | 330.71 |
| L6c | 172.14 | 331.80 |
| L8a | 197.58 | 243.69 |
| L9a | 200.57 | 244.49 |
| L5b | 97.53 | 128.33 |
| L6b | 99.13 | 132.65 |
| L8b | 95.94 | 138.95 |
| L4a | 66.55 | 97.53 |
| L5a | 69.46 | 99.13 |
| L6a | 70.81 | 95.94 |
| L1a | 45.84 | 68.42 |
| L2a | 46.24 | 68.54 |
| L3a | 47.63 | 70.50 |
| L1b | 47.82 | 71.18 |
| L2b | 43.40 | 69.07 |
| L3b | 43.12 | 68.83 |
| L4b | 43.13 | 67.07 |
| Doxorubicin | 801 | 5120 |
| Cisplatin | 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 700 |
2230 |
3.7 Structure–activity relationship
Across both cell lines tested, compounds L4b, L2b, L3b, L1a, L2a, and L3a show the lowest IC50 values, indicating they are the most potent among the tested compounds. L1a has the lowest IC50 in both cell lines, suggesting significant antiproliferative activity. The chloroquinoxaline group at the 6-position and a 3-ethyl substituent on the thioxothiazolidinone ring may favor the activity. Comparing IC50 values of compounds with ethyl (L7a, L7c, L7b) versus methyl substituents (L4a, L5a, L6a) shows that, in general, ethyl-containing compounds possess better antiproliferative activity, suggesting that a slightly larger alkyl group may enhance potency. The presence of a halogen, particularly chlorine, on the quinoxaline ring (compare L7a, L7c with L7b; and L4a, L5a with L6a) generally improves the activity, which might be attributed to the electron-withdrawing nature of the halogens contributing to better interactions with the target. Compounds with a simple phenyl ring, such as L8a, and L2b, show moderate activity, but when this phenyl ring has additional substituents, such as NO2 in compound L2b, there's a notable increase in potency towards the MCF7 line compared to its counterpart without the nitro group (L8a). This suggests that certain para-substituents on the phenyl ring could positively affect inhibitory activity. When comparing compound pairs L3a/L1a, L8b/L8c, and L5b/L3b, where the only difference is the presence of thiazolidine versus imidazolidine, there isn't a consistent trend suggesting one ring system is superior to the other. Compounds tend to have a higher potency in the MCF7 cell line compared to the A549 cell line, as seen with compounds such as L5b, L6b, and L8b, which possess sub-100 nM potency against MCF7 cells but not A549 cells. Such selective cytotoxicity can be beneficial in reducing side effects during chemotherapy. Across both A549 and MCF7 cell lines, the compounds exhibit a broad spectrum of efficacies, with IC50 values ranging from 43.12 nM to 388.85 nM for the A549 cell line and 67.07 nM to 448.28 nM for the MCF7 cell line. Notably, compounds L1b, L2b, L3b, and L4b demonstrated potent activities in both cell lines, with IC50 values below 50 nM, highlighting their strong anticancer properties. A comparison between the two cell lines reveals that certain compounds demonstrate more pronounced cytotoxicity against one cell line over the other. For instance, compounds from the L5b–L8b series show lower IC50 values for the MCF7 cell line than the A549 cell line, suggesting a heightened sensitivity or potentially distinct biochemical interactions in the MCF7 cell line. It is particularly striking that all the tested compounds outperformed doxorubicin and cisplatin in lower IC50 values in the A549 cell line. In the case of MCF7, only three compounds have higher IC50 values than cisplatin, indicating differential cytotoxic profiles among cell lines and suggesting that these novel compounds could offer an advantage over traditional chemotherapy agents. Indeed, the series L5b–L8b and L4a–L6a show greater inhibitory effects in both cell lines. The molecular docking simulations provide a granular view of how ligands interact with proteins, which is crucial for predicting their biological activity. Our comparative analysis revealed that L1a might offer a higher protein inhibition due to its robust binding affinity (−9.9 kcal mol−1) to 3CD8. This is evidenced by its specific interactions with ARG1208 and unique hydrophobic contacts with ALA1108 and TYR1159. Such interactions likely contribute to a pharmacophore model that optimally positions key functionalities for target engagement. Conversely, L2a, while demonstrating a lower binding affinity, engages in a pi-stacking interaction with TYR1230, suggesting alternative interaction modes that could be vital when considering selectivity across different cancer cell types. This highlights the importance of pi interactions as potential pharmacophoric features for future optimization. Addressing ligand L4b's high affinity (−10.8 kcal mol−1) toward 2WGJ illustrates the significance of ASP1164 and ASN1167 in hydrogen bonding and the role of hydrophobic interactions in maintaining complex stability. Stereochemical influences, especially E and Z isomerism, are crucial due to their governing effects on the three-dimensional orientation of these molecules. This geometric effect directly impacts the capacity of the compounds to adapt to enzymatic active sites or protein pockets. Taking isomers L1a and L3a exhibit distinct activities, likely due to their stereo configuration around the double bond, signifying the relevance of conformational dynamics concerning biological activity. MD simulations reinforced the docking data by validating the stability of the ligand–protein complexes over time. The simulations depicted stable RMSD profiles for L1a–3CD8 and L4b–2WGJ complexes, suggesting a snug fit within the binding pocket. RMSF values shed light on residues that fluctuate significantly, often indicators of binding site flexibility or regions that could accommodate structural modifications without substantial activity loss.3.8 Docking results
Molecular docking plays a crucial role in silico drug design and discovery. It furnishes essential insights into the interaction between proteins and ligands by offering details such as binding affinity scores and generating the binding pose.31 The results of the molecular docking simulations offer a comprehensive understanding of the interactions between protein PDB: 3CD8 and the three ligands L1a, L2a, and L3a, Table 6. The comparative analysis of these ligands provides valuable insights into their respective binding characteristics and potential implications for drug design. The binding affinities of the ligands are a critical indicator of their interaction strength with protein 3CD8. L1a demonstrated the highest binding affinity with a score of −9.9, followed closely by L3a with a score of −9.8 and L2a with a slightly lower affinity of −8.8. This hierarchy in binding affinities suggests varying degrees of effectiveness in forming stable complexes with the protein. Fig. 10 shows the residues of 3CD8 protein that are interacting with the studied ligands. All three ligands engaged in hydrogen bond interactions with the protein. L1a and L3a formed hydrogen bonds with ARG1208, while L2a formed hydrogen bonds with ARG1086. All three ligands exhibited extensive hydrophobic contacts with common residues such as ILE1084, VAL1092, LEU1157, and TYR1230. However, L1a uniquely interacted with ALA1108 and TYR1159. These diverse hydrophobic interactions contribute to the ligands' distinct binding profiles. Interestingly, L2a exhibited a distinct pi-stacking interaction with TYR1230. Moreover, from our docking simulation studies, the binding affinities of the ligands indicate their strength in binding to protein PDB: 2WGJ, Table 7. Ligand L4b demonstrated the highest affinity with a score of −10.8, followed by L3b with −9.0, L2b with −8.9, and L1b with −8.8. This hierarchy in binding affinities suggests varying degrees of effectiveness in forming stable complexes with the protein. As shown in Fig. 11, ligands L1b, L2b, L3b, and L4b formed hydrogen bonds with specific residues. L1b formed hydrogen bonds with TYR1159 and ASN1171, L2b with ARG1166 and ASN1167, L3b with ASP1164 and ASN1167, and L4b with ASP1164 and ASN1167. Hydrophobic interactions are essential for stabilizing protein-ligand complexes. All four ligands exhibited significant hydrophobic contacts with common residues, including ILE1084, VAL1092, LEU1157, and TYR1230. Additionally, L1b interacted with ASN1167, L2b with ARG1208, and L4b with LEU1157, PHE1168, ALA1226. Ligands L2b and L3b formed water bridge interactions with TYR1230. All four ligands exhibited pi-stacking interactions with TYR1230. Additionally, L1b formed a pi-stacking interaction with ASN1167.
|
|||||
|---|---|---|---|---|---|
| Ligands | Docking score (kcal mol−1) | Hydrogen bonds | Hydrophobic interactions | Water bridge interactions | Pi stacking interactions |
| L1a | −9.9 | ARG1208 | ILE1084, VAL1092, ALA1108, LEU1157, TYR1159, TYT1230 | — | — |
| L2a | −8.8 | ARG1086 | ILE1084, VAL1092, LEU1157, TYR1230 | — | TYR1230 |
| L3a | −9.8 | ARG1208 | ILE1084, VAL1092, ALA1108, LEU1157, TYR1159, TYR1230 | — | — |
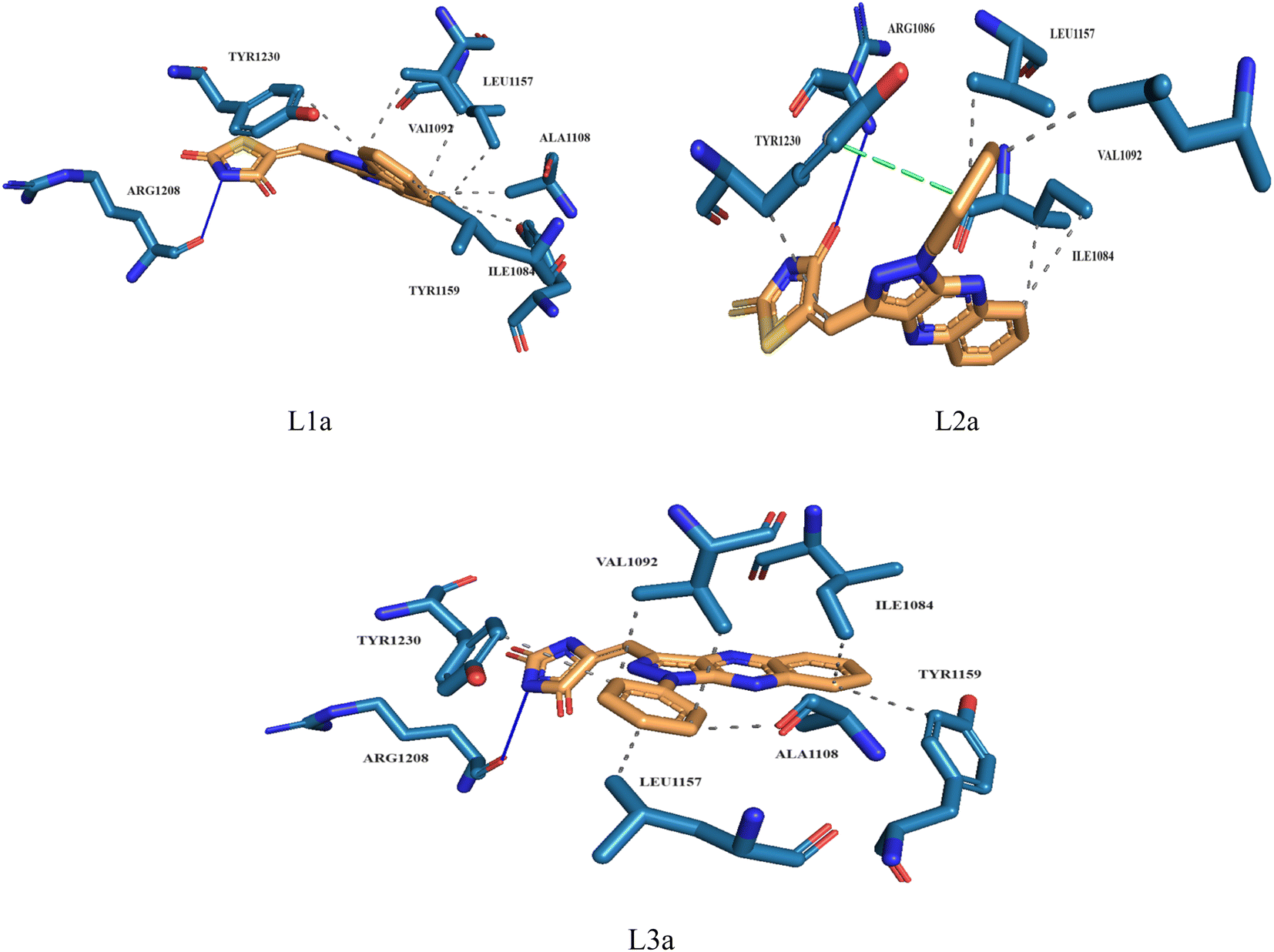 | ||
| Fig. 10 3D structure of ligands interactions with binding residues of 3CD8. | ||
| Ligands | Docking score (kcal mol−1) | Hydrogen bonds | Hydrophobic interactions | Water bridge interactions | Pi stacking interactions |
|---|---|---|---|---|---|
| L1b | −8.8 | TYR1159, ASN1171 | ILE1084, VAL1092, LEU1157, TYR1159, ASN1167 | ASN1167 | TYR1230 |
| L2b | −8.9 | ARG1166, ASN1167 | VAL1092, ARG1208 | — | TYR1230 |
| L3b | −9.0 | ASP1164, ASN1167 | PHE1168, TYR1230 | — | TYR1230 |
| L4b | −10.8 | ASP1164, ASN1167 | VAL1092, LEU1157, PHE1168, ALA1226, TYR1230 | — | TYR1230 |
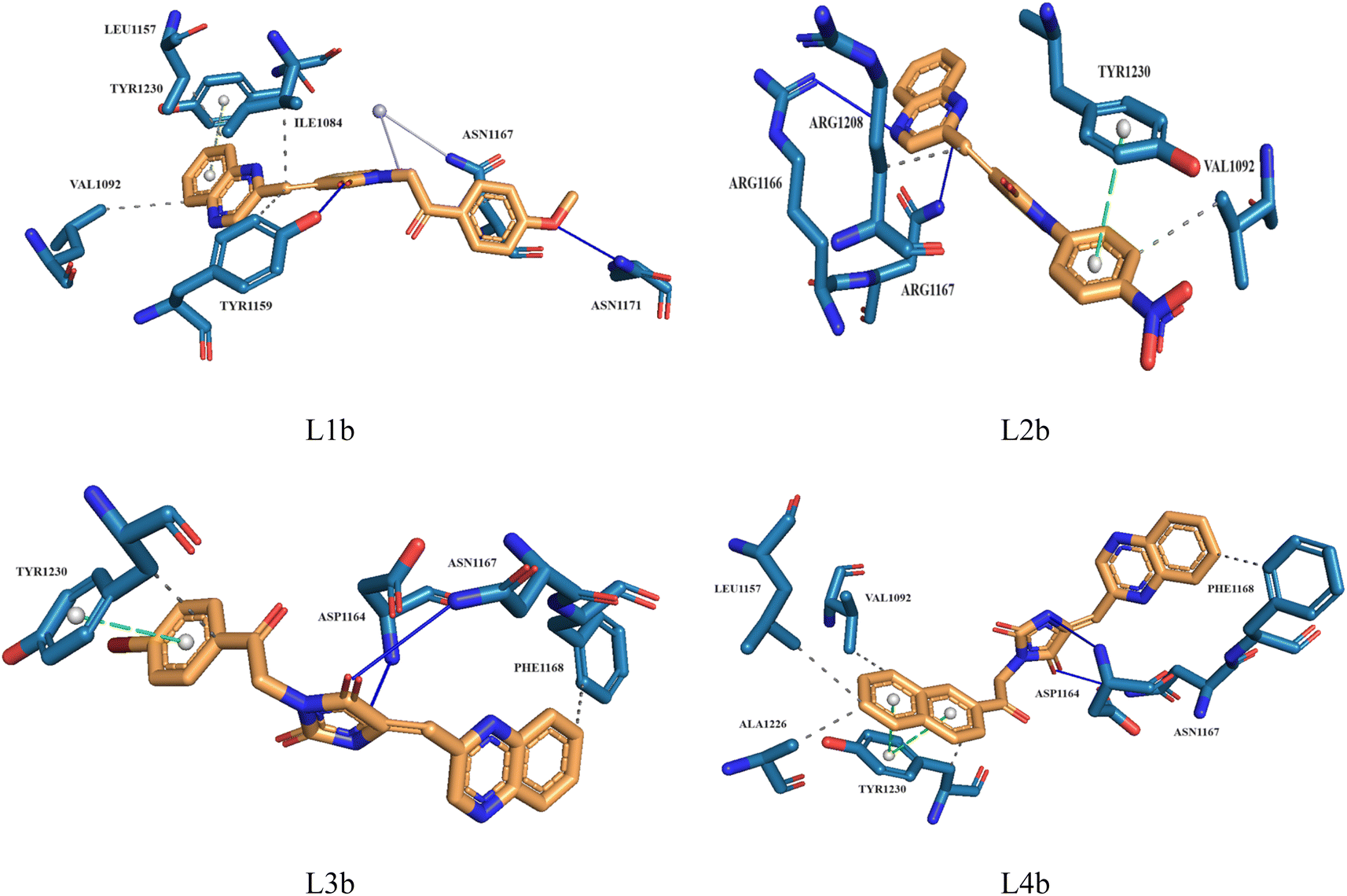 | ||
| Fig. 11 3D structure of ligands interactions with binding residues of 2WGJ. | ||
3.9 Molecular dynamic simulation results
In this study, we used molecular dynamic simulations to explore how stable and structurally sound complexes of L1a with 3CD8 and L4b with the 2WGJ are. The simulations provided insights into the detailed interactions at the atomic level and the overall structural stability of these complexes with the proteins. The Root Mean Square Deviation (RMSD) is a crucial metric in assessing the stability and convergence of molecular dynamic simulations. We tracked the Root Mean Square Deviation (RMSD) of Cα atoms in the protein backbone throughout a 100 ns simulation, Fig. 12. The RMSD values indicated that the complexes remained stable, with values around 0.25 for 3CD8 and 0.22 for 2WGJ in the presence of inhibitors. Looking at the dynamic simulation results, during the initial 50 ns, the RMSD values for the L1a–3CD8 protein gradually increased from 0.2 to 0.25 nm and stayed stable between 0.25 and 0.3 nm for the next 50 ns. On the other hand, for the L4b–2WGJ complex, the RMSD values increased from 0.15 to 0.22 nm and then started to relax after 80 ns.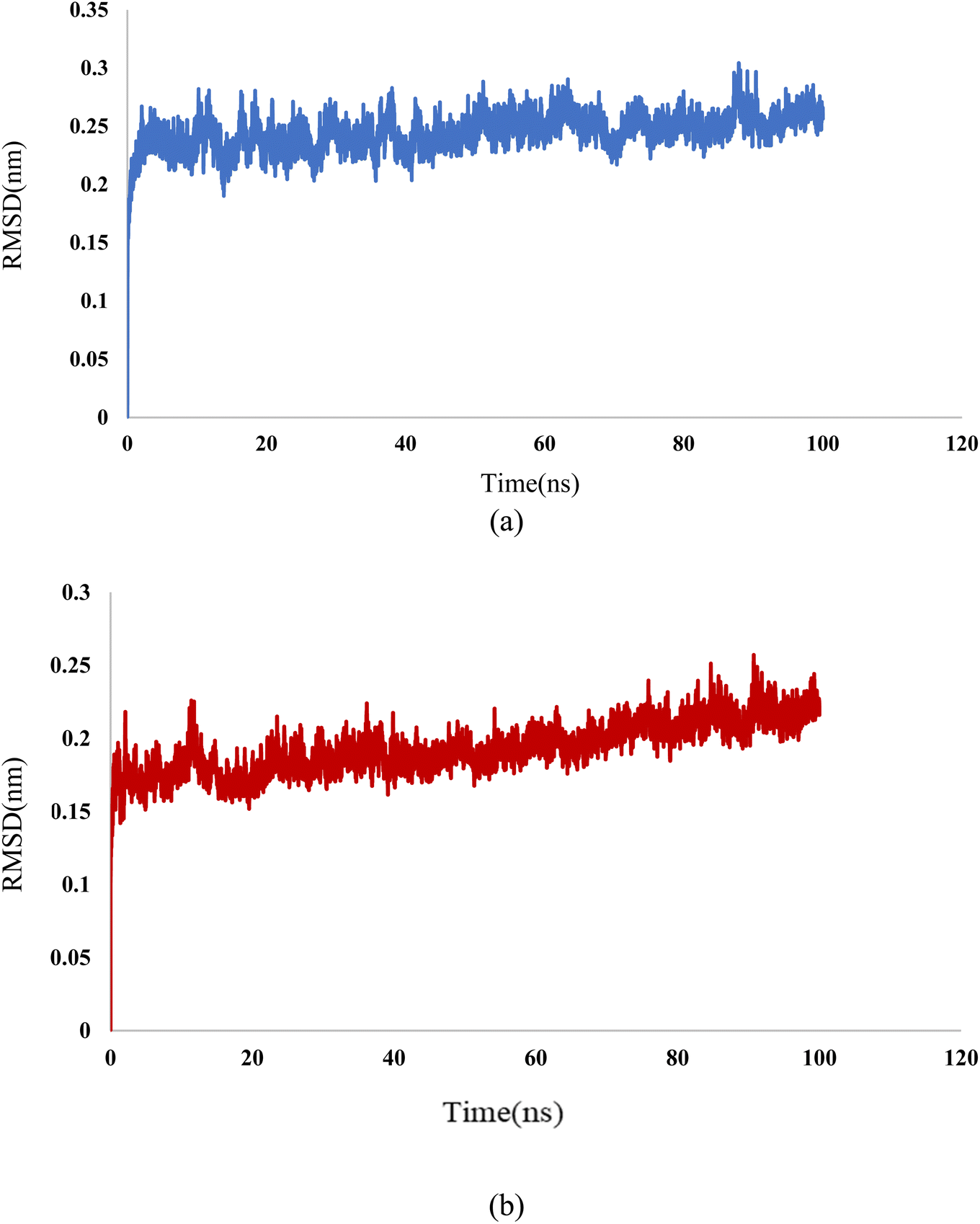
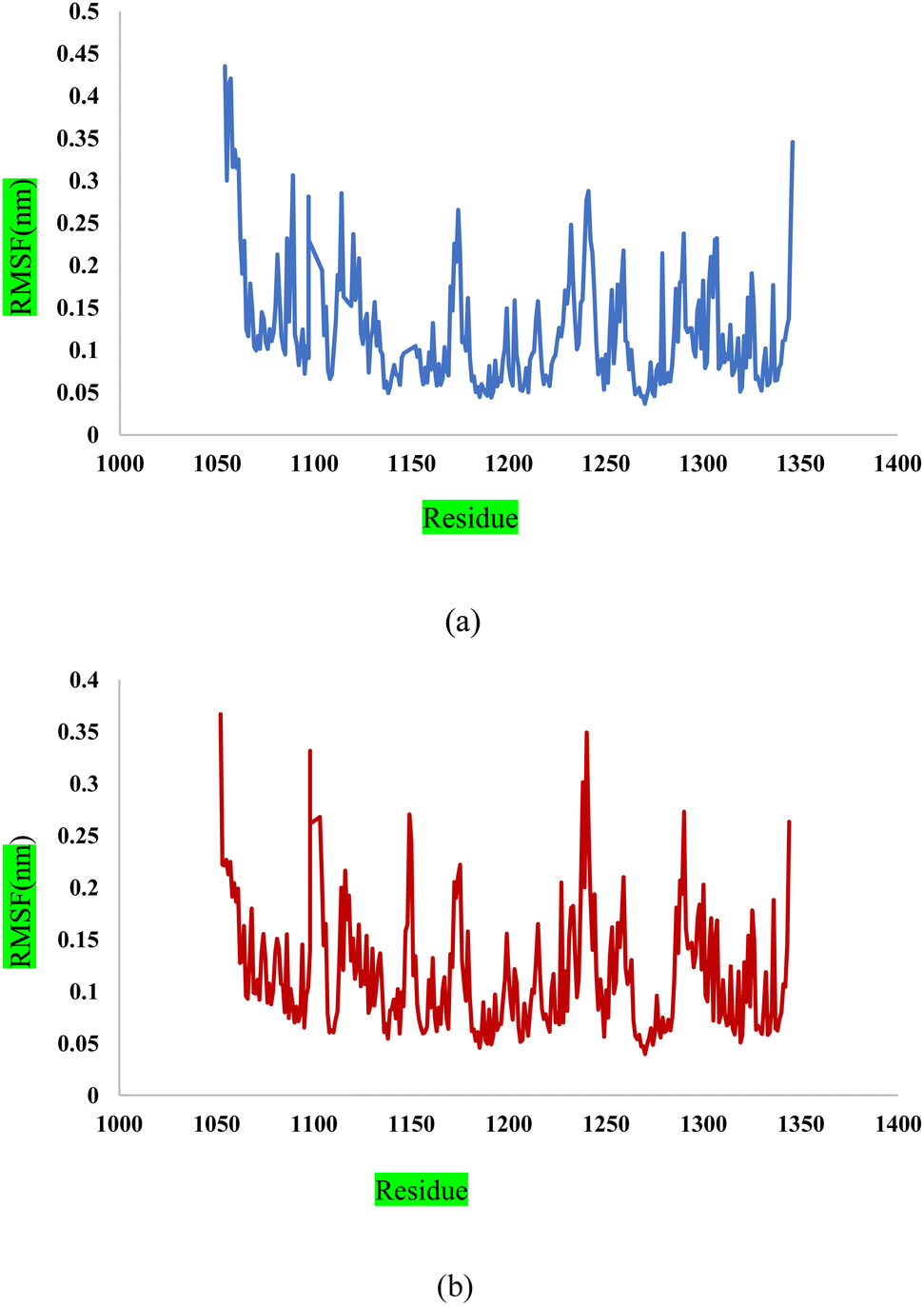
Fig. 13a shows the Root Mean Square Fluctuation (RMSF) values for different residues from the molecular dynamics simulation of 3CD8. The RMSF values range from 0.0368 to 0.4353. This indicates variability in the degree of fluctuation for different residues. The analysis revealed several peaks in the RMSF profile, indicating regions with higher flexibility. Notable peaks include residues 1054, 1086, 1097, 1104, 1114, and 1240. Each peak corresponds to a specific residue, and the corresponding RMSF values denote the extent of fluctuation exhibited by these residues during the simulation. From Fig. 13b, some regions in 2WGJ, such as residues 1086–1098, 1103–1130, and 1240–1259, consistently exhibit higher flexibility throughout the simulation. Peaks in flexibility, like those observed around residues 1086, 1103, 1124, 1140, 1149, 1150, and 1240, suggest specific points in the protein where dynamic structural changes or interactions may occur. These residues are close to the active site and play crucial roles in the protein's biological activity.
4 Conclusion
In this study, we successfully developed an efficient acidic nanocatalyst with magnetic properties, Fe3O4@CPTMS@guanidine–BuSO3H, significantly facilitating one-pot multicomponent reactions. This innovative approach addresses the limitations of traditional mineral acids and homogeneous organic catalysts by embodying the principles of green chemistry. The nanocatalyst offers a non-toxic, cost-effective, and environmentally friendly alternative, with the added advantage of simplifying the catalyst recovery process through magnetic separation. The catalytic capability of Fe3O4@CPTMS@guanidine–BuSO3H was extensively evaluated, demonstrating its ability to maintain high activity even after multiple recycling cycles. We synthesized a series of N-substituted (Z)-5-arylidene imidazolidine/thiazolidine-2,4-dione/4-thione derivatives using this nanocatalyst. L1a exhibited the lowest IC50 values among the synthesized compounds in both the A549 and MCF7 cell lines, indicating significant antiproliferative activity.Additionally, incorporating halogen atoms, especially chlorine, into the quinoxaline ring substantially enhanced the biological activity. The synthesized compounds displayed a broad spectrum of efficacy, with IC50 values ranging from 43.12 nM to 388.85 nM for the A549 cell line and 67.07 nM to 448.28 nM for the MCF7 cell line. Select compounds, including L1b, L2b, L3b, and L4b, demonstrated potent activity in both cell lines, with IC50 values below 50 nM. Remarkably, all tested compounds outperformed the established anticancer drugs doxorubicin and cisplatin in the A549 cell line, while only three compounds exhibited higher IC50 values than cisplatin in the MCF7 cell line. This suggests that the novel compounds synthesized in this study could offer a significant advantage over traditional chemotherapy agents. Molecular docking simulations provided further insights into the interactions between the synthesized compounds and their target proteins. Compound L1a exhibited a high binding affinity of −9.9 kcal mol−1 to the protein 3CD8, supported by specific interactions with ARG1208, ALA1108, and TYR1159.
Conversely, L2a demonstrated a lower binding affinity but engaged in pi-stacking interactions with TYR1230, highlighting alternative interaction modes that could be crucial for selectivity across different cancer cell types. Ligand L4b displayed a high-affinity binding (−10.8 kcal mol−1) to the protein 2WGJ, emphasizing the importance of hydrogen bonding and hydrophobic interactions in maintaining compound stability. Furthermore, molecular dynamic simulations validated the stability of the ligand–protein complexes, offering valuable insights into binding site flexibility and the potential for structural modifications to enhance activity. This research introduces a new class of potent anticancer agents and showcases the potential of Fe3O4-based MNPs as a versatile and sustainable catalytic platform in pharmaceutical research. The combined environmental compatibility and superior operational performance establish these nanoparticles as a valuable asset for future synthetic endeavors to discover novel therapeutic compounds. This innovative approach promises significant advancements in the development of more effective and sustainable cancer treatments.
Data availability
All data generated or analyzed during this study are included in this published article and data will be made available on request.Author contributions
Malihe Akhavan: conceptualization, methodology, validation, investigation, synthesis of derivatives, writing – original draft preparation. Zohreh Esam: methodology, validation, formal analysis, investigation. Maryam Lotfi: software, validation, formal analysis, molecular docking studies, computational work. Atefeh Mirshafa: investigation, biological assays, validation, writing – review & editing. Saeed Pourmand: software, validation, molecular dynamics simulations, computational work. Ahmadreza Bekhradnia: supervision, writing – review & editing, project administration, funding acquisition, Froug Ashori: synthesis of derivatives, Motahare Rabani: synthesis of derivatives, Golbahar Ekbatani: synthesis of derivatives, Saeed Tourani: synthesis of derivatives, Zahra Keshavarzian: synthesis of derivatives, Reza Beheshti: synthesis of derivatives, Zahra Ghanbarimasir: synthesis of derivatives.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The main part of this work was connected to the Postdoc Thesis/Research of Dr Malihe Akhavan from Faculty of Pharmacy, Mazandaran University of Medical Sciences.References
- P.-S. Wang, F.-S. Chou, M. Bloomston, M. S. Vonau, M. Saji, A. Espinosa and J. J. Pinzone, J. Surg. Res., 2009, 153, 210–216 CrossRef CAS.
- M. H. Jarrar and A. Baranova, J. Cell. Mol. Med., 2007, 11, 71–87 CrossRef CAS PubMed.
- A. Levitzki and A. Gazit, Science, 1995, 267, 1782–1788 CrossRef CAS PubMed.
- C. Flordellis, P. Papathanasopoulos, A. Lymperopoulos, J. Matsoukas and H. Paris, Cardiovasc. Hematol. Agents Med. Chem., 2007, 5, 133–145 CrossRef CAS PubMed.
- S. H. Lee, I. B. Jaganath, S. M. Wang and S. D. Sekaran, PLoS One, 2011, 6, e20994 CrossRef CAS.
- N. S. Sethi, D. N. Prasad and R. K. Singh, Mini-Rev. Med. Chem., 2020, 20, 308–330 CrossRef CAS.
- S. Kermorgant, D. Zicha and P. J. Parker, EMBO J., 2004, 23, 3721–3734 CrossRef CAS PubMed.
- M. Naufal, E. Hermawati, Y. M. Syah, A. T. Hidayat, I. W. Hidayat and J. Al-Anshori, ACS Omega, 2024, 9(4), 4186–4209 CrossRef CAS PubMed.
- T. Francis, S. R. Dixit and B. P. Kumar, Polycyclic Aromat. Compd., 2022, 42, 2483–2499 CrossRef CAS.
- A. Cavazzoni, R. R. Alfieri, C. Carmi, V. Zuliani, M. Galetti, C. Fumarola, R. Frazzi, M. Bonelli, F. Bordi and A. Lodola, Mol. Cancer Therapeut., 2008, 7, 361–370 CrossRef CAS PubMed.
- D. Dewangan, K. Nakhate, A. Mishra, A. S. Thakur, H. Rajak, J. Dwivedi, S. Sharma and S. Paliwal, J. Heterocycl. Chem., 2019, 56, 566–578 CrossRef CAS.
- Z. Ghanbarimasir, A. Bekhradnia, K. Morteza-Semnani, A. Rafiei, N. Razzaghi-Asl and M. Kardan, Spectrochim. Acta, Part A, 2018, 194, 21–35 CrossRef CAS.
- H. T. Abdel-Mohsen, M. M. Anwar, N. S. Ahmed, S. S. Abd El-Karim and S. H. Abdelwahed, Molecules, 2024, 29, 875 CrossRef CAS.
- M. K. Manickasamy, A. Sajeev, B. BharathwajChetty, M. S. Alqahtani, M. Abbas, M. Hegde, B. S. Aswani, M. Shakibaei, G. Sethi and A. B. Kunnumakkara, Cell. Mol. Life Sci., 2024, 81, 1–36 CrossRef.
- J. Zhang, M. Tang and J. Shang, Biomolecules, 2024, 14, 190 CrossRef CAS.
- M. Knura, W. Garczorz, A. Borek, F. Drzymała, K. Rachwał, K. George and T. Francuz, Cancers, 2021, 13, 1827 CrossRef CAS PubMed.
- V. Zuliani, C. Carmi, M. Rivara, M. Fantini, A. Lodola, F. Vacondio, F. Bordi, P. V. Plazzi, A. Cavazzoni and M. Galetti, Eur. J. Med. Chem., 2009, 44, 3471–3479 CrossRef CAS.
- M. Akhavan, N. Foroughifar, H. Pasdar and A. Bekhradnia, Comb. Chem. High Throughput Screening, 2019, 22, 716–727 CrossRef CAS.
- S. A. Shintre, D. Ramjugernath, M. S. Islam, R. Mopuri, C. Mocktar and N. A. Koorbanally, Med. Chem. Res., 2017, 26, 2141–2151 CrossRef CAS.
- S. Wei, J. Yang, S.-L. Lee, S. K. Kulp and C.-S. Chen, Cancer Lett., 2009, 276, 119–124 CrossRef CAS.
- V. Gopi, S. Subbiahraj, K. Chemmanghattu and P. C. Ramamurthy, Dyes Pigm., 2020, 173, 107887 CrossRef CAS.
- M. Akhavan, N. Foroughifar, H. Pasdar, A. Khajeh-Amiri and A. Bekhradnia, Transition Met. Chem., 2017, 42, 543–552 CrossRef CAS.
- Z. Esam, M. Akhavan, A. Mirshafa and A. Bekhradnia, RSC Adv., 2023, 13, 25229–25245 RSC.
- Z. Esam, M. Akhavan, A. Bekhradnia, M. Mohammadi and S. Tourani, Catal. Lett., 2020, 150, 3112–3131 CrossRef CAS.
- Z. M. M. Alzhrani, M. M. Alam, T. Neamatallah and S. Nazreen, J. Enzyme Inhib. Med. Chem., 2020, 35, 1116–1123 CrossRef CAS PubMed.
- J. T. Sims and R. Plattner, Cancer Chemother. Pharmacol., 2009, 64, 629–633 CrossRef CAS.
- A. Cimmino, V. Mathieu, M. Evidente, M. Ferderin, L. M. Y. Banuls, M. Masi, A. De Carvalho, R. Kiss and A. Evidente, Fitoterapia, 2016, 109, 138–145 CrossRef CAS.
- S. K. Burley, H. M. Berman, C. Bhikadiya, C. Bi, L. Chen, L. Di Costanzo, C. Christie, K. Dalenberg, J. M. Duarte and S. Dutta, Nucleic Acids Res., 2019, 47, D464–D474 CrossRef CAS.
- Y. Wang, E. Bolton, S. Dracheva, K. Karapetyan, B. A. Shoemaker, T. O. Suzek, J. Wang, J. Xiao, J. Zhang and S. H. Bryant, Nucleic Acids Res., 2010, 38, D255–D266 CrossRef CAS PubMed.
- A. Bekhradnia and P.-O. Norrby, Dalton Trans., 2015, 44(9), 3959–3962 RSC.
- M. F. Adasme, K. L. Linnemann, S. N. Bolz, F. Kaiser, S. Salentin, V. J. Haupt and M. Schroeder, Nucleic Acids Res., 2021, 49, W530–W534 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1, 19–25 CrossRef.
- P. J. Schmid, Exp. Fluids, 2011, 50, 1123–1130 CrossRef.
- D. M. York, A. Wlodawer, L. G. Pedersen and T. A. Darden, Proc. Natl. Acad. Sci., 1994, 91, 8715–8718 CrossRef CAS.
- Z. Esam, M. Akhavan and A. Bekhradnia, Appl. Organomet. Chem., 2021, 35, e6005 CrossRef CAS.
- R. Bhatti, M. Singh and J. Sandhu, Rasayan J. Chem., 2008, 5, 738–742 Search PubMed.
- L. Sonawane and S. Bari, Int. J. Biol. Chem., 2011, 5, 68–74 CrossRef CAS.
- A. Paul, A. Maji, A. Sarkar, S. Saha, P. Janah and T. K. Maity, Mini-Rev. Org. Chem., 2023, 20, 5–34 CrossRef CAS.
- K. F. Shelke, S. B. Sapkal, G. K. Kakade, S. A. Sadaphal, B. B. Shingate and M. S. Shingare, Green Chem. Lett. Rev., 2010, 3, 17–21 CrossRef CAS.
- J. Zhang, Y. Zhang and Z. Zhou, Green Chem. Lett. Rev., 2014, 7, 90–94 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra03881a |
| This journal is © The Royal Society of Chemistry 2024 |