
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering Escherichia coli with a symbiotic plasmid for the production of phenylpyruvic acid†
Tianzhen Xiong *a,
Qiuyue Gaob,
Jiting Zhanga,
Jiaguang Zhanga,
Can Zhanga,
Huidie Yuea,
Junling Liua,
Dingyuan Baia and
Jinying Lia
*a,
Qiuyue Gaob,
Jiting Zhanga,
Jiaguang Zhanga,
Can Zhanga,
Huidie Yuea,
Junling Liua,
Dingyuan Baia and
Jinying Lia
aCollege of Life Science, Xinyang Normal University, 237 Nanhu Road, Xinyang, Henan 464000, China. E-mail: tzxiong@xynu.edu.cn; Tel: +86-13939748578
bCollege of Social Science, Xinyang University, 7th New Avenue West, Xinyang, Henan 464000, China
First published on 22nd August 2024
Abstract
Plasmid-based microbial systems have become a major avenue for the production of pharmaceutical and chemical products; however, antibiotics are often required to maintain the stability of the plasmid. To eliminate the need for antibiotics, we developed a symbiotic system between plasmids and hosts by knocking out the essential gene of folP on the chromosome and placing it on the same plasmid as L-amino acid dehydrogenase (aadL); the resulting strain was named E. coli A06ΔfolP. To increase the copy number of aadL, different strengths of promoters were used for the expression of folP, resulting in the creation of a mutant E. coli A17ΔfolP. The yield of phenylpyruvic acid (PPA) from E. coli A17ΔfolP (4.1 ± 0.3 g L−1) was 1.9-fold that of E. coli A06ΔfolP (2.1 ± 0.2 g L−1). Next, the stability of plasmids was tested, and results showed that the plasmids could be maintained stably for 10 transfer numbers under antibiotic-free conditions. Finally, E. coli A17ΔfolP was used to produce PPA; the yield of PPA was 18.7 g L−1 within 14 h.
1. Introduction
Phenylpyruvic acid (PPA), an α-keto acid, is widely used in the food and pharmaceutical industries as well as agriculture. PPA is more important than phenylalanine in the formation of specific smells, tastes and textures.1 In the food industry, PPA can be used as a substrate to produce phenyllactic acid, which is an important preservative.2 In medicine, PPA is used to diagnose phenylketonuria, which is a genetic disorder involving phenylalanine metabolism.3 PPA is also used in the diet of kidney patients to replace amino acids and reduce the accumulation of urea in the body without altering the nutrient intake.4 In agriculture, excessive nitrogen excretion in chicken manure can have adverse effects on the environment, and PPA can be used as a supplement in poultry feed to prevent excessive nitrogen accumulation in manure. Therefore, the production of PPA has great market potential.PPA can be synthesized through chemical reactions. However, the chemical synthesis of PPA produces many toxic by-products5 or employs heavy metal catalysts, which are environmentally unfriendly.6 Therefore, developing a green and efficient method to produce PPA has great market prospects.7 PPA can be produced via fermentation from microorganisms, including Zygosaccharomyces rouxii, Corynebacterium glutamicum and Proteus vulgaris.8 Many types of enzymes can be used to produce PPA, including amino acid oxidase, amino acid deaminase and aminotransferase. Amino acid oxidase produces hydrogen peroxide, which disrupts enzyme activity.7 Aminotransferase requires the use of cosubstrates and the production of coproducts, thus increasing the difficulty of purification.9 Amino acid deaminase is widely used in the production of ketoacids.10 Hou et al. overexpressed L-AAD in Escherichia coli from Proteus mirabilis for the one-step production of PPA from L-phenylalanine. PPA with a yield of 31.4 g L−1 was obtained within 9 h.11 Xiong et al. constructed a chromosome engineering strain for the synthesis of PPA from L-phenylalanine. L-Amino acid deaminase (pmLAAD) from Proteus myxofaciens was incorporated into the Escherichia coli BL21 (DE3) chromosome and the copy numbers of pmLAAD were increased via chemically induced chromosomal evolution (CIChE). Finally, 19.14 g L−1 PPA was obtained within 24 h.12
Target genes can be efficiently expressed in plasmids. However, the stability of plasmids is vulnerable in the absence of antibiotics.13 As an alternative method, heterologous genes can be transferred to the host chromosome, and the genes on the chromosome can be steadily inherited and expressed. However, the expression level from chromosome-based strains is significantly lower than plasmid-based strains owing to an insufficient gene copy number.14 Although chemically inducible chromosomal evolution can increase heterologous genes to 40 copies on the host chromosome,15 this method destroys the repair system of genes. A previous study has described that gene expression can be maintained by engineering promoters at any copy number in Escherichia coli.16 However, this strategy was implemented by suppressing the strong promoter and eventually stabilizing a weak expression level of genes. The method makes it difficult to achieve efficient production. Several approaches have been pursued to replace antibiotics as selective markers for plasmid stability in bacteria. The symbiotic system was previously developed using essential genes. The stability of the plasmid can be maintained by integrating essential genes into the plasmid and deleting essential genes in chromosomes.17 This method has been used to replace antibiotic genes. These genes are involved in the biosynthesis of the amino acid (glyA),18 NAD+ (nadC)19 and glycerol-dependent growth (tpiA).20 Nevertheless, most of these stabilization systems have not been tested under bioprocess protein production conditions, nor tested to modify conventional production systems that are already in use to avoid antibiotics and antibiotic resistance genes, and further compared in terms of plasmid stability and process productivities.
In this study, we developed a “symbiotic system” between plasmid and host (Fig. 1). Engineering Escherichia coli contains an essential gene of folP on plasmids instead of chromosomes. This approach has several advantages; (1) the plasmids have extremely high stability because the cells would die without them. (2) This strain can maintain the stability of plasmids in any growth medium, and the growth of the strain does not require the addition of antibiotics. (3) In the large-scale culture, this system provides considerable advantage due to the elimination of environmental problems from antibiotic resistance genes and antibiotics.
 | ||
| Fig. 1 Schematic representation of the symbiosis system for the synthesis of phenylpyruvic acid from phenylalanine. folp, dihydropteroate synthase; CmR, chloramphenicol resistance gene; t7, IPTG-inducible t7/laco promoter; rbs, ribosome binding site; tt7, t7 terminator; RSF ori, RSF origin of replication. | ||
2. Materials and methods
2.1. Bacterial strains, plasmids, and chemicals
Escherichia coli DH5α was used for plasmid amplification and Escherichia coli BL21(DE3) was used for recombinant enzyme production. Antibiotics were added to the media of plasmid-bearing Escherichia coli as required. L-Phenylalanine and phenylpyruvic acid were obtained from Sigma-Aldrich (St. Louis, MO, USA). All restriction enzymes, T4 DNA ligase, and high-fidelity DNA polymerase were purchased from TaKaRa (Dalian, China). The gel extraction kit and plasmid extraction kit were procured from Axygen (Suzhou, China).2.2. Gene knockout and construction of plasmids and strains
All primers are listed in Table S1.† The strains and plasmids are listed in Table S2.† The aadL was obtained from previous study.21 The aadL was amplified using gene-specific primers aadL-BamHI (F) and aadL-EcoRI (R) and cloned into pRSFDuet-1 between restriction sites of BamHI and EcoRI. The resulting plasmids were named pRSF-aadL. The folP expression cassette with different promoters was amplified by primers of folP-J23100 (F) and folP (R), folP-J23101 (F) and folP (R), folP-J23106 (F) and folP (R), folP-J23107 (F) and folP (R), folP-J23116 (F) and folP (R), folP-J23117 (F) and folP (R), folP-J23119 (F) and folP (R). The amplified fragments were inserted into pRSF-aadL in SphI and XmaI. The recombinant plasmids were named pRSF-A00, pRSF-A01, pRSF-A06, pRSF-A07, pRSF-A16, pRSF-A17, and pRSF-A19 (Fig. S1A†).The folP expression cassette with the promoter of J23106 was amplified by primers of pKD46-folP (F) and pKD46-folP (R). The amplified fragments were inserted into pKD46 in the restriction sites of AarI. The recombinant plasmid was named pKD46-folP (Fig. S1B†). The pRSF-aadL and pKD46-folP were transferred into E. coli BL21 (DE3) and the resulting strains were named E. coli A and E. coli pKD46-folP, respectively. The chloramphenicol resistance gene (CmR) with homologous arms was amplified from pKD3 by using a primer of pOut-folP (F) and pOut-folP (R) and inserted into T-Vector pMD19. The recombinant plasmids were named pOut-folP (Fig. S1C†). The folP recombination cassette was recruited from pOut-folP by digestion (SmaI). To delete folP, the folP recombination cassette was inserted in the E. coli pKD46-folP genome by λ-Red recombination. The recombinant strain was named E. coli pKD46ΔfolP.22
All recombinant plasmids were transferred into E. coli pKD46ΔfolP or E. coli BL21 (DE3). Then, pKD-folP was eliminated at 42 °C for 12 h, and the resulting strains were named E. coli A, E. coli A00ΔfolP, E. coli A01ΔfolP, E. coli A06ΔfolP, E. coli A07ΔfolP, E. coli A16ΔfolP, E. coli A17ΔfolP, and E. coli A19ΔfolP.
2.3. Quantification of plasmid copy number
Quantitative real-time PCR (qPCR) was performed to determine plasmid copy number.23,24 The bacterial cells were lysed by 98 °C incubation for 10 min followed −20 °C incubation for 10 min. The qPCR was performed by targeting the plasmid and chromosome. The primers of qPCR-aadL (F) and qPCR-aadL (R) for plasmid were complementary to aadL and the primers of qPCR-16S (F) and qPCR-16S (R) for chromosomal were complementary to 16S ribosomal DNA from Escherichia coli (Table S1†). The qPCR and lysed mixture were performed on the CFX96 Real-Time PCR Detection System (BioRad, Hercules, CA, USA). The plasmid copy number was determined by comparing the 16S ribosomal DNA gene.2.4. Heterologous gene expression and whole-cell biotransformation
To express aadL, the medium inoculated with recombinant Escherichia coli was incubated overnight (37 °C) and then transferred to a fresh Terrific Broth medium to an OD600 of 0.1 and until the biomass of strains reached 0.6 (OD600). Isopropyl-β-D-thiogalactopyranoside (IPTG) was added to the cultures and the final concentration of IPTG reached 0.4 mM, then the mixture was cultured at 18 °C, using a rotary shaker at 220 rpm for 24 h.The reaction mixture (20 mL) contained 50 mM phosphate buffer (pH 7). The composition of the mixture was 3.1 g L−1 wet cells and 20 g L−1 L-phenylalanine. The mixture was cultured at 40 °C, with a rotary shaker at 200 rpm for 1 h and the reaction was terminated by boiling for 1 min. Then, the mixture was centrifuged at 8000×g for 5 min, and the supernatant was collected for analysis.
2.5. Plasmid stability assay
To test plasmid stability, E. coli A17ΔfolP and E. coli A were cultured in Terrific Broth medium. For the initial culture, a single colony was transferred to the flask with a 50 mL medium. The strain was inoculated into fresh medium until OD600 of 0.1 and the culture was performed for 12 h at 37 °C, with a rotary shaker at 220 rpm. The operation method was repeated ten times. The OD600 was determined using a UV-vis spectrophotometer (Lengguang, Shanghai, China) at 600 nm.2.6. Analytical procedure
The analysis of samples was performed on the HPLC system with the GALAK column. The temperature of the column was maintained at 35 °C. The mobile phase was 0.1% formic acid solution and methanol. A linear elution was performed from 10% to 100% methanol over 15 min, 100% methanol was maintained in 15–20 min and 10% methanol was maintained in 20–30 min. The flow rate of the solution was 1 mL min−1. Phenylalanine and PPA were detected at 254 nm wavelength in an ultraviolet detector (Fig. S2†).3. Results and discussion
3.1. Characterization of symbiosis plasmid system
The folP encodes a small protein, which is fundamental to tetrahydrofolate biosynthesis (NCBI Reference Sequence: NP_417644.4). Knockout of folP on chromosomes will lead to the death of Escherichia coli.17 The plasmid with folp was introduced into a strain with chromosome knockout folp, which could make the bacteria grow normally. To test the efficiency of the symbiotic system, we constructed E. coli A06ΔfolP (folP and aadL was expressed on pRSFDuet-1, and folP on E. coli chromosome was knocked out) with E. coli A (aadL was expressed on pRSFDuet-1) used as a control. Then, the whole-cell biotransformation was performed. The concentration of PPA from E. coli A was 4.3 g L−1, which was 2.05-fold higher than E. coli A06ΔfolP. The plasmid copy number of each strain was determined by qPCR (Fig. S3†). The results showed that the plasmid copy number from E. coli A was 946, which was 2.24-fold higher than E. coli A06ΔfolP (Fig. 2).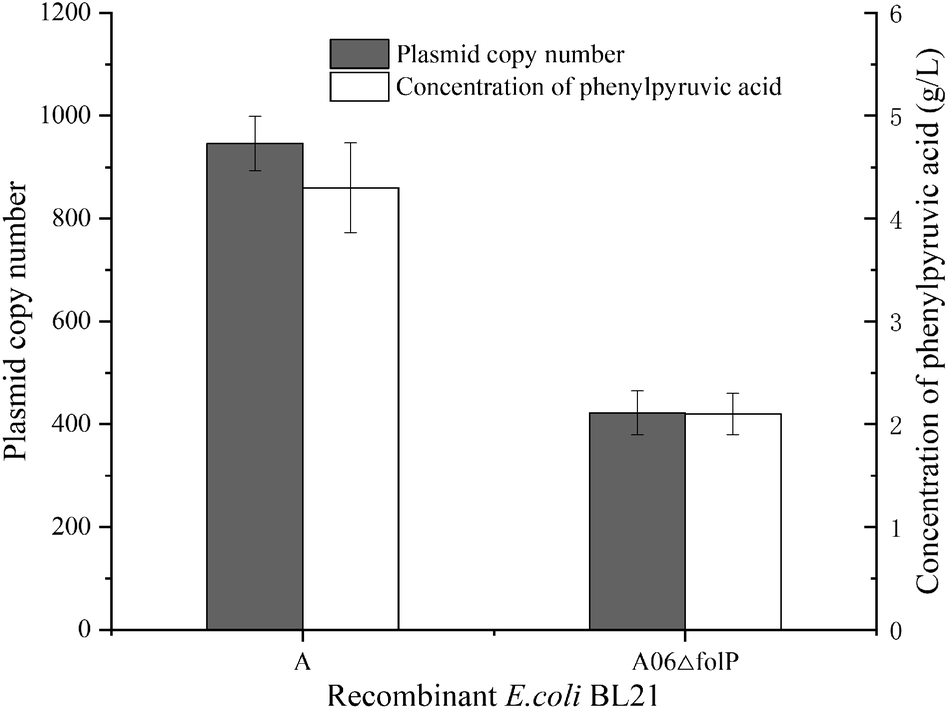 | ||
| Fig. 2 Plasmid copy number and concentration of phenylpyruvic acid from E. coli A and E. coli A06ΔfolP. | ||
3.2. Improving PPA production with varied folP expression
When folP from Escherichia coli chromosome is knocked out, folP is also attached to the plasmid. The folP expression level is related to cell viability.17 Plasmid copy number and promoter upstream of folP affect the expression of folP. Thus, we hypothesized that changing the folP expression levels would affect the plasmid copy number and production of PPA. Previously, many studies have shown that symbiotic systems can maintain plasmid stability.19,25,26 However, the main challenge lies in controlling the high-level expression of genes. Few studies elucidate the relationship between promoter strength for expressing basic genes and plasmid copy number.To verify the relationship between folP expression and plasmid copy number, seven different strengths promoters (Table. S3†) were screened for expression of folP.27 Then, the symbiotic system with different strength promoters was constructed (Fig. 3A). The plasmid copy number was determined by qPCR (Fig. S4†). This result shows that the plasmid copy number was inversely proportional to the promoter strength (Fig. 3B and Table. S3†). With the increase of promoter strength before folp, the plasmid copy number decreased gradually. Changing promoter strength before folP can successfully regulate the plasmid copy number. The plasmid copy number of E. coli A17ΔfolP was 923 copies/cell (Fig. 3B), which is close to E. coli A. The yield of PPA from E. coli A17ΔfolP (4.1 g L−1) was 1.9-fold that of E. coli A06ΔfolP (2.1 g L−1) and close to that of E. coli A (4.3 g L−1) (Fig. 3B). The plasmid copy number and production of PPA were successfully increased by adjusting the promoter strength before folp.
 | ||
| Fig. 3 Effect of recombinant Escherichia coli with different promoters on the expression strength of the plasmid copy number and concentration of phenylpyruvic acid. (A) Construction of symbiotic strains using promoters of different strengths; (B) plasmid copy number and phenylpyruvic acid concentration of the recombinant strain. | ||
3.3. Plasmid stability test of symbiotic system
Plasmids are often lost without antibiotics, greatly affecting the production of engineered bacteria. The maintenance of plasmid copy numbers is the key to the efficient production of engineered strains. The strains to achieve stable and efficient expression of target genes without the addition of antibiotics are greatly valuable.To test plasmid stability without the addition of antibiotics, we performed a serial dilution culture experiment. The plasmid copy number of continuously cultured E. coli A17ΔfolP was determined by qPCR (Fig. S5†), the result shows that E. coli A17ΔfolP has no significant influence on plasmid copy number within transfer number of 10 (Fig. 4A). At the same time, the plasmid copy number of the continuously cultured E. coli A was also determined by qPCR (Fig. S6†), the result shows that E. coli A gradually decreased on plasmid copy number within transfer number of 10 (Fig. 4B). E. coli A17ΔfolP and E. coli A convert phenylalanine to PPA. The plasmid copy number remained stable with the transformation of E. coli A17ΔfolP (Fig. 4A). The concentration of PPA from E. coli A decreased dramatically with the transfer number, a transfer number of 10 yielded a 37.2% decrease in PPA production compared to the initial strain (Fig. 4B). The plasmid copy number can be maintained without appreciably altering the natural system. No antibiotics were added during the culture of engineered Escherichia coli. The plasmid remained stable within the transfer number of 10. The folP-based system was able to maintain plasmid stability over a long time of cell propagation.
 | ||
| Fig. 4 Effect of recombinant Escherichia coli with different promoters on the plasmid copy number and concentration of phenylpyruvic acid. (A) E. coli A17ΔfolP in different transfer numbers. (B) E. coli A in different transfer numbers. | ||
3.4. Production of PPA
E. coli A17ΔfolP was used for whole-cell biotransformation. The biocatalytic performance was evaluated based on the concentration of L-phenylalanine and PPA. The time course of cascade biotransformation is shown. PPA was 9.1 g L−1 and yield of 95.5% within 14 h (Fig. 5). This research system can not only be used for the production of PPA but also can be combined with other genes to produce corresponding compounds. | ||
| Fig. 5 Time course of one-pot biotransformation for the production of phenylpyruvic acid from L-phenylalanine. ■ L-phenylalanine; ● phenylpyruvic acid. | ||
4. Conclusions
In summary, we develop a symbiotic system between plasmid and host to produce PPA from L-phenylalanine. It avoids the loss of plasmids without antibiotics. Future research can explore the development of a stable multi-plasmid system, with varied strengths of overexpressing heterologous genes for whole-cell biotransformation or metabolic engineering with the goal of producing natural products. The symbiotic system has great industrial application prospects.Data availability
The data supporting this study's findings are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Key scientific research projects of higher education institutions in Henan Province (24B180017), the Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX19_1841) and the Nanhu Scholars Program for Young Scholars of XYNU for financial support.References
- D. Zhang, T. Zhang, Y. Lei, W. Lin, X. Chen and M. Wu, Front. Bioeng. Biotechnol., 2022, 10, 846489 CrossRef PubMed.
- Y. Liu, R. Wang, D. Wang, Z. Sun, F. Liu, D. Zhang and D. Wang, Food Hydrocolloids, 2022, 127, 107546 CrossRef CAS.
- A. T. Tankeu, D. C. Pavlidou, A. Superti-Furga, K. Gariani and C. Tran, Orphanet J. Rare Dis., 2023, 18, 37 CrossRef PubMed.
- Z. Luo, S. Yu, W. Zeng and J. Zhou, Biotechnol. Adv., 2021, 47, 107706 CrossRef CAS PubMed.
- M. Villablanca and G. Cilento, Biochim. Biophys. Acta, 1987, 926, 224–230 CrossRef CAS PubMed.
- H. des Abbayes and J.-Y. Salaün, Dalton Trans., 2003, 1041–1052 RSC.
- K. Oike and H. Gröger, J. Biotechnol., 2020, 323, 203–207 CrossRef CAS PubMed.
- H. B. Coban, A. Demirci, P. H. Patterson and R. J. Elias, Bioprocess Biosyst. Eng., 2014, 37, 2343–2352 CrossRef CAS PubMed.
- X. Feng, J. Guo, R. Zhang, W. Liu, Y. Cao, M. Xian and H. Liu, ACS Omega, 2020, 5, 7745–7750 CrossRef CAS PubMed.
- J. Liu, J. Liu, B. Yang, C. Gao, W. Song, G. Hu, L. Liu and J. Wu, Biotechnol. Lett., 2022, 44, 635–642 CrossRef CAS PubMed.
- Y. Hou, G. S. Hossain, J. Li, H.-D. Shin, G. Du, J. Chen and L. Liu, Biotechnol. Bioeng., 2017, 114, 1928–1936 CrossRef CAS PubMed.
- T. Xiong, Y. Bai, T. P. Fan, X. Zheng and Y. Cai, Biotechnol. Appl. Biochem., 2022, 69, 1909–1916 CrossRef CAS PubMed.
- D. Sünderhauf, U. Klümper, W. H. Gaze, E. R. Westra and S. van Houte, ISME J., 2023, 17, 1765–1773 CrossRef PubMed.
- B. Ou, C. Garcia, Y. Wang, W. Zhang and G. Zhu, Biotechnol. Bioeng., 2018, 115, 2467–2478 CrossRef CAS PubMed.
- K. E. Tyo, P. K. Ajikumar and G. Stephanopoulos, Nat. Biotechnol., 2009, 27, 760–765 CrossRef CAS PubMed.
- T. H. Segall-Shapiro, E. D. Sontag and C. A. Voigt, Nat. Biotechnol., 2018, 36, 352–358 CrossRef CAS PubMed.
- R. Zhang, Y. Yang, J. Wang, Y. Lin and Y. Yan, Metab. Eng., 2019, 55, 85–91 CrossRef CAS PubMed.
- A. S. Markus Fiedler, Gene, 2001, 274, 111–118 CrossRef PubMed.
- W. R. Dong, L. X. Xiang and J. Z. Shao, Appl. Environ. Microbiol., 2010, 76, 2295–2303 CrossRef CAS PubMed.
- L. Vidal, J. Pinsach, G. Striedner, G. Caminal and P. Ferrer, J. Biotechnol., 2008, 134, 127–136 CrossRef CAS PubMed.
- T. Xiong, Y. Bai, T.-P. Fan, X. Zheng and Y. Cai, Biotechnol. Appl. Biochem., 2022, 69, 1909–1916 CrossRef CAS PubMed.
- K. A. Datsenko and B. L. Wanner, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6640–6645 CrossRef CAS PubMed.
- M. Skulj, V. Okrslar, S. Jalen, S. Jevsevar, P. Slanc, B. Strukelj and V. Menart, Microb. Cell Fact., 2008, 7, 6 CrossRef PubMed.
- C. W. Kang, H. G. Lim, J. Yang, M. H. Noh, S. W. Seo and G. Y. Jung, Metab. Eng., 2018, 48, 121–128 CrossRef CAS PubMed.
- M. Barroca, P. Rodrigues, R. Sobral, M. M. R. Costa, S. R. Chaves, R. Machado, M. Casal and T. Collins, Sci. Rep., 2016, 6, 39329 CrossRef CAS PubMed.
- M. Terrinoni, S. L. Nordqvist, S. Kallgard, J. Holmgren and M. Lebens, Appl. Environ. Microbiol., 2018, 84(4), e02143 CrossRef PubMed.
- S. Kosuri, D. B. Goodman, G. Cambray, V. K. Mutalik, Y. Gao, A. P. Arkin, D. Endy and G. M. Church, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 14024–14029 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra03707c |
| This journal is © The Royal Society of Chemistry 2024 |