
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterocycles as supramolecular handles for crystal engineering: a case study with 7-(diethylamino)coumarin derivatives†
Geraldyne Castro a,
Margarita Romero-Ávilaa,
Norberto Farfán*a,
Rafael Arcos-Ramos*b and
Mauricio Maldonado-Domínguez*a
a,
Margarita Romero-Ávilaa,
Norberto Farfán*a,
Rafael Arcos-Ramos*b and
Mauricio Maldonado-Domínguez*a
aFacultad de Química, Departamento de Química Orgánica, Universidad Nacional Autónoma de México, Ciudad de México, México. E-mail: imardio@comunidad.unam.mx
bDepartamento de Química de Radiaciones y Radioquímica, Instituto de Ciencias Nucleares, Universidad Nacional Autónoma de México, Ciudad de México, México
First published on 1st July 2024
Abstract
In this study, we present the synthesis and detailed solid-state structural characterization of a Schiff-base-bridged derivative of 7-(diethylamino)coumarin (7-DAC), a molecular block displaying repetitive aggregation modes in the solid state despite being attached to broadly different molecular frameworks. To map the supramolecular habits of this unconventional moiety, we carry out a comparative analysis of the crystal packing in a curated dataset of 50 molecules decorated with the 7-DAC group, retrieved from the literature. We uncover that self-recognition of the 7-DAC moiety has two main components: a set of directional C–H⋯O interactions between neighboring coumarins, and antiparallel dipole–dipole interactions, taking the form of distinct π-stacking modes. The pendant 7-diethylamino group is key to the behavior of 7-DAC, favoring its solubilization through its conformational flexibility in solution, while in the crystalline matrix, it acts as a structural spacer that favors π-stacking interactions. Our findings present a comprehensive analysis of the preferential arrangements of the 7-DAC fragment in various (supra)molecular scenarios, confirming that it is (i) a mobile but mostly planar group, (ii) a group prone to antiparallel aggregation, and (iii) up to 90% likely to pack via π-stacking supported by hydrogen-bonding interactions. These findings enrich the palette of supramolecular motifs available for the bottom-up design of organic materials and their programmed construction.
Introduction
Molecular materials are continuous phases whose smallest components are molecules. These materials, frequently encountered as glassy or crystalline solids, are held together mainly by non-covalent intermolecular interactions, weaker and less directional than the covalent and ionic bonds present in continuous materials such as diamond, glass, and table salt. Constructing a molecular material requires encoding in molecular structures the necessary information to assemble a macroscopic phase, ideally with precise characteristics and predictable behavior. This bottom-up approach to the design of molecular materials can strongly accelerate the advance of urgent technologies like cleaner light sources,1 ultradense storage of digital data,2,3 efficient sunlight harvesting,4 and the miniaturization of electronic devices.5Encoding macroscopic properties into microscopic and discrete molecular structures is one of the challenges in developing molecular materials.6–9 There are functional groups prone to dictate aggregation, like carboxylic acids which form H-bonded dimers in solution,10 and nucleobases, which readily associate via complementary H-bonding interactions to form DNA.11,12 As we will present, similar patterns are observed in heteroaromatic structures such as coumarin derivatives (Fig. 1).
 | ||
| Fig. 1 Hydrogen bonding motifs in (A) carboxylic acids, (B) 7-(diethylamino)coumarin (7-DAC). | ||
As these examples show, functional groups that can engage in H-bonding tend to tailor aggregation due to the strength and directionality of hydrogen bonds, which make them effective handles for supramolecular assembly.13 In addition to H-bonds, halogen bonds,14 and π-stacking between aromatic rings15,16 interplay to determine the final arrangement within organic solids. Since a material is often required to display a macroscopic response, an ideal molecular building block should display the desired electronic property or response, like luminescence or charge-carrier conductivity, and at the same time it must possess a molecular structure that directs the supramolecular assemblage of the material from the bottom-up, to display the desired property.
Within the field of programmable molecular aggregation, we currently investigate the usability of heterocyclic fragments to direct self-assembly through their hierarchical organization of non-covalent interactions, with a primary focus on H-bond patterning and the concomitant formation of π-stacked supramolecular assemblies. Heterocycles engage in π-stacking like their carbocyclic counterparts, and the heteroatoms in their structure imbue them with distinct polarities and H-bonding capabilities. From the vast ecosystem of heterocyclic rings, we focus on coumarins which, in our experience, tend to form crystalline solids effortlessly.17–19 Derivatives of 7-DAC, (Fig. 2) stands out in this respect, because (a) they readily crystallize, (b) the 7-DAC motif appears to self-recognize, displaying characteristic stacking patterns, and (c) 7-DAC-derivatives tend to be reasonably soluble in common organic solvents, such as: CH2Cl2, CHCl3, toluene, THF, acetone, EtOAc, ACN, EtOH, MeOH, and DMSO. These qualities are remarkable because functional groups displaying predictable aggregation modes can serve as building blocks for the design and construction of (supra)molecular materials, especially if such aggregation patterns are transferable between different molecular architectures.
 | ||
| Fig. 2 Molecular structure of 7-(diethylamino)coumarin (7-DAC), a functionalized heterocycle prone to self-assembly, primarily via coumarin–coumarin C–H⋯O bonds and dipole–dipole interactions of the π-stacking type. A secondary motif that is frequently observed comprises C–H⋯O bonding between the –NEt2 and carbonyl groups from neighboring molecules. | ||
As we previously found, coumarin-based molecules exhibit a propensity to self-assemble into one-dimensional (1D) or two-dimensional (2D) supramolecular π-stacked arrays; this characteristic makes them interesting candidates for organic electronic applications, where the predictable aggregation of π-conjugated molecules is paramount, and where the ideal candidate molecules are also processable in solution. With these requirements in mind, we have explored the viability of distinct covalent linkers to introduce the 7-DAC moiety to molecules with different native functionalities. Complementarily, we have elucidated the impact of such functionalization on the materials' electronic structure and on its packing arrangement in the solid state in several 7-DAC derivatives.
As part of our investigation, we currently explore the integration of the 7-DAC core to nitrogenated heterocycles, as plausible interfaces between 7-DAC and complexes of transition metal ions. In this context, the 1,2,4-triazole ring caught our attention due to its known effectiveness as part of corrosion inhibitors for low-carbon steel,20–26 and to its use as a building block of light-emitting devices in organic electronic applications due to the luminescent properties and ready thin-film formation of several of its derivatives.27–31 Moreover, it has recently become an important synthon for Metal–Organic Frameworks (MOFs) and coordination polymers. Since the molecular packing of 1,2,4-triazole derivatives tends to be directed by H-bonding interactions, it allows the controlled bottom-up construction of MOFs and it has been applied to Fe(II) coordination polymers that present spin-crossover behavior.32–37
In light of the aforementioned considerations, the impact of incorporating a 4-amino-1,2,4-triazole (51) moiety into the 7-DAC nucleus was investigated to assess its influence on the solid-state organization.27,31,38 The amino group (–NH2) is a plausible precursor to an imine or Schiff base π-bridge for the targeted derivative (Scheme 1). The imine linkage, R–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–R′, lacks the ability to be a classical hydrogen bond donor group.31–33 We hypothesize that this H-bond-impaired functionality will allow weaker (or less directional) forces, such as dipolar interactions and planar π-stacking arrangements, to dominate the crystal phase arrangement.39,40
N–R′, lacks the ability to be a classical hydrogen bond donor group.31–33 We hypothesize that this H-bond-impaired functionality will allow weaker (or less directional) forces, such as dipolar interactions and planar π-stacking arrangements, to dominate the crystal phase arrangement.39,40
 | ||
| Scheme 1 Synthesis of compound 11. | ||
In this work, we present the synthesis, characterization, and crystal structure of a novel Schiff-base-bridged 7-DAC derivative, and we compare its solid-state structure with the full set of 52 crystalline 7-DAC derivatives retrieved from the literature. This analysis aims to comprehensively evaluate the predictability of the 7-DAC group orientation and non-covalent interactions within molecular solids. At the same time, we computed frontier molecular orbital energies of all the studied compounds, using density functional theory, charting this way the structural and electronic landscape of the 7-DAC group in its known molecular contexts. With this study, we seek to expand the understanding of noncovalent interactions in polar π-systems, enriching the existing palette of molecular handles to tailor the bottom-up design and predictable construction of organic solids.
Materials and methods
Experimental section
All reagents used were obtained from commercial suppliers and used without further purification. Solvents were dried using standard methods or distilled prior to use. Reactions were monitored by TLC on precoated silica gel plates (Aldrich silica gel on TLC plates with 254 nm fluorescent indicator) and revealed by exposure to a UV lamp. 1H and 13C NMR spectra were recorded using a Bruker 400 MHz spectrometer; chemical shifts (δ, ppm) are reported relative to CDCl3. High-resolution mass spectra were acquired with Bruker micrOTOF-Q II spectrometer, and the UV-vis absorption data were acquired with a Thermo Scientific Evolution 220 spectrophotometer.Synthesis of 7-(diethylamino)coumarin-3-carbaldehyde, compound 2
Compound 2 was synthesized from 4-(diethylamino)salicylaldehyde in two steps according to a literature procedure,41,42 to give an orange crystalline solid. Yield: 79%. Rf = 0.2 (hexanes/ethyl acetate, 8

![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2). Melting point 163–165 °C. 1H-NMR [400 MHz, CDCl3] (δ, ppm): 10.10 (s, 1H, H-11), 8.23 (s, 1H, H-4), 7.40 (d, J = 9.0 Hz, 1H, H-5), 6.62 (dd, J = 9.0, 2.5 Hz, 1H, H-6), 6.47 (d, J = 2.5 Hz, 1H, H-8), 3.46 (q, J = 7.2 Hz, 4H, H-9), 1.24 (t, J = 7.2 Hz, 6H, H-10). 13C-NMR [75 MHz, CDCl3] (δ, ppm): 188.0 (C-11), 162.0 (C-2), 159.0 (C-8a), 153.6 (C-7), 145.5 (C-4), 132.6 (C-5), 114.4 (C-3), 110.3 (C-6), 108.3 (C-4a), 97.2 (C-8), 45.4 (C-9), 12.6 (C-10).
:2). Melting point 163–165 °C. 1H-NMR [400 MHz, CDCl3] (δ, ppm): 10.10 (s, 1H, H-11), 8.23 (s, 1H, H-4), 7.40 (d, J = 9.0 Hz, 1H, H-5), 6.62 (dd, J = 9.0, 2.5 Hz, 1H, H-6), 6.47 (d, J = 2.5 Hz, 1H, H-8), 3.46 (q, J = 7.2 Hz, 4H, H-9), 1.24 (t, J = 7.2 Hz, 6H, H-10). 13C-NMR [75 MHz, CDCl3] (δ, ppm): 188.0 (C-11), 162.0 (C-2), 159.0 (C-8a), 153.6 (C-7), 145.5 (C-4), 132.6 (C-5), 114.4 (C-3), 110.3 (C-6), 108.3 (C-4a), 97.2 (C-8), 45.4 (C-9), 12.6 (C-10).
Synthesis of (E)-3-(((4H-1,2,4-triazol-4-yl)imino)methyl)-7-(diethylamino)-2H-chromen-2-one, compound 11
In a round bottom flask equipped with a Dean–Stark distillation trap, 4-amino-4H-1,2,4-triazole (0.017 g, 0.2 mmol) and 7-(diethylamino)coumarin-3-carbaldehyde (2) (0.05 g, 0.2 mmol) were suspended in 5 mL of a 2
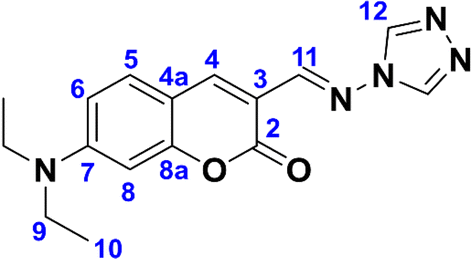 :1 mixture of EtOH and toluene. The mixture was stirred at reflux temperature for 150 h, frequently draining the Dean–Stark trap and adding more solvent mixture accordingly. The product was obtained as an orange precipitate. The solution was cooled in an ice bath, the solid was filtered and washed with cold toluene. The crude product was purified by chromatography on silica gel, starting with CH2Cl2 100%, followed by a linear gradient of CH2Cl2:acetone up to an 8:2 v/v ratio, to give the product as a bright orange solid that grows crystals from CH2Cl2 (0.16 mmol, 50 mg, 80%). Rf = 0.15 (CH2Cl2:acetone 9:1). Melting point (from CH2Cl2): 238–240 °C. FTIR-ATR (ν, cm−1): 3105, 3080, 2968, 2950, 2930, 1701, 1603, 1570, 1516, 1502, 1478, 1424, 1351, 1255, 1188, 1180, 1127. 1H-NMR [400 MHz, CDCl3] (δ, ppm): 8.75 (s, 1H, H-11), 8.57 (s, 2H, H-12), 8.40 (s, 1H, H-4), 7.40 (d, J = 9.0 Hz, 1H, H-5), 6.65 (dd, J = 9.0, 2.5 Hz, 1H, H-6), 6.49 (d, J = 2.5 Hz, 1H, H-8), 3.46 (q, J = 7.2 Hz, 4H, H-9), 1.24 (t, J = 7.2 Hz, 6H, H-10). 13C-NMR [75 MHz, CDCl3] (δ, ppm): 161.4 (C-2), 158.1 (C-8a), 153.0 (C-7), 151.8 (C-11), 142.2 (C-4), 138.3 (C-12), 131.5 (C-5), 110.3 (C-6), 110.1 (C-3), 108.5 (C-4a), 97.3 (C-8), 45.3 (C-9), 12.6 (C-10). HRMS (ESI): calculated for C16H17N5O2Na ([M + Na]+) 334.1274, found [C16H17N5O2 + Na]+ 334.1283. Error: 2.693583 ppm.
:1 mixture of EtOH and toluene. The mixture was stirred at reflux temperature for 150 h, frequently draining the Dean–Stark trap and adding more solvent mixture accordingly. The product was obtained as an orange precipitate. The solution was cooled in an ice bath, the solid was filtered and washed with cold toluene. The crude product was purified by chromatography on silica gel, starting with CH2Cl2 100%, followed by a linear gradient of CH2Cl2:acetone up to an 8:2 v/v ratio, to give the product as a bright orange solid that grows crystals from CH2Cl2 (0.16 mmol, 50 mg, 80%). Rf = 0.15 (CH2Cl2:acetone 9:1). Melting point (from CH2Cl2): 238–240 °C. FTIR-ATR (ν, cm−1): 3105, 3080, 2968, 2950, 2930, 1701, 1603, 1570, 1516, 1502, 1478, 1424, 1351, 1255, 1188, 1180, 1127. 1H-NMR [400 MHz, CDCl3] (δ, ppm): 8.75 (s, 1H, H-11), 8.57 (s, 2H, H-12), 8.40 (s, 1H, H-4), 7.40 (d, J = 9.0 Hz, 1H, H-5), 6.65 (dd, J = 9.0, 2.5 Hz, 1H, H-6), 6.49 (d, J = 2.5 Hz, 1H, H-8), 3.46 (q, J = 7.2 Hz, 4H, H-9), 1.24 (t, J = 7.2 Hz, 6H, H-10). 13C-NMR [75 MHz, CDCl3] (δ, ppm): 161.4 (C-2), 158.1 (C-8a), 153.0 (C-7), 151.8 (C-11), 142.2 (C-4), 138.3 (C-12), 131.5 (C-5), 110.3 (C-6), 110.1 (C-3), 108.5 (C-4a), 97.3 (C-8), 45.3 (C-9), 12.6 (C-10). HRMS (ESI): calculated for C16H17N5O2Na ([M + Na]+) 334.1274, found [C16H17N5O2 + Na]+ 334.1283. Error: 2.693583 ppm.
Database search, retrieval, and curation of experimental XRD data
A search on the Crystallography Open Database (05/02/2024),43–46 7-DAC as parent fragment, produced a total of 82 crystal structures. Solvates, cocrystals, and molecules not containing the 7-DAC fragment were excluded from our analysis. We found 50 distinct molecules and 52 distinct crystal structures (including two examples of polymorphism), and the corresponding structural data was retrieved as CIF files. Database IDs and structural parameters of all the retrieved cells are condensed in Table S2.†Single X-ray diffraction experiments
A suitable single crystal of compound 11 was mounted on a glass fiber. Crystallographic data were collected with an Oxford Diffraction Gemini Atlas diffractometer with a CCD area detector; the radiation using a monochromator of graphite with λMoKα = 0.71073 Å, at 122 K. The double pass method of scanning was used to exclude any noise. The collected frames were integrated by using an orientation matrix determined from the narrow frame scans. CrysAlisPro and CrysAlisRED software package47 were used for data collection and integration. Analysis of the integrated data did not reveal any decay. Collected data were corrected for absorption effects by a numerical absorption correction48 using a multifaceted crystal model based on expressions upon the Laue symmetry with equivalent reflections. The structure solution and refinement were carried out with the OLEX2 analysis program.49 Mercury50 was used to prepare artwork representation material for publication. Full-matrix least-squares refinement was carried out by minimizing (Fo2 − Fc2)2. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms attached to carbon atoms were placed in geometrically idealized positions and refined as riding on their parent atoms, with C–H = 0.95–0.99 Å with Uiso(H) = 1.2Ueq(C) for aromatic and methylene groups, and Uiso(H) = 1.5Ueq(C) for methyl groups. Crystallographic data have been deposited at the Cambridge Crystallographic Data Center as ESI CCDC: 2338583.† The structural cell parameters of compound 11 are condensed in Table 1. The ORTEP diagram is depicted in Fig. 3.| Chemical formula: C16H17N5O2 |
| Formula weight = 311.35 |
| T = 122 K |
| Crystal system: monoclinic, space group: P21/c |
| a = 14.3674(10)Å, α = 90° |
| b = 10.3669(7)Å, β = 103.365(7)° |
| c = 10.6054(6)Å, γ = 90° |
| V = 1538.12(18) Å3, Z = 4 |
| Dx = 1.344 g cm−3 |
| Radiation: Mo Kα (l = 0.71073 Å) |
| µ(Mo Kα) = 0.093 mm−1 F(0 0 0) = 656.3 |
| Crystal size = 0.1053 × 0.2728 × 0.4735 mm3 |
| No. of reflections collected = 4035 |
| No. of independent reflections = 3267 |
| 2θmax = 59.1° with Mo Kα |
| Theta range for data collection: 3.9530 to 29.249° |
| Index ranges: −18 ≦ h ≦ 19, −14 ≦ k ≦ 14, −14 ≦ l ≦ 14 |
| Completeness to theta = 25.2417°, 99.8% |
| Data/restraints/parameters = 3267/0/210 |
| Final R indices [I > 2sigma(I)] R1 = 0.0422, wR2 = 0.1308 |
| R indices (all data): R1 = 0.0563, wR2 = 0.1474 |
| Goodness-of-fit on F2 = 1.0953 |
| Largest diff. peak and hole (eÅ−3): 0.3835, −0.2745 |
| Refinement method: full-matrix least-squares on F2 |
| Measurement: Oxford Diffraction Gemini Atlas diffractometer |
| Data collection & cell refinement program: CrysAlisPro and CrysAlisRED |
| Structure solving & refinement program: OLEX2 |
| CCDC: 2338583 |
 | ||
| Fig. 3 ORTEP diagram of compound 11 (CCDC no. 2338583†). | ||
Density functional theory calculations
We optimized all molecular geometries using the MN15 density functional51 with the def2TZVP basis set.52 Convergence thresholds for this procedure were: 10−8 Ha for energy gradients, 1.8 × 10−4 Ha Å−1 for maximum force, and 4.5 × 10−4 Å for maximum atomic displacement. Vibrational frequencies were calculated with the same scheme, using the harmonic approximation, observing only real frequencies for all minima and a single imaginary frequency for transition states. The Conductor-like Polarizable Continuum Model, CPCM,52,53 was employed to approximate the polarity of a molecular crystal as a continuum of dielectric constant ε = 3.5, which is the average of the value range observed experimentally for polar molecules, from ε = 2 to ε = 5.54,55 For brevity, the DFT method just described, MN15(CPCM)/def2TZVP, will be referred to simply as the DFT protocol throughout this work. Frontier molecular orbital energies were calculated using time-dependent DFT with the DFT protocol, allowing 20 states in the configuration interaction singles (CIS) procedure.56–58 Results from calculations are condensed in the ESI file, in Table S4.†Results and discussion
Synthesis of a Schiff-base-bridged 7-DAC-derivative and analysis of its crystal structure
As depicted in Scheme 1, 7-(diethylamino)coumarin-3-carbaldehyde (2) and 4-amino-4H-1,2,4-triazole (51) were suspended in a 2:1 mixture of EtOH:toluene under reflux temperature, removing the wet solvent from a Dean–Stark trap and replacing it as needed. The reaction proceeded to obtain the Schiff-base-bridged 7-DAC-derivative (11) which after column chromatography purification was obtained with a yield of 80%.
Compound 11 was characterized by FTIR, 1H NMR, 13C NMR, UV/vis, and HRMS. Detailed information and the spectral data are provided in ESI.†
The FTIR spectrum exhibited several bands between 1400 and 3100 cm−1. The stretching frequency of the azomethine group (CN) in 11 is observed at 1701 cm−1 which agrees with the literature range for similar compounds.59,60 This well-defined band confirms the formation of the Schiff base. The structure of 11 was also confirmed by NMR. 1H NMR spectra in CDCl3 as a solvent, shows the azomethine proton (H–CN) as a singlet approximately at 8.75 ppm, and for the 1,2,4-triazol-4-yl protons (NC–H) a singlet appears at 8.57 ppm. The protons of the 7-DAC core exhibited no shifting with respect to the 7-(diethylamino)coumarin-3-carbaldehyde (2). For the 13C NMR studies, a signal commonly assigned to the azomethine carbon atom (CN) is observed at 151.8 ppm consistent with previous reports.59
On the other hand, the electronic absorption spectra [Fig. S7†] were measured in CHCl3, at room temperature. The compound showed one intense absorption band centered at 471 nm with a shoulder at 450 nm, and 3 overlapped low absorption bands in the UV region (between 250 and 300 nm). The low-intensity absorption band can be assigned to n → π* (azomethine group; –CN–) and π → π* transitions, respectively, according to previous reports.32,33 The maximum emission corresponds to π → π* transitions (λ = 471 nm, ε = 61973) due to the conjugation of the 7-DAC with the 1,2,4-triazol-4-yl fragment.
Compound 11 was obtained as a crystalline orange solid, soluble in polar solvents, protic and aprotic, like AcOEt and EtOH. Suitable crystals for single X-ray diffraction were obtained from slow evaporation of a CH2Cl2 dissolution. Relevant crystallographic data is summarized in Table 1. The crystal structure of 11 was solved in the monoclinic space group P21/c with one molecule per asymmetric unit (Z = 4, Z′ = 1). The crystal packing is discussed in terms of hydrogen bonding interactions and planar π-stacking between coumarin cores, ethyl residues, or substituent residues. As a visual guide, we employ a color code that describes each type of interaction found: H-bonding interactions between the carbonyl group CO of the 7-DAC and any C–H, N–H, or O–H unit is represented with orange dots ( ), i.e., [N–H
), i.e., [N–H O]. Any other type of supramolecular interaction (hydrogen-bonding or π-stacking) between any of the groups, cores, or residues previously mentioned, is represented with magenta dots (
O]. Any other type of supramolecular interaction (hydrogen-bonding or π-stacking) between any of the groups, cores, or residues previously mentioned, is represented with magenta dots ( ), i.e., [C–H
), i.e., [C–H N]. A full description of all interactions for each crystal is available in Table S3.†
N]. A full description of all interactions for each crystal is available in Table S3.†
The detailed analysis of the crystal packing of 11 reveals that the carbonyl group interacts with the triazole ring via cyclic hydrogen bonding interactions [C–H O, R21 (7)], producing the anti-parallel pairing of molecular units [Fig. 4A]. Anti-parallel pairs interact with their neighbors through (a) cyclic hydrogen-bonding involving the ethyl chains and the triazole ring [C–H
O, R21 (7)], producing the anti-parallel pairing of molecular units [Fig. 4A]. Anti-parallel pairs interact with their neighbors through (a) cyclic hydrogen-bonding involving the ethyl chains and the triazole ring [C–H N, R22 (8)] and (b) complementary hydrogen-bonding interactions [C–H
N, R22 (8)] and (b) complementary hydrogen-bonding interactions [C–H N] between triazole subunits [Fig. 4B], resulting in a multilayered self-assembled π-stacked structure [Fig. 4C].
N] between triazole subunits [Fig. 4B], resulting in a multilayered self-assembled π-stacked structure [Fig. 4C].
 | ||
Fig. 4 Crystal structure analysis of 11: relevant hydrogen-bonding interactions between the (A) carbonyl coumarin substituent, (B) ethyl substituent, (B) ethyl substituent, and (C) multilayered π-stacked arrangement. substituent, and (C) multilayered π-stacked arrangement. | ||
Analysis of molecular crystals containing the DAC moiety
The full set of 50 7-DAC-substituted coumarins considered in this study, labeled 1–50, is presented in Fig. 5. The crystal structure for compound 11 is reported in this work. Crystal structures for 1–10 and 12–50 were retrieved from the Crystallography Open Database. All crystals were inspected for the presence or absence of planar π-stacking, defined as the interplanar distance, dstack less or equal to 4.5 Å, between π-systems tilted at most 45 °C relative to each other. We found that 47 out of 52 (90%) examples present various forms of planar π-stacking. | ||
| Fig. 5 Molecular structures of 7-DAC-containing derivatives analyzed in this work and retrieved from the Crystallographic Open Database. | ||
The 7-DAC-H parent compound 1 displays a full anti-parallel π-stacking in both available polymorphs, derived from cyclic [C–H O] interactions between adjacent coumarins and supported by [C–H
O] interactions between adjacent coumarins and supported by [C–H O] bonding between carbonyl and ethyl chains [Fig. 2].17 The carbonyl group in position 2 of the 7-DAC-core is key to direct self-assembly, displaying synergistic interactions with various substituents at positions C-3 and C-4 of the coumarin skeleton. These interactions facilitate an anti-parallel coumarin alignment and promote the formation of various modes of planar π-stacking.
O] bonding between carbonyl and ethyl chains [Fig. 2].17 The carbonyl group in position 2 of the 7-DAC-core is key to direct self-assembly, displaying synergistic interactions with various substituents at positions C-3 and C-4 of the coumarin skeleton. These interactions facilitate an anti-parallel coumarin alignment and promote the formation of various modes of planar π-stacking.
Our studies on extending π-conjugation in 7-DAC derivatives using various π-linkers (amide, alkene, alkyne, phenylene, triazole)61 have evinced the impact of distinct molecular and electronic structures on the solid-state self-assembly in this family of compounds. During this continuing effort, we have produced a series of 7-DAC derivatives that exemplify how the inherent supramolecular information within the coumarin core dictates the establishment of primary hydrogen bonding interactions; these interactions, in turn, govern the formation of diverse π-stacked arrangements dependent upon the C-3 substituent. To illustrate this we can consider the crystal packing observed in representative 7-DAC-starting materials, such as 7-(diethylamino)coumarin-3-carbaldehyde (2) and 7-(diethylamino)coumarin-3-carboxylic acid (3), where the coumarin carbonyl group directs self-assembly through hydrogen-bonding interactions [C–H O, ethyl
O, ethyl carbonyl coumarin] [C–H
carbonyl coumarin] [C–H O, ethyl/coumarin
O, ethyl/coumarin substituent] and [C–H
substituent] and [C–H O, ethyl
O, ethyl carbonyl coumarin] [C–H
carbonyl coumarin] [C–H O, ethyl/coumarin
O, ethyl/coumarin substituent] respectively, regardless of the functional group in position 3 of the coumarin bicycle (Fig. 6).
substituent] respectively, regardless of the functional group in position 3 of the coumarin bicycle (Fig. 6).
 | ||
| Fig. 6 Selected examples illustrate how the coumarin carbonyl group acts as a versatile handle in different supramolecular assemblies, demonstrating a cooperative effect with various substituents. | ||
Functionalization of position 3 in the coumarin heterocycle allows the extension of the π-conjugated system through different π-connectors (triazole (19), amide (22), alkyne (53) and p-phenylene (54)). Remarkably, the carbonyl group in position 3 of these π-extended coumarins remains in control of self-assembly through H-bonding interactions. This prevalent motif is usually accompanied by antiparallel dipole–dipole interactions and weak contacts involving the pendant ethyl chains from the –NEt2 group in position 7 of the coumarin system and the different substituents at the C-3 position [Fig. S8†]: [C–H O, coumarin
O, coumarin carbonyl coumarin] [CO
carbonyl coumarin] [CO π, carbonyl coumarin
π, carbonyl coumarin substituent] for (19) [Fig. S8†]; [C–H
substituent] for (19) [Fig. S8†]; [C–H O, substituent
O, substituent carbonyl coumarin] [C–H
carbonyl coumarin] [C–H O, coumarin
O, coumarin substituent] for (22) [Fig. 7]; [C–H
substituent] for (22) [Fig. 7]; [C–H O, coumarin
O, coumarin carbonyl coumarin] [C–H
carbonyl coumarin] [C–H O, ethyl
O, ethyl coumarin] [C–H
coumarin] [C–H N, substituent
N, substituent substituent] for (53); [C–H
substituent] for (53); [C–H O, substituent
O, substituent carbonyl coumarin] [C–H
carbonyl coumarin] [C–H O, ethyl
O, ethyl substituent] for (54), respectively.
substituent] for (54), respectively.
 | ||
| Fig. 7 Selected examples (53 and 54) illustrate how the coumarin carbonyl group acts as a versatile handle in different supramolecular assemblies, demonstrating a cooperative effect with various substituents in a series of π-extended 7-DAC derivatives. | ||
Building upon the preceding discussion, we extended our analysis to 7-DAC derivatives obtained from the COD database taking into account the structural features of the substituent such as size, shape, and interaction capabilities. Data on the hierarchy of observed interactions, the alignment between coumarin cores, the π-stacking modes, and detailed analysis of the directing and non-directing functional groups present in each derivative are summarized in Table S3 of the ESI† file.
In general, the introduction of electron-donating or electron-withdrawing substituents, as well as the presence of fused rings or heterocycles, does not alter the ability of the coumarin carbonyl group to direct self-assembly via hydrogen bonding [C–H O] and dipolar interactions [CO
O] and dipolar interactions [CO CO]. Coumarin molecules are generally aligned in an anti-parallel fashion enabling diverse π-stacking modes governed by their planarity, polarity, and stackable surface [Fig. 8].
CO]. Coumarin molecules are generally aligned in an anti-parallel fashion enabling diverse π-stacking modes governed by their planarity, polarity, and stackable surface [Fig. 8].
 | ||
| Fig. 8 Selected examples (8, 15, 23, 47) to show how the carbonyl group acts as a building block within supramolecular assemblies, showcasing its cooperative effect with complex substituents like rings or fused cycles. | ||
In certain cases, structural features of substituents or rings attached to the 7-DAC nucleus can modify or override coumarin–coumarin interactions, as in compounds 14, 32, 40, 43, 48, and 50. Said structural features include size, shape, and π-stackable surface area, and can hinder the formation of the usual anti-parallel one-dimensional coumarin arrangements and in some cases promote the formation of parallel-aligned coumarins [Fig. S9†]. For these 7-DAC derivatives, self-assembly is primarily governed by substituent–mediated interactions, particularly through π-stacking between fragments with large-stackable surface areas. This effect is particularly pronounced in derivatives 40, 48 and 50, where interactions the 7-DAC-nucleus act as a spacer, complementing the substituent's ability to form two-dimensional π-stacked assemblies [Fig. 9].
 | ||
| Fig. 9 Selected examples where interactions from the 7-DAC-nucleus function as spacers, thereby complementing the substituent's ability to form two-dimensional π-stacked assemblies. | ||
Molecular and electronic structure of the DAC-H parent block
The 7-DAC-H parent compound 1 possesses a strong ground-state permanent dipole, µ7-DAC = 6.97 D (Fig. 10A), calculated with the DFT protocol described in Materials and methods. For comparison, the dipole moments of water and dimethylsulfoxide, two archetypal polar molecules, estimated with the same method are µH2O = 1.85 D and for µDMSO = 4.06 D. Therefore, the frequent appearance of antiparallel π-stacking in crystalline 7-DAC-derivatives (Fig. 10B), is a combination of electrostatic and geometric features. The electrostatic potential map on the molecular surface of 1 displays a complex pattern of locally positive and negative regions (Fig. 10A), which are self-complementary as evinced by the propensity of the 7-DAC fragment to form stacked supramolecular dimers. As shown in Fig. S10,† the molecular dipole moments µ calculated with the DFT protocol for a set of reference molecules lie within 10% of their experimental values, supporting our choice of DFT protocol. | ||
| Fig. 10 (A) The strong dipole moment of the 7-DAC-H parent compound 1, and the electrostatic potential (ESP) map on the surface of its molecular electron density. (B) The antiparallel π-stacking of 7-DAC fragments, a common motif in 7-DAC containing molecular crystals, can be seen as a combination of dipole–dipole interaction and planar intermolecular contacts. | ||
The dynamic nature of the pendant 7-diethylamino group in 7-DAC is as important as its electronic properties for the observed trends. Besides contributing to the polarity of this building block through the push–pull effect,18,19 the pendant Et2N– group contributes entropically to the solubility of a compound incorporating it, following:
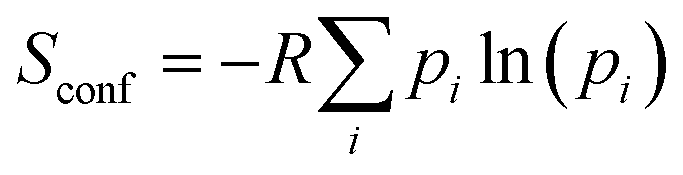 | (1) |
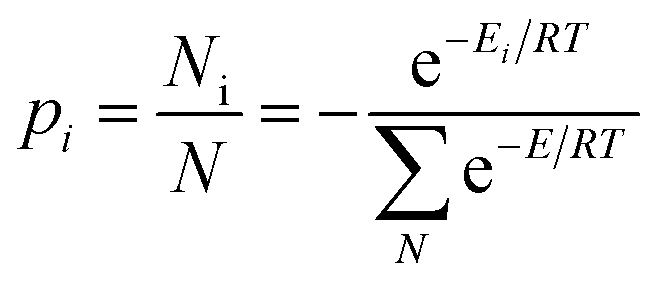 | (2) |
 | (3) |
 | ||
| Fig. 11 The conformational space of 7-DAC features seven accessible microstates, separated by low free-energy barriers to the torsion of the pendant ethyl chains. | ||
The result from eqn (3) implies that the sevenfold increase in the total number of conformational microstates from the solid state to the solution phase triples the equilibrium constant towards dissolution, compared to a rigid group with no flexibility. Importantly, the size and shape of the ethyl chains are sufficiently small to allow dense packing in the solid state in all 7-DAC-containing molecular crystals, a potentially useful balance between solubility and unhindered dense stacking.
As a final remark to this section, the highest torsional barrier in Fig. 11 has a height of 3.4 kcal mol−1 which, approximated using Eyring's transition state theory:
 | (4) |
Frontier molecular orbital energies of DAC-containing molecules
We have focused our interest on the solid state of organic heterocycles, among them coumarins, because their propensity for dense packing with offset π–π stacking makes them attractive to investigate their capability as charge carrier phases in the context of organic materials for optoelectronics. Electron energy levels in continuous materials are delocalized as bands and electronic transitions occur mainly near the highest-energy occupied and the lowest-energy unoccupied bands, known as the valence and conduction bands, respectively. In conductors, these bands overlap, while semiconductor materials display an energy gap between the valence and conduction bands. In contrast, discrete molecular materials localize most of their electron density on individual molecules, and their corresponding electron transitions tend to be similarly localized. Such a scenario typically involves the highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO), apart by a HOMO–LUMO gap akin to the valence-conduction gap in inorganic semiconductors.62 Importantly, the energy eigenvalues of the frontier molecular orbitals, HOMO and LUMO, are accessible through quantum chemical calculation.In optoelectronic materials, the luminescent or semiconducting response of a given molecular material tends to correlate with frontier molecular orbital (FMO) energy values, which serve as proxies of the ionization potential (IP) and electron affinities (EA) of the molecule; IP and EA are critical parameters for candidate discrimination and for the selection of suitable electrodes for the implementation of a given material into an electronic devices such as a diode or a transistor.63–66 Frontier molecular orbital energies were calculated using a DFT protocol based on an accurate density functional from the Minnesota family (MN15), a global hybrid functional with excellent performance in scenarios with electron degeneracy, as is the case of organic molecules with extensive π conjugation. Details are provided in the Materials and methods section and the scatter of FMO values for all compounds 1–50 are presented in Fig. 12. All numerical values for FMO energies and optimized molecular geometries are provided in ESI.†
 | ||
| Fig. 12 (A) Frontier molecular orbital (FMO) energies of compounds 1–50, calculated at the MN15(CPCM)/def2TZVP level using density functional theory. Average FMO energies are represented by continuous lines, in colors cyan for EHOMO and magenta for ELUMO. Deviations from the mean are shown as colored areas around each average line, enclosing ±σ, one standard deviation. All energies are expressed in electronvolts. (B) Isocontours for the HOMO and LUMO of compound 11, highlighting the delocalized nature of these molecular orbitals. Surfaces were rendered on an isovalue of 0.02. | ||
As shown in Fig. 12A, the FMO energies for the existing 7-DAC derivatives are in most of the cases centered around ĒHOMO ± σHOMO = −6.29 ± 0.17 eV, which is in the range of values (−6.6 to −4.4 eV) reported for n-type semiconductors.66 Moreover, the average LUMO energy is ĒLUMO ± σLUMO = −1.51 ± 0.28 eV, within the range of values (−1.2 to −3.6 eV) reported for p-type semiconductors. This ambivalent behavior is illustrated in Fig. 12B with the frontier molecular orbitals (FMOs) of compound 11 which, as seen, are delocalized over the entire π-system. Such delocalization suggests that the 7-DAC fragment, in addition to being a supramolecular element, can also contribute through most of its molecular surface to the active orbitals of a semiconducting molecular material built upon it, be it an N-type electron-conducting material or a P-type hole-conducting material.
Summarizing, we presented a detailed structural and electronic characterization of 7-diethylaminocoumarin (7-DAC), and compared its impact on the crystal structure of a molecular solid containing it. The rigidity, planarity, and strong electric dipole of 7-DAC make it an unconventional structure-directing functional group, that can be readily incorporated as a substituent into different molecular architectures. A common result is aggregation by a combination of antiparallel stacking and H-bonding in 90% of the known experimental examples. We synthesized and crystallized a highly nitrogenated 7-DAC derivative 11, which followed the general observed trend. Heterocyclic molecular fragments are a mostly uncharted space of structure-directing groups, that will be available to the crystal engineer when their preferred aggregation and the physical determinants of it, are known. We contribute to this understanding, focused on the 7-DAC fragment.
Conclusions
The coumarin heterocycle displays a strong propensity for planar π-stacking, a feature that can be employed to direct the aggregation of molecules into self-assembled molecular materials. In this work, we synthesize the highly nitrogenated aminotriazole derivative 11, covalently tethered to the 7-diethylaminocoumarin (7-DAC) fragment via a Schiff-base bridge. We then subject compound 11, and its corresponding molecular crystal structure, to a comparative analysis of all available molecular crystals containing the 7-DAC fragment, which we have consistently observed as a group that dictates the supramolecular arrangement in molecular crystals. In general hydrogen-bonding interactions between the carbonyl group in position 3 of the coumarin heterocycle in 7-DAC and C–H, N–H, or O–H involving either the coumarin core, ethyl chains or substituent residues could play a crucial role in this tendency. We found that 90% of the 52 reported molecular crystal structures containing the 7-DAC fragment display planar π-stacking, imposed by the coumarin planar structure and its strong electric dipole moment. We obtained a new molecular crystal containing the 7-DAC moiety, displaying the same general pattern of aggregation in the crystal phase showcased by 7-DAC-decorated compounds. The 7-diethylamino group is a key component of DAC, contributing to (i) the strong electric dipole present in DAC, and (ii) the solubility of DAC-containing compounds due to its favorable conformational entropy. Electronically, DAC-derivatives can behave as n-type and p-type semiconductors and can, therefore, be versatile building blocks for the design of supramolecular arrangements with precisely tuned electronic structures.Data availability
Crystallographic data for the structures reported in this article are available in Crystallography Open Database at https://www.crystallography.net/cod/. The corresponding structural data was retrieved as CIF files. Database IDs and structural parameters of all the retrieved cells are condensed in Tables S2 and S3.† All other relevant data generated and analyzed during this study, which include experimental, spectroscopic, crystallographic, and computational data supporting the findings of this study are available within the paper and its ESI† files.Author contributions
Geraldyne Castro: investigation, methodology, and writing – original draft. Margarita Romero-Ávila: investigation, methodology. Norberto Farfán: funding acquisition. Rafael Arcos-Ramos: investigation, methodology, formal analysis, data curation, and writing – review and editing. Mauricio Maldonado-Domínguez: conceptualization, software, investigation, data curation, and writing – review and editing, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. L. C. H. acknowledges CONAHCYT for the granted PhD scholarship (CVU 72652). R. A. R. acknowledges IN-100722 (DGAPA-PAPIIT). M. M. D. acknowledges funding to projects 5000-9220 (FQ-PAIP) and IA-201024 (DGAPA-PAPIIT). We thank M. Flores-Álamo for collecting the X-ray diffraction data.References
- J. Song, H. Lee, E. G. Jeong, K. C. Choi and S. Yoo, Adv. Mater., 2020, 32, 1907539 CrossRef CAS PubMed.
- R. Achal, M. Rashidi, J. Croshaw, T. R. Huff and R. A. Wolkow, ACS Nano, 2020, 14, 2947–2955 CrossRef CAS PubMed.
- L. Ceze, J. Nivala and K. Strauss, Nat. Rev. Genet., 2019, 20, 456–466 CrossRef CAS PubMed.
- M. Vasilopoulou, A. Fakharuddin, A. G. Coutsolelos, P. Falaras, P. Argitis, A. R. bin, M. Yusoff and M. K. Nazeeruddin, Chem. Soc. Rev., 2020, 49, 4496–4526 RSC.
- Y. Zhao, W. Liu, J. Zhao, Y. Wang, J. Zheng, J. Liu, W. Hong and Z.-Q. Tian, Int. J. Extreme Manuf., 2022, 4, 022003 CrossRef CAS.
- A. R. Oganov, C. J. Pickard, Q. Zhu and R. J. Needs, Nat. Rev. Mater., 2019, 4, 331–348 CrossRef.
- J. G. P. Wicker and R. I. Cooper, CrystEngComm, 2015, 17, 1927–1934 RSC.
- T. R. Walsh, Acc. Chem. Res., 2017, 50, 1617–1624 CrossRef CAS PubMed.
- G. R. Desiraju, Angew Chem. Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS.
- J. Chen, C. L. Brooks and H. A. Scheraga, J. Phys. Chem. B, 2008, 112, 242–249 CrossRef CAS PubMed.
- H. M. Berman, W. K. Olson, D. L. Beveridge, J. Westbrook, A. Gelbin, T. Demeny, S. H. Hsieh, A. R. Srinivasan and B. Schneider, Biophys. J., 1992, 63, 751–759 CrossRef CAS PubMed.
- N. B. Leontis, Nucleic Acids Res., 2002, 30, 3497–3531 CrossRef CAS PubMed.
- L. J. Prins, D. N. Reinhoudt and P. Timmerman, Angew. Chem., Int. Ed., 2001, 40, 2382–2426 CrossRef CAS PubMed.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- J. Ma, N. Han, H. Yu, J. Li, J. Shi, S. Wang, H. Zhang and M. Wang, Small, 2022, 18, 2202167 CrossRef CAS PubMed.
- J.-H. Deng, J. Luo, Y.-L. Mao, S. Lai, Y.-N. Gong, D.-C. Zhong and T.-B. Lu, Sci. Adv., 2020, 6, eaax9976 CrossRef CAS PubMed.
- R. Arcos-Ramos, M. Maldonado-Domínguez, J. Ordóñez-Hernández, M. Romero-Ávila, N. Farfán and M. d. P. Carreón-Castro, J. Mol. Struct., 2017, 1130, 914–921 CrossRef CAS.
- E. González-Rodríguez, B. Guzmán-Juárez, M. Miranda-Olvera, M. d. P. Carreón-Castro, M. Maldonado-Domínguez, R. Arcos-Ramos, N. Farfán and R. Santillan, Spectrochim. Acta, Part A, 2022, 267, 120520 CrossRef PubMed.
- M. Maldonado-Domínguez, R. Arcos-Ramos, M. Romero, B. Flores-Pérez, N. Farfán, R. Santillan, P. G. Lacroix and I. Malfant, New J. Chem., 2014, 38, 260–268 RSC.
- Sheetal, R. Batra, A. K. Singh, M. Singh, S. Thakur, B. Pani and S. Kaya, Corros. Eng., Sci. Technol., 2023, 58, 73–101 CrossRef CAS.
- M. E. Belghiti, F. Benhiba, N. Benzbiria, C. H. Lai, S. Echihi, M. Salah, A. Zeroual, Y. Karzazi, A. Tounsi, K. Abbiche, S. Belaaouad, H. Elalaoui-Elabdallaoui and Y. Naimi, J. Mol. Struct., 2022, 1256, 132515 CrossRef CAS.
- A. K. Singh, M. Singh, S. Thakur, B. Pani, S. Kaya, B. EL Ibrahimi and R. Marzouki, Surf. Interfaces, 2022, 33, 102169 CrossRef CAS.
- Sheetal, S. Sengupta, M. Singh, S. Thakur, B. Pani, P. Banerjee, S. Kaya and A. K. Singh, J. Mol. Liq., 2022, 354, 118890 CrossRef CAS.
- Y. G. Avdeev, T. A. Nenasheva, A. Y. Luchkin, A. I. Marshakov and Y. I. Kuznetsov, Coatings, 2023, 13, 1221 CrossRef CAS.
- S. John, A. Joseph, T. Sajini and A. James Jose, Egypt. J. Pet., 2017, 26, 721–732 CrossRef.
- B. El Mehdi, B. Mernari, M. Traisnel, F. Bentiss and M. Lagrenée, Mater. Chem. Phys., 2002, 77, 489–496 CrossRef.
- V. Nazarov, D. Miroshnichenko, O. Ivakh, S. Pyshyev and B. Korchak, Mini-Rev. Org. Chem., 2022, 20, 394–402 CrossRef.
- M. Irfan, H. A. Khan, S. Bibi, G. Wu, A. Ali, S. G. Khan, N. Alhokbany, F. Rasool and K. Chen, Sci. Rep., 2024, 14, 2732 CrossRef CAS PubMed.
- H. Xu, P. Sun, K. Wang, Y. Miao, T. Yang, H. Wang, B. Xu and W. Y. Wong, Tetrahedron, 2016, 72, 4408–4413 CrossRef CAS.
- Y. Tao, Q. Wang, L. Ao, C. Zhong, C. Yang, J. Qin and D. Ma, J. Phys. Chem. C, 2010, 114, 601–609 CrossRef CAS.
- I. Matulková, I. Němec, K. Teubner, P. Němec and Z. Mička, J. Mol. Struct., 2008, 873, 46–60 CrossRef.
- H. Benaissa, M. Wolff, K. Robeyns, G. Knör, K. Van Hecke, N. Campagnol, J. Fransaer and Y. Garcia, Cryst. Growth Des., 2019, 19, 5292–5307 CrossRef CAS.
- H. Benaissa, N. N. Adarsh, K. Robeyns, J. J. Zakrzewski, S. Chorazy, J. G. M. Hooper, F. Sagan, M. P. Mitoraj, M. Wolff, S. Radi and Y. Garcia, Cryst. Growth Des., 2021, 21, 3562–3581 CrossRef CAS.
- A. D. Naik, M. M. Dîrtu, A. Léonard, B. Tinant, J. Marchand-Brynaert, B.-L. Su and Y. Garcia, Cryst. Growth Des., 2010, 10, 1798–1807 CrossRef CAS.
- A. P. Railliet, A. D. Naik, P. Castanho-Vaz, A. Rotaru, M. Grigoras, N. Lupu, J. Marchand-Brynaert and Y. Garcia, Hyperfine Interact., 2013, 217, 67–72 CrossRef CAS.
- S. I. Vasylevs'kyy, G. A. Senchyk, A. B. Lysenko, E. B. Rusanov, A. N. Chernega, J. Jezierska, H. Krautscheid, K. V. Domasevitch and A. Ozarowski, Inorg. Chem., 2014, 53, 3642–3654 CrossRef PubMed.
- M. M. Dîrtu, N. N. Adarsh, A. D. Naik, K. Robeyns and Y. Garcia, New J. Chem., 2016, 40, 9025–9029 RSC.
- Y. R. Xi, X. K. Chen, Y. T. Wang, G. M. Tang, X. M. Chen, Y. S. Wu and S. N. Wang, J. Mol. Struct., 2021, 1243, 130893 CrossRef CAS.
- G. R. Desiraju, Acc. Chem. Res., 2002, 35, 565–573 CrossRef CAS PubMed.
- C. Janiak and T. G. Scharmann, Polyhedron, 2003, 22, 1123–1133 CrossRef CAS.
- J.-S. Wu, W.-M. Liu, X.-Q. Zhuang, F. Wang, P.-F. Wang, S.-L. Tao, X.-H. Zhang, S.-K. Wu and S.-T. Lee, Org. Lett., 2007, 9, 33–36 CrossRef CAS PubMed.
- J. Ordóñez-Hernández, A. Jiménez-Sánchez, H. García-Ortega, N. Sánchez-Puig, M. Flores-Álamo, R. Santillan and N. Farfán, Dyes Pigm., 2018, 157, 305–313 CrossRef.
- A. Vaitkus, A. Merkys, T. Sander, M. Quirós, P. A. Thiessen, E. E. Bolton and S. Gražulis, J. Cheminf., 2023, 15, 123 Search PubMed.
- A. Merkys, A. Vaitkus, A. Grybauskas, A. Konovalovas, M. Quirós and S. Gražulis, J. Cheminf., 2023, 15, 25 Search PubMed.
- A. Vaitkus, A. Merkys and S. Gražulis, J. Appl. Crystallogr., 2021, 54, 661–672 CrossRef CAS PubMed.
- S. Gražulis, D. Chateigner, R. T. Downs, A. F. T. Yokochi, M. Quirós, L. Lutterotti, E. Manakova, J. Butkus, P. Moeck and A. Le Bail, J. Appl. Crystallogr., 2009, 42, 726–729 CrossRef PubMed.
- Agilent, CrysAlis PRO and CrysAlis RED, Agilent Technologies Ltd, 2013 Search PubMed.
- R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A: Found. Adv., 1995, 51, 887–897 CrossRef.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- H. S. Yu, X. He, S. L. Li and D. G. Truhlar, Chem. Sci., 2016, 7, 5032–5051 RSC.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- H. Cui, K. Takahashi, Y. Okano, H. Kobayashi, Z. Wang and A. Kobayashi, Angew. Chem., Int. Ed., 2005, 44, 6508–6512 CrossRef CAS PubMed.
- J. Harada, M. Ohtani, Y. Takahashi and T. Inabe, J. Am. Chem. Soc., 2015, 137, 4477–4486 CrossRef CAS PubMed.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS.
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218–8224 CrossRef CAS.
- K. C. Ko, J.-S. Wu, H. J. Kim, P. S. Kwon, J. W. Kim, R. A. Bartsch, J. Y. Lee and J. S. Kim, Chem. Commun., 2011, 47, 3165–3167 RSC.
- K. Ambroziak, Z. Rozwadowski, T. Dziembowska and B. Bieg, J. Mol. Struct., 2002, 615, 109–120 CrossRef CAS.
- J. E. De La Cerda-Pedro, R. Arcos-Ramos, M. Maldonado-Domínguez, S. Rojas-Lima, M. Romero-Ávila, M. P. Carreón-Castro, R. Santillan, N. Farfán and H. López-Ruiz, CrystEngComm, 2016, 18, 5562–5571 RSC.
- R. A. K. Yadav, D. K. Dubey, S.-Z. Chen, T.-W. Liang and J.-H. Jou, Sci. Rep., 2020, 10, 9915 CrossRef CAS PubMed.
- J. F. Janak, Phys. Rev. B: Solid State, 1978, 18, 7165–7168 CrossRef CAS.
- P. Politzer and F. Abu-Awwad, Theor. Chem. Acc., 1998, 99, 83–87 Search PubMed.
- S. Hamel, P. Duffy, M. E. Casida and D. R. Salahub, J. Electron Spectrosc. Relat. Phenom., 2002, 123, 345–363 CrossRef CAS.
- D. Cagardová and V. Lukeš, Acta Chim. Slovaca, 2017, 10, 6–16 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2338583. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra03656e |
| This journal is © The Royal Society of Chemistry 2024 |