DOI:
10.1039/D4RA03637A
(Paper)
RSC Adv., 2024,
14, 24828-24837
Development and in vitro evaluation of ursolic acid-loaded poly(lactic-co-glycolic acid) nanoparticles in cholangiocarcinoma†
Received
17th May 2024
, Accepted 11th July 2024
First published on 8th August 2024
Abstract
Cholangiocarcinoma (CCA), an epithelial biliary tract malignancy, is a significant health concern in the Greater Mekong Subregion, particularly in northeastern Thailand. Prior to the development of advanced stages, CCA is typically asymptomatic, thereby limiting treatment options and chemotherapeutic effectiveness. Ursolic acid (UA), a triterpenoid derived from plants, was previously discovered to inhibit CCA cell growth through induction of apoptosis. Nevertheless, the therapeutic effectiveness of UA is limited by its poor solubility in water and low bioavailability; therefore, dimethyl sulfoxide (DMSO) is utilized as a solvent to treat UA with CCA cells. Enhancing cellular uptake and reducing toxicity, the utilization of polymeric nanoparticles (NPs) proves beneficial. In this study, UA-loaded PLGA nanoparticles (UA-PLGA NPs) were synthesized using nanoprecipitation and characterized through in silico formation analysis, average particle size, surface functional groups and ζ-potential measurements, electron microscopic imaging, drug loading efficiency and drug release studies, stability, hemo- and biocompatibility, cytotoxicity and cellular uptake assays. Molecular dynamics simulations validated the loading of UA into PLGA via hydrogen bonding. The synthesized UA-PLGA NPs had a spherical shape with an average size of 240 nm, a negative ζ-potential, good stability, great hemo- and bio-compatibility and an encapsulation efficiency of 98%. The NPs exhibited a characteristic of a simple diffusion-controlled Fickian process, as predicted by the Peppas–Sahlin drug release kinetic model. UA-PLGA NPs exhibited cytotoxic effects on KKU-213A and KKU-055 CCA cells even when dispersed in media without organic solvent, i.e., DMSO, highlighting the ability of PLGA NPs to overcome the poor water solubility of UA. Rhodamine 6G (R6G) was loaded into PLGA NPs using the same approach as UA-PLGA NPs, demonstrating effective delivery of the dye into CCA cells. These findings suggest that UA-PLGA NPs showed promise as a potential phytochemical delivery system for CCA treatment.
Introduction
Cholangiocarcinoma (CCA), often known as bile duct cancer, is a highly malignant condition characterized by the neoplasm of the epithelium that lines the biliary tract. Although classified as an uncommon type of cancer, the global incidence has increased significantly during the past few decades.1 CCA presents a significant public health concern in Thailand, mostly in its northeastern region (85 cases per 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 individuals per year).1 As evidenced by several studies, the high incidence of CCA in the region has been associated with Opisthorchis viverrini, a carcinogenic liver fluke.2–4 Infection with O. viverrini can lead to chronic inflammation, malignant cholangiocyte transformation, oxidative stress, and CCA development.5 Early-stage CCA is typically asymptomatic; as a result, late detection is common, which further complicates treatment options.6 When feasible, liver surgical resection offers the best outcome; however, only one-third of patients diagnosed with CCA are eligible for surgical resection.7 In the case of patients with very early-stage intrahepatic CCA, liver transplants are considered a viable option.8 Combining gemcitabine and cisplatin (GC) is the conventional method for managing patients with advanced unresectable CCA; however, the prognosis is dismal in such particular cases, possibly due to low effectiveness and undesirable off-target effects, which contributes to a poor prognosis and mortality rate for CCA patients.9,10 Additionally, the GC regimen may give rise to hematological effects, fatigue, and nausea.11,12 Therefore, there is an urgent need for the development of an alternative therapeutic formulation with fewer adverse effects for the treatment of CCA.
000 individuals per year).1 As evidenced by several studies, the high incidence of CCA in the region has been associated with Opisthorchis viverrini, a carcinogenic liver fluke.2–4 Infection with O. viverrini can lead to chronic inflammation, malignant cholangiocyte transformation, oxidative stress, and CCA development.5 Early-stage CCA is typically asymptomatic; as a result, late detection is common, which further complicates treatment options.6 When feasible, liver surgical resection offers the best outcome; however, only one-third of patients diagnosed with CCA are eligible for surgical resection.7 In the case of patients with very early-stage intrahepatic CCA, liver transplants are considered a viable option.8 Combining gemcitabine and cisplatin (GC) is the conventional method for managing patients with advanced unresectable CCA; however, the prognosis is dismal in such particular cases, possibly due to low effectiveness and undesirable off-target effects, which contributes to a poor prognosis and mortality rate for CCA patients.9,10 Additionally, the GC regimen may give rise to hematological effects, fatigue, and nausea.11,12 Therefore, there is an urgent need for the development of an alternative therapeutic formulation with fewer adverse effects for the treatment of CCA.
In recent decades, nanotechnology-based drug delivery methods have enhanced the effectiveness of conventional chemotherapy.13 Poly(lactic-co-glycolic acid) (PLGA) is a type of copolymer that has been approved by the U.S. Food and Drug Administration (FDA) to function as a carrier for various substances – including genes, drugs, peptides, and proteins – due to its controlled release capabilities, biodegradability, and biocompatibility.14 PLGA was used to conjugate TAT cell penetrating peptide and transferrin onto selenocysteine-loaded microspheres (MSNs) for enhanced cervical cancer chemotherapy and radiation therapy.15 PLGA was also utilized to develop an injectable PLGA-core hydrogel device that delivered microRNA-222 and aspirin, with the aim of enhancing bone regeneration and innervation.16 PLGA nanoparticles (NPs) have the potential to accumulate passively in the tumor vascularized area and deliver their contents into cells through the enhanced permeability and retention (EPR) effect.17 For the treatment of CCA, few specific nanodrug delivery strategies based on PLGA have been developed. For instance, atractylodin, a major bioactive hydrophobic compound identified in Atractylodes lancea (Thunb) D.C., was encapsulated into PLGA NPs using the solvent displacement method.18 PLGA NPs loaded with atractylodin demonstrated enhanced cytotoxicity against CCA cells and improved water solubility. Additionally, these NPs exhibited high blood dispersion, substantial accumulation, extensive distribution in the gastrointestinal tract, and a comparatively slower clearance rate in comparison to free atractylodin.19 Moreover, PLGA NPs encapsulated with α-mangostin, a major xanthone mainly present in the pericarp of the mangosteen (Garcinia mangostana Linn.) fruit, inhibited CCA cell proliferation, induced apoptosis and improved water solubility of α-mangostin.20
Ursolic acid (UA), alternatively referred to as 3-β-3-hydroxy-urs-12-ene-28-oic acid, is a pentacyclic triterpenoid found in numerous fruits and herbs, such as apple peels and holy basil leaves.21 UA has been reported to exhibit a wide range of biological properties, including anti-inflammatory, anti-cancer and antioxidant activities,22 making it a promising candidate for therapeutic applications. Anti-cancer properties of UA have been demonstrated against a variety of cancers, including hepatocellular carcinoma, breast cancer, gallbladder cancer, prostate cancer and CCA, through numerous mechanisms, including inhibiting tumor angiogenesis and metastasis, causing apoptosis and cell cycle arrest, and decreasing cell proliferation.23–27 Furthermore, UA was found to suppress tumor development of gallbladder cancer and hepatocellular carcinoma, according to in vivo investigations.27,28 As a Bioavailability Classification System (BCS) class IV compound, UA has limited water solubility, resulting in low bioavailability and therapeutic efficacy. Previous studies have demonstrated the anti-cancer efficacy of PLGA NPs encapsulated with UA produced by nanoprecipitation or solvent evaporation technique to treat melanoma29 and pancreatic ductal adenocarcinoma.30 Furthermore, it has been recently demonstrated that UA-loaded chitosan-coated PLGA NPs possess anti-breast cancer potential.31 Despite these promising findings, the bioavailability of UA remains a challenge due to its poor water solubility. This study aimed to develop UA-loaded PLGA NPs (UA-PLGA NPs) and evaluate its anti-cancer efficacy against CCA cells. The UA-PLGA NPs were synthesized via nanoprecipitation. Molecular dynamics (MD) simulations were used to investigate NPs formation and interactions between UA and PLGA. The fabricated NPs were characterized in terms of particle size, ζ-potential, polydispersity index (PDI), surface functional groups and stability using transmission electron microscopy (TEM), scanning electron microscopy (SEM), dynamic light scattering (DLS) and Fourier-transform infrared (FTIR) spectroscopy. Encapsulation efficiency, drug loading and release profile of UA-PLGA NPs were determined using high performance liquid chromatography (HPLC). Cytotoxic effects, cellular uptake, hemo- and biocompatibility studies of PLGA-based NPs were performed using MTT assays, fluorescence microscopy and UV-visible spectroscopy.
Materials and methods
Chemicals
PLGA with lactic acid:glycolic acid ratio of 50:50 (Resomer RG 502H, Mw 7000–17000 Da), poly(vinyl alcohol) (PVA; Mw 30000–70000), UA, paraformaldehyde, Triton X-100 and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Merck KGaA (Darmstadt, Germany). Acetone, acetonitrile and methanol, all HPLC grade, were acquired from RCI Labscan Limited (Bangkok, Thailand). Dimethyl sulfoxide (DMSO, HPLC grade) was acquired from Altmann Analytical GmbH & Co (Munich, Germany). 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI) was obtained from Thermo Scientific (MA, USA). Rhodamine 6G (R6G) was purchased from Tokyo Chemical Industry Co. Ltd (Tokyo, Japan). Ham's F12 nutritional combination, fetal bovine serum (FBS), and penicillin-streptomycin were acquired from Gibco (MA, USA). Sheep blood was purchased from clinical diagnostics limited partnership (Bangkok, Thailand).
Cell lines and cell culture
KKU-213A (JCRB1557) and KKU-055 (JCRB1551) CCA cell lines were obtained from the Japanese Collection of Research Bioresources (JCRB) Cell Bank and Cholangiocarcinoma Research Institute (CARI), Khon Kaen University, Khon Kaen, Thailand. Cells were cultured in Ham's F12 complete medium (Gibco, MD, USA) supplemented with 10% fetal bovine serum, 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin, and maintained at 37 °C in a humidified atmosphere containing 5% CO2. The culture medium was replaced every other day and subjected to subculturing upon reaching 80% confluence.
Preparation of polymeric NPs
PLGA (200 mg) and UA (20 mg) were dissolved in acetone (20 mL) and gradually added dropwise to a 0.1% (w/v) PVA aqueous solution (20 mL) using a YSP-101 syringe pump (YMC Co. Ltd, Kyoto, Japan) with a flow rate of 0.4 mL min−1. Following overnight stirring at ambient temperature to allow the organic solvent to evaporate, the UA-PLGA NPs were collected using centrifugation method at 14000 rpm for 30 min. The resulting pellet was redispersed in DI water, washed twice to remove residual PVA, freeze-dried, and stored at 4 °C until use. Blank PLGA NPs were prepared using the same procedure, omitting the addition of UA during the synthesis. For the preparation of fluorescent NPs, PLGA NPs were loaded with R6G by combining R6G (20 mg) with PLGA (200 mg) in the presence of 0.1% (w/v) PVA under identical conditions as previously described for UA, employing the nanoprecipitation method.
Characterization of NPs
The UA-PLGA NPs were characterized based on size, PDI, surface charge, surface morphology, surface functional groups, the efficacy of UA loading, cellular uptake, and in vitro release of UA as per standard procedure mentioned below.
Measurement of particle size, PDI, and ζ-potential. The UA-PLGA NPs suspension was diluted and the hydrodynamic diameter (Dh), PDI and ζ-potential were measured at 25 °C by DLS using a Malvern Zetasizer Nano ZS (Malvern Panalytical Ltd, UK). Each result was measured in triplicate.
Transmission electron microscopy (TEM). The actual size and uniformity of NPs were evaluated by TEM (Tecnai G2 20 FEI, CR). The sample was prepared by placing a drop of sample suspension on a carbon-coated copper grid, which was dried thereafter in air and examined under TEM. The diameter of the UA-PLGA NPs was measured from TEM images using ImageJ software.
Scanning electron microscopy (SEM). The morphology of UA-PLGA NPs was determined using SEM (Carl Zeiss, Stuttgart, Germany) at 5.0 kV. The NPs were fixed on SEM sample stubs by placing UA-PLGA NPs onto double-sided carbon tape and coated with a thin layer of gold/palladium for 180 s in a vacuum prior to measurement.
Fourier-transform infrared (FTIR) spectroscopy. FTIR was used to detect surface functional groups and encapsulation of UA into PLGA NPs. The lyophilized samples were analyzed on a Tensor 27 FT-IR Spectrometer (Bruker, USA). With air as the background, the FTIR spectra of the samples were obtained over the wavenumber range from 4000–500 cm−1.
UV-visible spectroscopy. The UV-visible spectra of R6G, R6G-PLGA NPs, and blank PLGA NPs were measured in the wavelength range of 250 to 650 nm using a UV-1800 spectrophotometer (Shimadzu, Tokyo, Japan). R6G solution (5 μg mL−1), R6G-PLGA NPs suspension (2 mg mL−1), and blank PLGA NPs suspension (2 mg mL−1) were dissolved in a mixture of acetonitrile and methanol (1:1, v/v).
Drug loading content and encapsulation efficiency. The concentration of UA in NPs was determined using a HPLC Instrument Shimadzu LC-20A system (Shimadzu, Tokyo, Japan) and an Agilent C-18 analytical column (5 μm, 150 × 4.6 mm). The mobile phase consisted of methanol:acetonitrile (80:20), the column was eluted at a flow rate of 0.6 mL min−1, and monitoring was performed at 210 nm. An aliquot of UA-PLGA NPs suspension was added to a mixture of acetonitrile:methanol (1:1), vortexed, sonicated, and centrifuged at 14000 rpm for 15 min. The supernatant was filtered and analyzed using HPLC. UA was measured using a standard solution of 0–80 μg mL−1 UA in methanol. After determining the concentration (in μg mL−1) of UA encapsulated in UA-PLGA NPs, we calculated the total amount of UA from the weight of UA-PLGA NPs used to prepare different concentrations for cell treatment. Thus, we assessed the equivalent concentrations of free UA and UA encapsulated in the NPs (Table S1†). The R6G loading content was determined by dissolving R6G-PLGA NPs with a mixture of acetonitrile and methanol (1:1, v/v) and spectroscopically analyzed using a UV-visible microplate reader (EZ Read 2000, Biochrom, Cambridge, UK) at 540 nm. A standard curve of R6G (0–30 μg mL−1) in the same solvent mixture was used to calculate the R6G content in the NPs. Drug loading and encapsulation efficiency were calculated using the following equations:
| Drug loading content (%) = weight of drug in NPs/weight of NPs × 100 |
| Encapsulation efficiency (%) = total amount of drug in NPs/amount of drug used in preparation × 100 |
Storage stability of UA-PLGA NPs. The stability of UA-PLGA NPs was investigated using DLS. UA-PLGA NPs were assessed for Dh and ζ-potential after being kept at 4 °C in DI water for a duration of 9 weeks.
Atomistic MD simulations
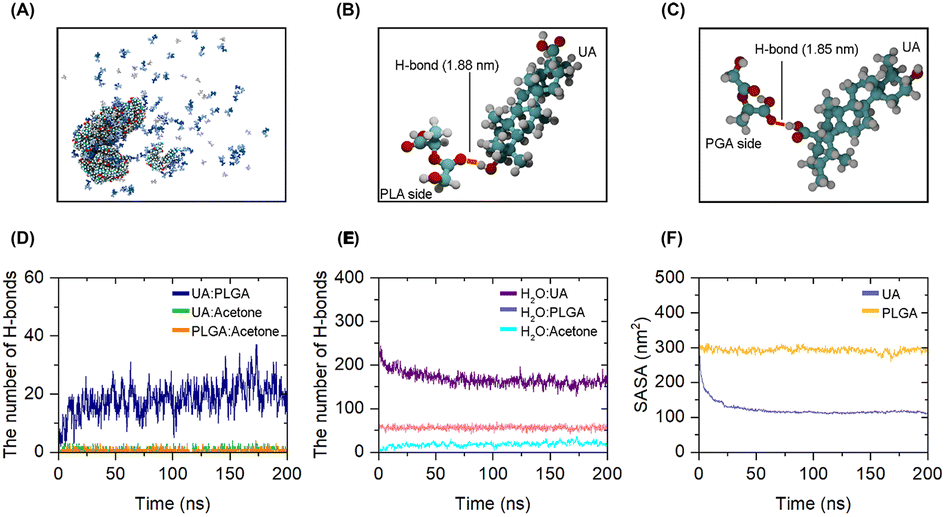
The overall objective of this MD simulation was to gain an insight into the process by which UA was loaded and to determine the molecular interactions that occurred between UA and PLGA copolymer. MD simulations at the atomistic level were conducted using GROMACS version 5.0.2.32 The generalized amber force field (GAFF) was employed to generate full-atom representations of UA, PLGA, and acetone. The UA and polymer model structures were generated and optimized using the Gaussian 09 software package and the HF/6-31G* method.33 The TIP3P model was used to represent water molecules.34 To observe UA loading, the substances were placed randomly within a 10 × 10 × 10 nm3 cubic simulation box filled with water. The simulation was carried out for 200 ns. Each simulation box contained 50 UA, 100 PLGA, and 40 acetone molecules with the positions of each molecule assigned randomly. Solvent accessible surface area (SASA), which is a metric that measures how much of a molecule's surface is accessible to a solvent, was calculated to provide information about the molecular environment. The number of H-bonds formed during a polymeric NP's self-assembly was investigated to study their self-assembly dynamics.
In vitro release studies
This in vitro drug release study was performed to evaluate UA release kinetics and profile from UA-PLGA NPs. The amount of UA released was determined using HPLC. UA-PLGA NPs (10 mg) were dispersed in 10 mL of freshly prepared PBS (pH 7.4) containing 1% (v/v) Tween 80. The solution was placed in shaker incubator (Innova 3100, Eppendorf, Germany) adjusted at 37 °C and 120 rpm. One mL of sample was collected at predetermined intervals (0.5–360 h) and 1 mL of PBS was added to solution after each sampling. The supernatant obtained after centrifugation at 14000 rpm for 15 min was utilized to determine the concentration of released UA via HPLC equipped with a UV detector set at 210 nm. To explore the kinetics of drug release, the following mathematical models (eqn (1)–(6)) were constructed using data from in vitro release studies and DDsolver, an add-in software for Microsoft Excel:35| | |
Zero-order model F = k0t
| (1) |
| | |
First-order model F = 100[1 − e(−k1t)]
| (2) |
| | |
Higuchi model F = kHt0.5
| (3) |
| | |
Korsmeyer-Peppas model F = kKPtn
| (4) |
| | |
Peppas–Sahlin F = k1tm + k2t2m
| (5) |
| | |
Weibull F = 100[1 − e−(t−T)β/α]
| (6) |
where F is the fraction of cumulative drug release at time t, t is the time, k0, k1, kH and kKP represent the release rate constant in the zero order, first order, Higuchi and Korsmeyer-Peppas model, respectively. The release exponent n indicates the mechanism of drug release. The Peppas–Sahlin equation is represented by the diffusional exponent m and the release rate constants, k1 and k2, which correspond to the Fickian and Case-II relaxation kinetics, respectively. In the case of Weibull equation, T is the lag time, α is the scale parameter at time dependent and β is the shape of curve. Determining that the model with the highest R2 and MSC, as well as the lowest AIC, was the most suitable for describing the drug release mechanism.
Cytotoxicity assay
To evaluate the cytotoxicity of free UA and UA-loaded PLGA NPs and to assess the biocompatibility of blank PLGA NPs, the MTT assays were performed against KKU-213A and KKU-055 CCA cells. Cells at a density of 8 × 103 cells per well were cultured in 96-well plates in Ham′s F12 complete media, supplemented with 10% FBS and 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin for 16 h under standard incubation conditions (37 °C and 5% CO2). After the incubation period, the complete medium was withdrawn and various concentrations of UA-PLGA NPs in serum-free medium were added in triplicate for 24 and 48 h. Negative controls included serum-free medium for UA-PLGA NPs treatment and serum-free medium containing 0.25% DMSO for UA treatment. UA solutions were prepared in serum-free medium containing 0.25% DMSO. After 24 and 48 h incubation periods, cells were washed with PBS, pH 7.4 and treated with 100 μL of 0.5 mg mL−1 MTT solution in complete medium for 2 h at 37 °C in the dark. The MTT solution was removed and 100 μL of DMSO was added to dissolve formazan crystals. The absorbance at 540 nm was measured using a microplate reader (Tecan Sunrise, Männedorf, Switzerland). To assess the biocompatibility of blank PLGA NPs in CCA cells, a suspension of blank PLGA NPs (1 mg mL−1) was prepared and treated to both types of CCA cell line using the identical methods used in the UA-PLGA NPs cytotoxic study.
Cell imaging and uptake study
The cell imaging and uptake study of R6G-PLGA NPs was conducted to visualize the internalization of fluorescent dye-encapsulated NPs. CCA cells were seeded at a density of 3 × 104 cells per mL in an 8-well chamber slide. After 2 h-incubation with 300 μL of 300 μg mL−1 R6G-PLGA NPs, the cells were washed with PBS, fixed with 4% paraformaldehyde in PBS, pH 7.4 for 30 min at room temperature. Subsequently, the adherent cells were stained with DAPI solution and washed with PBS, pH 7.4. The images were taken at 20× magnification under a fluorescence microscope (Eclipse TiU, Nikon, Tokyo, Japan).
Hemocompatibility assay
To assess blood compatibility and safety, hemolysis rates of UA-PLGA NPs and blank PLGA NPs were assessed using spectrophotometry and microscopy, following the established approach described by Zhou A. and co-workers.36 Initially, the sheep blood was centrifuged at 2000 rpm for 10 min to remove the supernatant serum and to obtain concentrated red blood cells. The condensed red blood cells were washed with 5 mL of sterile PBS (pH 7.4), and then centrifuged at 3000 rpm for 5 min. After repeating the washing and centrifugation procedures thrice, the cells were diluted to the final density of 20% (v/v). Equal volumes (500 μL) of red blood cell solution and UA-PLGA NPs (0.125, 0.25, 0.5, 1, 2, and 4 mg mL−1) in PBS (pH 7.4) and blank PLGA NPs (0.25, 2, and 4 mg mL−1) in PBS (pH 7.4) were mixed in a 1.5 mL centrifuge tube and incubated at 37 °C for 60 min with gentle agitation using an orbital shaker incubator (S-100D, Comecta, Barcelona, Spain). Photographs of the samples were taken. A solution of 0.1% (v/v) Triton X-100 in PBS (pH 7.4) was used as a positive control and PBS (pH 7.4) was used as a negative control. After centrifugation at 3000 rpm for an additional 10 min, the supernatant (100 μL) was collected and transferred into a 96-well plate. The absorbance at 540 nm was recorded using a UV-visible microplate reader (EZ Read 2000, Biochrom, Cambridge, UK) to determine the hemoglobin content in the supernatant. The proportion of hemolysis was calculated using the following equation:
| % Hemolysis = (Asample/Apositive control) × 100% |
where Asample is the absorbance of the copolymer solution, and Apositive control is the absorbance of 0.1% Triton X-100 solution. Each PLGA NPs sample was tested in duplicate. The microscopic images of red blood cells were captured using a light microscope (Eclipse NiU, Nikon, Tokyo, Japan).
Statistical analyses
The statistical analyses were performed using the DDsolver add-in program, Microsoft Excel and GraphPad Prism 8.0.2 (GraphPad Software Inc., USA). The results were reported as the mean ± standard deviation (SD). The Student's t-test was employed to assess differences between groups. A p-value below 0.05 was considered to be statistically significant.
Results and discussion
Synthesis and characterization of UA-PLGA NPs
UA-PLGA NPs were synthesized via nanoprecipitation as outlined in Scheme 1. The mixture of UA and PLGA was added to the PVA solution, which served as a stabilizer to reduce aggregation and increase the stability of the NPs.37 Afterwards, the organic chemicals and PVA were removed by centrifugation, and the NPs were obtained. The NPs were assessed using various characterization parameters, including size, charge, PDI, surface morphology, and functional groups. UA-PLGA NPs exhibited a Dh of 239.6 ± 7.7 nm, as determined by DLS (Fig. 1A). TEM analysis revealed that the synthesized UA-PLGA NPs were spherical and measured 112.1 ± 31.7 nm in size (Fig. 1B). SEM image also confirmed the spherical shape and exhibited smooth surface of UA-PLGA NPs (Fig. 1C). Moreover, the functional groups of PLGA, UA, and UA-PLGA NPs were characterized using FTIR spectroscopy. As shown in Fig. 1D, PLGA displayed characteristic peaks at 2995 cm−1 (for C–H stretching), 1752 cm−1 (for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), and 1089 cm−1 (for C–O stretching), which were consistent with the literature.38,39 UA exhibited FTIR peaks at 3519 cm−1, 2955 cm−1 and 1713 cm−1, corresponding to –OH stretching, C–H stretching and CO stretching, respectively (Fig. 1E). The spectrum of UA-PLGA NPs contained peaks that aligned with the FTIR spectra of PLGA (Fig. 1F). However, the FTIR peaks of UA were of relatively low intensity, which might have been attributed to the insignificant quantities of UA that were detectable by FTIR spectroscopy.29 In the UA-PLGA NPs, the lack of peak shifts or losses in the FTIR spectrum indicated that the substance and polymer did not engage in any chemical interaction during NP formation.29 DLS measurement showed an average ζ-potential of −16.4 ± 0.6 mV. The ζ-potential distribution graph of UA-PLGA NPs is shown in Fig. S1.† The PDI measurement resulted in 0.07 ± 0.02, suggesting a relatively narrow size dispersity.40 The negative surface charge of UA-PLGA NPs could potentially be attributed to the –COOH group, which repeled particles and prevents aggregation.41,42 Moreover, the stability of UA-PLGA NPs stored in DI water at 4 °C exhibited good Dh and surface charge for 9 weeks, as shown in Fig. S2.† Fluorescent PLGA NPs were successfully produced by loading R6G using the same procedure as for UA-PLGA NPs. The successful incorporation of R6G into PLGA NPs was evident from the pink color of the R6G-PLGA NPs, while the UA-PLGA NPs appeared as a white powder (Fig. S3A†). The UV-visible spectrum of R6G-PLGA NPs further confirmed R6G loading, displaying both the characteristic absorption peak of free R6G at 530 nm and the distinctive peak of blank PLGA NPs at 270 nm (Fig. S3B†).
O stretching), and 1089 cm−1 (for C–O stretching), which were consistent with the literature.38,39 UA exhibited FTIR peaks at 3519 cm−1, 2955 cm−1 and 1713 cm−1, corresponding to –OH stretching, C–H stretching and CO stretching, respectively (Fig. 1E). The spectrum of UA-PLGA NPs contained peaks that aligned with the FTIR spectra of PLGA (Fig. 1F). However, the FTIR peaks of UA were of relatively low intensity, which might have been attributed to the insignificant quantities of UA that were detectable by FTIR spectroscopy.29 In the UA-PLGA NPs, the lack of peak shifts or losses in the FTIR spectrum indicated that the substance and polymer did not engage in any chemical interaction during NP formation.29 DLS measurement showed an average ζ-potential of −16.4 ± 0.6 mV. The ζ-potential distribution graph of UA-PLGA NPs is shown in Fig. S1.† The PDI measurement resulted in 0.07 ± 0.02, suggesting a relatively narrow size dispersity.40 The negative surface charge of UA-PLGA NPs could potentially be attributed to the –COOH group, which repeled particles and prevents aggregation.41,42 Moreover, the stability of UA-PLGA NPs stored in DI water at 4 °C exhibited good Dh and surface charge for 9 weeks, as shown in Fig. S2.† Fluorescent PLGA NPs were successfully produced by loading R6G using the same procedure as for UA-PLGA NPs. The successful incorporation of R6G into PLGA NPs was evident from the pink color of the R6G-PLGA NPs, while the UA-PLGA NPs appeared as a white powder (Fig. S3A†). The UV-visible spectrum of R6G-PLGA NPs further confirmed R6G loading, displaying both the characteristic absorption peak of free R6G at 530 nm and the distinctive peak of blank PLGA NPs at 270 nm (Fig. S3B†).
 |
| | Scheme 1 Schematic representation of UA-PLGA NPs preparation via nanoprecipitation method. | |
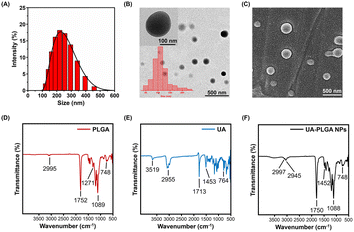 |
| | Fig. 1 (A) Hydrodynamic size distribution; (B) TEM image of UA-PLGA NPs (insets: size distribution histogram and zoomed image); (C) SEM image of UA-PLGA NPs; FTIR spectra of (D) PLGA, (E) UA and (F) UA-PLGA NPs. Scale bar: 500 nm. | |
Molecular interactions during drug loading
MD simulations were used to investigate the loading dynamics of UA molecules into PLGA cavities and their adsorption on the carrier surface. The assembly of PLGA and UA molecules subsequent to their attainment of equilibrium (t = 200 ns; Fig. 2A). It was discovered that hydrogen bonding plays an important role in complex formation. As a result of their hydrophobic nature, UA molecules tend to aggregate to reduce their exposure to the surrounding solvent.43 During the assembly process, two distinct hydrogen bonding patterns between UA and PLGA molecules were observed.44 Polylactic acid (PLA) is composed of lactic acid units. Each unit of lactic acid contains a hydroxyl group (–OH) and a carbonyl group (CO). The oxygen atoms in both the hydroxyl and carbonyl groups can form hydrogen bonds (H-bonds). Our simulation demonstrated that UA could form H-bonds with the PLA side of PLGA via the hydrogen atom of UA bonding to the oxygen atom of PLA (Fig. 2B). In addition, UA formed a complex with the polyglycolic acid (PGA) side of PLGA. In addition, the hydroxyl groups in UA can act as a H-bond donor. As depicted in Fig. 2C, when UA molecules approached the PGA side of PLGA molecules in close proximity, H-bonds can form between the oxygen atoms on the PGA side of PLGA and the hydroxyl groups in UA.
 |
| | Fig. 2 Self-assembly of UA-PLGA NPs. (A) Snapshot of UA-PLGA NPs, which was observed at t = 200 ns; (B) and (C) are H-bonds formed between PLGA and UA during self-assembly. Red, cyan, white represent oxygen, carbon, and hydrogen atoms, respectively; (D) the number of H-bonds as a function of time during complex formation; (E) the number of H-bonds as a function of time between water molecules and UA, PLGA, and acetone molecules as a function of simulation times; (F) SASA as a function of simulation time during complex formation. | |
To gain insights into H-bonds formation, the number of H-bonds formed during complex formation was monitored for 200 ns. It was discovered that UA and PLGA can form complexes via H-bonds. As shown in Fig. 2D, an increase in the number of H-bonds over time may be due to a desire for thermodynamic stability. It should also be noted that UA and acetone did not form H-bonds during assembly process, implying that acetone might not have played a role in strengthening the H-bonds between UA and PLGA. In addition, we discovered that the presence of multiple H-bond donor and acceptor groups on UA can interact with water molecules, as illustrated in Fig. 2E. SASA analysis was used to evaluate the formation of polymeric NPs.45 A decrease in SASA indicated the formation of NPs as well as the interaction and accumulation of system components. Fig. 2F shows how nanoparticle formation reduces the SASA value. The reduction in SASA for UA exceeds that for the PLGA. This could be due to the limited solubility of UA in water, resulting in a higher amount of UA within the inner parts of polymeric PLGA NPs.
Drug loading, encapsulation efficiency and release profile
Using HPLC, the encapsulation and loading efficiencies of UA were determined to be 98 and 11%, respectively. For R6G-PLGA NPs, the R6G loading content was determined to be 0.13% using the standard curve shown in Fig. S3C.† The drug release profile of UA-PLGA NPs was investigated using HPLC under physiological conditions (37 °C and a pH of 7.4; Fig. 3A). A slow release of UA was observed following a 360 h incubation period. During the initial 4 h, a burst release phase of encapsulated UA was observed, which accounted for 17%. This was succeeded by a sustained release of 42% over the course of 24 h. At 360 h, prolonged release persisted at 52%. UA was released from the NPs in accordance with sustained release kinetics. A comparable pattern of release was found in the previously reported study on docetaxel-loaded pegylated (PEG)-PLGA NPs.46 Moreover, six different release models, including zero order, first order, Higuchi, Korsmeyer-Peppas, Peppas–Sahlin and Weibull were fitted to the in vitro UA release data (Fig. 3B). In the process of identifying the best-fit model, evaluation parameters, including the coefficient of determination (R2), Akaike Information Criterion (AIC), and model selection criterion (MSC) values, were determined (Table 1).35 The degree to which the predicted model matches the observed data is indicated by R2 values; values approaching 1 suggest that the predictions are an ideal fit for the data. Additionally, the AIC and MSC were utilized as statistical parameters in the process of selecting the most suitable model. Preferred is the model with the greatest MSC value and the lowest AIC.35,47 Peppas–Sahlin model was the best-fit kinetic model for UA-PLGA NPs due to its lowest AIC and high R2, elevated MSC values, and greater kmodel than k2, implying that UA was released through Fickian diffusion.48,49
 |
| | Fig. 3 In vitro release studies and kinetic release models for UA-PLGA NPs. (A) In vitro release studies of UA at pH 7.4, 37 °C for 360 h, highlighting the burst release pattern over the 24 h; (B) fitting curve from various kinetic drug release models using the DDsolver program, a Microsoft Excel add-in software package. | |
Table 1 Release kinetics parameters (R2, AIC, MSC, kmodel and k2) calculated for UA-PLGA NPsa
| Model |
R2 |
AIC |
MSC |
kmodel |
k2 |
| R2 is the coefficient of determination, AIC is the Akaike Information Criterion, MSC indicates the model selection criteria, kmodel is the kinetic constants of each model and k2 is rate constants for relaxation in Peppas–Sahlin model. |
| Zero-order |
−0.825 |
98.213 |
−1.118 |
0.180 |
— |
| First-order |
−0.521 |
96.211 |
−0.936 |
0.004 |
— |
| Higuchi |
0.208 |
89.036 |
−0.284 |
3.591 |
— |
| Korsmeyer-Peppas |
0.870 |
71.162 |
1.341 |
18.034 |
— |
| Peppas–Sahlin |
0.955 |
61.610 |
2.209 |
13.403 |
−0.819 |
| Weibull |
0.917 |
68.221 |
1.608 |
— |
— |
Cytotoxicity of UA-PLGA NPs and biocompatibility of blank PLGA NPs
The anti-cancer activity of UA-PLGA NPs was investigated in two CCA cell lines (i.e., KKU-213A and KKU-055) using the MTT assay. Free UA was insoluble in culture media and needed to be dissolved in serum-free media with 0.25% DMSO to treat cells.24 However, the organic solvent was not necessary for the dispersion of the UA-PLGA NPs. It should be noted that the comparison of the two conditions was different: free UA was dissolved in serum-free media with 0.25% DMSO, whereas UA-PLGA NPs were solely dissolved in serum-free media. It was found that UA-PLGA NPs inhibited the proliferation of KKU-213A and KKU-055 CCA cells (Fig. 4A). The UA content in the NPs was utilized to determine the half-maximal inhibitory concentration (IC50) values for UA-PLGA NPs. In KKU-213A cells, the IC50 values of UA in UA-PLGA NPs, were 87.6 ± 2.9 and 82.2 ± 19.9 μg mL−1, at 24 and 48 h, respectively (Table S2†). The IC50 values for KKU-055 CCA cells were 65.4 ± 6.2 and 60.1 ± 2.5 μg mL−1 after 24 and 48 h post-treatment, respectively. Following 24 and 48 h of treatment, the IC50 values for UA alone in KKU-213A and KKU-055 CCA cells appeared to be superior to those for UA encapsulated in PLGA NPs: 12.1 ± 0.3 and 8.4 ± 0.2 μg mL−1 and 10.4 ± 0.3 and 10.0 ± 0.2 μg mL−1, respectively. To confirm that the cytotoxicity of UA-PLGA NPs was solely due to UA, the biocompatibility of blank PLGA NPs was assessed (Fig. S4†). The results showed that the viability of CCA cells treated with 1 mg mL−1 of blank PLGA NPs was above 88% at both incubation times, indicating biocompatibility, as viability above 80% compared to control is considered acceptable.50
 |
| | Fig. 4 (A) Cytotoxic effects of UA-PLGA NPs and free UA on KKU-213A and KKU-055 CCA cells. The UA concentration (μg mL−1) was calculated by converting the UA content to μg mL−1 for comparison with free UA. (B) The cellular uptake of RG6-PLGA NPs was examined in KKU-213A and KKU-055 CCA cell lines using fluorescence microscopy. Cell nuclei were stained with DAPI and visualized by blue fluorescence, whereas R6G in NPs was visualized by red fluorescence. Scale bar: 50 μm. | |
This could potentially be attributed to the inability to compare UA content exclusively in serum-free media, i.e., UA dissolved in 0.25% DMSO, as well as the requirement for UA in PLGA-NPs to diffuse gradually from the NPs. These findings revealed that UA-PLGA NPs exhibited anti-cancer properties, a slow-released characteristic and an increase in the solubility of UA in water. This was achieved by encapsulating UA in PLGA NPs, which eliminated the need for prior dissolution in organic solvents.30 These results suggest that UA-PLGA NPs have the potential to deliver UA to CCA cells, sustain the release of UA, and effectively inhibit the CCA cells.
Cellular uptake of R6G-PLGA NPs
R6G, a fluorescent dye was encapsulated within the PLGA NPs using the same procedure as UA-PLGA NPs to enable the tracking of NP internalization into CCA cells.51 The cellular uptake of R6G-PLGA NPs in KKU-213A and KKU-055 CCA cells were investigated using fluorescence microscopy. R6G-PLGA NPs were successfully delivered into CCA cells as shown in Fig. 4B. Both the cytoplasm and nucleus exhibited the red fluorescence signal of R6G. Notably, R6G accumulation was greater in the cytoplasm than in the nucleus. Consequently, the PLGA-based NPs produced via nanoprecipitation in this study exhibited the capability of carrying the encapsulated agents into CCA cells.
Hemocompatibility
The assessment of hemocompatibility is essential for determining the safety and effectiveness of nanomaterials designed for biological purposes. In this study, hemolysis assays were conducted using sheep blood. Fig. 5a demonstrates that the supernatants of both UA-PLGA NPs and blank PLGA NPs, at various concentrations (0.125, 0.25, 0.5, 1, 2, and 4 mg mL−1 for UA-PLGA NPs and 0.25, 1, and 4 mg mL−1 for blank PLGA NPs), had a pale-yellow color resembling that of the PBS group. Conversely, when red blood cells were treated with Triton X-100, they exhibited hemolytic properties and appeared red, indicating that the hemoglobin had leaked out. The hemolysis rate (%) of various concentrations of UA-PLGA NPs and blank PLGA NPs were determined. To be suitable for clinical applications, the hemolysis rate of biomedical materials is required to be lower than 5%.36 Fig. 5b shows the supernatants of the UA-PLGA NPs all showed a low hemolytic effect with the percentage that was below 5%, indicating satisfactory hemocompatibility. The morphology of red blood cells upon incubation with different samples was further studied by using microscopy (Fig. 5c). Microscopic images showed that the red blood cells incubated with UA-PLGA NPs and blank PLGA NPs at both 4 mg mL−1 were intact and had a smooth structure, indicating a similar morphology to that of the negative control. On the other hand, the red blood cells incubated with PBS exhibited fragmentation and leakage, lacking intact cellular structures. These results suggested that UA-PLGA NPs and blank PLGA NPs did not cause hemolysis and showed excellent biocompatibility in blood.
 |
| | Fig. 5 Hemolysis assay of UA-PLGA NPs and blank PLGA NPs on sheep red blood cells. The photographs (A) and percentages of hemolysis (B) upon incubation of red blood cells with various concentrations of UA-PLGA NPs (0.125, 0.25, 0.5, 1, 2 and 4 mg mL−1) and blank PLGA NPs (0.25, 1, and 4 mg mL−1). Triton X-100 was used as a positive control and PBS was used as a negative control; (C) microscopic images showing the morphology of red blood cells upon incubation with negative control, 4 mg mL−1 UA-PLGA NPs, 4 mg mL−1 blank PLGA NPs and positive control. Data represent ± SD (n = 2). Scale bar: 25 μm. | |
Conclusions
The development of UA-PLGA NPs as a phytochemical delivery system using nanoprecipitation exhibits considerable potential for enhancing the therapeutic efficacy of UA in CCA. Atomic MD simulations confirmed the interactions between UA and the biocompatible copolymer PLGA through hydrogen bonding. Synthesized UA-PLGA NPs, with suitable size and surface charge, exhibited good stability, cytotoxic properties, controlled UA release, and effective internalization into CCA cells. These findings indicate that UA-PLGA NPs have the potential to function as a platform for enhanced UA delivery to CCA cells, thereby potentially increasing its therapeutic effectiveness. Additionally, the improved solubility provided by the UA-PLGA system may overcome the inherent solubility challenges of UA, which might contribute to improved treatment outcomes. Moreover, UA-PLGA NPs demonstrated hemocompatibility, indicating their potential as a safe alternative treatment with no toxicity in blood. Effective implementation of this strategy may result in a new method of administering CCA treatment, thereby facilitating the development of enhanced UA delivery strategies that can further improve the efficacy of CCA treatment.
Data availability
All data generated in this study are included within the main article and/or ESI† accompanying this paper.
Author contributions
Pornpattra Maphanao: investigation, visualization, data curation, formal analysis and writing – original draft preparation. Yaowaret Phothikul: investigation, visualization and formal analysis. Cherdpong Choodet: investigation, visualization and formal analysis. Theerapong Puangmali: formal analysis and writing – review and editing. Kanlaya Katewongsa: conceptualisation, methodology, formal analysis and writing – review and editing. Somchai Pinlaor: writing – review and editing. Raynoo Thanan: supervision and writing – review and editing. Umaporn Yordpratum: investigation, writing – review and editing. Chadamas Sakonsinsiri: conceptualisation, methodology, visualisation, data curation, validation, formal analysis, supervision, writing – original draft preparation, writing – review and editing and funding acquisition.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the Basic Research Fund of Khon Kaen University under Cholangiocarcinoma Research Institute (CARI), Khon Kaen University, Thailand (CARI-BRF64-24) and the Cholangiocarcinoma Research Institute (CARI), Khon Kaen University, Thailand (CARI 03/2563).
References
- J. M. Banales, J. J. G. Marin, A. Lamarca, P. M. Rodrigues, S. A. Khan, L. R. Roberts, V. Cardinale, G. Carpino, J. B. Andersen, C. Braconi, D. F. Calvisi, M. J. Perugorria, L. Fabris, L. Boulter, R. I. R. Macias, E. Gaudio, D. Alvaro, S. A. Gradilone, M. Strazzabosco, M. Marzioni, C. Coulouarn, L. Fouassier, C. Raggi, P. Invernizzi, J. C. Mertens, A. Moncsek, S. Rizvi, J. Heimbach, B. G. Koerkamp, J. Bruix, A. Forner, J. Bridgewater, J. W. Valle and G. J. Gores, Nat. Rev. Gastroenterol. Hepatol., 2020, 17, 557–588 CrossRef PubMed.
- P. Srivatanakul, H. Ohshima, M. Khlat, M. Parkin, S. Sukaryodhin, I. Brouet and H. Bartsch, Int. J. Cancer, 1991, 48, 821–825 CrossRef CAS PubMed.
- B. Sripa, S. Kaewkes, P. Sithithaworn, E. Mairiang, T. Laha, M. Smout, C. Pairojkul, V. Bhudhisawasdi, S. Tesana, B. Thinkamrop, J. M. Bethony, A. Loukas and P. J. Brindley, PLoS Med., 2007, 4, e201 CrossRef PubMed.
- S. Sriamporn, P. Pisani, V. Pipitgool, K. Suwanrungruang, S. Kamsa-ard and D. M. Parkin, Trop. Med. Int. Health, 2004, 9, 588–594 CrossRef CAS PubMed.
- P. Yongvanit, S. Pinlaor and H. Bartsch, Parasitol. Int., 2012, 61, 130–135 CrossRef CAS PubMed.
- M. Alsaleh, Z. Leftley, T. A. Barbera, P. Sithithaworn, N. Khuntikeo, W. Loilome, P. Yongvanit, I. J. Cox, N. Chamodol, R. R. Syms, R. H. Andrews and S. D. Taylor-Robinson, Int. J. Gen. Med., 2019, 12, 13–23 CrossRef CAS PubMed.
- J. M. Banales, V. Cardinale, G. Carpino, M. Marzioni, J. B. Andersen, P. Invernizzi, G. E. Lind, T. Folseraas, S. J. Forbes, L. Fouassier, A. Geier, D. F. Calvisi, J. C. Mertens, M. Trauner, A. Benedetti, L. Maroni, J. Vaquero, R. I. Macias, C. Raggi, M. J. Perugorria, E. Gaudio, K. M. Boberg, J. J. Marin and D. Alvaro, Nat. Rev. Gastroenterol. Hepatol., 2016, 13, 261–280 CrossRef PubMed.
- A. Elvevi, A. Laffusa, M. Scaravaglio, R. E. Rossi, R. Longarini, A. M. Stagno, L. Cristoferi, A. Ciaccio, D. L. Cortinovis, P. Invernizzi and S. Massironi, Ann. Hepatol., 2022, 100737 CrossRef PubMed.
- K. R. Eckmann, D. K. Patel, A. Landgraf, J. H. Slade, E. Lin, H. Kaur, E. Loyer, J. M. Weatherly and M. Javle, Gastrointest Cancer Res., 2011, 4, 155–160 Search PubMed.
- T. Okusaka, K. Nakachi, A. Fukutomi, N. Mizuno, S. Ohkawa, A. Funakoshi, M. Nagino, S. Kondo, S. Nagaoka, J. Funai, M. Koshiji, Y. Nambu, J. Furuse, M. Miyazaki and Y. Nimura, Br. J. Cancer, 2010, 103, 469–474 CrossRef CAS PubMed.
- D. H. Ahn, J. Reardon, C. W. Ahn, M. Bupathi, S. Mikhail, C. S.-Y. Wu and T. Bekaii-Saab, Cancer Prev. Res., 2018, 142, 1671–1675 CAS.
- O. Maeda, T. Ebata, T. Shimokata, A. Matsuoka, M. Inada-Inoue, S. Morita, Y. Takano, H. Urakawa, Y. Miyai, M. Sugishita, A. Mitsuma, M. Ando, T. Mizuno, M. Nagino and Y. Ando, Nagoya J. Med. Sci., 2020, 82, 725–733 CAS.
- D. Nierenberg, O. Flores, D. Fox, Y. Y. L. Sip, C. Finn, H. Ghozlan, A. Cox, K. K. McKinstry, L. Zhai and A. R. Khaled, ACS Omega, 2021, 6, 5591–5606 CrossRef CAS PubMed.
- H. K. Makadia and S. J. Siegel, Polymers, 2011, 3, 1377–1397 CrossRef CAS PubMed.
- L. He, H. Lai and T. Chen, Biomaterials, 2015, 51, 30–42 CrossRef CAS PubMed.
- L. Lei, Z. Liu, P. Yuan, R. Jin, X. Wang, T. Jiang and X. Chen, J. Mater. Chem. B, 2019, 7, 2722–2735 RSC.
- S. Acharya and S. K. Sahoo, Adv. Drug Deliv. Rev., 2011, 63, 170–183 CrossRef CAS PubMed.
- N. Muhamad, T. Plengsuriyakarn, C. Chittasupho and K. Na-Bangchang, Asian Pac. J. Cancer Prev. APJCP, 2020, 21, 935–941 CrossRef CAS PubMed.
- A. I. Omar, T. Plengsuriyakarn, C. Chittasupho and K. Na-Bangchang, Clin. Exp. Pharmacol. Physiol., 2021, 48, 318–328 CrossRef CAS PubMed.
- A. Tahir, T. Plengsuriyakarn, C. Chittasupho and K. Na-Bangchang, Polymers, 2022, 14, 4444 CrossRef CAS PubMed.
- A. Batra and V. G. Sastry, Pteridines, 2013, 24, 191–199 CrossRef CAS.
- L. Wozniak, S. Skapska and K. Marszalek, Molecules, 2015, 20, 20614–20641 CrossRef CAS PubMed.
- C. Tang, Y. H. Lu, J. H. Xie, F. Wang, J. N. Zou, J. S. Yang, Y. Y. Xing and T. Xi, Anti-Cancer Drugs, 2009, 20, 249–258 CrossRef CAS PubMed.
- P. Maphanao, R. Thanan, W. Loilome, S. Chio-Srichan, M. Wongwattanakul and C. Sakonsinsiri, Biochim. Biophys. Acta, Gen. Subj., 2020, 1864, 129708 CrossRef CAS PubMed.
- D. Mu, G. Zhou, J. Li, B. Su and H. Guo, Oncol. Lett., 2018, 15, 3202–3206 Search PubMed.
- J. S. Wang, T. N. Ren and T. Xi, Med. Oncol., 2012, 29, 10–15 CrossRef CAS PubMed.
- H. Weng, Z. J. Tan, Y. P. Hu, Y. J. Shu, R. F. Bao, L. Jiang, X. S. Wu, M. L. Li, Q. Ding, X. A. Wang, S. S. Xiang, H. F. Li, Y. Cao, F. Tao and Y. B. Liu, Cancer Cell Int., 2014, 14, 96 CrossRef PubMed.
- X. Wang, F. Zhang, L. Yang, Y. Mei, H. Long, X. Zhang, J. Zhang, S. Qimuge and X. Su, J. Biomed. Biotechnol., 2011, 2011, 419343 Search PubMed.
- R. Baishya, D. K. Nayak, D. Kumar, S. Sinha, A. Gupta, S. Ganguly and M. C. Debnath, Pharm. Res., 2016, 33, 2691–2703 CrossRef CAS PubMed.
- A. Markowski, P. Migdał, A. Zygmunt, M. Zaremba-Czogalla and J. Gubernator, Materials, 2021, 14, 4917 CrossRef CAS PubMed.
- P. Payomhom, N. Panyain, C. Sakonsinsiri, P. Wongtrakoongate, K. Lertsuwan, D. Pissuwan and K. P. Katewongsa, ACS Appl. Nano Mater., 2024, 7, 5383–5395 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, 2016 Search PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- Y. Zhang, M. Huo, J. Zhou, A. Zou, W. Li, C. Yao and S. Xie, AAPS J., 2010, 12, 263–271 CrossRef CAS PubMed.
- A. Zhou, Y. Zhang, X. Zhang, Y. Deng, D. Huang, C. Huang and Q. Qu, Int. J. Biol. Macromol., 2022, 201, 448–457 CrossRef CAS PubMed.
- B. Song and C.-W. Cho, J. Pharm. Investig., 2024, 54, 249–266 CrossRef CAS.
- K. Kızılbey, ACS Omega, 2019, 4, 555–562 CrossRef.
- G. Singh, K. Tanurajvir, K. Ravinder and A. Kaur, Int. J. Res. Pharm. Sci., 2014, 1, 30–42 CAS.
- Y. Takechi-Haraya, T. Ohgita, Y. Demizu, H. Saito, K.-i. Izutsu and K. Sakai-Kato, AAPS PharmSciTech, 2022, 23, 150 CrossRef CAS PubMed.
- C. Albert, N. Huang, N. Tsapis, S. Geiger, V. Rosilio, G. Mekhloufi, D. Chapron, B. Robin, M. Beladjine, V. Nicolas, E. Fattal and F. Agnely, Langmuir, 2018, 34, 13935–13945 CrossRef CAS PubMed.
- H. Gupta, M. Aqil, R. K. Khar, A. Ali, A. Bhatnagar and G. Mittal, Nanomed. Nanotechnol. Biol. Med., 2010, 6, 324–333 CrossRef CAS PubMed.
- L. Lopez-Hortas, P. Perez-Larran, M. J. Gonzalez-Munoz, E. Falque and H. Dominguez, Food Res. Int., 2018, 103, 130–149 CrossRef CAS PubMed.
- A. Luzar and D. Chandler, Nature, 1996, 379, 55–57 CrossRef CAS.
- F. Eisenhaber, P. Lijnzaad, P. Argos, C. Sander and M. Scharf, J. Comput. Chem., 1995, 16, 273–284 CrossRef CAS.
- M. Noori Koopaei, M. R. Khoshayand, S. H. Mostafavi, M. Amini, M. R. Khorramizadeh, M. Jeddi Tehrani, F. Atyabi and R. Dinarvand, Iran. J. Pharm. Res., 2014, 13, 819–833 Search PubMed.
- G. Yurtdas Kirimlioglu and A. A. Ozturk, Turk. J. Pharm. Sci., 2020, 17, 27–35 CrossRef CAS PubMed.
- A. Albisa, E. Piacentini, V. Sebastian, M. Arruebo, J. Santamaria and L. Giorno, Pharm. Res., 2017, 34, 1296–1308 CrossRef CAS PubMed.
- P. L. Ritger and N. A. Peppas, J. Control. Release, 1987, 5, 23–36 CrossRef CAS.
- C. Poulios, V. Karagkiozaki, D. Kapoukranidou, Z. Chakim, T. Zarampoukas, N. Foroglou and S. Logothetidis, Virchows Arch., 2023, 483, 775–786 CrossRef CAS PubMed.
- A. Markowski, A. Jaromin, P. Migdał, E. Olczak, A. Zygmunt, M. Zaremba-Czogalla, K. Pawlik and J. Gubernator, Int. J. Mol. Sci., 2022, 23, 5536 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Kanlaya Katewongsa
c,
Kanlaya Katewongsa