DOI:
10.1039/D4RA03538K
(Paper)
RSC Adv., 2024,
14, 24898-24909
2-Chloro-3-cyano-4-nitrobenzyl pyridinium bromide as a potent anti-lung cancer molecule prepared using a single-step solvent-free method†
Received
14th May 2024
, Accepted 11th July 2024
First published on 8th August 2024
Abstract
Mono-/dimeric-substituted pyridinium and pyrazolium bromides were prepared under conventional and solvent-free silica-supported domestic microwave conditions. The atom economy, environmental product mass intensity and product mass intensity for solvent-free reactions showed significant importance for the synthesis of target molecules. 4-Nitrobenzyl-substituted pyridinium bromide showed potent anticancer properties compared with mono-/dimeric-substituted pyridinium and pyrazolium bromides against a lung cancer cell line (A-549). Molecular simulation studies were carried out for mono-/dimeric-substituted pyridinium and pyrazolium bromide against protein human CDK1/cyclinB1/CKS2 using the AutoDock program.
1. Introduction
Osteosarcoma is a common cancer that begins in the bone and affects people under 25 years of age.1,2 Imidazole, thiazole, pyrazole, triazole and pyridine derivatives play a crucial role in medicinal chemistry and biomedical applications. Almost 80–90 percent of novel drug molecules are made of derivatives of heterocyclic compounds.3–6 Mohamed et al. reported on derivatives of pyridone, thiazolidinone and pyrazolone and studied their anticancer properties against A-549 (lung) and MDA-MB-231 (breast) cancer cell lines using MTT assay. They tested the anticancer properties of nitro-, N,N-dimethyl amino-, methoxy-, and methyl-substituted pyridone derivatives. Among these, methyl-substituted pyridone showed a significant response against breast and lung cancer cell lines with IC50 values.7 Amide-substituted pyrazolo pyrimidine derivatives showed a potent anticancer response against HepG2, MCF7, HCT116 and HeLa cancer cell lines among other derivatives of pyrazolo pyrimidine and thione pyridone.8 The anticancer activities of naturally occurring communesin alkaloid and synthesized derivatives of communesin against five human cancer cell lines were compared using cell Titer-G10 method. Methyl-substituted communesin showed a ten fold greater anticancer response against the tested cancer cell lines than the other derivatives and naturally occurring communesin.9
A pH-responsive PEGylated paclitaxel prodrug is mixed with a 3D printed titanium scaffold, which is used for bone stem cell transplantation under physiological conditions for the eradication of osteosarcoma cancer in mice models.10 A bromo-substituted 1,2,3-triazole derivative showed significant anticancer activities against the MCF-7 and Caco-2 cancer cell lines compared to the fluoro-, chloro- and ester-substituted triazole–triazole derivatives.11 Cytotoxic properties of amide-based pyridine and amide-based pyridine iodide have been demonstrated against glioma cells using short term cell culture. Amide-based pyridinium iodide showed significant anticancer properties compared with amide-based bipyridines.12 IC50 values of steroid-substituted pyridine derivatives showed a significant anticancer response when compared with its parent compounds.13–15 Phenyl amino pyrimidine and hydroxy-substituted phenyl amino pyrimidine showed a selective src kinase inhibitory response and excellent anticancer response against the HT29CRC and HCT-116 cell lines.16 Mohammed et al. reported on in vitro anti-proliferative studies of trimethoxy phenyl-substituted pyridine against the MCF7, HEPG-2 and HCT116 cancer cell lines. 4-Nitro-substituted trimethoxy phenyl pyridine showed a significant anticancer response against the HCT116, HEPG-2 and MCF7 cancer cell lines compared with hydroxy-, methoxy-, methyl-substituted and chloro trimethoxy phenyl pyridine.17 Substituted pyrido pyrimidithione and substituted pyrazo pyridine showed interesting anticancer responses against the human hepatoma (Huh-7), lung cancer adenocarcinoma (A549) and Michigan cancer foundation 7 (MCF-7) cell lines with IC50 values of 2.48 μm and 2.23 μm, respectively.18 Antiproliferative screening of steroid-substituted pyridine moieties was performed against human tumor cell lines and some selected protein targets for breast cancer.19 Rajavenkatesh and coworkers reported on the synthesis of simple and substituted imidazopyridine derivatives via one-pot multicomponent cycloaddition reactions. They have tested the anticancer properties of imidazopyridine derivatives against Hep-2, HepG2, MCF-7 and A375 cancer cell lines. 4-Aminophenyl-substituted imidazopyridine showed an excellent anticancer response against the tested cancer cell lines.20 Substituted imidazopyridine was synthesized under moderate reaction conditions, and its cytotoxic screening was performed against human lung cancer, human breast cancer marine skin cancer cell lines.21 Based on the available literature, we wish to prepare novel amide-based mono-/dimeric pyridinium and pyrazolium bromide under conventional/muffle furnace solid-supported solvent-free methods and study the anticancer, molecular docking and DFT properties of mono-/dimeric pyridinium pyrazolium bromides.
2. Results and discussion
2.1 Chemistry
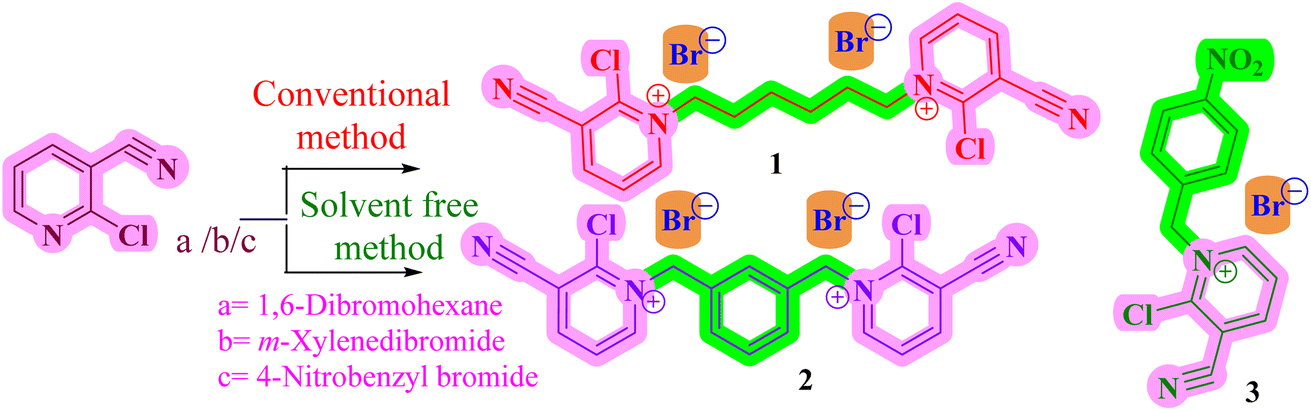
2.1.1 Synthesis of mono-/dimeric pyridinium/pyrazolium bromides. 4-Nitrobenzylbromide/1,6-dibromohexane/m-xylene dibromide treated with 2-chloropyridine-3-carbonitrile in the presence of dry CH3CN under refluxing condition afforded the mono- and dimeric pyridinium derivatives 1/2/3 in quantitative yield. We wished to avoid the environmental pollution caused by the use of volatile organic solvents, so we tried to perform the reaction under a greener synthetic method. In this method, we completely avoided volatile organic solvents. The required equivalents of 2-chloropyridine-3-carbonitrile and 4-nitrobenzylbromide/1,6-dibromohexane/m-xylene dibromide were mixed with 2 g of 60 – 120 mesh silica gel, and transferred into a mortar and pestle, followed by simple manual grinding for uniform mixing and absorption. The solvent-free silica-supported reaction mixture was divided into five portions for optimization of the reaction condition and percentage of conversion. Fortunately, we obtained the mono- and dimeric derivatives of pyridinium bromide in excellent yield with a shorter reaction time without any volatile organic solvents. The results are summarized in Table 1 (Scheme 1 and 2).
Table 1 Reaction condition and percentage of yield for mono-/dimeric bromo derivatives 1–5
| Dimeric pyridinium bromide 1 |
Dimeric pyridinium bromide 2 |
Monomeric pyridinium bromide 3 |
Dimeric pyridinium bromide 4 |
Dimeric pyridinium bromide 5 |
| Reflux |
MW |
Reflux |
MW |
Reflux |
MW |
Reflux |
MW |
Reflux |
MW |
| Min |
% |
Min. |
% |
Min |
% |
Min |
% |
Min |
% |
Min |
% |
Min |
% |
Min |
% |
Min |
% |
Min |
% |
| 60 |
90 |
04 |
92 |
30 |
92 |
02 |
95 |
45 |
90 |
03 |
94 |
120 |
91 |
07 |
93 |
90 |
92 |
05 |
95 |
 |
| | Scheme 1 Conventional method: CH3CH, reflux, 30–60 min: 90–92% microwave method: silica gel, 2–4 min; 92–95%. | |
 |
| | Scheme 2 Conventional method: CH3CN, reflux, 90–120 min: 91–92%; microwave method: silica gel, 5–7 min; 93–95%. | |
The rate of the reaction is much greater with 2-chloropyridine-3-carbonitrile than with 5-amino-1-pheneyl-1H pyrazole-4-carbonitrile due to steric hindrance. The microwave-assisted solvent-free silica-supported method has more merits compared with the conventional refluxing condition, such as the complete avoidance of volatile organic solvents, the greater percentage yields, and a reaction time that is nearly fifteen times shorter. Hence, the microwave-assisted solvent-free silica-supported synthetic method is most suitable for the preparation of mono- and dimeric-substituted pyridinium bromide/substituted pyrazolium bromides 1–5. The preparation of the longer alkyl chain linked pyridinium bromide 1 requires 60 min reaction time under the refluxing condition, but the formation of the m-xylene linked dimeric pyridinium bromide 2 is requires only 30 min under the refluxing condition.
2.1.2 Atom economy (AE). The atom economy of a chemical reaction is defined as the number of atoms of the reactant converted into the product (Scheme 3). The atom economy of a reaction is determined as follows:
 |
| | Scheme 3 The model chemical reaction used for the atom economy calculation. | |
Atom economy = molecular weight of product ÷ ∑ (molecular weight of reactants) × 100
AE = 0.5210 g mmol−1 ÷ (0.1385 g mmol−1 + 0.1385 g mmol−1 + 0.243.9 g mmol−1) × 100
AE = 100%
2.1.3 Environmental factor. The environmental factor is defined as the ratio between the mass value of waste/byproduct and the mass value of the product (Scheme 4).
| Environmental factor = [mass value of waste]/mass value of product |
 |
| | Scheme 4 The model chemical reaction used for the environmental factor calculation. | |
Here, the mass of waste is calculated as follows:
| E-factor = [mass of waste]/mass of product |
2.1.4 Product mass intensity (PMI). The PMI is the ratio between the total amount of mass utilized in a reaction and the mass of product in a reaction. In the product mass intensity, the solvent is also considered. Hence, acetonitrile also adds value to the PMI (Scheme 5).
| PMI = ∑(mass of reactants + solvent)/mass of product |
| PMI = (2 × 0.1842 g mmol−1 + 0.2439 g mmol−1 + 0.4110 g mmol−1)/0.6123 g mmol−1 |
 |
| | Scheme 5 The chemical reaction used for the product mass intensity calculation. | |
2.1.5 Product mass intensity solvent-free (PMISF). The product mass intensity solvent-free (PMISF) is the ratio between the total amount of mass utilized in a reaction and the mass of product in a reaction (Scheme 6).
| PMISF = ∑(mass of reactants + solvent free)/mass of product |
| PMISF = (2 × 0.1842 g mmol−1 + 0.2639 g mmol−1 + 0)/0.6323 g mmol−1 |
 |
| | Scheme 6 The chemical reaction used for the product mass intensity solvent-free calculation. | |
Similarly, we have calculated the atom economy, environmental factors and the product mass intensity for the remaining reactions (Table 2). We used non-toxic and low boiling solvents for our syntheses. The mass value is given in Table 2, which is used for all of the reactions. We did not used any expensive, toxic and environmental hazardous catalysts for the preparation of our target drug molecules.
Table 2 Atom economy, environmental factors and product mass intensity for mono-/dimeric pyridinium/pyrazolium bromides 1–5
| Compound |
No of steps |
Overall yield (%) |
Overall AE (%) |
E-factor |
E-solvent |
E-solvent free |
E-catalyst |
Product mass intensity with solvent (PMI) total |
Product mass intensity solvent-free (PMISF) total |
| 1 |
1 |
90 |
100 |
0 |
41.01 |
0 |
0 |
1.0 |
1.0 |
| 2 |
1 |
92 |
100 |
0 |
41.01 |
0 |
0 |
1.0 |
1.0 |
| 3 |
1 |
90 |
100 |
0 |
41.01 |
0 |
0 |
1.0 |
1.0 |
| 4 |
1 |
91 |
100 |
0 |
41.01 |
0 |
0 |
1.0 |
1.0 |
| 5 |
1 |
92 |
100 |
0 |
41.01 |
0 |
0 |
1.0 |
1.0 |
3. Anticancer activity
3.1 Materials and methods
3.1.1 Cell line maintenance. The lung cancer (A-549) cell lines were provided by NCCS, Pune. The T25 culture flask was used to sustain the cells, along with DMEM, 10% fetal bovine serum (FBS) (Gibco, Thermo Fisher Scientifc Inc.) 1% antibiotics. The cells were maintained at 37 °C in a humid atmosphere with 5% CO2. Trypsinization and subsequent passage were performed on confluent cells.
3.1.2 Cell viability (MTT) assay. Cell viability of the A-549 cells was assessed using the MTT test to assess how well they reacted to mono- and dimeric-substituted pyridinium bromide/substituted pyrazolium bromides 1–5. In order for the test to work, metabolically active cells must convert soluble yellow tetrazolium salt into insoluble purple formazan crystals. We placed A-549 cells in 96-well plates at a density of 5 × 103 cells per well. Subsequently, the cells were starved for three hours at 37 °C in the medium after being twice cleansed with 100 μL of serum-free medium after twenty-four hours. After starvation, mono- and dimeric-substituted pyridinium bromide/substituted pyrazolium bromides 1–5 were exposed to the cells for a duration of 24 hours. After treatment, the media from the cells treated with the rutin fraction and control were removed, and 100 μL of DMEM containing 0.5 mg ml−1 of MTT was added to each well. After that, the cells were maintained at 37 °C for 3 hours in the CO2 chamber. The cells were washed with 1× PBS after the MTT-containing solution was discarded. Subsequently, the formazan crystals were dissolved in 100 μL of dimethyl sulfoxide following an hour of dark incubation. A micro ELISA plate reader was then used to measure the color's intensity at 570 nm. The percentage of control cells cultured in serum-free medium representing the quantity of viable cells was determined by the proportion of cells that survived in the control media without receiving any medication. The following formula is used to calculate the cell viability:
| % cell viability = [A570 nm of treated cells/A570 nm of reference cells] × 100 |
3.1.3 Cell morphological analysis. For the study of morphology based on the MTT assay, the most effective doses of mono- and dimeric-substituted pyridinium bromide/substituted pyrazolium bromides 1–5 were chosen for additional study with IC50 concentrations flexible dimeric pyridinium bromide 1 (100 μg ml−1), m-xylene linked pyridinium bromide 2 (100 μg ml−1), monomeric pyridinium bromide 3 (20 μg ml−1), flexible dimeric pyrazolium bromide 4 (140 μg ml−1), and m-xylene linked pyrazolium bromide 5 (20 μg ml−1). Changes in the cell morphology were examined with a phase contrast microscope. For the study, 2 × 105 cells were plated in 6-well plates and treated with the IC50 concentrations of 1–5 for 24 hours. Following the incubation period, the media was removed, and the cells received one saline wash in phosphate buffer solution (pH = 7.4). The plates were examined using a phase contrast microscope.
3.1.4 Determination of nuclear morphological changes of cells (DAPI staining). For the nuclear morphological analysis, the monolayer of cells was washed with PBS and fixed with 3% paraformaldehyde for 10 min at room temperature. The fixed cells were permeabilized with 0.2% Triton X-100 in PBS for 10 min at room temperature, and incubated with 0.5 μg ml−1 of DAPI for 5 min. The apoptotic nuclei (intensely stained, fragmented nuclei, and condensed chromatin) were viewed under a fluorescence microscope.
3.2 Cytotoxic effect of mono-/dimeric substituted pyridinium/pyrazolium bromides on lung cancer cells (A-549)
Mono- and dimeric-substituted pyridinium bromide/substituted pyrazolium bromides 1–5 were evaluated for their cytotoxic characteristics in lung cancer cells with concentrations ranging from 10–140 μg ml−1 using the MTT technique during a 24 hours period. The lung cancer cells' vitality is significantly decreased by mono- and dimeric-substituted pyridinium bromide/substituted pyrazolium bromides 1–5 (Fig. 1). Mono- and dimeric-substituted pyridinium bromide/substituted pyrazolium bromides 1–5 were reported to have IC50 dosages at the following concentrations: compound 1 (100 μg ml−1), compound 2 (100 μg ml−1), compound 3 (20 μg ml−1), compound 4 (140 μg ml−1) and compound 5 (20 μg ml−1).
 |
| | Fig. 1 Cytotoxic effects of mono- and dimeric-substituted pyridinium bromide/substituted pyrazolium bromides 1–5 on lung cancer cells (A-549). Cells were treated with concentrations from 10 to 140 μg ml−1 for 24 h, and the cell viability was evaluated using MTT assay. | |
Bar 1: flexible dimeric pyridinium bromide 1; Bar 2: m-xylene linked pyridinium bromide 2; Bar 3: monomeric pyridinium bromide 3; Bar 4: flexible dimeric pyrazolium bromide 4; Bar 5: m-xylene linked pyrazolium bromide 5. Data are shown as means ± SD (n = 3). * Compared with the control blank group, p < 0.05.
3.3 Cell morphological analysis
The morphological analysis of A-549 cells was viewed and photographed using a phase contrast microscope. The A-549 cells treated with IC50 concentrations of flexible dimeric pyridinium bromide 1 (100 μg ml−1), m-xylene linked pyridinium bromide 2 (100 μg ml−1), monomeric pyridinium bromide 3 (20 μg ml−1), flexible dimeric pyrazolium bromide 4 (140 μg ml−1), and m-xylene linked pyrazolium bromide 5 (20 μg ml−1) showed significant morphological changes (Fig. 2), including cell shrinkage and decreased cell density, which are suggestive of apoptotic cells. Apoptotic cells also exhibited other morphological alterations, such as rounder cells that contract and become disconnected from neighboring cells. A few fragile cells were also observed to separate from the plate surface.
 |
| | Fig. 2 Effect of monomeric- and dimeric-substituted pyridinium bromide/substituted pyrazolium bromides 1–5 on the cell morphology of lung cancer cells (A-549). Cells were treated with monomeric- and dimeric-substituted pyridinium and pyrazolium bromides 1–5 of IC50 concentrations for 24 h, along with the control group. Images were obtained using an inverted phase contrast microscope. | |
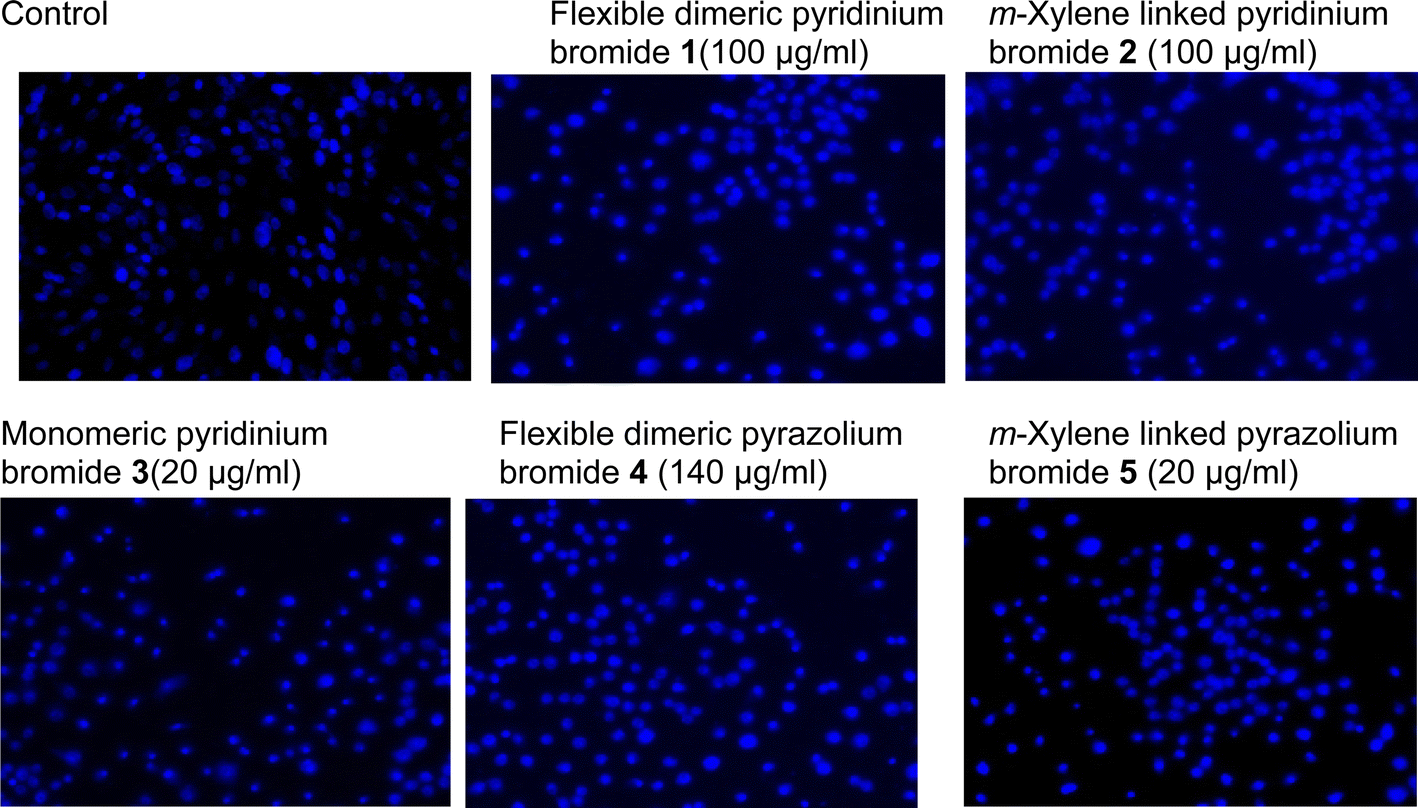
3.4 DAPI staining for apoptosis detection
The induction of apoptosis via the mono- and dimeric-substituted pyridinium and pyrazolium bromides 1–5 (100 μg ml−1, 100 μg ml−1, 140 μg ml−1, 20 μg ml−1, and 20 μg ml−1) treated cells was analyzed by DAPI staining. After a 24 h treatment period, the cells were stained with nuclear staining (DAPI) and observed via fluorescence microscopy. The treated cells clearly showed condensed chromatin and nuclear fragmentation, which are characteristics of apoptosis compared to the control, which showed clear round nuclei (Fig. 3).
 |
| | Fig. 3 Human lung cancer cells were treated with monomeric and dimeric-substituted pyridinium and pyrazolium bromides 1–5 IC50 concentrations for 24 h, along with the control group. After the treatment, the cells were incubated with DAPI staining. Images were obtained using an inverted fluorescence phase contrast microscope. | |
4 Molecular docking studies
Molecular docking studies were performed using the AutoDock Tools (ADT) version 1.5.6 and the AutoDock version 4.2.5.1 docking program. The energy calculations were achieved using genetic algorithms, which allowed for a comprehensive exploration of molecular interactions and binding affinities between the molecules of interest. The chemical structures from ChemDraw were transformed into energy-optimized PDB (Protein Data Bank) format using Chem 3D 17.0. This conversion allows the representation of the compounds in a suitable format for further computational analysis and modeling. Pymol software was used for the visualization of the docked molecules.
4.1 Ligand and protein preparation
The 3D structures of synthesized compounds were constructed by maestro builder panel. The ligand preparation wizard was used to add hydrogen atoms and regulate the rational bond angles, geometry and ring conformations. The most favourable ionization state of compounds was produced with the aid of force field OPLS-2005 (optimized potentials for liquid simulations), and geometry minimization was carried out until it reached a RMSD cutoff of 0.01 Å. The subsequent output structure of compounds was suitable for docking with protein Human CDK1/CyclinB1/CKS2. The crystal structure of the Human CDK1/CyclinB1/CKS2 (PDB ID: 4AGD) was downloaded from the protein data bank (http://www.rcsb.org/, https://www.rcsb.org/structure/4y72). The receptor was fully optimized in the protein preparation wizard, which involved adding polar hydrogen atoms, assigning bond orders, determining protonation states, and subsequently removing water molecules beyond a 5 Å distance. Thereafter, by using the force field (OPLS-2005), an energy minimization of 0.03 Å RMSD was performed to reduce the steric hindrance caused by the addition of hydrogen atoms. Now, the minimized Human CDK1/CyclinB1/CKS2 receptor22,23 was suitable for docking by the supplied x, y, and z coordinates 45.14, −7.99, and −8.72 Å, respectively, with the grid box diameter (xyz) of 30 × 30 × 30 Å. The output results were visualized with the help of the PyMol console for virtual examination of various binding modes between the compounds and receptor.
4.2 Molecular docking with Human CDK1/CyclinB1/CKS2
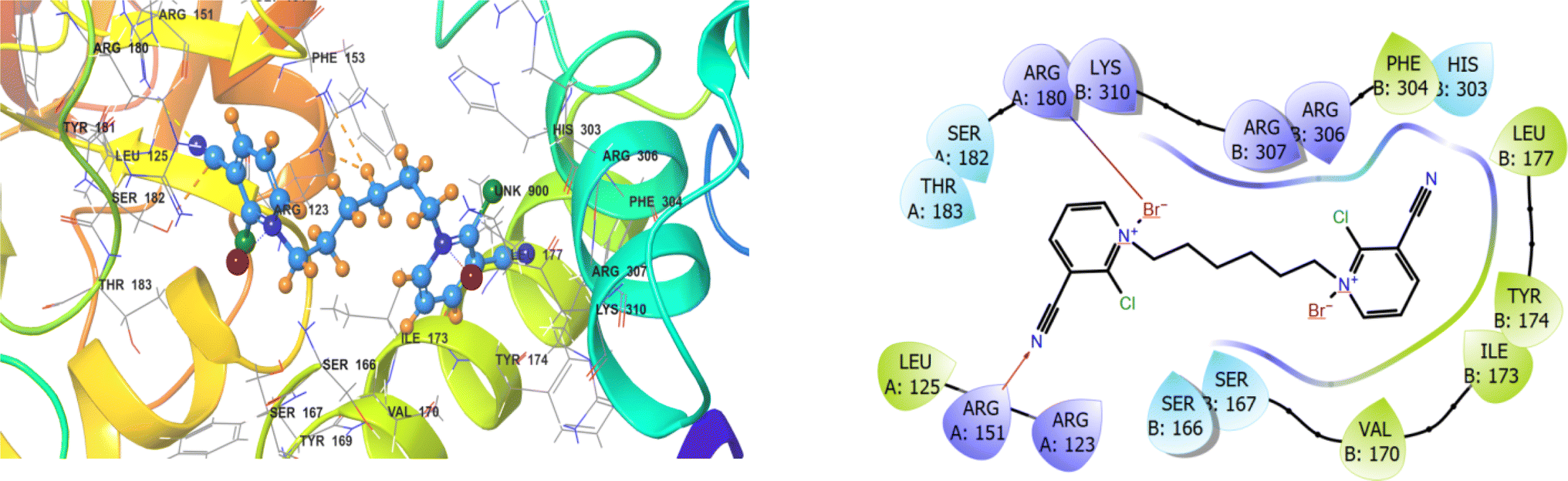
Molecular docking study is an attractive scaffold to understand the protein interactions with mono-/dimeric substituted pyridinium and pyrazolium bromides, which can corroborate our experimental results. The best interaction site of the mono-/dimeric substituted pyridinium and pyrazolium bromides with the target protein was visualized (Fig. 4–8 and Table 3). The observed docking score values of the mono-/dimeric substituted pyridinium and pyrazolium bromides are −3.668, −4.036, −4.458, −3.501 and −3.755 kcal mol−1, respectively. The docking score results revealed that mono-/dimeric substituted pyridinium and pyrazolium bromides were well located in the hydrophobic site and strongly interacted with Human CDK1/CyclinB1/CKS2 kinase receptor via π–π stacking, hydrophobic and hydrogen bonding interactions. The hydrogen bonding interaction of compounds 1–5 shows ARG A:151, SER B:167, TYR A:169, SER B:166 and TYR A:160 GLY A:154, respectively. Among the synthesized compounds, the mono-/dimeric substituted pyridinium and pyrazolium bromides showed the highest docking score, which was influenced by the hydrogen bond with residue TYR A:169 and numerous hydrophobic contacts like TYR A:160, VAL A:174, MET B:260, PRO A:156, ILE A:155, TYR A:181, and ALA A:179. Based on the aforementioned evidence, the mono-/dimeric substituted pyridinium and pyrazolium bromides exhibit robust binding, surpassing the affinity observed. The m-xylene linked dimeric pyrazolium bromide 3 effectively modulates CDK1/CyclinB1/CKS2 activity, showcasing its potential in therapeutic strategies and cancer prevention Table 3.
 |
| | Fig. 4 3D and 2D interactions of flexible dimeric pyridinium bromide 1 with the receptor human CDK1/cyclinB1/CKS2. | |
 |
| | Fig. 5 3D and 2D interactions of m-xylene linked pyridinium bromide 2 with the receptor human CDK1/cyclinB1/CKS2. | |
 |
| | Fig. 6 3D and 2D interactions of monomeric pyridinium bromide 3 with the receptor human CDK1/cyclinB1/CKS2. | |
 |
| | Fig. 7 3D and 2D interactions of flexible dimeric pyrazolium bromide 4 with the receptor human CDK1/cyclinB1/CKS2. | |
 |
| | Fig. 8 3D and 2D interactions of m-xylene linked pyrazolium bromide 5 with the receptor human CDK1/cyclinB1/CKS2. | |
Table 3 Molecular docking parameters of mono-/dimeric substituted pyridinium and pyrazolium bromides with VEGFR-2 kinase
| Compound |
Docking score kcal mol−1 |
Active sites with a mode of interaction |
| H-bond |
π–π stacking |
Salt bridge |
Hydrophobic contacts (at 5 Å) |
| 1 |
−3.668 |
ARG A:151 |
— |
ARG A:180 |
PHE B:304, LEU B:177, TYR B:174, ILE B:173, VAL B:170, LEU A:125 |
| 2 |
−4.036 |
SER B:167 |
ARG A:123 |
ARGA:180 |
TYR B:169, VAL B:170, ILE B:173, LEU B:177 |
| 3 |
−4.458 |
TYR A:169 |
— |
ARGA:151 |
TYR A:160, VAL A:174, MET B:260, PRO A:156, ILE A:155, TYR A:181, ALA A:179 |
| 4 |
−3.501 |
SER B:166 |
— |
LYS B:310 |
PHE A:153, ILE A:155, PRO B:299, LEU B:218, LEU B:177, TYR B:174, ILE B:173, VAL B:170 |
| GLUB:220 |
| 5 |
−3.755 |
TYR A:160 |
— |
|
TYR B:169, VAL B:170, ILE B:173, LEU A:125 |
| GLY A:154 |
4.3 Frontier molecular orbital analysis (FMO's)
Frontier molecular orbital analysis of mono-/dimeric substituted pyridinium and pyrazolium bromides focuses on the electron transition between the highest occupied molecular orbitals (HOMO) and the lowest unoccupied molecular orbitals (LUMO), with the former functioning as electron-donating species or nucleophiles and the latter as electron-accepting species or electrophiles in nature.24 The calculated energy values of HOMO and LUMO provide more information about the compound.
The FMO analysis, which is represented in the lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO), is crucial in identifying the molecule's optical and electric characteristics, in addition to its chemical activities. This technique produced a clear image of the HOMO and LUMO energies.25 The energy differences (ΔE) of HOMO–LUMO were considered an important parameter for enlightening the structure and reactivity (Fig. 9). An essential factor in assessing a compound's stability or activity is its band gap. A molecule known as the “soft molecule” has low kinetic stability, high reactivity, and greater polarizability when its ΔE value is low. In this study, the following values were used to calculate the quantum parameter ΔE of mono-/dimeric substituted pyridinium and pyrazolium bromides: (EHOMO values are −6.254, −6.023, −6.214, −5.397 and −3.837, while the ELUMO values are −2.020, −2.055, −3.089, −1.239 and −2.216 eV) (Table 4). The differences of the HOMO–LUMO (ΔE) for the mono-/dimeric substituted pyridinium and pyrazolium bromides molecules were determined as follows: ΔE = 4.234, 3.968, 3.125, 4.158 and 1.622 eV, respectively. The negative convention for the HOMO–LUMO is due to the zero point on the energy scale corresponding to an infinite separation of the electron from the molecule, so any electron in an orbital (HOMO or LUMO, bonding or anti-bonding) corresponds to a more stable state than an infinite separation, and therefore would have a negative energy. The m-xylene linked substituted pyrazolium bromide 5 was found to have the lowest band gap, with ΔE = 1.622 eV. The lesser band gap favored easy transitions of electrons from the ground state to the excited state. Moreover, the biological activity of the compound is influenced by the smaller band gap.
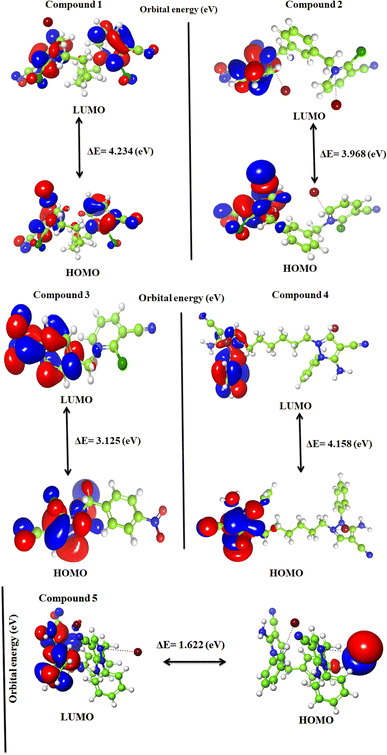 |
| | Fig. 9 Frontier molecular orbitals of the investigated mono-/dimeric substituted pyridinium and pyrazolium bromides 1–5 using the B3LYP/LACVP++ basis set. | |
Table 4 Calculated quantum parameters of the mono-/dimeric substituted pyridinium and pyrazolium bromides
| Compound |
HOMO (eV) |
LUMO (eV) |
ΔE (eV) |
| 1 |
−6.254 |
−2.020 |
4.234 |
| 2 |
−6.023 |
−2.055 |
3.968 |
| 3 |
−6.214 |
−3.089 |
3.125 |
| 4 |
−5.397 |
−1.239 |
4.158 |
| 5 |
−3.837 |
−2.216 |
1.622 |
5 Experimental section
5.1 Preparation of ionic salts
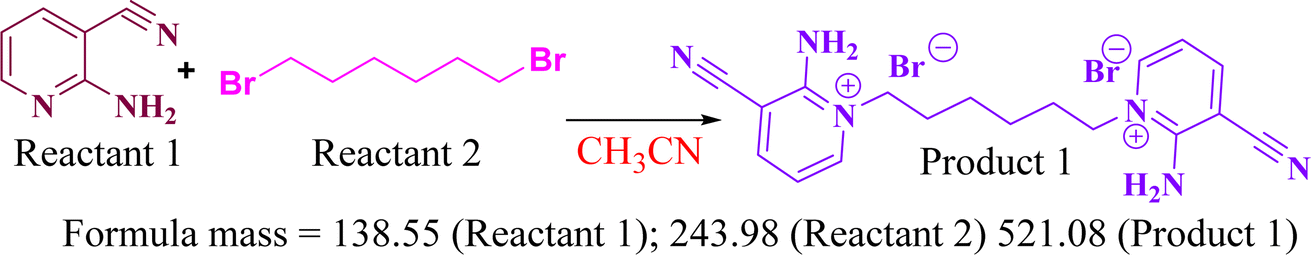
5.1.1 Preparation of 1,1′-(hexane-1,6-diyl)bis(2-chloro-3-cyano)pyridin-1-ium bromide (1). 1,6-Dibromohexane (0.00178 mmol; 1.0 equiv.) treated with 2-chloropyridine-3-carbonitrile (0.00360 mmol; 2.02 equiv.) in the presence of 30 ml of dry CH3CN under refluxing condition for 60 min afforded the 1,1'-(hexane-1,6-diyl)bis(2-chloro-3-cyano)pyridin-1-ium bromide 1 in 90% yield. Reactants were completely soluble in CH3CN when the reaction proceeded, and a colourless precipitate was obtained. After completion of the reaction, the unreacted slight excess amount of substituted pyridine was removed via filtration and washing with CH3CN. Colourless solid; yield 90%; Rf (EtOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Hex 2:3) 0.46; MP = 110–112 °C; 1H-NMR (400 MHz, CDCl3, δ ppm): 8.54–8.56 (d, J = 8 Hz, 2H), 7.95–7.98 (d, J = 12 Hz, 2H), 7.33–7.36 (dd, J = 12 Hz, 2H), 3.33–3.36 (t, J = 12 Hz, 4H), 1.79–1.82 (q, 4H), 1.39–1.41 (t, 4H); 13C-NMR (100 MHz, CDCl3, δ ppm): 152.8, 142.6, 142.3, 122.4, 114.5, 110.9, 33.7, 32.5, 27.2; HRMS: m/z: 180.0814; anal. calcd.: C18H18Br2Cl2N4: C: 41.45; H: 3.45; N: 10.74; found C: 41.42; H: 3.42; N: 10.70.
:Hex 2:3) 0.46; MP = 110–112 °C; 1H-NMR (400 MHz, CDCl3, δ ppm): 8.54–8.56 (d, J = 8 Hz, 2H), 7.95–7.98 (d, J = 12 Hz, 2H), 7.33–7.36 (dd, J = 12 Hz, 2H), 3.33–3.36 (t, J = 12 Hz, 4H), 1.79–1.82 (q, 4H), 1.39–1.41 (t, 4H); 13C-NMR (100 MHz, CDCl3, δ ppm): 152.8, 142.6, 142.3, 122.4, 114.5, 110.9, 33.7, 32.5, 27.2; HRMS: m/z: 180.0814; anal. calcd.: C18H18Br2Cl2N4: C: 41.45; H: 3.45; N: 10.74; found C: 41.42; H: 3.42; N: 10.70.
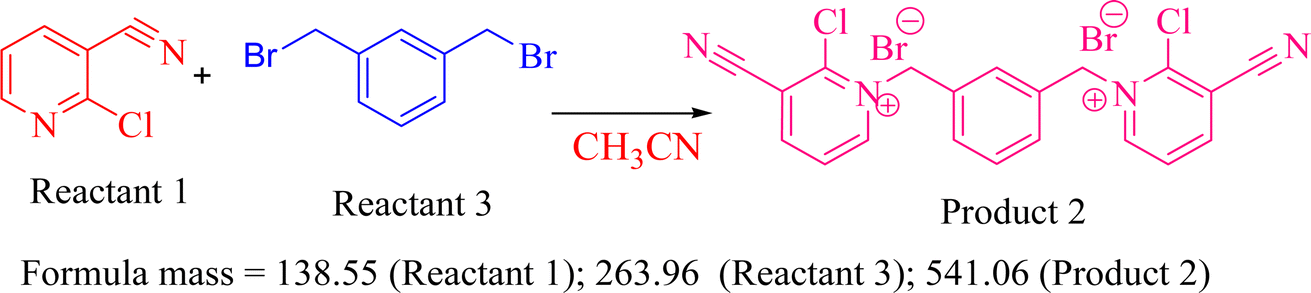
5.1.2 Preparation of 1,1′-(1,3-phenylenebis(methylene))bis(2-chloro-3-cyano)pyridin-1-ium bromide (2). m-Xylene dibromide (0.00192 mmol; 1.0 equiv.) was treated with 2-chloropyridine-3-carbonitrile (0.0038 mmol; 2.02 equiv.) in the presence of 30 ml of dry CH3CN under refluxing condition for 30 min to get 1,1′-(1,3-phenylenebis(methylene))bis(2-chloro-3-cyano)pyridin-1-ium bromide 2 in 92%. Reactants were completely soluble in CH3CN when the reaction proceeded, and a colourless precipitate was obtained. After completion of the reaction, the unreacted slight excess amount of substituted pyridine was removed via filtration and washing with CH3CN. Colourless solid; yield 92%; Rf (EtOH:Hex 2:3) 0.42; MP = 94–96 °C; 1H-NMR (400 MHz, CDCl3, δ ppm): 8.61–8.63 (d, J = 8 Hz, 2H), 8.02–8.04 (dd, J = 8 Hz, 2H), 7.44–7.46 (d, J = 8 Hz, 2H), 7.41–7.43 (d, J = 8 Hz, 1H), 7.36–7.39 (d, J = 12 Hz, 2H), 7.33 (s, 1H), 4.48 (s, 4H); 13C-NMR (100 MHz, CDCl3, δ ppm): 152.9, 152.8, 143.9, 142.6, 142.3, 129.0, 122.4, 122.2, 114.5, 110.9, 66.5; HRMS: m/z: 190.0835; anal. calcd.: C20H14N4Br2Cl2: C: 44.36; H: 2.58; N: 10.35; found C: 44.33; H: 2.54; N: 10.32.
5.1.3 Preparation of 2-chloro-3-cyano-1-(4-nitrobenzyl)pyridin-1-ium bromide (3). 2-Chloropyridine-3-carbonitrile (0.0042 mmol; 1.0 equiv.) was mixed with 4-nitrobenzylbromide (0.00433 mmol; 1.02 equi.) in the presence of 40 ml of dry CH3CN under refluxing condition for 45 min to afford 90% of 2-chloro-3-cyano-1-(4-nitrobenzyl)pyridin-1-ium bromide 3. Reactants were completely soluble in CH3CN when the reaction proceeded, and a colourless precipitate was obtained. After completion of the reaction, the unreacted slight excess amount of substituted pyridine was removed via filtration and washing with CH3CN. Colourless solid; yield 90%; Rf (EtOH:Hex 2:2) 0.36; MP = 84–86 °C; 1H-NMR (400 MHz, CDCl3, δ ppm): 8.62–8.60 (d, J = 8 Hz, 2H), 8.18–8.20 (d, J = 8 Hz, 2H), 8.03–8.05 (d, J = 8 Hz, 1H), 7.55–7.57 (dd, J = 8 Hz, 1H), 7.42–7.44 (d, J = 8 Hz, 1H), 4.52 (s, 2H); 13C-NMR (100 MHz, CDCl3, δ ppm): 152.8, 151.0, 144.8, 142.6, 129.9, 129.3, 124.0, 122.2, 114.6, 110.9, 55.1; HRMS: m/z: 274.0248; anal. calcd.: C13H9BrClN3O2: C: 44.06; H: 2.54: N: 11.86; found C: 44.02; H: 2.50: N: 11.83.
5.1.4 Preparation of 2,′-(hexane-1,6-diyl)bis(5-amino-4-cyano-1-phenyl)-1H-pyrazol-2-ium bromide (4). 1,6-Dibromohexane (0.00806 mmol; 1.0 equiv.) treated with 5-amino-1-pheneyl-1H-pyrazole-4- carbonitrile (0.0162 mmol; 2.02 equiv.) in the presence of 60 ml of dry CH3CN under refluxing condition for 2 hours afforded 2,2′-(hexane-1,6-diyl)bis(5-amino-4-cyano-1-phenyl)-1H-pyrazol-2-ium bromide 4 in 91% yield. Reactants were completely soluble in CH3CN when the reaction proceeded, and a colourless precipitate was obtained. After completion of the reaction, the unreacted slight excess amount of substituted pyridine was removed via filtration and washing with CH3CN. Colourless solid; yield 91%; Rf (EtOH:Hex 3:2) 0.40; MP = 156–158 °C; 1H-NMR (400 MHz, CDCl3, δ ppm): 7.86 (s, 2H), 7.78–7.81 (d, J = 12 Hz, 4H), 7.58–7.59 (dd, J = 4 Hz, 4H), 7.38–7.40 (d, J = 8 Hz, 2H), 3.53–3.56 (t, 4H), 1.75–1.84 (q, 4H), 1.29–1.32 (t, 4H), 13C-NMR (100 MHz, CDCl3, δ ppm): 151.7, 142.1, 137.9, 129.9, 128.3, 124.5, 115.3, 74.0, 35.3, 32.5, 27.1; HRMS: m/z: 226.1851; anal. calcd.: C26H28Br2N8: C: 50.98; H: 4.57: N: 18.30; found C: 50.95; H: 4.54; N: 18.26.
5.1.5 Preparation of 2,2′-(1,3-phenylenebis(methylene))bis(5-amino-4-cyano-1-phenyl)-1H-pyrazol-2-ium bromide (5). m-Xylenedibromide (0.0026 mmol; 1.0 equiv.) treated with 5-amino-1-pheneyl-1H-pyrazole-4-carbonitrile (0.0050 mmol; 2.02 equiv.) in the presence of 60 ml of dry CH3CN under refluxing condition for 90 min afforded 2,2′-(1,3-phenylenebis(methylene))bis(5-amino-4-cyano-1-phenyl)-1H-pyrazol-2-ium bromide 5 in 92% yield. Reactants were completely soluble in CH3CN when the reaction proceeded, and a colourless precipitate was obtained. After completion of the reaction, the unreacted slight excess amount of substituted pyridine was removed via filtration and washing with CH3CN. Colourless solid; yield 92%; Rf (EtOH:Hex 3:3) 0.36; MP = 102–104 °C; 1H-NMR (400 MHz, CDCl3, δ ppm): 7.54 (s, 2H), 7.45–7.47 (d, J = 8 Hz, 4H), 7.42–7.44 (dd, J = 8 Hz, 4H), 7.40–7.41 (d, J = 4 Hz, 2H), 7.38–7.39 (d, J = 4 Hz, 2H), 7.36–7.37 (d, J = 4 Hz, 1H), 7.34 (s, 1H), 4.86 (s, 4H); 13C-NMR (100 MHz, CDCl3, δ ppm): 154.4, 153.6, 150.2, 141.3, 136.9, 133.2, 131.0, 129.9, 128.8, 124.2, 117.8, 114.3, 75.6; HRMS: m/z: 236.0182; anal. calcd.: C28H24Br2N8: C: 53.16; H: 3.79: N: 17.72; found C: 53.13; H: 3.75; N: 17.68.
5.2 General procedures for the solid-supported solvent-free microwave reaction
A domestic 20 litre microwave oven (1350 watts, 230 volts) was used for all solvent-free silica-supported microwave reactions. We used column chromatography silica gel with 60–120 mesh size from suppliers to make the silica-supported reaction mixture without any further purification/treatment. A 1:2 equivalence of 2-chloropyridine-3-carbonitrile 1/5-amino-1-pheneyl-1H pyrazole-4-carbonitrile 2 and alkyl/aryl dibromide were mixed, along with 5 g of silica gel (80–120 mesh), followed by manual grinding. The reaction mixture was divided into two and three portions to optimize the reaction time and yield. Flexible dimeric pyridinium bromide 1 was obtained in 4 min with 92% yield. The m-xylene linked pyridinium bromide 2 was obtained in 2 min with 95% yield. The monomeric pyridinium bromide 3 was obtained in 3 min with 94% yield. The flexible dimeric pyrazolium bromide 4 was obtained in 7 min with 93% yield and the m-xylene linked pyrazolium bromide 5 was obtained in 5 min with 95% yield. The reaction mixture was dissolved in ethanol, followed by concentration, to obtain the colourless precipitate. The crude product was washed with CH3CN to remove the unreacted slight excess amount of substituted pyridine.
6 Conclusion
Mono/dimeric pyridinium and pyrazolium bromides were prepared under conventional refluxing conditions and solvent-free silica-supported domestic microwave conditions. The solvent-free silica supported method has more merits when compared with the conventional method, and volatile organic solvents were completely avoided. Pyridinium and pyrazolium salts were formed nearly fifteen time faster with a higher percentage of yield. We have calculated the atom economy for pyridinium and pyrazolium bromide formation, which was 100% for all reactions. The environmental factors for all of the reactions showed zero percent of environmental waste. In addition, the product mass intensity for the solvent-free reactions and PMISF value were 1. We have studied the anticancer properties of mono-/dimeric substituted pyridinium and pyrazolium bromide against lung cancer (A-549) lines at 10/20/40/80/100/140 μg ml−1 concentration under MTT assay for 24 hours. We found that the pyridinium and pyrazolium bromides showed significant cytotoxicity against the lung cancer cell lines. 4-Nitrobenzylpyridiniumbromide showed excellent anticancer properties when compared with the flexible aliphatic chain and m-xylene linker based dimeric pyridinium and pyrazolium bromides. We have performed computer-assisted molecular docking with human CDK1/CyclinB1/CKS2 kinase receptor. m-Xylene linked pyrazolium bromide showed the highest docking score when compared with other mono-/dimeric pyridinium and pyrazolium bromides. The docking outcome was in good agreement with the anticancer activity result. Further applications of this approach will be performed on the synthesis of various substituted mono-, di- and trimeric pyridinium bromides, and the anticancer activities will be studied.
Data availability
The data supporting this article have been included within the manuscript and its ESI.†
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
M. A. and A. S. thank the Deanship of Research, King Khalid University, Saudi Arabia, for the large research group under the grant number R. G. P. 2/121/1445.
References
- L. Alessandra, E. Costantino, D. Massimiliano, M. Mario and B. Gaetano, Primary bone osteosarcoma in the pediatric age: State of the art, Cancer Treat. Rev., 2006, 32, 423–436 CrossRef PubMed.
- L. B. Matthew, C. M. C. Jonathan, E. M. Damian, R. D. Crispin and F. M. C. Peter, The molecular pathogenesis of osteosarcoma, Sarcoma, 2011, 2011, 959236–959248 Search PubMed.
- Z. Qian, L. Jin-Li, F. Xiao-Xia and L. Jia-Cheng, Nitrile-containing copper(II) porphyrin coordination complexes for efficient anticancer activity and mechanism research, New J. Chem., 2021, 45, 5221–5227 RSC.
- S. Govindaraj, K. Ganesan, D. Mahendiran, R. Lakshmisundaram, M. M. Alam and M. Amanullah, Synthesis of potent MDA-MB 231 breast cancer drug molecules from single step, Sci. Rep., 2023, 13, 18241–18253 CrossRef CAS PubMed.
- S. Govindaraj, K. Ganesan, D. Mahendiran, R. Lakshmisundaram, M. K. Kalaivani, P. Viswanathan, M. M. Alam and M. Amanullah, Discovery of novel dimeric pyridinium bromide analogs; Inhibits cancer cell growth by activating caspases and downregulating Bcl-2 protein, ACS Omega, 2023, 8, 13243–13251 CrossRef CAS PubMed.
- P. Ganapathi and K. Ganesan, Anti-bacterial, catalytic and docking behaviours of novel di/trimeric imidazolium salts, J. Mol. Liq., 2017, 233, 452–464 CrossRef CAS.
- R. E. Mohamed, E. M. Samar, A. F. Ahmed, A. L. Ehab and A. A. Miral, Synthesis, biological evaluation and molecular docking of new triphenylamine-linked pyridine, thiazole and pyrazole analogues as anticancer agents, BMC Chem., 2022, 16, 68–88 CrossRef PubMed.
- M. A. Osama, A. M. Mahmoud, R. A. Rasha and A. A. Sayed, Synthesis and anti-tumor activities of some new pyridines and pyrazolo [1,5-a]pyrimidines, Eur. J. Med. Chem., 2009, 44, 3519–3523 CrossRef PubMed.
- M. P. Matthew, H. C. Jaime and M. Mohammad, Total synthesis and anti-cancer activity of all known communesin alkaloids and related derivatives, J. Am. Chem. Soc., 2019, 141, 14411–14420 CrossRef PubMed.
- F. Daoyang, Z. Chaoqi, W. Hufei, W. Qingguang, C. Hong, W. Feng, B. Zhilei, L. Weifeng, W. Xing and L. Zhongjun, Fabrication of a composite 3D-printed titanium alloy combined with controlled in situ drug release to prevent osteosarcoma recurrence, Mater. Today Bio, 2023, 20, 100672–100683 CrossRef PubMed.
- S.-A. Adeeb Al, K. Daoud, N. Arshi, A. F. Faleh, R. Nadjet, A. R. Mohamed and H. Mohamed, Design, synthesis, molecular modeling, anticancer studies, and density functional theory calculations of 4-(1,2,4-triazol-3- ylsulfanylmethyl)-1,2,3-triazole derivatives, ACS Omega, 2021, 6, 301–316 CrossRef PubMed.
- F. Sara, B. R. Farjana, R. D. Sarah, P. S. Yogita, W. Tracy and J. S. Timothy, Development of a novel, multifunctional, membrane-interactive pyridinium salt with potent anticancer activity, Bioorg. Med. Chem. Lett., 2014, 24, 3430–3433 CrossRef PubMed.
- J. A. R. Salvador, R. M. A. Pinto and S. M. Silvestre, Steroidal 5α-reductase and 17α-hydroxylase/17, 20-lyase (CYP17) inhibitors useful in the treatment of prostatic diseases, J. Steroid Biochem. Mol. Biol., 2013, 137, 199–222 CrossRef CAS PubMed.
- R. Ferraldeschi, N. Sharifi, R. J. Auchus and G. Attard, Molecular pathways: Inhibiting steroid biosynthesis in prostate cancer, Clin. Cancer Res., 2013, 19, 3353–3359 CrossRef CAS PubMed.
- E. A. Djurendić, M. P. Zaviš, M. N. Sakač, J. J. Čanadi, V. V. Kojić, G. M. Bogdanović and K. M. Penov Gaši, Synthesis and antitumor activity of new d-seco and d-homo androstane derivatives, Steroids, 2009, 74, 983–988 CrossRef PubMed.
- H. A. Henidi, A. M. Al-Abd, F. A. Al-Abbasi, H. A. BinMahfouza and I. M. El-Deeb, Design and synthesis of novel phenylaminopyrimidines with antiproliferative activity against colorectal cancer, RSC Adv., 2019, 9, 21578–21586 RSC.
- M. Hagras, A. A. Mandour, E. A. Mohamed, E. B. Elkaeed, I. M. M. Gobaara, A. B. M. Mehany, N. S. M. Ismail and H. M. Refaatb, Design, synthesis, docking study and anticancer evaluation of new trimethoxyphenyl pyridine derivatives as tubulin inhibitors and apoptosis inducers, RSC Adv., 2021, 11, 39728–39741 RSC.
- M. M. Abdelshaheed, H. I. El Subbagh, M. A. Tantawy, R. T. Attia and K. M. Yousseff, I.M. Fawzy Discovery of new pyridine heterocyclic hybrids; design, synthesis, dynamic simulations, and in vitro and in vivo breast cancer biological assays, RSC Adv., 2023, 13, 15689–15703 RSC.
- M. P. Savić, J. J. Ajduković, J. J. Plavša, S. S. Bekić, A. S. Ćelić, O. R. Klisurić, D. S. Jakimov, E. T. Petri and E. A. Djurendića, Evaluation of A-ring fused pyridine D-modified androstane derivatives for antiproliferative and aldo–keto reductase 1C3 inhibitory activity, Med. Chem. Commun., 2018, 9, 969–981 RSC.
- R. Krishnamoorthy and P. Anaikutti, Iodine catalyzed synthesis of imidazo[1,2-a] pyrazine and imidazo[1,2-a]pyridine derivatives and their anticancer activity, RSC Adv., 2023, 13, 36439–36454 RSC.
- G. S. Mani, S. P. Shaik, Y. Tangella, S. Bale, C. Godugua and A. Kamal, A facile I2-catalyzed synthesis of imidazo[1,2-a]pyridines via sp3 C-H functionalization of azaarenes and evaluation of anticancer activity, Org. Biomol. Chem., 2017, 15, 6780–6791 RSC.
- R. B. W. Nicholas, K. Svitlana, P. M. Mathew, A. S. Will, M. Rouslan, M. E. M. Noble and A. E. N. Jane, CDK1 structures reveal conserved and unique features of the essential cell cycle CDK, Nat. Commun., 2015, 6, 6769–6781 CrossRef PubMed.
- B. Martina, C. Luca, V. Sapthaswaran, C. Catherine, R. Camilla and P. Jonathon, Cell cycle-dependent binding between cyclin B1 and CDK1 revealed by time-resolved fluorescence correlation spectroscopy, Open Biol., 2022, 12, 220057–220070 CrossRef PubMed.
- A. Z. El-Sonbati, M. A. Diab, A. A. El-Bindary, A. M. Eldesoky and S. M. Morgan, Correlation between ionic radii of metals and thermal decomposition of supramolecular structure of azodye complexes, Spectrochim. Acta, Part A, 2015, 135, 774–791 CrossRef CAS PubMed.
- K. Daniel and N. Robert, ESR Studies on the Bonding in Copper Complexes, J. Chem. Phys., 1961, 35, 149–155 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Perumal Elumalaib,
Rajanathadurai Jeevithab,
Annadurai Subramanic,
Mohammed Amanullahd and
Awad Saeed Al-Samghane
*a,
Perumal Elumalaib,
Rajanathadurai Jeevithab,
Annadurai Subramanic,
Mohammed Amanullahd and
Awad Saeed Al-Samghane