
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and computational evaluation of the antioxidant activity of pyrrolo[2,3-b]quinoxaline derivatives†
Nguyen Tran Nguyen *a,
Vo Viet Daia,
Adam Mechlerb,
Luc Van Meerveltc,
Nguyen Thi Hoad and
Quan V. Vo*d
*a,
Vo Viet Daia,
Adam Mechlerb,
Luc Van Meerveltc,
Nguyen Thi Hoad and
Quan V. Vo*d
aThe University of Danang-University of Science and Education, Danang 550000, Vietnam. E-mail: ntnguyen@ued.udn.vn
bDepartment of Biochemistry and Chemistry, La Trobe University, Victoria 3086, Australia
cBiomolecular Architecture, Department of Chemistry, KU Leuven, Celestijnenlaan 200F, B-3001 Leuven, Belgium
dThe University of Danang – University of Technology and Education, Danang 550000, Vietnam. E-mail: vvquan@ute.udn.vn
First published on 5th August 2024
Abstract
Pyrrolo[2,3-b]quinoxaline derivatives are known to possess antioxidant, anticancer, and antibacterial properties. Here we report the successful synthesis of five derivatives of 3-hydroxy-3-pyrroline-2-one through substitution. The 2,2-diphenyl-1-picrylhydrazyl (DPPH) assay was employed to evaluate the antioxidant activity of the compounds. Out of these, ethyl 1,2-diphenyl-1H-pyrrolo[2,3-b]quinoxaline-3-carboxylate (3a) demonstrated the greatest potential as a radical scavenger. Thermodynamic and kinetic calculations of the radical scavenging activity indicated that 3a exhibited HO˙ radical scavenging activity with the overall rate constant of 8.56 × 108 M−1 s−1 in pentyl ethanoate; however, it was incapable of scavenging hydroperoxyl radicals in nonpolar media. In non-polar environments, the hydroxyl radical scavenging capability of 3a is fairly similar to that of reference antioxidants such as Trolox, melatonin, indole-3-carbinol, and gallic acid. Hence, in the physiological lipid environment, 3a holds promise as a scavenger of HO˙ radicals.
1. Introduction
The quinoxaline nucleus exists in numerous bioactive natural products. For instance, Lumiphenazine A isolated from Streptomyces sp. IFM 11204 showed biological activity against human gastric adenocarcinoma cells.1 Triostin A isolated from Streptomyces aureus S-2-210 exhibited antitumor and antimicrobial activities.2,3 In addition to natural products, several quinoxaline-containing synthetic drugs have been discovered.4–7 Bromonidine, for instance, is a commercially available α-2 adrenergic agonist which has been used to treat glaucoma and ocular hypertension, as well as facial erythema in rosacea.6 Varenicline is a quinoxaline-based α4β2 nicotinic receptor partial agonist drug available in the market and is used for smoking cessation (Fig. 1).5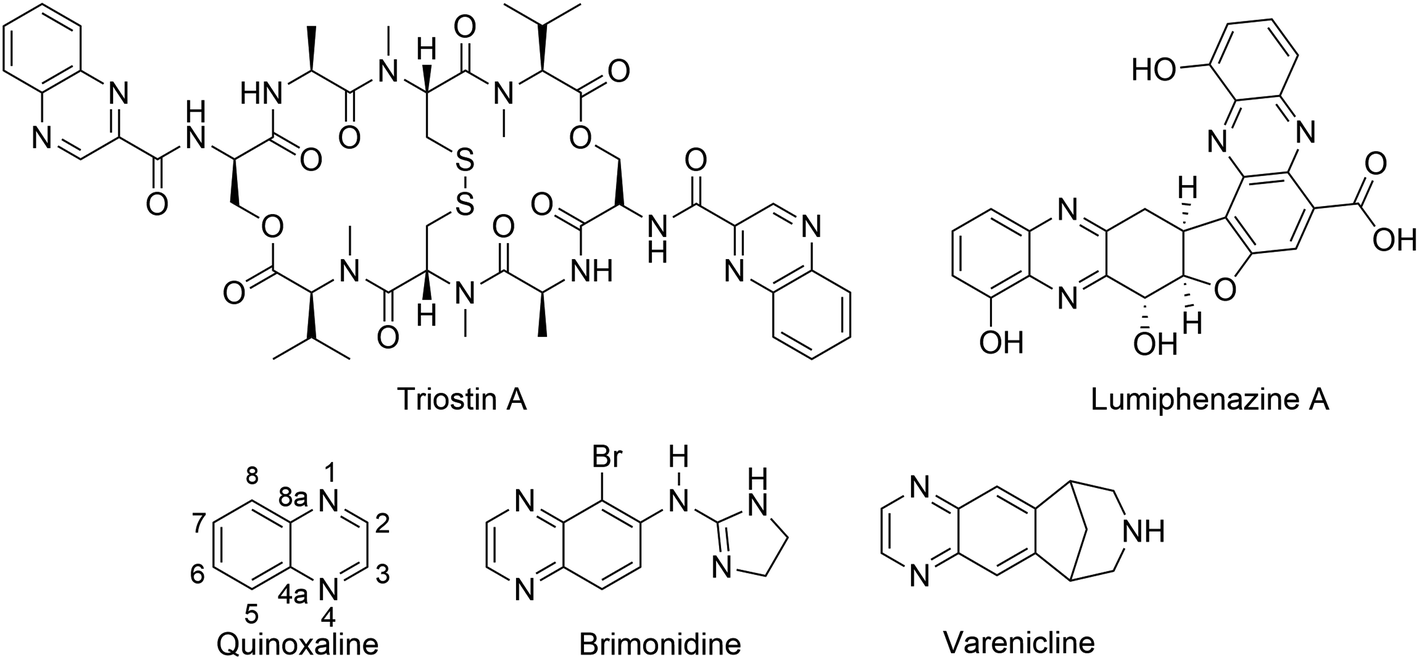 | ||
| Fig. 1 Representative examples of natural products and commercially available drugs containing quinoxaline moiety. | ||
It should be noted that most pyrrolo[2,3-b]quinoxalines have been synthesized from halogen-containing quinoxaline derivatives with palladium catalysts. For instance, reactions of 2-alkynyl-3-trifluoroacetamido quinoxalines with aryl and vinyl halides in the presence of Pd(PPh3)4 and K2CO3,8 Sonogashira coupling reaction of N-alkyl-3-chloroquinoxaline-2-amines with propargyl bromide in the presence of PdCl2(PPh3)2 and CuI,9 and Pd/C-catalyzed reactions of dichloroquinoxaline with hydrazine, phenylacetylene and aldehydes,10 Recently, bismuth salts Bi(OTf)3 were used as the catalyst for the condensation of 1H-pyrrole-2,3-diones and o-phenylenediamine to yield pyrrolo[2,3-b]quinoxalines.11 Biological and pharmacological activities of synthetic pyrrolo[2,3-b]quinoxalines were also reported identifying these compounds as anticancer agents,12,13 antibiotics,14,15 and antioxidants.16
Polysubstituted 3-hydroxy-3-pyrroline-2-one has recently appeared as an effective 1,2-diketone-containing building block for the synthesis of pyrrolo[2,3-b]quinoxalines.16 These 2-oxopyrrole-containing compounds also possess a wide array of biological activities on their own, e.g., anticancer,17,18 anti-bacterial,19,20 anti-HIV,21 anti-inflammatory,22 and antioxidant23,24 activities. Therefore, the synthesis of quinoxaline derivatives starting from polysubstituted 3-hydroxy-3-pyrroline-2-ones can yield pyrrolo[2,3-b]quinoxalines with potential biological activities. In this manuscript, we report the synthesis of pyrrolo[2,3-b]quinoxalines via the condensation of 1,5-disubstituted-4-ethoxycarbonyl-3-hydroxy-3-pyrroline-2-one with o-phenylenediamine. The 1,1-diphenyl-1-picrylhydrazyl (DPPH) free radical scavenging capacity of pyrrolo[2,3-b]quinoxalines is discussed. Furthermore, the interaction of the best-performing pyrrolo[2,3-b]quinoxaline with HO˙ and HOO˙ radicals is evaluated using well-established model chemistry based on the quantum mechanics-based test for the overall free radical scavenging activity (QM-ORSA) protocol.
2. Experimental and computational methods
2.1. Experimental
(a) Ethyl 1,2-diphenyl-1H-pyrrolo[2,3-b]quinoxaline-3-carboxylate (3a). 1H NMR (600 MHz, CDCl3) δ 8.41 (dd, 3J(H,H) = 8.28 Hz, 4J(H,H) = 1.32 Hz, 1H; Ar–H), 8.05 (dd, 3J(H,H) = 8.27 Hz, 4J(H,H) = 1.37 Hz, 1H; Ar–H), 7.71 (m, 2H; Ar–H), 7.38 (m, 6H; Ar–H), 7.27 (m, 2H; Ar–H), 4.32 (q, 3J(H,H) = 7.14 Hz, 2H; OCH2), 1.19 ppm (t, 3J(H,H) = 7.14 Hz, 3H; CH3). 13C NMR (150 MHz, CDCl3) 163.34, 155.14, 142.87, 142.27, 140.63, 139.84, 134.93, 130.70, 130.15, 130.07, 129.70, 129.21, 128.79, 128.72, 128.53, 128.45, 127.90, 127.75, 105.31, 60.54, 14.19 ppm. HRMS (ESI-quadrupole) m/z [M + H]+ calcd for C25H19N3O2: 394.1555; found: 394.1540.
(b) Ethyl 1,2-diphenyl-2,4-dihydro-1H-pyrrolo[2,3-b]quinoxaline-3-carboxylate (3a′). 1H NMR (600 MHz, CDCl3) δ 8.96 (sbr, 1H; NH), 7.77–6.90 (14H; Ar–H), 6.25 (s, 1H), 4.12 (q, 3J(H,H) = 7.05 Hz, 2H; O–CH2), 1.18 ppm (t, 3J(H,H) = 7.10 Hz, 3H; CH3). 13C NMR (150 MHz, CDCl3) 155.75, 139.30, 138.61, 135.95, 128.85, 128.36, 127.90, 127.42, 127.39, 126.60, 124.95, 124.26, 123.71, 121.54, 114.60, 96.61, 67.43, 59.86, 14.39 ppm. HRMS (ESI-TOF MS/MS) m/z [M + H]+ calcd for C25H21N3O2: 396.1712; found: 396.1682.
(c) Ethyl 1-(3-nitrophenyl)-2-(p-tolyl)-1H-pyrrolo[2,3-b]quinoxaline-3-carboxylate (3b). 1H NMR (600 MHz, CDCl3) δ 8.41 (dd, 3J(H,H) = 8.22 Hz, 4J(H,H) = 1.60 Hz, 1H; Ar–H), 8.22 (m, 2H; Ar–H), 8.03 (dd, 3J(H,H) = 8.23 Hz, 4J(H,H) = 1.65 Hz, 1H; Ar–H), 7.74 (m, 2H; Ar–H), 7.59 (m, 2H; Ar–H), 7.27 (d, 3J(H,H) = 8.12 Hz, 2H; Ar–H), 7.18 (d, 3J(H,H) = 7.86 Hz, 2H; Ar–H), 4.37 (q, 3J(H,H) = 7.13 Hz, 2H; OCH2), 2.37 (s, 3H; CH3), 1.25 ppm (t, 3J(H,H) = 7.10 Hz, 3H; CH3). 13C NMR (150 MHz, CDCl3) 163.14, 154.44, 148.58, 142.53, 142.46, 140.62, 140.60, 139.62, 136.25, 134.45, 130.66, 130.17, 129.94, 129.10, 128.98, 128.59, 128.13, 126.29, 123.85, 122.96, 106.46, 60.81, 21.65, 14.27 ppm. HRMS (ESI-quadrupole) m/z [M + H]+ calcd for C26H20N4O4: 453.1562; found: 453.1555.
(d) Ethyl 2-(4-nitrophenyl)-1-phenyl-1H-pyrrolo[2,3-b]quinoxaline-3-carboxylate (3c). 1H NMR (600 MHz, CDCl3) δ 8.41 (dd, 3J(H,H) = 8.22 Hz, 4J(H,H) = 1.27 Hz, 1H; Ar–H), 8.20 (d, 3J(H,H) = 8.79 Hz, 2H; Ar–H), 8.06 (dd, 3J(H,H) = 8.16 Hz, 4J(H,H) = 1.01 Hz, 1H; Ar–H), 7.75 (m, 2H; Ar–H), 7.59 (d, 3J(H,H) = 8.82 Hz, 2H; Ar–H), 7.42 (m, 3H; Ar–H), 7.26 (m, 2H; Ar–H), 4.36 (q, 3J(H,H) = 7.12 Hz, 2H; OCH2), 1.26 ppm (t, 3J(H,H) = 7.12 Hz, 3H; CH3). 13C NMR (150 MHz, CDCl3) 162.91, 151.86, 148.37, 142.76, 142.55, 140.11, 139.90, 136.78, 134.36, 131.95, 130.19, 129.63, 129.13, 129.05, 128.78, 128.70, 128.20, 123.05, 106.27, 60.94, 14.31 ppm. HRMS (ESI-TOF MS/MS) m/z [M + H]+ calcd for C25H18N4O4: 439.1406; found: 439.1388.
(e) Ethyl 1-phenyl-2-(p-tolyl)-1H-pyrrolo[2,3-b]quinoxaline-3-carboxylate (3d). 1H NMR (600 MHz, CDCl3) δ 8.38 (dd, 3J(H,H) = 8.59 Hz, 4J(H,H) = 1.56 Hz, 1H; Ar–H), 8.03 (dd, 3J(H,H) = 8.41 Hz, 4J(H,H) = 1.30 Hz, 1H; Ar–H), 7.68 (m, 2H; Ar–H), 7.39 (m, 3H; Ar–H), 7.25 (m, 4H; Ar–H), 7.12 (d, 3J(H,H) = 7.59 Hz, 2H; Ar–H), 4.35 (q, 3J(H,H) = 7.09 Hz, 2H; OCH2), 2.35 (s, 3H; CH3), 1.24 ppm (t, 3J(H,H) = 7.15 Hz, 3H; CH3). 13C NMR (150 MHz, CDCl3) 163.51, 155.49, 142.90, 142.23, 140.73, 139.93, 139.76, 135.06, 130.65, 130.01, 129.20, 128.81, 128.70, 128.63, 128.40, 128.36, 127.67, 126.97, 105.10, 60.55, 21.61, 14.29 ppm. HRMS (ESI-TOF MS/MS) m/z [M + H]+ calcd for C26H21N3O4: 408.1712; found: 408.1691.
(f) Ethyl 1-benzyl-2-(4-nitrophenyl)-1H-pyrrolo[2,3-b]quinoxaline-3-carboxylate (3e). 1H NMR (600 MHz, CDCl3) δ 8.43 (m, 1H; Ar–H), 8.28 (d, 3J(H,H) = 8.67 Hz, 2H; Ar–H), 8.14 (m, 1H; Ar–H), 7.77 (m, 2H; Ar–H), 7.47 (d, 3J(H,H) = 8.67 Hz, 2H; Ar–H), 7.17 (m, 3H; Ar–H), 6.83 (dd, 3J(H,H) = 8.26 Hz, 4J(H,H) = 1.02 Hz, 2H; Ar–H), 5.48 (s, 2H; CH2), 4.26 (q, 3J(H,H) = 7.11 Hz, 2H; OCH2), 1.16 ppm (t, 3J(H,H) = 7.11 Hz, 3H; CH3). 13C NMR (150 MHz, CDCl3) 162.72, 152.89, 148.71, 142.51, 142.09, 139.91, 139.85, 137.09, 136.39, 131.16, 130.27, 129.07, 128.91, 128.60, 128.18, 128.02, 127.11, 123.27, 105.58, 60.63, 46.39, 29.84, 14.22 ppm. HRMS (ESI-quadrupole) m/z [M + H]+ calcd for C26H20N4O4: 453.1562; found: 453.1552.
2.2. Determination of crystal structure of 3a
Single crystals of 3a suitable for X-ray diffraction were obtained by evaporating a solution in dichloromethane/n-hexane mixture at room temperature. X-ray diffraction patterns were collected at 293(2) K on an Agilent SuperNova diffractometer, equipped with an Eos CCD detector, using Mo Kα radiation (λ = 0.71073 Å). The images were interpreted and integrated with the CrysAlisPro and the implemented absorption correction was applied.27 Using Olex2,28 the structure was solved with the ShelXT29 structure solution program using intrinsic phasing and refined with the ShelXL30 refinement package using full-matrix least-squares minimization on F2. Non-hydrogen atoms were anisotropically refined and the hydrogen atoms in the riding mode with isotropic temperature factors were fixed at 1.2 times Ueq of the parent atoms (1.5 for methyl groups). Table S2† gives crystal data and structure refinement details for 3a.2.3. Computational details
All DFT calculations were carried out with the Gaussian 09 suite of programs.31 M06-2X functional32 and 6-311++G(d,p) basis set were used for all calculations. The M06-2X functional offers one of the most reliable methods to study the thermodynamics and kinetics of radical reactions.32–37 The kinetic calculations were performed following the QM-ORSA protocol,38,39 following the literature.36,37,40–43 This method has been repeatedly benchmarked against experimental data, delivering results with low errors (kcalc/kexp ratio = 1–2.9), particularly in lipid medium.34,38,39 As a reference all details of the calculations are shown in Table S1, ESI.†3. Results and discussion
3.1. Synthesis of pyrrolo[2,3-b]quinoxalines
The reaction between 1,5-diphenyl-4-ethoxycarbonyl-3-hydroxy-3-pyrroline-2-one (1a)24 and o-phenylenediamine (2) was chosen as the model to optimize the reaction conditions, such as the reactant ratio and solvent. Heating 3-pyrroline-2-one derivative 1a (1 equiv.), o-phenylenediamine (2) (1 equiv.) and citric acid catalyst (2 equiv.) in 1 mL of absolute ethanol at 80 °C resulted in the formation of pyrrolo[2,3-b]quinoxaline 3a with a yield of only 12% (Table 1). However, keeping the equimolar amounts of starting materials 1a (1 equiv., 0.16 mmol) and 2 (1 equiv., 0.16 mmol) in 1 mL of glacial acetic acid solvent at 90 °C brought a dramatic increase in the yield of ethyl 1-phenyl-2-phenyl-1H-pyrrolo[2,3-b]quinoxaline-3-carboxylate (3a), 50%. Therefore, acetic acid was chosen as the solvent to optimize the concentration of reactants as well as the reactant ratio. The decrease in the concentration of both reactants 1a and 2 to 0.10 M led to a downward trend in the yield of pyrrolo[2,3-b]quinoxaline 3a, 40%. In contrast, there was a slight increase in the yield of desired product 3a, 56%, when the concentration of starting materials in acetic acid increased to 0.31 M.| Entry | Solvent | Volume (mL) | Ratio 1a : 2 (equiv.) | Concentration 1a : 2 (mmol mL−1) | Temperature (°C) | Time (hour) | Yield (%) |
|---|---|---|---|---|---|---|---|
| 1 | Ethanol | 1.0 | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
0.16:0.16 |
80 °C | 5 | 12 |
| 2 | AcOH | 1.0 | 1:1 |
0.16:0.16 |
90 °C | 5 | 50 |
| 3 | AcOH | 0.5 | 1:1 |
0.31:0.31 |
90 °C | 5 | 56 |
| 4 | AcOH | 0.31 | 1:1 |
0.5:0.5 |
90 °C | 5 | 53 |
| 5 | AcOH | 1.5 | 1:1 |
0.10:0.10 |
90 °C | 5 | 40 |
| 6 | AcOH | 0.5 | 1:1.5 |
0.31:0.47 |
90 °C | 5 | 71 |
| 7 | AcOH | 0.5 | 1:2.0 |
0.31:0.62 |
90 °C | 5 | 78 |
| 8 | AcOH | 0.5 | 1:3.0 |
0.31:0.93 |
90 °C | 5 | 83 |
| 9 | AcOH | 0.5 | 1:3.0 |
0.31:0.93 |
110 °C | 5 | 60 |
The product 3a could be obtained in 83% yield when the concentration of aromatic amine 2 in the solvent increased to 0.93 M, while the amount of 3-pyrroline-2-one derivative 1a remained unchanged at 0.31 M. Therefore, the ratio 1:3.0 of reactants 1,5-disubstituted-4-ethoxycarbonyl-3-hydroxy-3-pyrroline-2-one and o-phenylenediamine, respectively, was used to synthesize other pyrrolo[2,3-b]quinoxalines in glacial acetic acid (Scheme 1 and Table 2).
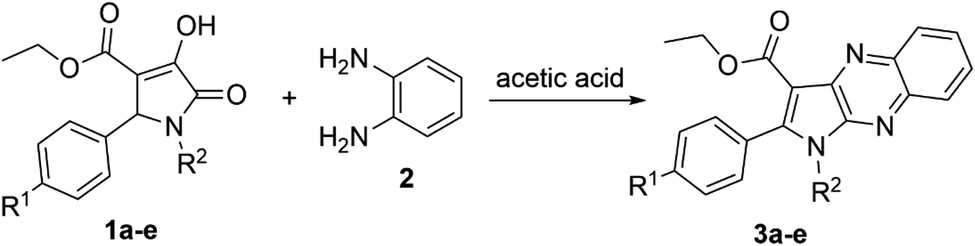 | ||
| Scheme 1 Synthesis of pyrrolo[2,3-b]quinoxalines (3a–e). | ||
| Entry | R1 | R2 | Time (hour) | Product | Yield (%) |
|---|---|---|---|---|---|
| 1 | H | C6H5 | 5 | 3a | 83 |
| 2 | CH3 | 3-NO2C6H4 | 8 | 3b | 70.1 |
| 3 | NO2 | C6H5 | 2 | 3c | 50 |
| 4 | CH3 | C6H5 | 3 | 3d | 65.2 |
| 5 | NO2 | C6H5CH2 | 3 | 3e | 45 |
The enol tautomer of 1,5-disubstituted-4-ethoxycarbonyl-3-hydroxy-3-pyrroline-2-ones (1a–e) could be converted to the more stable keto form 1a′–e′ in a polar solvent such as ethanol or acetic acid.44 The reactions between 1a–e/1a′–e′ and o-phenylenediamine (2) are reversible and occur at the 3-position of pyrrolin-2,3-dione heterocyclic ring to yield imine intermediate 4 (ref. 16) Intermediate 4 exists predominantly in the enamine form 5 due to resonance stabilization via intramolecular hydrogen bonding45 and the intramolecular nucleophilic addition reaction between the amino (NH2) and carbonyl group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in intermediate 5 results in products 3a′–e′. Finally, oxidative dehydrogenation of compounds 3a′–e′ by oxygen molecules in fresh air will result in the formation of pyrrolo[2,3-b]quinoxalines 3a–e (Scheme 2).46
O) in intermediate 5 results in products 3a′–e′. Finally, oxidative dehydrogenation of compounds 3a′–e′ by oxygen molecules in fresh air will result in the formation of pyrrolo[2,3-b]quinoxalines 3a–e (Scheme 2).46
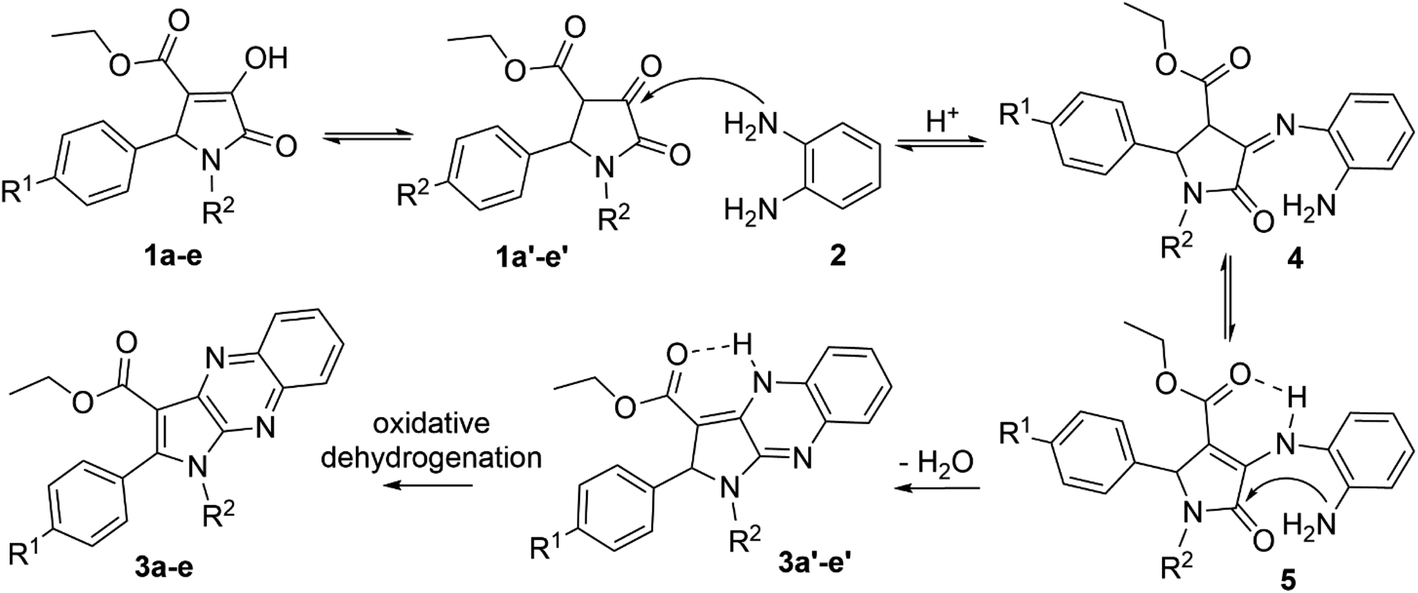 | ||
| Scheme 2 Proposed mechanism for the synthesis of pyrrolo[2,3-b]quinoxalines. | ||
All new compounds were characterized by NMR and high resolution mass spectrometry (HRMS). In the 1H NMR spectrum of ethyl 1,2-diphenyl-2,4-dihydro-1H-pyrrolo[2,3-b]quinoxaline-3-carboxylate (3a′), there is a characteristic broad and low-intensity peak corresponding to the proton of the secondary amino group (NH) at the chemical shift of 8.97 ppm. Undoubtedly, the intramolecular hydrogen bonding induced the higher chemical shift of the secondary amino proton. Furthermore, the proton at the 2-position of the pyrrole moiety of compound 3a′ appears as a singlet at 6.24 ppm. In contrast, this peak disappears in the 1H NMR spectrum of ethyl 1,2-diphenyl-1H-pyrrolo[2,3-b]quinoxaline-3-carboxylate (3a). Moreover, 1H NMR spectra of pyrrolo[2,3-b]quinoxalines 3a–e all showed two doublets of doublets (dd) at chemical shifts around 8.41 ppm and 8.05 ppm, representing two protons at the 5,8-positions of the quinoxaline moieties. In addition, resonance signals of two methylene protons and three methyl protons of the ethoxycarbonyl group (–C(O)OCH2CH3) in compounds 3a–e were characterized by a quartet (q) and a triplet (t) at chemical shifts around 4.36 ppm and 1.26 ppm, respectively.
3.2. X-ray study of ethyl 1,2-diphenyl-1H-pyrrolo[2,3-b]quinoxaline-3-carboxylate (3a)
The structure of 3a was verified through single-crystal X-ray diffraction (Fig. 2). The central pyrrolo[2,3-b]quinoxaline ring is planar (r.m.s. deviation 0.039 Å) and makes an angle of 57.83(13)° with phenyl ring C14–C19 and of 63.63(13)° with phenyl ring C25–C30.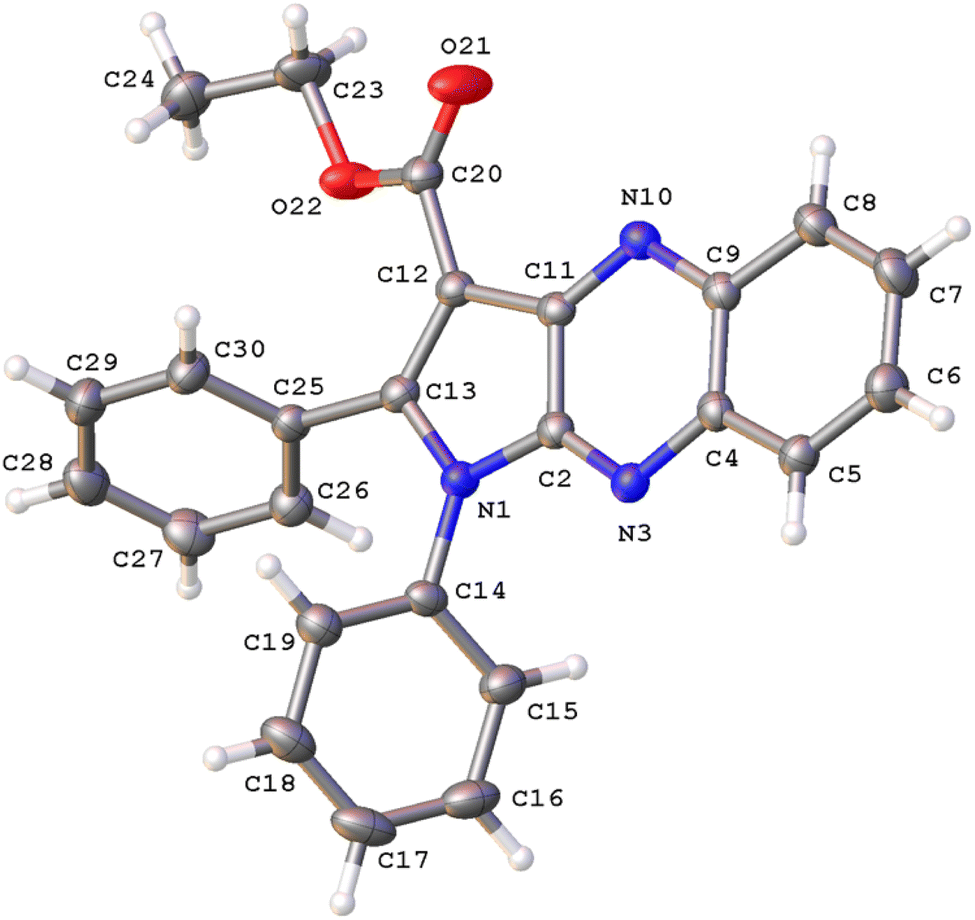 | ||
| Fig. 2 A view of the molecular structure of 3a, with atom labels and displacement ellipsoids drawn at the 30% probability level. H atoms are shown as small circles of arbitrary radii. | ||
Both phenyl rings make a mutual angle of 57.21(17)°. The best plane through ester group C12, C20, O21, O22 makes an angle of 18.13(14)° with the central ring system. The crystal packing of 3a (Fig. S1†) is characterized by C17–H17⋯O21i hydrogen bonds resulting in chain formation along the c direction [C17–H17: 0.93 Å, H17⋯O21: 2.35 Å, C17⋯O21: 3.383(5) Å, C17–H17⋯O21: 150°; symmetry code: (i) 3/2 − x, 1 − y, 1/2 + z]. Neighboring chains connect by C–H⋯π interactions between C24–H24A and the central pyrazine ring [H24A⋯Cgii: 2.81 Å; Cg is the centroid of ring N3, C4, C9, N10, C11, C2; symmetry code: (ii) 1 − x, −1/2 + y, 1/2 − z].
3.3. The radical scavenging activity of pyrrolo[2,3-b]quinoxalines 3a–e
| Compound | DPPH (%) | EC50 (μg mL−1) | |||
|---|---|---|---|---|---|
| 265 (μg mL−1) | 128 (μg mL−1) | 32 (μg mL−1) | 8 (μg mL−1) | ||
| 3a | 30 | 24 | 0 | 0 | >128 |
| 3b | 9 | 7 | 0 | 0 | >128 |
| 3c | 23 | 20 | 0 | 0 | >128 |
| 3d | 10 | 9 | 0 | 0 | >128 |
| 3e | 21 | 18 | 0 | 0 | >128 |
| Quercetin | 100 | 100 | 45.5 | 0 | 9.97 ± 0.25 |
(a) Structure evaluation. The hexagon rings of 3a are capable of undergoing rotation in order to generate an extensive range of conformers. Since the conformer with the highest probability of engaging in a radical scavenging reaction is also the most stable, the initial step involved assessing the electron energy levels of every conceivable conformers of 3a.50 Following this, the conformer exhibiting the least amount of electronic energy was optimized utilizing the M06-2X/6-311++G (d,p) level of theory. The resulting data, including the angles (°) and bond length (Å), was subsequently compared to the crystal structure and is displayed in Fig. 3, S19†, Table S2 and ESI.† It was found that the deviations in bond lengths and angles between the optimized and crystal structures were less than 0.025 Å and 2°, i.e. less than 1.7% and 1.6%, respectively. This internally confirms the high accuracy of the model chemistry used in this work.
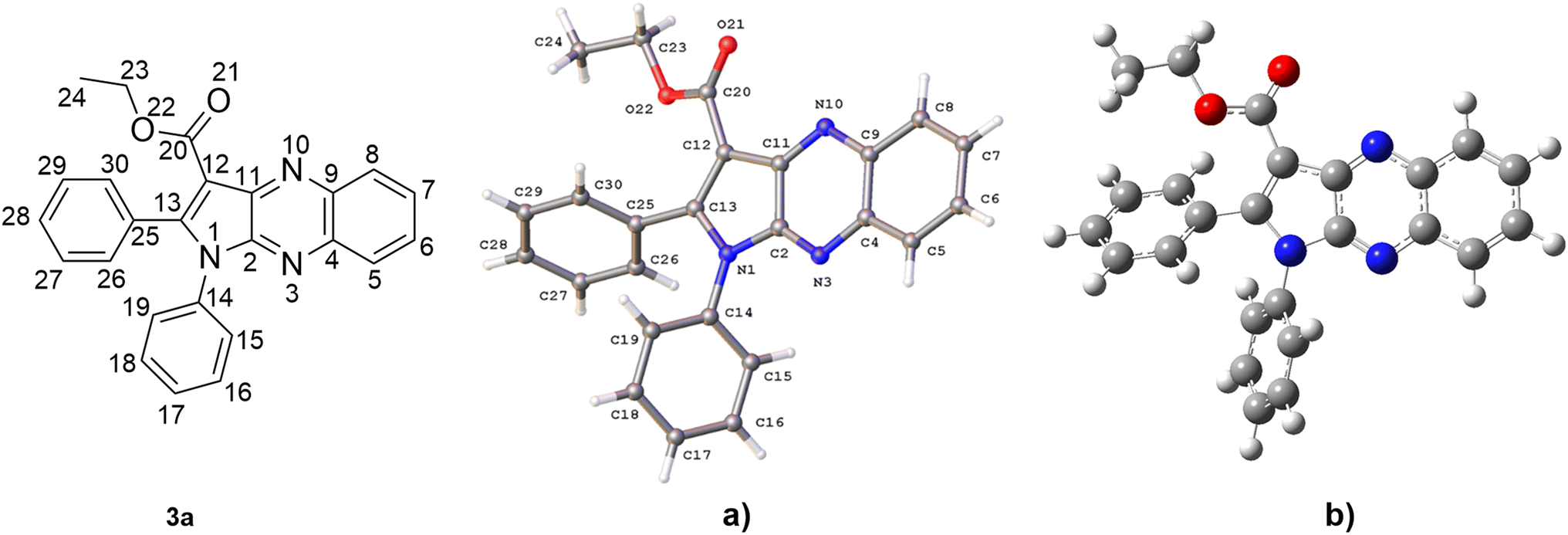 | ||
| Fig. 3 Crystal (a) and optimized (b) structures of 3a. | ||
(b) The antiradical activity in the gas phase. To identify the primary mechanisms by which 3a would function under the more complex physiological conditions, its antioxidant properties were initially evaluated in the gas phase. Depending on the molecular structures involved, various pathways may be followed by the 3a − HO˙/HOO˙ reactions. These are formal hydrogen transfer (FHT), radical adduct formation (RAF), and single electron transfer (SET) pathways.33,36,38,51–54 To identify the preferred radical quenching mechanism (FHT, SET, and RAF) followed by 3a to eliminate the HO˙ and HOO˙ radicals, first the gas phase Gibbs free energy changes (ΔG°) were computed. Table 4 displays the results.
| Mechanisms | Positions | HO˙ | HOO˙ |
|---|---|---|---|
| FHT | C23–H | −21.7 | 9.6 |
| C24–H | −16.4 | 14.8 | |
| RAF | C2 | −5.0 | |
| C4 | 4.2 | ||
| C5 | −19 | ||
| C6 | −9.3 | ||
| C7 | −10.9 | ||
| C8 | −15.5 | ||
| C9 | 1.5 | ||
| C11 | −6.9 | ||
| C12 | −20.7 | ||
| C13 | −24.5 | ||
| C14 | −9.5 | ||
| C15 | −10.2 | ||
| C16 | −6.1 | ||
| C17 | −7.2 | ||
| C18 | −5.2 | ||
| C19 | −9.8 | ||
| C25 | −2.7 | ||
| C26 | −8.3 | ||
| C27 | −5.8 | ||
| C28 | −8.2 | ||
| C29 | −5.5 | ||
| C30 | −9.4 | ||
| N3 | 13.8 | ||
| N10 | −2.5 | ||
| SET | 155.6 | 165.5 |
The radical scavenging activity for the HO˙ radicals are thermodynamically spontaneous (ΔG° < 0) at most positions in 3a, with the exception of the SET reaction (ΔG° = 155.6 kcal mol−1) and the RAF reactions at C4, C9, and N13 (ΔG° > 0). On the contrary, the radical scavenging of the HOO˙ radical is not spontaneous in any of the mechanisms that have been investigated. Thus, the radical scavenging of HOO˙ was excluded from the investigation. The kinetic evaluation of the possible positions (ΔG° < 0, Table 4) was conducted in a consistent manner to assess the scavenging of HO˙ radical by 3a in the gas phase. The outcomes of this analysis are displayed in Table 5, Fig. 4 and 5.
| Mechanisms | Positions | ΔG‡ | κ | kEck | Γ |
|---|---|---|---|---|---|
| FHT | C23 | 4.1 | 2.5 | 4.94 × 1010 | 18.3 |
| C24 | 11.0 | 5.2 | 7.83 × 105 | 0.0 | |
| RAF | C2 | 10.1 | 1.5 | 3.67 × 105 | 0.0 |
| C4 | 13.4 | 1.4 | 1.26 × 103 | 0.0 | |
| C5 | 7.2 | 1.2 | 3.98 × 107 | 0.0 | |
| C6 | 7.6 | 1.2 | 1.87 × 107 | 0.0 | |
| C7 | 7.3 | 1.2 | 3.31 × 107 | 0.0 | |
| C8 | 6.5 | 1.2 | 1.23 × 108 | 0.0 | |
| C9 | 13.1 | 1.4 | 2.35 × 103 | 0.0 | |
| C11 | 10.0 | 1.4 | 4.04 × 105 | 0.0 | |
| C12 | 2.3 | 1.2 | 1.45 × 1011 | 53.5 | |
| C13 | 2.8 | 1.3 | 7.60 × 1010 | 28.1 | |
| C14 | 12.7 | 1.5 | 4.52 × 103 | 0.0 | |
| C15 | 6.8 | 1.0 | 6.63 × 107 | 0.0 | |
| C16 | 8.8 | 1.3 | 3.13 × 106 | 0.0 | |
| C18 | 8.4 | 1.4 | 6.63 × 106 | 0.0 | |
| C19 | 13.4 | 1.4 | 1.14 × 103 | 0.0 | |
| C25 | 12.7 | 1.3 | 3.85 × 103 | 0.0 | |
| C26 | 6.8 | 1.2 | 7.95 × 107 | 0.0 | |
| C27 | 9.0 | 1.3 | 2.05 × 106 | 0.0 | |
| C28 | 7.4 | 1.3 | 3.01 × 107 | 0.0 | |
| C29 | 8.0 | 1.3 | 1.02 × 107 | 0.0 | |
| C30 | 6.8 | 1.2 | 7.95 × 107 | 0.0 | |
| N10 | 10.3 | 1.7 | 2.95 × 105 | 0 | |
| koverall | 2.70 × 1011 | ||||
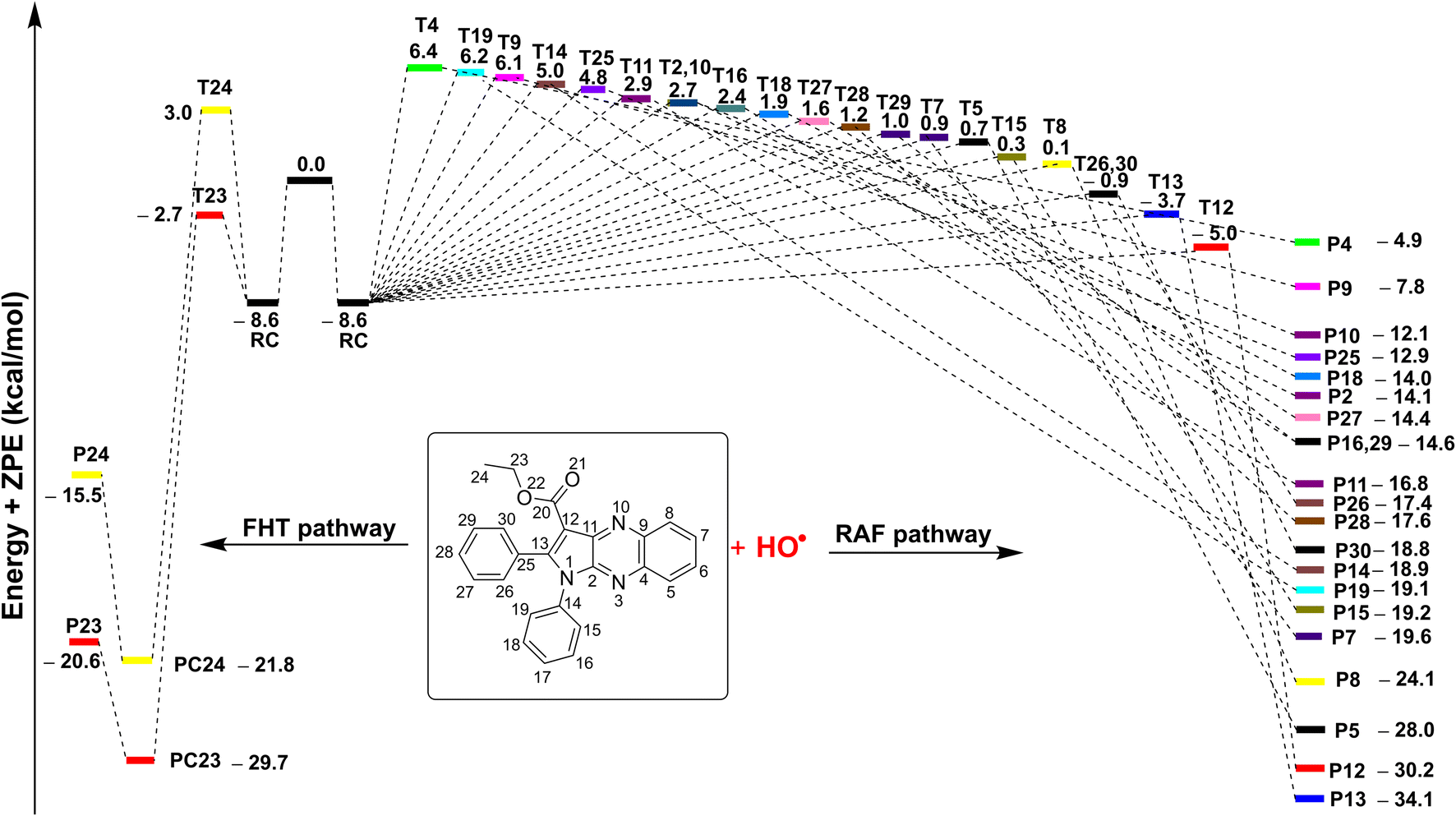 | ||
| Fig. 4 The potential energy surfaces (PES) of the 3a + HO˙ according to FHT and RAF reactions (RC: pre-complex; TS: transition state; PC: post-complex; P: product). | ||
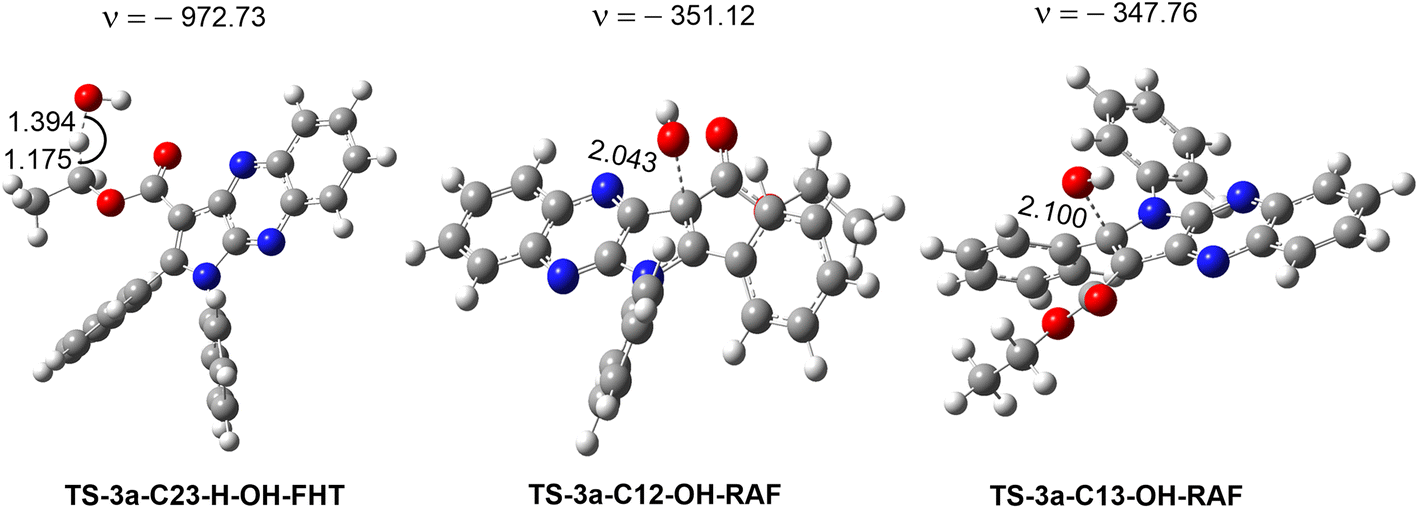 | ||
| Fig. 5 Optimized geometries of the main transition states between 3a and HO˙ radicals according to FHT and RAF processes. | ||
The results presented in the PES (Fig. 4) indicate that the C12 position has the lowest reaction barrier value of 3.6 kcal mol−1. In contrast, the C4 position of 3a displayed the highest reaction barrier value of 15.0 kcal mol−1. The FHT reaction of C23–H and the RAF at position C13 exhibited the second and third lowest reaction barriers, measuring 5.9 and 4.9 kcal mol−1, respectively. In contrast, the 3a + HO˙ reaction has comparatively lower reaction barriers, ranging from 7.7 to 15.0 kcal mol−1 in the rest of the reactions. According to the PES analysis, the HO˙ radical scavenging activity of 3a is dominated by the FHT reaction at the C23–H bond and the RAF reaction at the C12/13.
Table 5 indicates that the overall rate constant (koverall) for the gas phase scavenging of the HO˙ radical was 2.70 × 1011 M−1 s−1. The rate constant for the 3a + HO˙ reaction was determined by the RAF reaction at positions C12 and C13, with rate constants of 1.45 × 1011 (Γ = 53.5%) and 7.60 × 1010 M−1 s−1 (Γ = 28.1%), respectively, while the H-abstraction contributed approximately 18.2% (kEck = 4.94 × 1010 M−1 s−1) to the overall rate constant. This result is consistent with the thermodynamic and the PES analysis (Table 4 and Fig. 4), which determined that the RAF-C12/13 and FHT-C23 reactions had the lowest ΔG° values (ΔG° = −20.7, −24.5 and −21.7 kcal mol−1 for C12, C13 and C23, respectively). No other reaction pathways make any notable contributions.
(c) The antiradical activity in the lipid medium. Previous studies established that pyrrolo[2,3-b]quinoxaline derivatives have good radical scavenging activity in biologically consequential contexts, including lipid peroxidation and resistance to HO˙ radicals.47–49 Therefore, the QM-ORSA protocol was employed here to assess the kinetics of the HO˙-scavenging reactions taking place in the lipid medium following the main reactions in nonpolar media i.e., the RAF reaction at C12/13 and FHT reaction in the C23–H bond. The findings are presented in Table 6.
| Mechanisms | Pentyl ethanoate | ||||
|---|---|---|---|---|---|
| ΔG‡ | κ | kapp | Γ | ||
| FHT | C23–H | 6.0 | 2.1 | 4.10 × 108 | 47.9 |
| RAF | C12 | 5.6 | 1.1 | 4.40 × 108 | 51.4 |
| C13 | 8.3 | 1.1 | 5.60 × 106 | 0.7 | |
| koverall | 8.56 × 108 | ||||
The koverall for the 3a + HO˙ reaction in pentyl ethanoate was 8.56 × 108 M−1 s−1. The C12 position RAF mechanism (Γ = 51.4%) and the FHT pathway at the C23–H bond (Γ = 47.9%) were found to define the HO˙ antiradical activity. In contrast, the C13 position RAF reaction accounted for a mere 0.7% of the overall reaction. The hydroxyl radical scavenging capability of 3a in a non-polar environment is analogous to that of widely used antioxidants including Trolox, melatonin,55 indole-3-carbinol,56 and gallic acid.57 3a is therefore an effective hydroxyl radical scavenger in lipid environments.
4. Conclusion
Successful synthesis of five pyrrolo[2,3-b]quinoxaline derivatives was achieved. 3a demonstrates the greatest DPPH radical scavenging activity, as determined by the DPPH assay. The structure of the synthesized lead compound 3a was verified by X-ray analysis. In pentyl ethanoate, the thermodynamic and kinetic calculations also revealed that 3a exhibited HO radical scavenging activity with koverall = 8.56 × 108 M−1 s−1. In contrast, in nonpolar media, 3a demonstrated only a negligible capacity to scavenge hydroperoxyl radicals. The capacity of 3a to scavenge hydroxyl radicals in non-polar environments is comparable to that of conventional antioxidants including Trolox, melatonin, indole-3-carbinol, and gallic acid. Therefore in physiological lipid environments 3a holds potential as a HO˙ radical scavenger.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for [3a] has been deposited at the [CCDC] under [2310877].Conflicts of interest
There are no conflicts to declare.Acknowledgements
LVM thanks the Hercules Foundation for supporting the purchase of the diffractometer through project AKUL/09/0035.References
- M. S. Abdelfattah, T. Kazufumi and M. Ishibashi, J. Nat. Prod., 2010, 73, 1999–2002 CrossRef CAS PubMed.
- M. Shin, K. Inouye and H. Otsuka, Bull. Chem. Soc. Jpn., 1984, 57, 2203–2210 CrossRef CAS.
- K. Hattori, K. Koike, K. Okuda, T. Hirayama, M. Ebihara, M. Takenaka and H. Nagasawa, Org. Biomol. Chem., 2016, 14, 2090–2111 RSC.
- H. Nagasawa, M. Yamashita, N. Mikamo, M. Shimamura, S. Oka, Y. Uto and H. Hori, Comp. Biochem. Physiol., Part A: Mol. Integr. Physiol., 2002, 132, 33–40 CrossRef PubMed.
- C. Oncken, D. Gonzales, M. Nides, S. Rennard, E. Watsky, C. B. Billing, R. Anziano, K. Reeves and V. S. Group, Arch. Intern. Med., 2006, 166, 1571–1577 CrossRef CAS PubMed.
- J. C. Adkins and J. A. Balfour, Drugs Aging, 1998, 12, 225–241 CrossRef CAS PubMed.
- K. Koike, M. Nagano, M. Ebihara, T. Hirayama, M. Tsuji, H. Suga and H. Nagasawa, J. Med. Chem., 2020, 63, 4022–4046 CrossRef CAS PubMed.
- A. Arcadi, S. Cacchi, G. Fabrizi and L. M. Parisi, Tetrahedron Lett., 2004, 45, 2431–2434 CrossRef CAS.
- A. Keivanloo, M. Bakherad and A. Rahimi, Synthesis, 2010, 1599–1602 CrossRef CAS.
- M. Bakherad, A. Keivanloo and S. Jajarmi, Tetrahedron, 2012, 68, 2107–2112 CrossRef CAS.
- A. K. Soda, K. P. Chinthapally, P. Krishna, S. K. Chilaka and S. Madabhushi, RSC Adv., 2023, 13, 15001–15005 RSC.
- B. Prasad, K. S. Kumar, P. V. Babu, K. Anusha, D. Rambabu, A. Kandale, G. Vanaja, A. M. Kalle and M. Pal, Tetrahedron Lett., 2012, 53, 6059–6066 CrossRef CAS.
- P. V. Babu, S. Mukherjee, G. S. Deora, K. S. Chennubhotla, R. Medisetti, S. Yellanki, P. Kulkarni, S. Sripelly, K. V. Parsa and K. Chatti, Org. Biomol. Chem., 2013, 11, 6680–6685 RSC.
- T. Besharati-Seidani, A. Keivanloo, B. Kaboudin, A. Yoshida and T. Yokomatsu, Tetrahedron, 2018, 74, 2350–2358 CrossRef CAS.
- R. Chemboli, R. Kapavarapu, K. Deepti, K. Prasad, A. G. Reddy, A. N. Kumar, M. V. B. Rao and M. Pal, J. Mol. Struct., 2021, 1230, 129868 CrossRef CAS PubMed.
- S. Manta, D.-N. Gkaragkouni, E. Kaffesaki, P. Gkizis, D. Hadjipavlou-Litina, E. Pontiki, J. Balzarini, W. Dehaen and D. Komiotis, Tetrahedron Lett., 2014, 55, 1873–1876 CrossRef CAS.
- X. Del Corte, A. López-Francés, A. Maestro, I. Villate-Beitia, M. Sainz-Ramos, E. Martínez de Marigorta, J. L. Pedraz, F. Palacios and J. Vicario, Pharmaceuticals, 2021, 14, 782 CrossRef CAS PubMed.
- N. Joksimović, J. Petronijević, N. Janković, D. Baskić, S. Popović, D. Todorović, S. Matić, G. A. Bogdanović, M. Vraneš and A. Tot, Bioorg. Chem., 2019, 88, 102954 CrossRef PubMed.
- A. Q. Cusumano and J. G. Pierce, Bioorg. Med. Chem. Lett., 2018, 28, 2732–2735 CrossRef CAS PubMed.
- A. López-Pérez, S. Freischem, I. Grimm, O. Weiergräber, A. J. Dingley, M. P. López-Alberca, H. Waldmann, W. Vollmer, K. Kumar and C. Vuong, Antibiotics, 2021, 10, 529 CrossRef PubMed.
- K. Ma, P. Wang, W. Fu, X. Wan, L. Zhou, Y. Chu and D. Ye, Bioorg. Med. Chem. Lett., 2011, 21, 6724–6727 CrossRef CAS PubMed.
- N. V. O. Zacarías, J. P. van Veldhoven, L. Portner, E. van Spronsen, S. Ullo, M. Veenhuizen, W. J. van der Velden, A. J. Zweemer, R. M. Kreekel and K. Oenema, J. Med. Chem., 2018, 61, 9146–9161 CrossRef PubMed.
- S. Zykova, M. Danchuk, V. Talismanov, N. Tokareva, N. Igidov, I. Rodin, A. Koshchaev, N. Gugushvili and O. Karmanova, Int. J. Pharm. Sci. Res., 2018, 10, 164–166 CAS.
- N. T. Nguyen, V. V. Dai, A. Mechler, N. T. Hoa and Q. V. Vo, RSC Adv., 2022, 12, 24579–24588 RSC.
- A. I. Elshamy, T. Yoneyama, N. Van Trang, N. T. Son, Y. Okamoto, S. Ban, M. Noji and A. Umeyama, J. Mol. Struct., 2020, 1200, 127061 CrossRef CAS.
- K. Marxen, K. H. Vanselow, S. Lippemeier, R. Hintze, A. Ruser and U.-P. Hansen, Sensors, 2007, 7, 2080–2095 CrossRef CAS PubMed.
- O. Rigaku, CrysAlis PRO, Rigaku Corporation, Oxford, England, 2018 Search PubMed.
- O. Dolomanov, L. Bourhis, R. Gildea, J. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, A. F. I. H. P. Hratchian, J. Bloino, G. Zheng, M. H. J. L. Sonnenberg, M. Ehara, K. Toyota, J. H. R. Fukuda, M. Ishida, T. Nakajima, Y. Honda, H. N. O. Kitao, T. Vreven, J. A. Montgomery Jr, F. O. J. E. Peralta, M. J. Bearpark, J. Heyd, K. N. K. E. N. Brothers, V. N. Staroverov, R. Kobayashi, K. R. J. Normand, A. P. Rendell, J. C. Burant, J. T. S. S. Iyengar, M. Cossi, N. Rega, N. J. Millam, J. E. K. M. Klene, J. B. Cross, V. Bakken, C. Adamo, R. G. J. Jaramillo, R. E. Stratmann, O. Yazyev, R. C. A. J. Austin, C. Pomelli, J. W. Ochterski, K. M. R. L. Martin, V. G. Zakrzewski, G. A. Voth, J. J. D. P. Salvador, S. Dapprich, A. D. Daniels, J. B. F. Ö. Farkas, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2008, 112, 1095–1099 CrossRef CAS PubMed.
- A. Galano and J. R. Alvarez-Idaboy, J. Comput. Chem., 2014, 35, 2019–2026 CrossRef CAS PubMed.
- J. R. l. Alvarez-Idaboy and A. Galano, J. Phys. Chem. B, 2012, 116, 9316–9325 CrossRef CAS PubMed.
- M. E. Alberto, N. Russo, A. Grand and A. Galano, Phys. Chem. Chem. Phys., 2013, 15, 4642–4650 RSC.
- E. Dzib, J. L. Cabellos, F. Ortíz-Chi, S. Pan, A. Galano and G. Merino, Int. J. Quantum Chem., 2019, 119, e25686 CrossRef.
- Q. V. Vo, M. V. Bay, P. C. Nam, D. T. Quang, M. Flavel, N. T. Hoa and A. Mechler, J. Org. Chem., 2020, 85, 15514–15520 CrossRef CAS PubMed.
- A. Galano and J. Raúl Alvarez-Idaboy, Int. J. Quantum Chem., 2019, 119, e25665 CrossRef.
- A. Galano and J. R. Alvarez-Idaboy, J. Comput. Chem., 2013, 34, 2430–2445 CrossRef CAS PubMed.
- M. G. Evans and M. Polanyi, Trans. Faraday Soc., 1935, 31, 875–894 RSC.
- H. Eyring, J. Chem. Phys., 1935, 3, 107–115 CrossRef CAS.
- D. G. Truhlar, W. L. Hase and J. T. Hynes, J. Phys. Chem., 1983, 87, 2664–2682 CrossRef CAS.
- E. Dzib, J. L. Cabellos, F. Ortiz-Chi, S. Pan, A. Galano and G. Merino, Eyringpy 1.0.2, Cinvestav, Mérida, Yucatán, 2018 Search PubMed.
- F. A. Rashid, M. Mohammat, F. Bouchamma, Z. Shaameri and A. Hamzah, Russ. J. Org. Chem., 2020, 56, 1082–1088 CrossRef.
- F. Jourdan, J. T. Kaiser and D. J. Lowe, Synth. Commun., 2005, 35, 2453–2466 CrossRef CAS.
- J. Li, J. Zhang, H. Yang, Z. Gao and G. Jiang, J. Org. Chem., 2017, 82, 765–769 CrossRef CAS PubMed.
- W.-H. Zhang, J. Liu, G. Xu, Q. Yuan and L. M. Sayre, Chem. Res. Toxicol., 2003, 16, 512–523 Search PubMed.
- K. Itakura, T. Osawa and K. Uchida, J. Org. Chem., 1998, 63, 185–187 CrossRef CAS PubMed.
- L. Tsai, P. A. Szweda, O. Vinogradova and L. I. Szweda, Proc. Natl. Acad. Sci. U.S.A., 1998, 95, 7975–7980 CrossRef CAS PubMed.
- W. Hehre, J. Yu, P. Klunzinger and L. Lou, Spartan Software, Wavefunction Inc., Irvine, 2000 Search PubMed.
- B. N. Ames, M. K. Shigenaga and T. M. Hagen, Proc. Natl. Acad. Sci. U.S.A., 1993, 90, 7915–7922 CrossRef CAS PubMed.
- A. Galano, G. Mazzone, R. Alvarez-Diduk, T. Marino, J. R. Alvarez-Idaboy and N. Russo, Annu. Rev. Food Sci. Technol., 2016, 7, 335–352 CrossRef CAS PubMed.
- N. T. Hoa and Q. V. Vo, Chemosphere, 2023, 314, 137682 CrossRef CAS PubMed.
- M. Missioui, S. Mortada, W. Guerrab, G. Serdaroğlu, S. Kaya, J. T. Mague, E. M. Essassi, M. E. A. Faouzi and Y. Ramli, J. Mol. Struct., 2021, 1239, 130484 CrossRef CAS.
- A. Galano, Phys. Chem. Chem. Phys., 2011, 13, 7178–7188 RSC.
- Q. V. Vo, M. Van Bay, P. C. Nam and A. Mechler, ACS Omega, 2019, 4, 19375–19381 CrossRef CAS PubMed.
- T. Marino, A. Galano and N. Russo, J. Phys. Chem. B, 2014, 118, 10380–10389 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2310877. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra03108c |
| This journal is © The Royal Society of Chemistry 2024 |